
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCLiB – a novel cardiolipin-binder isolated via data-driven and in vitro screening†
Isabel
Kleinwächter
a,
Bernadette
Mohr
b,
Aljoscha
Joppe
c,
Nadja
Hellmann
a,
Tristan
Bereau
 b,
Heinz D.
Osiewacz
c and
Dirk
Schneider
*ad
b,
Heinz D.
Osiewacz
c and
Dirk
Schneider
*ad
aDepartment of Chemistry, Biochemistry, Johannes Gutenberg University Mainz, Hanns-Dieter-Hüsch-Weg 17, 55128 Mainz, Germany. E-mail: Dirk.Schneider@uni-mainz.de
bVan ‘t Hoff Institute for Molecular Sciences and Informatics Institute, University of Amsterdam, Amsterdam, The Netherlands
cInstitute for Molecular Biosciences, J. W. Goethe University, Frankfurt am Main, Germany
dInstitute of Molecular Physiology, Johannes Gutenberg University Mainz, Hanns-Dieter-Hüsch-Weg 17, 55128 Mainz, Germany
First published on 10th June 2022
Abstract
Cardiolipin, the mitochondria marker lipid, is crucially involved in stabilizing the inner mitochondrial membrane and is vital for the activity of mitochondrial proteins and protein complexes. Directly targeting cardiolipin by a chemical-biology approach and thereby altering the cellular concentration of “available” cardiolipin eventually allows to systematically study the dependence of cellular processes on cardiolipin availability. In the present study, physics-based coarse-grained free energy calculations allowed us to identify the physical and chemical properties indicative of cardiolipin selectivity and to apply these to screen a compound database for putative cardiolipin-binders. The membrane binding properties of the 22 most promising molecules identified in the in silico approach were screened in vitro, using model membrane systems finally resulting in the identification of a single molecule, CLiB (CardioLipin-Binder). CLiB clearly affects respiration of cardiolipin-containing intact bacterial cells as well as of isolated mitochondria. Thus, the structure and function of mitochondrial membranes and membrane proteins might be (indirectly) targeted and controlled by CLiB for basic research and, potentially, also for therapeutic purposes.
Introduction
Cellular membranes separate intracellular processes from the environment and surround defined intracellular structures within cells, resulting in formation of defined intracellular reaction compartments, the organelles, a basis for the evolution of complex life.1,2 Biomembranes consist of a scaffold of multiple membrane lipids, albeit the concentration of membrane-attached and/or integrated protein can be high. Biological membranes can contain more than 1000 different lipid species, where the main membrane constituents belong to three classes of lipids: phospholipids, glycolipids and cholesterol, with glycerophospholipids being the major class.2,3 The exact lipid composition varies significantly between the diverse intracellular membrane systems in eukaryotes, with the membrane lipid composition of organelles differing from each other as well as from the composition of the plasma membrane.3 Eukaryotic membranes are typically asymmetric, with the lipid composition of an inner membrane leaflet differing from that of the outer.2E.g., in highly curved membrane regions, such as the cristae of the inner mitochondrial membranes, the phospholipid cardiolipin (CL) is enriched in the inner membrane leaflet as it promotes and/or stabilizes negative membrane curvature.4 Even within a lipid bilayer, regions with diverse lipid compositions can co-exist, such as the basolateral and apical regions of epithelial plasma membranes, and in eu- as well as in prokaryotes the formation of defined lipid domains is discussed, regions where distinct lipid species segregate. It is assumed that specific proteins diffuse into such domains and thereby form signalling platforms. (Targeted) disturbance of such lipid domains can severely affect cellular functions, resulting in mis-signaling.5While the relative concentrations of individual lipid species within the eukaryotic membrane systems varies, and thus the exact lipid composition appears to define the membranes’ physico-chemical properties, several lipids are restricted to chloroplasts and mitochondria, the organelles with an endosymbiotic origin. e.g., galactolipids are solely found in chloroplasts, as expected due to their cyanobacterial origin.6 Many bacteria contain significant amounts of CL (around 10% in E. coli),5 whereas in eukaryotes this “bacterial lipid” can only be found in significant amounts in mitochondria.7,8 In the inner membrane of mitochondria, CL typically constitutes 10–20% of the total lipid,9,10 whereas in the outer mitochondrial membrane only minor amounts of CL are found.11,12 Consequently, CL is a marker for the mitochondrial inner membrane. In contrast, in fungi the CL content in mitochondria can be much lower,13–15 and especially in mitochondria from P. anserina the CL level is very low (1.5%).16
Besides its unique origin and subcellular localization, also the structure of CL is uncommon (Fig. 1): two phosphatidyl backbones are linked via a glycerol head group, and thus one CL molecule contains two negative charges.8,17,18 Four fatty acid chains are attached to this backbone, which typically differ in their respective length and saturation.8,19,20
 | ||
| Fig. 1 Molecular structures of (A) CL, (B) PG and (C) NAO. In (A) and (B), R represents the fatty acid chains. | ||
Because of its overall cone-shaped structure, CL is primarily found in high curvature membrane regions, for example at the poles of rod-shaped bacteria21,22 or at highly-curved regions of the mitochondrial inner membrane.8 Consequently, alterations in the mitochondrial CL content can result in an altered morphology of the mitochondrial inner membrane.23 Mitochondria are the dominant producer of adenosine triphosphate (ATP) in most eukaryotic cells and crucially involved in several metabolic processes.8 CL is essential for the activity of several mitochondrial enzymes,8,19 such as the glycerol-3-phosphate dehydrogenase,24 creatine kinase or phosphate carriers,25,26 and the activity of the respiratory chain complexes depends to some extent on CL.19 Additionally, during apoptosis, interaction of CL with cytochrome c appears to trigger intracellular signal propagation.27,28 Accordingly, CL deficiency or alterations in the mitochondrial CL levels have pathophysiological consequences in humans, such as ischemia or reperfusion, and result in diabetes, heart disease or the Barth Syndrome.19,23,29 All these observations indicate that molecular engineering of drugs that target CL is an innovative chemical concept that potentially allows controlled analysis of physiological consequences caused by CL abnormalities. In general, when a membrane-active compound binds a defined lipid and/or affects lipid heterogeneity, this can (indirectly) affect the activity of proteins or protein networks that need the specific lipid and/or lipid-containing membrane-platforms to perform their cellular functions.19 Based on this concept, the structure and function of mitochondrial membrane proteins might be indirectly controlled for basic research and, potentially, also for therapeutic purposes using engineered, CL-binding drugs (“CL-binders”).
Presently, molecules that specifically bind CL are still rare. The antibiotic Daptomycin appears to bind more specifically to CL than to the glycerophospholipid phosphatidylcholine (PC), yet with even higher affinity to phosphatidylglycerol (PG).30 TTAPE-Me has been introduced as a promising candidate for binding CL,31 yet here binding also appears to be rather non-specific.32 Similarly, the synthetic tetrapeptide SS-31 binds to CL via hydrophobic and electrostatic interactions, yet it has been shown that it also interacts with phosphatidylserine (PS).33
10-N-Nonylacridine orange (NAO) (Fig. 1) has already been introduced decades ago as a mitochondrial surface marker, as it was suggested to specifically interact with CL.34
NAO intercalates into the inner mitochondrial membrane in a membrane potential independent manner and thereby poisons mitochondrial respiration.35 At low concentrations, it inhibits ATP synthesis and at increased concentrations it additionally inhibits the respiratory electron transfer, potentially via competitively capturing CL.34 In line with this, at high doses, NAO alters the structure of mitochondrial membranes.36,37 The positively charged NAO likely electrostatically interacts with the negatively charged CL head group, and the CL fatty acid composition appears not to affect this binding specificity.38,39 Upon CL-binding, a shift in the NAO fluorescence emission spectrum is observed. NAO is able to diffuse spontaneously across membranes, and can thus be well used in situ, in whole cells.38 Consequently, NAO is used to visualize and quantify the CL content of and/or clustering within bacterial cells and/or mitochondria. Nevertheless, while a CL specificity has been postulated many times, it has turned out in recent years that NAO is not CL-specific. In fact, NAO still properly accumulates in mitochondria in CL-deficient yeast mutants,40 and using model membranes it has been shown that NAO binds with high affinity to PG, which is chemically similar to CL (Fig. 1A and B). In summary, CL-selective molecules are urgently needed for future research.
In the present study, we set up an efficient pipeline to screen for novel specific CL-binders. Computational screening methods are gaining increasing recognition in materials modelling and drug design for efficient identification of molecular candidates that feature desired properties.41 A microscopic picture not only provides the means to resolve the main physicochemical interactions at play, it can also directly connect chemical structure with property.42 This connection allows the identification of a number of candidate molecules with structural features linked to desired properties that can be investigated in more detail. Molecular dynamics (MD) simulations can provide microscopic insight into macromolecular structure-property relationships that are difficult to gain experimentally.43 Unfortunately, atomistic simulations remain prohibitive in the context of compound screening and rational design. Instead, we turned to coarse-grained (CG) models. Coarse-graining reduces the complexity of molecular representations by averaging over the chemical and physical properties of adjacent heavy (non-hydrogen) atoms represented by a single bead. The result is a considerable reduction in computational complexity, while still reproducing the underlying chemical and physical properties sufficiently well.44–47 Here we show that CG modelling is an efficient strategy to discover CL-binding small molecules (SM). Starting from the reference compound NAO, we employ a series of CG free-energy calculations to unravel the physicochemical interactions that lead to specific binding to CL. Free energy calculations via alchemical transformations explicitly compare differences in stability between CL and phosphatidylglycerol (PG) membranes. The calculations lead to the establishment of physicochemical design rules. In a separate study, we employed a more elaborate combination of CG simulations, free-energy calculations, deep representation learning, and Bayesian optimization to tackle the same CL-binding small-molecule discovery problem.48 The results of this parallel study provide a more detailed description of the chemical space of putative CL-binders yet resulted in consistent physicochemical design rules. We subsequently applied these design rules to identify via computational screening 22 small molecules which potentially bind CL with high specificity. Subsequent in vitro analyses of these small molecules, using model membranes, resulted in the identification of a single molecule, CLiB (CardioLipin-Binder) with a high preference for CL over PG. In vivo analyses indicate that interaction of CLiB with CL affects respiration in bacteria as well as in mitochondria. Thus, we here introduce CLiB as a new chemical biology tool for studying the physiological role of CL in cells and/or organelles.
Results and discussion
Typically, the impact of CL on the physiology of mitochondria is analyzed in organisms or isolated mitochondria using strains in which CL synthesis is inhibited due to deletion of genes coding for CL-synthesizing enzymes. Yet, it remains an open question as to how far any physiological consequences observed in such deletion strains de facto reflect CL-dependent processes or are due to an altered membrane, the composition of which has been adjusted in the organisms when CL is absent. Thus, it clearly is desirable to manipulate the cellular CL content in a more controlled way using a chemical biology approach.CL, the mitochondrial marker lipid, is crucially involved in stabilization of the inner mitochondrial membrane as well as in the stabilization and activity of mitochondrial proteins and protein complexes. Furthermore, the CL content of mitochondria is altered during cellular adaptation to defined stress conditions, as well as during aging.11,49 Directly targeting CL by a chemical biology approach and thereby altering the cellular concentration of “free” CL eventually allows to systematically study the dependence of mitochondrial processes on CL availability. Therefore, we aimed at identifying a previously unrecognized small molecule that interacts specifically with CL and can be used in future research on mitochondrial physiology. To do so, we set up a workflow involving two initial computational steps, (i) the identification of physical and chemical properties crucial for CL selectivity of a small molecule and (ii) screening of a vendor database for possible candidates. This was done by applying a physics-based CG model of a known CL probe, reducing the complexity of chemical space, and thereby allowing us to systematically observe the change in selectivity caused by the introduction of new interaction types into the CG model. The observations were formulated as design rules describing which substructures in candidate molecules are indicative of the properties required for CL selectivity. The pre-selected candidate molecules were subsequently in vitro screened to identify promising candidate substances. Finally, the activity of the top CL-binders identified in the present study on the physiology of a CL-containing bacterium as well as on mitochondria was evaluated.
Rational identification of 22 candidate molecules with a putative CL-binding propensity
Due to its claimed CL-selectivity, NAO is commonly used to stain CL-containing membranes, or CL-containing membrane domains. Yet, its specificity and sensitivity towards CL is not high (as often proposed), as it also interacts with other negatively charged lipids, such as PG, phosphatidylserine (PS) and phosphatidylinositol (PI).35,50 Nevertheless, the chemical structure of NAO can serve as a reasonable starting point to search for new CL-binders. Our approach then consisted of locally probing chemical space to identify more selective compounds. The underlying assumption was the presence of local regions of chemical space, or basins, that offer high selectivity—a hypothesis that we checked in a recent publication.48Via free-energy calculations, and in particular alchemical transformations, changes in chemical interactions were directly linked to selectivity improvements. The physical and chemical properties of the CG representation of NAO were systematically varied by exchanging individual bead types, for instance by altering the number of hydrophobic sites or charged groups and calculating the relative partitioning free energy ΔΔG of the altered CG NAO from the water phase into each lipid membrane. This is accomplished through two different free-energy methods, performing alchemical transformations51–53 and calculating the potential of mean force (PMF), see Fig. 8B. Negative ΔΔG and minima of the PMF curve represent energetically favorable processes, i.e., insertion at the membrane-water interface and selectivity to the targeted CL. We monitored the influence of the bead-type exchanges on the preferential stability for CL compared to PG. The resulting partitioning free energy differences ΔΔGPG→CL = ΔΔGCL − ΔΔGPG between the same bead-type transformation in both the CL membrane and the PG membrane are shown in (Fig. 2A). The modified NAO structures are represented on the horizontal axis, the color code indicates the position in the CG structure a specific bead was assigned to (Fig. 2B). Due to the symmetry of the reference NAO structure along its vertical axis, for the pink and blue bead positions, no distinction was made whether a specific bead was placed on the left or on the right side of the molecule. The CL-selectivity ΔΔGPG→CL is represented on the y-axis. The compounds are sorted by their resulting ΔΔGPG→CL, from left (low selectivity) to right (high selectivity). The Martini coarse-grained force field56 defines 14 neutral bead types sorted by increasing polarity, from C1–C5 (very apolar) via N-types (neutral) to P-types (polar). Additionally, it provides four charged bead types (Q). From here on, we denote all CG Martini bead types in bold and italics. It becomes clear that increased hydrophobicity brought on by replacing the two C5 beads on the outer sides of the original CG NAO structure (blue) by C1 or C2 beads shifted the selectivity of the resulting CG structure away from CL and towards PG (ΔΔGPG→CL > 0). Adding a neutral Na bead with hydrogen-bond acceptor properties instead of the original N0 beads to the top of the structure (pink) had no noticeable effect on CL-selectivity. In contrast, introducing hydrogen-bond donor properties to the pink beads (Nda/Nd) together with weak hydrophobicity (C5) in the blue beads resulted in an increase in CL-selectivity. This trend was increased further by replacing the hydrophobic C-types at the blue positions with neutral beads with added hydrogen-bond donor sites (Nda/Nd). Finally, the biggest increase in CL-selectivity was obtained in the calculations by replacing the central charged bead (yellow) with a neutral hydrogen-bond donor bead type, keeping the same bead types at the pink bead positions, and introducing two positively charged hydrogen-bond donor beads (Qd/Qda) at the blue positions on the outside of the molecule.
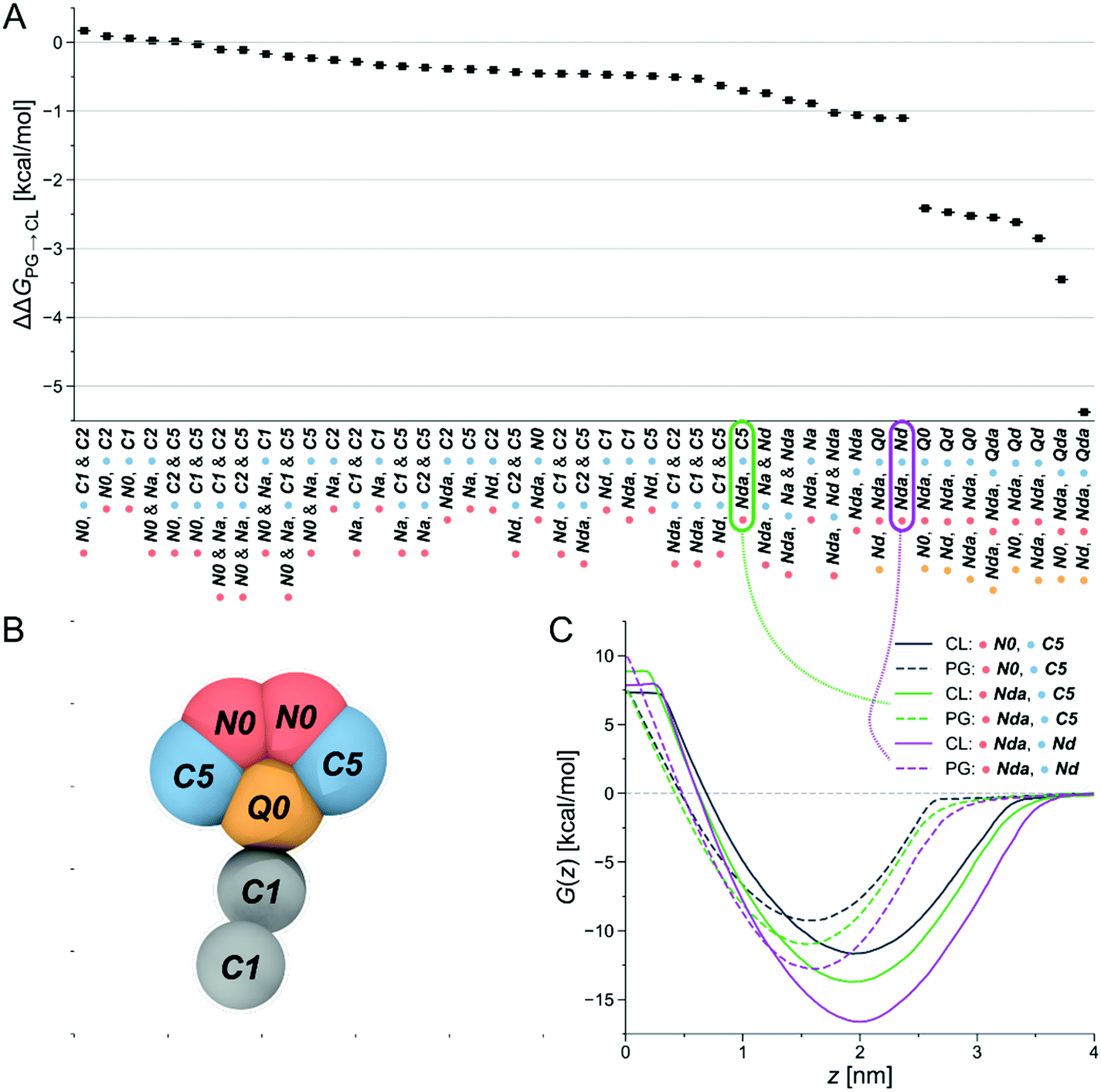 | ||
| Fig. 2 Computational selection of SM1–22. (A) Differences in partitioning free energies ΔΔGPG→CL from alchemical transformations of one or more beads in the CG-representation of NAO. (B) Original CG representation of NAO color-coded to visualize the locations of bead changes in the free energy calculations. (C) Exemplary PMFs of CG structures with different bead types in the CL membrane (solid lines) and the PG membrane (dashed lines). Connections show which PMF curve corresponds to which partitioning free energy difference ΔΔGPG→CL. | ||
In summary, slightly shifting the overall hydrophobicity of the CG NAO representation towards a more amphiphilic character, adding hydrogen bond donor properties, and doubling the positive charge while simultaneously moving the charged beads from the center to the outsides of the CG structure led to a larger negative ΔΔGPG→CL, indicating increased CL selectivity. These design rules can be directly translated to binder-lipid intermolecular interactions: electrostatics with the phosphate group of the lipid head, and amphiphilic character to anchor the compound at the water-membrane interface. Example PMFs for three of those molecules are shown in Fig. 2C. Here, the solid lines represent PMFs calculated in the CL membrane, the dashed lines PMFs in the PG membrane. The grey PMF curve resulted from the original NAO representation to provide a reference for our introduced bead-type changes. The purple and the green lines represent the bead-type conformations circled in the same colors in Fig. 2A. The increased depths of the minima of the PMF curves indicate an increased selectivity of the candidate compounds for both the PG and the CL membranes, and the increased distance at the minima of the PMF curves for the same compound indicate an increased selectivity for CL over PG. Overall, the umbrella sampling results show the same trend as found in the alchemical transformation results shown in Fig. 2A. Due to the increase in computational cost, umbrella sampling calculations with more than one charged bead were not performed.
Our observations now allowed us to express three design rules of molecules that selectively bind to CL and prefer CL over PG:
1.Positively charged beads: at least one, ideally two sites that will carry a positive charge at physiological pH (pH ≈ 7.3).
2.Neutral and charged beads (N- and Q-types) with hydrogen-bond donor properties: Functional groups able to form hydrogen bonds with the CL headgroup.
3.Apolarity (represented by the C1 beads in the tail): Hydrophobic areas in the molecule that induce alignment with or insertion into the lipid bilayer.
Those findings are in good agreement with the interaction types described as being crucial in a comprehensive study on CL-binding proteins, where positively charged amino acids have been identified to be overrepresented in sites binding to the CL headgroup, and hydrophobic amino acids were dominant in sites binding the acyl chains.57
The design rules allowed us to select candidate molecules for CL selectivity from the MCULE database (Fig. 8C).58 As NAO is described as a CL probe in the literature, the Tanimoto similarity coefficient59,60 to NAO at the level of fingerprint representations of the molecules was used for comparison. Candidate molecules with a Tanimoto coefficient >0.39 (lower threshold for similarity, identity is 1.0) were extracted. We further screened the candidates for the presence of positively ionizable sites and aromaticity.61 From the resulting 93 molecules, a subset of 16 candidates with a Tanimoto coefficient >0.44 were subjected to experimental validation (SM1 to SM7, SM10 to SM12, SM16 and SM18 to SM22). Eight of the 16 molecules had at least one hydrogen-bond acceptor site identified using the RDKit62 chemical features definition, and one of those eight had both hydrogen-bond donor and acceptor sites. Six molecules with a lower Tanimoto coefficient were also added to cover the aspects of the design rules in more detail (SM8, SM9, SM13 to SM15 and SM17). Only hydrogen-bond donor- and no acceptor sites were identified in SM13 to SM15, SM8 showed five positively ionizable sites, SM9 four hydrogen-bond acceptor sites and one donor site. Additionally, SM14 and SM15 are structurally very similar to SM16, even though their Tanimoto similarity to NAO is lower. SM17 follows the design rules, even though the similarity to NAO was lower than that of the 16 initially selected candidates. It was included to probe the role of the structural similarity to the detected CL selectivity. The 22 molecules are shown in Fig. 3, all descriptors used for their selection can be found in Table S1 in the ESI.†
 | ||
| Fig. 3 Structures of 22 SMs selected in silico and further analyzed. The chemical names of the substances can be found in Table S2 in the ESI.† | ||
We successfully applied a combination of physics-based coarse-grained representations of molecules to reduce the combinatorial complexity of chemical space together with efficient free-energy calculation methods, thereby greatly increasing the efficiency of examining the influence of different physical and chemical properties on our targeted CL selectivity. We were able to formulate design rules describing the identified properties on the CG level and linking back to related characteristics in molecular structures. The design rules were successfully used to screen the MCULE database of purchasable, in-stock compounds for potential CL-binders (Fig. 3).
Membrane binding properties of SM1–22
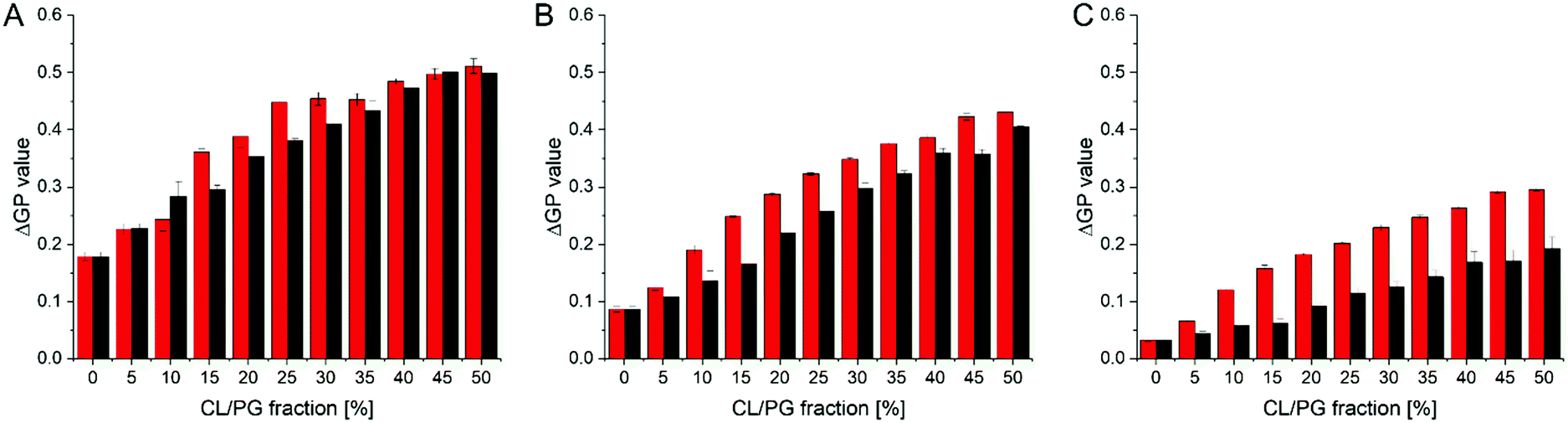
The CG free energy calculations allowed us to identify the physical and chemical properties indicative of CL selectivity, to formulate design rules and to apply those to screen a database for candidate molecules conforming to those design rules. To experimentally verify our in silico results, we next screened the membrane binding properties of the 22 molecules identified in the in silico approach in vitro, using liposomal systems.When a small molecule integrates into a hydrophobic lipid bilayer due to its amphipathic character, this will affect lateral lipid packing, an effect often observed e.g. with alcohols or other small, hydrophobic molecules.63 Additional specific interactions with certain lipid headgroups will lead to an increase in the effective substance concentration at the surface of the bilayer, and therefore to an increased effective partitioning coefficient (see e.g. ref. 64). Furthermore, the effect on order and packing of the hydrophobic part might be altered, e.g. due to a different orientation of the small molecule when specifically binding to a defined lipid species. Thus, comparing the impact of a small molecule on the membrane structure in a system where the molecules merely partition into the membrane with a system where the molecules additionally (more or less) specifically interact with a defined lipid species will allow parallel screening of multiple molecules for their potential to bind to CL. We rationalized that the molecules pre-selected in the computational screen will “only” integrate into net uncharged pure phosphatidylcholine (PC) membranes but will (in the best of all cases) specifically interact with CL in CL-containing membranes. To distinguish general membrane partitioning from specific lipid binding, we additionally tested the structure of a model membrane system where PG is present to isolate selectivity to CL by direct comparison. For monitoring changes in the membrane lipid structure, we utilized Laurdan, an environment-sensitive fluorescent dye that incorporates into lipid bilayers. From the Laurdan fluorescence emission spectra the generalized polarization (GP) values were calculated (eqn (5))65 for membranes in absence vs. presence of the 22 substances identified in our computational screening. Besides pure PC and the PG- or CL-containing PC membranes, respectively, we also monitored the Laurdan fluorescence emission in PC liposomes containing phosphatidylethanolamine (PE), another net-uncharged phospholipid. PE introduces membrane curvature stress, and these measurements enabled us to test whether any observed effects on the Laurdan fluorescence spectra were mainly due to changes in the lateral membrane pressure. This would result in changes observed in the PC/PE system being unequal to the pure PC system. The Laurdan GP value is largely independent of the lipid head group and its acyl chain chemistry, and therefore any changes of this value upon the addition of a substance provides information about changes in the lipid order.66 Thus, if the GP value in presence of a substance is altered, one can infer that binding/incorporation of the substance has occurred. Indeed, for a number of substances large positive ΔGP values were observed (Fig. 4).
 | ||
| Fig. 4 ΔGP values determined in various model membrane systems in presence of the preselected 22 SMs. GP values were measured using pure PC liposomes, as well as liposomes containing PC/PG, PC/PE and PC/CL, all in a molar ratio of 80/20 (mol%/mol%) at 300 μM total lipid. Low ΔGP values indicate no influence of the respective SM on the membrane structure. The more a ΔGP value deviates from 0.0, the higher is the respective impact of a given SM on the membrane structure. Positive ΔGP values indicate a decreased polarity in the environment of Laurdan, as e.g. the case upon increased lipid ordering due to the presence of a SM within a membrane. | ||
Addition of SM4, SM5, SM11, SM13, SM17 and SM19 resulted in substantial changes in the GP values (high ΔGP values) in all lipid systems, which clearly indicates considerable interaction of these SMs with the membranes. Noteworthy, while we did not observe ΔGP changes for the remaining SMs, this does not finally exclude that these SMs do interact with membranes. We only limited the selection and further analysis in the screening pipeline developed and presented here, to substances which had a measurable impact on membrane lipid packing, as sensed by Laurdan. While all six molecules affected the Laurdan fluorescence emission spectrum, for SM5, SM13 and SM17 the results did not indicate a specificity towards charged lipids, as anticipated, as the ΔGP values were similarly affected in membranes containing solely zwitterionic or additionally negatively charged lipids. Thus, a specific interaction with negatively charged lipids is unlikely. In contrast, SM4, SM11 and SM19 showed higher ΔGP values in CL- and PG-containing PC membranes, indicating a specificity towards negatively charged lipids.
The objective of our research was to identify membrane-active compounds with a putative specificity towards CL. Thus far, our initial computational and in vitro screens limited our further analyses to three compounds, for which we next analyzed in greater detail their respective impact on the GP values of CL-containing liposomes compared to PG-containing liposomes. The magnitude of a GP value change observed upon SM binding might be specific for the substance and the lipid environment. Yet, with increasing amounts of negatively charged lipids but at a constant SM concentration, the ΔGP values are affected more substantially when an observed change is due to a specific interaction of the substance with the lipid and thus information about the relative specificity can be extracted: a steeper relative increase of ΔGP values is an indicator for a higher affinity. Therefore, we next monitored how the GP values determined in a (neutral) PC background are differentially affected when the mole fractions of the two negatively charged lipids PG or CL, respectively, increase (Fig. 5), yielding a binding isotherm. In presence of all three SMs, increasing ΔGP values were observed with increasing amounts of negatively charged membrane lipids. Yet, in case of SM4 the shape of the relative increase is similar for CL- and PG-containing membranes, and also the ΔGP values are similar. Thus, SM4 (Fig. 5A) does not significantly discriminate between liposomes containing PG or CL, indicating about similar interaction with the negatively charged membrane lipids. For SM11 (Fig. 5B), a difference between liposomes containing PG and those containing CL was observed at all PG/CL concentrations. Furthermore, the curve is slightly steeper at the initial part in case of CL, indicating a specificity of SM11 for CL. Finally, in case of SM19, the ΔGP values are overall lower than for the other two substances, yet the difference in the values obtained for CL- vs. PG-containing liposomes is larger. More importantly, the binding isotherm is steeper in case of CL-containing liposomes. Thus, SM19 (Fig. 5C) clearly discriminates best between CL- or PG-containing liposomes. In order to visualize the differences in shape of the binding isotherms more clearly, the ratio between the ΔGP values obtained in presence of CL or PG, respectively, was calculated (Fig. S3, ESI†). If the shapes of the binding isotherms were identical, yet differ in their ΔGP value, the same ratio (within error limits) is expected at all mole fractions of anionic lipids. The data obtained in presence of SM19 clearly show the largest deviation from a constant ratio, and thus, based on these analyses, SM19 has the highest specificity towards CL (compared to PG).
 | ||
| Fig. 5 ΔGP values in presence of the three most promising CL-binders determined in CL- or PG-containing model membranes. ΔGP values were determined in liposomes, containing increasing amounts of the negatively charged lipids PG (black) or CL (red), respectively, in a PC background (300 μM total lipid) at a constant concentration (3 μM) of SM4 (A), SM11 (B) and SM19 (C). All SMs induce an increase in ΔGP values with increasing amounts of the negatively charged lipids. The most significant difference between PG- vs. CL- containing membranes was observed for SM19. | ||
To obtain an estimate of the SM19 affinity for CL, changes in the membrane lipid order were monitored at a constant lipid- but at varying SM19 concentrations (Fig. 6A). The CL content in the liposomes was 10%, approximately mirroring the CL concentration in E. coli and mitochondrial membranes.11 In agreement with the above presented results, addition of increasing amounts of SM19 lead to higher ΔGP values in case of CL-containing membranes when compared to PG-containing membranes. However, the shape of the curves does not allow to estimate the affinity constant. In agreement with the data in Fig. 4 and 5, the ΔGP value is consistently higher in case of CL compared to PG. As SM19 is intrinsically fluorescent (Fig. 6B, inlet), we next additionally analyzed SM19 interaction with negatively charged lipids via following the SM19 fluorescence. In both PG- as well as CL-containing PC liposomes, the intensity of the SM19 fluorescence is rising with increasing concentrations of negatively charged lipids, which is perfectly in line with the Laurdan measurements (Fig. 6A). Already when SM19 was added to pure PC liposomes, the fluorescence emission increased (Fig. 6B), indicating that membrane incorporation of SM19, i.e., placing the dye in a more hydrophobic environment, affects the SM19 fluorescence properties. When the liposome surface charge was increased via increasing the amount of PG or CL, respectively, the fluorescence emission intensity steadily increased. Thus, interaction of SM19 with negatively charged lipids can also be monitored via the substances’ inherent fluorescent properties and, as observed before using Laurdan as a probe, this effect is more pronounced in CL-containing liposomes.
 | ||
| Fig. 6 Specificity of SM19-binding to CL-containing PC membranes. ΔGP values determined in PG- or CL- containing model membranes in presence of increasing SM19 concentrations. ΔGP values were measured using liposomes containing a fixed lipid composition of 90% PC and 10% CL (red) or PG (black), respectively, at increasing amounts of SM19. At low SM19 concentrations no significant differences were observed. (B) The inherent SM19 fluorescence properties are affected by the membrane lipid composition. The fluorescence emission at 592 nm increases with increasing fractions of negatively charged lipids (PC/PG (black), PC/CL (red)). The intensity increase is consistently higher in CL-containing membranes. (B inlet) Emission spectrum of pure SM19 in buffer (green), or in pure PC (blue), PC/PG (50/50) (black) and PC/CL (50/50) liposomes (red) (SM19 concentration: 3 μM). | ||
Impact of SM19 on CL-containing cellular membranes
In the above presented in silico and in vitro analyses, SM19 has emerged as the most promising CL-binder. Yet, as we aimed to establish SM19 as a chemical tool allowing to directly target CL-containing membranes in biological systems, we next tested the impact of SM19 on the respiration of bacterial (E. coli) cells as well as of isolated mitochondria (P. anserina).Similar to mitochondria (10–20%), E. coli cells contain ∼10% CL in their membrane as well as around 20% PG.5 When SM19 was added to E. coli cells at increasing concentrations, the SM19 fluorescence emission intensity at 592 nm increased (ESI,† Fig. S1), indicating membrane incorporation. To next study the impact of a SM19-induced CL tethering on the physiology of E. coli cells, the impact of SM19 addition on E. coli respiration was analyzed. In fact, SM19 addition significantly affected the respiration rates of E. coli cells. Without the substance the respiration rate was around -19 ± 0.7 μmol oxygen per min per L, which decreased in the presence of SM19 to −15 ± 0.5 μmol oxygen per min per L.
As these initial observations indicated a direct impact of SM19 on the activity of proteins embedded in CL-containing membranes, we then ultimately analyzed the impact of SM19 on mitochondrial respiration determined using mitochondria isolated from the fungus P. anserina. In presence of SM19, the relative oxygen consumption was decreased by around 40% in wild type mitochondria (Fig. 7), indicating that SM19 efficiently tethers CL in mitochondrial membranes.
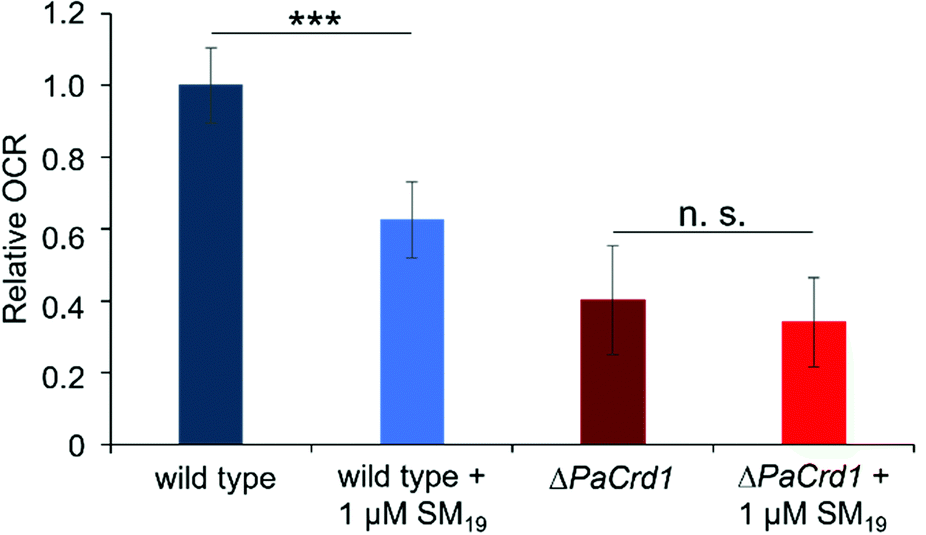 | ||
| Fig. 7 Oxygen consumption measured with mitochondria isolated from 6 days old wild-type and ΔPaCrd1 isolates. Measurements were performed in absence vs. presence of 1 μM SM19. Oxygen consumption rate (OCR) in the phosphorylating respiration state (in the presence of ADP) of the wild type was set to 1. In the wild-type, SM19 treatment significantly reduced phosphorylating respiration by 40% compared to the control. In contrast, in the mutant, addition of SM19 had only marginal effects. (Measurements of the phosphorylating respiration of wild type (n = 15), of wild type + 1 μM SM19 (n = 8), of ΔPaCrd1 (n = 14) and of ΔPaCrd1 + 1 μM SM19 (n = 9); mean values ± standard deviation are shown; significant differences are marked with “***”, p < 0.001); n. s.: not significant). | ||
To finally test whether this impact of SM19 on mitochondrial respiration was due to a specific interaction of the compound with CL, we next used a CL-free P. anserina strain16 and tested the impact of SM19 on mitochondrial oxygen consumption using CL-free mitochondria. While the respiration rate is already reduced in CL-free mitochondria, addition of SM19 showed only marginal effects. To also exclude that the non-responsiveness of CL-free mitochondria is due to their general impairment in respiration, we additionally tested whether they respond to inhibition of the two terminal oxidases present in P. anserina.67 While complex IV (COX) is inhibited by KCN, SHAM inhibits the alternative oxidase AOX. Thus, mitochondrial respiration should be completely blocked when both inhibitors are given in combination. In fact, both in wild type and ΔPaCrd1 mitochondria the respiration rate was significantly decreased after addition of KCN/SHAM (Supplementary data, Fig. S2, ESI†). Thus, the non-responsiveness of ΔPaCrd1 to SM19 was indeed due to the lack of CL in this strain. These observations strongly indicate that SM19 specifically interacts with CL as the substance clearly has an impact on the oxygen consumption rates (OCR) in wild type but not in CL-free mitochondria.
SM19 clearly affects respiration of intact E. coli cells as well as of isolated P. anserina mitochondria. Importantly, a similar effect on respiration of CL-containing mitochondria was not observed with SM18, again underlaying the specificity of SM19 (see Fig. S4, ESI†). As an impact of SM19 was not observed when CL-free mitochondria were analyzed, the observed effects on respiration were very likely caused by CL-tethering via SM19.
Thus, after identifying 22 putative CL-binders in silico and further analyzing them in vitro, we finally identified a single small molecule, 2-[(E)-2-[4-(dimethylamino)phenyl]ethenyl]-3-ethyl-1,3-benzothiazol-3-ium iodide (SM19) (Fig. 3), which interacts with negative charged lipids and prefers CL over PG. Therefore, we will from now on name this molecule CLiB (CardioLipin-Binder).
The interaction of CLiB with CL might be due to its quaternary ammonium ion (Fig. 1), an interaction known to drive binding of NAO to CL and PG.38 As the lipid composition in bacteria and eukaryotic cells can substantially differ in the chemistry of the acyl chains,68–72 the specificity of CLiB towards CL appears to be mainly based on the interaction with the lipid headgroup. While the structures of PG vs. CL are very similar (Fig. 1), the main difference is their respective charge. Thus, a specificity of a given substance would probably be based on a change in entropy: binding to a single CL molecule might be thermodynamically more favorable than binding two PG molecules due to a reduced loss in entropy. This is reflected in the design rules calling for two positively charged sites in the candidate structures to increase selectivity. The two positive charges correspond to the two negatively charged phosphate groups present in a CL molecule. Yet, this effect is likely very small, and thus it is potentially a deleterious task trying to isolate a substance with 100% specificity towards CL.
In summary, we have identified and introduce here CLiB, a novel small molecule able to bind to negatively charged phospholipids, with a clear preference for CL (over PG). However, incorporating CLiB into a membrane and CL-binding could have unspecific toxic effects on a living organism, for example due to the induced change in the membrane structure caused by the interaction between CLiB and CL (see Fig. 4). Yet, CLiB now allows to systematically analyze the role of CL in biological membranes and in membrane-harbored physiological processes, at least in vitro, as with this chemical biology tool CL can be specifically targeted. We are optimistic that the identification of CLiB will now trigger new analyses of mitochondrial processes, involving aging and dysfunction.
Conclusions
To find a drug/molecule that specifically targets CL, potentially allows controlled analysis of physiological consequences caused by CL abnormalities. So far, such specific CL-binders are rare. For this reason, we set up an efficient pipeline to screen for novel cardiolipin-binders. Starting with CG free-energy calculation and computational screening methods, followed by in vitro and finally some in vivo studies, we discovered a molecule, showing a clear preference for binding to cardiolipin. This workflow and especially the in silico screening can, in our specific use-case, be applied to other databases and lead to the identification of possibly more specific CL binders. Furthermore, we thus far limited our search for natural compounds. Yet, the here identified design principles might even serve to design a novel molecule with increased CL-binding propensity. Finally, the method of identifying design rules can also be generalized to discover candidate small molecules with a high binding affinity to any given molecular target.Experimental section
Chemicals
All lipids were purchased at Avanti Polar Lipids (Alabaster, ALA, US). The substances 1–22 (further described in the text) were purchased at MCULE (Budapest, HUN). All other chemicals were purchased at Sigma Aldrich (Munich, GER). Structures were drawn with the program ChemSketch V5 (Freeware from ACD/Labs, Toronto, Ontario, Canada).Molecular representations
Coarse-grained (CG) representations of each lipid from the Martini 2 force field were used.56 The bead types defined in the Martini force field link to specific physicochemical interaction types, considering four main types of interactions: polar (P), nonpolar (N), apolar (C), and charged (Q). Within a main type, subtypes are distinguished either for hydrogen-bonding capability (d ≡ donor, a ≡ acceptor, da ≡ both, 0 ≡ none) or by a number indicating the degree of hydrophobicity (from 1 ≡ strongly hydrophobic to 5 ≡ weakly hydrophobic).73 CL was modelled from di-anionic tetraoleoyl-cardiolipin (TOCL).74–76 We chose the Cardiolipin (CL) representation carrying two negative charges, following evidence of CL likely being fully ionized at physiological conditions.7,17,18 The CG force field of PG represents 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG). While bilayers consisting of only one lipid type are not found in nature, we used homogeneous membranes as proxy systems to isolate the contributions of individual lipid types to the investigated interactions. Homogeneous bilayers for each lipid were created using the CHARMM-GUI Martini Maker77 with the Martini56 force field version 2.0, 06/2015. We solvated the membranes with the Martini polarizable water.78 The net charges of the systems were brought to zero by adding the appropriate number of negatively charged counter ions per head group (one ion for PG, two for CL). This led to the CL simulation system containing 98 lipid representations (49 per membrane leaflet), 3609 water particles, and 196 counter ions. The PG system contained 118 lipids, 1957 water particles and 118 counter ions. The CG force field of NAO was generated using the auto-martini algorithm79 and then manually refined to balance the overestimated hydrophobicity of the aromatic groups. Subsequently, new chemical interactions were introduced by systematically changing individual bead types and monitoring the resulting change in free energy (Fig. 8A). | ||
| Fig. 8 The computational workflow. (A) Introduction of new chemical interactions by systematically changing of individual bead types. (B) The different areas of a membrane system where free energies are calculated. The black compounds indicate the two regions for the alchemical transformations, the PMFs show the free energy change of a molecule moving along the membrane normal z. The deeper the minimum of the PMF at the interface, the more selective the candidate is for the lipid headgroup. (C) Screening of a database of small molecules for candidates conforming to our identified design rules. The structures were drawn using RDKit (Landrum), the images were obtained from https://Flaticon.com. | ||
Molecular dynamics (MD) simulations
MD simulations were performed in Gromacs 5.1.80 The integration time step was δt = 0.02τ, where τ is the time constant defined as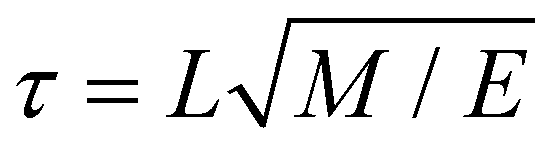 with the units of length L, mass M and energy E. Temperature and pressure of the system (T = 300 K and P = 1 bar) were controlled by means of a velocity rescaling thermostat81 and a Parrinello–Rahman barostat,82 with coupling constants τT = τ and τP = 12τ. Long-range electrostatic interactions were calculated using Particle-mesh Ewald summation.83 Other parameters, such as cut-off range and dielectric constant, follow recommendations for the Martini force field.84 The small molecule was inserted at the appropriate position in the system, either at the membrane–water interface or in the water phase (see Fig. 8B). The whole system was subsequently minimized and equilibrated for 8 ns. The free energy calculations were performed in a production run for 40 ns following the protocol described below.
with the units of length L, mass M and energy E. Temperature and pressure of the system (T = 300 K and P = 1 bar) were controlled by means of a velocity rescaling thermostat81 and a Parrinello–Rahman barostat,82 with coupling constants τT = τ and τP = 12τ. Long-range electrostatic interactions were calculated using Particle-mesh Ewald summation.83 Other parameters, such as cut-off range and dielectric constant, follow recommendations for the Martini force field.84 The small molecule was inserted at the appropriate position in the system, either at the membrane–water interface or in the water phase (see Fig. 8B). The whole system was subsequently minimized and equilibrated for 8 ns. The free energy calculations were performed in a production run for 40 ns following the protocol described below.
Free-energy calculations
A change in free energy ΔG between two states of a system indicates whether the transitioning process between those states is thermodynamically favorable (ΔG < 0) or not (ΔG > 0). We compared the difference in partitioning free energies of different NAO-derived CG candidates into a CL membrane and a PG membrane at the interface region (Fig. 8B). Thereby, we identified which types of physicochemical interaction introduced into the CG candidate structures positively influence CL selectivity. This is measured as the difference in partitioning free energy of a candidate compound between the two different lipid membranes, ΔΔGPG→CL (Fig. 8B(c)).The free energy change was calculated by two different methods; by the transformation of a parameter of the potential energy function (alchemical transformations61–63) or as a potential of mean force (PMF) along a reaction coordinate defined on atomic coordinates shown as red, blue and yellow curves in Fig. 8B) (umbrella sampling54,55).
We used the less computationally expensive alchemical transformation approach for an initial screening, the promising compounds were validated using umbrella sampling.
Alchemical transformations
Alchemical transformations51–53 allow us to estimate the free-energy difference of physical processes by transforming an initial state A of a molecular system to an end state B via a succession of intermediate steps. This facilitates the calculation of free energy differences between states not necessarily connected by actual chemical or physical processes. Using this method, we were able to determine the influence of introducing different chemical interactions by turning individual CG beads from one bead type into another. State A is the free energy calculated for the CG representation of the original NAO molecule; state B is the corresponding free energy for a modified version of NAO. The transformation free energy was calculated by defining a thermodynamic path between the states and integrating over the ensemble average of the potential energy changes relative to a coupling parameter λ along this path (eqn (1)).| U(λ) = UX + λ(UY − UX) | (1) |
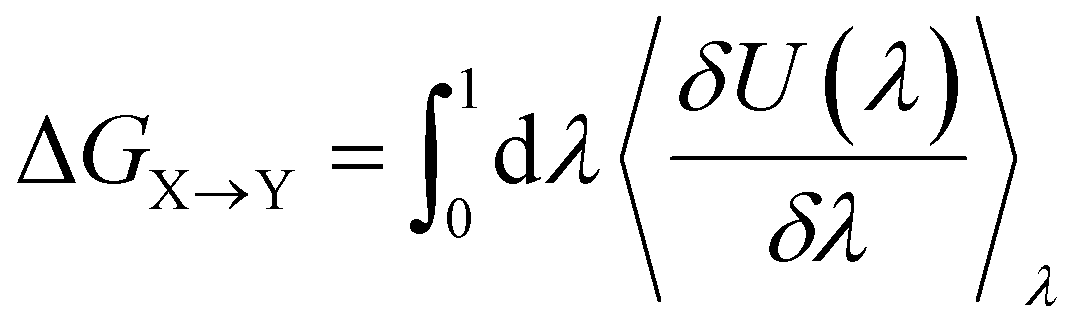 | (2) |
To investigate the influence of van der Waals interactions modeled by the Lennard-Jones potential (LJ), 20 intermediate steps between state A and state B were calculated with successively increasing values for λ ∈ [0, 1]. The influence of electrostatic interactions was evaluated by introducing additional charged particles or moving the position of existing charges involved. The interactions of charged beads were transformed first through 18 LJ steps followed by 18 Coulomb steps, adding up to 36 intermediate steps. The net charge of the system was kept at zero by simultaneously transforming the appropriate number of ions in the solution into water particles.85,86 The individual free energy contributions of the intermediate steps were integrated into the overall free energy change ΔG between states X and Y using the MBAR method,87,88 which also gives the statistical uncertainty of the calculated free energy.
We calculated the transfer free energy ΔGW→I of a candidate compound from bulk water to the membrane interface to determine whether this compound freely partitions into the membrane. This partitioning free energy ΔGW→I comprises the transformation free energy (eqn (2)) ΔGIX→Y of this compound at the interface region and the transformation free energy ΔGWX→Y of the same compound in the bulk water phase, following eqn (3):
| ΔGW→I = ΔGIX→Y − ΔGWX→Y = (ΔGIY − ΔGIX) − (ΔGWY − ΔGWX) | (3) |
This necessitated three individual series of calculations for each NAO variant, one at the interface of each membrane and one in bulk water (Fig. 8B(a)). To keep the small molecules in the interface region of the membrane-containing systems, a small constraint force of 500 kJ mol−1 nm−2 was applied.
To evaluate the selectivity of the modified NAO structures for CL over PG, we used the free energy difference ΔΔGPG→CL (eqn (4)) between the partitioning free energies of the candidate compounds into each membrane
| ΔΔGPG→CL = ΔGCLW→I − ΔGPGW→I | (4) |
Umbrella sampling
Umbrella sampling54,55 is a free-energy technique that allows the calculation of the potential of mean force (PMF) from moving a molecule along defined reaction coordinates (Fig. 8B, colored curves). We applied it to a selection of the modified candidate structures to show the difference in partitioning free energy between NAO representations with replaced bead types (Fig. 8B(b)).The reaction coordinate was the normal of the membrane midplane and the PMF describes how the free energy of a compound changes as a function of the normal distance z between its center of mass and the membrane midplane. The reaction coordinate was split into 24 individual sampling windows and the molecule was kept close to the center zi of each window i along the reaction coordinate z by a biasing potential of 1000 kJ mol−1 nm−2. Unbiased potentials of mean force were extracted from the biased Umbrella simulations by means of the weighted histogram analysis method89–91 and the corresponding errors via bootstrapping.92
Virtual screening
Molecules were selected from the MCULE database of purchasable, in-stock compounds (https://mcule.com/, version 18-08-17,58 (Fig. 8C)). This was done using Morgan circular fingerprints93 implemented in the RDKit62 with feature-based atom invariants and a bit vector length of 2084 bits. The interaction radius was set to two, considering the two nearest neighbors of each atom, the setting best suited for comparing diverse structures for similarity.94 The chemical features defined in the RDKit for feature-based circular fingerprints (FCFP) are hydrogen bond donor and acceptor sites, aromaticity, the presence of a halogen and acidic or basic properties. The resulting FCFP representations of the molecules were used to calculate the Tanimoto similarity coefficient95 to the FCFP representation of NAO. The similarity score reads as follows: 0.0 ≡ no similarity at all to 1.0 ≡ identity.Liposome preparation
For liposome preparation, the lipid or lipid mixture of interest (dissolved in chloroform) was mixed with Laurdan (dissolved in methanol) in a 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. If needed, a substance (dissolved in methanol) was added at the indicated concentration. The solvents were removed under a gentle nitrogen stream and vacuum desiccation overnight. The next day, 300 μL of 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffer (pH 7.4, 150 mM NaCl) was added and the lipid film was rehydrated, resulting in a solution with 300 μM total lipid. The mixture was vortexed two times for 1 minute. To prepare large unilamellar liposomes (LUVs), five freeze–thaw cycles were performed.
:1 molar ratio. If needed, a substance (dissolved in methanol) was added at the indicated concentration. The solvents were removed under a gentle nitrogen stream and vacuum desiccation overnight. The next day, 300 μL of 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-buffer (pH 7.4, 150 mM NaCl) was added and the lipid film was rehydrated, resulting in a solution with 300 μM total lipid. The mixture was vortexed two times for 1 minute. To prepare large unilamellar liposomes (LUVs), five freeze–thaw cycles were performed.
Laurdan fluorescence spectroscopy and generalized polarization (GP) values
Laurdan spectra were recorded at 25 °C using a Jasco Spectrofluorometer FP-8500 (Jasco Deutschland GmbH, Pfungstadt, GER). The excitation and emission band widths were set to 2.5 nm. Upon excitation at 360 nm, spectra were recorded from 400 to 600 nm. Each lipid composition was measured at least three times using freshly prepared liposomes. Generalized polarization (GP) values were calculated from the fluorescence emission spectra using the following equation:96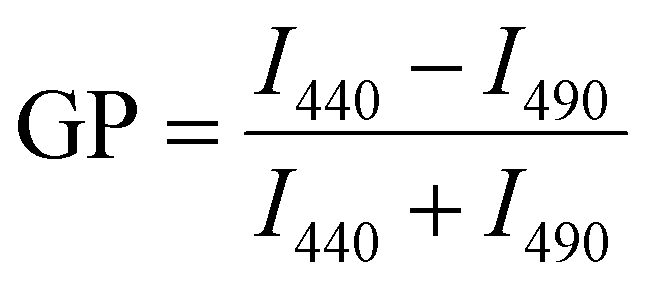 | (5) |
| ΔGP = GP+SMXX − GP−SMXX | (6) |
For each mean value, the standard error of the mean (SEM) was calculated, and the error for ΔGP and ΔGP_CL/ΔGP_PG (Fig. S3, ESI†) was calculated based on Gaussian error propagation.
Fluorescence measurements using the emission of SM19
LUVs containing different amounts of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG) or cardiolipin (CL, 1,3-bis[1,2-dioleoyl-sn-glycero-3-phospho]-glycerol (TOCL)), respectively, were prepared as described. 3 μM SM19 (2-[(E)-2-[4-(dimethylamino)phenyl]ethenyl]-3-ethyl-1,3-benzothiazol-3-ium iodide) was mixed with the lipids during liposome preparation. Fluorescence spectra were measured using a Jasco Spectrofluorometer FP-8500 (Jasco Deutschland GmbH, Pfungstadt, GER). The sample was excited at 500 nm and spectra were recorded from 520 to 680 nm. The band widths were set to 2.5 nm.Fluorescence measurements of SM19 in presence of E. coli
Escherichia coli strain MC4100 was grown in terrific broth (TB)-medium buffered with 10% K2HPO4/KH2PO4 (0.17 M/0.72 M). From an overnight culture, containing 10 mL TB medium with 1:1000 streptomycin (50 mg mL−1), E. coli MC4100 were pelleted. The cells were washed three times with 10 mM HEPES buffer (pH 7.4, 150 mM NaCl) and resuspended in buffer to a final OD600 of 2. Different concentrations of SM19 (dissolved in water) were added to 500 μL of cells and incubated for 5 minutes. The emission spectra of SM19 were recorded between 520 and 680 nm with a Jasco Spectrofluorometer FP-8500 (Jasco Deutschland GmbH, Pfungstadt, GER), upon excitation at 500 nm. The intensity at 592 nm was plotted.
Oxygen consumption of E. coli
For E. coli oxygen consumption measurements, a PreSens Fibox 3 Minisensor and an Oxygen Sensitive Optrode (DP-PSt3-L2.5-5510-YOP, PreSens Precision Sensing GmbH, Regensburg, Germany) was used. E. coli MC4100 cells from an overnight culture (prepared as described above) were diluted and grown until an OD600 of around 0.6 was reached. The cells were harvested, and the pellet was resuspended in 200 μL PBS Puffer (137 mM NaCl, 2.7 mM KCl, 12 mM H3PO4, pH = 7.4) containing 10 mM EDTA. Upon incubation for 5 minutes, cells were pelleted and resuspended in TB media (OD600 = 0.2). SM19 was added up to a final concentration of 10 μM. 3 mL cells were placed in the reaction chamber, the chamber was closed airtight and the measurement was started at 37 °C. The measurement was stopped when the oxygen amount reached 0 μmol L−1.Generation and cultivation of P. anserina strains
The wild-type strain “s”97 and the ΔPaCrd1-mutant16 of P. anserina was used in this study. The ΔPaCrd1-mutant lacking the gene encoding PaCRD1 (UniProt B2AX19) was generated as described in ref. 16. Monokaryotic ascospores were allowed to germinate at BMM medium containing 80 mM ammonium acetate for 2 days at 27 °C in the dark. For mitochondria isolation, monokaryotic strains of wild-type and ΔPaCrd1 were grown on cellophane foil covered solid M2 agar under constant light for 2 days at 27 °C. After 2 days, grown mycelia were transferred to CM for 2 days at 27 °C with shaking.98Isolation of mitochondria from P. anserina
Isolation of mitochondria was performed as described in.98 Briefly, grown mycelia of 6 days old strains were disrupted in isotonic mitochondria buffer with 0.2% (w/v) of bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, Missouri, USA, A6003). The homogenate was filtered through nettle cloth and centrifuged at 600g for 10 min at 4 °C. Subsequently, the supernatant was filtered through glass wool and centrifuged at 12000g for 20 min at 4 °C. The pellet was resuspended in isotonic mitochondrial buffer without BSA and centrifuged at 15000g for 20 min at 4 °C. The mitochondria were enriched in the exterior circle of the pellet. The interior part (consisting of vacuoles) was removed, and the mitochondria were resuspended in isotonic mitochondrial buffer without BSA. The freshly isolated mitochondria were immediately used for respirometry.
Respirometry of P. anserina mitochondria
For measurement of the mitochondrial oxygen consumption rate, 150 μg of freshly isolated mitochondria was used and measured by high-resolution respirometry at 27 °C (Oxygraph-2k series C and G, OROBOROS Instruments, Innsbruck, Austria). Mitochondria were injected into a chamber with 2 ml of air-saturated buffer (0.3 M sucrose, 10 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, 10 mM KCl and 0.1% BSA; pH 7.2) in presence vs. absence of 1 μM SM19. To stimulate complex I-dependent phosphorylating respiration in the presence of ADP, 10 mM pyruvate (Sigma-Aldrich, P2256), 2 mM malate (Sigma-Aldrich, M1000) and 1.5 mM ADP (Sigma-Aldrich, A5285) was added.99 When oxygen consumption reached a constant level, 1 mM potassium cyanide (KCN, inhibitor of cytochrome c oxidase (COX)) and 1 mM salicylhydroxamic acid (SHAM, inhibitor of the alternative oxidase (AOX)) were added to completely block respiration. Wild-type and ΔPaCrd1 mitochondria were analyzed in presence of absence of 1 μM SM19. For analyzing the data, the manufacturer's software DatLab 6 was used.Author contributions
Conceptualization: T. B., H. D. O. and D. S.; methodology: I. K., B. M. and A. J.; software: B. M. and T. B.; validation: I. K., B. M., A. J., N. H., T. B., H. D. O. and D. S.; formal analysis: I. K., N. H., A. J. and B. M.; investigation: I. K., A. J. and B. M.; resources: T. B., H. O., D. S.; data curation: H. D. O., T. B. and D. S.; writing – original draft: I. K., A. J., B. M.; writing – review & editing: N. H., T. B., H. D. O., D. S.; visualization: I. K., A. J., B. M.; supervision: T. B., H. D. O., D. S.; project administration: D. S., T. B., H. D. O.; funding acquisition: T. B., H. D. O. and D. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Andrea Hamann (Frankfurt) for discussions. We thank Christian Hoffmann for the initial parameterization of the coarse-grained NAO. B. M. and T. B. acknowledge support from the Sectorplan Bèta & Techniek of the Dutch Government. D. S. acknowledges support from DynaMem (State of Rhineland-Palatinate); H. D. O. was funded by the German Federal State of Hesse as part of the LOEWE Main Research Focus DynaMem.References
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef CAS PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2009, 101, 1–4 Search PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Stryer Biochemie, Springer Berlin Heidelberg, Berlin, Heidelberg, 7th edn, 2013 Search PubMed.
- G. Paradies, V. Paradies, F. M. Ruggiero and G. Petrosillo, Cells, 2019, 8, 728 CrossRef CAS PubMed.
- I. Barák and K. Muchová, Int. J. Mol. Sci., 2013, 14, 4050–4065 CrossRef.
- E. R. Moellering and C. Benning, Trends Plant Sci., 2011, 16, 98–107 CrossRef CAS PubMed.
- M. C. Pangborn, J. Biol. Chem., 1942, 143, 247–256 CrossRef CAS.
- J. Dudek, Front. Cell Dev. Biol., 2017, 5, 1–17 Search PubMed.
- M. Schlame, M. Ren, Y. Xu, M. L. Greenberg and I. Haller, Chem. Phys. Lipids, 2005, 138, 38–49 CrossRef CAS PubMed.
- L. E. Stefanyk, N. Coverdale, B. D. Roy, S. J. Peters and P. J. LeBlanc, J. Membr. Biol., 2010, 234, 207–215 CrossRef CAS PubMed.
- M. Schlame and M. L. Greenberg, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 3–7 CrossRef CAS PubMed.
- A. I. P. de Kroon, D. Dolis, A. Mayer, R. Lill and B. de Kruijff, Biochim. Biophys. Acta, Biomembr., 1997, 1325, 108–116 CrossRef CAS.
- G. Hallermayer and W. Neupert, Biol. Chem., 1974, 355, 279–288 CAS.
- S. C. Kushwaha, M. Kates, J. K. G. Kramer and R. E. Subden, Lipids, 1976, 11, 778–780 CrossRef CAS.
- E. Gryz, U. Perlińska-Lenart, K. Gawarecka, A. Jozwiak, S. Piłsyk, A. Lipko, M. Jemiola-Rzeminska, P. Bernat, A. Muszewska, K. Steczkiewicz, K. Ginalski, J. Długoński, K. Strzalka, E. Swiezewska and J. S. Kruszewska, Int. J. Mol. Sci., 2019, 20, 3043 CrossRef CAS PubMed.
- T. Löser, A. Joppe, A. Hamann and H. D. Osiewacz, Cells, 2021, 10, 2775 CrossRef PubMed.
- J. LeCocq and C. E. Ballou, Biochemistry, 1964, 3, 976–980 CrossRef CAS PubMed.
- E. E. Kooijman, L. A. Swim, Z. T. Graber, Y. Y. Tyurina, H. Bayır and V. E. Kagan, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 61–68 CrossRef CAS PubMed.
- R. H. Houtkooper and F. M. Vaz, Cell. Mol. Life Sci., 2008, 65, 2493–2506 CrossRef CAS PubMed.
- E. Kraffe, P. Soudant, Y. Marty, N. Kervarec and P. Jehan, Lipids, 2002, 37, 507–514 CrossRef CAS PubMed.
- P. M. Oliver, J. A. Crooks, M. Leidl, E. J. Yoon, A. Saghatelian and D. B. Weibel, J. Bacteriol., 2014, 196, 3386–3398 CrossRef PubMed.
- K. C. Huang, R. Mukhopadhyay and N. S. Wingreen, PLoS Comput. Biol., 2006, 2, e151 CrossRef PubMed.
- A. J. Chicco and G. C. Sparagna, Am. J. Physiol., 2007, 292, C33–C44 CrossRef CAS PubMed.
- Z. Beleznai and V. Jancsik, Biochem. Biophys. Res. Commun., 1989, 159, 132–139 CrossRef CAS.
- F. Bisaccia and F. Palmieri, Biochim. Biophys. Acta, Bioenerg., 1984, 766, 386–394 CrossRef CAS.
- M. Müller, R. Moser, D. Cheneval and E. Carafoli, J. Biol. Chem., 1985, 260, 3839–3843 CrossRef.
- V. E. Kagan, Y. Y. Tyurina, H. Bayir, C. T. Chu, A. A. Kapralov, I. I. Vlasova, N. A. Belikova, V. A. Tyurin, A. Amoscato, M. Epperly, J. Greenberger, S. DeKosky, A. A. Shvedova and J. Jiang, Chem. – Biol. Interact., 2006, 163, 15–28 CrossRef CAS.
- F. Gonzalvez and E. Gottlieb, Apoptosis, 2007, 12, 877–885 CrossRef CAS PubMed.
- B. A. Wilson, A. Ramanathan and C. F. Lopez, Biophys. J., 2019, 117, 429–444 CrossRef CAS PubMed.
- T. Zhang, J. K. Muraih, N. Tishbi, J. Herskowitz, R. L. Victor, J. Silverman, S. Uwumarenogie, S. D. Taylor, M. Palmer and E. Mintzer, J. Biol. Chem., 2014, 289, 11584–11591 CrossRef CAS.
- C. W. T. Leung, Y. Hong, J. Hanske, E. Zhao, S. Chen, E. V. Pletneva and B. Z. Tang, Anal. Chem., 2014, 86, 1263–1268 CrossRef CAS PubMed.
- K. Pyrshev, S. Yesylevskyy and M. Bogdanov, Biophys. J., 2021, 120, 3776–3786 CrossRef CAS PubMed.
- H. H. Szeto, Br. J. Pharmacol., 2014, 171, 2029–2050 CrossRef CAS PubMed.
- A. Maftah, J. M. Petit and R. Julien, FEBS Lett., 1990, 260, 236–240 CrossRef CAS PubMed.
- A. Maftah, J. M. Petit, M.-H. Ratinaud and R. Julien, Biochem. Biophys. Res. Commun., 1989, 164, 185–190 CrossRef CAS PubMed.
- M. Septinus, T. Berthold, A. Naujok and H. W. Zimmermann, Histochemistry, 1985, 82, 51–66 CAS.
- M. H. Ratinaud, P. Leprat and R. Julien, Cytometry, 1988, 9, 206–212 CrossRef CAS PubMed.
- J.-M. Petit, A. Maftah, M.-H. Ratinaud and R. Julien, Eur. J. Biochem., 1992, 209, 267–273 CrossRef CAS PubMed.
- P. F. Gallet, A. Maftah, J.-M. Petit, M. Denis-Gay and R. Julien, Eur. J. Biochem., 1995, 228, 113–119 CrossRef CAS PubMed.
- V. M. Gohil, J. Gvozdenovic-Jeremic, M. Schlame and M. L. Greenberg, Anal. Biochem., 2005, 343, 350–352 CrossRef CAS PubMed.
- J. D. Durrant and J. A. McCammon, BMC Biol., 2011, 9, 71 CrossRef CAS PubMed.
- T. Bereau, D. Andrienko and K. Kremer, APL Mater., 2016, 4, 053101 CrossRef.
- S. Curtarolo, G. L. W. Hart, M. Buongiorno Nardelli, N. Mingo, S. Sanvito and O. Levy, Nat. Mater., 2013, 12, 191–201 CrossRef CAS PubMed.
- W. G. Noid, J. Chem. Phys., 2013, 139, 090901 CrossRef CAS PubMed.
- R. Menichetti, K. H. Kanekal and T. Bereau, ACS Cent. Sci., 2019, 5, 290–298 CrossRef CAS.
- D. Mohammadyani, N. Yanamala, A. K. Samhan-Arias, A. A. Kapralov, G. Stepanov, N. Nuar, J. Planas-Iglesias, N. Sanghera, V. E. Kagan and J. Klein-Seetharaman, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1057–1068 CrossRef CAS PubMed.
- K. J. Boyd, N. N. Alder and E. R. May, Biophys. J., 2018, 114, 2116–2127 CrossRef CAS PubMed.
- B. Mohr, K. Shmilovich, I. Kleinwächter, D. Schneider, A. L. Ferguson and T. Bereau, Chem. Sci., 2022, 13, 4498 RSC.
- L. A. Luévano-Martínez, M. F. Forni, V. T. dos Santos, N. C. Souza-Pinto and A. J. Kowaltowski, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 587–598 CrossRef.
- E. Mileykovskaya, W. Dowhan, R. L. Birke, D. Zheng, L. Lutterodt and T. H. Haines, FEBS Lett., 2001, 507, 187–190 CrossRef CAS.
- J. D. Chodera, D. L. Mobley, M. R. Shirts, R. W. Dixon, K. Branson and V. S. Pande, Curr. Opin. Struct. Biol., 2011, 21, 150–160 CrossRef CAS PubMed.
- T. P. Straatsma, Free Energy by Molecular Simulation, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley, 1996, vol. 9 DOI:10.1002/9780470125861.ch2.
- P. Kollman, Chem. Rev., 1993, 93, 2395–2417 CrossRef CAS.
- G. M. Torrie and J. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 Search PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- J. Planas-Iglesias, H. Dwarakanath, D. Mohammadyani, N. Yanamala, V. E. Kagan and J. Klein-Seetharaman, Biophys. J., 2015, 109, 1282–1294 CrossRef CAS.
- R. Kiss, M. Sandor and F. A. Szalai, J. Cheminf., 2012, 4, P17 Search PubMed.
- T. T. Tanimoto, An elementary mathematical theory of classification and prediction, International Business Machines Corp., 1985 Search PubMed.
- P. Jaccard, New Phytol., 1912, 11, 37–50 CrossRef.
- A.-R. Nekoei and M. Vatanparast, Phys. Chem. Chem. Phys., 2019, 21, 623–630 RSC.
- G. Landrum, RDKit: Open-Source Cheminformatics Software, https://www.rdkit.org, accessed 14 July 2021.
- I. S. Kleinwächter, S. Pannwitt, A. Centi, N. Hellmann, E. Thines, T. Bereau and D. Schneider, Membranes, 2021, 11, 254 CrossRef.
- J. Seelig, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 40–50 CrossRef CAS PubMed.
- T. Parasassi, G. De Stasio, G. Ravagnan, R. M. Rusch and E. Gratton, Biophys. J., 1991, 60, 179–189 CrossRef CAS.
- T. Parasassi, M. Di Stefano, M. Loiero, G. Ravagnan and E. Gratton, Biophys. J., 1994, 66, 120–132 CrossRef CAS PubMed.
- R. Gredilla, J. Grief and H. D. Osiewacz, Exp. Gerontol., 2006, 41, 439–447 CrossRef CAS PubMed.
- G. Oemer, J. Koch, Y. Wohlfarter, M. T. Alam, K. Lackner, S. Sailer, L. Neumann, H. H. Lindner, K. Watschinger, M. Haltmeier, E. R. Werner, J. Zschocke and M. A. Keller, Cell Rep., 2020, 30, 4281.e4–4291.e4 Search PubMed.
- T. Romantsov, Z. Guan and J. M. Wood, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 2092–2100 CrossRef CAS PubMed.
- M. Schlame, S. Brody and K. Y. Hostetler, Eur. J. Biochem., 1993, 212, 727–733 CrossRef CAS PubMed.
- E. R. Pennington, K. Funai, D. A. Brown and S. R. Shaikh, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 1039–1052 CrossRef CAS PubMed.
- C. Sohlenkamp and O. Geiger, FEMS Microbiol. Rev., 2015, 40, 133–159 CrossRef PubMed.
- X. Periole and S.-J. Marrink, Methods in Molecular Biology, 2013, pp. 533–565 Search PubMed.
- M. Dahlberg, J. Phys. Chem. B, 2007, 111, 7194–7200 CrossRef CAS PubMed.
- M. Dahlberg and A. Maliniak, J. Phys. Chem. B, 2008, 112, 11655–11663 CrossRef CAS PubMed.
- M. Dahlberg and A. Maliniak, J. Chem. Theory Comput., 2010, 6, 1638–1649 CrossRef CAS PubMed.
- S. Jo, X. Cheng, J. Lee, S. Kim, S. Park, D. S. Patel, A. H. Beaven, K. Il Lee, H. Rui, S. Park, H. S. Lee, B. Roux, A. D. MacKerell, J. B. Klauda, Y. Qi and W. Im, J. Comput. Chem., 2017, 38, 1114–1124 CrossRef CAS PubMed.
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, e1000810 CrossRef PubMed.
- T. Bereau and K. Kremer, J. Chem. Theory Comput., 2015, 11, 2783–2791 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- D. H. de Jong, S. Baoukina, H. I. Ingólfsson and S. J. Marrink, Comput. Phys. Commun., 2016, 199, 1–7 CrossRef CAS.
- W. Chen, Y. Deng, E. Russell, Y. Wu, R. Abel and L. Wang, J. Chem. Theory Comput., 2018, 14, 6346–6358 CrossRef CAS PubMed.
- A. Pohorille, C. Jarzynski and C. Chipot, J. Phys. Chem. B, 2010, 114, 10235–10253 CrossRef CAS PubMed.
- P. V. Klimovich, M. R. Shirts and D. L. Mobley, J. Comput. – Aided Mol. Des., 2015, 29, 397–411 CrossRef CAS PubMed.
- M. R. Shirts and J. D. Chodera, J. Chem. Phys., 2008, 129, 124105 CrossRef PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- T. Bereau and R. H. Swendsen, J. Comput. Phys., 2009, 228, 6119–6129 CrossRef CAS.
- J. S. Hub, B. L. de Groot and D. van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- T. F. Liao, C. Z. Mooney, R. D. Duval and S. R. Eliason, Contemp. Sociol., 1995, 24, 269 CrossRef.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- P. Baldi and R. Nasr, J. Chem. Inf. Model., 2010, 50, 1205–1222 CrossRef CAS PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- T. Parasassi, G. De Stasio, A. D’Ubaldo and E. Gratton, Biophys. J., 1990, 57, 1179–1186 CrossRef CAS PubMed.
- G. Rizet, C. R. Hebd. Seances Acad. Sci., 1953, 237, 838–840 CAS.
- H. D. Osiewacz, A. Hamann and S. Zintel, Methods in Molecular Biology, 2013, vol. 965, pp. 439–462 Search PubMed.
- V. Warnsmann, S. Hainbuch and H. D. Osiewacz, Front. Genet., 2018, 9, 1–14 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00125j |
| This journal is © The Royal Society of Chemistry 2022 |