
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective methane photooxidation into methanol under mild conditions promoted by highly dispersed Cu atoms on crystalline carbon nitrides†
Marcos A. R.
da Silva
 ab,
Jéssica C.
Gil
ab,
Nadezda V.
Tarakina
c,
Gelson T. S. T.
Silva
ab,
José B. G.
Filho
d,
Klaus
Krambrock
e,
Markus
Antonietti
c,
Caue
Ribeiro
b and
Ivo F.
Teixeira
*ac
ab,
Jéssica C.
Gil
ab,
Nadezda V.
Tarakina
c,
Gelson T. S. T.
Silva
ab,
José B. G.
Filho
d,
Klaus
Krambrock
e,
Markus
Antonietti
c,
Caue
Ribeiro
b and
Ivo F.
Teixeira
*ac
aDepartment of Chemistry, Federal University of São Carlos, 13565-905, São Carlos, SP, Brazil. E-mail: ivo@ufscar.br
bNational Nanotechnology Laboratory for Agriculture, Brazilian Agriculture Research Corporation (Embrapa), XV de Novembro Street – 1452, São Carlos, Brazil
cDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, D-14476 Potsdam, Germany
dDepartment of Chemistry, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais 35400-000, Brazil
eDepartment of Physics, Federal University of Minas Gerais, 31270-901, Belo Horizonte, Minas Gerais, Brazil
First published on 31st May 2022
Abstract
Here we report a photocatalytic system based on crystalline carbon nitrides (PHI) and highly dispersed transition metals (Fe, Co and Cu) for controlled methane photooxidation to methanol under mild conditions. The Cu-PHI catalyst showed a remarkable methanol production (2900 μmol g−1) in 4 hours, with a turnover number of 51 moles of oxygenated liquid product per mole of Cu. To date, this result is the highest value for methane oxidation under mild conditions (1 bar, 25 °C).
Primarily found in natural gas, methane (CH4) is the most abundant existing hydrocarbon, and it is often used as a heat source and energy carrier, and in the steam reforming process.1,2 Conventional and unconventional methane reserves, especially in their hydrate form, are extensive and could supply global energy demand, with an estimated volume twice that of other fuels.3 At the same time, methane is a greenhouse gas with a global warming potential 25 times higher than carbon dioxide (CO2).1 Thus, methane conversion to other chemical products is essential to avoid emissions and exploit its vast reserves. Currently, methane conversion involves high-temperature processes to produce syngas. Liquid products, such as methanol, are made under high pressures through the Fischer-Tropsch process.1 Direct methods are necessary, without the syngas intermediate, especially those that proceed under lower temperatures and pressures saving energy and resources. Several previous studies attempted to convert methane into methanol with different oxidants, e.g., H2O2, O2 and NO. Nevertheless, low concentrations of products are obtained in ambient conditions, and harsh conditions (high temperatures and/or pressures) are required to obtain high yields of oxygenated liquid products.
Photocatalysis is a pivotal pathway to overcome the energy barrier of challenging oxidation reactions, using light to promote electrons from reagents or catalysts.2,4 Much effort has been recently invested in synthesising photocatalysts to convert methane into methanol. However, the best performances reported in the literature still are under high pressures or UV-excited photocatalysts (TiO2).5–7 Carbon nitrides are suitable photocatalysts with desirable features, i.e., low cost and active under visible light, adequate for solar light utilisation. Some reports have applied polymeric carbon nitrides in methane oxidation with elevated production of oxygenated liquids,8–10 but these materials lack crystallinity and reproducibility, it being difficult to control and understand the metal site role. A family of carbon nitride ions was recently discovered through a synthetic procedure with C/N precursor and alkali chlorides.11 Our group have synthesised crystalline carbon nitride, in its poly(heptazine imide) form (PHI), through thermal treatment of melamine and NaCl, yielding Na-PHI. These sodium cations can be exchanged with other elements, and it was verified that transition metals could coordinate in the carbon nitride structure bonded to N atoms.12 This step enables the control of metal sites and boosts the photocatalytic activity per mole of metal.
Here we synthesised high crystalline carbon nitrides with long 2D extension (Na-PHI). Through exchanging the Na cations, different first-row transition metals (i.e., Fe, Co and Cu) were stabilized in the CN scaffold. The obtained materials were investigated in methane photooxidation reactions. We observed that Cu-PHI has an extraordinary activity for oxygenated liquid products (especially methanol). The production of methanol in this study (2900 μmol g−1) is, to the best of our knowledge, the highest value for this reaction under mild conditions (1 bar, 25 °C).
Synthesis of crystalline carbon nitride was performed through thermal treatment of melamine with sodium chloride (NaCl), as reported by Chen et al.11 In this case, a chemical reaction between melamine and the salt results in a highly crystalline material formed by poly(heptazine imide) units (PHI). This as-prepared material was called Na-PHI due to the stabilised Na+ ions between the layers of PHI. Crystalline Na-PHI was employed as a scaffold for coordinating transition metal ions (Fig. S1, ESI†). Furthermore, Na+ is replaced by metal ions (i.e. Cu2+, Co2+ and Fe3+). For instance, an aqueous suspension of Na-PHI and the metal chloride precursor are mixed.13,14 After the metal coordination, the material was washed, dried and named M-PHI (M stands for the metal symbol).
The XRD patterns (Fig. 1a) show that the majority of the reflections present in the Na-PHI structure are retained in all M-PHI samples, indicating that the cation exchange reactions do not considerably change the structure of carbon nitride. The main difference between the XRD patterns is the intensity distribution of the peaks in the 2θ = 20–30° range, reflecting changes in the interactions between PHI layers, depending on the size and charge of transition metal cations. The peak between 45° is related to the NaCl crystal lattice, which disappears after metal addition. HRTEM images of the Na-PHI, Fe-PHI and Cu-PHI samples confirm the high crystallinity of the obtained compounds (Fig. S2, ESI†). Further evidence of structure preservation comes from the Raman spectra in Fig. S3 (ESI†), revealing the characteristic bands from the heptazine units (Table S1, ESI†) with the primary breathing mode at 733 cm−1, and the strong intensity of this band indicates the long 2D extension. Infrared spectroscopy first revealed the coordination of metal atoms in carbon nitride (Fig. S4, ESI†), whereby shifts in wavenumbers are observed after metal insertion. UV-Vis diffuse reflectance (Fig. S5, ESI†) and Mott–Schottky analyses provide the materials' band potentials (Fig. S6, ESI†). We noticed a shift to positive potentials in the following order: Na < Fe < Cu. In addition, Co-PHI shows a pronounced shift to negative potentials, indicating that this photocatalyst has a different oxidation ability than the others.
 | ||
| Fig. 1 (a) Powder XRD patterns of M-PHI; (b) distribution of products of methane photooxidation with Na, Co, Fe and Cu-PHI (10% of metal loading); (c) influence of metal loading in Cu-PHI samples for methanol production; (d) methane conversion in different Cu loadings in PHI. | ||
Methane photooxidation reactions were carried out in a quartz tube under visible-light irradiation. All tests were performed during 4 hours. The distribution of formed products (gaseous and liquid phases) is exhibited in Fig. 1b–d. The nature of the metal sites has strongly influenced the catalyst photoactivity. Na-PHI does not produce any methanol or even CO2, indicating that the band structure is not active for CH4 oxidation. However, all the modified M-PHIs displayed better photoactivities (Fig. 1c), showing that the presence of the active metal site is critical for methane oxidation. Between M-PHIs, Co-PHI exhibits the lowest activity, possibly due to the position of its energy bands (Fig. S6, ESI†), which has a lower oxidation potential. Cu-PHI and Fe-PHI showed the highest methane conversions. However, Fe-PHI produces significant CO2 quantities, implying that this photocatalyst promotes methane overoxidation. Cu-PHI, in contrast, generates high amounts of methanol and other C2 products, while yields negligible evolution of carbon dioxide.
The copper loading influence was evaluated for methanol production (Fig. 1c), whereby an increasing generation from 10% to 0.5% of Cu is noted. This indicates that we can significantly improve the methanol yield using only 0.5% of metal. For Cu-PHI 0.5%, we obtained an impressive methanol production of 2900 μmol g−1, and that result is the best performance of a heterogeneous catalyst reported in the literature so far (Table S2, ESI†). It is worth highlighting that the total oxygenated liquid production of Cu-PHI 0.5% is even more stunning, with an activity of 4060 μmol g−1 (Fig. S7a, ESI†) and a remarkable turnover number of 51 moles of liquid oxygenate products per mole of Cu. In all Cu-PHI samples, CO2 evolution is negligible (Table S3, ESI†), while methanol and formaldehyde are the only C1 products. Interestingly, when the loading of Cu drops to 0.1%, the methanol yield dramatically falls (220 μmol g−1), indicating that a maximum is achieved with 0.5% of copper.
Methane conversion in photooxidation tests with Cu-PHI was also calculated (see ESI† for details) (Fig. 1d). Our best catalyst (Cu-PHI 0.5%) led to a methane conversion of 11%, an excellent result under mild conditions (1 bar and 25 °C) and low concentrations of hydrogen peroxide.
The influence of the metal support was also discussed (Fig. S7b, ESI†). PHI is suitable to stabilise Cu species, which can selectively convert methane into oxygenated liquids. Tests without light and in the absence of H2O2 were performed to investigate the effect of the reaction conditions on methanol yield (Fig. S7c, ESI†). The reaction without H2O2 yields 410 μmol G−1 of methanol, indicating that hydrogen peroxide despite being important to promote the reaction is not essential. At the same time, light seems to be a crucial factor in activating methane molecules since only 130 μmol g−1 of methanol is formed without visible light irradiation. Hydrogen peroxide is not excited by visible light, and methanol production without catalyst does not occur (Fig. S7c, ESI†).
We conducted experiments with an atmosphere of CO2 (Fig. S7d, ESI†) to confirm that methanol is produced from methane oxidation rather than CO2 reduction. A small methanol production of 140 μmol g−1 was observed, demonstrating that most methanol is actually from methane oxidation. The possibility of homogenous catalysis is excluded when we compare the metal loading of Cu-PHI before and after the reaction (Table S4, ESI†). The Cu amount remains unaltered even after 3 cycles. In addition, the stability of the photocatalyst was verified, and after 4 cycles, methanol production does not change (Fig. S8, ESI†).
The chemical composition of each material reveals the origin of Fe and Cu-PHI activity. Fig. S9 (ESI†) shows the high-resolution XPS spectra for M2p of the corresponding metals. The XPS analyses of Cu 2p3/2 (Fig. S9a, ESI†), and Fe 2p3/2 (Fig. S9b, ESI†) indicate that metal-N bonds are predominant in these catalysts, with binding energies of 935.1 eV and 711 eV for Cu–N15 and Fe–N16 bonds, respectively.
The presence of metal-N bonds along PHI indicates a high dispersion of active sites. TEM images confirm the dispersion of Cu and Fe species (Fig. 2) in Cu-PHI and Fe-PHI, respectively. A better distribution of metal atoms in the material enables a more specific interaction between reactants and catalysts.
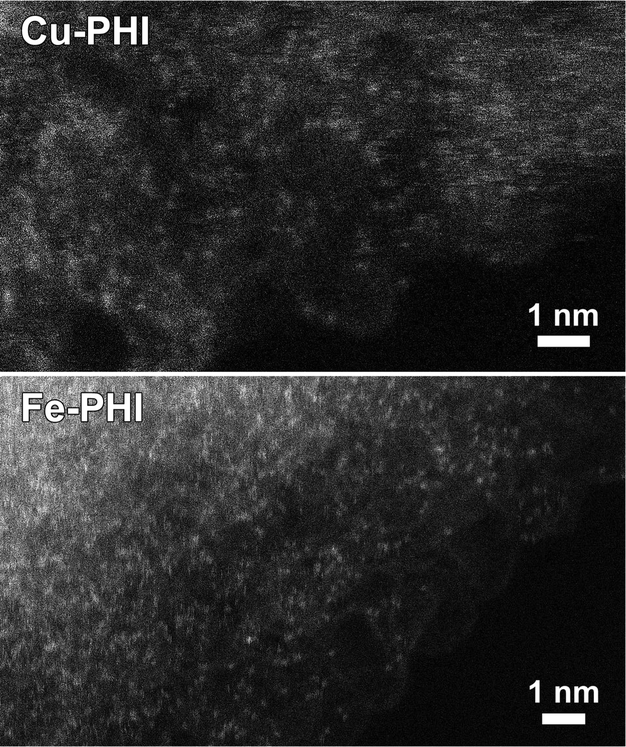 | ||
| Fig. 2 Annular dark-field scanning transmission electron microscopy images of Cu-PHI and Fe-PHI, where bright dots correspond to highly dispersed metal atoms. | ||
We perform time-resolved photoluminescence and Raman analyses to investigate the interaction between metal sites and hydrogen peroxide molecules. Fluorescence lifetimes (TR-PL) of the photocatalysts are shown in Table S5 (ESI†). An intriguing feature occurs when H2O2 is added: while all catalysts present higher lifetimes, Cu-PHI with hydrogen peroxide has a shorter lifetime than Cu-PHI. This result shows that nonradiative decay may be opened through the electron transfer from Cu atoms to hydrogen peroxide, suppressing electron-hole recombination.17 Thus, a specific interaction between Cu and H2O2 is observed.
In the presence of Cu-PHI with H2O2 and light, Raman spectroscopy analyses indicate the influence of these components on the material structure. Fig. S10a and b (ESI†) show the Raman of Cu-PHI 0.5% with H2O2 and H2O2 under light, respectively. We noticed significant differences mainly at 200–800 cm−1 (Fig. 3a). The band at 707 cm−1 increases in intensity in the presence of peroxide, indicating a break in the symmetry of the heptazine groups present in carbon nitride. This outcome shows that a change in the Cu chemical environment occurs when peroxide is added. The region of 450–500 cm−1 has a significant change. Three bands are shown in the spectrum: the central band at 471 cm−1 is present in the initial spectrum and is related to in-plane ring torsion. Its increase corroborates the proposition that a change in symmetry occurs within the heptazine rings. Furthermore, two new bands in this region, 455 and 487 cm−1, are related to Cu–OH bonds (Fig. 3a),18,19 suggesting that hydroxyl groups are connected to the catalyst. The region between 800–900 cm−1 (Fig. S10a and b, ESI†) has not changed with the addition of peroxide. This region is characteristic of O–O bonds,20 indicating that peroxo groups are not linked to Cu. It agrees with NMR results, which did not identify CH3OOH species, commonly reported in CH4 oxidation with H2O2.21
 | ||
| Fig. 3 (a) Raman spectroscopy analyses of Cu-PHI 0.5% in the presence of H2O2 and light at 380–580 cm−1; (b) EPR spectra of DMPO adducts in water with H2O2 in the presence and absence of visible light radiation for Cu-PHI 0.5%; (c) experimental and calculated EPR spectra of spin adducts of DMPO under visible light using Cu-PHI 0.5%; (d) EPR spectra of the PBN-methoxy adduct at the point of highest intensity for both Cu-PHI 0.5% and Fe-PHI photocatalysts (5 min illumination). | ||
EPR experiments (Fig. 3(b and c) and Fig. S11–13, ESI†) allied with the spin trapping method investigate the radical formation in the photocatalytic system. Cu-PHI and Fe-PHI show distinct behaviours; while Cu-PHI 0.5% produces high amounts of OH* radicals (Fig. 3b and Fig. S11c, ESI†) detected by the formation of the hydroxyl adduct of DMPO in water, Fe-PHI generates oxygen superoxide (O2˙−) species (Fig. S12b, ESI†) seen by the formation of the DMPO-O2˙− adduct in oxygenated acetonitrile. The kinetic curves as a function of illumination OH time are shown in Fig. S12c and d (ESI†), respectively. The formation of a high amount of O2˙− radicals is intricately related to overoxidation of methane products,5 which explains carbon dioxide evolution with Fe-PHI. On the other hand, Cu-PHI yields more ˙OH radicals than Fe-PHI (Fig. S12c, ESI†), according to EPR, explaining the higher yield in methanol production. In addition, oxygenate liquids, such as methanol, can be found in the solution for prolonged periods, as described by EPR assays with methanol and PBN,22 which detects the adduct methoxy-PBN only for Cu-PHI (Fig. 3d and Fig. S13, ESI†). This feature explains the high production of methanol with Cu-PHI.
EPR experiments in the solid state at different temperatures indicate that Cu ions are in the (2+) charge state with nearly axial spin Hamiltonian parameters (Fig. S14, ESI†). Such parameters are consistent with a nearly axially elongated symmetry. Also, under illumination the line intensity is slightly reduced, probably by forming Cu+ species, that return to the (2+) ground state immediately after the light is turned off. All these results suggest a rapid transfer of photogenerated electron charges to the solution, as depicted in photoluminescence results.
According to the obtained results, adsorption of hydroxyl groups occurs on the Cu atom, distorting the PHI symmetry due to the increase in the coordination number and the unpaired electrons of oxygen. Cu atoms have a unique interaction with peroxide molecules, creating Cu–OH bonds on the catalyst's surface.18,19 This process is promoted by electrons, as shown in Fig. 4(I). At the same time, the formation of Cu–OH from H2O2 by one-electron transfer leads to the formation of hydroxyl radicals (as evidenced by EPR), which are commonly responsible for activating methane in radical-based mechanisms (Fig. 4(II)).9 Considering that the concentration of oxygenated products (203 μmol) is greater than the amount of peroxide (80 μmol), it is reasonable to assume that the oxidation of water to H2O2 occurs. Thus, hydrogen peroxide generation seems to be controlled, avoiding overoxidation to CO2 by an excess of hydroxyl radicals. Also, carbon nitride structure can adsorb methyl radicals and transfer them to Cu–OH, producing methanol. C2 products are formed when two or more methyl radicals adsorb onto the catalyst surface.9
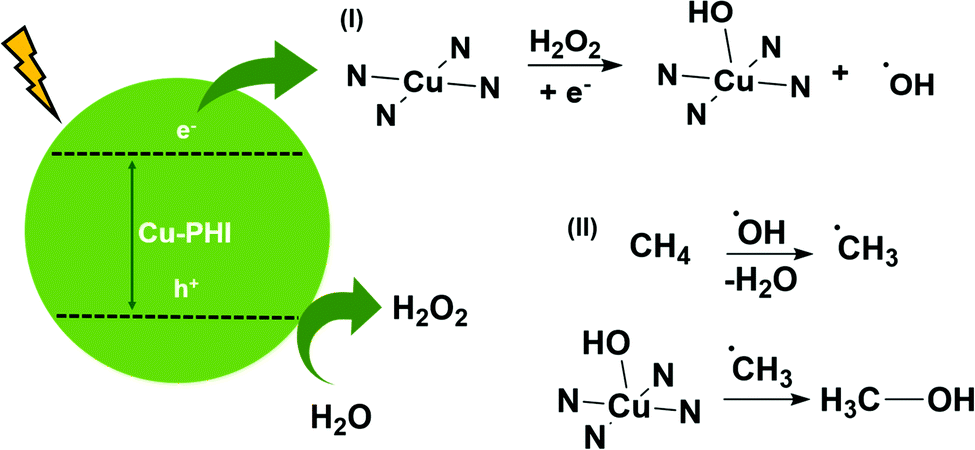 | ||
| Fig. 4 Proposed mechanism of methane photo-oxidation by the Cu-PHI catalyst. | ||
In summary, we synthesized highly crystalline poly(heptazine imides) (PHI) compounds with first-row transition metals (i.e. Fe, Co, and Cu) coordinated to the carbon nitride framework. These visible-light-active photocatalysts were employed on methane oxidation reactions with hydrogen peroxide, whereby Cu-PHI generates high amounts of oxygenated liquids, especially methanol (2900 μmol g−1). EPR experiments in the absence of water showed that H2O2 interaction with Cu-PHI primarily leads to OH radicals (started by H2O2 reduction) while oxygen superoxide (O2˙−) radicals are present in Fe-PHI (by H2O2 oxidation), promoting CO2 evolution. Raman analyses indicate that hydroxyl groups are bonded to Cu sites (Cu–OH). These groups are highly specific for methane conversion and can produce oxygenated liquids, such as methanol. This result is the best performance reported so far for methane conversion reaction under mild conditions (1 bar, 25 °C).
This research was financially supported by CAPES, CNPq and FAPESP. Dr Carlos Ospina Ramirez and LNNano are greatly acknowledged for their support with TEM data collection and EDX mapping.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- C. Sahu, R. Kumar and J. S. Sangwai, Energy Fuels, 2021, 35, 11740–11760 CrossRef CAS.
- H. Song, X. Meng, Z.-J. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar and D. Ma, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- Y. Fan, W. Zhou, X. Qiu, H. Li, Y. Jiang, Z. Sun, D. Han, L. Niu and Z. Tang, Nat. Sustainability, 2021, 4, 509–515 CrossRef.
- H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507–20515 CrossRef CAS PubMed.
- B. Wu, R. Yang, L. Shi, T. Lin, X. Yu, M. Huang, K. Gong, F. Sun, Z. Jiang and S. Li, Chem. Commun., 2020, 56, 14677–14680 RSC.
- Y. Zhou, L. Zhang and W. Wang, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- Z. Yang, Q. Zhang, L. Ren, X. Chen, D. Wang, L. Liu and J. Ye, Chem. Commun., 2021, 57, 871–874 RSC.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- M. A. da Silva, I. F. Silva, Q. Xue, B. T. Lo, N. V. Tarakina, B. N. Nunes, P. Adler, S. K. Sahoo, D. W. Bahnemann and N. López-Salas, Appl. Catal., B, 2022, 304, 120965 CrossRef CAS.
- F. M. Colombari, M. A. R. da Silva, M. S. Homsi, B. R. L. de Souza, M. Araujo, J. L. Francisco, G. T. S. T. da Silva, I. F. Silva, A. F. de Moura and I. F. Teixeira, Faraday Discuss., 2021, 227, 306–320 RSC.
- I. F. Teixeira, N. V. Tarakina, I. F. Silva, N. López-Salas, A. Savateev and M. Antonietti, Adv. Sustainable Syst., 2022, 6, 2100429 CrossRef CAS.
- W. Li, C. Min, F. Tan, Z. Li, B. Zhang, R. Si, M. Xu, W. Liu, L. Zhou and Q. Wei, ACS Nano, 2019, 13, 3177–3187 CrossRef CAS PubMed.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang, J. Li, S. Jiao, Y. Kuang and S. Luo, Adv. Funct. Mater., 2021, 31, 2011289 CrossRef CAS.
- C. Li, Y. Du, D. Wang, S. Yin, W. Tu, Z. Chen, M. Kraft, G. Chen and R. Xu, Adv. Funct. Mater., 2017, 27, 1604328 CrossRef.
- G. Niaura, Electrochim. Acta, 2000, 45, 3507–3519 CrossRef CAS.
- H. Y. H. Chan, C. G. Takoudis and M. J. Weaver, J. Phys. Chem. B, 1999, 103, 357–365 CrossRef CAS.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS PubMed.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock and S. H. Taylor, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- B. Jose Filho, R. D. Rios, C. G. Bruziquesi, D. C. Ferreira, H. F. Victoria, K. Krambrock, M. C. Pereira and L. C. Oliveira, Appl. Catal., B, 2021, 285, 119814 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc01757a |
| This journal is © The Royal Society of Chemistry 2022 |