
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stereoselective hydride transfer reaction with contributions from attractive dispersion force control†
D. Christopher
Braddock
 *,
Natnicha
Limpaitoon
,
Krzysztof
Oliwa
,
Daniel
O’Reilly
,
Henry S.
Rzepa
* and
Andrew J. P.
White
*,
Natnicha
Limpaitoon
,
Krzysztof
Oliwa
,
Daniel
O’Reilly
,
Henry S.
Rzepa
* and
Andrew J. P.
White
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK. E-mail: c.braddock@imperial.ac.uk; h.rzepa@imperial.ac.uk; Tel: +44(0)2075945772
First published on 24th March 2022
Abstract
The experimentally determined stereochemical outcome of an unprecedented hydride transfer from a lithium alkoxide to an aldehyde is reported, as deconvoluted by the combined use of a single enantiomer alkoxide in conjunction with a deuterium label. The stereoselective outcome is consistent with a computationally predicted transition state model stabilised by contributions from attractive dispersion forces.
Recently, we reported the addition of the lithium acetylide of alkyne 1 to adamantane carboxaldehyde 2 unexpectedly produced quantities of ynone 3 and adamantane methanol 4 (Fig. 1).1 We invoked a hydride transfer in a six-membered chair-like transition state (TS A) from the initially formed secondary lithium alkoxide AdCH(C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)OLi to still unreacted AdCHO to explain this observation. In line with this explanation, isolation of the expected addition product 5, and reformation of the lithium alkoxide in the presence of AdCHO 2 gave the same products.1 While the transfer of hydride from LiCCH (e.g., tBuLi)2 and LiNCH (e.g., LDA)3 motifs to carbonyl compounds are well precedented, to the best of our knowledge the above example constitutes the only known example of hydride transfer from a LiOCH atomic tetrad to an aldehyde.4 We had noted that this outcome could be linked to the particular structure of this secondary alkoxide whereby steric compression of the adamantyl bearing sp3 centre is simultaneously relieved as the formation of conjugated ynone 3 acts as a thermodynamic driving force. While these two factors seem intuitive to the organic chemist, and where we invoked TS A with the adamantyl groups of both components in equatorial positions, herein we provide computational and experimental evidence for a third factor at work: attractive dispersion forces that help stabilise a transition state that we initially formulated with the adamantyl grouping of the aldehyde as occupying an axial position (TS B).
CtBu)OLi to still unreacted AdCHO to explain this observation. In line with this explanation, isolation of the expected addition product 5, and reformation of the lithium alkoxide in the presence of AdCHO 2 gave the same products.1 While the transfer of hydride from LiCCH (e.g., tBuLi)2 and LiNCH (e.g., LDA)3 motifs to carbonyl compounds are well precedented, to the best of our knowledge the above example constitutes the only known example of hydride transfer from a LiOCH atomic tetrad to an aldehyde.4 We had noted that this outcome could be linked to the particular structure of this secondary alkoxide whereby steric compression of the adamantyl bearing sp3 centre is simultaneously relieved as the formation of conjugated ynone 3 acts as a thermodynamic driving force. While these two factors seem intuitive to the organic chemist, and where we invoked TS A with the adamantyl groups of both components in equatorial positions, herein we provide computational and experimental evidence for a third factor at work: attractive dispersion forces that help stabilise a transition state that we initially formulated with the adamantyl grouping of the aldehyde as occupying an axial position (TS B).
 | ||
| Fig. 1 Unexpected products of hydride transfer (boxed left). Transition states A and B (boxed right). | ||
Prior to undertaking experimental studies, transition state computational modelling was undertaken. DFT procedures were based on the following four models: (i) Li coordination with 2 explicit thf solvent molecules; 2thf@B3LYP + GD3BJ/Def2-SVPP/SCRF = thf; (ii) 2thf@B3LYP + GD3BJ/Def2-TZVPP/SCRF = thf; (iii) model (i) with 3thf and; (iv) model (ii) with 3thf (Fig. 2, top).5 These models are all consistent in predicting that using the lithium alkoxide of (S)-5 results in a transition state for the (S,Si)-diastereoisomer (TS B) for which ΔG‡298 is ∼0.3–0.8 kcal mol−1 lower than the (S,Re)-transition state diastereoisomer (TS A) using the 2thf solvent model and ∼3 kcal mol−1 lower (∼3.3 kcal mol−1@195K) using the 3thf solvent model;6 the dispersion stabilisation contribution (B3LYP + GD3BJ) for this latter value is ∼1.2 kcal mol−1, which suggests a significant contribution from dispersion effects. Non-covalent interaction (NCI) analysis6 indicates this originates at least in part from the region where the alkynyl-t-butyl and adamantyl (from the aldehyde) groups are proximate and which are absent from TS A (Fig. 2, bottom). Further inspection of the calculated transition state geometry for TS B – originally formulated as a chair-like transition state (c.f.Fig. 1) – reveals instead that the six atoms of the cycle are essentially coplanar. For TS A, a distorted half-chair conformation is evident, but also with significant co-planarity in the ring. In these situations, axial and equatorial descriptors clearly do not apply.‡ Notwithstanding the conformations adopted by each of the two transition state geometries as revealed by computations, we sought to devise an experiment that could discriminate between them.
 | ||
| Fig. 2 (top) Calculated geometries of TS A and TS B with three explicit THF solvent molecules showing the distorted half-chair and coplanar nature of the six atoms of the cycles respectively; (bottom) NCI plots derived from the total electron density (B3LYP + GD3BJ/Def2-TZVP) for TS A and TS B showing the weakly attractive regions (green) between the alkynyl-t-butyl and adamantyl groups in TS B (red circle), which are not present in TS A. See reference 6 for a clickable link to interactive 3D transition state models and NCI isosurfaces. | ||
To investigate this experimentally we proposed that transition states A and B could be differentiated by judicious combined use of a single enantiomer alcohol and a deuterium label (Fig. 3). Thus, we envisaged tracking the stereochemical course of reaction of single enantiomer deuterated alcohol 5-d (after deprotonation to the lithium alkoxide) with AdCHO 2 and/or the reaction of single enantiomer alcohol 5 (after deprotonation to the lithium alkoxide) with AdCDO 2-d.
 | ||
| Fig. 3 Proposed combined single enantiomer-deuterium labelling experiments to differentiate transitions states A and B. | ||
Accordingly, our studies commenced by the reduction of readily available adamantanecarboxyaldehyde 21 with inexpensive sodium borodeuteride to give monodeuterated alcohol (±)-4-d (Scheme 1).§ To our delight, simple reoxidation with pyridinium chlorochromate-silica7 provided aldehyde 2-d with high deuterium content evidently under the control of a primary kinetic isotope effect.8 To the best of our knowledge this tactic has not previously been reported in the literature for the preparation of deuterated aldehydes and it is our expectation that this should prove to be a general approach.9 Subsequent lithiation of terminal alkyne 1 and addition to aldehyde 2-d under the conditions reported for the formation of (±)-51 gave novel propargylic alcohol (±)-5-d.¶
 | ||
| Scheme 1 Synthesis of labelled propargylic alcohol (±)-5-d. | ||
Prior to investing effort in enantiomeric resolution of secondary alcohol (±)-5-d, a preliminary attempted deuteride transfer by treating (±)-5-d with n-BuLi and AdCHO 2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3, 0 °C to r.t., 16h), showed no evidence for the formation of alcohol (±)-4-d by deuteride transfer. Instead, non-deuterated alcohol 4 was observed as the major product in a complex mixture where all of aldehyde 2 had been consumed. We therefore invoke faster Tischenko-type hydride transfers (consuming the aldehyde) over the proposed and evidently KIE-retarded deuteride transfer.||
:1:3, 0 °C to r.t., 16h), showed no evidence for the formation of alcohol (±)-4-d by deuteride transfer. Instead, non-deuterated alcohol 4 was observed as the major product in a complex mixture where all of aldehyde 2 had been consumed. We therefore invoke faster Tischenko-type hydride transfers (consuming the aldehyde) over the proposed and evidently KIE-retarded deuteride transfer.||
Undaunted by the failure to transfer deuteride anion to AdCHO, attention turned to the other possible experiment, viz. transfer of hydride to AdCDO (cf., Fig. 3). Accordingly, derivatisation of previously reported propargylic alcohol (±)-51 with (S)-camphanic acid chloride gave the expected 1:1 mixture of diastereomeric esters 6 and 7 in high yield, from which ester 7 could be obtained directly as a single diastereoisomer by fractional crystallisation (Scheme 2) as confirmed by X-ray crystallography (Fig. 4).** Ester hydrolysis gave single enantiomer propargylic alcohol (S)-5, as confirmed by HPLC studies of its benzoate ester on a chiral stationary phase by comparison to a racemic sample.**
 | ||
| Scheme 2 Resolution of propargylic alcohol (±)-5via fractional crystallisation of camphanic esters 6 and 7 and subsequent hydrolysis. | ||
 | ||
| Fig. 4 X-Ray crystal structure of ester 7. | ||
Much to our delight, subsequent treatment of single enantiomer (S)-5 with n-BuLi and AdCDO 2-d allowed isolation of deuterated alcohol 4-d (Scheme 3).†† Isolated alcohol 4-d showed a small but negative optical rotation [−2.41, c 1.87, CHCl3; −1.39 ± 0.55σ (16 measurements), c 1.08, cyclopentane] allowing assignment as enriched in the (R)-enantiomer by comparison with the previously reported positive rotation for (S)-4-d (+0.21, c 2.33 in cyclopentane, e.e. 34 ± 5).10 This assignment as enriched in (R)-enantiomer of 4-d was also supported by measurement of the VCD spectrum and comparison with DFT calculations (B3LYP + GD3BJ/Def2-QZVPP/SCRF = CCl4)11 and also by (R)-O-methylmandelate derivatisation of alcohol 4, which showed a ca. 2:1 ratio of diastereomeric esters (R,R)-8 and (S,R)-9 for which the major isomer can also be assigned as (R,R)-8 on the basis of a previous assignment (Fig. 5 and 6).‡‡12
 | ||
| Scheme 3 Hydride transfer from single enantiomer (S)-5 to labelled aldehyde 2-d and derivatisation as (R)-O-mandelate esters 8 and 9. | ||
 | ||
| Fig. 5 1H NMR spectra of diastereomeric mandelate esters (R,R)-8 and (S,R)-9 showing the resolved AdCHDO2CCH(OMe)Ph protons in the 3.50–3.90 ppm region from (a, left) (±)-alcohol 4 and (b, right) hydride transfer derived alcohol 4. The additional peaks in the spectra (*) are diastereotopic methylene protons of AdCH2O2CCH(OMe)Ph. | ||
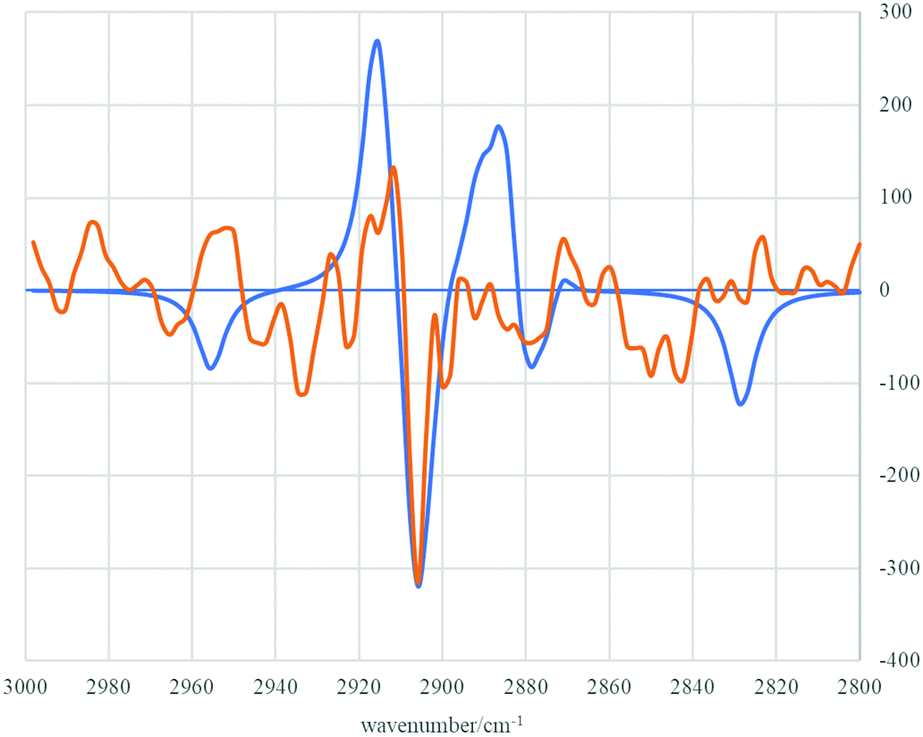 | ||
| Fig. 6 Experimental (orange line) and calculated (blue line) VCD spectra of (R)-4-d in the region 3000–2800 cm−1. | ||
The experimental observation of the (R)-4-d enantiomer configuration as the major isomer formed in this experiment is therefore consistent with transition state model TS B (Fig. 1). In turn, this is in agreement with our computations which predicted stabilising contributions from attractive dispersion forces operating between the tBu-alkynyl and adamantyl groups. That such attractive dispersion forces can play a role in controlling transition state assemblies and thence stereochemical outcome is notable and may be more widespread than is currently appreciated.13,14
NL, KO and HSR performed the calculations, KO and DO performed the synthetic procedures, DCB and HSR wrote the text and all authors edited to the final version.
We thank the Royal Society of Chemistry for a Undergraduate Research Bursary (to K. O.) and the EPSRC for a studentship (to D. O’R.).
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- D. C. Braddock, A. Mahtey, H. S. Rzepa and A. J. P. White, Chem. Commun., 2016, 52, 11219–11222 RSC.
- For an early example involving the reduction of hexamethyl acetone to di-tert-butyl carbinol with tBuLi see: P. D. Bartlett, C. G. Swain and R. B. Woodward, J. Am. Chem. Soc., 1941, 63, 3229–3230 CrossRef CAS.
- For reductions with lithium dialkylamines see: M. Majewski and D. M. Gleave, J. Organomet. Chem., 1994, 470, 1–16 CrossRef CAS.
- For hydride transfer from lithium alkoxides to perfluoroalkyl ketones see: (a) Y. S. Sokeirik, K. Sato, M. Omote, A. Ando and I. Kumadaki, J. Fluorine Chem., 2006, 127, 150–152 CrossRef CAS; (b) Y. S. Sokeirik, K. Sato, M. Omote, A. Ando and I. Kumadaki, Tetrahedron Lett., 2006, 47, 2821–2824 CrossRef CAS . For hydride transfer from lithium alkoxides to diaryl ketones see: ; (c) E. C. Ashby, A. B. Goel and J. N. Argyropolous, Tetrahedron Lett., 1982, 23, 2273–2276 CrossRef CAS.
- For all managed computational and experimental FAIR research data, including input and output files, see: D. C. Braddock, N. Limpaitoon, K. Oliwa, D. O’Reilly, H. S. Rzepa and A. J. P. White, Imperial College Research Data Repository, 2021, DOI: 10.14469/hpc/9237 and sub-collections referenced therein.
- Free energies, 3D transition state models and NCI (non-covalent-analysis) plots can be found at D. C. Braddock, N. Limpaitoon, K. Oliwa, D. O’Reilly, H. S. Rzepa and A. J. P. White, Imperial College Research Data Repository, DOI: 10.14469/hpc/9869.
- F. A. Luzzio, R. W. Fitch, W. J. Moore and K. J. Mudd, Angew. Chem., Int. Ed., 1999, 76, 974–975 CAS.
- This effectively constitutes an example of ‘Experiment C’ as in this excellent essay: E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- For an excellent overview of pre-existing methods of deuterated aldehyde synthesis, and for RCDO preparation directly from RCHO, see: H. Geng, X. Chen, J. Gui, Y. Zhang, Z. Shen, P. Qian, J. Chen, S. Zhang and W. Wang, Nat. Catal., 2019, 2, 1071–1077 CrossRef CAS PubMed.
- C. J. Reich, G. R. Sullivan and H. S. Mosher, Tetrahedron Lett., 1973, 17, 1505–1508 CrossRef.
- For measured and computed VCD spectra, see D. C. Braddock, N. Limpaitoon, K. Oliwa, D. O’Reilly, H. S. Rzepa and A. J. P. White, Imperial College Research Data Repository, 2022, DOI: 10.14469/hpc/9239 We thank Jordan Nafie, BioTools Inc, Florida, USA for the measurement (CCl4, 75000 scans, 72 h).
- S. H. Liggero, R. Sustmann, P. von and R. Schleyer, J. Am. Chem. Soc., 1969, 91, 4571–4573 CrossRef CAS.
- For a report of dispersive stabilisation controlling stereoselectivity see: (a) E. H. Krenske, K. N. Houk, A. G. Lohse, J. E. Antoline and R. P. Hsung, Chem. Sci., 2010, 1, 387–392 RSC; (b) For a rate acceleration through dispersive interactions see: R. Warmuth, J.-L. Kerdelhué, S. Sánchez Carrera, K. J. Langenwalter and N. Brown, Angew. Chem., Int. Ed., 2002, 41, 96–99 CrossRef CAS; (c) For cooperative weak dispersive interactions actuating catalysis see: Y. Wang, M. Rickhaus, O. Blacque, K. K. Baldridge, M. Juríček and J. S. Siegel, J. Am. Chem. Soc., 2022, 144, 2679–2684 CrossRef PubMed.
- For a review entitled ‘London Dispersion in Molecular Chemistry – Reconsidering Steric Effects’ see: J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterising data; X-ray crystallographic details for 7; copies of 1H and 13C NMR spectra; HPLC e.r. determinations; VCD spectrum for (R)-4-d. CCDC 2151865. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2cc01136k |
| ‡ We also considered and computed transitions states C and D (below) with the alkynyl substituent in the equatorial position by inverting the configuration of the alkoxide stereocentre from (S) to (R). Minimisation of these transition state geometries resulted in their convergence to TS B and A respectively with identical energies but now with (R,Re) and (R,Si) descriptors (i.e., as their enantiomeric forms). These calculations therefore reveal that there are only two diastereomeric transition states for the system regardless of whether the alkyne or adamantyl substituents are initially designated axial or equatorial. |
| § Atom % D for deuterated compounds were assessed by quantitative NMR methods integrating relevant signals in 1H NMR and/or inverse-gated 13C spectra. See ESI† for details. |
| ¶ A secondary kinetic isotope effect is evident here in the low isolated yield and reduced atom % D of product 5-d. |
| || We invoke nucleophilic O-attack of the lithium alkoxide of (±)-5-d on AdCHO followed by hydride transfer from the so-formed tetrahedral intermediate to another AdCHO molecule to initiate this process, but the expected adamantanoate ester product could not be identified in the product mixture. In support of a Tischenko-type process ester AdC(O)OCH2Ad could be identified as another component in the mixture as confirmed by comparison with an authentic sample. |
| ** See ESI† for details. |
| †† Benzoylation of the recovered unreacted alcohol 5 after chromatography and HPLC analysis on a chiral stationary phase and comparison with an authentic racemic sample showed that it was single enantiomer, thus ruling out a reversible reaction for the hydride transfer. |
‡‡ A secondary kinetic isotope effect is again in evidence here in the reduced atom % D of product 4-d. This subsequently results in the observation of diastereomeric methylene protons of AdC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2O2CCH(OMe)Ph in the 1H NMR spectrum of 8 and 9 as a pair of doublets integrating for approximately 24% of the mixture (cf., 22% AdC2OH in 4-d). 2O2CCH(OMe)Ph in the 1H NMR spectrum of 8 and 9 as a pair of doublets integrating for approximately 24% of the mixture (cf., 22% AdC2OH in 4-d). |
| This journal is © The Royal Society of Chemistry 2022 |