
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isothiourea-catalysed enantioselective radical conjugate addition under batch and flow conditions†
Roberto
del Río-Rodríguez‡
 a,
Matthew T.
Westwood‡
b,
Marina
Sicignano
a,
Martin
Juhl
b,
Jose A.
Fernández-Salas
ac,
José
Alemán
*acd and
Andrew D.
Smith
*b
a,
Matthew T.
Westwood‡
b,
Marina
Sicignano
a,
Martin
Juhl
b,
Jose A.
Fernández-Salas
ac,
José
Alemán
*acd and
Andrew D.
Smith
*b
aDepartamento de Química Orgánica (Módulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049-Madrid, Spain. E-mail: jose.aleman@uam.es; Web: https://www.uam.es/jose.aleman
bEaStCHEM, School of Chemistry, University of St Andrews, North Haugh, Fife, Scotland, KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
dCenter for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad Autónoma de Madrid, 28049, Madrid, Spain
First published on 25th May 2022
Abstract
The photocatalytic generation of α-amino radicals is combined with chiral isothiourea derived α,β-unsaturated acyl ammonium intermediates. The reaction proceeds VIA a [3+2] radical-polar crossover mechanism to generate γ-lactams in good yields and enantioselectivities. The enantioselective radical conjugate addition was carried out under batch and flow conditions.
The selective synthesis of enantioenriched molecules is a fast-moving research area in organic synthesis and the development of methodologies for their preparation still plays a crucial role for the synthesis of prevalent structures in natural and synthetic products.1 Michael additions (or conjugate additions) are among the most widely studied transformations in organic synthesis and are a powerful and appealing synthetic tool for the direct and rapid modification of activated alkenes.2 In this context, the addition of radicals to electron poor alkenes (Giese reaction) is a well-established alternative for the construction of C–C bonds.2b,3 Despite modern organic synthesis relying heavily upon radical species,4 efficient catalytic enantioselective versions are still limited,3,5 which can be pinpointed to the high background (uncatalyzed) reactivity of the highly reactive radicals involved.5 In this area, Sibi and coworkers6 described one of the first enantioselective conjugate radical additions using chiral Lewis acid catalysis. Inspired by these pioneering studies, and taking advantage of the development of dual catalytic systems for the efficient construction of stereochemically defined motifs,7 a dual Lewis acid/photoredox catalysed enantioselective addition of α-amino radicals to Michael acceptors has been reported (Scheme 1A, left).8 In addition, other radical precursors and Lewis acid-based catalytic systems have been described in enantioselective photoredox Giese-type reactions.9 In these processes, these Lewis acid catalysed systems all require two-point binding of an acyl auxiliary bearing Michael acceptor to ensure bidentate coordination with chiral Lewis acids to achieve high enantioinduction (Scheme 1A, left).9 During the last decade, organocatalysis has proved well suited to be combined with photochemical processes for the enantioselective construction of complex molecules.7,10 As an overview, enantioselective Giese-type reactions of transient iminium ions of enals11 and enones12 as chiral platforms have been studied (Scheme 1A, right). Despite these advances, enantioselective organocatalytic Giese-type reactions are still underdeveloped. At the onset of these studies, to the best of our knowledge, versatile carboxylic acid derivatives had not been reported as prochiral templates in enantioselective radical conjugate additions.13
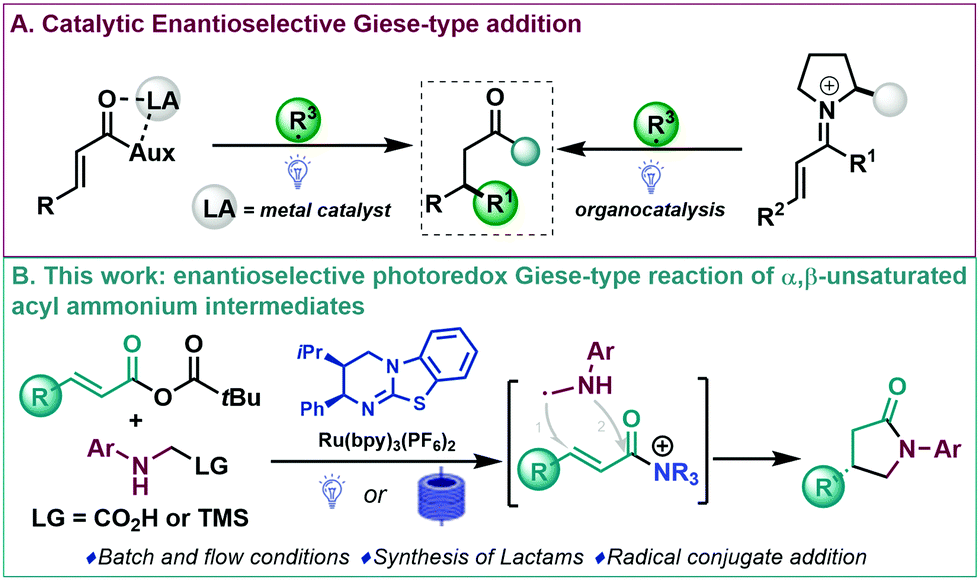 | ||
| Scheme 1 Previous works (A) and present work (B). | ||
The use of chiral tertiary amine derived α,β-unsaturated acyl ammonium intermediates has proved an increasingly popular strategy over the past decade.14 Various ionic Michael addition processes have been developed with a wide array of structural diversity accessible but radical derived approaches have remaining underexplored. Based upon our experience,14,15 the use of chiral isothiourea derived α,β-unsaturated acyl ammonium intermediates for the stereoselective addition of α-amino radicals was considered. In this process, the proposed conjugate radical addition would yield interesting γ-lactams through a [3+2] radical-polar crossover mechanism that would allow catalyst turnover by N-acylation that should be amenable in both batch and flow (Scheme 1B). Enantioselective routes to γ-lactams are an important and appealing synthetic goal,16 with the targeted γ-lactam motif a widespread heterocyclic scaffold found in an array of natural products, pharmaceuticals and agrochemicals.17
Initial exploration used the reaction of mixed α,β-unsaturated anhydride 1a, accessible in a high yielding single step from the corresponding acid, and N-phenylglycine 2a as the radical precursor (Table 1 and ESI†). Using (2R,3S)-hyperBTM isothiourea catalyst (3a) in the presence of TBAB (tetrabutylammonium bromide) as an additive and [Ru(bpy)3](PF6)2], under 23W CFL irradiation, the corresponding lactam (4a) was isolated in good yield and enantioselectivity (entry 1). Other isothiourea catalysts were tested (see ESI†), but product enantioselectivity was significantly decreased (entry 2 and ESI†). The use of TBAB as an additive was found to be decisive, with substitution by a base leading to diminished yield and enantioselectivity of the desired lactam (entry 3). Various photocatalysts were also tested (see ESI†), with for example fac-Ir(ppy)3 giving 4a in substantially lower yield and enantioselectivity (entry 4). The use of only acetonitrile as solvent, or as a mixture with other aromatic solvents or TBME, was detrimental to reaction efficiency (entries 5–7 and ESI†). Control experiments ensured that in absence of light or photocatalyst the reaction did not take place (entries 8 and 9).
|
|
|||
|---|---|---|---|
| Entry | Deviation from optimized conditions | Yieldsb (%) | e.r.c |
a Reaction conditions: 1a (0.15 mmol) and 2a (0,18 mmol), TBAB (0.15 mmol), Ru(bby)3(PF6)2 (2 mol%), (2R,3S)-hyperBTM (20 mol%), CH3CN:PhCH3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.05 M), r.t, under N2, 23W CFL, 20 °C, 4 h.
b
1H-NMR yields using 1,3,5-trimethoxybenzene as internal standard. Isolated yield in brackets.
c Enantiomeric excess was measured by Supercritical Fluid Chromatography (SFC) using chiral columns. :1, 0.05 M), r.t, under N2, 23W CFL, 20 °C, 4 h.
b
1H-NMR yields using 1,3,5-trimethoxybenzene as internal standard. Isolated yield in brackets.
c Enantiomeric excess was measured by Supercritical Fluid Chromatography (SFC) using chiral columns.
|
|||
| 1 | No deviation | 65(59) | 81:19 |
| 2 | (S)-BTM (3b) catalyst instead of (2R,3S)-HyperBTM (3a) | 24 | 55:45 |
| 3 | K2HPO4 | 40 | 75:25 |
| 4 | fac-Ir(ppy)3 instead of Ru(bby)3(PF6)2 | 30 | 70:30 |
| 5 | CH3CN instead of CH3CN:PhCH3 | 70 | 70:30 |
| 6 | CH3CN:PhCF3 instead of CH3CN:PhCH3 | 59 | 81:19 |
| 7 | CH3CN:TBME instead of CH3CN:PhCH3 | 35 | 81:19 |
| 8 | No light | n.r. | — |
| 9 | No Ru(bby)3(PF6)2 | n.r. | — |
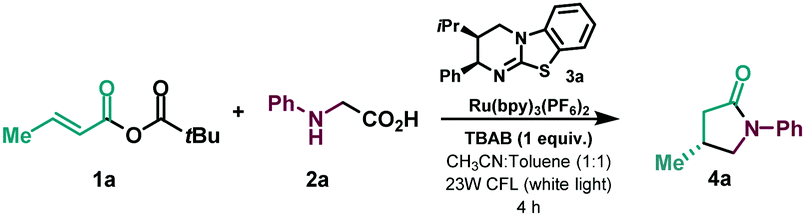
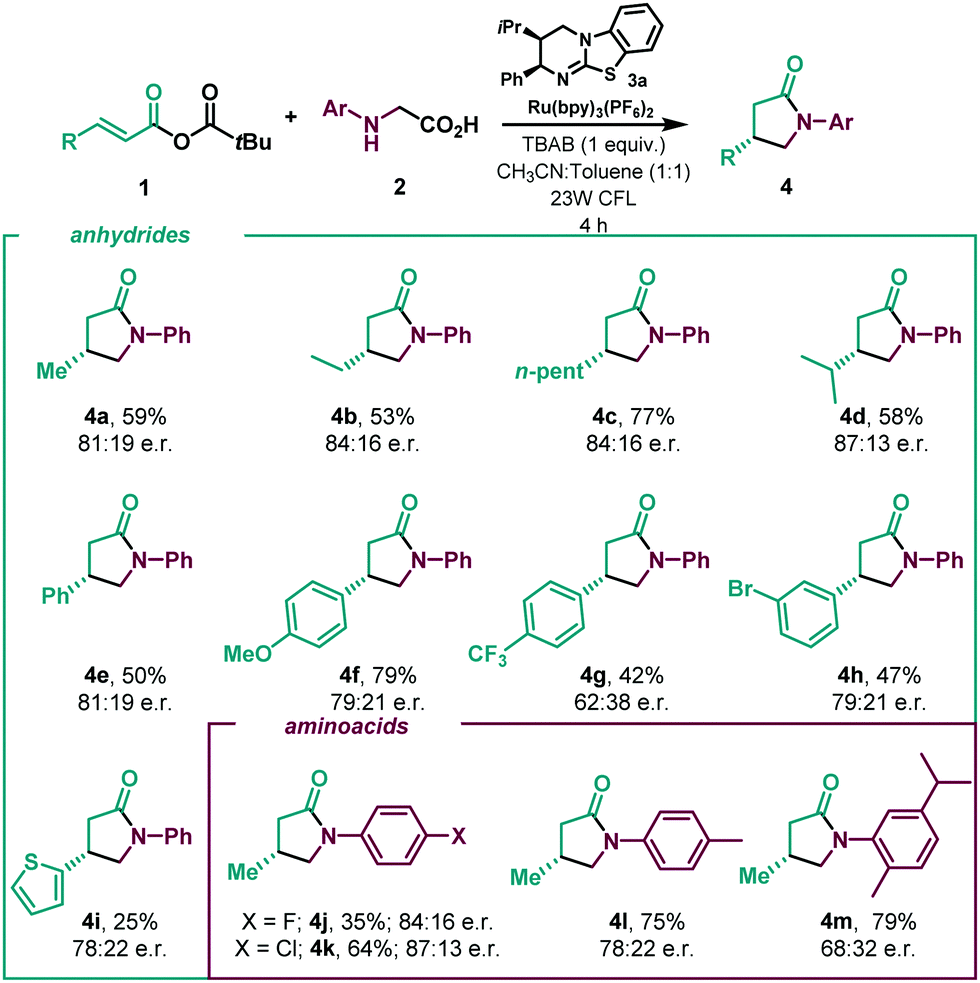
With the optimized reaction in hand (Table 1, entry 1), we next investigated the scope of the enantioselective radical conjugate addition to α,β-unsaturated anhydrides using amino acids as the α-amino radical source (Table 2). Firstly, the scope of the α,β-unsaturated component was evaluated. Aliphatic α,β-unsaturated anhydrides were very well tolerated, leading to the desired γ-lactams in good yields and enantioselectivities (4a–4d). Notably, β-phenyl substitution in the anhydride showed a similar performance in the catalytic system (4e). Various β-aryl substituted α,β-unsaturated systems were then tested. While electron donating para-methoxy (4f) and meta-bromo (4h) substituents were well tolerated, an electron deficient para-CF3 substituent delivered the lactam with diminished enantioselectivity (4g). A β-2-thiophene substituent led to the desired cyclised product in slightly lower yield and enantioselectivity (4i). A series of N-aryl glycines were then subjected to the optimised reaction conditions. While para-F (4j) and para-Cl (4k) substituents showed moderate yields and good enantioselectivities, para-methyl displayed enhanced efficiency, yielding lactam 4l in good yield with a slight erosion in the enantiomeric ratio. The use of a sterically hindered disubstituted N-aryl glycine gave rise to the desired product in high yield and low enantioselectivity (4m).
| a All reactions performed using 2 (0.15 mmol scale). Er determined by SFC analysis on a chiral stationary phase. |
|---|
|
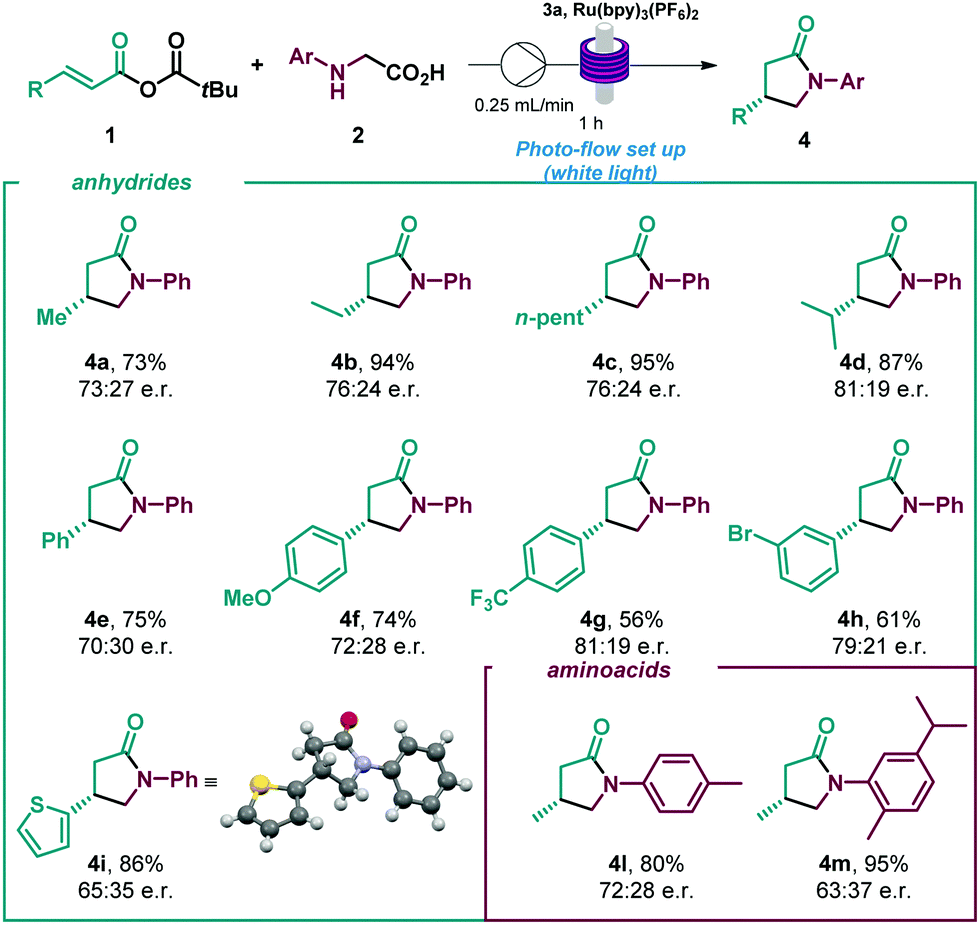
Given the encouraging results achieved and the synthetic potential of the developed method, we decided to perform the enantioselective reaction under flow conditions (Table 3, see ESI†). The synthetic method proved compatible using a flow-photoreactor (Table 3, see ESI†), and applying similar reaction conditions when compared with the batch conditions (Table 2), 4a was obtained in a significantly higher yield, although diminished enantioselectivity. The robustness of the flow system was proved as high yields of the γ-lactams were obtained in all cases in a shortened reaction time (1 h), which provides a reaction throughput up to 30 times higher than the batch conditions. Surprisingly, a β-para-CF3 substituent delivered the lactam with improved enantioselectivity to batch (4g). Considering other potential α-amino radical precursors,18 the applicability of this isothiourea catalysed system was expanded to radicals generated from α-silyl secondary amines (Scheme 2). In this system, an acid additive proved essential to minimise competitive 1,2-amine addition. Using the hydrochloride salt of the corresponding isothiourea HyperBTM organocatalyst under slightly modified reaction conditions (see ESI† for more details), γ-lactam 4a was obtained in similar yield with slightly enhanced enantioselectivity. β-Alkyl substitution was well tolerated, giving the corresponding γ-lactams with good yields and enantioselectivities (4b and 4c, 4n and 4o). β-Aryl substituted anhydrides showed increased competitive 1,2-addition leading to reduced product yield and enantioselectivity (4f and 4g). N-aryl substitution within the amine component was tolerated, with the ortho-CH3 α-silyl amine giving γ-lactam 4p in good yield and enantioselectivity. However, ortho-phenyl substitution gave negligible reactivity under the optimised conditions and a solvent mixture with a greater proportion of MeCN was necessary to deliver 4q with good yield but reduced enantioselectivity. These conditions gave the meta-CF3 substituted lactam 4r in good yield and enantioselectivity.
| a All reactions performed using 2 (0.15 mmol scale) in presence of K2HPO4 (0.075 mol). Er determined by SFC analysis on a chiral stationary phase. |
|---|
|
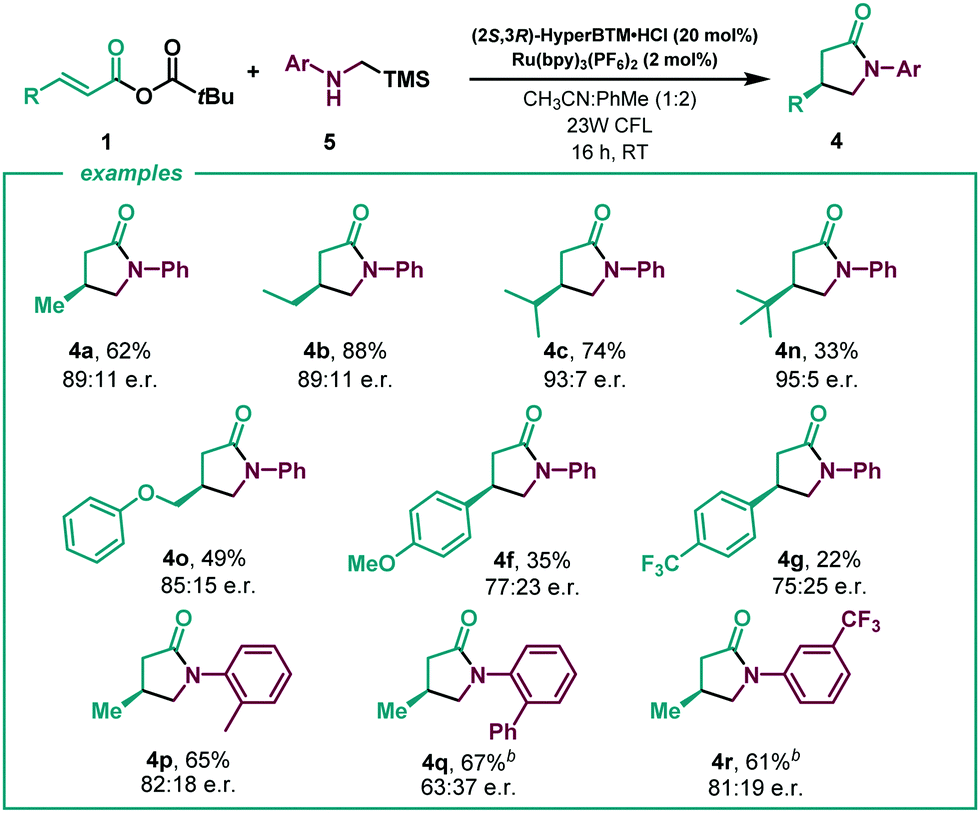 | ||
| Scheme 2 Scope of the conjugate addition of α-amino radicals generated from α-silyl amines.a a All reactions performed using 5 (0.15 mmol scale). b Using PhMe:MeCN (1:2). Er determined by GC or HPLC analysis on a chiral stationary phase. | ||
The absolute configuration of 4i (Table 3) was unequivocally assigned as (R) by X-ray crystallographic analysis19 and further corroborated by the specific rotation of (R)-4a8 (see ESI†). The same stereochemical outcome was assigned by analogy for all the compounds included in Tables 2 and 3 and Scheme 2.
A mechanism is proposed for the transformation (Scheme 3A). N-acylation of the mixed anhydride 1 by the isothiourea leads to the α,β-unsaturated acyl ammonium ion pair I, adopting an s-cis conformation with a syn-coplanar 1,5 S⋯O chalcogen bonding interaction providing a conformational lock.20 Meanwhile, the excited state photocatalyst oxidises a molecule of the α-amino radical precursor. Fluorescence quenching experiments of the excited photocatalyst prove that hypothesis. The data (Scheme 3B) confirm the lack of interaction between the Ru(bpy)3(PF6)2 and the anhydride, and indicate slight potential oxidation of the organocatalyst.
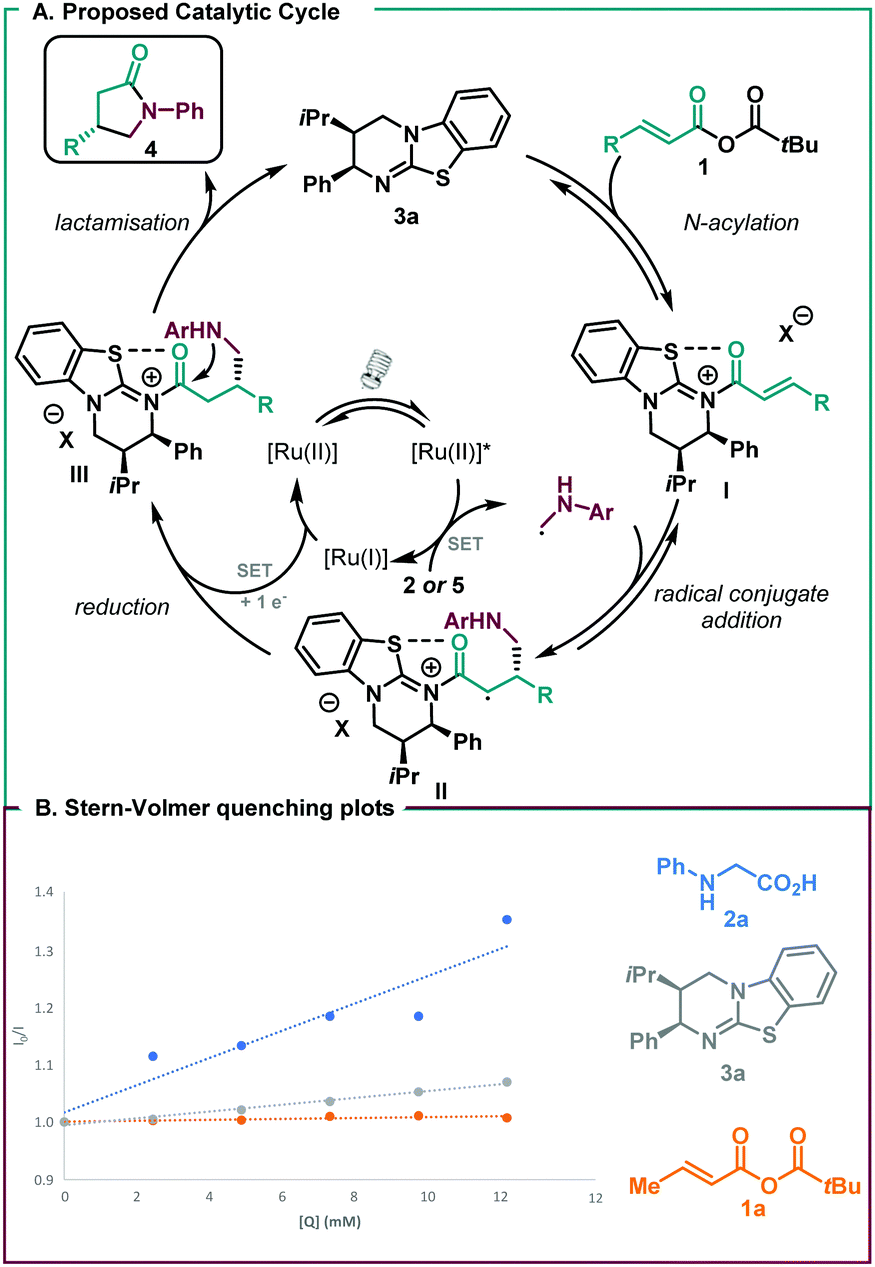 | ||
| Scheme 3 Proposed mechanism of the [3+2] radical-polar crossover reaction to generate γ-lactams. | ||
This presents the possibility of the α-amino radical precursor quenching the excited state of Ru(bpy)3(PF6)2, suggesting that the initial step of the photocatalytic cycle would be the oxidation of the α-amino acid or α-trimethylsilyl amine by the excited state photocatalyst. Subsequent removal of the electrofugal substituent (CO2 or TMS) generates the α-amino radical that subsequently undergoes enantioselective radical conjugate addition into the α,β-unsaturated acyl ammonium intermediate, with facial selectivity dictated by the stereodiscriminating phenyl substituent. The reduced state photocatalyst (Ru(I)) then reduces the α-carbonyl radical II to the corresponding C(1)-ammonium enolate, regenerating the ground state photocatalyst, which upon protonation leads to the acyl ammonium III. Intramolecular N-acylation gives the corresponding γ-lactam 4 with concomitant release of the isothiourea catalyst.
In summary, we have described the combination of an isothiourea organocatalyst and a photocatalyst for the development of an enantioselective [3+2] radical-polar crossover process to generate γ-lactams under batch and flow conditions. The developed system relies on the conjugate addition of α-amino radicals to transient isothiourea derived α,β-unsaturated acyl ammonium intermediates formed in situ.21
Financial support was provided by the Spanish Government (RTI2018-095038-B-I00), “Comunidad de Madrid” for European Structural Funds (S2018/NMT-4367) and proyectos sinergicos I + D (Y2020/NMT-6469). J. A. F.-S. thanks the Spanish Government for a Ramón y Cajal contract. The research leading to these results has received funding from the EaSI-CAT centre for Doctoral Training (M. T. W.) and Carlsberg Foundation (M. J.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) In Fundamentals of Asymmetric Catalysis, ed. P. J. Walsh and M. C. Kozlowski, University Science Books, York, 2009 Search PubMed; (b) In Multicatalyst System in Asymmetric Catalysis, ed. J. Zhou, John Wiley & Sons Inc., Hoboken, 2015 Search PubMed; (c) E. L. Eliel, S. H. Wilen and L. N. Mander in Stereochemistry of Organic Compounds, Wiley-Interscience, New York, 1994, pp 1–10 Search PubMed.
- For selected reviews (a) J. L. Vicario, D. Badia, L. Carrillo and E. Reyes, Organocatalytic Enantioselective Conjugate Addition Reactions, Royal Society of Chemistry, Cambridge, 2010 Search PubMed; (b) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586 CrossRef CAS PubMed.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116 CrossRef CAS PubMed.
- G. J. Rowlands, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2011, 107, 19 RSC.
- For a review on enantioselective radical reactions, see: J. Zimmerman and M. P. Sibi, Top. Curr. Chem., 2006, 263, 107 CrossRef CAS.
- (a) M. P. Sibi, J. Ji, J. H. Wu, S. Gürtler and N. A. Porter, J. Am. Chem. Soc., 1996, 118, 9200 CrossRef CAS; (b) M. P. Sibi and J. Ji, J. Org. Chem., 1997, 62, 3800 CrossRef CAS.
- For selected reviews on dual catalysis, see: (a) C. K. Prier and D. W.-C. MacMillan in Visible Light Photocatalysis in Organic Chemistry ed. C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, 2018, pp. 299 Search PubMed; (b) C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363 CrossRef PubMed; (c) S. C. Coote, and T. Bach, Enantioselective photocatalysis, in Visible Light Photocatalysis in Organic Chemistry, ed. C. Stephenson, T. Yoon and D. W. C. MacMillan, Wiley-VCH, Germany, 2018, pp. 335–361 Search PubMed.
- L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320 CrossRef CAS PubMed; (b) X. Dong, Q. Y. Li and T. P. Yoon, Org. Lett., 2021, 23, 5703 CrossRef CAS PubMed; (c) X. Huang, S. Luo, O. Burghaus, R. D. Webster, K. Harmsa and E. Meggers, Chem. Sci., 2017, 8, 7126 RSC; (d) C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495 CrossRef CAS PubMed; (e) H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936 CrossRef CAS PubMed; (f) S.-X. Lin, G.-J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665 RSC.
- For selected reviews on organocatalysis in photochemical processes, see: (a) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed; (b) Y. P. Rey, H. B. Hepburn and P. Melchiorre, Organocatalysis with amines in photocatalysis, in Science of Synthesis: Photocatalysis in Organic Synthesis, ed. B. König, Thieme, 2018, pp. 243–270 Search PubMed; (c) Y.-Q. Zou, F. M. Hörmann and T. Bach, Chem. Soc. Rev., 2018, 47, 278 RSC.
- E. Le Saux, D. Ma, P. Bonilla, C. M. Holden, D. Lustosa and P. Melchiorre, Angew. Chem., Int. Ed., 2021, 60, 5357 CrossRef CAS PubMed.
- (a) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed; (b) A. Bahamonde, J. J. Murphy, M. Savarese, É. Brémond, A. Cavalli and P. Melchiorre, J. Am. Chem. Soc., 2017, 139, 4559 CrossRef CAS PubMed; (c) Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- During the submission of the presented studies, Melchiorre and coworkers have reported a similar process: W. C. Hartley, F. Schiel, E. Ermini and P. Melchiorre, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202204735.
- (a) J. Bitai, M. T. Westwood and A. D. Smith, Org. Biomol. Chem., 2021, 19, 2366 RSC For selected recent examples, see: ; (b) J. Bitai, A. J. Nimmo, A. M.-Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2022, 61, e202202621 Search PubMed; (c) T. Kurihara, M. Kojima, T. Yoshino and S. Matsunaga, J. Am. Chem. Soc., 2022, 144, 7058 CrossRef CAS PubMed.
- (a) R. Rodríguez, M. Sicignano and J. Alemán, Angew. Chem., Int. Ed., 2022, 61, e2021126 CrossRef PubMed; (b) A. Casado-Sánchez, P. Domingo-Legarda, S. Cabrera and J. Alemán, Chem. Commun., 2019, 55, 11303 RSC; (c) C. Shu, H. Liu, A. M.-Z. Slawin, C. Carpenter-Warren and A. D. Smith, Chem. Sci., 2020, 11, 241 RSC; (d) M. D. Greenhalgh, S. Qu, A. M.-Z. Slawin and A. D. Smith, Chem. Sci., 2018, 9, 4909 RSC.
- Few selected enantioselective synthesis of γ-lactams, see: (a) H. Wang, Y. Park, Z. Bai, S. Chang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 7194 CrossRef CAS PubMed; (b) X. Wu, J. Qu and Y. Chen, J. Am. Chem. Soc., 2020, 142, 15654 CrossRef CAS PubMed; (c) Q. Xing, C.-M. Chan, Y.-W. Yeung and W.-Y. Yu, J. Am. Chem. Soc., 2019, 141, 3849 CrossRef CAS PubMed; (d) Y. Li, F.-P. Zhang, R.-H. Wang, S.-L. Qi, Y.-X. Luan and M. Ye, J. Am. Chem. Soc., 2020, 142, 19844 CrossRef CAS PubMed.
- J. Caruano, G. G. Muccioli and R. Robiette, Org. Biomol. Chem., 2016, 14, 10134 RSC.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946 CrossRef CAS PubMed.
- The X-ray analysed was crystallised from a 78:22 er mixture (Table 2). CCDC 2168909 (4i)†.
- (a) E. R.-T. Robinson, C. Fallan, C. Simal, A. M.-Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193 RSC; (b) E. R.-T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919 RSC; (c) C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. de la Houpliere, K. B. Ling, T. K. Smith, A. M.-Z. Slawin, P. H. Willoughby, S. L. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705 CrossRef CAS PubMed.
- The research data underpinning this publication can be accessed at https://doi.org/10.17630/2522a5e8-3d9f-4b24-8aec-46c8a455b394.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc02432b |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2022 |