
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeteroatom oxidation controls singlet–triplet energy splitting in singlet fission building blocks†
J. Terence
Blaskovits
 ,
Maria
Fumanal
,
Sergi
Vela
,
Yuri
Cho
and
Clémence
Corminboeuf
*
,
Maria
Fumanal
,
Sergi
Vela
,
Yuri
Cho
and
Clémence
Corminboeuf
*
Laboratory for Computational Molecular Design (LCMD), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: clemence.corminboeuf@epfl.ch
First published on 24th December 2021
Abstract
Singlet fission (SF) is a promising multiexciton-generating process. Its demanding energy splitting criterion – that the S1 energy must be at least twice that of T1 – has limited the range of materials capable of SF. We propose heteroatom oxidation as a robust strategy to achieve sufficient S1/T1 splitting, and demonstrate the potential of this approach for intramolecular SF.
Singlet fission (SF) has shown potential to improve the power conversion efficiency in photovoltaic devices beyond the Shockly–Queisser limit by promoting the splitting of a photon-absorbing singlet exciton into two triplet excitons.1 SF involves the excitation of a ground state (S0) chromophore to an excited singlet state (S1) upon absorption of light, followed by energy transfer to a second chromophore. The initial S1 state is coupled to a triplet pair (1TT) state, a process which may be mediated by low-lying charge transfer (CT) states or may proceed directly, via a resonance mechanism.2,3 The triplet pair then evolves into physically separate and energetically independent triplets (T1), one on each chromophore.
Among the requirements necessary for a system to be capable of SF, the most stringent is that the process be thermodynamically possible, meaning that the energy of the S1 state must be no less than twice that of the T1 state: ΔEST = E(S1) − 2E(T1) ≥ 0. This energy splitting term ΔEST is therefore the most relevant target property in the discovery of new SF materials.1,4,5 It has been shown that (i) extending the conjugation, and increasing the (ii) biradicaloid4 or (iii) quinoidal6 character of a chromophore can improve ΔEST, as summarized in Scheme 1. These strategies have drawbacks. For instance, compounds with high diradicaloid and quinoidal character tend to suffer from chemical instability.7
 | ||
| Scheme 1 Proposed strategies to increase singlet–triplet splitting (ΔEST) in organic chromophores. | ||
A particular challenge arises in designing materials which fall into the ΔEST ≥ 0 regime: the S1 and T1 energies tend to move in parallel. When the excitation energies are stabilized to the point that ΔEST is fulfilled, T1 is often too low to be of value for device applications. A historically relevant example of this is the acene family, in which the excited state energies decrease with an increasing number of fused rings. In early reports of SF, in anthracene (3 rings), SF was not favored due to a negative ΔEST and, therefore, the endothermicity of SF.8 This was also the case for tetracene (4 rings),9 while pentacene (5 rings) became the poster child for SF due to it being the first acene in which SF is exergonic (ΔEST > 0), although its T1 energy is already somewhat lower (0.9 eV) than desirable.10 The next acene, hexacene,11 exhibits much more favorable ΔEST for SF, but has a far too low T1 energy (0.4 eV).
A moiety which stabilizes T1 incrementally to the point that it can be tuned to remain above 1 eV (for exciton injection into silicon for instance, whose band gap is 1.1 eV) without also lowering S1 substantially would be greatly beneficial (Scheme 1, right panel). Here, we identify a chemical functionality, heteroatom oxidation, which modulates the ΔEST in potential SF chromophores in a foreseeable way (Scheme 1 (iv)). This approach is motivated by the experimental observation that the oxidized form of thiophene (thiophene-S,S-dioxide) is an effective acceptor in donor–acceptor copolymers capable of intramolecular SF (iSF),12–14 and that nitrone/N-oxide groups were found in large numbers in a recent screening of thousands of crystal structures for compounds with high ΔEST.5 The present results show that, indeed, heteroatom oxidation governs the S1 and T1 energies and thus can be used to improve the singlet–triplet splitting of potential SF chromophores.
To establish if a systematic improvement in ΔEST can be achieved through heteroatom oxidation, we constructed a dataset consisting of 11 heteroatom-containing building blocks found widely in the organic electronics literature (see Fig. S1, ESI† for full dataset). These are classified by the number of heteroatoms in the conjugated system, as shown in Scheme 2a. All oxidized derivatives of these compounds were generated by placing one oxygen atom at the electron pair of all sp2-hybridized nitrogen atoms, thereby forming N-oxide (nitrone) moieties, and one or two oxygen atoms at the electron pairs of all sulfur atoms, forming S-oxide or S,S-dioxide moieties, respectively (as shown for bithiophene in Scheme 2b). This produced a total of 67 oxidized compounds (all structures shown in ESI†). The oxidation of sp3 nitrogens was not considered, as this would lead to charged or radical species. Compound geometries were relaxed using density functional theory (ωB97X-D/6-31G*), and the S1 and T1 exited-state energies were computed both vertically and at their minima using time-dependent DFT within the Tamm–Dancoff approximation at the same level of theory (see ESI† for details).
 | ||
| Scheme 2 (a) Examples of building blocks studied in this work. (b) Five oxidized derivatives of 2,2′-bithiophene. | ||
Representative results for the effect of oxidation on the vertical and adiabatic S1, T1 and ΔEST energies of bithiophene are shown in Fig. 1a and results for all other compounds are given in the ESI.† We observe a constant difference between the vertical and adiabatic excitation energies. This allows us to extend our previous observation in dimers13 – that the adiabatic energy splitting cutoff (ΔEadiaST ≥ 0 eV) can be expressed as ΔEvertST ≥ −1 eV in the Franck–Condon regime – to smaller (monomer) building blocks, due to the linear relationship between the two ΔEST values (Fig. S2, ESI†). Although it is immediately clear that increasing the number of oxygens (regardless of their position) stabilizes T1 in an additive fashion across all compounds, the effect on S1 is less evident. While mono-oxidation of sulfur leads to a sharp reduction in both S1 and T1 energies, which has little positive effect on ΔEST, a second oxidation of the same sulfur increases the S1 energy while further stabilizing T1, leading to a strong improvement in ΔEST. For example, the dioxide derivatives of bithiophene have ΔEadiaST ≥ 0 eV (above the grey line in Fig. 1a) while bare bithiophene and its mono-oxidized derivatives do not. The same conclusions can be drawn with all other sulfur-containing units (Fig. S3–S6, ESI†): as outlined in Fig. 1b, single sulfur oxidations have little effect on ΔEST as they stabilize S1 more than T1, while double oxidations of sulfur are invariably beneficial to ΔEST due to their similar stabilization of T1 but smaller impact on S1. N-Oxidation systematically improves ΔEST through a robust stabilization of T1 (Fig. S7 and S8, ESI†). These trends are observed regardless of the degree of oxidation of all others heteroatoms, resulting in a remarkably simple cumulative effect across all compounds: the most highly oxidized structures have the highest ΔEST of all combinations of oxidation products (Fig. 1 and Section S2 of ESI†).
 | ||
| Fig. 1 (a) Vertical and adiabatic S1, T1, and ΔEST energies of bithiophene and its oxidized derivatives. Grey line indicates the ΔEST cut-off. (b) Summary of the change of adiabatic S1, T1, and ΔEST upon S-, S,S- and N-oxidation for all compounds, showing averages (white points), 1st–3rd quartiles (black bars), and maximal/minimal values (whiskers). See ESI† for details. | ||
To understand the effect of oxidation on S1 and T1 energies, we turned to the nature of the excitations in the simplest sub-unit, thiophene (Table 1). The T1 state of thiophene is dominated by a local HOMO → LUMO transition in the carbon backbone, while the S1 state involves predominantly charge transfer (CT) from sulfur (HOMO−1) into the backbone. In thiophene-S-oxide, the T1 excitation is a localized HOMO−1 → LUMO transition on the backbone, as the HOMO is located on the oxygen. It is instead the S1 excitation which corresponds to a HOMO → LUMO CT state from oxygen into the backbone π* orbital, which explains the significant stabilization of S1 upon mono-oxidation. Finally, in thiophene-S,S-dioxide both S1 and T1 are characterized by backbone HOMO → LUMO (π → π*) transitions, as the O and S orbitals are much lower in energy.
| Compound | State | State character | Orbitals | E (eV) | ΔESTvert (eV) | |
|---|---|---|---|---|---|---|

|
Th | T1 | Local BB | H → L | 3.90 | −1.47 |
| S1 | CT S-to-BB | H−1 → L | 6.33 | |||

|
Th-1O | T1 | Local BB | H−1 → L | 3.01 | −2.24 |
| S1 | CT O-to-BB | H → L | 3.78 | |||
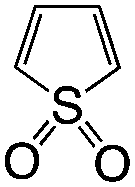
|
Th-2O | T1 | Local BB | H → L | 2.80 | −0.94 |
| S1 | Local BB | H → L | 4.66 | |||
Similarly, in benzodithiophenedione (BDO) and thienopyrroledione (TPD, see Fig. S12, ESI†), S1 is stabilized by CT states from the oxygen n orbitals into the π* orbital of the backbone. The difference compared to thiophene is that BDO and TPD already contain oxygens in their conjugated systems by virtue of their carbonyls, such that S1 in non-oxidized TPD and BDO is described by CT from the carbonyls into the heterocycle. A first S-oxidation stabilizes S1 through CT from the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, as with thiophene-S-oxide, while a second oxidation shifts the source of CT back to the carbonyls. Therefore, the nature of the CT (i.e. the n orbitals involved) changes, depending on the structure of the unit, but the stabilizing effect of a single S-oxidation on S1 remains constant across all compounds. And yet, the oscillator strength of S1 tends to drop significantly for S-mono-oxidized compounds (see Fig. S14, ESI†), which may have consequences on the SF decay pathway and overall mechanism.15 Less impact is expected on the photophysical properties of the S1 state of S,S- and N-oxidized compounds.
O moiety, as with thiophene-S-oxide, while a second oxidation shifts the source of CT back to the carbonyls. Therefore, the nature of the CT (i.e. the n orbitals involved) changes, depending on the structure of the unit, but the stabilizing effect of a single S-oxidation on S1 remains constant across all compounds. And yet, the oscillator strength of S1 tends to drop significantly for S-mono-oxidized compounds (see Fig. S14, ESI†), which may have consequences on the SF decay pathway and overall mechanism.15 Less impact is expected on the photophysical properties of the S1 state of S,S- and N-oxidized compounds.
Recent work has rationalized ΔEST based on ground- and excited-state aromaticity.16,17 To explain the effect of these substitutions on aromaticity, we computed the nucleus independent chemical shifts (NICS) of thiophene, TPD, and thiazole (Fig. S15, ESI†). While thiophene is aromatic in the ground state and anti-aromatic in the first triplet state, thiophene-S-oxide is much less aromatic in the ground state, but still significantly anti-aromatic in the triplet. The absence of lone pairs on the sulfur atom of thiophene-S,S-dioxide leads to non-aromatic character of both the ground state singlet and first triplet. This is reflected in the bond order of the backbone which, like butadiene, is reversed in the triplet (CH–CHCH–CH) compared to the singlet (CHCH–CHCH). This is not the case for non-oxidized or mono-oxidized thiophene (Fig. S16, ESI†). The TPD ring aromaticity is similarly suppressed upon the double oxidation of sulfur. Put together, these results suggest that the destabilization of S1 upon S,S-dioxidation, and its consequently beneficial effect on ΔEST, originate from the SO2 moiety eliminating the aromatic character of the heterocycle and instead inducing a polarized butadiene-like behavior to the backbone.18 In this way, T1 is sufficiently stabilized, while the stabilizing effect of CT from S (in thiophene rings) or O (in thiophene-S-oxide rings) into the π-system observed in S1 is eliminated. This is similar to the ‘breaking’ of conjugation in polycyclic hydrocarbons through boron-doping, an approach proposed to build molecules that fulfill ΔEST.19
CT, primarily from oxygen into sulfur (HOMO → LUMO+1), also accounts for the stabilization of S1 in N-oxidized benzothiadiazole (BT, see Fig. S13, ESI†), compared to non-oxidized BT, which has a local π → π* (HOMO → LUMO) character. The T1 states are also described by a π → π* transition in both non-oxidized and N,N′-dioxidized BT. The inclusion of the N–O moiety in the conjugated system reduces the π/π* energy gap, leading to an extreme lowering of both the S1 and T1 energies by approximately 2 eV. N-Oxidation has the effect of strongly reducing the antiaromatic character of the triplet (Fig. S15, ESI†), while retaining ground state aromaticity, explaining the significant T1 stabilization and consequent increase in ΔEST.
iSF has been demonstrated experimentally in donor–acceptor (D–A) polymers,12,20,21 in which the triplet pair formation is mediated through low-lying donor-to-acceptor CT states, while the spatial separation of the acceptors by the absorbing donor leads to a weakly bound 1TT state. We have recently proposed a protocol with which to identify potential polymer candidates for iSF based on the ΔEST of the constituent monomers and their relative frontier molecular orbital (FMO) energies.13,14 To assess the performance of these new oxidized units to form iSF-capable D–A pairs, we treat all those in the dataset that fulfill ΔEadiaST ≥ 0 (and ΔEvertST ≥ −1 eV, vide supra) as acceptor monomers (34 compounds). The FMOs of each acceptor were compared to all other buildings blocks (2244 monomer pairs), and only those whose FMO arrangement is conducive to CT (i.e. donor HOMO higher than acceptor HOMO and donor LUMO higher than acceptor LUMO; see earlier work for details14) were retained. For these 631 D–A combinations, the dimers were generated, their ground state geometries optimized, and their vertical excited states were evaluated at the same level of theory as the monomers. All dimers exhibit energy splitting above the vertical threshold ΔEvertST ≥ −1 eV (Fig. S17, ESI†), which is consistent with our observation14 that the dimer ΔEST originates from the monomer with the higher (i.e. more positive) ΔEST.
We have previously outlined two other requirements beyond ΔEST for iSF to be possible in D–A systems: S1 must have significant donor-to-acceptor CT character to drive triplet-pair formation (S1 → 1TT), and T1 must be located on the acceptor to promote dissociation of the triplet states (1TT → T1 + T1).13,14 Deactivation of S1 towards higher energy triplet states is neglected but the S1 → 1TT transition required for iSF is expected to be the most efficient decay pathway (see Section S6). Quantum chemical descriptors were introduced to quantify these criteria using the character of excited states (see ESI†). Fig. 2 shows the fraction of CT from donor to acceptor in S1 (ΩS1D→A) and the fraction of local T1 character on the acceptor (ΩT1A→A). The upper righthand corner corresponds to the ‘ideal’ region in which these two criteria are fulfilled simultaneously. The majority of the present dimers display a highly localized T1 (ΩT1A→A = 0.5–1.0) and non-negligible S1 CT character (ΩS1D→A = 0.1–0.4), but are nonetheless not in the ideal region.
 | ||
| Fig. 2 Donor-to-acceptor charge-transfer character of S1 (ΩS1D→A, x-axis) and local acceptor character of T1 (ΩT1A→A, y-axis) in dimers, colored by the dihedral between the donor and acceptor (φD–A). | ||
A striking exception are dimers containing N,N-dioxidized benzothiadiazole along the top of Fig. 2, indicating pure localization of T1 on the acceptor (Fig. S17, ESI†). In addition to stabilizing T1 to achieve positive ΔEST, this acceptor induces dihedral torsion to the D–A linkage (φD–A ≈ 50°), thereby contributing to very high CT in S1 (up to 0.8). The best of these dimers is shown in Fig. 2 (compound A), and is revealed to have a donor partner which differs from the acceptor only with regard to the sulfur oxidation. However, the N-oxidation of compound A stabilizes T1 too much for it to be of practical use if extracted (0.41 eV). All other dimers constructed with this acceptor suffer from this problem (T1 = 0.3–0.9 eV). Two other dimers (B and C) have a similar acceptor, albeit without N-oxidation, which leads to promising excited state behavior in the dimer and appropriate ΔEST (as with A), but importantly, they retain attractive T1 energies (1.47 eV and 1.24 eV, respectively; see Table S2, ESI†). The absence of nitroxides leads to smaller dimer dihedrals (30° and 19°) and therefore slightly lower CT compared to A, but are still near the ideal region. This analysis demonstrates that through judicious chromophore oxidation, both ΔEST and T1 can be fine-tuned without losing the CT character which mediates the SF process in D–A copolymers.
We have disclosed heteroatom oxidation as a convenient handle through which to modulate singlet–triplet splitting in SF building blocks. Beneficial ΔEST through double oxidation of sulfur is obtained by suppressing aromaticity while maintaining overall conjugation, thereby stabilizing T1 compared to non-oxidized analogs, while having a smaller impact on S1. A higher number of heteroatom oxidations stabilizes T1 additively, making it possible to drive the T1 energy down as far as necessary to achieve exergonic splitting. The utility of this approach is demonstrated using new S- and N-oxidized compounds to construct D–A materials for iSF, although this method is equally valid in intermolecular SF materials design. D–A systems based on a new benzothiadiazole-S,S-dioxide acceptor may be excellent candidates, as sulfur oxidation modulates the excited state energies for SF to be thermodynamically possible while ensuring that the resulting T1 is appropriate for injection into silicon (1.1–1.7 eV). N-Oxidations, on the other hand, also systematically improve ΔEST, but at the expense of an attractive T1 energy. While these specific units have not been described in the literature, previously reported preparation of S-oxidized22 and S,S-(di)oxidized22,23 analogs of benzothiadiazole, as well as N-oxidized thiazoles,24 bithiazoles,25 and thiadiazoles26 suggest that they are synthesizable.
The authors are grateful to the EPFL for financial support. M. F. acknowledges funding from European Union's H2020 research and innovation, under MSCA-IF-2018 (G.A. #836849).
Conflicts of interest
There are no conflicts to declare.References
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- E. G. Fuemmeler, S. N. Sanders, A. B. Pun, E. Kumarasamy, T. Zeng, K. Miyata, M. L. Steigerwald, X.-Y. Zhu, M. Y. Sfeir and L. M. Campos, ACS Cent. Sci., 2016, 2, 316–324 CrossRef CAS PubMed.
- D. Casanova, Chem. Rev., 2018, 118, 7164–7207 CrossRef CAS PubMed.
- I. Paci, J. C. Johnson, X. Chen, G. Rana, D. Popović, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546–16553 CrossRef CAS PubMed.
- D. Padula, Ö. H. Omar, T. Nematiaram and A. Troisi, Energy Environ. Sci., 2019, 12, 2412–2416 RSC.
- B. C. Streifel, J. L. Zafra, G. L. Espejo, C. J. Gómez-García, J. Casado and J. D. Tovar, Angew. Chem., Int. Ed., 2015, 54, 5888–5893 CrossRef CAS PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- S. Singh and B. Stoicheff, J. Chem. Phys., 1963, 38, 2032–2033 CrossRef.
- C. E. Swenberg and W. T. Stacy, Chem. Phys. Lett., 1968, 2, 327–328 CrossRef CAS.
- S. Yoo, B. Domercq and B. Kippelen, Appl. Phys. Lett., 2004, 85, 5427–5429 CrossRef CAS.
- N. R. Monahan, D. Sun, H. Tamura, K. W. Williams, B. Xu, Y. Zhong, B. Kumar, C. Nuckolls, A. R. Harutyunyan, G. Chen, H.-L. Dai, D. Beljonne, Y. Rao and X. Y. Zhu, Nat. Chem., 2017, 9, 341–346 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426 CrossRef CAS PubMed.
- J. T. Blaskovits, M. Fumanal, S. Vela and C. Corminboeuf, Chem. Mater., 2020, 32, 6515–6524 CrossRef CAS.
- J. T. Blaskovits, M. Fumanal, S. Vela, R. Fabregat and C. Corminboeuf, Chem. Mater., 2021, 33, 2567–2575 CrossRef CAS.
- M. Fumanal and C. Corminboeuf, J. Phys. Chem. Lett., 2021, 12, 7270–7277 CrossRef CAS PubMed.
- O. El Bakouri, J. R. Smith and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett and C. Risko, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS PubMed.
- M. M. Oliva, J. Casado, J. T. L. Navarrete, S. Patchkovskii, T. Goodson, M. R. Harpham, J. S. Seixas de Melo, E. Amir and S. Rozen, J. Am. Chem. Soc., 2010, 132, 6231–6242 CrossRef CAS PubMed.
- J. Stoycheva, A. Tadjer, M. Garavelli, M. Spassova, A. Nenov and J. Romanova, J. Phys. Chem. Lett., 2020, 11, 1390–1396 CrossRef CAS PubMed.
- G. Grancini, M. Maiuri, D. Fazzi, A. Petrozza, H. Egelhaaf, D. Brida, G. Cerullo and G. Lanzani, Nat. Mater., 2013, 12, 29 CrossRef CAS PubMed.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- T. Linder, E. Badiola, T. Baumgartner and T. C. Sutherland, Org. Lett., 2010, 12, 4520–4523 CrossRef CAS PubMed.
- D. Pinkowicz, Z. Li, P. Pietrzyk and M. Rams, Cryst. Growth Des., 2014, 14, 4878–4881 CrossRef CAS.
- E. Amir and S. Rozen, Chem. Commun., 2006, 2262–2264 RSC.
- R. A. Mirabal, L. Vanderzwet, S. Abuadas, M. R. Emmett and D. Schipper, Chem. – Eur. J., 2018, 24, 12231–12235 CrossRef CAS PubMed.
- L. S. Konstantinova, E. A. Knyazeva, N. V. Obruchnikova, Y. V. Gatilov, A. V. Zibarev and O. A. Rakitin, Tetrahedron Lett., 2013, 54, 3075–3078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, dataset, excited state energies for individual compounds, projected density of states, nucleus-independent chemical shifts. See DOI: 10.1039/d1cc06755a |
| This journal is © The Royal Society of Chemistry 2022 |