DOI:
10.1039/D2CB00126H
(Paper)
RSC Chem. Biol., 2022,
3, 1260-1275
Extending the in vivo persistence of synthetic glycoconjugates using a serum-protein binder†
Received
19th May 2022
, Accepted 22nd August 2022
First published on 23rd August 2022
Abstract
Synthetic glycoconjugates are used in the development of vaccines and the design of inhibitors for glycan–protein interactions. The in vivo persistence of synthetic glycoconjugates is an important factor in their efficacy, especially when prolonged interactions with specific cell types may be required. In this study, we applied a strategy for non-covalent association of an active compound with serum proteins for extension of glycoconjugate half-life in serum. The small molecule, AG10, has previously been used to extend the half-life of small molecules through its high affinity for transthyretin (TTR), a serum protein. Using a tetravalent polyethylene glycol (PEG)-based scaffold we developed a synthetic strategy for glycoconjugates that allowed for controlled addition of multiple tags, such as a TTR affinity tag or fluorophore. We designed a version of AG10 modified at the pyrazole core, named GD10, amenable to our conjugation strategy and introduced to glycoconjugates using a tri-functional linker. This approach allowed for attachment of GD10 and fluorophore tags, as well as carbohydrate antigens. We then tested the influence of the GD10 tag on glycoconjugate half-life in vivo using a mouse model. Our results suggest that the combination of the GD10 tag and the PEG scaffold extended the half-life of glycoconjugates by as much as 10-fold when compared to proteins of similar molecular weight. The GD10 tag was able to extend the half-life of similar glycoconjugates by as much as 2-fold. We observed a role for the terminal saccharide residue of the carbohydrate antigen and confirmed that conjugates were able to penetrate multiple compartments in vivo including bone marrow, lymph nodes, and other organs. The introduction of the GD10 tag did not obstruct the ability of conjugates to interact with lectin receptors. We conclude that serum protein binders can be used to extend the persistence of glycoconjugates in vivo.
Introduction
Glycans in biological systems are often found as components of high-molecular weight glycoproteins, proteoglycans, and polysaccharides; as a class, these molecules are referred to as glycoconjugates. Synthetic glycoconjugates are commonly used in targeting biomolecules which recognize the presentation of native glycans.1–3 Synthetic glycoconjugates are used as vaccines and therapeutic proteins in a variety of applications.4–7 High molecular weight glycoconjugates have some inherent advantages as biological therapeutics. First, glycoconjugates of sufficient size (1–50 kDa) have increased persistence due to reduced elimination through renal glomerular filtration.8–10 Furthermore, the presence of certain terminal saccharide residues (e.g. galactose or sialic acid) can directly modulate the half-life of glycoconjugates by modifying their interaction with receptor-mediated clearance mechanisms (e.g. the asialoglycoprotein receptor, ASGPR).8,11,12 Challenges associated with glycoconjugate therapeutics include synthesis of defined high-molecular weight targets and discrete copy numbers of glycans, attachment of pendant groups (e.g. fluorophores, toxins, adjuvants, or other labels), as well as purification and characterization.13 Our group is interested in the design of glycoconjugates which can be used to target glycan-recognizing immune receptors in vivo.14
Previous reports have employed glycoconjugates containing CD22 ligands and B cell receptor (BCR)-specific small molecule antigens as a strategy to suppress B cell activation.15,16 We have extended this approach using human ABO blood group antigens to investigate the interaction of glycoconjugates with cells bearing a carbohydrate-binding BCR.14 We selected a commercially available four-armed polyethylene glycol (PEG) scaffold that had been previously used for in vivo tolerance of protein antigens.17 Part of our motivation for constructing ABO antigen-containing glycoconjugates was to explore their application as tolerogens – compounds that inactivate or suppress the B cell response to specific antigens.18–20 Effective tolerogens must be able to penetrate into compartments where they can interact with immune cells, and must be persistent in serum.17 Our previous strategy was able to generate glycoconjugates with the ability to bind and cluster BCRs and CD22 receptors in vitro, but we did not investigate their persistence in vivo.
To enable in vivo applications of these conjugates, we sought to establish new methods for significant extension of their half-life in serum. The PEG scaffold we selected has a molecular weight of 11.2 kDa (PDI, 1.025). Increased persistence of the compounds in serum is desirable as it is likely to enhance their function as tolerogens.17 We considered an established strategy for extending the half-life of small molecules in serum which exploits small molecule binders of serum proteins.21 This strategy non-covalently links the therapeutic molecule to serum proteins, which are found in high concentration and are cleared slowly. The most notable example of this approach used a small molecule transthyretin (TTR) binder, known as AG10, which has nanomolar affinity for the TTR complex.21,22 The essential features for recognition of AG10 by TTR is a 3,5-dimethylpyrazole and a pendant benzoic acid group. The binding site for AG10 is located between two monomer units of the TTR tetramer.23 Previous modifications of the AG10 binding epitope included changing the identity of the aromatic group or its substitution, as well as attachment of pendant groups from the pyrazole nitrogen or the aromatic side chain.24 Symmetric modification of the 3,5-dimethylpyrazole ring to a 3,5-diethylpyrazole resulted in a major loss of activity.24 Analogs that linked biorthogonal groups from the aromatic side chain have been pursued as TTR ligands for half-life extension (known as TLHEs).21,22,25 The attachment of TLHEs to peptides resulted in large increases in circulating half-life by as much as 13-fold (from 3.5 to 46 min).21 To the best of our knowledge, this approach has not been explored for the extension of glycoconjugate half-life in serum.
We hypothesized that an appropriate AG10 analog linked to a large glycoconjugate could significantly extend the persistence of these molecules in serum, a property likely to improve their activity as tolerogens. Herein, we report the development of a modification to our glycoconjugate synthesis that allows incorporation of two pendant groups (e.g. fluorophore and TTR-ligand) in addition to multiple copies of a carbohydrate antigen (e.g. ABO A-type II antigen) (Scheme 1). We describe an AG10 analog, referred to as GD10, containing a free amine linked through the C5 methyl group of the 3,5-dimethylpyrazole that maintained affinity for TTR. The GD10 analog, when attached to a synthetic glycoconjugate scaffold, was able to significantly extend persistence of the compound in serum in a murine model. When combined with optimal glycan ligands on a PEG scaffold, our conjugates showed increases of as much as 2-fold of their persistence in mouse serum as compared to glyconjugates with similar functionalization. Furthermore, we investigated the fate of glycoconjugates using fluorescence imaging of whole animals; excised organs confirmed these materials were able to penetrate organs including lymph nodes and bone marrow.
 |
| | Scheme 1 Strategy for the synthesis of TTR-complexing glycoconjugates. | |
Results and discussion
Synthesis of an amine-terminated AG10 analog, GD10
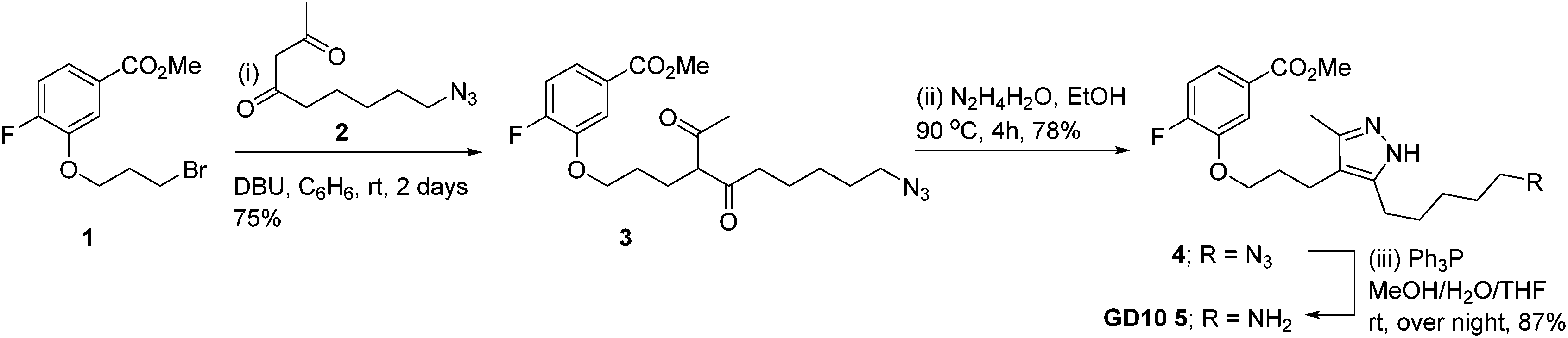
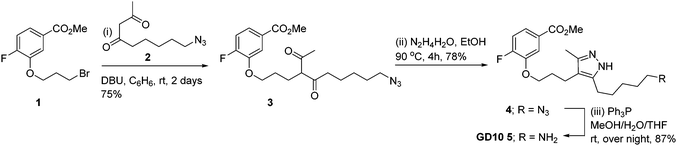
Co-crystal structures of AG10 analogs with WT-transthyretin (TTR or pre-albumin) have been reported.22–24 Previous structure activity work found that the free acid, fluorine, and 3,5-dimethyl-1H-pyrazole groups are essential for binding.24 While modification of both methyl groups of the pyrazole ring (3,5-diethyl-1H-pyrazole) showed poor activity, we reasoned that modification of a single position could maintain binding affinity. We planned a synthesis for an AG10 analog, GD10 5, containing a single modification of the pyrazole ring at C5 to introduce an amine while maintaining the fluorobenzoic acid moiety. The route began from compound 1 (Scheme 2), bearing a bromopropyl moiety, and was synthesized as previously reported.22 Alkylation of compound 1 with DBU and azido derivative 2 in anhydrous benzene at room temperature afforded compound 3 in 75% yield. Treatment of a solution of compound 3 in ethanol with hydrazine hydrate under refluxing conditions for 4 h provided azido derivative 4 in a 78% yield after purification. Reduction of the azide moiety with triphenylphosphine in a mixture of methanol and water in THF at room temperature afforded amine-terminated GD10 5 (Scheme 2) in good yield (50% overall).
 |
| | Scheme 2 Synthesis of amine-terminated analog 5 (GD10). | |
Synthesis of amine-terminated carbohydrate antigens
Carbohydrate antigens suitable for attachment to a conjugate scaffold were prepared as free amines. For this study we generated lactose 6, ABO A-type II 7, and 6′-sialyllactose 8 (see ESI†). Amine-terminated carbohydrate antigens 6, 7, and 8 were synthesized first as alkene terminated glycosides, followed by a UV-promoted (λ = 254 nm) thiol–ene coupling (TEC)26 with cysteamine as previously reported.14
Synthesis of AlexaFluor-labeled glycoconjugates
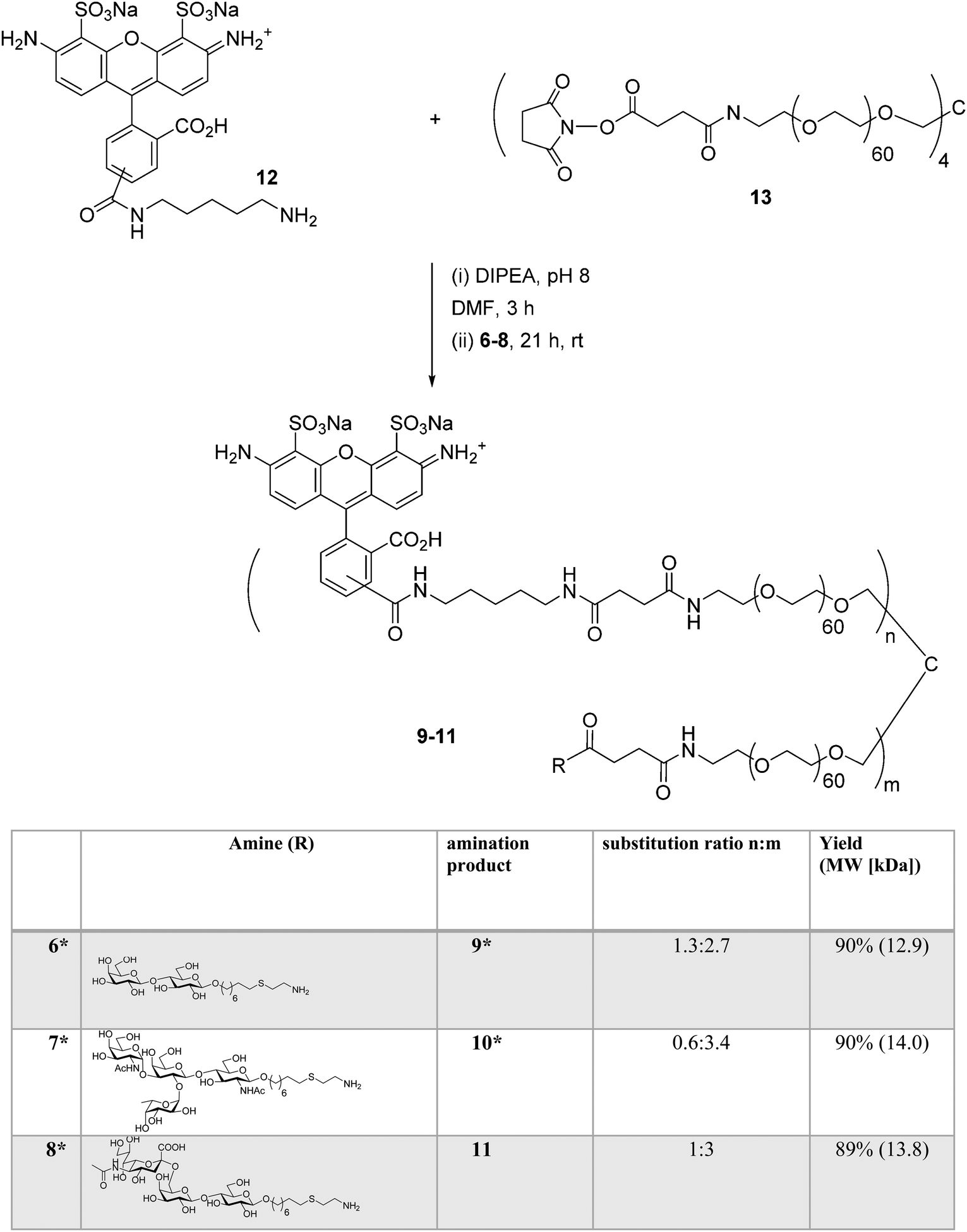
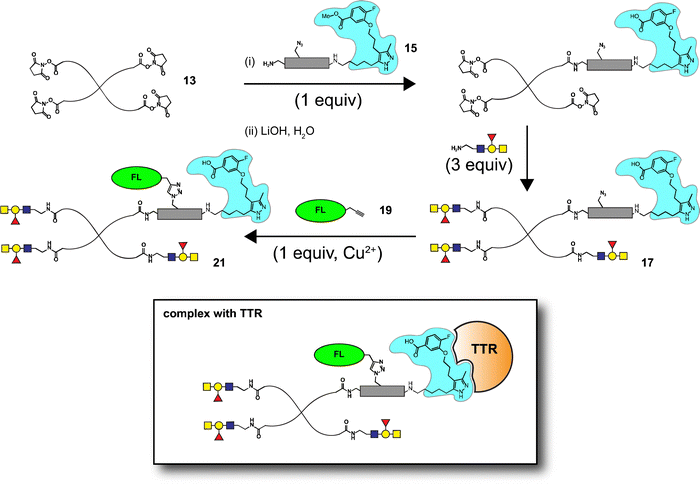
Synthesis of AlexaFluor 488-labeled (AF) lactose conjugate 9 (AF/lactose/PEG = 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8:1, 13.0 kDa) and A-type II conjugate 10 (AF/A-type II/PEG = 0.6:3.4:1, 14.0 kDa) were reported previously.14 A similar procedure was followed to prepare 6'-sialyllactose (6'-SL) conjugate 11 (AF/6'-SL/PEG = 1:3:1, 13.9 kDa). The conjugation strategy consisted of sequential amine coupling of AF-cadaverine 12 (λex = 493 nm, λem = 516 nm) and carbohydrate antigen 8 with an NHS-activated PEG scaffold 13 (11.2 kDa) in the presence of DIPEA in DMF for one day at room temperature (Scheme 3). The ratio of substitution was determined using NMR and confirmed by mass spectrometry (see Experimental procedures).
:2.8:1, 13.0 kDa) and A-type II conjugate 10 (AF/A-type II/PEG = 0.6:3.4:1, 14.0 kDa) were reported previously.14 A similar procedure was followed to prepare 6'-sialyllactose (6'-SL) conjugate 11 (AF/6'-SL/PEG = 1:3:1, 13.9 kDa). The conjugation strategy consisted of sequential amine coupling of AF-cadaverine 12 (λex = 493 nm, λem = 516 nm) and carbohydrate antigen 8 with an NHS-activated PEG scaffold 13 (11.2 kDa) in the presence of DIPEA in DMF for one day at room temperature (Scheme 3). The ratio of substitution was determined using NMR and confirmed by mass spectrometry (see Experimental procedures).
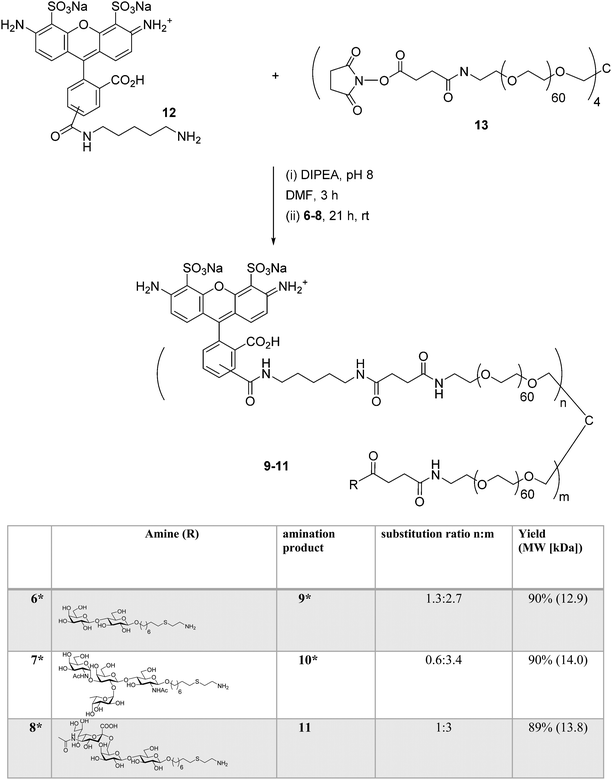 |
| | Scheme 3 Synthesis of AF-labeled glycoconjugate 11 using iterative amine coupling. *Previously reported.14 | |
Synthesis of tetravalent glycoconjugates containing GD10
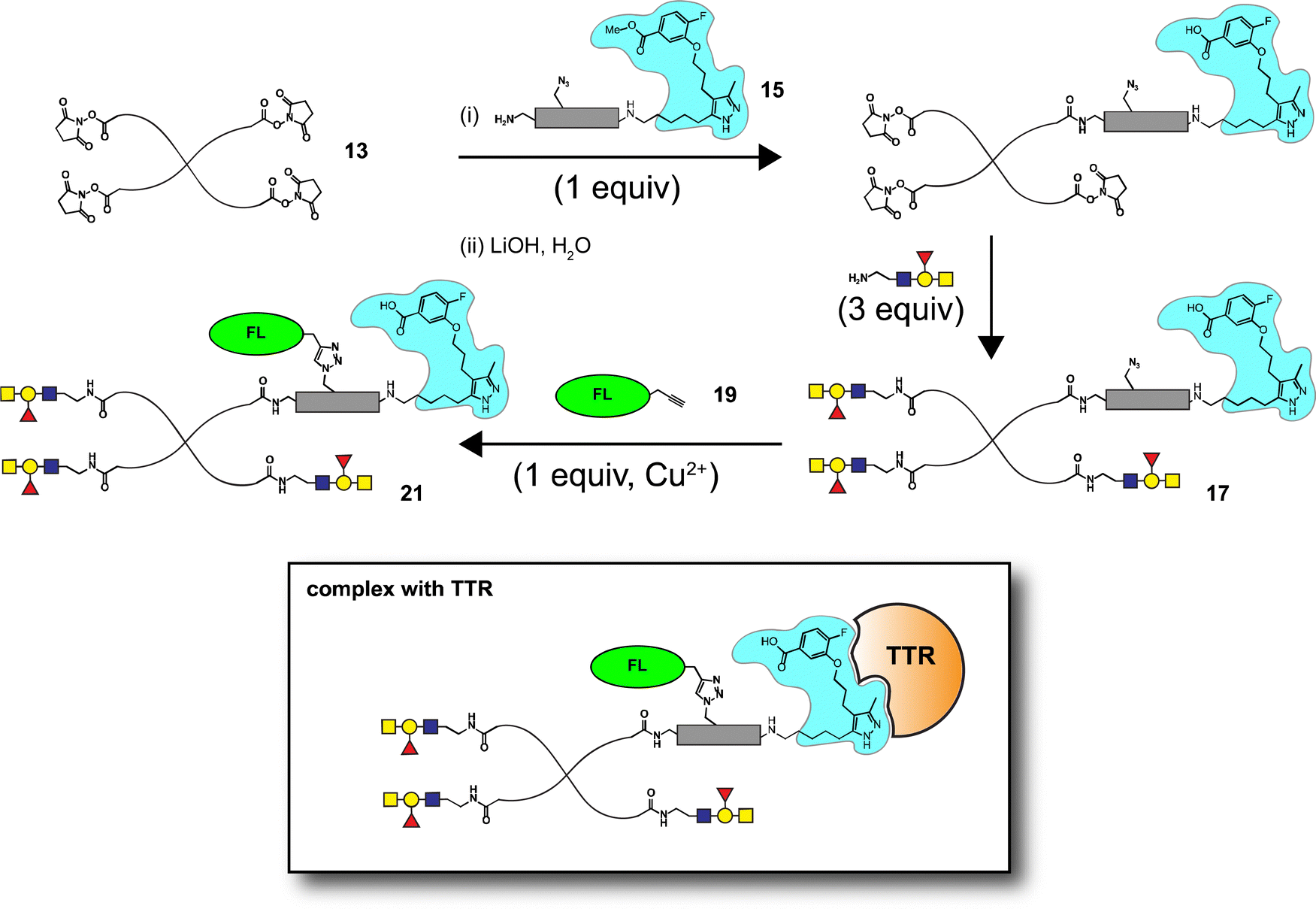
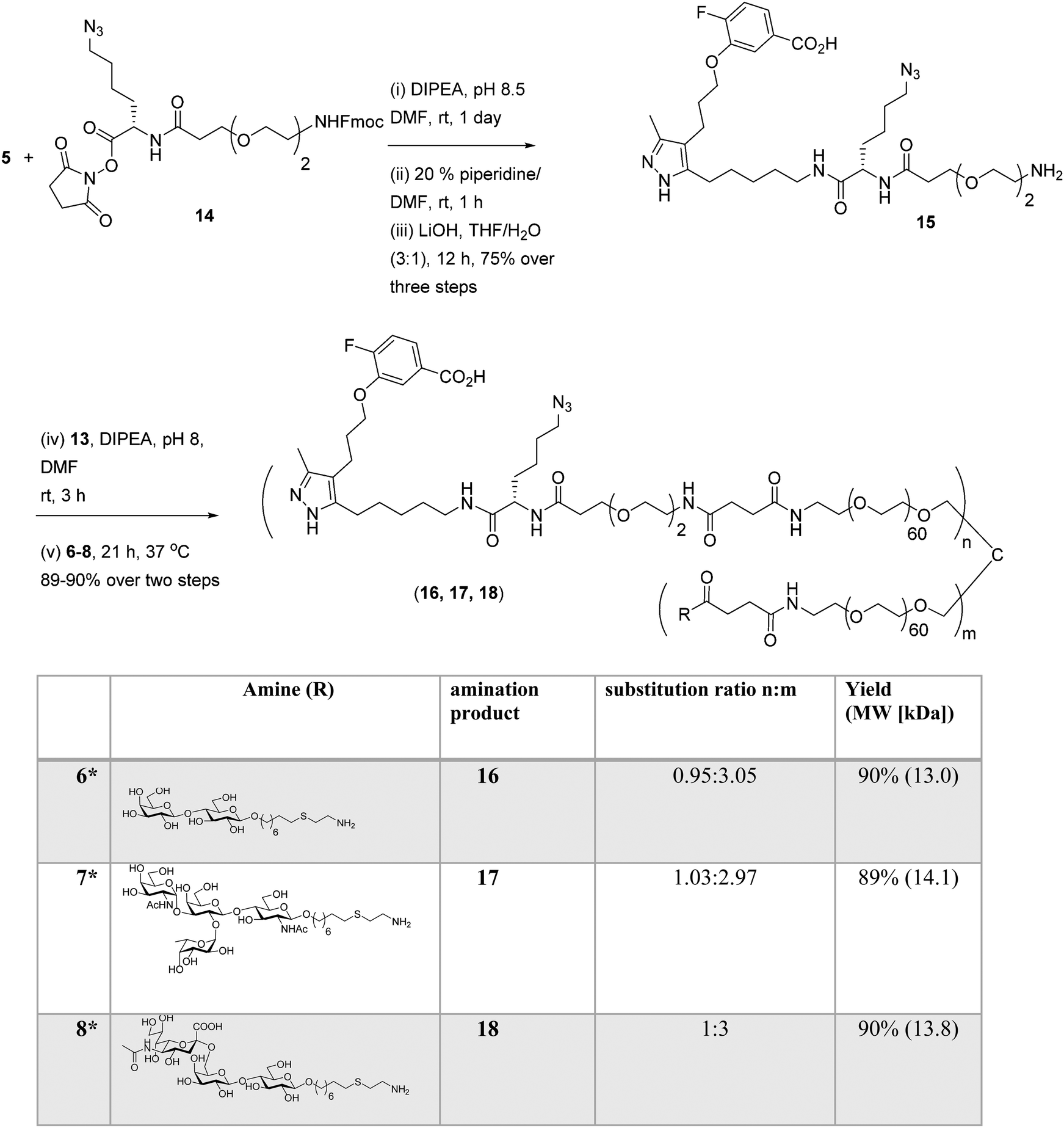
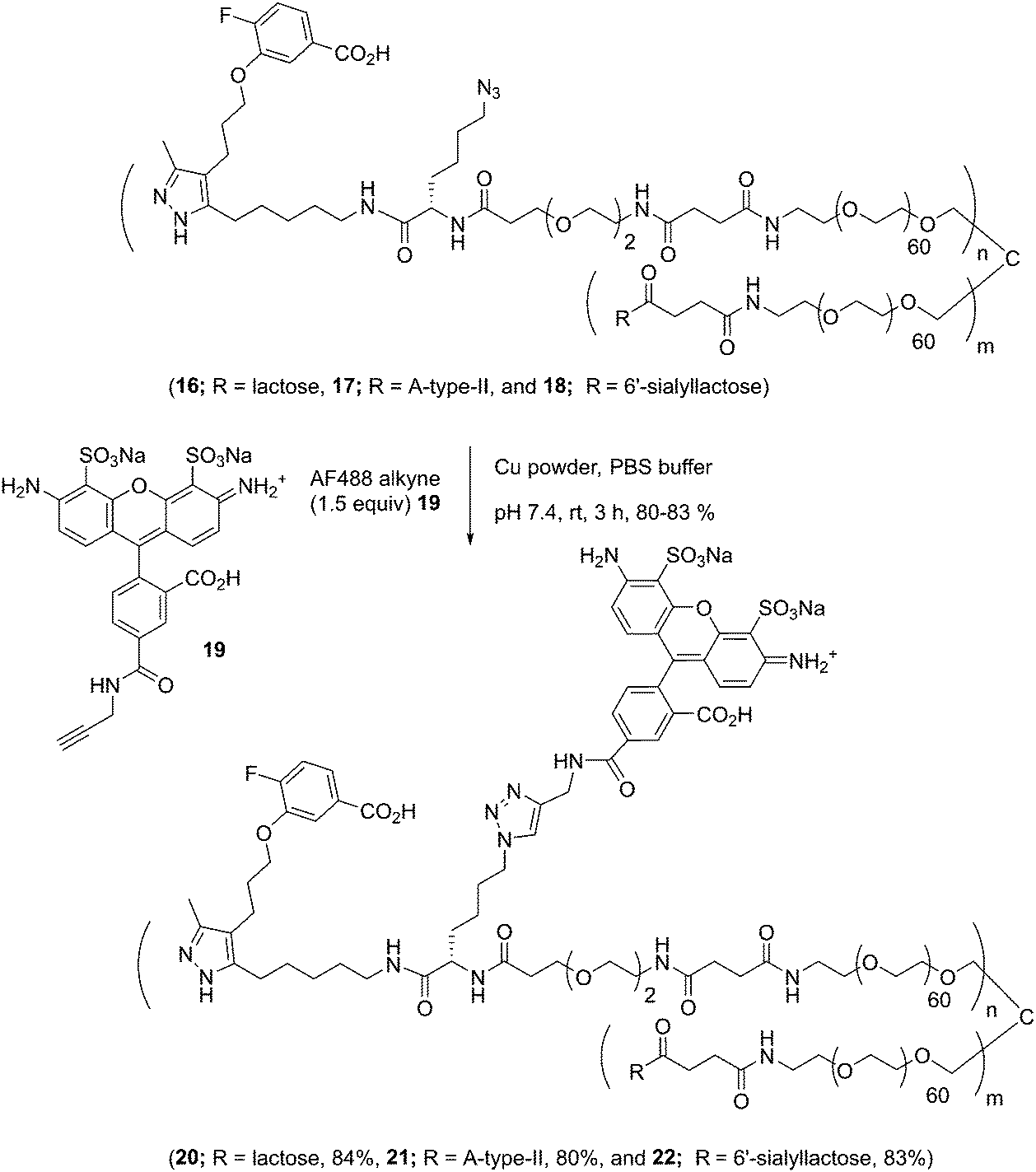
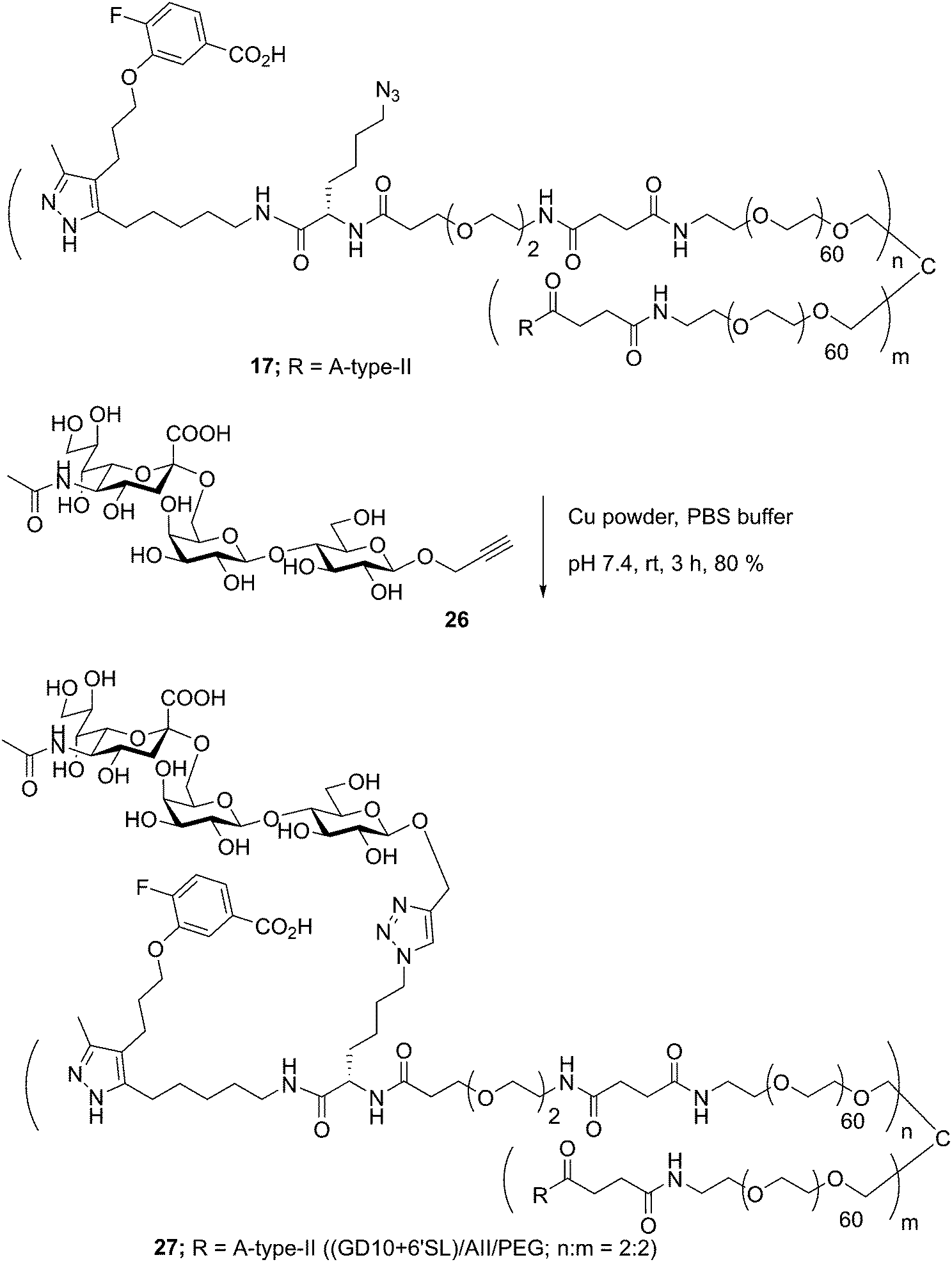
Well-defined tetravalent glycoconjugates with GD10 were generated by developing a stoichiometrically-controlled conjugation strategy (Scheme 4). We note that this strategy results in a statistical mixture of different copy numbers on individual conjugates; however, we treated the final conjugates as an average based on NMR characterization (vide infra). Taking advantage of the compatibility of trifunctional linker 14 in bioconjugation, we decided to explore a sequential iterative amine coupling strategy for all three components to generate the target glycoconjugates. We previously used linker 14 for the synthesis of multivalent homo- and hetero-functional glycoconjugates presenting both BCR-specific ABO blood group antigens and sialosides as Siglec ligands that were able to co-cluster BCR and CD22 receptors in vitro.14 We adapted our previous strategy by coupling of GD10 5 with linker 14 to generate adduct 15 bearing an amine. This was followed by successive attachment of 15 and the carbohydrate amines (6, 7, or 8) onto the tetravalent NHS-activated PEG scaffold 13 under stoichiometric control. In the first step, GD10 5 (1.25 equiv.) was coupled with NHS-activated linker 14 in the presence of DIPEA in anhydrous DMF at room temperature for one day. Subsequently, removal of the Fmoc-protecting group with 20% piperidine in DMF at room temperature followed by hydrolysis of the methyl ester was achieved with LiOH in a mixture of THF and water for 12 h. This one-pot, three-step reaction provided compound 15 in a 75% yield after purification by semipreparative RP-HPLC. The free amine 15 (1.0–1.25 equiv. per NHS-ester) was conjugated with NHS-activated PEG scaffold 13 (11.2 kDa) under similar conditions to those above. After ∼3 h, consumption of 15 was observed by TLC, subsequently the lactose amine 6 (3.5 equiv.) was immediately coupled with an intermediate NHS-PEG-GD10 adduct via amine coupling, and the reaction mixture was incubated at 37 °C for an additional 21 h.14 After 24 h, the crude product was purified by C-18 chromatography to provide lactose-GD10 conjugate 16 (GD10/Lac/PEG = 0.95:3.05:1; 13.0 kDa) in 90% yield and purity over two-steps as a colorless powder after lyophilization. Substitution of GD10 and formation of the tetravalent structure with an azide moiety was confirmed by 1H NMR and 19F spectra. While the MALDI-TOF MS spectrum could suggest a loss of the azide group, the azide group was confirmed to be intact through subsequent CuAAC couplings (vide infra).14
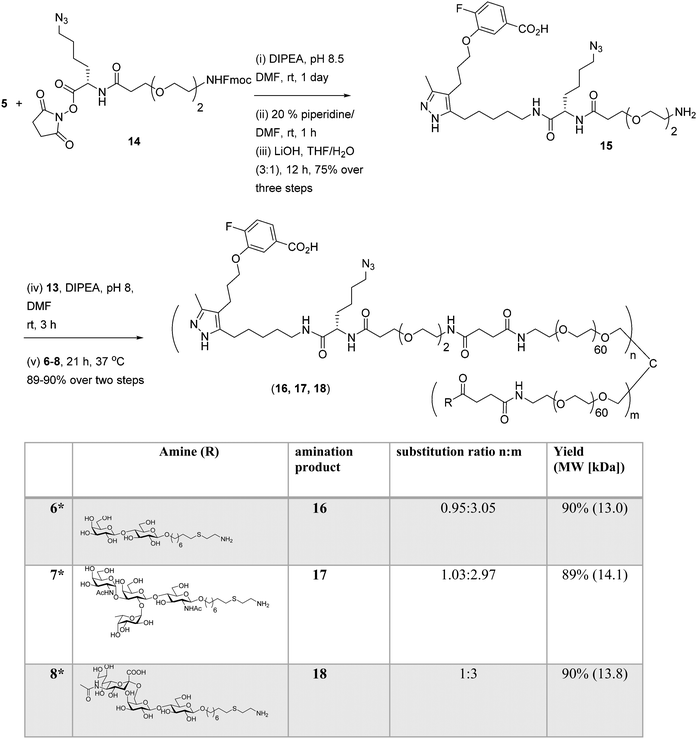 |
| | Scheme 4 Synthesis of tetravalent glycoconjugates (16, 17, and 18) with GD10 using iterative amine coupling. | |
The A-type II and 6'-sialyllactose amine-terminated compounds 7 and 8 were coupled with an intermediate NHS-PEG-GD10-adduct under similar coupling conditions described for conjugate 16 to provide A-type II-GD10 conjugate 17 (GD10/A-type II/PEG = 1.03:2.97:1, 14.1 kDa) and 6'-sialyllactose-GD10 conjugate 18 (GD10/6'-SL/PEG = 1:3:1; 13.8 kDa) in 89% and 90% respective yields after C-18 purification and lyophilization.14 Analysis of the 1H and 19F NMR spectra, and MALDI-TOF mass spectra of 17 and 18 established the structure of the final targets (see Experimental procedures).
Synthesis of AF-labeled GD10-containing glycoconjugates
The glycoconjugates (16–18, 13.0–14.1 kDa) bearing GD10 and an azide moiety are primed for introducing additional tags for use in biological experiments using CuAAC ligation. To illustrate this, we prepared a new series of AF-labeled glycoconjugates with GD10 (20–22, 13.7–14.8 kDa). We adopted an established ligand-free Cu-power catalyzed CuAAC ligation method originally described by Danishefsky and co-workers,27 with some modification (Scheme 5).14 The CuAAC ligation of conjugate 16 was performed with commercially available AF alkyne 19 (1.5 equiv. per N3) in the presence of Cu-powder (10 equiv. per N3) in phosphate-buffered saline (PBS pH 7.4) under an N2 atmosphere for ∼3 h. The solid was removed by filtration and the crude product was purified by C-18 chromatography to afford conjugate 20 (13.7 kDa) in good yield. The appearance of triazole peaks at 8 ppm in 1H NMR, and analysis of the MALDI-TOF mass spectrometry confirmed the structure of glycoconjugate 20. A similar CuAAC ligation strategy was adopted for labeling of conjugates 17 and 18 with alkyne 19 under CuAAC ligation conditions to provide the desired AF-labeled glycoconjugates 21 (14.8 kDa) and 22 (13.7 kDa) in 80% and 83% respective yields after C-18 purification and lyophilization.
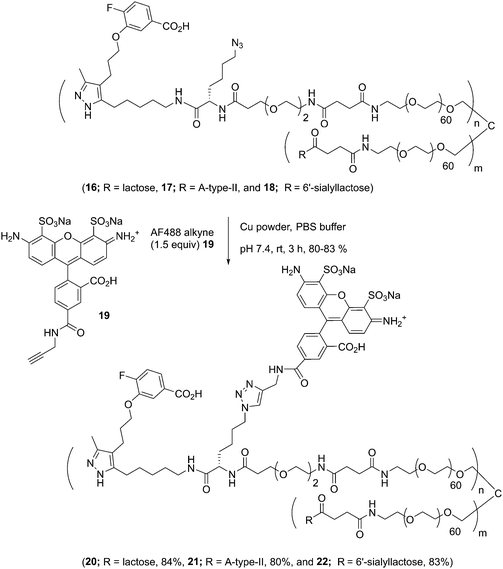 |
| | Scheme 5 Conjugation of AF to tetravalent scaffolds (16, 17, and 18) using CuAAC. | |
GD10 analogs bind to TTR with high affinity
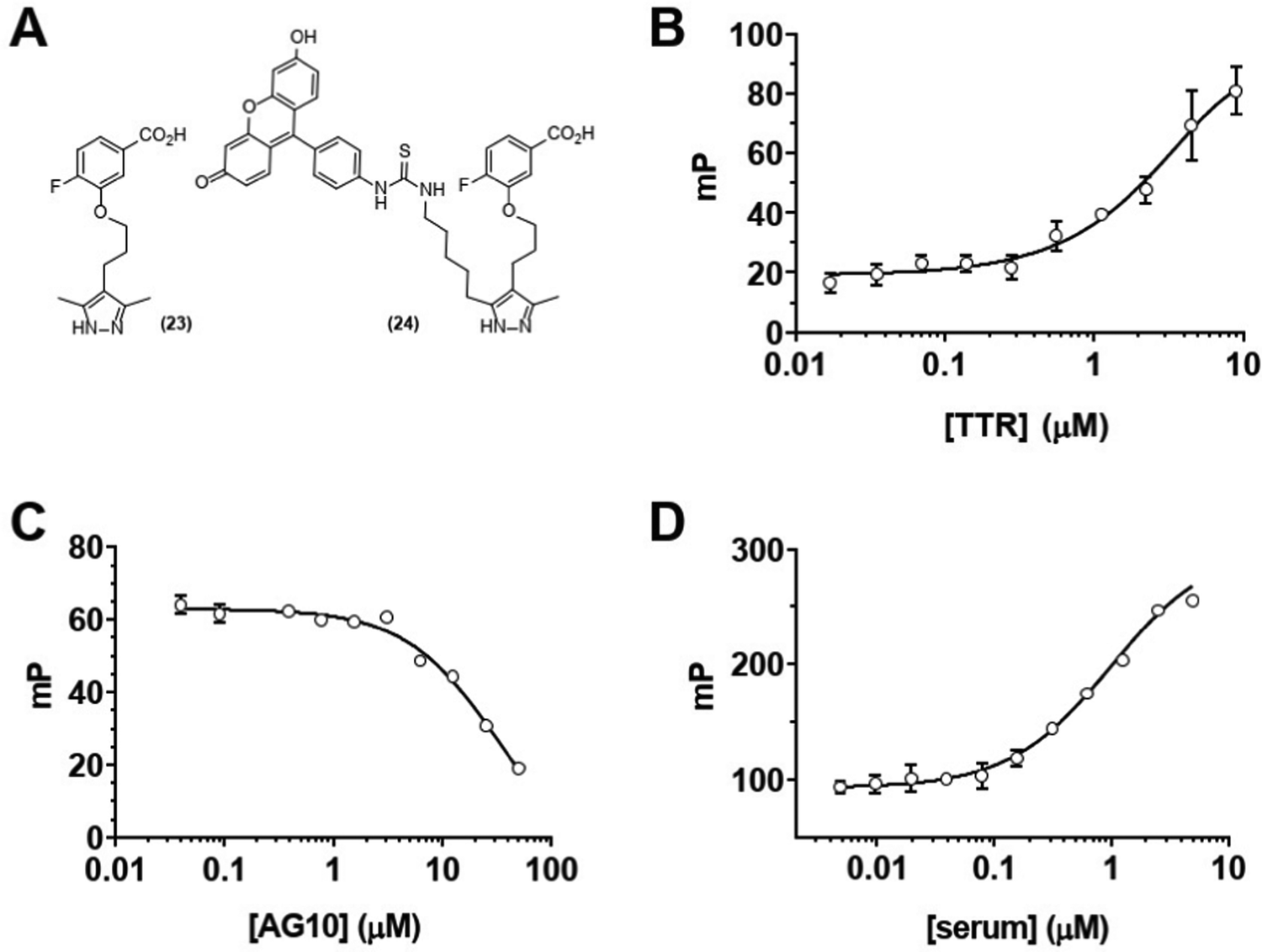
We next sought to confirm that GD10 maintained affinity for its TTR protein target. The modification of the AG10 binding epitope in GD10 involved attachment through one of the methyl groups of the pyrazole ring. We investigated the binding of these analogs to TTR in PBS (pH 7.4) using a fluorescence polarization (FP) assay (Fig. 1). We first tested the direct binding of 24 to TTR by titrating a solution of the compound with increasing concentration of protein (Fig. 1B). These data confirmed that 24 bound directly to TTR (kapp = 5 μM). We note that this is lower than the reported affinity for AG10 (5 nM) and closer to that of diethylpyrazole analogs of AG10.22 We next confirmed that complex formation between TTR and 24 could be disrupted by competition with the AG10 analog 23. TTR was first pre-complexed to a GD10 analog with a fluorescent tag (24), followed by titration of unlabelled AG10 analog 23. The loss of polarization observed was consistent with competitive binding of GD10 on TTR with a kapp = 30 μM (Fig. 1C). Finally, we considered that the hydrophobic structure of GD10 could interact with native serum proteins. We performed an analogous experiment by titrating mouse serum into a solution of 24 (Fig. 1D). We observed an increase in polarization consistent with direct binding of 24 to mouse serum proteins, similar to that of TTR alone (kapp = 1 μM). The observed lower kapp for 24 to serum over purified TTR may indicate that interactions with other serum proteins could affect clearance.
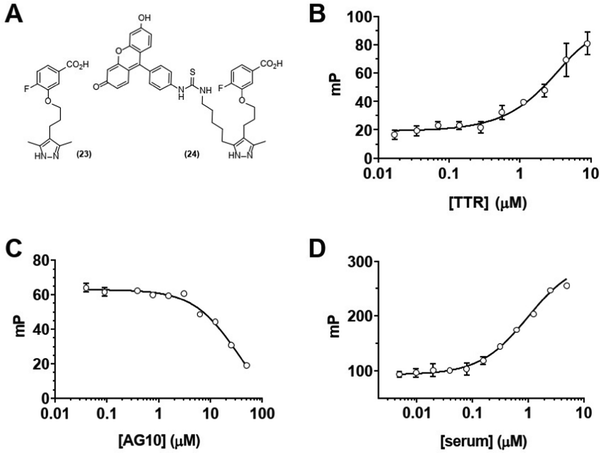 |
| | Fig. 1 Analogs of AG10 (23) and GD10 (24) bind to TTR and serum proteins. (A) Chemical structures of TTR ligands. (B) Direct binding of ligand 24 to TTR (0.017–9 μM) in buffer by FP (kapp = 5 μM); (C) competition of the 24-TTR complex with increasing concentrations (0.04–50 μM) of AG10 analog 23 (kapp = 30 μM); (D) binding of compound 24 to mouse serum proteins (0.005–5 μM) (kapp = 1 μM). | |
Glycoconjugates with GD10 have extended half-life in mouse serum
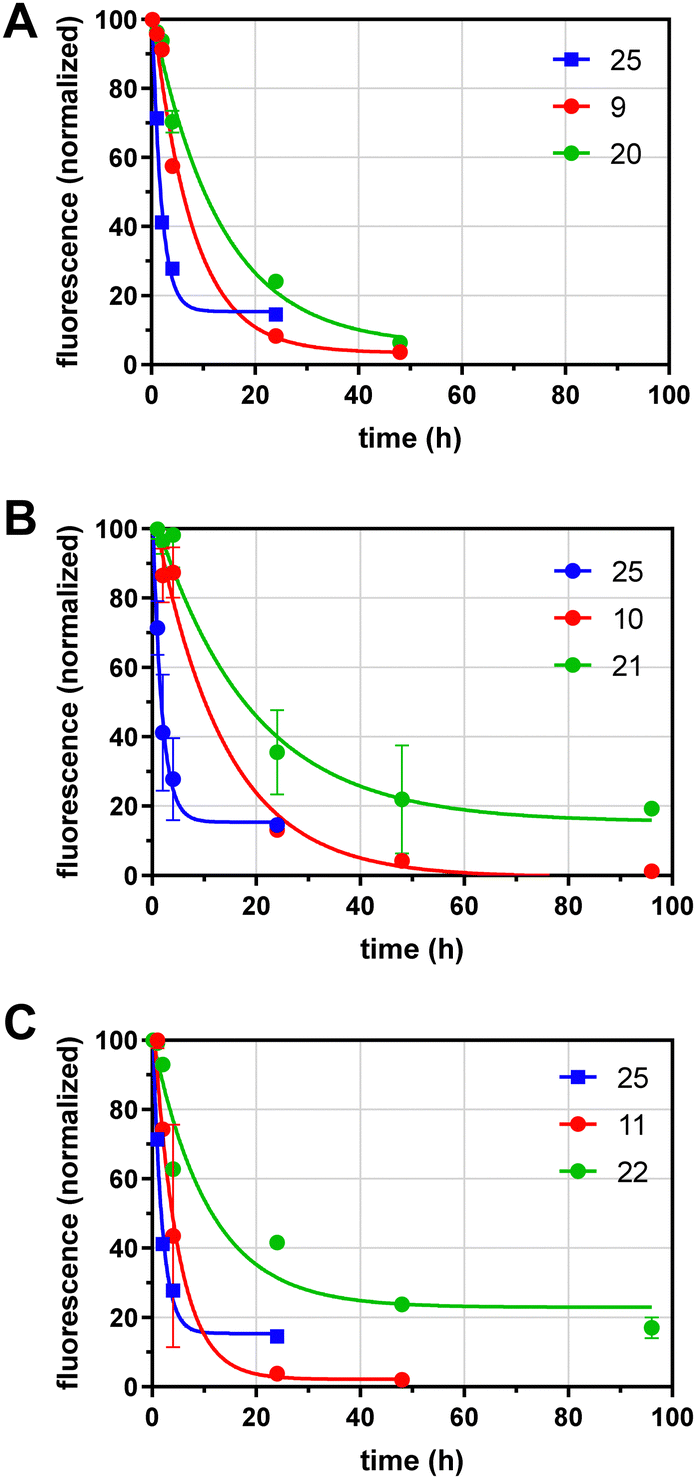
We next analyzed the effects of GD10 and glycoside incorporation on the clearance of glycoconjugates from mouse serum in vivo. To determine the half-life of conjugates in serum, fluorescent conjugates (100–400 μg) were injected into the saphenous vein in adult wild-type mice under isoflurane anesthesia and samples of serum were obtained over 96 h. The fluorescence remaining in serum was determined and normalized to the earliest timepoint (10 min) and the data fit to a single exponential decay (Fig. 2 and Table 1). We compared the clearance of our glycoconjugates to a protein of similar molecular weight (alpha-1-acidic glycoprotein, AGP) that was also labelled with AF 25 (dye:protein ratio of 1.2), which had a t1/2 of 1.2 h. Conjugates containing lactose and an AF tag on the tetravalent PEG scaffold (9) had a longer t1/2 of 5.3 h. A conjugate with 6′-SL and AF 11 had relatively rapid clearance with a t1/2 of 3.3 h. Modification of the lactose conjugate with GD10 extended the t1/2 to 9.1 h in the case of the lactose conjugate 20, and we observed a similar effect for the AII conjugate 10 (t1/2 of 9.3 h) which was extended to t1/2 of 12.8 h after attachment of GD10 in conjugate 21. Modification of the 6′-sialyllactose conjugate with GD10 in conjugate 22 extended the t1/2 to 7.5 h. In all, we observed that 6′-sialylactose and lactose conjugates were cleared most rapidly, while AII conjugates were more persistent. The attachment of GD10 extended the t1/2 in all cases, with increases of 2-fold or more. The most persistent conjugate in this series was 21, which had more than 10-fold longer t1/2 as compared to protein conjugate 25. We note that the metabolic stability of the conjugates was not investigated in this study. Furthermore, we cannot rule out that a portion of the conjugates tested may have more than one copy of GD10 due to the statistical mixture that results from our strategy.
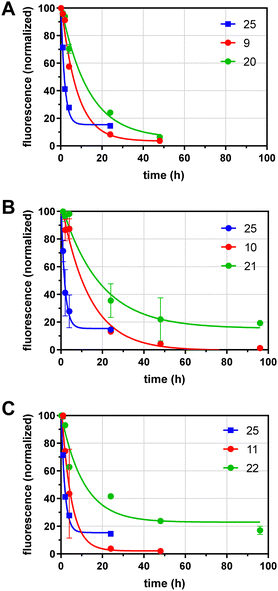 |
| | Fig. 2
In vivo half-life determinations for glycoconjugates. Compounds were injected i.v. into mice, and serum samples taken at indicated time points. The fluorescence of serum samples was compared to the initial reading (10 min) and fit to a single-phase exponential decay. Fluorophore labeled glycoconjugates are plotted in red, with the corresponding GD10 conjugate shown in green. Data for protein conjugate 25 (blue) are shown for comparison in all panels relative to (A) compound 9 and 20; (B) compounds 10 and 21; and (C) compounds 11 and 22. | |
Table 1 The in vivo half-life in mouse serum of glycoconjugates with and without serum protein binders
| Entry |
Conjugate |
Condensed name |
Stoichiometry (n:m) |
M
W (kDa) |
In vivo half-life (h) |
|
Compounds previously reported.14
|
| 1 |
9
|
AF/Lac/PEG |
1.2:2.8 |
13.0 |
5.3 |
| 2 |
20
|
(GD10 + AF)/Lac/PEG |
1:3 |
13.7 |
9.1 |
| 3 |
10
|
AF/AII/PEG |
0.6:3.4 |
14.0 |
9.3 |
| 4 |
21
|
(GD10 + AF)/AII/PEG |
1:3 |
14.7 |
12.8 |
| 5 |
11
|
AF/6′SL/PEG |
1:3 |
13.9 |
3.3 |
| 6 |
22
|
(GD10 + AF)/6′SL/PEG |
1:3 |
14.5 |
7.5 |
| 7 |
25
|
AF-protein |
1.2 (Dye:protein) |
18.4 |
1.2 |
Distribution of fluorescent glycoconjugates in whole animals
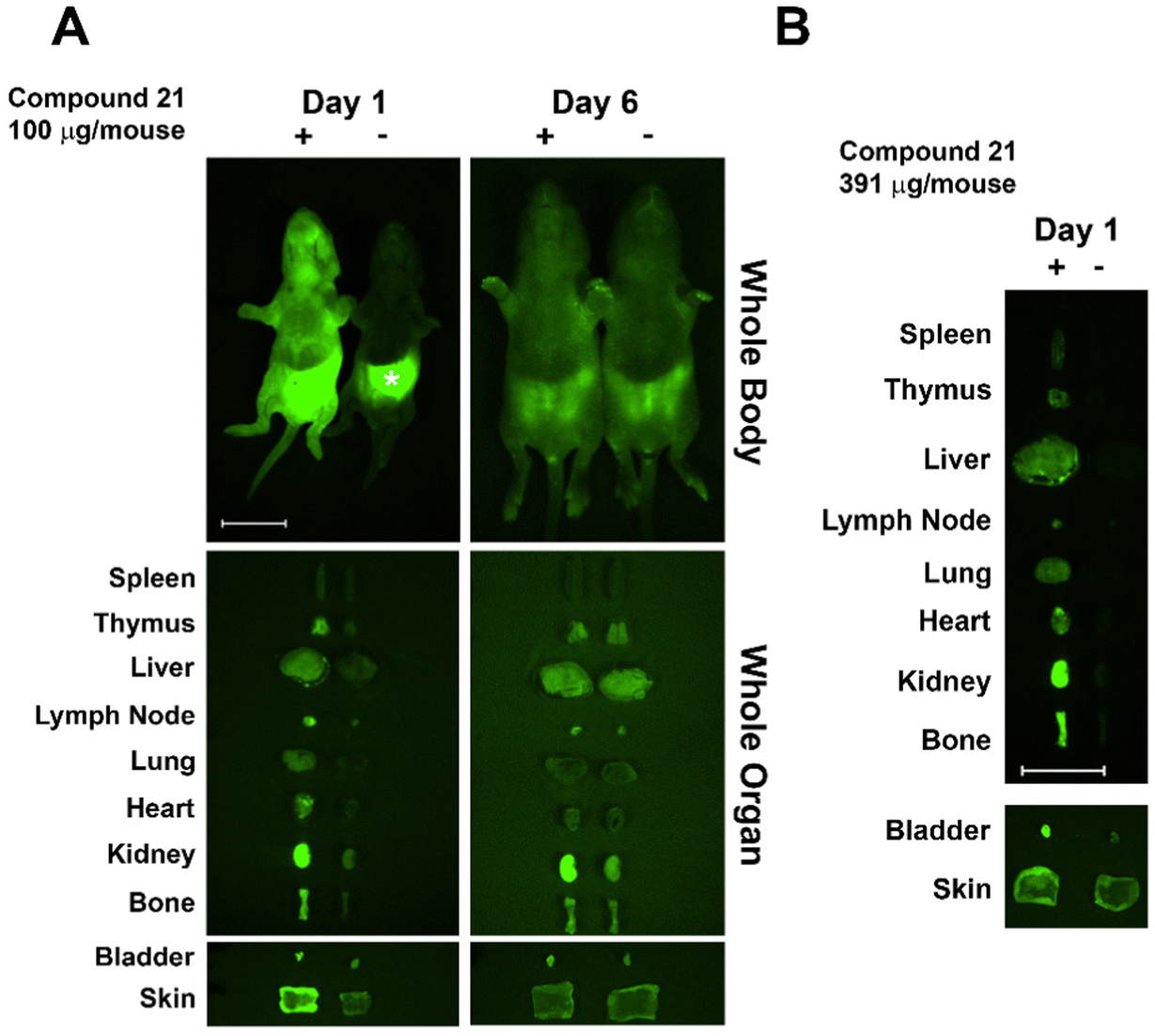
Based on previous studies in C3H mice, we used newborn mice for clearance studies as newborns have decreased background autofluorescence in imaged tissues/whole animals vs. adults.28 Glycoconjugate 21 was injected i.v. into newborn mice (within 24 h of birth) and its trafficking and clearance monitored. The glycoconjugate was injected at a concentration of 100 μg per mouse, similar to the dose used in previous studies.29 Injected mice were euthanized at days one and six post-injection, and internal organs dissected and imaged next to those from non-injected littermate controls. The injected AF labeled-glycoconjugate was detected in host organs using an Olympus OV100 small animal imaging system. At day one post-injection 21 was detected in multiple organs with accumulation/sequestering in specific organs. Conjugate 21 sequestered mainly in kidney and bladder, followed by bone, with lower amounts detected throughout the mouse (Fig. 3). Sequestration in kidney and bladder indicate conjugate 21 was targeted for secretion. Skewed distribution in bone at day one may facilitate antigen exposure and tolerance induction, though additional higher resolution studies are needed. At day 6 post-injection almost all remaining AF signal was in kidney; however, a weaker signal was detected in other organs including bone. Injection of 21 at a higher dose (400 μg per mouse) resulted in higher fluorescence in bone at day one (Fig. 3B).
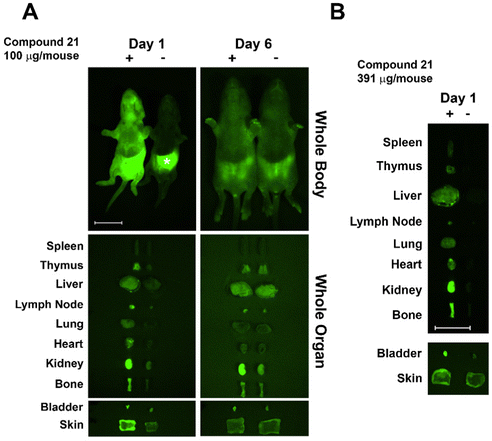 |
| | Fig. 3 Trafficking, fate, and clearance of injected A-type II antigen conjugates (21) in neonatal mice. Whole body and organ imaging of mice injected with fluorescent conjugate 21 or control animals was performed. Background autofluorescence was observed in controls due to stomach milk (*). Samples are labeled in the figure as (−) non-injected littermate control, and (+) animals injected with 21. Scale bars: 10 mm (whole-body and organ imaging). | |
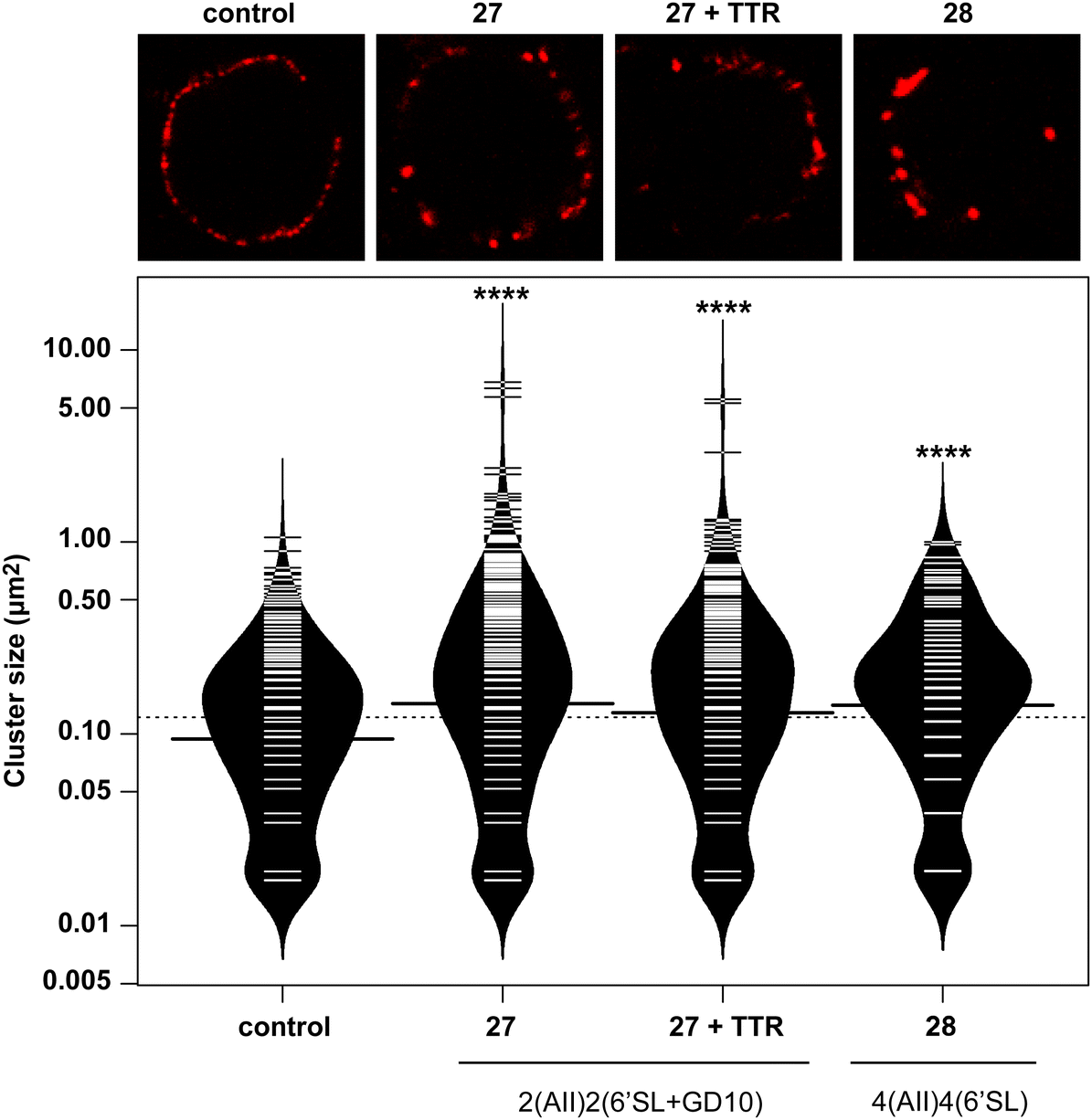
Glycoconjugates with GD10 maintain the ability to cluster BCR on ABL cells
In previous work, we had designed and tested glycoconjugates similar to those developed here featuring two carbohydrate epitopes that each engaged different receptors on B cells (BCR and CD22).14 In that study, we found that four copies of a CD22 ligand were not sufficient to cluster the receptor, but that inclusion of four copies of a BCR ligand (the ABO A-type II antigen) resulted in co-clustering of both receptors. To investigate if the inclusion of a TTR ligand would interfere with clustering of a target receptor, we used 17 to synthesize a conjugate bearing GD10, the A-type II antigen, and 6'SL (27, Scheme 6). We then analyzed the cluster size of CD22 on B cells using confocal microscopy, and compared its activity to a previously reported conjugate 28 that was able to cluster CD22 (Fig. 4).14 We observed that conjugate 28 was able to cluster CD22 on B cells as before. Treatment of the cells with conjugate 27, or conjugate 27 in the presence of TTR, resulted in similar levels of CD22 clustering. The addition of TTR did not lead to a significant increase relative to treatment with 27 alone. These results confirm that the trivalent linker strategy developed here can provide conjugates that are able to engage cell surface receptors.
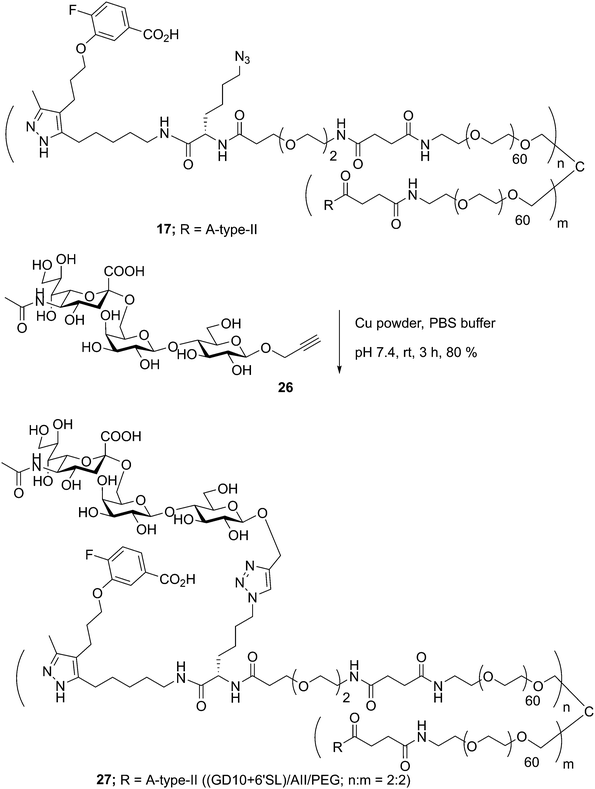 |
| | Scheme 6 Conjugation of 6'SL to tetravalent scaffold 17 using CuAAC ligation. | |
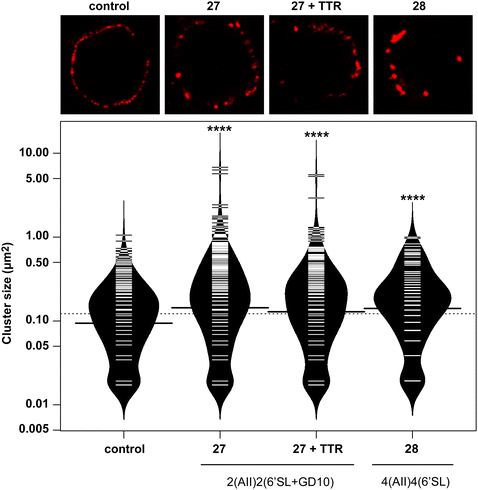 |
| | Fig. 4 Clustering of CD22 receptors by glycoconjugates and glycoproteins. | |
A-BCL cells were treated with 25 ng mL−1 PBS, 27, 27 with TTR, or 28,11 then fixed and stained with anti-CD22 (mouse) and visualized using confocal microscopy. (A) Fluorescence images of representative cells are shown. (B) Cluster size on 20 individual cells was determined using particle analysis in ImageJ and plotted using the beanplot statistical package in R.30 Each individual white horizontal line within the beanplot represents one single data point, the black horizontal line indicates the mean for each condition, and the dotted line indicates mean of all conditions within the plot. Populations which were statistically different from control are indicated where p < 0.0001 with ****, as calculated using a student's t-test in GraphPad Prism.
Conclusions
In this study, we developed a TTR-binding small molecule, GD10, which is easily prepared as an amine in three steps (overall yield 50%). Our results indicate that asymmetric modification of the pyrazole ring of AG10 resulted in ligands with moderate affinity for TTR. This is the first demonstration of the extension of glycoconjugate half-life in vivo using this strategy. The inclusion of the GD10 tag was able to extend the serum half-life of conjugates by as much as 2-fold over the parent structure. PEG-based conjugates featuring human ABO blood group antigens and GD10 had half-lives that were extended by as much as 10-fold in serum relative to protein conjugates of similar molecular weight. Finally, we confirmed that these conjugates were able to penetrate multiple organs of animals including lymph nodes and bone and maintained the ability to engage cellular receptors. These results suggest this strategy may have enhanced capacity for induction of immune tolerance to carbohydrate structures.
Experimental procedures
General methods
Reagents were purchased from commercial sources as noted and used without additional purification. Compounds 6, 7, 8, and 26 as well as conjugates 9, 10, and 28 were previously reported (see ESI†)14 Compound 23 was prepared as reported. NHS-activated PEG scaffold 13 was obtained from a commercial source (Laysan Bio Inc, Arab, Alabama). AF488 alkyne 19 was obtained from ThermoFischer Scientific. Cu-powder (<425 μm) and DIPEA were purchased from Sigma-Aldrich, Canada. Chloroform-d1 and D2O were purchased from Deutero GmbH. Other solvents (analytical and HPLC grade) and reagents were purchased from Aldrich and were used as received. Reactions were monitored by analytical TLC on silica gel 60-F254 (0.25 nm, Silicycle, QC, Canada). Developed TLC plates were visualized under UV lamp (λmax = 254 nm) and charred by heating plates that were dipped in ninhydrin solution in ethanol, and acidified anisaldehyde solution in ethanol. Reaction products were purified by silica gel column chromatography (230–400 mesh, Silicycle, QC, Canada) or C-18 Sep-pak chromatography using ethyl acetate/hexane, or CH2Cl2/MeOH, and MeOH/H2O as the elution solvents, respectively. Purifications were also performed by semi-preparative RP-HPLC with a Waters Delta 600 pump, and a Waters 600 controller with Empower 2 software. Eluted peaks were detected with a Waters 2420 evaporative light scattering (ELS) detector or a Waters 2996 photodiode array (PDA) detector (Waters Ltd, Mississauga, ON, Canada). Purifications were performed using solvent A: 0.01% TFA in water and solvent B: in CH3CN at flow rate of 8.0 mL min−1 using a linear gradient of 2–50% solvent B in 20 min and UV detection at 214 nm. NMR experiments were conducted on a Varian 400, 500, or 600 MHz instruments in the Chemistry NMR Facility, University of Alberta. Chemical shifts are reported relative to the deuterated solvent peak or 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt as an internal standard and are in parts per million (ppm). Coupling constants (J) are reported in Hz and apparent multiplicities were described in standard abbreviations as singlet (s), doublet (d), doublet of doublets (dd), triplet (t), broad singlet (bs), or multiplet (m). The ratio of the incorporated GD10, carbohydrate structures and AF onto the PEG scaffold was determined by integration of the aromatic peaks of GD10 (7.63–7.07 ppm), anomeric peaks (5.30–4.40 ppm), PEG–COCH2CH2CO–(s, 2.49 or 2.50 ppm, 4H/per site), NHAc moiety of sialic acid (s, 1.98 or 1.99 or 2.00 ppm, 3H) and aromatic peaks of the fluorophore protons in 1H NMR spectra. Electrospray mass spectra (ESI-MS) were recorded on Agilent Technologies 6220 TOF.
BALB/c and C3H mice (Charles River Laboratories, Sherbrooke, Québec, Canada) were housed in facilities at University of Alberta and cared for under guidelines of the Canadian Council of Animal Care. Animal protocols were approved by UofA Health Sciences Animal Care and Use Committee.
Synthesis of compound 2
To a suspension of NaH (270 mg, 60% mineral oil) in dry THF (12 mL) in a 50 mL flask, was added dropwise 2,4-diketopentane (0.58 mL, 5.8 μmol) at 0 °C under a nitrogen atmosphere. The solution was stirred for 10 min and to this suspension was added dropwise nBuLi (2.2 mL, 2.5 M in hexane) and the resulting yellow solution was stirred for additional 15 min at 0 °C under an argon atmosphere. The reaction mixture was cooled to −30 °C and then was added 1-bromo-4-chlorobutane (0.67 mL, 5.8 μmol) slowly by syringe and the reaction mixture was stirred for additional ∼1 h at −25 °C. The reaction mixture was quenched with aqueous HCl (2N, 1 mL) and the solvents were removed under vacuum, diluted with diethyl ether (2 × 100 mL), washed with brine solution, dried over Na2SO4 and concentrated under vacuum to afford 9-chloro-4-oxo-2-nonanone as yellow oil which was used for the next reaction directly. NaN3 (207 mg, 3.18 mmol) and NaI (10 mg) were added to a solution of 9-chloro-4-oxo-2-nonanone in dry DMF (5 mL) and the reaction mixture was heated to 60 °C overnight under an argon atmosphere. The solvent was removed, and the crude product was purified by silica gel chromatography using gradient elution (hexane to 1:9 v/v EtOAc/hexane) to afford compound 2 (867 mg, 76%) as a colorless oil. 1H NMR (600 MHz, CDCl3): δ 5.49 (s, 1H), 3.27 (t, J = 10.2 Hz, 2H), 2.89 (t, J = 10.8 Hz, 2H), 2.05 (s, 3H), 1.66–1.58 (m, 4H), 1.43–1.39 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 193.82, 191.30, 99.83, 44.78, 38.04, 32.30, 26.46, 24.93, 24.87; (ESI): m/z [M + H]+ calcd for C9H15N3O2 − H: 196.1092, found: 196.1091.
Synthesis of methyl-3-(3-(3-methyl-5-(5-azido pentanyl)-1H-pyrazol-4-fluorobenzoate 4
To a solution of compound 2 (299 mg, 1.51 mmol) and DBU (0.22 mL, 1.48 mmol) in dry benzene (2 mL) was added slowly a solution of 1 (220 mg, 0.75 mmol) in benzene (1 mL) under an argon atmosphere. The reaction mixture was stirred at room temperature for 2 days and filtered through Celite pad and solvent was concentrated under vacuum. The crude product was purified by silica gel chromatograph using gradient elution (hexane to 1:4 v/v EtOAc/hexane) to afford 3 as a colorless oil, which was directly used for the next reaction. To a solution of 3 in EtOH (2 mL) was added hydrazine monohydrate (73 μL, 2.27 mmol) and the reaction mixture was refluxed at 92 °C for 4 h. The reaction mixture was concentrated and purified by silica gel chromatograph using gradient elution (hexane to 1:4 v/v MeOH/CHCl3) to afford desired compound 4 as a colorless oil. Rf = 0.38 (EtOAc/hexane = 2:3); 1H NMR (400 MHz, CDCl3): δ 7.40–7.38 (m, 1H), 7.28–7.25 (m, 1H), 7.11–7.07 (m, 1H), 3.99 (t, J = 6.0 Hz, 2H), 3.23–3.19 (m, 2H), 2.59–2.50 (m, 4H), 2.16 (S, 3H), 1.96–1.95 (m, 2H), 1.62–1.53 (m, 4H), 1.37–1.35 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 167.67, 155.79, 153.79, 147.31, 147.23, 129.21, 129.18, 119.30, 119.24, 116.20, 116.05, 113.91, 113.76, 67.84, 51.34, 29.67, 28.78, 28.60, 26.56, 25.43, 18.89, 10.77; (ESI): m/z [M + H]+ calcd for C20H26FN5O3 + H: 404.2092, found: 404.2204.
Synthesis of ethyl-3-(3-(3-methyl-5-(5-amino pentanyl)-1H-pyrazol-4-fluorobenzoate 5
To a solution of compound 4 (100 mg, 0.24 mmol) in a mixture of MeOH–H2O–THF (3 mL, 1:0.5:1.5 v/v) was added PPh3 (325 mg, 1.24 mmol) at room temperature and the reaction mixture was vigorously stirred for overnight. The solvents were removed and the crude product was purified by silica–gel chromatography using elution gradient (CHCl3 to 2:3 v/v MeOH/CHCl3) to afford compound 5 (81.4 mg, 87%) as a colorless oil. Rf = 0.28 (MeOH/CHCl3 = 2:3); 1H NMR (400 MHz, CDCl3): δ 7.63–7.59 (m, 2H), 7.13–7.09 (m, 1H), 4.02 (t, J = 5.6 Hz, 2H), 3.89 (s, 3H), 2.90 (bs, 2H), 2.55–2.53 (m, 3H), 2.16 (s, 3H), 1.96 (bs, 6H), 1.63–1.62 (m, 4H), 1.38 (bs, 2H); 13C NMR (125 MHz, CDCl3): δ 166.17, 156.66, 154.64, 146.93, 146.85, 126.55, 126.52, 123.09, 123.03, 116.06, 115.91, 115.62, 115.59, 67.95, 52.26, 38.94, 29.82, 27.21, 26.82, 25.17, 24.56, 18.89, 10.45; (ESI): m/z [M + H]+ calcd for C20H28FN3O3 + H: 378.2187, found: 378.2186.
Synthesis of conjugate 11
A mixture of AlexaFluor 488 cadaverine 12 (0.6 mg, 0.93 μmol, 1.05 equiv.) and NHS-activated PEG 13 (10 mg, 0.89 μmol, 1 equiv.) were dissolved in dry DMF (80 μL) and DIPEA and placed in a 0.5 mL centrifuge tube at room temperature. After ∼3 h, the 6′-sialyllactose amine 8 (2.37 mg, 2.90 μmol, 3.25 equiv.) was added and the reaction mixture was purged with argon and incubated at 23 °C for an additional ∼21 h. The solvent was removed and the crude product was purified by C-18 Sep-pak chromatography using gradient elution of H2O to MeOH/H2O (8:2 v/v) to provide conjugate 11 (11.0 mg, 89%) as a reddish-colored powder after lyophilization. Substitution of the PEG scaffold was determined by 1H NMR integration of the underlined peaks to be Alexa Fluor:lactose:PEG = 1:3:1. 1H NMR (500 MHz, D2O): δ ![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif)
![[. with combining low line]](https://www.rsc.org/images/entities/char_002e_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) (bs, 0.15H),
(bs, 0.15H), ![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) (bs, 0.6H),
(bs, 0.6H), ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif)
![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) (d, J = 7.2 Hz, 0.3H), 8.09–8.05 (m, 0.15H),
(d, J = 7.2 Hz, 0.3H), 8.09–8.05 (m, 0.15H), ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif)
![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) (s, 0.3H),
(s, 0.3H), ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) (s, 0.3H), (d, J = 9.6 Hz, 0.6H), (d, J = 9.0 Hz, 0.6H),
(s, 0.3H), (d, J = 9.6 Hz, 0.6H), (d, J = 9.0 Hz, 0.6H), ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif)
![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) (d, J = 8.0 Hz, 1H), (dd, J = 7.8 Hz, 1H), 4.41 (dd, J = 1.8, 12.0 Hz, 1H), 4.03–4.01 (m, 2H), 3.98–3.67 (bs, 273H), 3.60 (bs, 3H), 3.40–3.35 (m, 2H), 2.72 (t, J = 6.6 Hz, 2H), 2.64 (t, J = 6.6 Hz, 2H), 2.55–2.63 (m, 1H), (bs, PEG–COCH2CH2CO–, 4H), (s, 3H, NHAc), 1.82 (t, J = 12.0 Hz, 1H), 1.68–1.62 (m, 6H), 1.50–1.49 (m, 3H), 1.32–1.31 (m, 13H); 13C NMR (125 MHz, D2O): δ 175.85, 175.63, 175.57, 173.69, 164.73, 156.70, 156.66, 142.98, 120.80, 111.70, 111.52, 104.21, 102.89, 80.69, 75.69, 75.59, 74.60, 73.73, 73.59, 73.35, 72.52, 71.75, 71.63, 71.55, 70.56 (PEG CH2–), 69.81, 69.46, 69.37, 69.09, 64.48, 63.72, 61.28, 52.75, 46.03, 40.81, 39.95, 39.70, 32.19 (2C), 32.04, 31.34, 29.71 (2C), 29.34, 29.16, 29.01, 28.85, 28.20, 25.97, 23.05; the MALDI-TOF spectrum showed the average mass centered at 13.8 kDa and expected average mass was 13.8 kDa.
(d, J = 8.0 Hz, 1H), (dd, J = 7.8 Hz, 1H), 4.41 (dd, J = 1.8, 12.0 Hz, 1H), 4.03–4.01 (m, 2H), 3.98–3.67 (bs, 273H), 3.60 (bs, 3H), 3.40–3.35 (m, 2H), 2.72 (t, J = 6.6 Hz, 2H), 2.64 (t, J = 6.6 Hz, 2H), 2.55–2.63 (m, 1H), (bs, PEG–COCH2CH2CO–, 4H), (s, 3H, NHAc), 1.82 (t, J = 12.0 Hz, 1H), 1.68–1.62 (m, 6H), 1.50–1.49 (m, 3H), 1.32–1.31 (m, 13H); 13C NMR (125 MHz, D2O): δ 175.85, 175.63, 175.57, 173.69, 164.73, 156.70, 156.66, 142.98, 120.80, 111.70, 111.52, 104.21, 102.89, 80.69, 75.69, 75.59, 74.60, 73.73, 73.59, 73.35, 72.52, 71.75, 71.63, 71.55, 70.56 (PEG CH2–), 69.81, 69.46, 69.37, 69.09, 64.48, 63.72, 61.28, 52.75, 46.03, 40.81, 39.95, 39.70, 32.19 (2C), 32.04, 31.34, 29.71 (2C), 29.34, 29.16, 29.01, 28.85, 28.20, 25.97, 23.05; the MALDI-TOF spectrum showed the average mass centered at 13.8 kDa and expected average mass was 13.8 kDa.
Synthesis of compound 15
DIPEA (∼5.5 μL, 31.90 μmol) was added to a solution of GD10 amine 5 (10.0 mg, 26.51 mmol) and NHS-activated linker 14 (21.5 mg, 33.13 mmol) in anhydrous DMF (150 μL) at room temperature. The pH of the solution was adjusted to between 7.5–8.0 and the reaction mixture was incubated for one day at room temperature under anhydrous conditions. The solvent was removed, and the crude product was dried and treated with a 20% solution of piperidine–DMF (1 mL, 1:4) for 2 h. The solvent was removed under vacuum, and the crude product was treated with LiOH (1.0 mg, 39.76 μmol) in H2O/THF (2 mL, v/v = 3:1) for overnight at room temperature. The solvents were removed, and the crude product was purified by semipreparative RP-HPLC to afford compound 15 (13.8 mg, 75%) as colorless oil after lyophilization. Analytical RP-HPLC tR = 12.7 min (gradient 5 to 100% B in 30 min). 1H NMR (600 MHz, D2O): δ 7.67 (q, J = 1.8 Hz, 1H), 7.65–7.62 (m, 1H), 7.23–7.19 (m, 1H), 4.26 (q, J = 6.0 Hz, 1H), 4.11 (t, J = 6.0 Hz, 2H), 3.75–3.73 (m, 2H), 3.68 (t, J = 6.0 Hz, 1H), 3.65–3.62 (m, 4H), 3.33 (s, 1H), 3.30 (bs, PEG–CH2–, 8H), 3.17–3.09 (m, 2H), 2.75–2.71 (m, 3H), 2.53–2.50 (m, 2H), 2.36 (s, 3H), 2.05–2.02 (m, 2H), 1.74–1.66 (m, 1H), 1.65–1.59 (m, 2H), 1.58–1.51 (m, 2H), 1.49–1.40 (m, 2H), 1.37–1.33 (m, 2H); 13C NMR (125 MHz, D2O): δ 174.09 (2CO), 173.92, 163.14, 162.87, 147.49, 135.83, 123.36, 121.74, 119.41, 117.08, 114.82, 71.31, 71.24, 68.86, 68.60, 68.23, 54.69, 52.28, 40.33, 37.40, 32.87, 31.11, 30.77, 30.25, 30.10, 29.49, 27.71, 26.49, 24.17, 19.77, 12.57; 19F–C NMR (400 MHz, D2O): δ −75.50; (ESI): m/z [M + H]+ calcd for C33H51FN8O7 + H: 691.3938, found: 691.3933.
General procedure for conjugation of compound 15 and amine-terminated carbohydrate with PEG scaffold 13
A mixture of compound 15 (1.0–1.25 equiv.) and NHS-activated PEG scaffold (13, 10 mg, 1 equiv.) were dissolved in anhydrous DMF (∼80–90 μL) and placed in a 0.5 mL centrifuge tube at room temperature. DIPEA (1–1.25 equiv.) was added to adjust pH of the solution ∼7.5–8.0 and the reaction mixture was incubated at room temperature. Consumption of compound 15 and progress of conjugation was monitored by TLC and LC-MS/MS analysis. After ∼3 h, the carbohydrate amine (3.0–3.5 equiv.) was added and the solution was incubated at 23 °C for additional ∼21 h. The solvent was removed under vacuum, and the crude product was purified by C-18 Sep-pak chromatography using H2O to H2O/MeOH (∼3:7, v/v) as an eluent to afford azido-terminated GD10 conjugate as a colorless powder after lyophilization.
General procedure for CuAAC ligation
A solution of an azido-terminated GD10-PEG-conjugate (1 equiv.) and AlexaFluor 488 alkyne 19 (1.5 equiv. per N3) in PBS buffer (10 mM, pH 7.4) was degassed under an argon atmosphere for ∼15 min. Cu-power (10 equiv. per N3) was added under an argon atmosphere and the reaction mixture was vigorously stirred for additional ∼3 h at room temperature. The solution was filtered through a two-micron filter and the crude product was purified by C-18 Sep-pak chromatography using H2O to MeOH/H2O eluent to afford the desired conjugate as a reddish-colored powder after lyophilization.
Synthesis of conjugate 16
Conjugate 16 was obtained through coupling of compound 15 (0.60 mg, 0.89 μmol, 1.0 equiv.) and lactose amine 6 (1.65 mg, 3.12 μmol, 3.5 equiv.) with NHS-activated PEG 13 (10 mg, 0.89 μmol, 1 equiv.) following the general procedure for conjugation of compound 15 and amine-terminated carbohydrate. Yield: (10.54 mg, 90%). Substitution of compound 15 and amine 6 with the PEG scaffold was determined by 1H NMR integration of the underlined peaks shown below to be GD10/lactose/PEG = 0.95:3.05:1. 1H NMR (500 MHz, D2O): δ – (m, 2H), – (m, 1H), (d, J = 8.0 Hz, 1H), (d, J = 8.0 Hz, 1H), 4.25 (q, J = 6.0 Hz, 1H), 4.01 (t, J = 6.0 Hz, 2H), 3.98–3.90 (m, 4H), 3.90–3.61 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 274H), 3.45–3.43 (band, PEG–CH2–, 8H), 3.37–3.34 (m, 2H), 3.16–3.09 (m, 2H), 3.07–3.03 (m, 2H), 2.97–2.90 (m, 2H), 2.74 (t, J = 7.8 Hz, 2H), 2.64 (t, J = 6.0 Hz, 2H), 2.70–2.65 (m, 2H), (bs, PEG–COCH2CH2CO–, 4H), 2.34–2.26 (m, 2H), 2.10–2.00 (m, 1H), 1.80–1.73 (m, 2H), 1.68–1.61 (m, 8H), 1.42–1.31 (bs, 14H); 13C NMR (125 MHz, D2O): δ 182.48, 175.48 (2CO), 174.92, 164.73, 163.87, 147.49, 103.91, 103.02, 79.42, 76.33, 75.73, 75.46, 73.83, 73.52, 72.70, 71.93, 71.65, 71.56 (2C), 70.57 (PEG–CH2–), 69.82 (2C), 69.52, 69.32, 67.99, 67.80, 67.64, 65.41, 64.33, 61.99 (2C), 61.34, 61.10, 54.91, 51.82, 51.64, 49.84, 46.04, 39.95, 39.71, 38.88, 36.69, 32.19, 32.04, 31.85, 31.35, 29.71, 29.34, 29.16, 29.02, 28.85, 28.59, 25.97, 23.36, 19.77, 12.57; 19F NMR (400 MHz, D2O): δ −75.51; the MALDI-TOF spectrum showed the average mass centered at 13.0 kDa and expected average mass was 12.9 kDa.
Synthesis of conjugate 17
Conjugate 17 was obtained through coupling of amine 15 (0.81 mg, 1.20 μmol, 1.35 equiv.) and A-type II amine 7 (2.87 mg, 3.12 μmol, 3.5 equiv.) with NHS-activated PEG 13 (10 mg, 0.89 μmol, 1 equiv.) following the general procedure for conjugation of compound 16. Yield: (11.30 mg, 89%). Substitution of the compound 15 and amine 7 with the PEG scaffold was determined by 1H NMR integration of the underlined peaks shown below to be GD10/A-type II/PEG = 1.03:2.97:1. 1H NMR (500 MHz, D2O): δ – (m, 1H), – (m, 1H), – (m, 1H), (d, J = 3.6 Hz, 1H), (d, J = 3.0 Hz, 1H), (d, J = 7.6 Hz, 1H), (d, J = 8.0 Hz, 2H), 4.42–4.40 (m, 1H), 4.37 (d, J = 6.6 Hz, 1H), 4.31–4.28 (m, 4H), 4.04 (t, J = 6.0 Hz, 2H), 3.97–3.94 (m, 4H), 3.89–3.67 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 271H), 3.64 (bs, 4H), 3.56 (bs, 4H), 3.51–3.48 (m, 2H), 3.45–3.43 (band, PEG–CH2–, GD10–CH2, 10H), 3.16–3.09 (m, 2H), 3.07–3.03 (m, 2H), 2.97–2.90 (m, 2H), 2.74 (t, J = 7.8 Hz, 2H), 2.64 (t, J = 7.0 Hz, 2H), 2.70–2.65 (m, 2H), (bs, PEG–COCH2CH2CO–, 4H), 2.30–2.26 (m, 2H), 2.10–2.00 (m, 1H), (s, 6H, NHAc), 1.81–1.72 (m, 2H), 1.65–1.60 (m, 6H), 1.43 (bs, 2H), 1.42–1.31 (bs, 14H), 1.30 (d, J = 6.0 Hz, 3H); 13C NMR (125 MHz, D2O): δ 175.75, 174.62 (2CO), 175.57 (2CO), 175.23, 164.74, 163.73, 147.46, 135.63, 102.08, 101.04, 99.61, 92.30, 79.17, 77.25, 76.65, 76.33, 76.14, 75.72, 73.38 (2C), 72.68, 72.05, 71.55, 71.65 (2C), 70.57 (PEG–CH2–), 69.82 (2C), 69.46, 68.77, 68.65, 67.82, 65.42, 64.01, 62.28 (2C), 62.14 (2C), 61.19, 56.41 (2C), 51.82, 50.49 (2C), 46.04, 39.95 (PEG–CH2–), 39.71 (2C), 38.88, 36.69, 32.18, 31.35 (2C), 29.72, 29.30, 28.88, 26.04, 22.93, 19.17, 16.16; 19F NMR (400 MHz, D2O): δ −75.54; the MALDI-TOF spectrum showed the average mass centered at 14.1 kDa and expected average mass was 14.0 kDa.
Synthesis of conjugate 18
Conjugate 18 was obtained through coupling of compound 15 (0.68 mg, 1.01 μmol, 1.05 equiv.) and 6′-sialyllactose amine 8 (2.76 mg, 3.37 μmol, 3.5 equiv.) with NHS-activated PEG 13 (10.8 mg, 0.96 μmol, 1 equiv.) following the general procedure for conjugation of compound 16. Yield: (12.0 mg, 90%). Substitution of the compound 15 and amine 8 with the PEG scaffold 13 was determined by 1H NMR integration of the underlined peaks shown below to be GD10/6′-sialyllactose/PEG = 1.0:3.0:1. 1H NMR (500 MHz, D2O): δ – (m, 2H), – (m, 1H), (d, J = 8.4 Hz, 1H), (d, J = 8.0 Hz, 1H), 4.01 (t, J = 6.0 Hz, 2H), 3.88–3.82 (m, 2H), 3.84–3.82 (m, 4H), 3.66–3.57 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 282H), 3.48–3.45 (m, 4H), 3.34 (t, J = 7.0 Hz, 1H), 3.30 (band, PEG–CH2–, GD10–CH2, 8H), 3.18–3.10 (m, 2H), 3.06–3.00 (m, 2H), 2.98–2.91 (m, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.54 (t, J = 7.0 Hz, 2H), 2.70–2.65 (m, 1H), (bs, PEG–COCH2CH2CO–, 4H), 2.32–2.24 (m, 2H), (s, 3H, NHAc), 1.86–1.82 (m, 2H), 1.69 (t, J = 15 Hz, 1H), 1.58–1.50 (m, 6H), 1.29 (bs, 14H); 13C NMR (125 MHz, D2O): δ 175.88 (2CO), 175.65 (2CO), 175.58 (2CO), 174.40, 164.04, 162.73, 147.43, 135.60, 104.20, 102.89, 101.28, 80.68, 75.70, 74.60, 74.65, 73.72, 73.52, 73.35, 72.73, 71.77, 71.63, 71.55 (2C), 70.56 (PEG–CH2–), 69.81 (2C), 69.48, 69.30, 64.51, 63.63, 61.29, 52.77, 49.83 (2C), 46.04, 41.06, 39.95 (2C), 39.70, 32.19 (2C), 32.04, 31.34, 29.70 (2C), 29.33, 29.15, 28.85, 25.97, 23.04, 16.17; 19F NMR (400 MHz, D2O): δ −75.54; the MALDI-TOF spectrum showed the average mass centered at 13.8 kDa and expected average mass was 13.7 kDa.
Synthesis of conjugate 20
Conjugate 20 was obtained through CuAAC coupling of compound 16 (8.0 mg, 0.61 μmol, 1 equiv.) and commercially available AF alkyne 19 (0.67 mg, 0.92 μmol, 1.5 equiv.) following the general procedure for CuAAC ligation. Yield: (7.10 mg, 84%). Substitution of compound 20 was determined by 1H NMR integration of the underlined peaks shown below to be (GD10 + AF)/Lac/PEG = 1:3. 1H NMR (500 MHz, D2O): δ (s, 0.15H), (s, 0.30H), – (m, 0.1H), (s, 0.5H, triazole H), (m, 0.15 H), – (m, 0.5H), (d, J = 7.2 Hz, 0.30 H), (d, J = 7.2 Hz, 0.30H), – (m, 0.30H), (d, J = 7.2 Hz, 0.10H), (d, J = 7.2 Hz, 0.50H), (d, J = 8.0 Hz, 1H), (d, J = 8.0 Hz, 1H), 4.28 (q, J = 6.0 Hz, 1H), 3.98 (dd, J = 3.6, 8.0 Hz, 1H), 3.93–3.91 (m, 2H), 3.81–3.79 (m, 4H), 3.76–3.62 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 286H), 3.56–3.54 (m, 2H), 3.51(band, PEG–CH2–, 4H), 3.40–3.39 (m, 4H), 3.31 (t, J = 6.0 Hz, 2H), 3.12–3.09 (m, 1H), 3.01–2.99 (m, 1H), 2.96–2.90 (m, 1H), 2.80 (s, 1H), 2.70 (t, J = 4.6 Hz, 2H), 2.60 (t, J = 6.0 Hz, 2H), 2.60–2.57 (m, 2H), (bs, PEG–COCH2CH2CO–, 4H), 2.08–2.02 (m, 1H), 1.90–1.86 (m, 2H), 1.79–1.74 (m, 2H), 1.64–1.61 (m, 8H), 1.42–1.31 (bs, 14H); 13C NMR (125 MHz, D2O): δ 175.64 (2CO), 175.62, 174.58 (2C), 103.91, 103.01, 79.41, 76.33, 75.73, 75.45, 73.83, 73.51, 72.69, 71.93, 71.67, 71.55 (2C), 70.56 (PEG–CH2–), 70.40 (2C), 69.82 (2C), 69.52, 69.32, 67.99, 67.80, 67.64, 65.41, 64.34, 61.99 (2C), 61.34, 61.10, 54.91, 51.65, 51.64, 49.83, 46.04, 39.96 (2C), 39.70, 38.88, 36.69, 34.27, 32.18, 32.16, 32.04, 31.35, 29.70 (2C), 29.32, 29.14, 29.02, 28.83, 28.59, 25.96, 22.80, 19.78; 19F–C NMR (400 MHz, D2O): δ −75.58; the MALDI-TOF spectrum showed the average mass centered at 13.6 kDa and expected average mass was 13.7 kDa.
Synthesis of conjugate 21
Conjugate 21 was obtained through CuAAC coupling of compound 17 (8.4 mg, 0.60 μmol, 1 equiv.) and commercially available AF alkyne 19 (0.66 mg, 0.91 μmol, 1.5 equiv.) following the general procedure for CuAAC ligation. Yield: (7.10 mg, 80%). Substitution of compound 21 was determined by 1H NMR integration of the underlined peaks shown below to be (GD10 + AF)/AII/PEG = 1:3. 1H NMR (500 MHz, D2O): δ (s, 0.15H), – (m, 0.1H), 8.00 (s, 0.5H,triazole H), (d, J = 7.2 Hz, 0.15 H), – (m, 0.5H), (d, J = 7.2 Hz, 0.30 H), – (m, 1H), – (m, 0.3H), – (m, 0.15H), (d, J = 4.0 Hz, 1H), (d, J = 3.6 Hz, 1H), 4.65 (d, J = 7.6 Hz, 1H), 4.55 (d, J = 8.0 Hz, 1H), 4.44–4.40 (m, 1H), 4.37 (d, J = 6.6 Hz, 1H), 4.30 (d, J = 3.6 Hz, 1H), 4.28 (bs, 2H), 4.18 (q, J = 6.0 Hz, 1H), 4.05 (bs, 2H), 4.02 (bs, 2H), 3.97–3.94 (m, 4H), 3.89 (bs, 4H), 3.83–3.66 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 269H), 3.64–3.62 (m, 4H), 3.56 (bs, 4H), 3.50–3.48 (m, 2H), 3.45–3.43 (m, 6H), 3.28–3.24 (m, 2H), 3.25–3.21 (m, 1H), 3.14–3.13 (m, 1H), 3.07–3.05 (m, 1H), 2.98–2.2.96 (m, 1H), 2.92–2.90 (m, 1H), 2.85 (s, 1H), 2.74 (t, J = 6.5 Hz, 2H), 2.70–2.65 (m, 2H), 2.64 (t, J = 7.0 Hz, 2H), (bs, PEG–COCH2CH2CO–, 4H), 2.33–2.26 (m, 2H), (s, 6H, NHAc), 1.82–1.80 (m, 1H), 1.69–1.60 (m, 6H), 1.38–1.36 (m, 14H), 1.32 (bs, 18H); 13C NMR (125 MHz, D2O): δ 175.76, 174.64 (2CO), 175.58 (2CO), 175.25, 164.74, 163.73, 102.08, 101.04, 99.61, 92.30, 79.17, 77.24, 76.65, 76.33, 76.13, 75.72, 73.38 (2C), 72.69 (2C), 72.05, 71.55 (2C), 71.65 (2C), 70.56 (PEG–CH2–), 69.81 (2C), 69.46, 69.32, 68.76, 68.65, 67.82, 65.42, 64.01, 62.28 (2C), 62.15 (2C), 61.34, 61.19, 56.41 (2C), 51.82, 51.81, 51.80, 50.48 (2C), 46.04, 39.95 (PEG–CH2–), 39.70 (2C), 38.89, 36.69, 32.18 (2C), 32.04, 31.35, 30.76, 29.71, 29.53, 29.29, 29.25, 29.02, 28.87, 26.03, 22.26, 16.16; 19F–C NMR (400 MHz, D2O): δ −75.50; the MALDI-TOF spectrum showed the average mass centered at 14.2 kDa and expected average mass was 14.7 kDa.
Synthesis of conjugate 22
Conjugate 22 was obtained through CuAAC coupling of compound 18 (7.0 mg, 0.50 μmol, 1 equiv.) and commercially available AF alkyne 19 (0.55 mg, 0.76 μmol, 1.5 equiv.) following the general procedure for CuAAC ligation. Yield: (6.12 mg, 83%). Substitution of compound 22 was determined by 1H NMR integration of the underlined peaks shown below to be (GD10 + AF)/6′SL/PEG = 1:3. 1H NMR (500 MHz, D2O): δ – (m, 2H), – (m, 1H), (d, J = 8.0 Hz, 1H), (d, J = 8.0 Hz, 1H), 4.42 (q, J = 6.0 Hz, 1H), 3.90–3.88 (m, 2H), 3.86–3.82 (m, 4H), 3.80–3.65 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 279H), 3.64–3.60 (m, 4H), 3.59 (bs, 4H), 3.34 (t, J = 7.0 Hz, 2H), 3.18–3.10 (m, 2H), 3.44 (t, J = 7.6 Hz, 2H), 3.28–3.24 (m, 2H), 3.25–3.21 (m, 1H), 3.14–3.13 (m, 1H), 3.07–3.05 (m, 1H), 2.98–2.2.97 (m, 1H), 2.92–2.90 (m, 1H), 2.85 (s, 1H), 2.80–2.75 (m, 1H), 2.74 (t, J = 7.6 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.65–2.62 (m, 1H), (bs, PEG–COCH2CH2CO–, 4H), 2.32–2.24 (m, 2H), (s, 3H, NHAc), 1.80 (t, J = 15 Hz, 1H), 1.68–1.60 (m, 6H), 1.39–1.33 (m, 12H), 1.32 (bs, 14H); 13C NMR (125 MHz, D2O): δ 179.89, 176.23, 175.66, 174.59 (2CO), 171.43, 175.25, 164.74, 163.74, 123.35, 122.75, 116.78, 109.97, 104.20, 102.89, 80.67, 79.17, 75.70, 75.611, 73.72, 73.51, 73.35, 72.76, 72.74, 71.77, 71.76, 71.55 (2C), 70.56 (PEG–CH2–), 69.81 (2C), 69.48, 69.32, 64.52, 64.50, 63.62, 62.34 (2C), 52.77 (2C), 48.21, 47.65, 46.04, 41.10, 39.95 (PEG–CH2–), 39.70, 32.19 (2C), 32.04, 31.35, 30.78, 29.70 (2C), 29.33, 29.15, 29.85, 25.97, 23.03, 14.91, 9.19; 19F–C NMR (400 MHz, D2O): δ −75.57; the MALDI-TOF spectrum showed the average mass centered at 14.2 kDa and expected average mass was 14.5 kDa.
MALDI-TOF mass spectrometry analysis
Samples were dissolved in water and an aliquot of 1 μL of sample was spotted onto a Bruker Daltonics MTP ground steel target and air-dried. A 1 μL aliquot of matrix solution was spotted on top of the sample spot and air-dried. The matrix solution consisted of a saturated solution of CHCA (α-cyano-4-hydroxycinnamic acid) or DHB (dihydroxybenzoic acid) in 50% water acetonitrile with 0.1% TFA. Mass spectra were obtained in the positive or negative linear mode of ionization using a Bruker Daltonics (Bremen, Germany) ultrafleXtreme MALDI TOF/TOF mass spectrometer. Data analysis software packages provided by the manufacturer were used for analysis.
Fluorescence polarization (FP) binding assay
The affinity of AG10 analog 23 and the labelled GD10 compound 24 to human TTR in PBS at pH 7.4 in assay buffer was evaluated by direct binding or displacement of an FP probe from the protein using an assay similar to that reported by Penchala et al.21 The affinity of GD10 conjugate 24 to TTR was determined by addition of 24 (2 μL, 1 nM) to serial dilutions of TTR (23 μL, 9.01 μM to 0.017 μM) in freshly prepared assay buffer (PBS pH 7.4, 0.01% Triton, 1% DMSO) in a black 384-well plate at room temperature. Samples were allowed to equilibrate by agitation for 25 min in the dark by covering an aluminum foil. Fluorescence polarization (λex 485 nm, λem 525 nm) was measured using a Cytation 5 cell imaging multi-mode micro plate reader. To evaluate the ability of AG10 analog 23 to act as a competitor to GD10, a complex between hTTR and GD10 conjugate 24 was formed by incubation of a mixture of hTTR (4.0 μM, 18 μL) and GD10 conjugate 24 (1 nM, 2 μL) in a black 384-well plate for 10 min. Serial dilutions of AG10 analog 23 (50 μM to 0.04 μM) were added in aliquots (5 μL) to the hTTR-GD10 complex (25 μL final volume) and allowed to incubate for 25 min at room temperature in the dark. The assay solutions were then briefly centrifuged to disperse air bubbles and then measured for fluorescence polarization. For serum binding experiments, mouse serum was assumed to contain 230 nM murine TTR for the purposes of calculating kapp.31
Synthesis of fluorescently labelled protein 25
To make the fluorescently labelled alpha-1-acidic glycoprotein (AGP), 2 mg mL−1 of the protein solution was prepared in 0.1 M NaCO3 (pH 8.5–9). N-Hydroxylsuccinimide-AF dye (Molecular Probes) dissolved in DMSO was then added to the protein solution to a final concentration of 0.05 μg mL−1 and the reaction left to proceed at room temperature for 1 h in dark. To quench the reaction, ethanolamine was added to the final concentration of 0.1 M (pH 9–10) for 2 h at 4 °C. To remove the excess unreacted dye, ultracentrifugation with molecular cut-off of 10 kDa was used. The protein solution was diluted to 200 μL using PBS, the solution was spun at 14000×g at 4 °C for 10 min; this process was repeated three times. The final concentration of the proteins and level of labeling was determined by UV absorbance to be 1 mg mL−1 protein with ∼1.2 dye:protein ratio.
Murine in vivo clearance model
Wild-type BALB/c mice (8–18 weeks of age, 4 male and 3 female) were injected i.v. via the saphenous vein with either conjugate 9, 10, 11, 20, 21, 22, or 25 at 300 μg/100 μL PBS buffer (pH 7.4). The concentration used for the glycoconjugates was based on previous ABO tolerance studies in BALB wild-type mice using A-antigen expressing erythrocytes.29 For replicate measurements mice were injected either with conjugates 10 or 21. Blood samples were collected from tail bleeds pre-injection and at 10 min, 1, 2, 4, 24, 48 and 96 h post-injection.
Blood samples were centrifuged at 4000×g for 5–10 min and 10 μL aliquots of serum diluted with 220 μL of buffer (PBS pH 7.4) at room temperature. Fluorescence (emission scan: λex = 494 nm and λem = 502–700 nm and excitation scan: λex = 400–500 nm and λem = 518 nm) was measured using a spectrophotometer reader (PTI QM-400 and PTI Horiba Scientific systems) for respective conjugates (9–11, 20, 21, and 25) and (11 and 22). For non-injected mouse serum samples, a fluorescence signal was not observed and these were considered as ‘blank’ samples. All half-life measurements were performed on two separate animals, with triplicate samples at each time point. Fluorescence changes were determined by height of the emission signal at 519–520 nm. Half-life of the AF labeled (λex = 496 nm and λem = 519 nm) conjugates was determined from plotting the post-injection time points and intensity of the emission signal. Half-life was derived through plotting the data on GraphPad prism and fitting the results to a non-linear regression and exponential one-phase decay.
Whole body/organ imaging of GC in mice
C3H/He mice were injected within 24 h of birth in the superficial temporal vein (30-gauge needle) with compound 21 dissolved in PBS at the indicated doses. At days one and six post-injection, mice were anesthetized on ice for whole body imaging. Injected mice were imaged next to non-injected littermate controls to distinguish compound 21 signal from autofluorescence. Mice were imaged with an Olympus OV100 small animal imaging system using a Hamamatsu ImagEM camera with automatic exposure. The objective lens for image acquisition was 0.14X/0.039 and the excitation and emission filters were Ex BP460-490/Em BA510-550. Images were acquired using the native OV100 software. As overlying tissue blocked fluorescent signals, freshly dissected organs were imaged in the unfixed state next to those from non-injected littermate controls.
Synthesis of conjugate 27
Conjugate 27 was obtained through CuAAC coupling of compounds 17 (10.0 mg, 0.71 μmol, 1 equiv.) and 6′-sialyllactoside 2614 (0.71 mg, 1.07 μmol, 1.5 equiv.) following the general procedure for CuAAC ligation. Yield: (8.95 mg, 80%). 1H NMR (500 MHz, D2O): δ 8.00 (s, 1H, triazole H), 7.67 (d, J = 7.6 Hz, 1H), 7.59 (bs, 1H), 7.59 (m, 1H), 5.40 (d, J = 4.0 Hz, 1H), 5.23 (d, J = 3.6 Hz, 1H), 4.65 (d, J = 7.6 Hz, 1H), 4.55 (d, J = 8.0 Hz, 1H), 4.44–4.40 (m, 1H), 4.43 (d, J = 12.6 Hz, 1H, H-1'), 4.38 (d, J = 12.0 Hz, 1H, H-1), 4.37 (d, J = 6.6 Hz, 1H), 4.30 (d, J = 3.6 Hz, 1H), 4.28 (bs, 2H), 4.18 (q, J = 6.0 Hz, 1H), 4.05 (bs, 2H), 4.02 (bs, 2H), 3.97–3.94 (m, 4H), 3.89 (bs, 4H), 3.83–3.66 (band, PEG–CH2–, GD10 CH2–, sugar–CH, 264H), 3.64–3.62 (m, 4H), 3.56 (bs, 4H), 3.50–3.48 (m, 2H), 3.45–3.43 (m, 6H), 3.28–3.24 (m, 2H), 3.25–3.21 (m, 1H), 3.17–3.11 (m, 1H), 3.07–3.03 (m, 1H), 2.98–2.2.94 (m, 1H), 2.92–2.90 (m, 1H), 2.85 (s, 1H), 2.74 (t, J = 6.5 Hz, 2H), 2.70–2.65 (m, 2H), 2.66 (dd, J = 6.6, 18.6 Hz, 1H, Heq-3), 2.64 (t, J = 7.0 Hz, 2H), 2.59 (bs, PEG–COCH2CH2CO–, 4H), 2.33–2.26 (m, 2H), 2.09 (s, 6H, NHAc), 1.98 (s, 3H, NHAc), 1.82–1.80 (m, 1H), 1.69–1.67 (m, 6H), 1.68 (t, J = 18.6 Hz, 1H, Hax-3), 1.38–1.36 (m, 14H), 1.32 (bs, 18H); 13C NMR (125 MHz, D2O): δ 175.76, 175.64 (2CO), 175.58 (2CO), 175.26 (2CO), 174.34, 161.19, 163.73, 145.69, 134.26, 104.23, 102.08 (2CO), 101.04 (2CO), 99.61, 92.30, 84.92, 80.55, 79.24, 77.24, 76.65, 76.33, 76.14, 75.66, 75.55, 74.48, 75.80, 75.77, 73.87, 73.61, 73.58, 73.38 (2C), 72.69 (2C), 72.05, 71.88, 71.65 (2C), 71.55 (2C), 71.49, 70.92 (2C), 70.56 (PEG–CH2–), 69.81 (2C), 69.47, 69.36, 69.23, 68.76, 68.65, 67.82, 64.42, 64.01, 62.27, 62.14, 62.0, 61.34, 61.19, 61.00, 56.41 (2C), 52.63, 51.89, 51.63, 51.01, 50.48 (2C), 46.04, 40.94, 39.95 (PEG–CH2–), 39.70 (2C), 38.89, 36.69, 32.18 (2C), 32.03, 31.34, 29.71, 29.53, 29.25, 29.02, 29.02, 28.87, 26.03, 23.26, 22.93, 22.26, 16.15; 19F–C NMR (400 MHz, D2O): δ −75.49; the MALDI-TOF spectrum showed the average mass centered at 14.6 kDa and expected average mass was 15.2 kDa.
Fluorescence microscopy and clustering analysis on B cells
A-BCL cells were prepared and grown as previously described.32 Cells were grown in R10 media supplemented with glutamax and 1% beta-mercaptoethanol added weekly in 12-well plates (Corning, Inc.). Plates were kept in a humidified incubator at 37 °C and 5% CO2. For single-color fluorescence microscopy experiments, 2 × 106 cells were washed and treated with buffer, conjugate 27 (25 ng mL−1), conjugate 27 and TTR (50 ng mL−1), or conjugate 28 (25 ng mL−1)14 on ice for 1 h, then fixed using 1% PFA on ice for 20 min. Samples were treated with 1 μL mL−1 mouse anti-human CD22 (clone HIB22, BD Pharmingen) at 4 °C overnight, washed, and stained with goat anti-mouse IgG (polyclonal, Sigma-Aldrich) conjugated with AF647 at room temperature for 1 hour. The loading of the fluorophores was approximately 2 dye per protein as determined by spectrophotometry. After washing, samples were transferred to 24-well plates (Corning, Inc.) with 12 mm circular cover glass slides pre-treated with 0.001% poly-L-lysine and spun at 300 × g for 15 min. Cover glass slides with samples were washed, mounted onto microscopy slides with Slowfade Antifade (ThermoFisher), and sealed with Cytoseal 60. Samples were imaged on a laser scanning confocal microscope (Olympus IX81) at 60X. Twenty cells from each condition were chosen for analysis based on transmitted and fluorescence images, in which each image was subjected to similar thresholding levels, and each cluster was analyzed using the particle analysis function on ImageJ.33 The area of a single pixel in these images was 0.053 μm2. The areas of each cluster from the analyses were plotted using beanplot in the R statistical package.30 Analysis of the means and Student's t-test were performed in Graphpad Prism.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Prof. Todd L. Lowary and Dr Peter Meloncelli for providing advanced synthetic intermediates for the synthesis of human ABO blood group antigens. This work was supported by grants from the Alberta Glycomics Centre (CWC, LJW) and the Natural Sciences and Engineering Research Council of Canada (NSERC; CWC RGPIN-2020-04371).
References
- Y. M. Chabre and R. Roy, Multivalent glycoconjugate syntheses and applications using aromatic scaffolds, Chem. Soc. Rev., 2013, 42, 4657–4708 RSC.
- P. H. Seeberger and D. B. Werz, Synthesis and medical applications of oligosaccharides, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- N. Jayaraman, Multivalent ligand presentation as a central concept to study intricate carbohydrate-protein interactions, Chem. Soc. Rev., 2009, 38, 3463–3483 RSC.
- Y. M. Chabre and R. Roy, Design and creativity in synthesis of multivalent neoglycoconjugates, Adv. Carbohydr. Chem. Biochem., 2010, 63, 165–393 CrossRef CAS PubMed.
- L. L. Kiessling, J. E. Gestwicki and L. E. Strong, Synthetic Multivalent Ligands as Probes of Signal Transduction, Angew. Chem., Int. Ed., 2006, 45, 2348–2368 CrossRef CAS PubMed.
- O. Renaudet and R. Roy, Multivalent scaffolds in glycoscience: an overview, Chem. Soc. Rev., 2013, 42, 4515–4517 RSC.
- P. Costantino, R. Rappuoli and F. Berti, The design of semi-synthetic and synthetic glycoconjugate vaccines, Expert Opin. Drug Discovery, 2011, 6, 1045–1066 CrossRef CAS PubMed.
-
R. Braeckman, Pharmacokinetics/ADME of large molecules, Preclinical Drug Development, CRC Press, 2nd edn, 2016, pp. 127–151 Search PubMed.
- B. Haraldsson, J. Nyström and W. M. Deen, Properties of the Glomerular Barrier and Mechanisms of Proteinuria, Physiol. Rev., 2008, 88, 451–487 CrossRef CAS PubMed.
- F. Yamashita and M. Hashida, Pharmacokinetic considerations for targeted drug delivery, Adv. Drug Delivery Rev., 2013, 65, 139–147 CrossRef CAS PubMed.
-
R. Braeckman and B. Meibohm, Pharmacokinetics and pharmacodynamics of peptide and protein drugs, Pharmaceutical biotechnology, CRC Press, 2016, pp. 107–135 Search PubMed.
- E. I. Park, Y. Mi, C. Unverzagt, H.-J. Gabius and J. U. Baenziger, The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acidα2, 6GalNAc, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17125–17129 CrossRef CAS PubMed.
- C. I. C. Crucho, P. Correia-da-Silva, K. T. Petrova and M. T. Barros, Recent progress in the field of glycoconjugates, Carbohydr. Res., 2015, 402, 124–132 CrossRef CAS PubMed.
- G. C. Daskhan, H.-T. T. Tran, P. J. Meloncelli, T. L. Lowary, L. J. West and C. W. Cairo, Construction of Multivalent Homo- and Heterofunctional ABO Blood Group Glycoconjugates Using a Trifunctional Linker Strategy, Bioconjugate Chem., 2018, 29, 343–362 CrossRef CAS PubMed.
- A. H. Courtney, E. B. Puffer, J. K. Pontrello, Z. Q. Yang and L. L. Kiessling, Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2500–2505 CrossRef CAS PubMed.
- M. K. O'Reilly, B. E. Collins, S. Han, L. Liao, C. Rillahan, P. I. Kitov, D. R. Bundle and J. C. Paulson, Bifunctional CD22 Ligands use multimeric immunoglobulins as protein scaffolds in assembly of immune complexes on B cells, J. Am. Chem. Soc., 2008, 130, 7736–7745 CrossRef PubMed.
- D. S. Jones, M. J. Branks, M. A. Campbell, K. A. Cockerill, J. R. Hammaker, C. A. Kessler, E. M. Smith, A. Tao, H. T. Ton-Nu and T. Xu, Multivalent Poly(ethylene glycol)-Containing Conjugates for in Vivo Antibody Suppression, Bioconjugate Chem., 2003, 14, 1067–1076 CrossRef CAS PubMed.
- H. Borel and Y. Borel, A novel technique to link either proteins or peptides to gammaglobulin to construct tolerogens, J. Immunol. Methods, 1990, 126, 159–168 CrossRef CAS PubMed.
- M. D. Mannie and I. I. A. D. Curtis, Tolerogenic vaccines for Multiple Sclerosis, Hum. Vaccines Immunother., 2013, 9, 1032–1038 CrossRef CAS PubMed.
- K. A. Cockerill, G. M. Iverson, D. S. Jones and M. D. Linnik, Therapeutic Potential of Toleragens in the Management of Antiphospholipid Syndrome, BioDrugs, 2004, 18, 297–305 CrossRef CAS PubMed.
- S. C. Penchala, M. R. Miller, A. Pal, J. Dong, N. R. Madadi, J. Xie, H. Joo, J. Tsai, P. Batoon, V. Samoshin, A. Franz, T. Cox, J. Miles, W. K. Chan, M. S. Park and M. M. Alhamadsheh, A biomimetic approach for enhancing the in vivo half-life of peptides, Nat. Chem. Biol., 2015, 11, 793–798 CrossRef CAS PubMed.
- S. C. Penchala, S. Connelly, Y. Wang, M. S. Park, L. Zhao, A. Baranczak, I. Rappley, H. Vogel, M. Liedtke, R. M. Witteles, E. T. Powers, N. Reixach, W. K. Chan, I. A. Wilson, J. W. Kelly, I. A. Graef and M. M. Alhamadsheh, AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9992–9997 CrossRef CAS PubMed.
- M. M. Alhamadsheh, S. Connelly, A. Cho, N. Reixach, E. T. Powers, D. W. Pan, I. A. Wilson, J. W. Kelly and I. A. Graef, Potent Kinetic Stabilizers That Prevent Transthyretin-Mediated Cardiomyocyte Proteotoxicity, Science Translational Medicine 3, 2011, 97ra81 Search PubMed.
- M. Miller, A. Pal, W. Albusairi, H. Joo, B. Pappas, M. T. Haque Tuhin, D. Liang, R. Jampala, F. Liu, J. Khan, M. Faaij, M. Park, W. Chan, I. Graef, R. Zamboni, N. Kumar, J. Fox, U. Sinha and M. Alhamadsheh, Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis, J. Med. Chem., 2018, 61, 7862–7876 CrossRef CAS PubMed.
- A. Pal, W. Albusairi, F. Liu, M. T. H. Tuhin, M. Miller, D. Liang, H. Joo, T. U. Amin, E. A. Wilson, J. S. Faridi, M. Park and M. M. Alhamadsheh, Hydrophilic Small Molecules That Harness Transthyretin To Enhance the Safety and Efficacy of Targeted Chemotherapeutic Agents, Mol. Pharm., 2019, 16, 3237–3252 CrossRef CAS PubMed.
- A. Dondoni, The Emergence of Thiol–Ene Coupling as a Click Process for Materials and Bioorganic Chemistry, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS PubMed.
- Q. Wan, J. Chen, G. Chen and S. J. Danishefsky, A Potentially Valuable Advance in the Synthesis of Carbohydrate-Based Anticancer Vaccines through Extended Cycloaddition Chemistry. The, J. Org. Chem., 2006, 71, 8244–8249 CrossRef CAS PubMed.
- R. Bascom, K. Tao, S. Tollenaar and L. West, Imaging Tolerance Induction in the Classic Medawar Neonatal Mouse Model: Active Roles of Multiple F1-Donor Cell Types, Am. J. Transplant., 2015, 15, 2346–2363 CrossRef CAS PubMed.
- B. Motyka, J. Fersovich, B. Lamarche, M. Sosniuk, I. Adam, J. Pearcey, K. Tao, C. W. Cairo, P. J. Cowan and L. J. West, MHC-Matched A-Expressing Blood Cells Induce ABO Tolerance in Infant and Adult Mice, Transplantation, 2018, 102, S292 CrossRef.
- P. Kampstra, Beanplot: A boxplot alternative for visual comparison of distributions, J. Stat. Softw., 2008, 28, 1–9 Search PubMed.
- I. Gonçalves, C. H. Alves, T. Quintela, G. Baltazar, S. Socorro, M. J. Saraiva, R. Abreu and C. R. A. Santos, Transthyretin is up-regulated by sex hormones in mice liver, Mol. Cell. Biochem., 2008, 317, 137–142 CrossRef PubMed.
- A. M. Slaney, I. E. Dijke, M. Jeyakanthan, C. Li, L. Zou, P. Plaza-Alexander, P. J. Meloncelli, J. A. Bau, L. L. Allan, T. L. Lowary, L. J. West, C. W. Cairo and J. M. Buriak, Conjugation of A and B Blood Group Structures to Silica Microparticles for the Detection of Antigen-Specific B Cells, Bioconjug. Chem., 2016, 27, 705–715 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of compound 8, structure of 28, and characterization data for intermediates and conjugates. See DOI: https://doi.org/10.1039/d2cb00126h |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Bruce
Motyka
a,
Bruce
Motyka