
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mn(II) sub-nanometric site stabilization in noble, N-doped carbonaceous materials for electrochemical CO2 reduction†
Janina
Kossmann
 ab,
Maria Luz Ortiz
Sánchez-Manjavacas
ab,
Jessica
Brandt
ab,
Tobias
Heil
ab,
Nieves
López-Salas
*ab and
Josep
Albero
*ab
ab,
Maria Luz Ortiz
Sánchez-Manjavacas
ab,
Jessica
Brandt
ab,
Tobias
Heil
ab,
Nieves
López-Salas
*ab and
Josep
Albero
*ab
aMax Planck Institute of Colloids and Interfaces, Research Campus Golm, D-14424 Potsdam, Germany
bUniversity of Potsdam, D-14424 Potsdam, Germany. E-mail: nieves.lopezsalas@mpikg.mpg.de; josep.albero@mpikg.mpg.de
First published on 21st March 2022
Abstract
The preparation of stable and efficient electrocatalysts comprising abundant and non-critical row-materials is of paramount importance for their industrial implementation. Herein, we present a simple synthetic route to prepare Mn(II) sub-nanometric active sites over a highly N-doped noble carbonaceous support. This support not only promotes a strong stabilization of the Mn(II) sites, improving its stability against oxidation, but also provides a convenient coordination environment in the Mn(II) sites able to produce CO, HCOOH and CH3COOH from electrochemical CO2 reduction.
The electrochemical CO2 reduction reaction (CO2RR) has been proposed as an appealing approach to directly convert CO2, as a chemical feedstock, into high value-added products and fuels.1,2 However, CO2 is a very stable molecule, with an extremely high dissociation energy of the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (∼750 kJ mol−1),3 and therefore, very high electrochemical overpotentials are needed for conversion at practical current densities. Currently, transition metal-based heterogeneous electrocatalysts have been widely investigated for the CO2RR due to economic and sustainability aspects.4 However, the activity and selectivity of CO2RR greatly depend on the valence state, crystal phase, size, and shape of the active sites, together with further factors, such as the electrolyte composition.4–7 For instance, Cu-based electrocatalysts have been reported to promote different C1 and C2 hydrocarbons and oxygenated products in complex mixtures, depending on the Cu size or crystal facet.8 Similarly, it has been reported that the oxidation state of some catalysts can influence the activity and selectivity of the CO2RR, promoting reactant activation, key intermediates adsorption or enabling C–C coupling.9
O bond (∼750 kJ mol−1),3 and therefore, very high electrochemical overpotentials are needed for conversion at practical current densities. Currently, transition metal-based heterogeneous electrocatalysts have been widely investigated for the CO2RR due to economic and sustainability aspects.4 However, the activity and selectivity of CO2RR greatly depend on the valence state, crystal phase, size, and shape of the active sites, together with further factors, such as the electrolyte composition.4–7 For instance, Cu-based electrocatalysts have been reported to promote different C1 and C2 hydrocarbons and oxygenated products in complex mixtures, depending on the Cu size or crystal facet.8 Similarly, it has been reported that the oxidation state of some catalysts can influence the activity and selectivity of the CO2RR, promoting reactant activation, key intermediates adsorption or enabling C–C coupling.9
Manganese is the third most abundant transition metal in the Earth's crust. Thus, several Mn-electrocatalysts have been reported, such as oxides (i.e. Mn4CaO5, MnO, Mn3O4, MnO2),10,11 organo-metallic complexes,12 and, more recently, supported single sites and small clusters.13–16 However, the large variety of Mn valence states, polymorphs and crystal structures can largely affect its catalytic activity and selectivity.17 In addition, these characteristics can be altered in operating conditions and, for instance, the Mn-based electrode valence state can be modified electrochemically upon the application of a bias potential.18 Hence, the design of convenient supports exhibiting strong metal–support interactions has been found to be of paramount importance for the CO2RR, since the activity and selectivity can be greatly influenced by the reaction conditions.
In 2018, Oschatz and Antonietti presented the concept of noble heteroatom doped-carbonaceous materials. Those materials are very resistant against oxidation.19 Among other properties that justify the interest of these materials as supports, the most relevant ones are their high resistance against oxidation, CO2 absorption capacity, conductivity, high heteroatom content, and large specific surface area. This makes them very convenient supports for energy conversion and storage, besides others.20 In this regard, nitrogenous bases or ionic liquids have been proposed as very convenient precursors of noble, nitrogen-doped-carbonaceous materials.21 The high nitrogen content in these carbonaceous materials can favor a strong interaction with transition metals. Moreover, the nobility of these materials provides high stability against oxidation or combustion.
In this communication, uracil has been used as precursor to produce a noble, N-doped carbonaceous material and subsequently stabilize sub-nanometric Mn sites on its surface using an easy and scalable procedure. The Mn-containing noble, N-doped porous carbonaceous materials were used as an electrocatalyst for the CO2RR. CO, formic acid, and acetic acid were detected as CO2 reduction products. More importantly, the obtained carbonaceous support demonstrates a strong interaction with the sub-nanometric Mn particles, stabilizing the Mn(II) sites with no evident changes in the Mn valence state after electrochemical CO2RR. Moreover, the coordination environment provided by the support has been found to strongly influence the CO2RR selectivity. These results open up a new avenue in the design of carbon-based supports stabilizing transition metals.
The preparation of the N-doped carbonaceous support is described in detail in the experimental section of the ESI.† In brief, the condensation of uracil at 800 °C in a N2 atmosphere produced a porous and highly N-doped carbonaceous material, hereafter named URA. Mn was incorporated with different loadings (URA–MnX, where X stand for the final Mn loading) through URA wet impregnation of Mn(II) acetate solutions of increasing concentrations, and subsequent calcination at 350 °C in air. The chemical composition of the prepared samples was analyzed by scanning electron microscopy, coupled with energy dispersive X-ray analysis (SEM-EDX), and inductively coupled plasma (ICP) measurements. The obtained results are presented in Table 1. The carbon content in all samples remained nearly constant, while the nitrogen content decreased slightly with Mn loading. All URA–MnX samples present a high content of nitrogen homogeneously distributed along the carbonaceous material (see Fig. S1, ESI†). It is worth noticing that the oxygen content of URA increased significantly upon Mn loading, probably due to the oxygenated functionalities of the Mn precursor (Mn(II) acetate), but does not increase further with different Mn loadings, pointing at the oxygen origin being mainly from the oxidative annealing conditions.
| Sample | EDX (at%) | ICP (wt%) | S BET (m2 g−1) | V T (m3 g−1) | ||
|---|---|---|---|---|---|---|
| C | N | O | Mn | |||
| URA | 73.8 | 21.8 | 4.3 | — | 1330 | 0.51 |
| URA–Mn2 | 74.8 | 15.5 | 9.7 | 1.8 | 1449 | 0.61 |
| URA–Mn4 | 74.4 | 16.0 | 9.6 | 3.7 | 1388 | 0.55 |
| URA–Mn8 | 73.0 | 18.2 | 8.8 | 7.7 | 997 | 0.39 |
The textural properties of URA–MnX were analyzed using N2 physisorption at 77 K. The results are presented in Table 1 and Fig. S2A (ESI†). All samples are mainly microporous. The obtained specific surface area (SBET) and total pore volume (VT) of URA, URA–Mn2 and URA–Mn4 are very similar (ca. 1360 m2 g−1 and ca. 0.55 cm3 g−1), while URA–Mn8 showed 997 m2 g−1 and 0.39 cm3 g−1, respectively. This can be rationalized by the porosity of the samples collapsing or clogging when large amounts of the manganese precursor are loaded on top of the URA surface. NLDFT was used to calculate the pore size distribution of all URA–MnX (Fig. S2B, ESI†). The results show that all the samples have defined mesopores with a pore width of 2.3 nm. CO2 adsorption isotherms at 273 K (Fig. S2C, ESI†) show a similar trend to that of the N2 isotherms.
Fourier transform infrared (FTIR) spectra of URA–MnX show no significant changes (Fig. S3A, ESI†). A broad band from ca. 2700–3700 cm−1, ascribed to OH stretching vibration, slightly increases upon Mn loading, which is in good agreement with the increase in O content observed from SEM-EDX. The peaks at ca. 1500 cm−1 and 1200 cm−1 have been previously assigned to C–N/C–C conjugated structures.22 Powder X-ray diffraction (PXRD) patterns of URA–MnX (Fig. S3B, ESI†) show no diffraction peaks related to Mn crystal phases. The broad peak at 26.5° can be attributed to the weak graphitic stacking of the carbonaceous support. The peak slightly decreases in intensity with the Mn loading due to the scattering effect of the manganese atoms.
Scanning electron microscopy images in Fig. S4 (ESI†) show the colloidal structure of URA and how its morphology remained unchanged upon increasing Mn loading. Dark-field and bright-field scanning transmission electron microscopy (STEM) images (Fig. 1A, B and Fig. S5, ESI†) revealed the presence of sub-nanometric Mn sites homogeneously distributed in URA–Mn2, URA–Mn4, and URA–Mn8 samples.
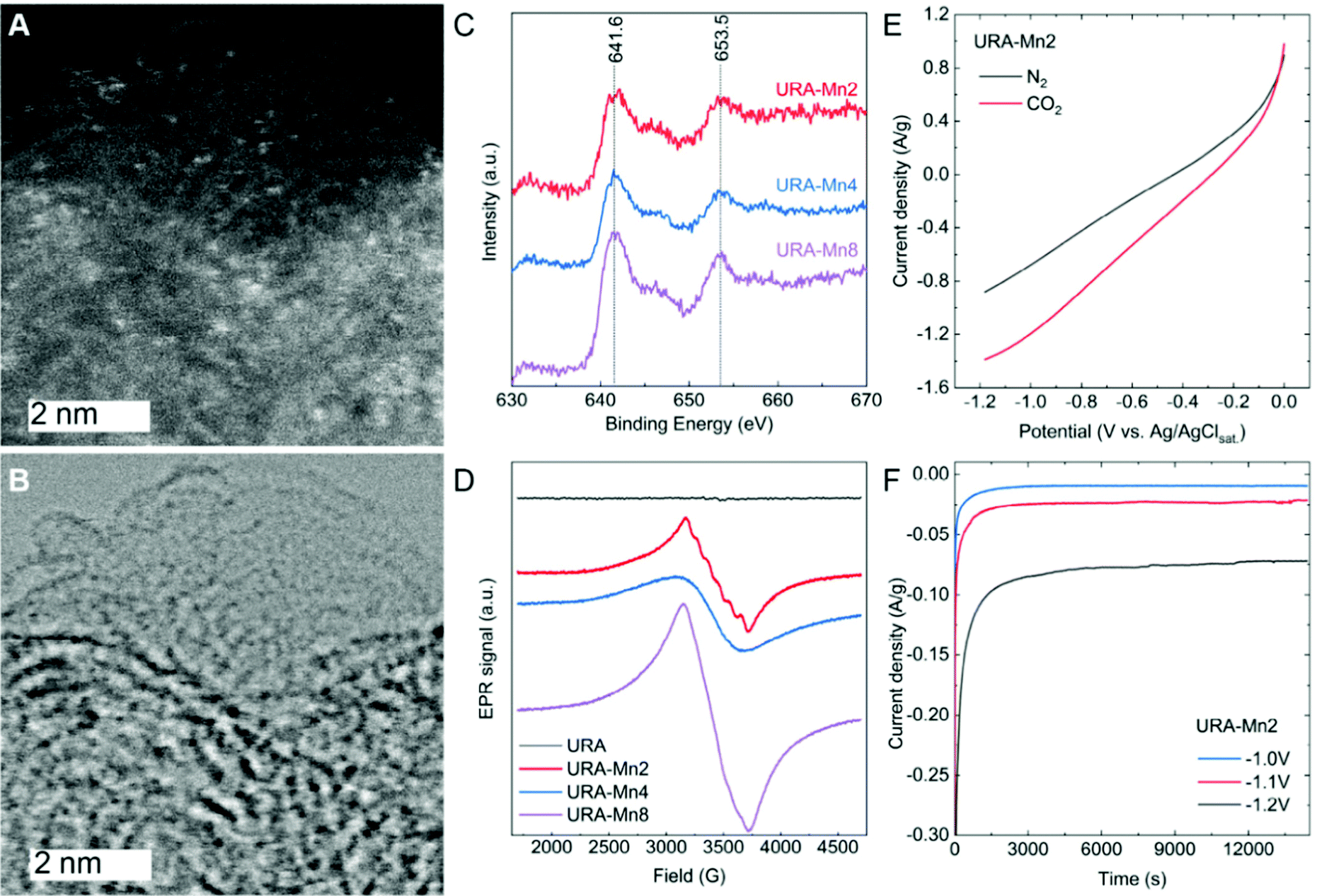 | ||
| Fig. 1 (A) Dark- and (B) bright-field STEM of URA–Mn2 with a scale bar of 2 nm. (C) Mn 2p XPS spectra and (D) EPR signals of URA–Mn2, URA–Mn4 and URA–Mn8. (E) LSV curves of URA–Mn2 in nitrogen (black) and CO2 (red) saturated electrolyte and (F) chronoamperometry measurements over 4 h applying different potentials. NaOH was used as an electrolyte. | ||
The deconvolution of high-resolution X-ray photoelectron spectroscopy (XPS) C 1s and N 1s spectra (Fig. S6, ESI†) did not evidence significant changes between them, indicating that the carbonaceous substrate suffered negligible changes upon Mn loading. The C 1s spectra of the samples showed a main component at 284.6 eV, attributed to sp2 carbon, together with components related to C–N, CO, and –CO bonds centered at 286.0 eV, 288.7 eV, and 291.3 eV. The N 1s XPS spectra of URA–MnX were deconvoluted in four different components, corresponding to pyridinic-N (397.9 eV), pyrrolic (399.7 eV), quaternary-N (401.2 eV), and oxide-N (404.6 eV). The lack of change in the C 1s and N 1s signals of the different samples highlights the support stability.
The high-resolution XPS Mn 2p spectra of the URA–MnX samples (Fig. 1C) display a main peak centered at 641.6 eV and a second peak at 653.5 eV, attributed to Mn 2p 3/2 and Mn 2p 1/2, respectively.23 As it can be observed, the position of these peaks does not change with increasing Mn loading. The positions of the Mn 2p 3/2 peaks have been previously attributed to Mn(II) or Mn(III) species, while the presence of Mn(IV) can be excluded, since the main peak is typically centered at a larger binding energy.24 Although the presence of Mn(III) in minor amounts cannot be completely disregarded, the presence of a satellite peak at ca. 647 eV is characteristic of Mn(II) species (and is not present in Mn(III) species XPS spectra). Therefore, we speculate that Mn(II) is the main species in URA–MnX.25
Electron paramagnetic resonance (EPR) measurements were run to further investigate variations of the Mn valence state of the samples. As can be seen in Fig. 1D, URA–Mn2 shows the typical sextet pattern of Mn(II) ions,26 corroborating a good dispersion of Mn(II) sites in this sample. However, the URA–Mn4 and URA–Mn 8 signals are broader and do not show these features, indicating the presence of less dispersed Mn(II) sites. The absence of the defined sextet pattern with increasing Mn loading points at larger cluster size Mn on the support. Moreover, the presence of Mn(III) can be excluded as the characteristic signal at 1800 G was not observed in any of the samples. Overall, STEM, XPS, and EPR results point to the formation of sub-nanometric clusters of oxidized Mn(II). Here, it is important to highlight that the presence of single atoms cannot be excluded, especially in sample URA–Mn2.
The electrocatalytic activity of URA–Mn2 towards the CO2RR has been first evaluated by linear sweep voltammetry (LSV) curves in CO2 and N2-saturated NaOH solutions (Fig. 1E). As it can be seen, the higher current density in the CO2-saturated NaOH compared to that of the N2-saturated one is indicative of URA–Mn2 electrocatalytic activity for the CO2RR. For comparison purposes, LSV of URA and URA–Mn2 after acid washing (0.036 wt% of Mn according to ICP) were recorded in CO2 and N2-saturated NaOH solutions (Fig. S7A and B, ESI†). Interestingly, both demonstrated a lower current density in the CO2-saturated NaOH solution, which points at the Mn(II) sub-nanometric clusters of the Mn loaded samples being the electroactive sites and not the support or Mn single atoms. In a similar way, the LSV of URA–Mn4 was measured in CO2-saturated NaOH solutions (Fig. S7C, ESI†). In this case, URA–Mn4 shows a lower current density pointing out the importance of dispersed Mn(II) species in URA–Mn2, indicating that Mn(II) is more active for the CO2RR than the samples with less dispersed Mn(II) sites.
Chronoamperometric measurements in CO2-saturated NaOH solutions at different potentials were carried out for 4 h (Fig. 1F). As can be seen, the URA–Mn2 electrodes show nearly constant current density with time at the tested potentials, which is indicative of the stability of the electrodes. High-resolution URA–Mn2 C 1s, N 1s, and Mn 2p XPS spectra after the chronoamperometry experiments revealed that both the support and Mn sites remained nearly unchanged (Fig. 2). Here it is important to point out that the XPS spectra after the CO2RR in Fig. 2 contain also Nafion and therefore the relative intensity of the Mn 2p signal in slightly decreased. In addition to this, ICP measurements of the electrolyte after 4 h of reaction do not show Mn leaching, hence demonstrating the suitability of this support stabilizing Mn(II) active sites.
 | ||
| Fig. 2 Mn 2p XPS of URA–Mn2 before (black) and after (red) the CO2RR. | ||
It is known that the interaction between the Mn species and the support have a strong influence on CO2RR selectivity. The products obtained in the gas and liquid phases from the URA–Mn2 electrodes were analyzed by on-line GC and off-line HPLC, respectively (see Experimental Section in SI for further details). H2 and CO were the only gas products detected by GC. Notably, HCOOH and CH3COOH were detected by HPLC and verified by 1H NMR (Fig. S8, ESI†). Control experiments in N2-saturated NaOH solutions were also carried out, obtaining H2 as the only detectable product, and confirming that CO, HCOOH, and CH3COOH are formed from the electrochemical reduction of CO2. CO2RRs were performed at potentials ranging from −0.9 V to −1.2 V to find the optimum working conditions. While the FE towards formic acid and acetic acid is significantly higher at lower working potentials (Fig. S9A, ESI†), the concentrations of the products decrease dramatically (Fig. S9B, ESI†) when decreasing the working potential. The FE of the different products at −1.2 V vs. Ag/AgClSat. was calculated, obtaining ∼60% FE towards CO, ∼ 23% FE towards CH3COOH, ∼ 2% FE towards HCOOH, and ∼15% FE towards H2. When increasing the loading of Mn to URA–Mn4 the concentrations of AA and FA are similar to those obtained by URA–Mn2 (0.07 mmol g−1 and 0.03 mmol g−1, respectively). We ascribe this to the larger cluster size with increasing Mn loading seen by EPR measurements.
It is worth pointing out that the existing literature reports CO as the main product obtained from Mn single atoms or clusters supported on N-doped graphitic carbon-based electrodes.15,16,27 However, theoretical calculations have predicted that Mn-coordinated to bis(imino thiolate)-based monolayers are more selective to HCOOH rather than CO.28 Also, MnO2 nanoparticles between 0.5 and 2 nm supported on g-C3N4 have been demonstrated to be electrocatalytically active towards formate production, indicating that the coordination environment between the support and the Mn active sites plays a key role in the product selectivity.
In conclusion, a noble, N-doped carbonaceous material was prepared from uracil condensation and used as a support for sub-nanometric Mn(II) clusters. Stabilization of Mn(II) sites was corroborated by STEM, XPS, and EPR. Electrochemical CO2 reduction experiments have shown that homogeneously dispersed Mn(II) species are very active for this reaction. 4 h chronoamperometry tests at different potentials show no apparent electrocatalyst deactivation and XPS analysis shows that the Mn valence state remains unchanged after the tests, confirming the strong interaction between the support and Mn(II) active sites. Interestingly, the CO2RR at −1.2 V vs. Ag/AgClSat. using URA–Mn2 shows CO, CH3COOH, and HCOOH as the main products. Hence, this support not only strongly stabilizes the active sites, but also shifts the CO2RR selectivity towards CO, CH3COOH, and HCOOH. These preliminary results introduce a new avenue in the design of transition metal carbon-based supports, which not only must provide a strong interaction with sub-nanometric and well-dispersed sites but also selectivity control through accurate composition control. Further investigations are currently ongoing in order to elucidate the origin of the strong interaction between the Mn species and the support as well as the selectivity-coordination environment relationship.
Janina Kossmann: investigation, writing – original draft; Maria Luz Ortiz Sanchez-Manjavacas: investigation; Jessica Brandt: investigation; Tobias Heil: investigation; Nieves Lopez-Salas: conceptualization, validation, writing – review & editing, supervision, project administration; Josep Albero: conceptualization, validation, writing – review & editing, supervision.
The Max Planck Society is gratefully acknowledged for financial support.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- S. Park, Y. H. Lee, S. Choi, H. Seo, M. Y. Lee, M. Balamurugan and K. T. Nam, Energy Environ. Sci., 2020, 13, 2310–2340 RSC.
- J. Heese-Gärtlein, A. Rabe and M. Behrens, Z. Anorg. Allg. Chem., 2021, 647, 1363–1372 CrossRef.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef CAS.
- B. B. Mulik, A. V. Munde, B. D. Bankar, A. V. Biradar and B. R. Sathe, Catal. Today, 2021, 370, 104–113 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- H. Tian, L. Zeng, Y. Huang, Z. Ma, G. Meng, L. Peng, C. Chen, X. Cui and J. Shi, Nano-Micro Lett., 2020, 12, 161 CrossRef CAS PubMed.
- M. Antonietti and M. Oschatz, Adv. Mater., 2018, 30, 1706836 CrossRef PubMed.
- J. Kossmann, M. L. Ortíz Sánchez-Manjavacas, H. Zschiesche, N. V. Tarakina, M. Antonietti, J. Albero and N. López-Salas, J. Mater. Chem. A, 2022, 10, 6107–6114 RSC.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- J. Kossmann, D. Piankova, N. V. Tarakina, J. Heske, T. D. Kühne, J. Schmidt, M. Antonietti and N. López-Salas, Carbon, 2021, 172, 497–505 CrossRef CAS.
- B. R. Strohmeier and D. M. Hercules, J. Phys. Chem., 1984, 88, 4922–4929 CrossRef CAS.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- M. A. Langell, C. W. Hutchings, G. A. Carson and M. H. Nassir, J. Vac. Sci. Technol., 1996, 14, 1656–1661 CrossRef CAS.
- G. Er, S. Ishida and N. Takeuchi, J. Mater. Sci., 1999, 34, 4265–4270 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- G. Xing, L. Cheng, K. Li, Y. Gao, H. Tang, Y. Wang and Z. Wu, New J. Chem., 2020, 44, 12299–12306 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, chemical composition, scanning and transmission electron microscopy, N2 and CO2 physisorption traces, Fourier transformed infrared spectroscopy, X-ray diffraction and X-ray photoelectron spectroscopy, 1H NMR and linear sweep voltammetry data are provided. See DOI: 10.1039/d2cc00585a |
| This journal is © The Royal Society of Chemistry 2022 |