
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient CO2 conversion to CO using chemical looping over Co–In oxide†
Jun-Ichiro
Makiura
a,
Sota
Kakihara
a,
Takuma
Higo
 a,
Naoki
Ito
b,
Yuichiro
Hirano
b and
Yasushi
Sekine
*a
a,
Naoki
Ito
b,
Yuichiro
Hirano
b and
Yasushi
Sekine
*a
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: ysekine@waseda.jp
bENEOS, 1-1-2 Otemachi, Chiyoda, Tokyo 100 8162, Japan
First published on 17th March 2022
Abstract
CO2 conversion to CO by reverse water-gas shift using chemical looping (RWGS-CL) can be conducted at lower temperatures (ca. 723–823 K) than the conventional catalytic RWGS (>973 K), and has been attracting attention as an efficient process for CO production from CO2. In this study, Co–In2O3 was developed as an oxygen storage material (OSM) that can realize an efficient RWGS-CL process. Co–In2O3 showed a high CO2 splitting rate in the mid-temperature range (723–823 K) compared with previously reported materials and had high durability through redox cycles. Importantly, the maximum CO2 conversion in the CO2 splitting step (ca. 80%) was much higher than the equilibrium conversion of catalytic RWGS in the mid-temperature range, indicating that Co–In2O3 is a suitable OSM for the RWGS-CL process.
Limiting anthropogenic global warming to a specific level requires reaching at least net zero CO2 emissions.1,2 For this purpose, syntheses of liquid fuels using renewable electricity and captured CO2, which are, respectively, designated as Sun-to-Fuel (STF) and Power-to-Liquid (PtL),3 have been proposed as technologies to replace fossil fuels by carbon-neutral energy resources. The CO2-based fuels produced by PtL processes can supply renewable energy to the transportation sector.4,5 One feasible route for PtL processing from captured CO2 is the Fischer–Tropsch (FT) method.6 Nevertheless, technologies for direct synthesis from CO2 are currently in an early stage. Consequently, in the near future, CO converted from captured CO2 might be a useful intermediate for PtL fuel processing. Reverse water-gas shift (RWGS) is a representative method of converting CO2 to CO. It is an endothermic equilibrium-limited reaction.7
| CO2 + H2 ↔ CO + H2O ΔH0298 = 42.1 kJ mol−1 | (1) |
| CO + 3H2 ↔ CH4 + H2O ΔH0298 = −206.5 kJ mol−1 | (2) |
| CO2 + 4H2 ↔ CH4 + 2H2O ΔH0298 = −165.0 kJ mol−1 | (3) |
| Reduction of OSM: MOx + δH2 ↔ MOx−δ + δH2O | (4) |
| Oxidation of OSM: MOx−δ + δCO2 ↔ MOx + δCO | (5) |
Here we propose a novel material for this purpose. The most desirable OSM for this purpose was found to be Co-modified In2O3 (Co–In2O3). In fact, Co–In2O3 showed a higher CO2 splitting rate, even at low temperatures (723–823 K) under an atmosphere with high CO concentration.
To elucidate the mechanism of CO2 splitting on this OSM, various characterizations were undertaken using powder X-ray diffraction (XRD), scanning transmission electron microscopy with energy-dispersive X-ray spectrometry (STEM-EDX) and in situ X-ray absorption fine structure (XAFS) measurements.
Co-supported indium-based oxides used for this study were prepared using a complex polymerization method and an impregnation method. The procedures are described in the ESI.† The procedures for redox tests, i.e. CO2 splitting and regeneration, are also described in the ESI.† The series of reduction and oxidation steps was defined as one cycle for isothermal RWGS-CL. The amount of reduction and oxidation (redox) was defined as the amount of oxygen atoms released or backfilled in each step. The maximum CO2 conversion of the oxidation reaction (eqn (5)) was also evaluated. Electronic states of samples were evaluated using in situ In and Co K-edge X-ray absorption fine structure (XAFS) spectroscopy at the BL14B2 beamline of SPring-8 in Japan. XAFS measurements were conducted in transmission mode. Details of these characterisations are described in the ESI.†
First, we conducted screening tests for oxides that achieve high RWGS-CL performance. Subsequently, Co–In oxide (Co–In2O3) was developed. Fig. 1 presents a comparison of the CO2 splitting rate with previously reported materials (please see also ESI†). The Co–In2O3 showed very high oxidation rates of 127.6, 191.0, 280.2, 365.7, and 429.5 μmol min−1 g−1, respectively, in the middle-temperature and low-temperature ranges of 723, 748, 773, 798, and 823 K, exceeding those of previously reported perovskite oxides,11,13–19 Fe-based oxides,20–26 and In-based oxides.12 Therefore, it is considered that Co–In2O3 is kinetically the most promising oxide for RWGS-CL.
 | ||
| Fig. 1 Comparison of average CO2 splitting rates on Co–In2O3 (this study) and on various oxides in earlier reports: The purple triangle plot represents the maximum CO2 conversion rate during temperature-programmed oxidation by CO2 over La0.6Ca0.4Fe0.4Mn0.6O3(15). | ||
The results of RWGS-CL cycling tests for 10 cycles by Co–In2O3 are presented in Fig. 2. The detailed RWGS-CL results and the confirmation of the outlet gas in the re-oxidation step are shown in Table S1 and Fig. S1 (ESI†). Co–In2O3 showed a reduction amount of 6.39 mmol g−1 and oxidation amount of 2.73 mmol g−1 in the first cycle and a redox amount of 3.25 ± 0.05 mmol g−1 after the second cycle. The amount of reduction that was not backfilled by CO2 in the first cycle was found to be the reduction of Co by crystal structure analysis, as discussed in detail later, which indicates that in Co–In2O3, the redox of Co is not involved in the reaction after the reduction of the first cycle. The only redox species is indium. In contrast to the high redox performance of Co–In2O3, In2O3 reportedly has a redox amount of about 1.64 mmol g−1 and an oxidation rate of 93.4 μmol min−1 g−1 at 773 K.12 Although the redox species is indium in both oxides, Co–In2O3 shows higher redox capacity and a higher oxidation rate than those of In2O3. Consequently, the results indicate that Co can enhance the redox performance of indium. Moreover, in terms of stability, Cu–In2O3, which has high RWGS-CL performance and the same redox mechanism as Co–In2O3, shows an oxidation rate of 161.8 μmol min−1 g−1 in the first cycle, but the rate decreases to 85.5 μmol min−1 g−1 in the fifth cycle.12 However, Co–In2O3 showed an oxidation rate of 293.2 μmol min−1 g−1 in the second cycle, and still shows a high oxidation rate of 281.8 μmol min−1 g−1 in the tenth cycle, with no noticeable decrease in the rate, maintaining high stability. These results demonstrate that Co can improve the redox performance of indium, especially its oxidation performance, and can stabilize the redox of indium.
 | ||
| Fig. 2 RWGS-CL stability test for Co–In2O3. | ||
Next, the maximum CO2 conversion in the oxidation step of Co–In2O3 and previously reported In-based oxides was compared with the equilibrium conversion of conventional RWGS.27 The results are depicted in Fig. 3. Actually, Cu–In2O3, which is reported to have high redox kinetics, was found to be thermodynamically disadvantageous when compared to conventional RWGS. This finding indicated that Cu–In2O3 as unsuitable for industrial applications. Regarding In2O3, a maximum CO2 conversion of 70–90% was achieved in the medium temperature region (at 673 K), which is much higher than the equilibrium conversion of conventional RWGS. Nevertheless, concern persists that In(0), which is derived from the reduction of In2O3, is liquefied in the medium temperature region (at 673 K) because of its low melting point. This issue engenders challenges for the industrialization of the RWGS-CL process. However, Co–In2O3 also achieved a high maximum CO2 conversion of 70–80% in the medium temperature region, while suppressing the melting of indium because the redox of indium progresses with the formation and decomposition of Co–In alloy (as discussed later) with a high melting point.28 To summarize the discussion presented above, Co–In2O3 can improve the redox kinetics of indium and can achieve high stability, while maintaining the maximum CO2 conversion. Additionally, compared to materials reported earlier, Co–In2O3 has higher performance and is an industrially more promising material.
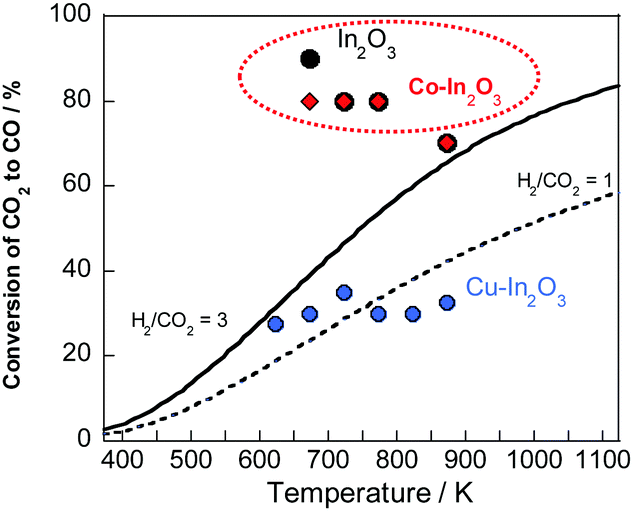 | ||
| Fig. 3 Comparison between equilibrium conversion of conventional RWGS and maximum CO2 conversion of RWGS-CL materials during the oxidation step. | ||
Next, the structural and electronic states of the Co–In2O3 in RWGS-CL were evaluated using XRD, STEM-EDX, and XAFS measurements. In the XRD of the as-prepared Co–In2O3 (see Fig. S2 in ESI†), the diffraction peaks of Co3O4 and In2O3 were observed, respectively, with no peak shift in each representative diffraction peak, indicating that the complexation of Co and In did not occur. The SEM image (Fig. S3 in ESI†) showed particles of several hundred nanometres. The EDS image showed non-uniform dispersion of Co species and uniform dispersion of In species. The XRD profile of the reduced sample (Fig. S2, ESI†) shows diffraction peaks of Co–In alloy (CoIn2), Co metal, and In2O3. The results demonstrated that a part of the Co reduced by hydrogen formed an alloy with indium, which was also reduced. The XRD profile of the re-oxidized sample (Fig. S2, ESI†) showed diffraction peaks of Co metal and In2O3, indicating that CO2 oxidizes only indium in the Co–In alloy and not in Co metal. In other words, the RWGS-CL in the Co–In2O3 is established by the redox of indium with the formation and decomposition of the Co–In alloy. The SEM images obtained after reduction and re-oxidation (Fig. S3, ESI†) showed particles of several thousand nanometres in size. The EDS images showed sparsely and heterogeneously dispersed Co on the In species. The Co species dispersed sparsely and heterogeneously on the indium species were regarded as involved in the alloy formation with indium.
The Co K-edge and In K-edge XANES spectra of Co–In2O3 during RWGS-CL are shown in Fig. 4. Several spectral changes at the In K-edge (Fig. 4(A and B)) represent the redox of indium during RWGS-CL. In addition, it is particularly noteworthy that the Co K-edge XANES spectra of Co–In2O3 (Fig. 4(C)) were peculiar. After the reduction, the spectrum was similar to that of Co-foil, but showed slightly different features (blue line in Fig. 4(C)). This spectrum indicated the formation of Co–In alloy in addition to Co metal. When the reduced Co–In2O3 was exposed to CO2, the spectrum shifted to that of Co-foil (red lines in Fig. 4(C)). Results of XAFS measurements support the redox of indium with the formation and decomposition of Co–In alloys (detailed in the ESI† text). This redox mechanism might have contributed to the high redox performance of indium, especially its oxidation performance.
 | ||
| Fig. 4 The XANES spectra of Co–In2O3 during (A) H2 reduction and (B) CO2 oxidation on In K-edge, and (C) H2 reduction and CO2 oxidation on the Co K-edge. | ||
To investigate details of the difference in oxidation performance of Co–In2O3, kinetic analysis of the isothermal solid-state reaction proposed by Hancock and Sharp29–31 was applied for the RWGS-CL oxidation step. Details of the method based on the kinetic model are described along with the fitting process in the ESI.† For In2O3, oxidation by CO2 is known to proceed in the nucleation model.12 In this model, the nuclei of the reaction are generated in the particles at the initial stage. The reaction proceeds as the nuclei grow and mutually collide continuously.32–34 For In2O3, a rapid decrease in the reaction rate has been reported because of a decrease in the nucleation region in the initial stage of oxidation. Regarding the Co–In2O3, the oxidation reaction kinetics fitted best to the zero-order model.29–31 This model is a reaction-order model in which the differential oxidation rate is kept constant irrespective of the degree of oxidation. Fig. 5 shows the differential oxidation rate behaviour of Co–In2O3. As presented in Fig. 5, the oxidation of Co–In2O3 progresses, while maintaining the differential oxidation rate up to solid conversion of about 50–60%, as expressed by the zero-order model. These results suggest that, in Co–In2O3, surface reaction sites with CO2 do not decrease with the progress of oxidation, i.e., the oxide ions on the surface oxidized by CO2 are regarded as migrating quickly to the bulk.
 | ||
| Fig. 5 Transition of differential oxidation rate on Co–In2O3. | ||
The rate constant k on the oxidation side at each temperature was calculated for the oxidation reaction of Co–In2O3. An Arrhenius plot was obtained (Fig. S6, ESI†). The reported apparent activation energy on the oxidation side of In2O3 is 85.9 kJ mol−1,12 whereas 48.5 kJ mol−1 in Co–In2O3, indicating that the co-existence of Co reduces the activation barrier of the oxidation reaction of indium by about half.
Completion of quick oxidation in the Co–In2O3 might be realized by fast oxygen ion migration in the Co–In alloy. The schematic diagrams are portrayed in Fig. S7 (ESI†). Specifically, as found from structural analyses, Co–In2O3 gives Co–In alloys dispersed on In2O3 after the reduction. When the oxidation starts, CO2 is adsorbed and dissociated on the Co–In alloy. Then oxide ions are supplied to the Co–In alloy surface. They quickly migrate from the Co–In alloy surface to the interface between the Co–In alloy and In2O3. This rapid migration of oxide ions into the bulk might engender the rapid growth of the In2O3. This peculiar oxidation mechanism gives Co–In2O3 a higher oxidation rate than those of materials described in earlier reports.
Conflicts of interest
The authors have no conflicts of interest.References
- IPCC AR6 Climate Change 2021: The Physical Science Basis.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- F. V. Vázquez, J. Koponen, V. Ruuskanen, C. Bajamundi, A. Kosonen, P. Simell, J. Ahola, C. Frilund, J. Elfving, M. Reinikainen, N. Heikkinen, J. Kauppinen and P. Piermartini, J. CO2 Util., 2018, 28, 235–246 CrossRef.
- I. Dimitriou, P. G. Gutiérrez, R. H. Elder, R. M. C. Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 RSC.
- S. Perathoner and G. Centi, ChemSusChem, 2014, 7, 1274–1282 CrossRef CAS.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC.
- D. S. Mallapragada, N. R. Singh, V. Curteanu and R. Agrawal, Ind. Eng. Chem. Res., 2013, 52, 5136–5144 CrossRef CAS.
- M. Wenzel, L. Rihko-Struckmann and K. Sundmacher, AIChE J., 2015, 61, 2–22 CrossRef.
- M. Keller and J. Otomo, J. CO2 Util., 2020, 40, 101191 CrossRef CAS.
- Y. A. Daza, D. Maiti, R. A. Kent, V. R. Bhethanabotla and J. N. Kuhn, Catal. Today, 2015, 258, 691–698 CrossRef CAS.
- J. Makiura, T. Higo, Y. Kurosawa, K. Murakami, S. Ogo, H. Tsuneki, Y. Hashimoto, Y. Sato and Y. Sekine, Chem. Sci., 2021, 12, 2108–2113 RSC.
- Y. A. Daza, R. A. Kent, M. M. Yung and J. N. Kuhn, Ind. Eng. Chem. Res., 2014, 53, 5828–5837 CrossRef CAS.
- Y. A. Daza, D. Maiti, B. J. Hare, V. R. Bhethanabotla and J. N. Kuhn, Surf. Sci., 2016, 648, 92–99 CrossRef CAS.
- D. Maiti, B. J. Hare, Y. A. Daza, A. E. Ramos, J. N. Kuhn and V. R. Bhethanabotla, Energy Environ. Sci., 2018, 11, 648–659 RSC.
- B. J. Hare, D. Maiti, Y. A. Daza, V. R. Bhethanabotla and J. N. Kuhn, ACS Catal., 2018, 8, 3021–3029 CrossRef CAS.
- B. L. Hare, D. Maiti, S. Ramani, A. E. Ramos, V. R. Bhethanabotla and J. N. Kuhn, Catal. Today, 2019, 323, 225–232 CrossRef CAS.
- A. E. Ramos, D. Maiti, Y. A. Daza, J. N. Kuhn and V. R. Bhethanabotla, Catal. Today, 2019, 338, 52–59 CrossRef CAS.
- B. J. Hare, D. Maiti, A. J. Meier, V. R. Bhethanabotla and J. N. Kuhn, Ind. Eng. Chem. Res., 2019, 58, 12551–12560 CrossRef CAS.
- M. Wenzel, N. V. R. A. Dharanipragada, V. V. Galvita, H. Poelman, G. B. Marin, L. Rihko-Struckmann and K. Sundmacher, J. CO2 Util., 2017, 17, 60–68 CrossRef CAS.
- M. Wenzel, L. Rihko-Struckmann and K. Sundmacher, Chem. Eng. J., 2018, 336, 278–296 CrossRef CAS.
- M. Najera, R. Solunke, T. Gardner and G. Veser, Chem. Eng. Res. Des., 2011, 89, 1533–1543 CrossRef CAS.
- N. V. R. A. Dharanipragada, L. C. Buelens, H. Poelman, E. D. Grave, V. V. Galvita and G. B. Marina, J. Mater. Chem. A, 2015, 3, 16251–16262 RSC.
- Y. Qiu, L. Ma, D. Zeng, M. Li, D. Cui, Y. Lv, S. Zhang and R. Xiao, J. Energy Chem., 2020, 46, 123–132 CrossRef.
- L. Ma, Y. Qiu, M. Li, D. Cui, S. Zhang, D. Zeng and R. Xiao, Ind. Eng. Chem. Res., 2020, 59, 6924–6930 CrossRef CAS.
- V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, Ind. Eng. Chem. Res., 2013, 52, 8416–8426 CrossRef CAS.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Chem. Ing. Tech., 2013, 85, 489–499 CrossRef CAS.
- J. P. Bros, M. Gaune-Escard, D. El Allam, R. Haddad and E. Hayer, J. Alloys Compd., 1996, 233, 264–271 CrossRef CAS.
- J. D. Hancock and J. H. Sharp, J. Am. Ceram. Soc., 1972, 55, 74–77 CrossRef CAS.
- K. Piotrowski, K. Mondal, H. Lorethova, L. Stonawski, T. Szymanski and T. Wiltowski, Int. J. Hydrogen Energy, 2005, 30, 1543–1554 CrossRef CAS.
- Z. Zhou, L. Han and G. M. Bollas, Int. J. Hydrogen Energy, 2014, 39, 8535–8556 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103–1112 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1940, 8, 212–224 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1941, 9, 177–184 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00208f |
| This journal is © The Royal Society of Chemistry 2022 |