
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rationally designed helical peptidomimetics disrupt α-synuclein fibrillation†
Clementine E.
Bavinton‡
a,
Rebecca
Sternke-Hoffmann‡
b,
Tohru
Yamashita
c,
Peter C.
Knipe
 cd,
Andrew D.
Hamilton
ce,
Jinghui
Luo
*b and
Sam
Thompson
*ac
cd,
Andrew D.
Hamilton
ce,
Jinghui
Luo
*b and
Sam
Thompson
*ac
aSchool of Chemistry and the Institute for Life Sciences, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: st3a15@soton.ac.uk
bPaul Scherrer Institute, Forschungsstrasse 111, 5232 Villigen PSI, Switzerland. E-mail: Jinghui.luo@psi.ch
cDepartment of Chemistry, University of Oxford, Oxford, OX1 3TA, UK
dSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Belfast, BT9 5AG, UK
eDepartment of Chemistry, New York University, 100 Washington Square East, NY 10003, USA
First published on 30th March 2022
Abstract
Misfolding of the human protein α-synuclein results in toxic fibrils and the aggregation of Lewy bodies, which are a hallmark of Parkinson's disease in brain tissue. Here we disclose a supramolecular approach where peptidomimetics are rationally designed and pre-organised to recognize the surface of native helical α-Syn by forming complementary contacts with key patches of protein surface composed of charged and hydrophobic residues. Under lipid-catalyzed conditions the mimetics slow the rate of aggregation (thioflavin-T assay) and disrupt the misfolding pathway (electron microscopy of aggregates). This hypothesis is supported by comparison with a series of negative control compounds and with circular dichroism spectroscopy. Given the approach relies on selective recognition of both amino acid sequence and conformation (helical secondary structure) there is potential to develop these compounds as tools to unravel the currently intractable structure–function relationships of (i) missense mutation, and (ii) amyloid polymorphism with disease pathogenesis.
Fibrillation of the 140 amino acid intrinsically disordered protein (IDP) α-synuclein (α-Syn) is critical to the neurotoxic pathway resulting ultimately in the deposition of β-sheet-rich Lewy bodies characteristic of Parkinson's disease.1 Extensive biophysical analysis of native α-Syn reveals transient population of a membrane-bound α-helical conformation,2,3 with recent studies supporting the presence of helical assemblies, the “tetramer hypothesis”, that may serve a protective role in preventing misfolding.4 Several early-onset familial forms of the disease are associated with point mutations, including A30P, E46K, H50Q, G51D, A53E/T, in which initiation of aggregation and subsequent accumulation are altered [Fig. 1].5 This suggests that amino acid substitution in these regions plays an important role in predisposing misfolding. Given the scale of the societal and economic cost of Parkinson's disease6 extensive efforts have led to the discovery of numerous small molecule mediators of misfolding,7,8 although it is often difficult to establish the nature of the interaction with α-Syn at a molecular level and how the molecules exert their effects. Thus, it is appealing to explore rationally designed molecules for the recognition of specific native conformational states of α-Syn in which sequence-structure-function can be interrogated.
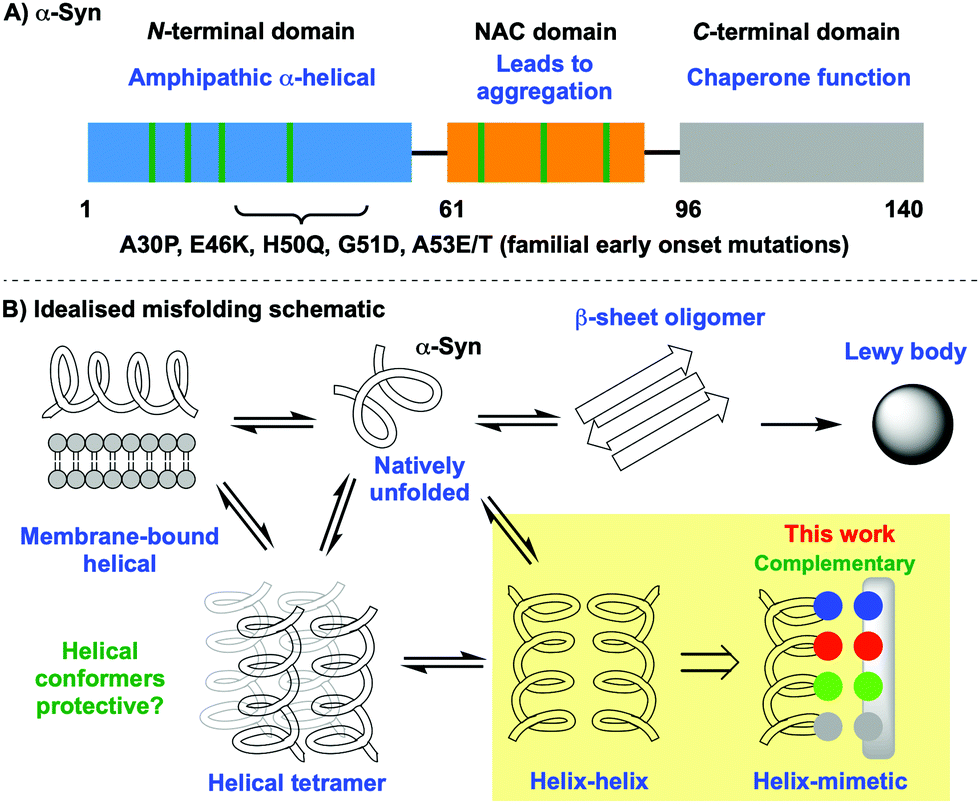 | ||
| Fig. 1 Outline of α-Syn structure and folding landscape: (A) domains, characteristics and location of point mutations, (B) introduction of a peptidomimetic to bias conformational equilibria towards helices. | ||
IDPs present a particular challenge from a molecular recognition perspective because they are moving targets.9 Molecules that selectively bind to one of many conformations require multiple points of contact orchestrated over large surface areas.10 Nature uses biomacromolecules such as peptides, nucleic acids and oligosaccharides for these purposes, achieving exquisite selectivity and affinity. Taking inspiration from these oligomers, we, and others, have shown that rationally designed conformationally pre-organised, non-peptidic scaffolds bind protein surfaces competitively with their endogenous ligands.11 Many of these scaffolds are mimetics of secondary structural elements such as the α-helix,12–15 β-strand16–19 and β-sheet.20–22 In addition to the work of several groups demonstrating mediation of therapeutically relevant protein–protein interactions (PPIs)23 we have shown that rationally designed peptidomimetics disrupt fibrillation of the IDPs islet amyloid polypeptide (IAPP)24–27 and amyloid beta (Aβ),28–31 which are implicated in the pathogenesis of type-II diabetes and Alzheimer's disease respectively.
Our hypothesis is that α-helical peptidomimetics can stabilise the α-helical conformation of α-Syn, which is transiently populated in the presence of lipid,23via the formation of a helix-mimic interaction analogous to that of the helix-helix. In seeking to stabilise the native helical form of the protein it was our hope that the equilibrium might be biased away from non-native fibrillar states that are toxic and lead to aggregation [Fig. 1B]. For this purpose, we selected the oligobenzamide scaffold because it performed well in our previous work with IAPP24,25,32 and Wilson has disclosed extensive synthetic routes towards mimetics for a broad range of successfully mediated targets.33–35 α-Syn contains many hydrophilic residues including those in the imperfect KTKEGV repeat sequence [Fig. 2]. Since familial point mutations occur at E46K (i), H50Q (i + 4), and A53E/T (i + 7), and these drastically increase early-onset Parkinson's, we reasoned that these residues appear to be important and that making complementary contacts at these positions might impact fibrillation kinetics. The choice of E20, Q24, E28 was more subtle: here we noted that Q24 was an exception to the KTKEGV repeat sequence, instead reading KTKQGV. Based on this information we designed a library of tribenzamides to provide complementary contacts along a face of the α-Syn helix displaying either E20, Q24, E28 (residues i, i + 4, i + 8) or E46, H50, A53 (residues i, i + 4, i + 7). For example, at neutral pH, mimetic 3 displays positive (RNH3+), negative (RCO2−) and hydrophobic (ROi-Pr) groups at the i, i + 4 and i + 7 positions with the potential to form salt bridges with E46 and H50, and hydrophobic interactions with A53. We also prepared a series of negative controls with random sequences of R groups that do not provide a sequence complementary to α-Syn [Fig. 3, see the ESI† for compound synthesis and spectral data].
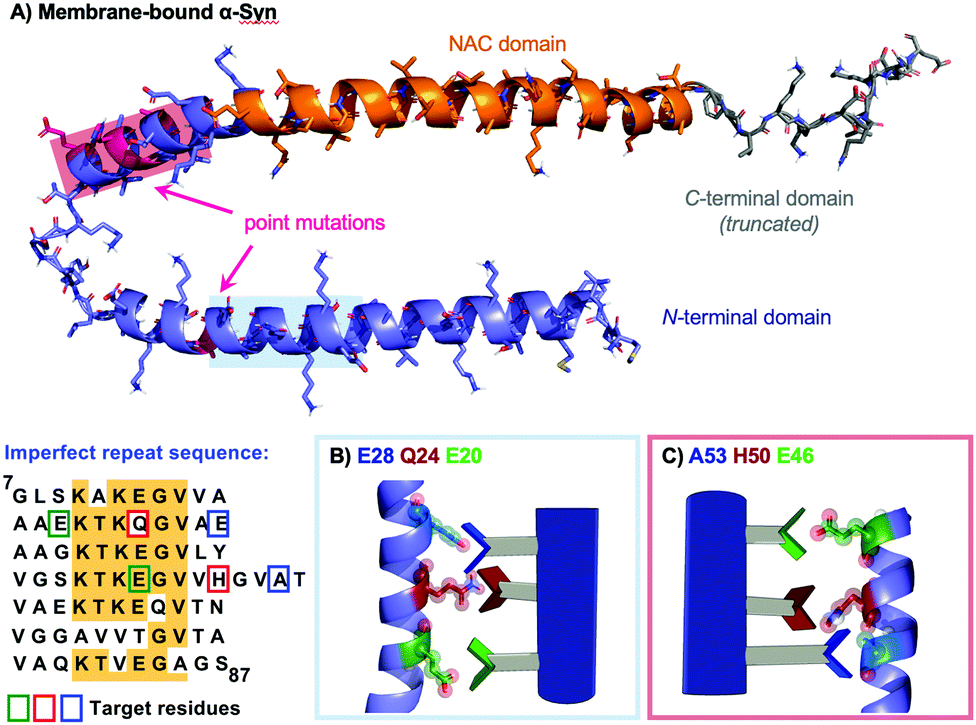 | ||
| Fig. 2 Peptidomimetics designed to present complementary non-covalent contacts with specific protein sequences and conformations. (A) Membrane-bound αSyn (C-term grey, NAC domain orange) with the two helical regions of the N-terminal domain (purple) to be targeted shown with light red and light blue overlays. These regions are coincident with the indicated point mutations (pink). The two sets of target residues are also shown on a sequence map highlighted as green, red and blue boxes. Schematic of mimetics targeting: (B) E20, Q24, E28, (C) E46, H50, A53. | ||
 | ||
| Fig. 3 Peptidomimetics designed to bind specific regions of helical α-Syn through presentation of complementary surfaces: [A] tribenzamide scaffold conformationally pre-organised to reproduce spatial and angular projection of side chains (i, i + 3/4, i + 7/8) along one face of α-helix; [B] library of oligobenzamides displaying various side chains to target specific surfaces. | ||
The focused library of peptidomimetics were evaluated for their ability to slow fibrillation kinetics at a 50 μM concentration using a thioflavin-T (ThT) assay in the presence of a model membrane system. This concentration was chosen based on our previous work in a related study,26 in which this concentration of mimetic successfully stabilised the helical conformation of IAPP in a lipidic environment and prevented fibril formation [Fig. 4 and ESI† for protein expression and purification].36 An increase in the fluorescence intensity is an indication for the formation of ThT-positive aggregates, in particular amyloid fibrils. Mimetics 3 and 4, targeting E46, H50, A53, and 1 and 2, targeting E20, Q24, E28, had a significant effect on fibril formation based on end-point fluorescence relative to α-Syn alone. In the presence of 1–4, in particular 1 and 2, the fluorescence signal showed almost no change over time, which means the monomeric α-Syn did not form ThT-positive aggregates. In contrast, the five negative control compounds demonstrated limited disruption of fibrillation with similar kinetics to those in the absence of compound (black curve). This is also presented as normalised intensity increase [Fig. 4b], relative to protein only (dashed line) and as time to 50% maximum fluorescence, or t1/2 [Fig. 4c], determined by sigmoidal fitting (see the ESI†). These show that in the presence of negative control peptidomimetics 5–9 the final fluorescence intensity approaches, but does not reach, that of α-Syn/lipid alone. Values of t1/2 for the negative control peptidomimetics 5–9 are similar to that for protein/lipid only, whilst for mimetic 3 it slowed t1/2 to around 60 hours. For mimetics 1, 2, and 4 the suppression of intensity increase was so great that it was not possible to produce meaningful t1/2 values via sigmoidal fitting.
 | ||
| Fig. 4 ThT fibrillation kinetics in the presence of 150 μM DMPS liposomes with 10 μM α-Syn: (A) raw data. Absence (black curve) or presence (various colours) of 50 μM peptidomimetics. Each curve represents the average of three replicates, (B) normalised increase in intensity of the raw data (see the ESI†), and (C) halftime of the aggregation (t1/2). The horizontal dotted line represents the control (protein without mimetic) and the arrows mark the compounds where a sigmoidal fitting was not feasible due to a lack of intensity increase. | ||
Encouraged by the ThT data we used electron microscopy to gain insights into the effect of the mimetics on the nature of α-Syn fibril formation under lipid catalysed conditions [Fig. 5]. Both in the absence of compounds [panel A], and in the presence of negative control 5 [panel C], fibrils of differing morphologies are evident. Conversely with compounds 1 and 3 [panels B and D respectively], two mimetics showing pronounced disruption by ThT, no fibrils are apparent.
 | ||
| Fig. 5 Electron microscopy of fibrils formed in the absence [A] or presence of peptidomimetics [B] 1, [C] 5, [D] 3. The EM samples were prepared after incubation from the ThT kinetic assays. | ||
Further evidence to support the hypothesis that peptidomimetics stabilise the α-helical conformation of α-Syn comes from circular dichroism experiments. After an incubation of 16 h at 37 °C in the presence of one of the most promising disruptors of fibrillation, mimetic 1, an α-helical signature (purple curve with local minima at ∼205 and 225 nm) is apparent, whereas this is absent for the analogous experiment with negative control 5 (green), or without added compound [black, Fig. 6]. These two latter curves are indicative of β-sheet conformation, which would be consistent with the onset of fibrillation and the ThT data.
 | ||
| Fig. 6 CD spectra of α-Syn in the presence of DMPS large unilamellar liposomes/vesicles (LUVs) after 16 h incubation (black) and in the presence of peptidomimetic 1 (purple) and negative control 5 (green). | ||
In conclusion, we have shown that α-helical peptidomimetics that are rationally designed to present complementary contacts with helical α-Syn disrupt fibrillation kinetics in the presence of a model membrane. Future work will explore structure/activity optimisation, use of the mimetics at lower concentration and membrane leakage and toxicity. It will also be interesting to explore selective recognition of familial forms of the protein bearing point mutations, in particular: A30P, E46K, H50Q, G51D, A53E/T. Further molecular characterisation of protein:peptidomimetic complexes, including their thermodynamic and kinetic parameters will be investigated using wild-type and conformationally constrained analogues.
We acknowledge financial support from The Universities of Oxford and Southampton, the EPSRC (EP/S028722/1, S. T.) and a studentship for C. E. B. (EP/R513325/1), the Swiss national scientific foundation (310030_197626, J. L.), the Brightfocus foundation (A20201759S, J. L.) and Takeda Pharmaceutical Company Limited for their generosity in providing financial and logistical support during a sabbatical position for T. Y. as a visiting Scientist in The University of Oxford.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. H. Shahmoradian, A. J. Lewis, C. Genoud, J. Hench, T. E. Moors, P. P. Navarro, D. Castaño-Díez, G. Schweighauser, A. Graff-Meyer, K. N. Goldie, R. Sütterlin, E. Huisman, A. Ingrassia, Y. de Gier, A. J. M. Rozemuller, J. Wang, A. D. Paepe, J. Erny, A. Staempfli, J. Hoernschemeyer, F. Großerüschkamp, D. Niedieker, S. F. El-Mashtoly, M. Quadri, W. F. J. Van IJcken, V. Bonifati, K. Gerwert, B. Bohrmann, S. Frank, M. Britschgi, H. Stahlberg, W. D. J. Van de Berg and M. E. Lauer, Nat. Neurosci., 2019, 22, 1099 CrossRef CAS PubMed.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595 CrossRef CAS PubMed.
- C. C. Jao, B. G. Hegde, J. Chen, I. S. Haworth and R. Langen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19666 CrossRef CAS PubMed.
- T. Bartels, J. G. Choi and D. J. Selkoe, Nature, 2011, 477, 107 CrossRef CAS PubMed.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10328 CrossRef CAS PubMed.
- W. Yang, J. L. Hamilton, C. Kopil, J. C. Beck, C. M. Tanner, R. L. Albin, E. Ray Dorsey, N. Dahodwala, I. Cintina, P. Hogan and T. Thompson, npj Parkinson's Dis., 2020, 6, 1 CrossRef PubMed.
- L. S. Shihabuddin, P. Brundin, J. T. Greenamyre, D. Stephenson and S. P. Sardi, J. Neurosci., 2018, 38, 9375 CrossRef CAS PubMed.
- M. Teil, M.-L. Arotcarena, E. Faggiani, F. Laferriere, E. Bezard and B. Dehay, Biomolecules, 2020, 10, 391 CrossRef CAS PubMed.
- J. Yang, M. Gao, J. Xiong, Z. Su and Y. Huang, Protein Sci., 2019, 28, 1952 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef PubMed.
- H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 4130 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161 CrossRef CAS PubMed.
- M. K. P. Jayatunga, S. Thompson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2014, 24, 717 CrossRef CAS PubMed.
- N. Sawyer, A. M. Watkins and P. S. Arora, Acc. Chem. Res., 2017, 50, 1313 CrossRef CAS PubMed.
- T. Flack, C. Romain, A. J. P. White, P. R. Haycock and A. Barnard, Org. Lett., 2019, 21, 4433 CrossRef CAS PubMed.
- W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn, T. A. Hill and D. P. Fairlie, Chem. Rev., 2010, 110, PR32 CrossRef CAS PubMed.
- E. A. German, J. E. Ross, P. C. Knipe, M. F. Don, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2015, 54, 2649 CrossRef CAS PubMed.
- T. Yamashita, P. C. Knipe, N. Busschaert, S. Thompson and A. D. Hamilton, Chem. – Eur. J., 2015, 21, 14699 CrossRef CAS PubMed.
- A. M. Watkins and P. S. Arora, ACS Chem. Biol., 2014, 9, 1747 CrossRef CAS PubMed.
- J. S. Nowick, Acc. Chem. Res., 2008, 41, 1319 CrossRef CAS PubMed.
- R. Sonti, H. N. Gopi, U. Muddegowda, S. Ragothama and P. Balaram, Chem. – Eur. J., 2013, 19, 5955 CrossRef CAS PubMed.
- H. Lingard, J. T. Han, A. L. Thompson, I. K. H. Leung, R. T. W. Scott, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2014, 53, 3650 CrossRef CAS PubMed.
- H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 1 CrossRef PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736 CrossRef CAS PubMed.
- O. V. Kulikov, S. Kumar, M. Magzoub, P. C. Knipe, I. Saraogi, S. Thompson, A. D. Miranker and A. D. Hamilton, Tetrahedron Lett., 2015, 56, 3670 CrossRef CAS.
- H. Peacock, J. Luo, T. Yamashita, J. Luccarelli, S. Thompson and A. D. Hamilton, Chem. Sci., 2016, 7, 6435 RSC.
- D. Maity, S. Kumar, R. AlHussein, L. Gremer, M. Howarth, L. Karpauskaite, W. Hoyer, M. Magzoub and A. D. Hamilton, RSC Chem. Biol., 2020, 1, 225 RSC.
- S. Kumar and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 5744 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2018, 140, 6562 CrossRef CAS PubMed.
- D. Maity, M. Howarth, M. C. Vogel, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2021, 143, 3086 CrossRef CAS PubMed.
- J. Luo and J. P. Abrahams, Chem. – Eur. J., 2014, 20, 2410–2419 CrossRef CAS PubMed.
- J. A. Hebda, I. Saraogi, M. Magzoub, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2009, 16, 943 CrossRef CAS PubMed.
- P. Prabhakaran, A. Barnard, N. S. Murphy, C. A. Kilner, T. A. Edwards and A. J. Wilson, Eur. J. Org. Chem., 2013, 3504 CrossRef CAS.
- G. M. Burslem, H. F. Kyle, A. L. Breeze, T. A. Edwards, A. Nelson, S. L. Warriner and A. J. Wilson, ChemBioChem, 2014, 15, 1083 CrossRef CAS PubMed.
- A. Barnard, K. Long, H. L. Martin, J. A. Miles, T. A. Edwards, D. C. Tomlinson, A. Macdonald and A. J. Wilson, Angew. Chem., Int. Ed., 2015, 54, 2960 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. T. Michaels, M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cc00212d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |