
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Do 2-coordinate iodine(I) and silver(I) complexes form nucleophilic iodonium interactions (NIIs) in solution?†‡
Scott
Wilcox
 a,
Daniel
Sethio
a,
Jas S.
Ward
b,
Antonio
Frontera
c,
Roland
Lindh
a,
Kari
Rissanen
b and
Máté
Erdélyi
*a
a,
Daniel
Sethio
a,
Jas S.
Ward
b,
Antonio
Frontera
c,
Roland
Lindh
a,
Kari
Rissanen
b and
Máté
Erdélyi
*a
aDepartment of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. E-mail: mate.erdelyi@kemi.uu.se
bUniversity of Jyvaskyla, Department of Chemistry, 40014 Jyväskylä, Finland
cDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, Palma de Mallorca Baleares 07122, Spain
First published on 1st April 2022
Abstract
The interaction of a [bis(pyridine)iodine(I)]+ cation with a [bis(pyridine)silver(I)]+ cation, in which an iodonium ion acts as a nucleophile by transferring electron density to the silver(I) cation, is reinvestigated herein. No measurable interaction is observed between the cationic species in solution by NMR; DFT reveals that if there is an attractive interaction between these complexes in solution, it is dominantly the π–π interaction of pyridines.
Halonium ions’ halogen bond1,2 complexes receive increasing attention due to the unusual strength of their three-center, four-electron bond (ΔG ≥ −100 kJ mol−1),3 and to their utility in, for instance, supramolecular and synthetic chemistry.2,4–11 This interaction has primarily been studied using [bis(pyridine)halogen(I)]+-type complexes as model systems.2,5 In these complexes, the positive charge of the halogen(I) is to a large extent, ∼60–85%, transferred into the coordinating pyridines.12 Besides the charge-transfer contribution, the bond is dominantly electrostatic in nature.12,13 Similar to other non-covalent complexes, these species exist as dynamic mixtures of their associated and dissociated forms, Pyr2X+ ⇌ PyrX+ + Pyr, in solutions.14 [Bis(pyridine)halogen(I)]+ complexes are different in nature (coordination number, strength and kinetics) from the coordinative [bis(pyridine)silver(I)]+ complexes, and do not coordinate Lewis bases.2,15
Recently, some of us proposed16 that despite their cationic character, [bis(pyridine)iodine(I)]+ complexes act as nucleophiles towards [bis(pyridine)silver(I)]+ complexes (Fig. 1), and that such nucleophilic iodonium interactions (NIIs) would be detectable both in the solid state and in solution. This suggestion originated from the observation of the surprisingly short 3.57184(7) Å (95% of the sum of the van der Waals radii of I and Ag atoms) iodine(I)–silver(I) distance for these intercomplexes in the solid state. The 15N NMR coordination shifts (Δδ15Ncoord = δ15Nintercomplex − δ15Nmonomer) were suggested to be supportive for the formation of the iodine(I)–silver(I) interaction in solution; however, they manifested an ambiguous behavior by not correlating to the electron density of the complexes (Fig. 1). For instance, the intercomplexes possessing the electronically similar Me, Et and i-Pr functionalities were reported to provide Δδ15Ncoord of +0.9 ppm, −7.8 ppm, and +7.7 ppm upon nucleophilic iodonium interaction. This is unexpected as the δ15N values of [N–I–N]+ and [N–Ag–N]+ complexes are sensitive to electron density alterations.17–19 The proposal for the existence of NIIs16 was based on QTAIM, NCIplot and RDG analysis, whereas it was neither supported by energy decomposition analysis nor by the +39.5 kcal mol−1 (DFT) endothermic interaction energy.16
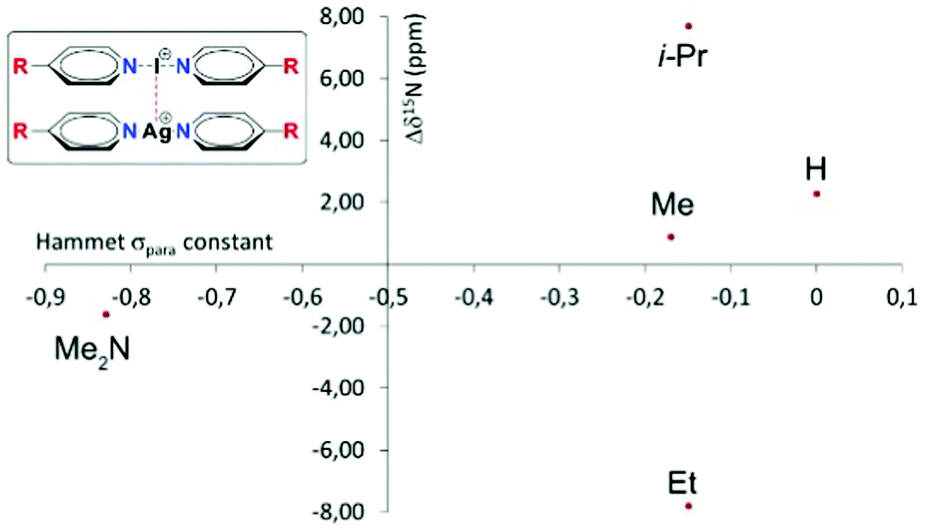 | ||
| Fig. 1 The 15N NMR coordination shifts of [bis(pyridine)iodine(I)]+ complexes (Δδ15Ncoord) from ref. 16 as a function of the Hammett σpara constant of the substituents. The interacting iodine(I) and silver(I) complexes are shown on the left, where the R group corresponds to the data points in the graph. | ||
Intrigued by the anomalous NMR and computational data, reported in ref. 16, we reinvestigated the nucleophilic iodonium interaction of the reported iodine(I) and silver(I) complexes in solution by NMR, DFT and CCSD(T). We acquired the 15N NMR chemical shifts (δ15N) and the translational diffusion coefficients (D) of [bis(4-methylpyridine)iodine(I)]+ (1-I) and [bis(4-methylpyridine)silver(I)]+ (1-Ag) complexes and their mixtures (Table 1). They were synthesized following a literature procedure, as PF6− complexes.13,18 To obtain reliable data, we measured 1H,15N HMBC with 0.3 ppm per point acquired and 0.08 ppm per point processed resolution in the 15N (indirect, f1) dimension. The spectra were acquired on solutions of equal overall bis(pyridine) complex concentration (44.4 mM), yet with varying molar ratios of the silver(I) and iodine(I) complexes. The δ15N of 1-I shows remarkable independence within the ∼0.1 ppm measurement error of the concentration of 1-Ag (Table 1). This is not compatible with an iodine(I)–silver(I) interaction that would be expected to yield a systematic concentration dependent δ15N alteration. The δ15N of 1-Ag shows a small and seemingly random variation that is larger than the measurement error and that does not correlate with the concentration of the added 1-I complex. This indicates that there is no nucleophilic iodonium interaction between these silver(I) and iodine(I) complexes in solution. The translational diffusion (DOSY) NMR data shown in Table 1 corroborates the above conclusion by the diffusion coefficients, D, of the iodine(I) and silver(I) complexes not showing a systematic concentration-dependent variation. Complex formation between the iodine(I) and silver(I) species would be expected to result in the simultaneous decrease of the diffusion coefficients of both due to the increased population-weighted Stokes radius; this is not observed. Similar to the δ15Ns, the diffusion coefficient of 1-Ag shows a small but measurably larger variation than that of the 1-I complex.
| # |
1-I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-Ag (eq.) :1-Ag (eq.) |
Added reagent (eq.) | δ15N (ppm) | D × 10−10 m s−2 | ||
|---|---|---|---|---|---|---|
| 1-I | 1-Ag | 1-I | 1-Ag | |||
| 1 | 4:0 |
−181.0 | 11.8 | |||
| 2 | 3:1 |
−181.1 | −131.2 | 11.9 | 12.0 | |
| 3 | 2:2 |
−181.0 | −130.4 | 12.1 | 11.8 | |
| 4 | 1:3 |
−181.1 | −130.8 | 11.8 | 11.5 | |
| 5 | 0:4 |
−130.8 | 11.6 | |||
| 6 | 0:4 |
4.0 H2O | −130.6 | |||
| 7 | 0:4 |
0.3 4-Me-Pyr | −125.2 | |||
| 8 | 0:4 |
0.3 4-Me-HPyr+ | −135.8 | |||
| 9 | 0:4 |
4.0 4-Me-HPyr+ | −152.5 | |||
| 10 | 0:4 |
1.0 TFA | −140.7 | |||
| 11 | 4:0 |
0.2 H2O | −181.0 | |||
| 12 | 2:2 |
1.0 H2O | −181.1 | −134.9 | ||
| 13 | 2:2 |
0.2 4-Me-Pyr | −181.0 | −132.3 | ||
The variation of the δ15N and D of the 1-Ag complex suggested that a further chemical process occurs in the studied solutions. The addition of 1 eq. water did not alter the δ15N of the silver(I) complex (Table 1, entry 6). This is in line with the literature19,20 as moisture is expected to induce the formation of a tricoordinate [bis(4-methylpyridine)silver(I)(H2O)]+ complex. The coordination of an oxygen donor ligand does not induce a larger change in the δ15N of bis(pyridine)-type complexes,20 which is in contrast to nitrogen-donor Lewis bases.19 Accordingly, the addition of 0.3 eq. of 4-methylpyridine to the 1-Ag complex induced a +5.6 ppm Δδ15N (Table 1, entry 7), whereas addition of 0.3 eq. of 4-methylpyridinium ion gave −4.6 ppm (Table 1, entry 8), and an aliquot of trifluoroacetic acid a −9.9 ppm (Table 1, entry 10) Δδ15N of the 1-Ag complex. This is in agreement with the dynamic nature of 1-Ag in solution, and hence the rapid equilibria shown in eqn (1) and (2), possibly accompanied by the pH-dependent process shown in eqn (3).
| (4-Me-Pyr)2Ag ⇌ 4-Me-PyrAg + 4-Me-pyr | (1) |
| (4-Me-Pyr)2Ag + H+ ⇌ 4-Me-PyrAg + 4-Me-pyrH+ | (2) |
| (4-Me-Pyr)2Ag + 4-Me-Pyr ⇌ (4-Me-Pyr)3Ag | (3) |
Despite several species being present in solution, a single 15N NMR signal is detected due to the rapid 4-Me-Pyr exchange between the species. The observed δ15N is hence the population-weighted average of the δ15N of pyridine in its free, protonated and silver-complexed forms. The opposite direction of the Δδ15N upon addition of 4-Me-Pyr (δ15N = −71.6 ppm) and 4-Me-PyrH+ (δ15N = −170.2 ppm) to the silver(I) complex is the result of their higher, and lower respective δ15N's as compared to that of the silver(I) complex, −130.8 ppm.
Upon addition of a small aliquot of water to the 1-I complex, partial decomposition to 4-methylpyridine and hypoiodous acid takes place, indicated by the emergence of a new set of peaks corresponding to protonated pyridine (Fig. S56, ESI‡). This shows a slow exchange process, and accordingly it does not cause a measurable Δδ15N of the 1-I complex itself (Table 1, entry 11).
The above observations suggest that in a solution of 1-Ag and 1-I complexes, moisture induces the partial decomposition of the iodine(I) complex that leads to the release of small amounts of protonated 4-methylpyridine (Fig. 2). This 4-methylpyridinium ion in turn is in rapid chemical exchange with the 1-Ag complex, influencing its δ15N by rapid chemical exchange and by the possible formation of [tris(4-methyl-pyridine)silver(I)]+ complex. Overall, the observed δ15N of the silver(I) complex is determined by the quantity of the water contaminant that induces the initial liberation of 4-methylpyridine and acid from the corresponding iodine(I) complex. The moister the solution, the larger |Δδ15N| of the silver(I) complex is expected. Accordingly, a small, −4.1 ppm, alteration of the δ15N of the 1-Ag complex but not of the 1-I complex is observed upon addition of 0.2 eq. water to the mixture (Table 1, entry 12). The varying amount of moisture also explains the δ15N variation of the 1-Ag complex in its mixtures with the analogous iodine(I) complex. The exchange of 4-methylpyridine between the silver(I) and iodine(I) complexes was further confirmed by the detection of EXSY cross-peaks between their signals (Fig. S36, ESI‡).
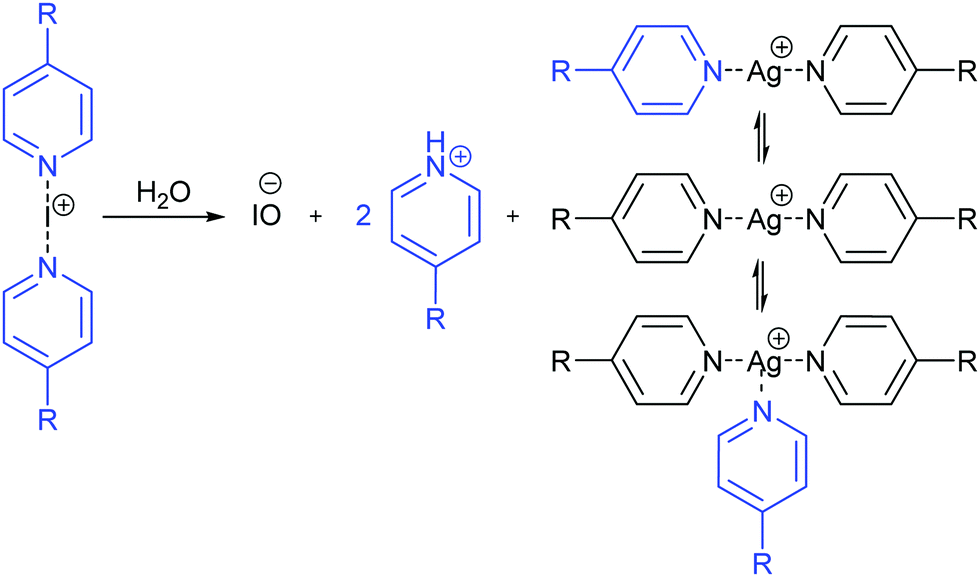 | ||
| Fig. 2 Chemical equilibria induced by moisture in a mixture of [bis(4-R-pyridine)silver(I)]+ and [bis(4-R-pyridine)iodine(I)]+ complexes. The water induced partial decomposition of the iodine(I) complex liberates protonated 4-R-pyridine. This enters a rapid ligand exchange with the coordinative silver(I) complex and forms [tris(4-R-pyridine)silver(I)]+ complex. In such a dynamic mixture, the NMR signals observed for the [bis(4-R-pyridine)silver(I)]+ complex are the population weighted average of those of all species that participate in the rapid dynamic exchange process. The observed chemical shift of the silver(I) complex is determined by the amount of moisture present in the sample. | ||
In line with this, the electron density independent variation of the δ15N disclosed in ref. 16 is in agreement with varying amounts of moisture present in the studied samples, rather than with a nucleophilic iodonium interaction. This presumption is corroborated by the reported synthesis16 under ambient, and non-dry conditions.
To exclude any influence of ligand exchange on the NMR data, we also measured the concentration dependence of Δδ15Ncoord and of the diffusion coefficients of mixtures of 1,2-bis((pyridine-2-ylethynyl)benzene)iodine(I)]+ and 1,2-bis((pyridine-2-ylethynyl)benzene)silver(I)]+ complexes that possess bidentate ligands. No EXSY cross-peaks were observed between these complexes (Fig. S64, ESI‡), confirming the absence of ligand exchange. The invariance of the δ15Ncoord and the diffusion coefficients of these complexes upon their mixing supports the conclusion that no nucleophilic iodonium interaction is detectable for these iodine(I) and silver(I) complexes in solution.
To gain further insight, the geometry of a hypothetical 1-Ag–1-I dimer was optimized (Table 2) at the M06-2X/def2-TZVP level of theory,21 starting from the previously reported16 X-ray structure. The CH2Cl2 solvation effects were included using the polarizable continuum (PCM) model, and the counterpoise correction was used to compensate for the basis set superposition error (BSSE). The optimized complex has a 3.376 Å Ag–I distance that is 91% of the sum of the van der Waals radii of silver and iodine, 3.70 Å,22,23 and is shorter than that observed by single crystal X-ray diffraction (3.5184(7) Å). A slightly longer yet comparable interionic distance was also computed at the B3LYP-D3/Def2-TZVP (3.739 Å) level of theory. QTAIM,24 RDG,25 IGM,26 DORI,27 IRI,28 local vibrational mode29 and van der Waals surface analyses all indicated the weak interaction of the iodine(I) and silver(I) complexes (Fig. S66–S70, ESI‡). However, the interaction energy is weak, −8.27 kcal mol−1 (DLPNO-CCSD(T)/Def2-TZVP) and is countered by the average kinetic (thermal) energy, ∼+5 kcal mol−1, and by the entropy contribution, ∼+16 kcal mol−1, at room temperature in solution (Table S5, ESI‡). Hence, we conclude that even if there is a weak attractive interaction between 1-Ag and 1-I it is by far too weak to be detectable in solution.
|
|
|||
|---|---|---|---|
| Computational method | E Int | E π–π | E Ag–I |
| B3LYP-D3/Def2-TZVP | −10.21 | −11.91 | +1.70 |
| wB97X-D3/Def2-TZVP | −7.74 | −10.57 | +2.83 |
| M06-2X/Def2-TZVP | −6.50 | −9.10 | +2.60 |
| MP2/Def2-TZVP | −13.97 | −15.89 | +1.92 |
| DLPNO-CCSD/Def2-TZVP | −6.50 | −10.02 | +3.52 |
| DLPNO-CCSD(T)/Def2-TZVP | −8.27 | −11.63 | +3.56 |
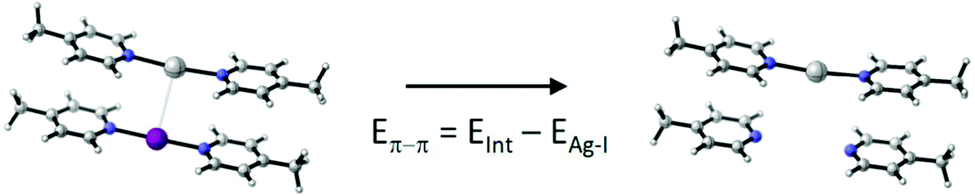
To understand whether the weak attractive force truly originates from the interaction of the silver(I) and iodine(I) cations, we decomposed the interaction energy into its main contributions, following the logic of ref. 16 (Table 2). Independent of the computational method used, the face-to-face π–π interaction of the pyridine rings is the dominant attractive force. This compensates for the silver(I)–iodine(I) repulsion, making the overall interaction slightly attractive. It should here be emphasized that in a [bis(pyridine)iodine(I)]+ complex the charge is not located on iodine(I), but is to a large extent, 60%, transferred to the pyridines through the halogen bond2,12 that is of a significant charge-transfer character.30 We expect charge-transfer also through the coordinative bond of the analogous silver(I) complex into its respective pyridines. Accordingly, it would be inaccurate to estimate the coulombic repulsion of the iodine(I) and silver(I) of these complexes by presuming them to be two positive charges 3.376 Å apart. Due to the charge transfer character of the complexes, the model shown in Table 2 inherently overestimates the energy of the π(δ+)–π(δ+) whereas underestimates that of the silver(I)–iodine(I) interaction. Hence, the removal of iodine(I) does not only eliminate a possible iodine(I)–silver(I) NII interaction, but also the charge–charge repulsion and provides non-optimized geometry for the π–π interactions. Whereas this may influence the estimated energy, it does not alter the overall conclusion.
In conclusion the NMR and DFT studies reveal a weak attractive interaction between 1-Ag and 1-I complexes in solution, driven by the π⋯π interaction of pyridines, compensating the coulombic repulsion of the cationic complexes, similarly to that observed for argentophilic interactions.31 The silver(I)–iodine(I) interaction of the complexes is not detectable in solution as the interaction energy is less than the average thermal energy and entropy contribution at room temperature. The moisture induced decomposition of the 1-I complex of the 1-Ag:1-I complex mixture liberates protonated pyridine. The rapid chemical exchange of the pyridinium ion with the 1-Ag complex results in a single set of population-averaged NMR signals. The misattribution of the spectroscopic changes upon the latter equilibrium led to an incorrect conclusion,16 and to the reporting of an attractive interaction between the iodine(I) and silver(I) complexes in solution. The earlier reported chemical shift changes are explained by the varying moisture content of the samples, and not by a nucleophilic iodonium interaction. [Bis(pyridine)iodine(I)]+ and [bis(pyridine)-silver(I)]+ complexes exist in solution as dynamic mixtures of their associated and dissociated forms. Ligand exchange takes place, which makes the interpretation of the NMR data challenging.14,32 Contrary to solution the 1-Ag:1-I complexes exist as dimers with a short silver(I)–iodine(I) contact in the solid state (Fig. 3, EROGIE33); however, it is unclear whether the components self-assemble into dimers prior to the final crystal lattice formation or if the 3-D intercomplex interactions in the crystal lattice are causing it.
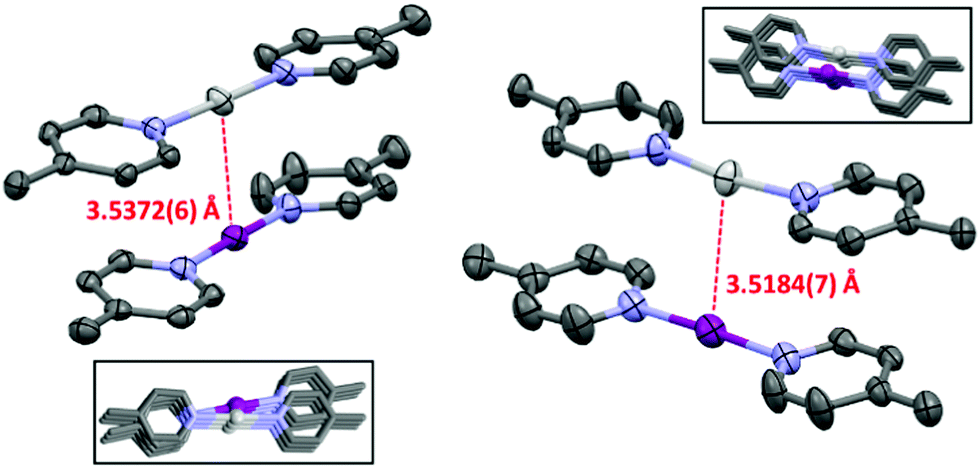 | ||
| Fig. 3 The asymmetric unit cell of the 1-Ag:1-I complex (NIIs indicated with dashed red lines; PF6− anions and hydrogen atoms omitted for clarity; thermal ellipsoids at 50% probability). Insets: The two different silver(I)–iodine(I) packing motifs observed in the crystal structure (CSD refcode EROGIE33). | ||
This work was supported by Vetenskapsrådet [2020-03431; 2020-03182] and made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry – BMC and the Disciplinary Domain of Medicine and Pharmacy. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) through project SNIC 2021-5-359.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CrossRef CAS.
- L. Turunen and M. Erdelyi, Chem. Soc. Rev., 2020, 49, 2688 RSC.
- A. C. Reiersolmoen, S. Battaglia, S. Oien-Odegaard, A. K. Gupta, A. Fiksdahl, R. Lindh and M. Erdelyi, Chem. Sci., 2020, 11, 7979 RSC.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831 CrossRef CAS PubMed.
- S. B. Hakkert and M. Erdelyi, J. Phys. Org. Chem., 2015, 28, 226 CrossRef CAS.
- L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033 CrossRef CAS PubMed.
- A. C. Reiersolmoen, D. Csókás, S. Oien-Odegaard, A. Vanderkooy, A. K. Gupta, A.-C. C. Carlsson, A. Orthaber, A. Fiksdahl, I. Papai and M. Erdelyi, J. Am. Chem. Soc., 2020, 142, 6439 CrossRef PubMed.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdelyi, Angew. Chem., Int. Ed., 2019, 58, 9012 CrossRef CAS PubMed.
- S. Lindblad, F. Boróka Németh, T. Földes, D. von der Heiden, H. G. Vang, Z. L. Driscoll, E. R. Gonnering, I. Pápai, N. Bowling and M. Erdelyi, Chem. – Eur. J., 2021, 27, 13748 CrossRef CAS PubMed.
- S. Lindblad, D. Sethio, O. B. Berryman and M. Erdelyi, Chem. Commun., 2021, 57, 6261 RSC.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Gräfenstein and M. Erdelyi, Chem. Sci., 2014, 5, 3226 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdelyi, J. Am. Chem. Soc., 2012, 134, 5706 CrossRef CAS PubMed.
- D. von der Heiden, K. Rissanen and M. Erdelyi, Chem. Commun., 2020, 56, 14431 RSC.
- M. Bedin, A. Karim, M. Reitti, A. C. Carlsson, F. Topic, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Grafenstein and M. Erdelyi, Chem. Sci., 2015, 6, 3746 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 5094 RSC . [retracted].
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdelyi, J. Am. Chem. Soc., 2018, 140, 13503 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdelyi, J. Am. Chem. Soc., 2016, 138, 9853 CrossRef CAS PubMed.
- R. Kleinmaier, S. Arenz, A. Karim, A.-C. C. Carlsson and M. Erdelyi, Magn. Reson. Chem., 2013, 51, 46 CrossRef CAS PubMed.
- S. Lindblad, F. B. Nemeth, T. Foldes, A. Vanderkooy, I. Papai and M. Erdelyi, Chem. Commun., 2020, 56, 9671 RSC.
- D. Sethio, G. Raggi, R. Lindh and M. Erdelyi, J. Chem. Theory Comput., 2020, 16, 7690 CrossRef CAS PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- R. F. Bader, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 13348 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef CAS PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-Garcia and E. Henon, Phys. Chem. Chem. Phys., 2017, 19, 17928 RSC.
- P. de Silva and C. Corminboeuf, J. Chem. Theory Comput., 2014, 10, 3745 CrossRef CAS PubMed.
- T. Lu and Q. Chen, Chem. Methodol., 2021, 1, 231 CrossRef.
- E. Kraka, W. L. Zou and Y. W. Tao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1480 CAS.
- L. Turunen, J. H. Hansen and M. Erdelyi, Chem. Rec., 2021, 21, 1252 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 CrossRef CAS PubMed.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428 RSC.
- The Cambridge Structural Database, C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
Footnotes |
| † This paper provides a revision for the article Chem. Commun., 2021, 527, 5094. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc00994c |
| This journal is © The Royal Society of Chemistry 2022 |