
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu(I)-mediated N2-elimination from triazaphospholes: a one-pot synthesis of novel N2P2-heterocycles†
Erlin
Yue‡
 ab,
Lea
Dettling‡
a,
Julian A. W.
Sklorz
a,
Selina
Kaiser
a,
Manuela
Weber
a and
Christian
Müller
*a
ab,
Lea
Dettling‡
a,
Julian A. W.
Sklorz
a,
Selina
Kaiser
a,
Manuela
Weber
a and
Christian
Müller
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Fabeckstr. 34/36, Berlin 14195, Germany. E-mail: c.mueller@fu-berlin.de; yueerlin@yau.edu.cn
bShaanxi Key Laboratory of Chemical Reaction Engineering, Key Laboratory of New Energy & New Functional Materials, College of Chemistry and Chemical Engineering, Yan’an University, Yan’an, Shaanxi 716000, People's Republic of China
First published on 29th November 2021
Abstract
Novel tosyl- and mesitylsulfonyl-substituted triazaphospholes were synthesized and structurally characterized. In an attempt to prepare the corresponding Au(I)-complexes with stoichiometric amounts of AuCl·S(CH3)2, cyclo-1,3-diphospha(III)-2,4-diazane-AuCl-complexes were obtained instead. Our here presented results offer a new strategy for preparing such coordination compounds selectively in a one-pot approach.
According to the isolobal relationship between a trivalent P-atom and a C–H fragment, the 3,5-disubstituted 3H-1,2,3,4-triazaphosphole derivatives of type B are the phosphorus congeners of the well-studied 1,2,3-triazoles A (Chart 1).
 | ||
| Chart 1 Triazaphosphole A, triazole B and possible coordination modes C. | ||
These λ3σ2 phosphorus heterocycles can be prepared in a modular [3+2] cycloaddition reaction, starting from organic azides and phosphaalkynes, as first reported independently by Carrié and Regitz in 1984.1 Generally, only one regioisomer is formed thermally and selectively, without the need of a copper-catalyst. 3H-1,2,3,4-triazaphosphole derivatives have a conjugated π-system with a high degree of aromaticity.2 Typically, a whole variety of alkyl- and aryl-substituted as well as donor-functionalized azides (R-N3) can be used for the preparation of triazaphospholes, but also TMS-N3 or even H–N3.3 On the other hand, the substituent R′ can only be varied to some extend due to the limited availability of the corresponding phosphaalkynes, although less sterically demanding phosphaalkynes can be generated in situ prior to the cycloaddition reaction.4
The first few reports on the coordination chemistry of triazaphospholes have only appeared in literature as recently as 2010.5 As ambidentate ligands the coordination to a metal center can proceed either via the phosphorus atom or the nitrogen donors N(1) or N(2) (Chart 1, C).6
Despite the few reported examples on the coordination chemistry of 3H-1,2,3,4-triazaphosphole derivatives, very little is known about their reactivity.7N-Aryl/alkyl-substituted triazaphospholes are thermally robust and do not show any sign of reactivity upon irradiation with UV light (λ ≥ 280 nm).7a We therefore anticipated that the hitherto unknown introduction of an electron-withdrawing substituent at the N(3)-atom might change the coordination properties and reactivity of the corresponding heterocycle considerably. As a matter of fact, the phosphorus-lacking N-sulfonyl-1,2,3-triazoles show interesting chemical transformations in the presence of [Rh2(OAc)4].8,9 Inspired by this fascinating reactivity, we started to transfer the chemistry of N-sulfonyl-1,2,3-triazoles to their phosphorus congeners and report here on our first results into this direction.
4-Methylbenzenesulfonylazide (1a) and mesitylsulfonylazide (1b) were prepared according to literature procedures.10 As anticipated, the 1,3-dipolar cycloaddition reaction of 1a/b with tBuC≡P afforded the desired N-arylsulfonyl-substituted triazaphospholes 2a/b, which were obtained as white solids in up to 85% yield after recrystallization from pentane (Scheme 1). Both compounds do not show any sign of decomposition when stored under inert conditions for several weeks.
 | ||
| Scheme 1 Synthesis of triazaphospholes 2a/b. | ||
The hitherto unknown N-arylsulfonyl-triazaphospholes show single resonances in the 31P{1H} NMR at δ(ppm) = 177.2 (2a) and δ(ppm) = 175.2 (2b) in DCM-d2. Although the N-arylsulfonyl group is supposed to be an electron withdrawing substituent, the resonances of 2a/b in the 31P{1H} spectra are only slightly shifted more downfield compared to the literature known benzyl-substituted triazaphosphole 2c (δ(ppm) = 171.4, DCM-d2, see Fig. 2).1b,6b
Single crystals of 2b suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into a dichloromethane solution of the compound at low temperature. The molecular structure is shown in Fig. 1 along with selected bond lengths and angles. Compound 2b crystallizes in the monoclinic space group P21/c. While the NMR spectroscopic data of 2a/b are very similar to triazaphosphole 2c, the crystallographic characterization of 2b reveals a clear influence of the N-arylsulfonyl group on the bond distances within the P-heterocycle (Fig. 2 and Table 1). As a matter of fact, the N(1)–N(2) distance in 2b is longer than in the known compound 2c, while the N(2)–N(3) distance is shorter. Moreover, both the C(1)–N(3) and P(1)–N(1) bond lengths in 2b are longer, while the C(1)–P(1) bond lengths is shorter compared to the situation in 2c.6b
 | ||
| Fig. 1 Molecular structure of 2b in the crystal. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): P(1)–N(1): 1.7047(16), N(1)–N(2): 1.364(2), N(2)–N(3): 1.298(2), N(3)–C(1): 1.369(2), C(1)–P(1): 1.715(2), N(1)–S(1): 1.7108(16), S(1)–O(1): 1.4232(14), S(1)–O(2): 1.4280(14). N(1)–P(1)–C(1): 85.35(9). | ||
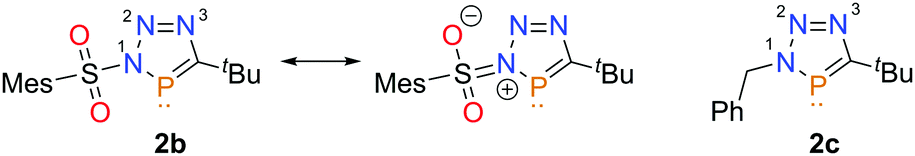 | ||
| Fig. 2 Resonance structures of 2b and comparison of 2b with 2c. | ||
| P(1)–C(1) | P(1)–N(1) | N(1)–N(2) | N(2)–N(3) | N(3)–C(1) | |
|---|---|---|---|---|---|
| 2b | 1.7047(16) | 1.7047(16) | 1.364(2) | 1.298(2) | 1.369(2) |
| 2c | 1.7128(17) | 1.6834(19) | 1.340(2) | 1.314(2) | 1.351(3) |
As also observed for N-sulfonamides, the N(1)–S(1) bond is with 1.7108(16) significantly shorter than the predicted value for pure S–N-single bonds, indicating the presence of a resonance structure with a partial S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (Fig. 2).11
N double bond (Fig. 2).11
Accordingly, the structural parameters are in line with a significant disruption of the aromaticity in 2b along with more localized bonds (Fig. 2).
Apparently, the electronic structures of the hitherto unknown N-sulfonyl-substituted phosphorus heterocycles 2a/b differ considerably from classical aryl- and alkyl-functionalized triazaphospholes. This should consequently also lead to a pronounced different chemical reactivity of 2a/b in comparison to 2c. As we were primarily interested in the coordination chemistry of aromatic λ3σ2-phosphorus compounds, also with respect to applications, we first considered the reaction of 2a/b with AuCl·S(CH3)2. It is well documented that phosphorus in low-coordination readily forms complexes with Au(I).12
Interestingly, a spontaneous and vigorous gas-evolution is observed when dichloromethane is added to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of either 2a or 2b and AuCl·S(CH3)2 at room temperature. The gas was identified as dinitrogen by means of GC-TCD. For triazaphosphole 2b (R = mesityl), the 31P{1H} NMR spectrum of the slightly yellow reaction mixture shows only two resonances at δ(ppm) = 133.9 and δ(ppm) = 11.6 in a ratio of approximately 4:1. Stirring the reaction solution for 2 h at T = 60 °C immediately after addition of the solvent leads, however, to a ratio of 20:1 (Fig. 3b). The isolation of the pure, air and moisture sensitive product 3b in 36% yield was achieved by washing the reaction mixture with toluene. For 2a (R = p-tolyl) the reaction seems to be less selective (see Fig. S10, ESI†).
:1 mixture of either 2a or 2b and AuCl·S(CH3)2 at room temperature. The gas was identified as dinitrogen by means of GC-TCD. For triazaphosphole 2b (R = mesityl), the 31P{1H} NMR spectrum of the slightly yellow reaction mixture shows only two resonances at δ(ppm) = 133.9 and δ(ppm) = 11.6 in a ratio of approximately 4:1. Stirring the reaction solution for 2 h at T = 60 °C immediately after addition of the solvent leads, however, to a ratio of 20:1 (Fig. 3b). The isolation of the pure, air and moisture sensitive product 3b in 36% yield was achieved by washing the reaction mixture with toluene. For 2a (R = p-tolyl) the reaction seems to be less selective (see Fig. S10, ESI†).
 | ||
| Fig. 3 31P{1H} NMR spectra of 2b (a), the reaction mixture (b) and of the obtained crystals (c). (*): unidentified species. | ||
Crystals of 3a and 3b, suitable for X-ray diffraction, could be obtained from both reaction mixtures. Dissolving the crystalline material of 3b in dichloromethane gave indeed the identical resonance of the major product observed in the 31P{1H} NMR spectrum of the reaction mixture (Fig. 3c). Much to our surprise, the crystallographic characterization of 3a and 3b reveals the formation of a cyclo-1,3-diphospha(III)-2,4-diazane, rather than the presence of a simple triazaphosphole-Au(I) complex. Moreover, the cyclo-diphosphadiazane serves as a ligand, which binds to a total of two Au(I)Cl fragments via both phosphorus donors. The molecular structure of 3b is depicted in Fig. 4, along with selected bond lengths and angles (for the single crystal X-ray structure of 3a see Fig. S2, ESI†). Based on the structural characterization of 3a/b, the novel and, in the case of 2b, highly selective “one-pot” reaction with stoichiometric amounts of AuCl·S(CH3)2 under formation of a dinuclear cyclo-diphosphadiazane–Au(I) complex is summarized in Scheme 2.
 | ||
| Fig. 4 Molecular structure of 3b in the crystal. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angels (°): P(1)–N(1): 1.730(3), N(1)–P(1)_i: 1.727(3), N(1)–S(1): 1.675(3), P(1)–Au(1): 2.2087(11), P(1)–C(10): 1.778(4), C(10)–C(12): 1.352(6). N(1)–P(1)–N(1)_i: 79.87(18), P(1)–N(1)–P(1)_i: 100.13(18). | ||
 | ||
| Scheme 2 Synthesis of cyclo-1,3-diphospha-2,4-diazane-Au(I)-complexes 3a/b. | ||
As a matter of fact, such N2P2 heterocylces are most commonly obtained as 1,3-dichloro-cyclo-1,3-diphospha(III)-2,4-diazanes of the type [ClP(μ-NR)2PCl] by reacting primary amines with PCl3.13 Subsequent reaction with appropriate nucleophiles leads to cyclo-diphosphadiazanes of the type [R′P(μ-NR)2PR′] (R′ = alkyl, aryl; OR, NR′′2, NHR′′), which can then be converted to the corresponding coordination compounds by reaction with an appropriate metal precursor.14 Importantly, there are no reports on cyclodiphospha(III)zanes featuring the exact susbstitution pattern of 3a/b, potentially due to synthetic difficulties.15 Therefore, our here described approach offers access to novel P2N2 heterocycles, which were so far not accessible.
3b crystallizes in the space group P21/c. In 3b (as well as in 3a, Fig. S2, ESI†) a perfectly planar P2N2-ring with both the R-groups and the Au(I)Cl-fragments at the phosphorus atoms pointing in opposite directions (trans isomer) is present. As observed for other cyclo-1,3-diphosphadiazanes, the nitrogen atoms are almost planar (sum of bond angles 359.3°), while the λ3,σ3-phosphorus atoms are pyramidally coordinated and bind each via the lone pair to the Au(I) center.14a The P–N bond lengths of 1.730(3) Å and 1.727(3) Å are slightly shorter than observed in other cyclo-diphosphadizanes, which might be due to a reduced electrostatic repulsion between the P- and N-lone pairs, which are involved in an interaction with the metal center and the –SO2R substituent, respectively.
The most striking feature of 3b (and 3a, Fig. S2, ESI†) is, however, that the tBu-group of the original triazaphosphole was converted into an iso-pentenyl substituent. Obviously, a CH3-shift took place during the conversion 2a/b → 3a/b, which implies the formation of a carbene intermediate. This has also been observed by Fokin and co-worker during the Rh-catalyzed denitrogenative transformation of a tBu-substituted 1-sulfonyl-1,2,3-triazole into a tetrasubstituted iminoalkene.8
The rather selective conversion 2a/b → 3a/b requires the presence of stoichiometric amounts of AuCl·S(CH3)2. We could not observe the formation of any cyclo-diphosphadiazane upon heating 2a/b in the absence of Au(I). Moreover, the presence of the electron withdrawing N-sulfonyl-group at N(3) is crucial for the dinitrogenative generation of 3a/b, as the PhCH2-substituted triazaphosphole 2c does not undergo the transformation to the corresponding N2P2-heterocycle.
Based on NMR-spectroscopic data, we propose the following mechanism for the conversion of the N-sulfonyl-triazaphosphole into the corresponding Au(I)-complex: the Au(I)Cl-fragment first coordinates to the donor-atoms of the phosphorus heterocycle in a dynamic exchange process (Scheme 3).16 Due to the electron-withdrawing nature of the N-sulfonyl-group, the aromaticity of the triazaphosphole is strongly disrupted and ring-opening to [(2a′/b′)AuCl] is facilitated. Loss of dinitrogen gives the zwitterionic species [(4a/b)AuCl], for which a neutral resonance structure exist. According to the HSAB concept, we anticipate that the Au(I)-fragment coordinates exclusively to the remaining soft phosphorus atom in [(4a/b)AuCl]. The neutral species is an iminophosphine-carbene, which undergoes a [1,2]-CH3-shift to the more stable iminophosphine [(5a/b)AuCl]. Iminophosphinines are known to form dimers and even trimers from the parent monomer depending on the substituents on both the phosphorus and nitrogen atom. Dimerization of [(5a/b)AuCl], especially in presence of electron-withdrawing sulfonyl groups then leads to the observed main product 3a/b (Scheme 3).17
 | ||
| Scheme 3 Proposed mechanism for the formation of 3a/b. | ||
In order to identify the reactive iminophosphine [(5a/b)AuCl] as an intermediate in the proposed mechanism, N-tosyl-triazaphosphole 2a and AuCl·SMe2 were cooled to T = −196 °C and a solution of dimethylbutadiene as a trapping reagent in dichloromethane was condensed into the reaction vessel. The solution was first stored at T = −78 °C and then slowly warmed to room temperature over 6–8 hours. Subsequent 31P{1H} NMR spectroscopy at room temperature showed only one major phosphorus resonance at δ(ppm) = 104.0. Analysis of the product by means of ESI-MS indeed provided evidence for the expected trapping product [(6a)AuCl] (Scheme 3). Further confirmation for cyclodiphosphazane formation via dimerization of two iminophosphines is provided by a cross-reaction of a 1:1 mixture of 2a and 2b with AuCl·SMe2 in DCM. In this case the 31P{1H} NMR of the reaction mixture showed the formation of 3a and 3b as well as a third species at δ(ppm) = 130.7, which we tentatively assigned to a mixed N-SO2-Tol/N-SO2-Mes substituted P2N2 ring. A similar cross reactivity in phosphazane chemistry has recently been described by Wright et al. as the authors also found evidence for the transient formation of monomeric phosphazane intermediates.18
We could demonstrate for the first time that 3H-1,2,3,4-triazaphosphole derivatives, containing electron-withdrawing N-sulfonyl-groups at the N3 atom, are synthetically accessible. These phosphorus heterocycles show a remarkable different reactivity compared to their classical alkyl- or aryl-substituted counterparts. Interestingly, the hitherto unknown N-sulfonyl-1,2,3,4-triazaphospholes undergo a highly selective and unprecedented transformation to cyclo-1,3-diphospha(III)-2,4-diazane-Au(I) complexes in the presence of stoichiometric amounts of AuCl·S(CH3)2 and loss of N2. Single crystal X-ray diffraction studies show, that the trans-isomer of the substituted N2P2 heterocycle has been generated, while NMR-spectroscopic and mass-spectrometric investigations give insight into the mechanism of its formation. Our results pave the way to explore the chemistry of N-sulfonyl-substituted triazaphospholes in detail and provide a first step in transferring the fascinating chemistry, reported for the phosphorus-lacking N-sulfonyl-1,2,3-triazoles, to their isolobal phosphorus congeners.
E. Y and C. M. are grateful for financial support provided by the Sino-German (CSC-DAAD) Postdoc Scholarship Program (57251553).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Y. Y. C. Yeung Lam Ko and R. Carrié, J. Chem. Soc., Chem. Commun., 1984, 1640 Search PubMed; (b) Y. Y. C. Yeung Lam, Ko, R. Carrié, A. Muench and G. Becker, J. Chem. Soc., Chem. Commun., 1984, 1634 Search PubMed; (c) W. Rösch and M. Regitz, Angew. Chem., 1984, 96, 898 CrossRef ; Angew. Chem. Int. Ed. Engl., 1984, 23, 900.
- L. Nyulászi, Chem. Rev., 2001, 101, 1229 CrossRef.
- See for example: (a) T. Allspach, M. Regitz, G. Becker and W. Becker, Synthesis, 1986, 31 CrossRef CAS; (b) M. Regitz and P. Binger, Angew. Chem., Int. Ed. Engl., 1988, 27, 1484 CrossRef; (c) J. A. W. Sklorz and C. Müller, Eur. J. Inorg. Chem., 2016, 595 CrossRef CAS.
- J.-C. Guillemin, T. Janati and J.-M. Denis, J. Org. Chem., 2001, 66, 7864 CrossRef CAS PubMed.
- (a) S. L. Choong, C. Jones and A. Stasch, Dalton Trans., 2010, 39, 5774 RSC; (b) S. L. Choong, A. Nafady, A. Stasch, A. M. Bond and C. Jones, Dalton Trans., 2013, 42, 7775 RSC.
- (a) J. A. W. Sklorz, S. Hoof, M. G. Sommer, F. Weißer, M. Weber, J. Wiecko, B. Sarkar and C. Müller, Organometallics, 2014, 33, 511 CrossRef CAS; (b) J. A. W. Sklorz, S. Hoof, N. Rades, N. De Rycke, L. Könczöl, D. Szieberth, M. Weber, J. Wiecko, L. Nyulászi, M. Hissler and C. Müller, Chem. – Eur. J., 2015, 21, 11096 CrossRef CAS.
- (a) W. Rösch, T. Facklam and M. Regitz, Tetrahedron, 1987, 43, 3247 CrossRef; (b) J. Kerth, U. Werz and G. Maas, Tetrahedron, 2000, 56, 35 CrossRef CAS; (c) M. Papke, L. Dettling, J. A. W. Sklorz, D. Szieberth, L. Nyulászi and C. Müller, Angew. Chem., 2017, 129, 16706 CrossRef ; Angew. Chem. Int. Ed., 2017, 56, 16484.
- N. Selander, B. T. Worrell and V. V. Fokin, Angew. Chem., 2012, 124, 13231 CrossRef ; Angew. Chem. Int. Ed., 2012, 51, 13054.
- For recent examples on rhodium(II)-catalyzed reactions of N-sulfonyl-1,2,3-triazoles: (a) K. Pal, R. K. Shukla and C. M. R. Volla, Org. Lett., 2017, 19, 5764 CrossRef CAS PubMed; (b) T. Miura, Q. Zhao and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 16645 CrossRef CAS; (c) J. O. Strelnikova, N. V. Rostovskii, G. L. Starova, A. F. Khlebnikov and M. S. Novikov, J. Org. Chem., 2018, 83, 11232 CrossRef CAS PubMed; (d) Y. Lv, A. A. Ogunlana, H. Li, D. Gao, C. Wang and X. Bao, Catal. Sci. Technol., 2018, 8, 3379 RSC; (e) C.-Z. Zhu, Y. Wei and M. Shi, Org. Chem. Front., 2019, 6, 2884 RSC; (f) J. Ge, X. Wu and X. Bao, Chem. Commun., 2019, 55, 6090 RSC; (g) N. Kahar, P. Jadhav, R. V. R. Reddy and S. Dawande, Chem. Commun., 2020, 56, 1207 RSC.
- J. Wang, J. Mei, E. Zhao, Z. Song, A. Qin, J. Z. Sun and B. Z. Tang, Macromolecules, 2012, 45, 7692 CrossRef CAS.
- F. A. Cotton and P. F. Stokely, J. Am. Chem. Soc., 1970, 92, 294 CrossRef CAS.
- See for example: (a) N. Mézailles, L. Ricard, F. Mathey and P. Le Floch, Eur. J. Inorg. Chem., 1999, 2233 CrossRef; (b) M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2017, 46, 86 RSC.
- (a) A. Michaelis and G. Schroeter, Ber. Dtsch. Chem. Ges., 1894, 27, 490 CrossRef; (b) A. Schulz, A. Villinger and A. Westenkirchner, Inorg. Chem., 2013, 52, 11457 CrossRef CAS PubMed; (c) J. Bresien, A. Hinz, A. Schulz, T. Suhrbier, M. Thomas and A. Villinger, Chem. – Eur. J., 2017, 23, 14738 CrossRef CAS PubMed.
- See for example: (a) M. M. Siddiqui, J. T. Mague and M. S. Balakrishna, Inorg. Chem., 2015, 54, 6063 CrossRef CAS PubMed; (b) M. S. Balakrishna, V. Sreenivasa Reddy and S. S. Krishnamurthy, Coord. Chem. Rev., 1994, 129, 1 CrossRef CAS; (c) G. G. Briand, T. Chivers and M. Krahn, Coord. Chem. Rev., 2002, 233–234, 237 CrossRef CAS; (d) M. S. Balakrishna, in Copper(I) Chemistry of Phosphines, Functionalized Phosphines, and Phosphorus Heterocycles, ed. M. S. Balakrishna, Elsevier, 2019, ch. 11, pp. 345–373 Search PubMed; (e) A. J. Plajer, J. Zhu, P. Pröhm, F. J. Rizzuto, U. F. Keyser and D. S. Wright, J. Am. Chem. Soc., 2020, 142, 1029 CrossRef CAS.
- For related species see: (a) A. Baceiredo, G. Bertrand, J.-P. Majoral and K. B. Dillon, J. Chem. Soc., Chem. Commun., 1985, 562 RSC; (b) F. L. Bowden, A. T. Dronsfield, R. N. Haszeldine and D. R. Taylor, J. Chem. Soc., Perkin Trans. 1, 1973, 516 RSC.
- Low-temperature 31P{1H} NMR measurements indicate a coordination of the Au(I)Cl fragment to the ambidentate triazaphosphole 2b. See ESI†.
- See for example: (a) E. Niecke, W. Flick and S. Pohl, Angew. Chem., Int. Ed. Engl., 1976, 15, 309 CrossRef; (b) E. Niecke, R. Rüger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1981, 20, 10346 Search PubMed; (c) N. Burford, T. S. Cameron, K. D. Conroy, B. Ellis, M. Lumsden, C. L. B. Macdonald, R. McDonald, A. D. Phillips, P. J. Ragogna, R. W. Schurko, D. Walsh and R. E. Wasylishen, J. Am. Chem. Soc., 2002, 124, 14012 CrossRef CAS; (d) M. Lehmann, A. Schulz and A. Villinger, Struct. Chem., 2011, 22, 35 CrossRef CAS; (e) M. Kuprat, M. Lehmann, A. Schulz and A. Villinger, Inorg. Chem., 2011, 50, 5784 CrossRef CAS; (f) E. Niecke, M. Nieger and F. Reichert, Angew. Chem., 1988, 100, 1783 Search PubMed.
- A. J. Plajer, K. Bold, F. J. Rizzuto, R. García-Rodríguez, T. K. Ronson and D. S. Wright, Dalton Trans., 2017, 46, 12775 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1983603, 1983601 and 1983602. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06205k |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |