
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the photophysical kinetics, electronic configurations and structural conformations of iridium–cobalt hydrogen evolution photocatalysts†
Jiangyun
Zhao
 a,
Simon
De Kreijger
b,
Ludovic
Troian-Gautier
b,
Jin
Yu
c,
Wenhui
Hu
d,
Xiaoyi
Zhang
c,
Benjamin
Elias
*b and
Dooshaye
Moonshiram
*a
a,
Simon
De Kreijger
b,
Ludovic
Troian-Gautier
b,
Jin
Yu
c,
Wenhui
Hu
d,
Xiaoyi
Zhang
c,
Benjamin
Elias
*b and
Dooshaye
Moonshiram
*a
aInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, Sor Juana Inés de la Cruz, 3, 28049, Madrid, Spain. E-mail: dooshaye.moonshiram@csic.es
bInstitute of Condensed Matter and Nanoscience, Molecular Chemistry, Materials and Catalysis Division, Place Louis Pasteur 1, Louvain-la-Neuve, Belgium. E-mail: benjamin.elias@uclouvain.be
cX-Ray Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Lemont, IL 60439, USA
dDepartment of Chemistry, Marquette University, Milwaukee, WI 53201, USA
First published on 26th May 2022
Abstract
Picosecond optical and X-ray absorption spectroscopies with time-dependent density functional theory revealed the reaction pathways, electronic and structural conformations of Ir–Co hydrogen evolution photocatalysts. The dyad bearing 2-phenylpyridine ancillary ligands produced more photoreduced Co(II) than its 2-phenylisoquinoline analogue. These findings are important for designs of earth-abundant photosensitizers for photocatalytic applications.
In the face of today's rapid depletion of fossil fuels and emerging climate change crisis, alternative sustainable means of storing, transporting and distributing energy are being devised.1 The abundant supply of solar energy provides an attractive means of storing energy through fuel forming reactions inspired by natural photosynthesis, such as the light induced splitting of water into molecular hydrogen and oxygen (2H2O + 4hν → O2 + 2H2).2 The prospect of using molecular hydrogen as a carbon-free fuel has led to the rationalization and design of sustainable artificial molecular photosensitizer (PS)/catalytic assemblies where visible light is absorbed at the photosensitizer unit and converted into a chemical potential through charge accumulation processes at the catalytic unit. Recently, Ir–Co dyads (Scheme 1) consisting of cobaloxime hydrogen evolution catalysts [Co(dmgH)(dmgH2)]Cl23 (dmgH2 = dimethylglyoxime) were chelated to Ir-based photosensitizers with 2-phenylpyridine ([Ir(ppy)2-L]) (dyad I) or 2-phenylisoquinoline ([Ir(piq)2-L]) ligands (dyad II) and found to display larger than 200 turnover rates of hydrogen generation upon light irradiation.4 However key insights on the photo-induced electron transfer dynamics, photogenerated intermediates and influence of the cyclometalating ligands on the Ir-based photosensitizers that are responsible for the dyads’ activities and essential for the fine-tuning of the light-harvesters’ electronic properties were not investigated, yet are of crucial importance. In this regard, time-resolved X-ray (tr-XAS) and optical transient absorption (OTA) spectroscopies in the picosecond to microsecond time regime coupled with time-dependent density functional theoretical calculations (TD-DFT) are employed as powerful tools at both Co and Ir K and L-edges to extract valuable information about the coordination sphere around the cobalt catalytic centre and resolve the kinetics, electronic and structure scheme of these Ir-based photosensitizers.
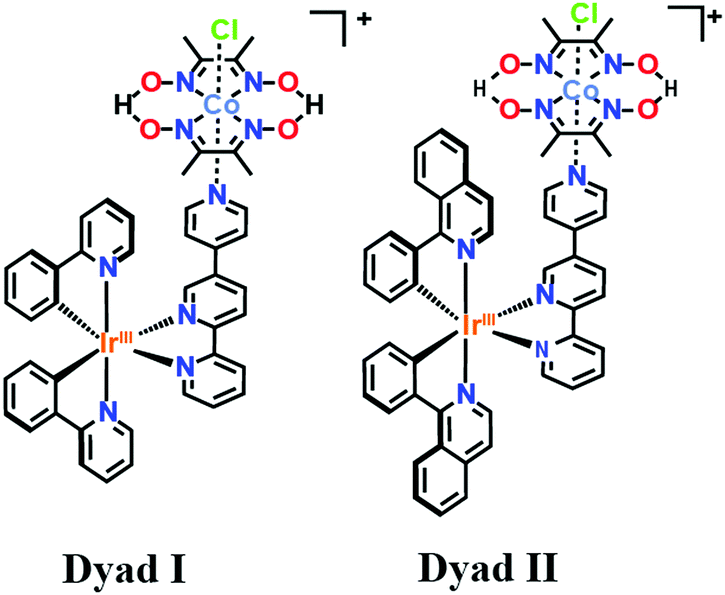 | ||
| Scheme 1 Structure of the Ir–Co based dyads I and II obtained respectively through chelation of the Ir-based photosensitizers [Ir(ppy)2-L] and [Ir(piq)2-L] with the Co-based catalyst. | ||
Prior to performing time-resolved measurements, the cobaloxime-based CoIII catalyst and Ir-based photosensitizers were studied by X-ray absorption near edge structure (XANES) and Extended X-ray absorption fine structure (EXAFS). The XANES, EXAFS and Fourier-transform (FT) of the k2-weighted Co EXAFS spectra are shown in Fig. 1A, Fig. S1A (ESI†) and Fig. 1B respectively. A prominent peak and shoulder denoted as Peaks I and II (Fig. 1B) are observed in the CoIII FT EXAFS spectrum corresponding to the distinctive Co–N and Co–Cl bond distances. EXAFS fits for the first coordination sphere and the entire spectrum are shown in Fig. S2 and Table S1 in the ESI.† Analysis of the first peak in the cobaloxime CoIII complex clearly resolves 4 Co–N distances at 1.85 Å, and inclusion of a Co–Cl distance at 2.16 Å improves the quality of the fit as shown by the decreased R-factors and χ2 value (Table S1, ESI,† Fits 1 and 3). The fitted Co–N and Co–Cl bond distances are in agreement with the reported XRD structure4 and relaxed structures from DFT geometry optimization. Changes detected in experimental FT of the CoIII EXAFS further correlate well with data from X-ray diffraction (XRD) analysis and EXAFS simulations of DFT optimized coordinates (Fig. S3, ESI†).
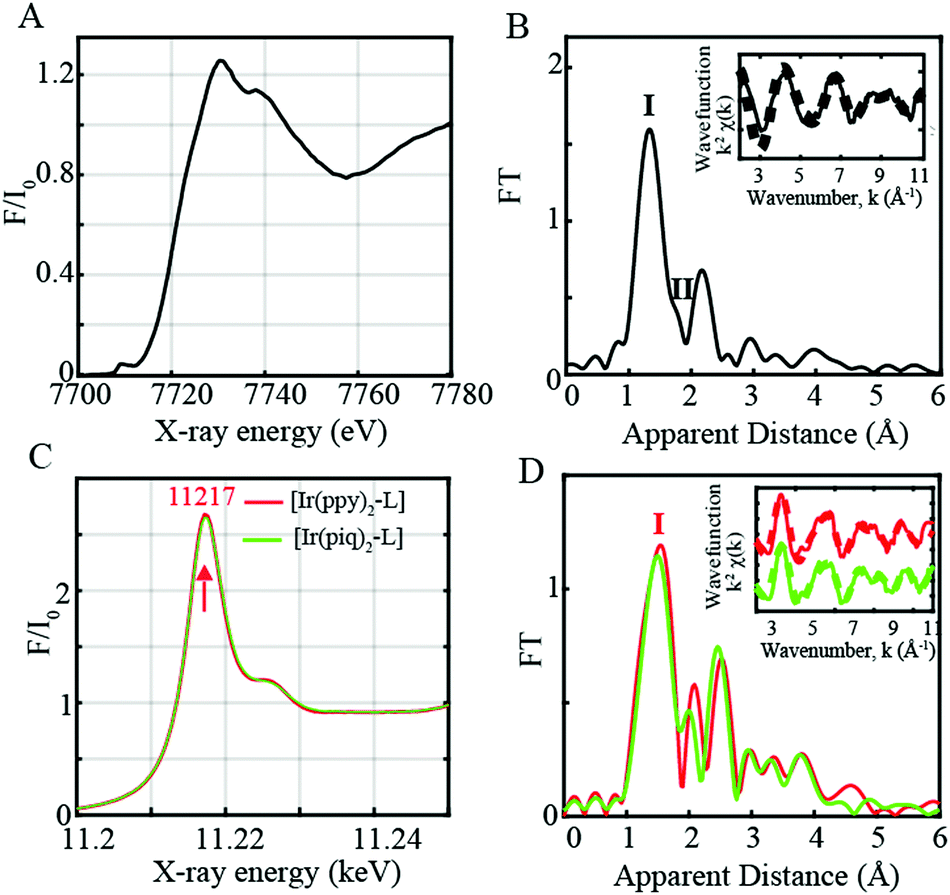 | ||
| Fig. 1 (A) Normalized Co K-edge XANES for CoIII complex in dyads I and II (black). (B) Fourier transforms of k2-weighted Co EXAFS for the Co catalyst (black). Inset: Back Fourier transformed experimental (solid lines) and fitted (dashed lines) k2[χ(k)]. (C) Normalized Ir L-edge XANES for [Ir(ppy)2-L] (red) and [Ir(piq)2-L] (green). (D) Fourier transforms of k2-weighted Ir EXAFS Inset. Back Fourier transformed experimental (solid lines) and fitted (dashed lines) k2[χ(k)] for Ir complexes in dyads I and II. Experimental spectra were calculated for k values of 2–11 Å−1. | ||
The two Ir-based photosensitizers [Ir(ppy)2-L] and [Ir(piq)2-L] were further assessed through steady-state L-edge XANES and EXAFS spectroscopy (Fig. 1C, D and Fig. S1B, ESI†). Ir(III) complexes have a formal d6 electronic configuration with filled t2g and empty eg levels. An intense peak corresponding to the Ir 2p to unoccupied 5d states with eg symmetry is consequently observed at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 217 eV (Fig. 1C) while no features are observed on the lower energy region below 11217 eV due to the lack of vacancy in the t2g states. The FT of the k2-weighted [Ir(ppy)2-L] and [Ir(piq)2-L] EXAFS spectra display a prominent peak (Peak I, Fig. 1D) corresponding to the averaged Ir–N/C distances at 2.00 Å and 2.01 Å respectively, in close agreement with the reported XRD structures with Ir–N distances of 2.06 Å (Table S1 and Fig. S3, ESI†).
217 eV (Fig. 1C) while no features are observed on the lower energy region below 11217 eV due to the lack of vacancy in the t2g states. The FT of the k2-weighted [Ir(ppy)2-L] and [Ir(piq)2-L] EXAFS spectra display a prominent peak (Peak I, Fig. 1D) corresponding to the averaged Ir–N/C distances at 2.00 Å and 2.01 Å respectively, in close agreement with the reported XRD structures with Ir–N distances of 2.06 Å (Table S1 and Fig. S3, ESI†).
Following the steady-state measurements, tr-XAS was used to directly monitor the electronic conformations, kinetics and structural dynamics of the photo-generated CoII and Ir triplet states in the supramolecular assemblies under photocatalytic conditions4 with triethanolamine (TEOA) as the sacrificial electron donor and aqueous tetrafluoroboric acid as the proton source (Fig. 2 and 3). The Ir–Co binuclear dyads are optically pumped at 400 nm in the mixed Metal–Ligand-to-Ligand Charge Transfer (MLLCT) range with a 10 kHz repetition-rate laser and probed with X-ray pulses at several time delays from 100 ps to ∼25 μs. XAS spectra of the supramolecular assemblies were collected before and after the laser excitation. By subtracting the laser-off from the laser-on spectrum, a transient signal is obtained for each pump-probe delay and provides information about the photo-induced dynamics, electronic and structural state of the catalyst's and photosensitizer's excited states.
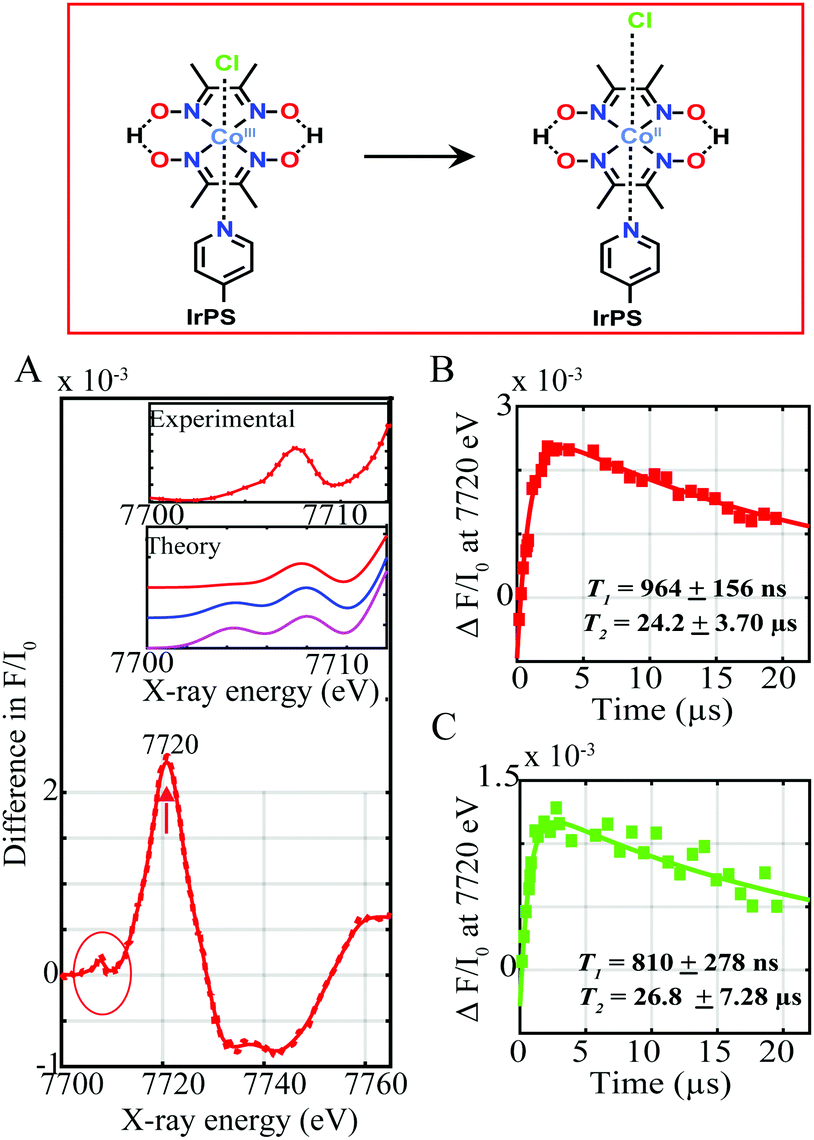 | ||
| Fig. 2 (A) Experimental difference spectrum (laser on-laser off) corresponding to the CoIII transient signal at a time delay of ∼11.3 μs between laser and X-ray pulses (red) in 1 mM dyads I and II with 1 M TEOA and 10 mM tetrafluoroboric acid. Inset. Zoom-in of the pre-edge CoII transient experimental spectrum (red) (top) together with the simulated difference spectra for [LCoIICl]2+ octahedral (red), [LCoIICH3CN]2+ octahedral (blue) and [LCoII]2+ square pyramidal (magenta) geometries (bottom). Pump-probe time delay scans recorded at 7720 eV for (B) dyad I and (C) dyad II, reflecting the formation and decay of the CoII photo-induced species. Kinetic fits T1 and T2 of the CoII formation and decay processes are shown. | ||
 | ||
| Fig. 3 Experimental difference spectra (laser on-laser off) corresponding to the 3MLLCT signals of (A) [Ir(ppy)2-L] and (B) [Ir(piq)2-L] in dyad I and II respectively. Pump-probe time delay scans recorded at 11.2 keV of (A inset) [Ir(ppy)2-L] in dyad I and (B inset) [Ir(piq)2-L] in dyad II corresponding to the decay of the 3MLLCT. Kinetic fits of the 3MLLCT state's reaction with TEOA (T1) with the excited-state relaxation time (T2) for [Ir(piq)2-L] due to inefficient quenching are indicated. | ||
Upon light excitation, mixed MLLCT from the 5 d orbital of Ir(III) to the π* orbital of the ligand followed by intersystem crossing (ISC) to a triplet excited state initially occurs.5 This process is followed by electron transfer from the excited photosensitizer to the CoIII complex, forming a CoII species. Fig. 2A shows the Co tr-XAS spectrum of both supramolecular assemblies at an averaged time delay of 11.3 μs between laser and X-ray pulses. A prominent peak at 7720 eV together with a broad dip at 7735 eV is obtained which corresponds to the formation of the reduced CoII species6 and the ground state bleaching of the CoIII, respectively. These energy transitions in turn show that the K-edge of the Co centre shifts to lower energy, indicating the reduction of CoIII and confirming formation of CoII through electron transfer from the Ir-based photosensitizers. Interestingly, clear changes are not only observed in the oxidation state of the Co catalytic metal site but also in its electronic configuration and coordination environment as indicated by the pre-edge region of the absorption spectrum at lower photon energies around 7710 eV (Fig. 2A inset, red trace).
Upon laser excitation, the occupation of the Co d shell changes from d6 to d7. The d level atomic rearrangements are accompanied by structure relaxation and hybridization of valence 3d states with N/O ligand p-orbitals.7 It should be mentioned that the fractions of CoII excited state between the laser pump excitation and X-ray probing were too low (less than 3%) to reconstruct the EXAFS and structural conformations of the excited states. Tr-XANES spectra were thus combined with TD-DFT XANES simulations to extract the electronic and geometric information about the photo-generated CoII (Fig. 2A inset). Several possible geometries of the intermediate CoII structure were theoretically explored. As shown from Fig. 2A inset, there is poor agreement between the experimental spectrum and theoretical simulation for a Co(II) square bipyramidal structure without any coordinated solvent molecules and an octahedral complex with a bonded acetonitrile molecule (compare red, blue and magenta traces). By contrast, the simulations for a distorted octahedral geometry with an elongated chlorido molecule at 2.4 Å resolve the pre-edge feature well (red trace) confirming the formation of a CoII derivative with a loosely coordinated chloride in both supramolecular assemblies upon photoexcitation.
The kinetics for the formation and decay of the CoII species were further investigated in both supramolecular assemblies by fixing the photon energy to 7720 eV, corresponding to the formation of CoII, and varying the time delays between the laser and X-ray pulses (Fig. 2B and C and Fig. S4, ESI†). The formation and decay kinetics for the CoII species are around the same in both dyads appearing promptly within 964 ± 154 ns and 810 ± 278 ns respectively (Fig. 2B and C), and decaying within 24.20 ± 3.70 μs and 26.80 ± 7.28 μs. Interestingly the Co transient signal of dyad I is around a factor of two greater than for dyad II, indicating more efficient formation of the CoII reduced species (Fig. 2B and C).
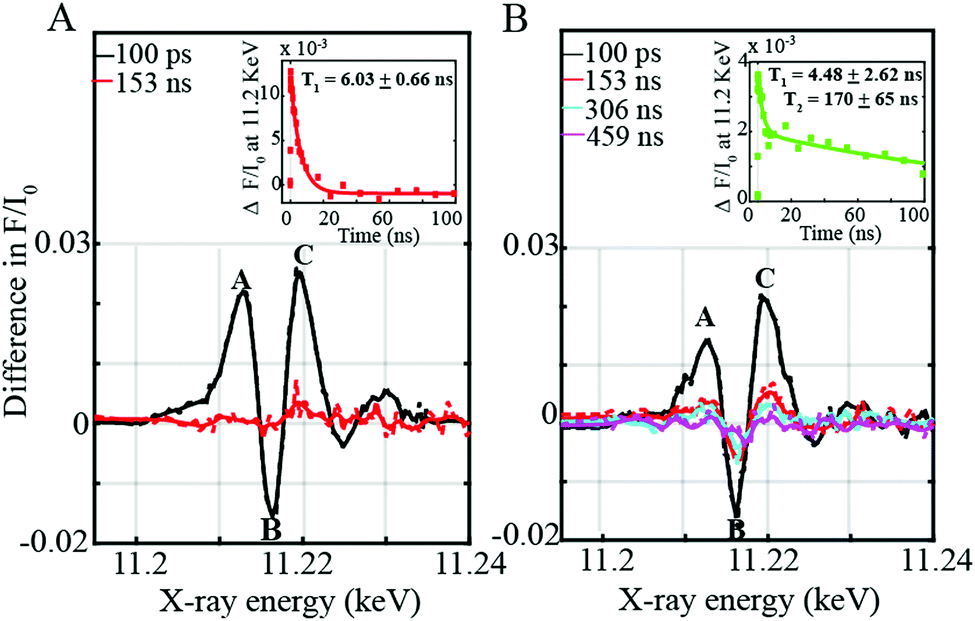
In order to investigate this process further, time-resolved XANES were subsequently performed to elucidate the photo-induced electron transfer dynamics of the supramolecular assemblies at the Ir L-edge site. Three prominent features I, II, and III at energies of 11213, 11216 and 11220 eV respectively are observed (Fig. 3A and B). Feature I (Fig. 3A and B) corresponds to a 2p to Ir unoccupied t2g state arising after electron transfer to the ligand.8 Features II and III (Fig. 3A and B) by contrast appear due to an increase in the energy of the XANES white line upon photoexcitation. As previously corroborated through DFT calculations,8 both the Ir 5d eg and 2p states move towards lower energies in the photo-excited configuration of the triplet state. The shift of the 2p levels is however more significant than that of the 5d states resulting in an increase in the XANES white line8 of the Ir-3MLLCT state compared to the ground state thereby leading to a more positive feature III with a lower minimum feature II (Fig. 3A and B).
On the one hand, the 3MLLCT state of the [Ir(ppy)2-L] within the Ir–Co dyad I appears promptly within the 100 ps pulse duration of the X-ray and decays within 6.03 ± 0.66 ns (Fig. 3A inset). On the other hand, the transient spectrum of the [Ir(piq)2-L] in Ir–Co dyad II persists within 100 ps to 459 ns time scales (Fig. 3B inset) and display 2 decay lifetimes of 4.48 ± 2.62 ns and 170 ± 65 ns. The Ir-based photosensitizers were further studied with TEOA in the absence of the Co-based catalyst and tetrafluoroboric acid, and similar time scales within error bars were obtained (Fig. S5, ESI†). Two electron-transfer pathways can exist for the photocatalytic cycle of the Ir–Co based supramolecular assemblies: (1) an oxidative quenching of the Ir photosensitizer 3MLLCT state by the Co catalyst (2) a reductive quenching of the Ir photosensitizer by TEOA followed by electron transfer to the Co-based complex (Scheme 2). However, the oxidative quenching pathway can be ruled out as the Co-based catalyst is formed at much longer time scales of 810–960 ns than the decay of the Ir-based 3MLLCT states as illustrated in Fig. 2B and C. Moreover, in the presence of excess TEOA employed as a sacrificial electron donor, the predominant pathway lies in the reductive quenching of the Ir 3MLLCT. The decay lifetime T1 of 4.48–6.02 ns observed in both dyads consequently corresponds to the formation of the reduced 3MLLCT state as illustrated in Scheme 2. Importantly, the 2nd decay lifetime T2 of 170 ± 65 ns obtained for the [Ir(piq)2-L] photosensitizer in both the absence and presence of the Co-based catalyst corresponds to the decay to the Ir(III) ground state due to inefficient quenching. This is further corroborated by the low excited state fraction obtained for the Ir–Co dyad II. Around 50% of the [Ir(piq)2-L] recombines back to the ground state configuration while only the other 50% participates in the efficient electron transfer reaction to the CoIII catalyst to generate the photo-reduced CoII species (Fig. 2C).
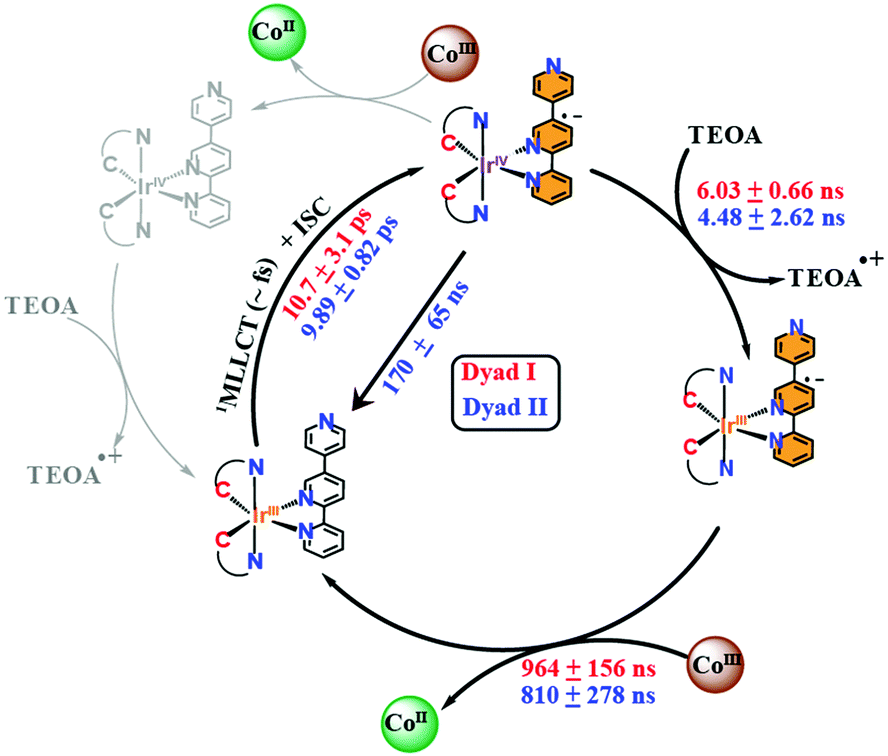 | ||
| Scheme 2 Mechanistic pathways of Ir–Co dyads I and II. Rate constants for dyads I and II are respectively shown in red and blue. | ||
Finally, limited by the 100 ps pulse duration of the X-rays, the ISC time frame from the singlet to the triplet excited states as well as the decay of the 3MLLCT states of the Ir-based photosensitizers within both dyads, were determined through OTA from 1 ps to 2 ns time scales (Fig. S6, ESI†).
Interestingly, [Ir(ppy)2-L] and [Ir(piq)2-L] display similar ISC times within experimental errors, i.e. 10.7 ± 3.1 ps and 9.89 ± 0.82 ps respectively (Fig. S6A and B, ESI†). Similarly, the 3MLLCT states for [Ir(ppy)2-L] and [Ir(piq)2-L] in dyads I and II decay within 7.74 ± 1.71 ns and 6.68 ± 0.19 ns (Fig. S6A and B, ESI†) respectively, consistent with the decay lifetimes of the 3MLLCT states of the Ir-based photosensitizers obtained through tr-XAS (Fig. 3A and B inset).
Despite their similarities, [Ir(ppy)2-L] and [Ir(piq)2-L] bear different electron densities leading to their varying photophysical properties. Previous TD-DFT calculations4 has showed that the lowest triplet excited state for [Ir(ppy)2-L] is a 3MLLCT state while that for [Ir(piq)2-L] consists of a combination of C^N centred and 3LC characters. The increased contribution of the 3LC transition on the excited state conformation of [Ir(piq)2-L] leads to a lesser efficient 3MLLCT state and a lower electron density in the excited state of the pendant pyridine moiety thus causing rapid decay to the ground state as revealed from tr-XAS measurements conducted in the presence and absence of the Co catalyst (Fig. 3B inset and Fig. S5B, ESI†). Hydrogen evolution measurements previously carried out4 under similar experimental conditions as tr-XAS with triethanolamine as sacrificial electron donor and tetrafluoroboric acid as the proton source further showed that dyads I and II displayed maximum turnover frequency (TOF) of 180 and 58 under green light excitation thus corroborating the decreased efficiency of [Ir(piq)2-L].
In summary, combined OTA and tr-XAS measurements at Co K-edge and Ir L-edge energies with DFT calculations were systematically employed to evaluate the influence of the 2-phenylpyridine and 2-phenylisoquinoline ligands on Ir-based photosensitizers when chelated to a Co-based hydrogen evolution catalyst (Scheme 2). Time-resolved XANES with TD-DFT simulations at the Co K-edge shows that upon photoexcitation, the CoIII octahedral catalyst is reduced to a CoII species and undergoes a Co–Cl bond elongation (Fig. 2A). We additionally monitored the rise time kinetics of the CoII intermediate in both dyads to be 810–964 ns followed by its decay to form the starting CoIII complex within 24.2–26.8 μs (Fig. 2B and C). The study of the supramolecular assemblies at the Ir L-edge showed the prompt formation of an Ir 3MLLCT state within the 100 ps pulse duration of the X-rays in both dyads (Fig. 3A and B). Further, combined kinetics studies through OTA and Ir L-edge tr-XAS conclusively reveal that both Ir-based photosensitizers in dyads I and II have similar ISC times of 9.89–10.7 ps and are reductively quenched by TEOA within 4.48–7.74 ns (Fig. 3A and B inset and Fig. S6, ESI†). Importantly, [Ir(piq)2-L] in dyad II is found to be less efficient than its [Ir(ppy)2-L] counterpart in dyad I as it not only displays decay to the IrIII ground state within 170 ± 65 ns (Fig. 3B and 2C) but additionally illustrates 50% less electron transfer efficiency to the CoIII catalyst to generate the photoreduced species. This study provides important insights on the reaction pathways and efficiencies of a family of highly versatile supramolecular Ir–Co dyads and holds future promise for the improved developments of both Ir-based and earth-abundant photosensitizers with lesser conjugated cyclometalated ligand derivatives for photocatalytic applications.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. G. Nocera, Acc. Chem. Res., 2011, 45, 767–776 CrossRef PubMed
.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed
- J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995–2004 CrossRef CAS PubMed
- C. Lentz, O. Schott, T. Auvray, G. S. Hanan and B. Elias, Dalton Trans., 2019, 48, 15567–15576 RSC
- R. Bevernaegie, S. A.-M. Wehlin, B. Elias and L. Troian-Gautier, ChemPhotoChem, 2021, 5, 217–234 CrossRef CAS
- D. Moonshiram, C. Gimbert-Suriñach, A. Guda, A. Picon, C. S. Lehmann, X. Zhang, G. Doumy, A. M. March, J. Benet-Buchholz, A. Soldatov, A. Llobet and S. H. Southworth, J. Am. Chem. Soc., 2016, 138(33), 10586–10596 CrossRef CAS PubMed
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS
- G. Smolentsev, K. M. van Vliet, N. Azzaroli, J. A. van Bokhoven, A. M. Brouwer, B. de Bruin, M. Nachtegaal and M. Tromp, Photochem. Photobiol. Sci., 2018, 17, 896–902 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, EXAFS fit analysis, XAS kinetic analysis, and results of DFT calculations. See DOI: https://doi.org/10.1039/d2cc02286a |
| This journal is © The Royal Society of Chemistry 2022 |