
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivities of allenic and olefinic Michael acceptors towards phosphines†
Feng
An
 a,
Harish
Jangra
a,
Yin
Wei
b,
Min
Shi
*bc,
Hendrik
Zipse
*a and
Armin R.
Ofial
*a
a,
Harish
Jangra
a,
Yin
Wei
b,
Min
Shi
*bc,
Hendrik
Zipse
*a and
Armin R.
Ofial
*a
aDepartment Chemie, Ludwig-Maximilians-Universtität München, Butenandtstr. 5-13, München 81377, Germany. E-mail: zipse@cup.uni-muenchen.de; ofial@lmu.de
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China. E-mail: mshi@mail.sioc.ac.cn
cKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 MeiLong Road, Shanghai 200237, P. R. China
First published on 21st February 2022
Abstract
The kinetics of the reactions of tributylphosphine with allenic and olefinic Michael acceptors in dichloromethane at 20 °C was followed by photometric and NMR spectroscopic methods. Combination with DFT-calculated methyl anion affinities revealed the relevance of retroaddition barriers in phosphine-catalysed reactions when mixtures of allenic and olefinic substrates are used.
In 1995, the Lu group discovered (3+2)-cycloadditions, in which Bu3P or Ph3P are used as Lewis-basic catalysts to furnish cyclopentenes from mixtures of alkyl allenoates and electron-deficient alkenes (Fig. 1).1 In recent years, the versatility of Lu cycloadditions was expanded by the development of several catalytic asymmetric versions, which were also applied to the synthesis of core units of natural products.2 Computational and kinetic studies showed that phosphonium–dienolate formation can be considered to be the rate-determining step in the catalytic cycle.2a,3
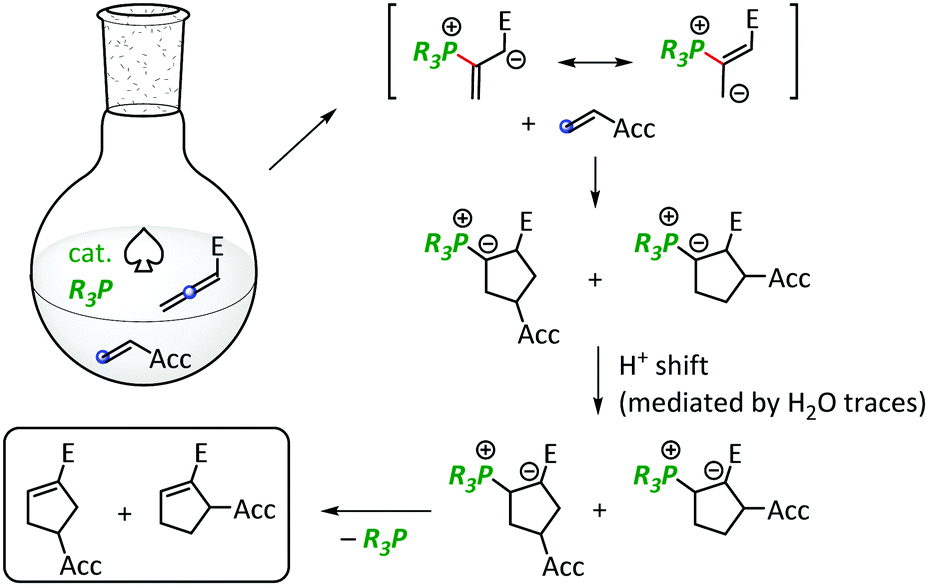 | ||
| Fig. 1 Mechanism of the phosphine-catalysed Lu cycloaddition (E = ester group, Acc = electron-accepting group). | ||
The rates of adduct formations, in which only one new bond is formed between an electrophile and a nucleophile, can be discussed in a broader context and beyond the limitations of structurally analogous compound classes when the Mayr–Patz eqn (1) is used.4–7 Thus, the nucleophilic reactivity of R3P catalysts in a certain solvent is described by the two parameters N (nucleophilicity) and sN (susceptibility) in eqn (1).8 The reactivities of Michael acceptors are characterised by electrophilicity parameters E. It has been shown that once E, N and sN of reaction partners in polar reactions are known the second-order rate constant k2 for adduct formation can be predicted with an accuracy within two orders of magnitude.5,6
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k2(20 °C) = sN(N + E) k2(20 °C) = sN(N + E) | (1) |
Many of the R3P-catalysed reactions are performed with mixtures of two competing electrophiles, which are often combined in a way that one of the electrophiles carries an sp-and the other one an sp2-hybridised carbon as the electrophilic centre.1,2,9 In this work, we set out to determine the kinetics of the adduct formation of R3P with electron-deficient olefins and a set of alkyl and phenyl allenoates to gain a deeper understanding of the factors that influence the initial step of the related R3P-catalysed organic reactions.1,2,9
First, we characterised the vinyl phosphonium triflates 3 obtained by Ph3P reactions with 1a, 1d, 1f, and 1i (Fig. 2A) by spectroscopic methods (ESI†). It is reasonable to assume that the entire set of R3P + 1a–1j reactions that we followed kinetically also yield vinyl phosphonium triflates 3. The kinetics of the carbon–phosphorus bond-formations between R3P and 1a–1j was followed by spectroscopic methods.
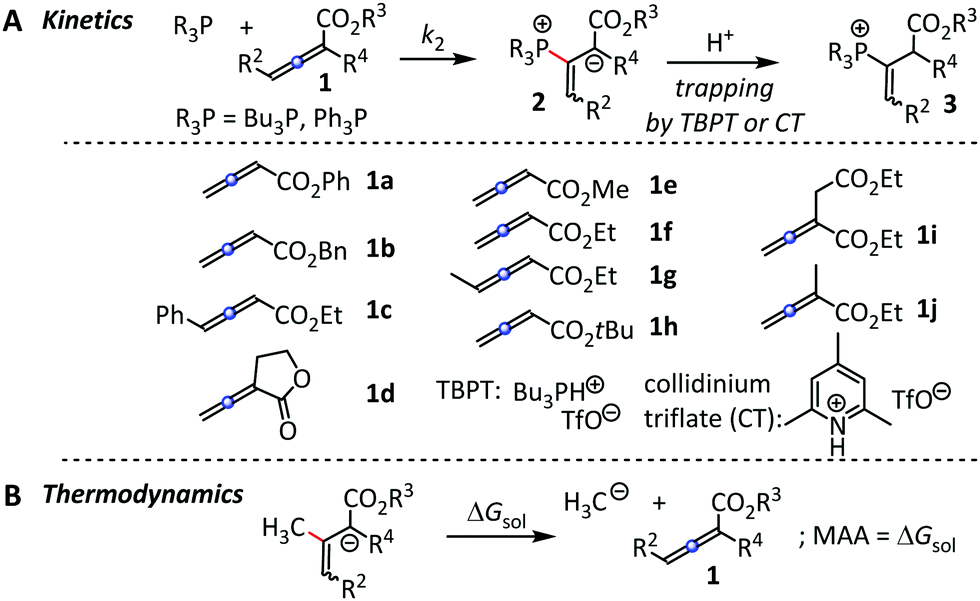 | ||
| Fig. 2 (A) R3P additions to 1 and (B) reference reaction for the determination of methyl anion affinities (MAA) by quantum-chemical methods. | ||
Reactions of Bu3P with alkyl allenoates 1a–1i in CH2Cl2 at 20 °C were monitored photometrically by following the decrease of the UV absorptions of 1 at or close to their absorption maximum (λmax = 242–262 nm). The Bu3P addition reactions to 1 give rise to polymerisation, which can be avoided by trapping the zwitterionic adducts 2 with tributylphosphonium triflate (TBPT) as the proton source ([TBPT]/[1]0 = 2–3). TBPT is a weak Brønsted acid in CH2Cl2 and does not activate the electrophiles 1 by hydrogen-bonding to an oxygen of the ester group (as shown for the combination of 1f + TBPT, ESI,† Fig. S7). To simplify the kinetic evaluation of the second-order reactions, we used the Bu3P in at least 10-fold excess relative to the initial electrophile concentrations [1]0. Hence, the decrease of absorptions A of 1 could be fitted by the mono-exponential decay function A = A0exp(−kobst) + C to determine the (pseudo) first-order rate constants kobs (s−1).
For each Bu3P + 1 pair, kobs was determined at four or five different Bu3P concentrations, which made it possible to calculate the second-order rate constants k2 (M−1 s−1) from the slope of the linear correlation of kobs with [Bu3P]0. Fig. 3 visualises this procedure for the Bu3P addition to 1f.
 | ||
| Fig. 3 (A) Kinetics of the reaction of Bu3P with 1f: mono-exponential decay of the absorbance A in the reaction of Bu3P (12.3 mM) with 1f (0.121 mM); (B) linear correlation of observed rate constants kobs with [Bu3P]0. | ||
Rate constants of the reactions of Ph3P with 1a–1j were determined by 1H NMR spectroscopy using mesitylene as an internal integration standard and collidinium triflate (CT) as a proton source ([CT]/[1]0 = 2). Generally, Ph3P additions to 1 are endergonic with retroadditions being faster than the addition reactions. Trapping of the zwitterionic intermediates 2 by CT is thus necessary to observe the kinetics of the addition (forward) reaction. By obeying the conditions for (pseudo)first-order kinetics, that is [Ph3P]0/[1]0 > 9, the time-dependent decrease of [1] could be fitted by the mono-exponential decay function to yield the rate constants kobs. NMR experiments at four different [Ph3P]0 made it possible to determine the second-order rate constants k2 for the Ph3P additions to the electrophiles 1a–1j from the slope of the linear correlation of kobs with [Ph3P]0. Rate constants (k2) for the reactions of Bu3P with 1j were determined analogously. Data of the individual kinetic measurements for the nucleophilic attack of Bu3P and Ph3P at the allenoates 1 are given in the ESI.† The experimentally determined second-order rate constants k2 are compiled in Table 1.
| Electrophiles 1 | k 2(Bu3P)a | k 2(Ph3P)b | MAAc | PAd |
|---|---|---|---|---|
| a Photometry, CH2Cl2, 20 °C. b 1H NMR spectroscopy, CD2Cl2, 20 °C. c MAA as defined in Fig. 2B [in kJ mol−1 at SMD(DMSO)/B3LYP/6-311++G(3df,2pd)//B3LYP/6-31G(d,p) level of theory, with Truhlar quasi-harmonic treatment]. d Phosphine affinities, PA, as defined in ESI, Table S42 [in kJ mol−1 at PCM(DCM,ua0)/B3LYP-D3/6-31+G(d,p) level of theory, with Truhlar quasi-harmonic treatment]. e MAA values from ref. 10. | ||||
| 1a | 5.52 | 1.19 × 10−1 | 182.5e | −15.0 |
| 1b | 1.16 | 1.70 × 10−2 | 163.1e | −22.6 |
| 1c | 1.10 | 1.90 × 10−2 | 165.5 | −23.7 |
| 1d | 9.55 × 10−1 | 2.33 × 10−2 | 171.2 | −15.9 |
| 1e | 8.40 × 10−1 | 1.10 × 10−2 | 167.6e | −26.5 |
| 1f | 6.35 × 10−1 | 7.67 × 10−3 | 163.4e | −29.8 |
| 1g | 5.00 × 10−1 | 8.05 × 10−3 | 152.7 | −29.5 |
| 1h | 2.47 × 10−1 | 2.39 × 10−3 | 153.9 | −33.3 |
| 1i | 2.01 × 10−1 | 4.59 × 10−3 | 142.5 | −26.0 |
| 1j | 1.96 × 10−2b | 3.49 × 10−4 | 133.6 | −38.3 |
The Bu3P-based reactivity scale (logk2) for 1a–1j is depicted in Fig. 4A. The phenyl ester 1a is the strongest electrophile of the studied allenoates without further substituents at the cumulated π-system and reacts about 5 to 9 times faster with Bu3P than the analogous benzyl (1b), methyl (1e), or ethyl esters (1f).11 Replacement of the ethyl by a t-butyl group (1f→1h) attenuates reactivity by a factor of 3. An additional methyl group (1g) at the 4-position of the alkyl allenoate has almost no influence on the reactivity of the electrophilic π-system, which remains at the level of the γ-unsubstituted 1f. Extending the π-system by a terminal phenyl group enhances the reactivity of 1c only slightly if compared to that of 1f, probably because the phenylvinyl moiety in 1c is perpendicular to the reactive π-system. In contrast, substituents in 2-position of 1 have a greater effect, and Bu3P reacts slower by a factor of 32 with 1j than with the parent 1f. This reactivity gap can be reduced by attaching an electron-accepting group to the C-2 substituent. Thus, 1i reacts 10-fold faster with Bu3P than 1j and is only 3 times less electrophilic (towards Bu3P) than 1f.
 | ||
| Fig. 4 (A) Relative reactivities of 1a–1j in reactions with Bu3P (in CH2Cl2, 20 °C). (B) Different stereoelectronic effects in 1d and the open-chain analogue 1j. | ||
Consistent with previous work on the relative reactivity of open chain esters and lactones,12 α-allenic γ-butyrolactone 1d is a significantly more reactive electrophile than 1j. The increase in electrophilic reactivity is explained by the different conformations of the CO–OR bonds, which is preferably in the s-(Z) conformation in 1j but fixed in the unfavourable s-(E) conformation in 1d (Fig. 4B).12,13 Ineffective nO→σ*CO interactions enhance the electron-deficiency of the reacting π-system, which enables the versatile use of allenic lactones in organic synthesis.14
Reactivity of 1 towards Bu3P is transferrable to other R3P as shown by the linear correlation (r2 = 0.9636, n = 10) with a slope (1.02) close to unity for the Ph3P vs. Bu3P comparison (Fig. 5).
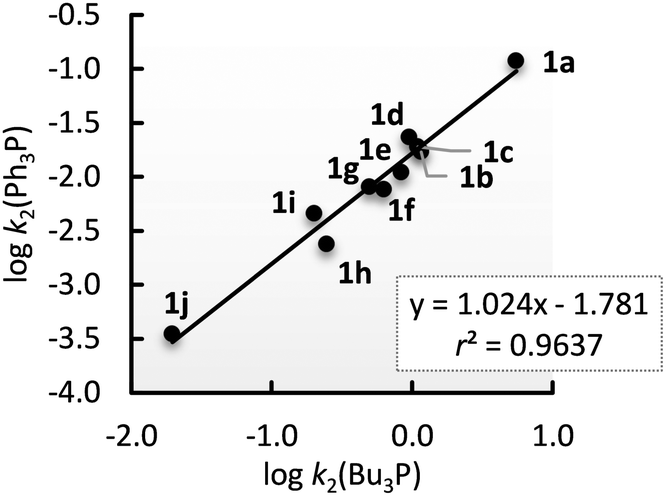 | ||
| Fig. 5 Linear relation of logk2(Bu3P) and logk2(Ph3P) for reactions with 1. | ||
To gain a better understanding of R3P-catalysed Lu reactions it is crucial to compare the R3P reactivities of 1 with those of competing electrophiles, which are typically olefinic Michael acceptors. It was previously shown that MAAs of olefinic Michael acceptors correlate linearly with their Mayr E parameters.15 MAA values have also been applied to rationalise R3P-catalysed (3+2) annulations of 1a, 1b, 1e, and 1f with 2-aminoacrylates.10 The data in Table 1 now show that the DFT-calculated MAA values for 1 (Table 1) are linearly related with their electrophilic reactivities towards the investigated R3P nucleophiles, that is, Bu3P and Ph3P (ESI,† Fig. S1 and S2). A linear correlation of similar quality was obtained when logk2 for 1 + Ph3P reactions were plotted against phosphine affinities, PA, which are defined analogously to MAA but use Ph3P instead of the methyl anion as the Lewis base (Fig. S3, ESI†).
The nucleophilic reactivity of Bu3P has previously been characterised by N = 15.49 (sN = 0.69) on the basis of the kinetics of its additions to benzhydrylium ions (E > −10.04).16 To avoid long range extrapolations for predicting reaction rates with Michael acceptors, we determined the kinetics of further reactions of Bu3P with the structurally diverse neutral and positively charged electrophiles 4a–4r of known Mayr E.7 Second-order rate constants k2 (CH2Cl2, 20 °C) were determined by applying the photometric methods described above for the 1 + Bu3P reactions (see ESI† for individual rate constants). Fig. 6 shows that logk2 for the additions of Bu3P to 4a–4r follow a linear correlation (r2 = 0.9635, n = 18) over a range of 16 units on the Mayr E scale, which gives N(Bu3P) = 19.11 and sN = 0.48. Applying the k2 values for reactions of 1 (Table 1) in the correlation depicted in Fig. 6 indicates that 1a–1j are located in the reactivity range −22.7 < E < −17.5.
 | ||
| Fig. 6 Linear correlation of logk2(Bu3P) with the Mayr E parameters of structurally diverse Michael acceptors 4a–o and benzhydrylium ions 4p–r (k2 in dichloromethane at 20 °C, see ESI† for details of the kinetic measurements, Table S1 (ESI†) gathers the data used to construct the correlation). | ||
In classical Lu reactions, the R3P catalyst first attacks the alkyl allenoate. The catalytic cycle continues with the reaction of a C-nucleophilic zwitterion with the second Michael acceptor in the reaction mixture. If the kinetics of the R3P reactions with the two competing electrophiles would be the decisive factor, Lu reactions could be expected to occur only if 1 is more reactive towards the R3P catalyst than the competing Michael acceptor (e.g., 4). This is not always the case, however. Already in the first publication on the R3P-catalysed cycloaddition, the Lu group1 used electrophiles with Mayr E < −16.8, that is, with comparable or even slightly higher electrophilicity than for 1. This indicates that the thermodynamics for R3P adduct formation is another crucial factor for the success of Lu reactions.
For reactions of vinyl cations with nucleophiles it has been observed that sp/sp2 rehybridisation occurs via higher Marcus intrinsic barriers than for reactions that involve sp2/sp3 rehybridisations.17,18 Analogously, in this work the higher MAA for 1 than for equally reactive olefinic Michael acceptors15a (Fig. 7) along with the experimental kinetic data indicate that the main difference between 1 and Michael acceptors, such as 4, are the Marcus intrinsic barriers for the R3P addition at the differently hybridised electrophilic centres.
 | ||
| Fig. 7 (A) Separate linear relationships between the electrophilicities E of acceptor-substituted olefins M1–M44 and allenes 1 with the calculated methyl anion affinities (MAA) (molecular structures of M1–M44 and data used to construct the linear correlation are given in ESI,† Table S2; electrophilicities E for allenoates 1 estimated by applying the k2(Bu3P) values from Table 1 in the correlation equation for Bu3P additions to Michael acceptors in Fig. 6). (B) MAA values and electrophilicities E for Michael acceptors 4s–4v. | ||
Even if the rate constants for the electrophile/R3P additions are of the same order of magnitude, the MAA values show that allenoates 1 are considerably stronger Lewis acids than olefinic Michael acceptors or imines, such as 4t. As a consequence, the differences in the barriers for the retroadditions differentiate the two competing classes of electrophiles. Owing to their higher energetic barrier for retroaddition, only the allenic electrophiles 1 generate sufficiently high concentrations of reactive zwitterions, which are the pivotal intermediates for the subsequent ring-forming reactions. Thus, allenoates 1 are capable to compete with much stronger electrophiles. For example, 1f (MAA = 163 kJ mol−1) can be used as a partner for the more electrophilic yet less Lewis acidic 4s, 4t, or 4u in Lu cycloadditions.19–21 Benzylidenemalononitrile (4v) seems to be an exception. However, despite of its high MAA, 4v reacts reversibly with Bu3P and does not form an adduct with Ph3P (ESI†). Yet, free 4v traps efficiently the zwitterion generated by Ph3P and 1f to yield cyclopentenes.22,23
The reactivities of allenic and olefinic Michael acceptors have been calibrated towards P-nucleophiles through determining the kinetics of their reactions with Bu3P. Allenoates 1 are weaker electrofuges as well as weaker electrophiles than Michael acceptors 4 of similar Lewis acidity because of the higher intrinsic barriers for sp/sp2 rehybridisation. The kinetic and thermodynamic data in this work will be instrumental for the design of novel R3P-catalysed reactions with alkyl allenoates.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG), project number 410831260, and the National Natural Science Foundation of China (NSFC) 21861132014.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS; (b) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (c) Y. Wei and M. Shi, Org. Chem. Front., 2017, 4, 1876 RSC.
- (a) Y. C. Fan and O. Kwon, in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH,Weinheim (Germany), 1st edn, 2016, ch. 15, p. 715 Search PubMed; (b) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed; (c) M. Shi, Y. Wei, M.-X. Zhao and J. Zhang, Organocatalytic Cycloadditions for Synthesis of Carbo- and Heterocycles, Wiley-VCH, Weinheim, 2018 CrossRef; (d) Y. Huang, J. Liao, W. Wang, H. Liu and H. Guo, Chem. Commun., 2020, 56, 15235 RSC; (e) Y. Wei and M. Shi, Chin. J. Chem., 2020, 38, 1395 CrossRef CAS; (f) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536 CrossRef CAS PubMed.
- (a) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem. – Eur. J., 2008, 14, 4361 CrossRef CAS PubMed; (b) G.-T. Huang, T. Lankau and C.-H. Yu, J. Org. Chem., 2014, 79, 1700 CrossRef CAS PubMed.
- H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938 CrossRef.
- (a) H. Mayr and A. R. Ofial, Pure Appl. Chem., 2005, 77, 1807 CAS; (b) H. Mayr, Angew. Chem., Int. Ed., 2011, 50, 3612 CrossRef CAS.
- (a) H. Mayr and A. R. Ofial, SAR QSAR Environ. Res., 2015, 26, 619 CrossRef CAS PubMed; (b) H. Mayr, Tetrahedron, 2015, 71, 5095 CrossRef CAS.
- For a freely accessible database of E, N, and sN parameters, see: https://www.cup.lmu.de/oc/mayr/DBintro.html.
- S. Lakhdar, in Lewis Base Catalysis in Organic Synthesis, ed. E. Vedejs and S. E. Denmark, Wiley-VCH, Weinheim (Germany), 1st edn, 2016, ch. 4, p. 85 Search PubMed.
- B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC.
- X.-Y. Wu, H.-Z. Gui, H. Jangra, Y. Wei, H. Zipse and M. Shi, Catal. Sci. Technol., 2020, 10, 3959 RSC.
- A ratio of k(1a)/k(1f) = 8 was reported for the reactions with pyrrolidine (in CHCl3): C. Kiattisewee, A. Kaidad, C. Jiarpinitnun and T. Luanphaisarnnont, Monatsh. Chem., 2018, 149, 1059 CrossRef CAS.
- (a) R. J. Mayer, P. W. A. Allihn, N. Hampel, P. Mayer, S. A. Sieber and A. R. Ofial, Chem. Sci., 2021, 12, 4850 RSC; (b) P. M. Jüstel, A. Stan, C. D. Pignot and A. R. Ofial, Chem. – Eur. J., 2021, 27, 15928 CrossRef PubMed.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil’, I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent’ev and M. A. Zolfigol, Chem. Soc. Rev., 2021, 50, 10253 RSC ; 10700; and ref. cited therein.
- (a) W. Yao, X. Dou, S. Wen, J. Wu, J. J. Vittal and Y. Lu, Nat. Commun., 2016, 7, 13024 CrossRef PubMed; (b) B. Mao, W. Shi, J. Liao, H. Liu, C. Zhang and H. Guo, Org. Lett., 2017, 19, 6340 CrossRef CAS PubMed; (c) W. Shi, B. Mao, J. Xu, Q. Wang, W. Wang, Y. Wu, X. Li and H. Guo, Org. Lett., 2020, 22, 2675 CrossRef CAS PubMed ; 6600.
- (a) D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2017, 139, 13318 CrossRef PubMed; (b) A. Mood, M. Tavakoli, E. Gutman, D. Kadish, P. Baldi and D. L. Van Vranken, J. Org. Chem., 2020, 85, 4096 CrossRef CAS PubMed.
- B. Kempf and H. Mayr, Chem. – Eur. J., 2005, 11, 917 CrossRef CAS PubMed.
- P. A. Byrne, S. Kobayashi, E.-U. Würthwein, J. Ammer and H. Mayr, J. Am. Chem. Soc., 2017, 139, 1499 CrossRef CAS PubMed.
- (a) R. A. Marcus, J. Phys. Chem., 1968, 72, 891 CrossRef CAS; (b) W. J. Albery, Annu. Rev. Phys. Chem., 1980, 31, 227 CrossRef CAS.
- Q. Zhao, X. Han, Y. Wei, M. Shi and Y. Lu, Chem. Commun., 2012, 48, 970–972 RSC.
- X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716 CrossRef CAS PubMed.
- L. Birbaum, L. Gillard, H. Gerard, H. Oulyadi, G. Vincent, X. Moreau, M. De Paolis and I. Chataigner, Chem. – Eur. J., 2019, 25, 13688 CrossRef CAS PubMed.
- X. Lu, Z. Lu and X. Zhang, Tetrahedron, 2006, 62, 457 CrossRef CAS.
- For [4+2] cycloadditions of 4v with 1i and 1j, see: Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tables and figures, procedures for the synthesis and spectroscopic characterisation of 1, product studies, details of the kinetic experiments and DFT calculations. See DOI: 10.1039/d1cc06786a |
| This journal is © The Royal Society of Chemistry 2022 |