
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A covalently linked nickel(II) porphyrin–ruthenium(II) tris(bipyridyl) dyad for efficient photocatalytic water oxidation†
Emmanouil
Nikoloudakis
 a,
Ajyal Z.
Alsaleh
b,
Georgios
Charalambidis
a,
Athanassios G.
Coutsolelos
*a and
Francis
D'Souza
*b
a,
Ajyal Z.
Alsaleh
b,
Georgios
Charalambidis
a,
Athanassios G.
Coutsolelos
*a and
Francis
D'Souza
*b
aDepartment of Chemistry, University of Crete, and Institute of Electronic Structure and Laser (IESL), Foundation for Research and Technology – Hellas (FORTH), Voutes Campus, Heraklion 70013, Crete, Greece. E-mail: acoutsol@uoc.gr
bDepartment of Chemistry, University of North Texas, 1155 Union Circle, #305070, Denton, TX 76203-5017, USA. E-mail: Francis.dsouza@unt.edu
First published on 5th October 2022
Abstract
Visible-light-induced oxidation of water to dioxygen, catalyzed by a newly synthesized NiP-Ru dyad consisting of a ruthenium tris(bipyridyl), [Ru(bpy)3]2+ as a photosensitizer, and a low-cost nickel porphyrin, NiP as a water oxidation catalyst is reported.
Photocatalytic water splitting into H2 and O2 has attracted significant scientific interest for solar energy conversion applications during the last two decades.1 One of the half-reactions of this process, water oxidation, is known to be the key step in natural and artificial photosynthesis to convert and store solar energy.2,3 Although plenty of material-based catalysts have been applied as water oxidation catalysts (WOCs), molecular catalysts have recently gained greater attention.3 Their versatile properties allow the rational design of chemical structures to optimize catalytic activity.4 While plenty of molecular catalysts based on noble metals such as Ru or Ir have been employed as WOCs and exhibited high performances,5–8 there is a need to switch to low-cost and earth-abundant first-row transition metals.9,10 Porphyrins have been extensively investigated for water oxidation due to their high chemical stability during catalysis, which is an important advantage among other molecular catalysts.11–13 Moreover, porphyrins can accommodate any first-row transition metal, which can serve as a catalytic center for water oxidation.11
Noteworthy, the examples of electrocatalytic water oxidation systems which utilize porphyrinoids as catalysts are numerous14–20 compared to the photocatalytic systems where no external bias is applied.21,22 Recently, Bonnet and co-workers prepared a series of water-soluble nickel porphyrin photocatalysts for water oxidation using [Ru(bpy)3]2+ as photosensitizer (PS) and [S2O8]2− as sacrificial electron acceptor.23 To the best of our knowledge, there is no report on a covalently linked Ni2+ porphyrin–ruthenium complex dyad that has been investigated towards photocatalytic water oxidation. Furthermore, in most of the reported photocatalytic systems utilizing porphyrins and ruthenium complexes for this application, the porphyrin was playing the role of a PS and not that of a water oxidation catalyst.20
One common drawback of photocatalytic systems is the need for a sacrificial agent, which is unavoidable for the completion of the catalytic cycle.24,25 One very promising approach to overcome this issue is the development of photoelectrochemical cells (PECs), where the catalyst and the PS are adsorbed onto an inorganic semiconductor electrode.26 In these devices, the adsorbed PS harvests solar energy and injects an electron into the conduction band of the semiconductor. Subsequently, the generated radical cation PS+· is reduced from the WOC, while repeating cycles of this process results in a highly oxidized catalytic moiety, able to convert water molecules into protons and molecular oxygen.20
Herein we synthesized a catalyst-photosensitizer dyad NiP-Ru (Scheme S1, ESI†), comprised of nickel porphyrin and ruthenium tris-bipyridine covalently linked via an amide bond, and investigated their ability to produce oxygen via water oxidation under visible light irradiation. The dyad was found active in organic solvent and demonstrated higher photocatalytic performance compared to the non-covalent two-component system. A simplified photocatalytic mechanism for the oxidation of water can be derived from the energy diagram shown in Scheme 1a (vide supra for additional details). In this system, the ruthenium complex acts as PS absorbing a photon and undergoes an intramolecular reductive electron transfer (or hole transfer) involving the NiP and intermolecular electron transfer involving the electron acceptor (EA), methyl viologen. Repetitions of the same sequence afford higher oxidation states of the NiP catalyst that can oxidize water, producing molecular oxygen and protons (see the ESI† for more detailed catalytic mechanism). It's worth mentioning that the homogeneous photocatalytic experiments described here, conducted in an organic solvent, could be helpful to couple this system with CO2 reduction catalysts for future tandem photocatalytic devices. Furthermore, for the first time, a photoelectrochemical cell utilizing a Ni2+ porphyrin moiety as a WOC and ruthenium complex as a PS is fabricated and applied for water oxidation in aqueous media. Scheme 1b illustrates the electron transfer processes on the photoanode of the PEC device. After the photoexcitation of the Ru moiety, an electron is injected into the conduction band of the TiO2 and travels through the external circuit to the Pt cathode. The holes are then moved from the ruthenium to the nickel porphyrin catalytic moiety, which gains the appropriate oxidation state to perform water oxidation. In a more appealing approach, the injected on the photoanode electrons can be transferred to an appropriately functionalized cathode containing a water reduction catalyst.27,28 This would result to the construction of a tandem photoelectrochemical system for overall water spliting.29
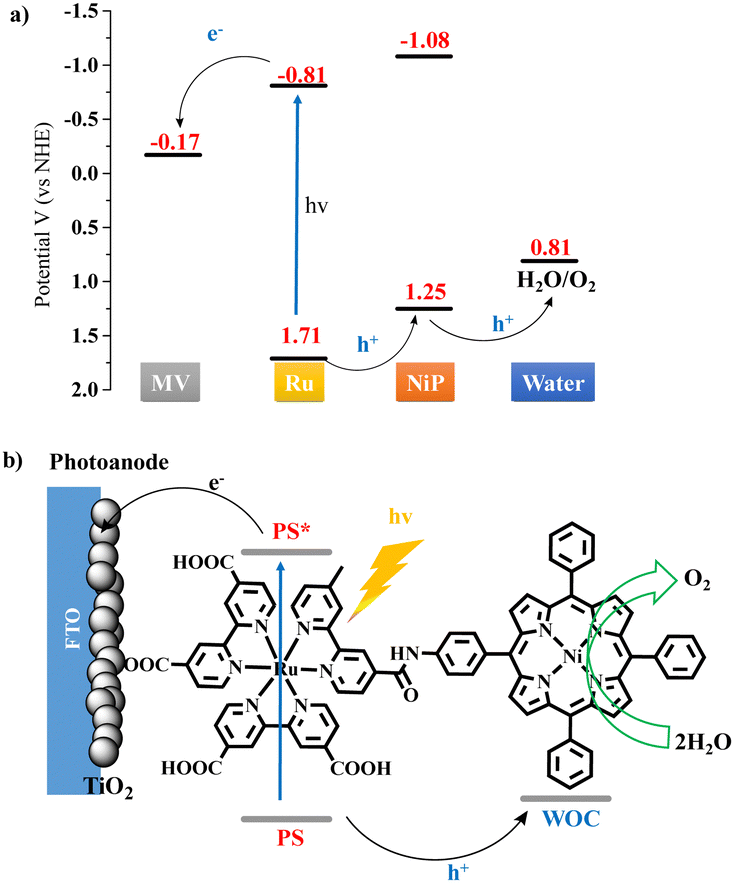 | ||
| Scheme 1 (a) Energy diagram of the NiP-Ru-bpy dyad derived photo-induced electron transfer process resulting in water oxidation. (b) Schematic diagram of the visible light-driven water oxidation by the NiP-Ru dyad modified TiO2/FTO photoanode. | ||
The synthetic procedure for the preparation of NiP-Ru dyad, is illustrated in Scheme S2 (ESI†) and the details are given the ESI.† The bis-pyridine intermediates, bpy-COOH30 and bpy(COOMe)2,31 were synthesized according to published procedures starting from commercially available 4,4′-dimethyl-2,2′-bipyridine and [2,2′-bipyridine]-4,4′-dicarboxylic acid, respectively. The next step was the amide coupling of TPP-NH2 and bpy-COOH via refluxing the carboxylic acid substituted bipyridine in SOCl2 and then introducing the amino porphyrin to afford TPP-bpy porphyrin derivative. Subsequently, the free-base H2P-Ru dyad was synthesized via a complexation reaction between the previous two intermediates, namely TPP-bpy and Ru(bpy(COOMe)2)2 following known experimental procedures.32 The final step was a metalation reaction to insert Ni(II) inside the porphyrin core affording the NiP-Ru dyad. The hydrolyzed dyad derivative NiP-Ru-COOH, bearing four carboxylic anchoring groups was obtained via basic hydrolysis. Moreover, the reference, nickel tetra-toluene porphyrin NiP and the ruthenium tris(bipyridyl) Ru-bpy and Ru-bpy-OOH were synthesized according to literature procedures.33,34 The synthesis of all the final and intermediate compounds was verified via1H and 13C NMR spectroscopies (Fig. S1–S13, ESI†) and MALDI-TOF mass spectrometry. The assignment of the corresponding peaks was achieved through 2D COSY, HMBC and HSQC NMR spectra analysis.
The porphyrin and ruthenium complexes investigated in this work were initially studied by UV-Vis absorption spectroscopy in dimethylformamide (DMF). NiP and NiP-Ru dyad exhibited typical porphyrin absorbance features in solution. NiP displays a Soret band at 416 nm and one Q band at 527 nm with a shoulder peak around 560 nm. Ru-bpy with a broad spectrum with peak maxima centered around 464 nm, attributed to MLCT transition.35 As depicted in Fig. 1a, the absorption spectrum of NiP-Ru was a superposition of the NiP and Ru-bpy spectra. Nickel porphyrins are not emissive and therefore only the ruthenium-based emission in the region 570–800 nm with peak maxima at 658 nm was observed. Fig. 1b illustrates the almost quantitative quenching of the ruthenium-based emission (∼98%) in the dyad due to the covalent attachment of the nickel porphyrin. This observation provides additional evidence of the interaction between the Ru complex and Ni porphyrin moieties in the excited state.
 | ||
| Fig. 1 (a) Absorption spectrum of NiP-Ru dyad, NiP and Ru-bpy in DMF, (b) Emission intensity of Ru-bpy (blue line) and NiP-Ru dyad (red line) after excitation at 480 nm, recorded in DMF solutions with a concentration of 0.01 mM of individual compounds. | ||
The electrochemical properties of NiP, Ru-bpy, and NiP-Ru dyad were studied using cyclic (CV) and differential pulse voltammetry (DPV) measurements in PhCN containing 0.1 M (TBA)ClO4 as the supporting electrolyte. Representative voltammograms are shown in Fig. S14 in ESI.† The NiP revealed two oxidations at 1.03 and 1.29 V and one reduction at −1.31 vs. Ag/AgCl. On the other hand, one oxidation at 1.56 V and three reductions at −1.03, −1.23 and −1.47 V vs. Ag/AgCl were observed for Ru-bpy. The NiP-Ru dyad exhibited three oxidation waves at 1.04, 1.31 and 1.50 V, and three reduction peaks at −1.02, −1.29, and −1.46 V vs. Ag/AgCl. By comparison with the NiP and Ru-bpy reference compounds, it was possible to assign all the redox waves in the NiP-Ru dyad. The first (−1.02 V) and the third (−1.46) reduction peaks correspond to the ruthenium part, while the wave at −1.23 V includes the porphyrin reduction and the ruthenium based reduction as well. On the other hand, to the cathodic side the two first oxidations (1.04 and 1.31 V) can be attributed to the porphyrin based oxidation and the third oxidation wave at 1.50 V can be assigned to the ruthenium oxidation. All the above results suggest that the covalent connection between the two components has little effect in the redox properties of the dyad. The CV studies revealed that the porphyrin macrocycle is oxidized more easily compared to the ruthenium chromophore. Importantly, the hole shift from the oxidized ruthenium complex to the porphyrin catalyst is thermodynamically favorable, as the calculated Gibbs free energy (ΔGh+s)36 for this reaction is –0.46 eV. Further, to assist in spectral characterization of the electron transfer products, spectroelectrochemical studies of NiP and Ru-bpy were performed, as shown in Fig. S15 (ESI†). One-electron oxidized product of NiP was characterized by a new peak at 620 nm while the reduced productd of Ru-bpy revealed increased absorption covering the visible range.
In order to establish the excited state electron transfer in the NiP-Ru dyad, femtosecond transient absorption (fs-TA) spectral studies were performed. Fig. S16a (ESI†) shows the fs-TA spectra at the indicated delay times of Ru-bpy in benzonitrile. In agreement with the literature report,37,38 in the visible region, the instanteneously formed excited state involving metal-to-ligand charge transfer revealed an excited state absorption (ESA) peak at 590 nm (see spectrum at 0.92 ps) and a negative signal at 658 nm. The peak maxima of the negative signal matched that of the emission of Ru-bpy (see inverted emission spectrum shown in green dash line) suggesting that it is due to stimulated emission (SE). With time this peak experienced a 6 nm blue-shift while the decay of the ESA peak was accompanied by a new broad signal in the 715 nm attributable to long-lived triplet state.
Although non-emissive, the fs-TA spectra of NiP was also recorded at the Soret band excitation to capture transient spectral details as shown in Fig. S16b (ESI†). ESA peaks at 553 and 584 nm, and a ground state bleach (GSB) at 525 nm was observed. As expected, no signal corresponding to SE was observed. Decay of the ESA and recovery of GSB peak did not result in any new peaks corresponding to 3NiP* within the monitoring time window of 3 ns.
The fs-TA spectra of NiP-Ru dyad was drastically different from that of Ru-bpy monomer, as shown in Fig. S16c (ESI†). Spectrum recorded at the earlier delay time (0.92 ps) revealed a broad ESA peak covering the 550–660 nm region with a maxima around 615 nm, expected for the NiP+-Ru− charge separated state from the earlier discussed spectroelectrochemical studies. Decay of this signal was fast, which resulted in a much less intense, new peak at 620 nm. In addition, a ESA peak at 553 nm and a GSB peak 525 were observed. The time constants for these signals were close to that of NiP suggesting this is due to simulteneous excitation of a small fraction of NiP of the dyad. From decay kinetics, a lifetime of 25 ps the NiP+–Ru− charge separated state (fast decay component) was possible to arrive. These results unequivocally provide charge separation in the NiP-Ru dyad.
In order to prove electron/hole migration from the initial NiP+-Ru− charge separated state, photocatalytic electron pooling experiments using methyl viologen (MV2+) as terminal electron acceptor and water as electron donor was performed. Here, the UV-Vis spectra of a mixture of NiP-Ru dyad, MV2+, in Ar-saturated DMF containing trace amounts of water, before and after irradiation was monitored, as shown in Fig. S17 (ESI†). The characteristic peak of MV˙+ appeared at 608 nm accompanied by absorption bands in the region of 370 nm–410 nm which were also attributed to the accumulation of MV˙+.39 This result suggests that the NiP+ of the charge separated product can indeed oxidize water and the electron from reduced ruthenium moiety is effectively shifting to MV2+. The final result is regeneration of the NiP-Ru photocatalyst, release of water oxidation product, viz. O2, and H+, and accumulation of reduced methyl viologen, MV+. The absorption changes of MV+ at 608 nm was plotted versus the irradiation time for NiP-Ru photocatalyst and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of NiP and Ru-bpy (see Fig. S18, ESI† for spectral data) with same amounts of MV2+ and H2O. Both systems were photocatalytically active and showed that the activity reached a plateau after about one hour of irradiation, and that the performance of the NiP-Ru dyad system was much superior than the non-covalent system.
:1 mixture of NiP and Ru-bpy (see Fig. S18, ESI† for spectral data) with same amounts of MV2+ and H2O. Both systems were photocatalytically active and showed that the activity reached a plateau after about one hour of irradiation, and that the performance of the NiP-Ru dyad system was much superior than the non-covalent system.
Next, photocatalytic irradiation experiments for O2 evolution were performed in DMF using methyl viologen as sacrificial electron acceptor and oxygen was detected via an optical oxygen sensor.40 In agreement with the electron pooling experiments, the NiP-Ru dyad exhibited significantly higher O2 evolution compared to the non-covalent system comprised of NiP and Ru-bpy as PS (Fig. 2a). The overall activity of NiP-Ru dyad reached a turn over number (TON) of 18 after one hour of continuous irradiation. The later system was additionally used to examine the impact of the PS concentration. After increasing PS concentration by a factor 10, no impact on the photocatalytic activity was observed. Control experiments with no catalyst, or no sacrificial agent or no water failed to reveal any photocatalytic O2 production. Varying the concentration of the of NiP-Ru dyad (Fig. 2b and c) showed a first order dependence of the oxygen evolution rate on the dyad concentration. Since the previous experiment showed no dependence on the PS concentration, the later result suggests that the rate-determining step of the water oxidation reaction involves one nickel center.
 | ||
| Fig. 2 (a) Oxygen evolution during photocatalytic H2O oxidation after irradiation with a 450 W xenon lamp (cut-off filter λ > 400 nm). All solutions contained 50 mM of Methyl Viologen in DMF and 4% H2O. (b) O2 evolution rate as a function of the dyad concentration. Conditions: 50 mM of Methyl Viologen in DMF and 4% H2O, (c) Photocatalytic oxygen evolution plots versus irradiation time using NiP-Ru dyad under different concentrations, (d) PEC performance illustrated by light on and off experiments from TiO2 (1 cm2) on FTO sensitized with NiP-Ru dyad, and Ru-bpy reference after applying 0 V versus Ag/AgCl reference electrode in phosphate buffer of pH = 7. | ||
Further investigation in aqueous media was also performed, since water is the greenest solvent and the ideal oxygen source for our photocatalytic system. In detail we conducted photoelectrochemical (PEC) studies in an aqueous phosphate buffer solution (pH = 7) and in the absence of any sacrificial agents. For these experiments, a 450 W Xe lamp equipped with a 400 nm cut-off filter was used as a light source. The photoelectrochemical performance of the hydrolyzed dyad was studied by using a standard three-electrode PEC system. The NiP-Ru-COOH/TiO2/FTO was utilized as photoanode while Pt mesh and Ag/AgCl were used as counter and reference electrodes, respectively. The photoelectrochemical responses were recorded at an applied bias potential of 0.0 V (Fig. 2d). The light switching experiments displayed that the photocurrent produced by the NiP-Ru/TiO2/FTO electrode was much higher than that compared to the reference anode where no NiP catalyst moiety was present. As it is observed in Fig. 2d the intensity of the photocurrent decreases over time. This can be attributed to the partial disorption of the dyad from the TiO2 electrode due to the hydrolysis in aqueous media (the color of the sensitized electrode faded during the catalysis). In order to suppress this effect, more appropriate anchoring groups must be applied (e.g. pyridyl) or different buffer electrolyte (e.g. borate buffer). The above results demonstrates a proof of concept, where the PEC device fabricated from a NiP-Ru dyad is able to perform light induced water oxidation in neutral aqueous media. Noteworthy, to the best of our knowledge, this is the first PEC device utilizing nickel porphyrin WOC and ruthenium PS.
In summary, successful utilization of a nickel(II) porphyrin–ruthenium tris(bipyridine), NiP-Ru dyad in photocatalytic water oxidation under visible light irradiation has been accomplished. Nickel porphyrin moiety was utilized as the water oxidation catalyst while the ruthenium complex had the role of the light-harvesting sensitizer. Initial photocatalytic experiments in organic solutions showed that the covalently connected dyad displayed improved performance compared to the non-covalent two-component system. This was verified both in an electron pooling experiment followed by UV-Vis spectral detection of electron transfer products as well as direct detection of generated dioxygen using an optical oxygen sensor. The overall activity of NiP-Ru dyad reached a TON of 18 after one hour of continuous irradiation. For the first time, a photoelectrochemical cell was fabricated using the NiP-Ru dyad anchored on TiO2 as a photoanode and was shown to perform water oxidation at neutral pH conditions. It may be mentioned here that the employment of organic solvent could serve a dual role of placing water oxidation and CO2 reduction catalysts in the same cell for CO2 conversion. Currently, we are in the process of designing such tandem catalytic devices.
General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI) (project code: 508) and the US-National Science Foundation (To FD) are gratefully acknowledged for the financial support. The European Commission's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI) and the Special Research Account of the University of Crete also supported this work. E. Nikoloudakis acknowledges the Fulbright Foundation for the visiting research scholarship to UNT.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- S. Ye, C. Ding, M. Liu, A. Wang, Q. Huang and C. Li, Adv. Mater., 2019, 31, 1902069 CrossRef CAS PubMed.
- L. Tong and R. P. Thummel, Chem. Sci., 2016, 7, 6591–6603 RSC.
- N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721–10724 CrossRef CAS PubMed.
- K. S. Joya, N. K. Subbaiyan, F. D'Souza and H. J. M. de Groot, Angew. Chem., Int. Ed., 2012, 51, 9601–9605 CrossRef CAS PubMed.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D’Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS PubMed.
- A. R. Parent and K. Sakai, ChemSusChem, 2014, 7, 2070–2080 CrossRef CAS PubMed.
- D. Wang and C. O. Bruner, Inorg. Chem., 2017, 56, 13638–13641 CrossRef CAS PubMed.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC.
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451–1467 CrossRef CAS.
- D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS PubMed.
- X. Li, X.-P. Zhang, M. Guo, B. Lv, K. Guo, X. Jin, W. Zhang, Y.-M. Lee, S. Fukuzumi, W. Nam and R. Cao, J. Am. Chem. Soc., 2021, 143, 14613–14621 CrossRef CAS PubMed.
- A. Han, H. Jia, H. Ma, S. Ye, H. Wu, H. Lei, Y. Han, R. Cao and P. Du, Phys. Chem. Chem. Phys., 2014, 16, 11224–11232 RSC.
- Q. Daniel, R. B. Ambre, B. Zhang, B. Philippe, H. Chen, F. Li, K. Fan, S. Ahmadi, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 1143–1149 CrossRef CAS.
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed.
- P. M. Usov, S. R. Ahrenholtz, W. A. Maza, B. Stratakes, C. C. Epley, M. C. Kessinger, J. Zhu and A. J. Morris, J. Mater. Chem. A, 2016, 4, 16818–16823 RSC.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC.
- T. Nakazono, A. R. Parent and K. Sakai, Chem. – Eur. J., 2015, 21, 6723–6726 CrossRef CAS PubMed.
- Y. S. Nam, A. P. Magyar, D. Lee, J.-W. Kim, D. S. Yun, H. Park, T. S. Pollom, D. A. Weitz and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 340–344 CrossRef CAS PubMed.
- C. Liu, D. van den Bos, B. den Hartog, D. van der Meij, A. Ramakrishnan and S. Bonnet, Angew. Chem., Int. Ed., 2021, 60, 13463–13469 CrossRef CAS PubMed.
- E. Nikoloudakis, P. B. Pati, G. Charalambidis, D. S. Budkina, S. Diring, A. Planchat, D. Jacquemin, E. Vauthey, A. G. Coutsolelos and F. Odobel, ACS Catal., 2021, 11, 12075–12086 CrossRef CAS.
- K. L. Materna, J. Jiang, K. P. Regan, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, ChemSusChem, 2017, 10, 4526–4534 CrossRef CAS PubMed.
- C. Decavoli, C. L. Boldrini, V. Trifiletti, S. Luong, O. Fenwick, N. Manfredi and A. Abbotto, RSC Adv., 2021, 11, 5311–5319 RSC.
- K. Akamine, K. Morita, K. Sakai and H. Ozawa, ACS Appl. Energy Mater., 2020, 3, 4860–4866 CrossRef CAS.
- K. Morita, K. Sakai and H. Ozawa, ACS Appl. Energy Mater., 2019, 2, 987–992 CrossRef CAS.
- K. Sakai and H. Ozawa, AsiaChem Magazine, 2021, 2, 80–97 Search PubMed.
- D. G. McCafferty, B. M. Bishop, C. G. Wall, S. G. Hughes, S. L. Mecklenberg, T. J. Meyer and B. W. Erickson, Tetrahedron, 1995, 51, 1093–1106 CrossRef CAS.
- G. J. Barbante, C. F. Hogan, D. J. D. Wilson, N. A. Lewcenko, F. M. Pfeffer, N. W. Barnett and P. S. Francis, Analyst, 2011, 136, 1329–1338 RSC.
- A. Charisiadis, E. Glymenaki, A. Planchat, S. Margiola, A.-C. Lavergne-Bril, E. Nikoloudakis, V. Nikolaou, G. Charalambidis, A. G. Coutsolelos and F. Odobel, Dyes Pigm., 2021, 185, 108908 CrossRef CAS.
- J. W. Buchler and G. Herget, Z. Naturforsch. B, 1987, 42, 1003–1008 CrossRef CAS.
- C. A. Kent, B. P. Mehl, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2010, 132, 12767–12769 CrossRef CAS PubMed.
- G. D. Hager and G. A. Crosby, J. Am. Chem. Soc., 1975, 97, 7031–7037 CrossRef CAS.
- Gibbs free energy for the hole shift reaction calculated from: ΔGh+s = EOx(NiP/NiP+) − EOx(Ru2+/Ru3+).
- S. Wallin, J. Davidsson, J. Modin and L. Hammarström, J. Phys. Chem. A, 2005, 109, 4697–4704 CrossRef CAS PubMed.
- R. Lomoth, T. Häupl, O. Johansson and L. Hammarström, Chem. – Eur. J., 2002, 8, 102–110 CrossRef CAS PubMed.
- M. Barrejón, L. M. Arellano, H. B. Gobeze, M. J. Gómez-Escalonilla, J. L. G. Fierro, F. D'Souza and F. Langa, Chem. Sci., 2018, 9, 8221–8227 RSC.
- S. Pintado, S. Goberna-Ferrón, E. C. Escudero-Adán and J. R. Galán-Mascarós, J. Am. Chem. Soc., 2013, 135, 13270–13273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, electrochemical and transient spectral data. See DOI: https://doi.org/10.1039/d2cc03563d |
| This journal is © The Royal Society of Chemistry 2022 |