
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetastable intermediate during hIAPP aggregation catalyzed by membranes as detected with 2D IR spectroscopy
Sidney S.
Dicke†
 a,
Michał
Maj†‡
ab,
Caitlyn R.
Fields
a and
Martin T.
Zanni
*a
a,
Michał
Maj†‡
ab,
Caitlyn R.
Fields
a and
Martin T.
Zanni
*a
aDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: zanni@chem.wisc.edu
bFormally at Department of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, WI 53706, USA
First published on 13th June 2022
Abstract
The aggregation of human islet amyloid polypeptide (hIAPP) into amyloid fibrils involves formation of oligomeric intermediates that are thought to be the cytotoxic species responsible for β-cell dysfunction in type 2 diabetes. hIAPP oligomers permeating or disrupting the cellular membrane may be one mechanism of toxicity and so measuring the structural kinetics of aggregation in the presence of membranes is of much interest. In this study, we use 2D IR spectroscopy and 13C18O isotope labeling to study the secondary structure of the oligomeric intermediates formed in solution and in the presence of phospholipid vesicles at sites L12A13, L16V17, G24A25 and V32G33. Pairs of labels monitor the couplings between associated polypeptides and the dihedral angles between adjacent residues. In solution, the L12A13 residues form an oligomeric β-sheet in addition to an α-helix whereas with the phospholipid vesicles they are α-helical throughout the aggregation process. In both solution and with DOPC vesicles, L16V17 and V32G33 have disordered structures until fibrils are formed. Similarly, under both conditions, G24A25 exhibits 3-state kinetics, created by an oligomeric intermediate with a well-defined β-sheet structure. Amyloid fibril formation is often thought to involve intermediates with exceedingly low populations that are difficult to detect experimentally. These experiments establish that amyloid fibril formation of hIAPP when catalyzed by membranes includes a metastable intermediate and that this intermediate has a similar structure at G24A25 in the FGAIL region as the corresponding intermediate in solution, thought to be the toxic species.
Introduction
Human islet amyloid polypeptide (hIAPP) is a hormone synthesized with insulin in the β-cells of pancreatic islets that plays a critical role in controlling glucose homeostasis and related metabolic processes.1,2 Despite its vital function, it is also known to form amyloid fibril aggregates that are deposited at high concentrations in pancreatic islets of type 2 diabetes (T2D) patients.3–7 The mechanism of cytotoxicity remains largely unknown, and several mechanisms may be at work. One of the proposed mechanisms for cytotoxicity involves hIAPP oligomers interacting with cellular membranes.8–13 Evidence has been found that hIAPP forms discrete pores in the membrane, similar to those formed by transmembrane ion channels.14–18 hIAPP may also damage membranes through a detergent-like effect or, in the process of forming fibrils, cause leakage by ripping lipids from the bilayers.19–21 Regardless of the mechanism, time-lapse toxicity studies suggest that it is oligomeric intermediates that are the toxic species and not the mature fibril.22–24There exists little direct information about the structure of hIAPP oligomers, and even less information about oligomer structure in the presence of membranes. Monomeric hIAPP is largely disordered in solution,25–27 but may adopt a helix conformation on phospholipid membranes or SDS micelles;28 residues 5–20 have been observed to form a helix upon introduction of HFIP29via NMR and CD spectroscopy; residues 8–18 have been predicted to form a helical core upon interacting with membranes preceding aggregation events;30 and residues 10–17 have the highest helical propensity31 in hIAPP according to MD simulations and EPR spectroscopy. Additional work by Miranker and co-workers suggest helices are a stable structure for rat IAPP (rIAPP), which they concluded using small molecule foldamers that stabilize helix dimers.32 There is also evidence of β-sheets in the oligomer, primarily observed via 2D IR spectroscopy, as discussed below. Recent work by Roderiguez Camargo et al. using nanodiscs to stabilize hIAPP intermediates has found further evidence of oligomeric β-sheets.33 In contrast to oligomer structure, the fibrillar structures are relatively well characterized with solid-state NMR and cryoEM. In fibrillar states, residues 20–29 have been observed as a disordered loop with flanking β-sheets.34 The structural properties also change with pH, and recent cryo-EM and NMR structures have identified β-sheets within the 20–29 region, at pH 6, whereas this region is a loop at pH 7.4.34–37
One reason that oligomers are difficult to study is because they are transiently populated, forming intermediates in the aggregation pathway. In classical nucleation theory, often used to describe amyloid fibril formation, oligomers are an unstable species that undergo a nucleation event when they are present above some critical concentration, upon which the fibrils are then grown. According to this model, as well as recent work on the secondary nucleation of hIAPP,38,39 intermediates should be difficult to identify because they grow into fibrils almost as soon as they form and so exist at low concentrations.40,41 Seemingly at odds with classical nucleation theory, we have experimentally identified an intermediate of hIAPP that is stable during the lag phase. It has a critical concentration of between 150 and 250 μM41 and a β-sheet-like structure between the FGAIL residues that span 23 to 27,42 and a helical structure at residues L12A13.43 The β-sheet-like region may extend as far as residues S20 to S29.44–48 The intermediate is an oligomer predicted to be formed by at least 5 associated polypeptides41 and correlates with cellular toxicity.22 The oligomers are kinetically stabilized because the FGAIL region must disorder to form the structure of the fibril, causing a free energy barrier between 3–20 kcal mol−1, depending on the size of the oligomer, which is the reason why this intermediate is observed throughout the lag phase.41 If proline mutations are introduced into the FGAIL region to disrupt the oligomer structure, fibrils do not form, consistent with an on-pathway intermediate.49
Here, we study hIAPP secondary structure in solution using Tris buffer at pH 7.37 to mimic the cytosol or extracellular space with and without the addition of phospholipid vesicles. The primary goal of our study is to identify differences in the aggregation pathway when aggregation is membrane catalyzed. The study is designed to test regions of hIAPP whose secondary structure we know is well defined in buffer. We are particularly interested in determining if the “FGAIL intermediate” described above also forms when aggregation takes place in solution with vesicles, since we think that the structure in the FGAIL region stabilizes the oligomers and is the cause of toxicity.
Materials and methods
2D IR spectroscopy
The 2D IR spectrometer setup used in this work consists of a 1 kHz amplifier (Solstice, Spectra-Physics) coupled to a commercial optical parametric amplifier (TOPAS, Light Conversion Ltd) that produces signal and idler beams in the near-IR. The signal and idler beams are then focused onto a 0.5 mm thick AgGaS2 crystal to generate mid-IR light at 6 μm through a difference-frequency generation process. The mid-IR light is split into pump and probe pulses with a beam-splitter and the pump light is directed into a Ge-based acousto-optic modulator (AOM) that enables phase stabilization, rapid data acquisition, and phase cycling. Rapid acquisition makes it possible to collect 2D IR spectra continuously during peptide aggregation with high signal-to-noise ratio while phase cycling effectively removes pump scatter.50,51 The pump beam leaving the AOM is focused with the probe beam onto a sample at a cross geometry and the self-heterodyned signal is measured with a LN2-cooled MCT array detector. For a more detailed description of a pulse-shaping assisted 2D IR spectrometer, we refer readers to previous publications.51–56Peptide synthesis and purification
Isotope-labeled hIAPP samples were synthesized by Fmoc-based chemistry using an automated microwave peptide synthesizer (LibertyBlue, CEM) on PAL–PEG–PS resin according to published protocols.51,57 Peptides were cleaved off resin with a microwave-assisted cleavage system (Accent, CEM) using TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:H2O (18:1:1 v/v/v) solution. To form a disulfide bond between the N-terminal cysteines, crude peptide was dissolved in DMSO and kept at room temperature for 24 hours. Following the oxidation, the peptide was purified with reversed-phase HPLC using C18 preparative column (Waters XSelect) using a two-buffer purification gradient. Buffer 1 was composed of 0.045% HCl in H2O, and buffer 2 of 80% CH3CN, 20% H2O and 0.045% HCl (v/v). Purification was run with a gradient, increasing the percentage of buffer 2 by 1% per minute. Purity of the peptides were assessed by integrating the 220 nm peptide backbone peak versus all other impurities visible on the HPLC chromatogram; peptide identity was confirmed via MALDI. All peptides used for experiments were >95% purity. All measurements were carried out at 1 mM peptide concentration.
:TIS:H2O (18:1:1 v/v/v) solution. To form a disulfide bond between the N-terminal cysteines, crude peptide was dissolved in DMSO and kept at room temperature for 24 hours. Following the oxidation, the peptide was purified with reversed-phase HPLC using C18 preparative column (Waters XSelect) using a two-buffer purification gradient. Buffer 1 was composed of 0.045% HCl in H2O, and buffer 2 of 80% CH3CN, 20% H2O and 0.045% HCl (v/v). Purification was run with a gradient, increasing the percentage of buffer 2 by 1% per minute. Purity of the peptides were assessed by integrating the 220 nm peptide backbone peak versus all other impurities visible on the HPLC chromatogram; peptide identity was confirmed via MALDI. All peptides used for experiments were >95% purity. All measurements were carried out at 1 mM peptide concentration.
Preparation of phospholipid vesicles
DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) was purchased from Avanti Polar Lipids and stored at −20 °C until use. Cholesterol was acquired from Sigma-Aldrich and used without further purifications. Liposome samples were prepared through the standard thin-film rehydration procedure. Initially, a mixture of DOPC and cholesterol was prepared in the ratio 75:25, dissolved in chloroform, and evaporated over a stream of nitrogen. Dry film was then placed under vacuum to remove any residual solvent. The film was rehydrated with a Tris buffer solution at pH 7.37 and vortexed at room temperature for 2 hours. The solution then underwent several freeze–thaw cycles and extruded 12 times through a 200 nm membrane. Dynamic light scattering measurements post-extrusion confirmed the average size of the vesicles to be between 140 and 160 nm. The final concentration of the liposomes was set to 75 mM.
Non-negative matrix factorization
2D IR spectra in this work were analyzed using a non-negative matrix factorization algorithm58 as previously applied by Maj et al.43 Briefly, a non-negative input matrix I consisting of time-resolved diagonal traces extracted from the 2D spectra is fit to a set of non-negative W and H matrices, containing vibrational eigenspectral and time-dependent intensity changes, respectively. Matrix multiplication of W by H yields a matrix similar to I, differing by a residual matrix U, giving the quality of the fit (eqn (1))| I = WH + U. | (1) |
Results
Amyloid aggregation is a difficult problem to study with standard structural biology techniques because it involves kinetics, large assemblies, and (in this case) DOPC vesicles. With two-dimensional infrared (2D IR) spectroscopy it is possible to monitor kinetically evolving structures via on-the-fly acquisition of 2D IR spectra.51,54,59,60 The information on the peptide structure is extracted from time-resolved vibrational spectra measured in the amide I band region (∼1610–1710 cm−1) and the 13C18O isotope-labeled region (∼1570–1610 cm−1). The amide I band arises primarily due to carbonyl stretching vibration of the peptide backbone, making it a sensitive probe of secondary structure, while introduction of 13C18O isotope labels is an efficient way of extracting structural information at a specific site in the peptide sequence.50,51,53 We utilize an isotope labeling method, called “dihedral indexing,”43 that increases the sensitivity to secondary structure by taking advantage of the vibrational coupling between adjacent amino acids, which was how we detected α-helices in the monomeric state of hIAPP at physiological pH.43In what follows, we apply 2D IR spectroscopy and the dihedral indexing approach to investigate the structure and kinetics of transient intermediates formed during the aggregation of hIAPP in neat Tris buffer and in the presence of phospholipid vesicles. We choose DOPC/cholesterol mixtures as our model membranes as mimics of the β-cell plasma environment although physiological plasma membranes are much more varied in composition and contain negatively-charged headgroups such as phosphoserine.61,62 We introduce isotope labels into four distinct sites of the hIAPP sequence, including the most amyloidogenic FGAIL region. As we show below, we resolve vibrational spectra of each species formed on the aggregation pathway, including α-helical vesicle-associated species, oligomers, and fully-formed fibrils. The data shows that the FGAIL intermediate formed with the presence of vesicles has the same secondary structure in the FGAIL region as to the one that forms in solution without vesicles at these locations. The results strengthen the hypothesis of the FGAIL being the most critical region for the amyloidogenic properties and toxicity of hIAPP.45
hIAPP in buffer: two- and three-state kinetics at different locations
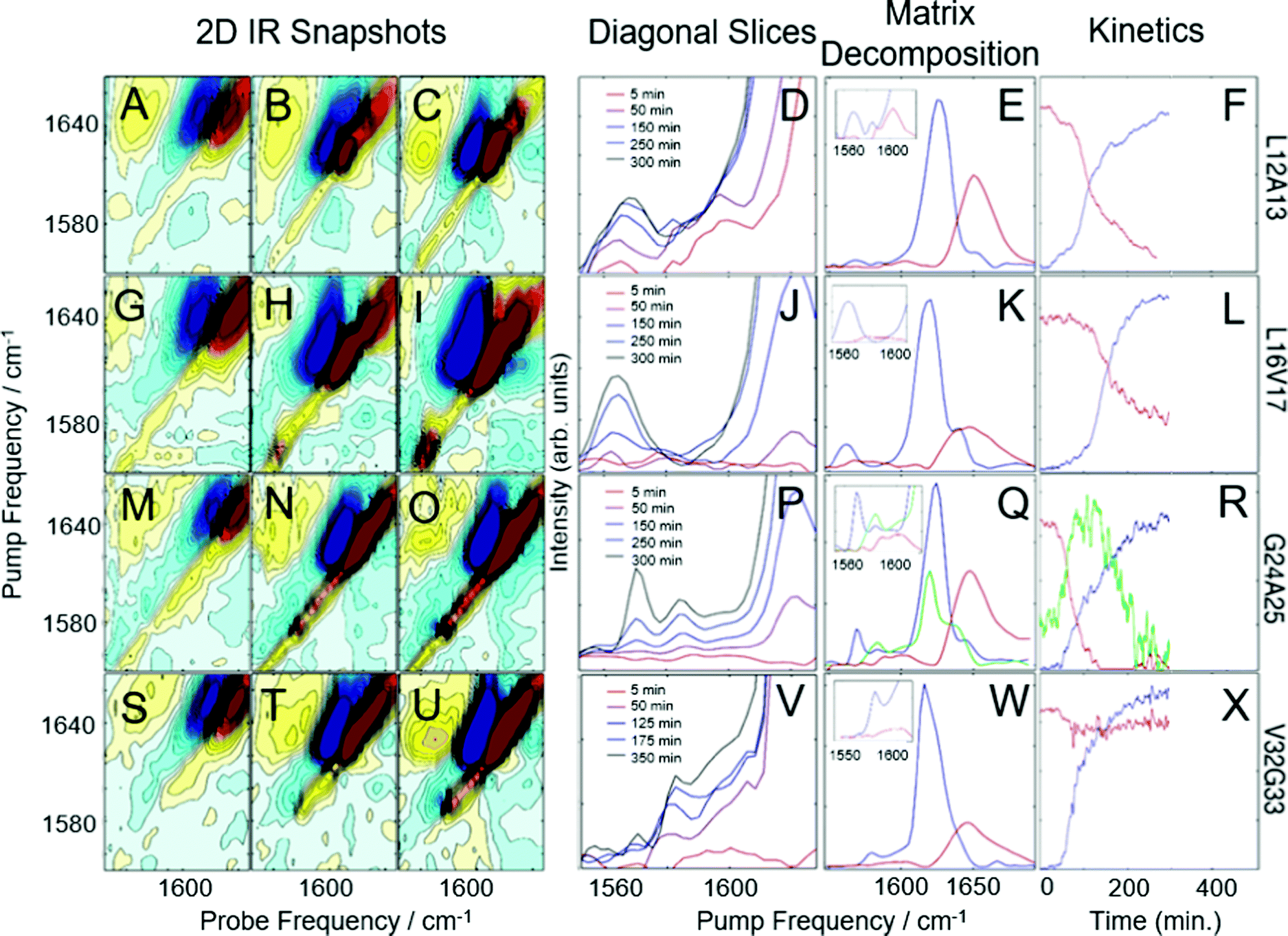
Shown in Fig. 1 are the results of 2D IR kinetics experiments of hIAPP labeled at L12A13, L16V17, G24A25, and V32G33, at a concentration of 1 mM in Tris buffer at pH 7.37, with no vesicles present. Data in Fig. 1A–L were previously published;43 we include them here for ease of reference. Representative 2D IR spectra are shown at the beginning of aggregation (5 minutes after initiating aggregation), at the end of aggregation whereafter no discernable changes to the spectra occurred, and at a mid-point between the two. A typical 2D IR spectrum consists of a diagonal signal arising due to transitions between the ground state and the first vibrationally excited states, and a negative excited state absorption signal that is shifted from the diagonal line due to anharmonicity.50,53 | ||
| Fig. 1 2D-IR spectra and kinetics of labeled hIAPP in Tris buffer. 2D IR snapshots of L12A13-labeled hIAPP aggregation at 5 minutes (A), 150 minutes (B), and 500 minutes (C). (D) The diagonal of 2D IR spectra in the isotope labeled region of L12A13-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 3 state, shown in red, green, and blue in (E). (F) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (E). 2D IR snapshots of L16V17-labeled hIAPP aggregation at 5 minutes (G), 250 minutes (H), and 500 minutes (I). (J) The diagonal of 2D IR spectra in the isotope labeled region of L16V17-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 2 states, shown as a red and blue trace in (K). (L) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (K). 2D IR snapshots of G24A25-labeled hIAPP aggregation at 5 minutes (M), 125 minutes (N), and 350 minutes (O). (P) The diagonal of 2D IR spectra in the isotope labeled region of G24A25-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 3 states, shown in red, green, and blue in (Q). (R) displays kinetics plots of the intensities of the matrix-decomposition traces shown in (Q). 2D IR snapshots of V32G33-labeled hIAPP aggregation at 5 minutes (S), 100 minutes (T), and 500 minutes (U). (V) The diagonal of 2D IR spectra in the isotope labeled region of V32G33-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time dependent diagonal trace reveals 2 states, shown as red and blue traces in (W). (X) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (W). Data presented in A–L was previously published by Maj et al.43 and is reproduced here for ease of comparison. | ||
The unlabeled residues will have absorptions between 1610 and 1710 cm−1, whereas the isotope labeled residues will fall between 1565 and 1605 cm−1.42,43,51 To help interpret the data, the unlabeled portion of the spectra in Fig. 1A–C are labeled “RC” for ‘random coil’ and “A-β sheet” for ‘amyloid B-sheet’, while the isotope labeled portion is labeled “13C18O”. The unlabeled residues are useful for monitoring the transition from monomer to fibrils. At 5 minutes, the spectrum in Fig. 1A is dominated by a broad peak at 1650 cm−1, indicating that hIAPP is largely random coil, as would be expected at the beginning of the lag phase.42,63 By 150 minutes, the amyloid β-sheet peak is the largest feature and continues to grow throughout the experiment (Fig. 1C). All of the samples measured, regardless of where hIAPP is isotope labeled, have similar unlabeled features because there are 35 unlabeled residues and only 2 labeled residues.
The more insightful portion of the spectrum is the 13C18O region encompassed by the black box in Fig. 1C for which there are clear differences between labeled amino acids. In Fig. 1D, a diagonal trace through the isotope labeled spectral region of the combination band is tracked over the course of the experiment.43,64 Diagonal slicing can be interpreted similarly to FTIR spectra, although 2D IR spectra are more sensitive to secondary structures.50,64–66 When a secondary structure forms, like an α-helix or a β-sheet, the backbone carbonyl (amide I) vibrations from multiple amino acids couple to one another, creating delocalized vibrational modes.43,50 That delocalization also increases the transition dipole strength, μ, of the delocalized mode. Higher sensitivity arises because 2D IR spectra scale as μ4, whereas FTIR scale as μ2.50,64,65 Thus, unlike an FTIR spectrum, one does not expect the integrated area of a 2D IR spectrum to remain constant.66,67 After 5 minutes (red trace), a peak is visible in the labeled region above 1600 cm−1, indicative of α-helical secondary structure. After 100–300 minutes (purple trace, blue trace, and grey trace), multiple features appear in the isotope region, with the lower-frequency peak increasing in intensity over time, consistent with a transition from α-helix to β-sheet structure. This data, determining that 27–38% of residues L12A13 form a helical structure during the lag phase of aggregation, prior to forming oligomeric β-sheets, was previously published.43 After 500 minutes (black trace), an isotope-labeled feature has grown in at 1570 cm−1, indicative of ordered β-sheet structure. Thus, L12A13 exhibits 3-states.
It is clear from the raw data that 3 states are present at L12A13, but their spectra are overlapping. To determine the spectra for each of the 3-states and better resolve their kinetics, we apply a non-negative matrix factorization (referred to as “matrix factorization” in the text) method previously applied by Maj et al.43 (See Methods). Matrix factorization uses the thousands of spectra measured during the kinetics to find either 2 or 3 eigenvectors corresponding to distinct spectra and their respective populations as a function of time. The raw experimental data are reproduced by carrying out matrix multiplication of the resulting vectors. Fig. 1E displays three state decomposition of the diagonal slices of L12A13 yielding the vibrational eigenspectra of monomers (red), intermediates (green), and fibrils (blue). Plotting the intensities of the eigenspectra over time yields the kinetics traces shown in Fig. 1F. The monomer trace (red) starts high during the lag phase, and decays throughout the experiment as amyloid fibrils form (blue). The monomer eigenspectrum corresponding to the monomer trace displays an isotope-region frequency above 1600 cm−1 (Fig. 1E, zoom, red), consistent with a population of monomeric hIAPP polypeptides with helices in this region. The oligomer trace (green) displays a lower frequency in the isotope region consistent with small β-sheets (Fig. 1E, zoom, green). The fibril formation eigenspectrum (Fig. 1E, blue) follows sigmoidal kinetics (Fig. 1F), as is typical for amyloid fibril formation. The intermediate state (green) rises as the monomers decay and falls as fibrils form, consistent with a transiently populated structure during the lag phase.
Fig. 1G–I present analogous spectra for isotope labeling at L16V17. For L16V17, only monomers or fibrils are observed; thus, as the fibrils form, the monomer decreases and the fibril signal increases, indicative of two-state kinetics. The matrix factorization identifies 2 states, consistent with the visual inspection of Fig. 1J. The intensities of the peaks corresponding to each state are plotted in Fig. 2L, showing the inverse correlation in intensities between monomers (red trace) and fibrils (blue trace) over time. In contrast to Fig. 1E, the monomeric trace does not display a prominent isotope-labeled peak indicating that unaggregated hIAPP is largely disordered in solution.
 | ||
| Fig. 2 2D-IR spectra and kinetics of each labeled peptide in the presence of DOPC vesicles prepared with cholesterol. 2D IR snapshots of L12A13-labeled hIAPP aggregation at 5 minutes (A), 150 minutes (B), and 350 minutes (C). (D) The diagonal of 2D IR spectra in the isotope labeled region of L12A13-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 2 states, shown in red and blue in (E). (F) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (E). 2D IR snapshots of L16V17-labeled hIAPP aggregation at 5 minutes (G), 150 minutes (H), and 350 minutes (I). (J) The diagonal of 2D IR spectra in the isotope labeled region of L16V17-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 2 states, shown as a red and blue trace in (K). (L) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (K). 2D IR snapshots of G24A25-labeled hIAPP aggregation at 5 minutes (M), 150 minutes (N), and 350 minutes (O). (P) The diagonal of 2D IR spectra in the isotope labeled region of G24A25-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time-dependent diagonal trace reveals 3 states, shown in red, green, and blue in (Q). (R) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (Q). 2D IR snapshots of V32G33-labeled hIAPP aggregation at 5 minutes (S), 125 minutes (T), and 350 minutes (U). (V) The diagonal of 2D IR spectra in the isotope labeled region of V32G33-labeled hIAPP at 5 time points during aggregation. Matrix decomposition of the time dependent diagonal trace reveals 2 states, shown as red and blue traces in (W). (X) Displays kinetics plots of the intensities of the matrix-decomposition traces shown in (W). | ||
The next set of labels, G24A25, is within the so-called “FGAIL” region of hIAPP. The FGAIL region spans residues 23 to 27 and has been a matter of extensive research because sequence comparisons between hIAPP variants distinguish it as the primary reason for the amyloidogenic properties of the peptide.45,68–71 We have studied the FGAIL region previously using 2D IR spectroscopy,41,42,56,72 but this is the first time that we use double isotope labels. The spectra of G24A25 peptide are presented in Fig. 1M–O. At 5 minutes, a small isotope signal is visible at ∼1580 cm−1. By 175 minutes, the 1580 cm−1 peak has become very intense. By the end of aggregation, the 1580 cm−1 isotope-labeled peak has lessened in intensity and a new intense spectral feature is seen at 1568 cm−1. Thus, G24A25 exhibits 3-state kinetics, which is confirmed by matrix decomposition in Fig. 1Q and R. In Fig. 1R, the intensity of the 3 states is plotted over time, showing the rise and fall of an intermediate, which is present during the lag time. The intensity of the intermediate peak indicates that it is caused by coupling between multiple polypeptides, consistent with an oligomer adopting a regular secondary structure. The oligomeric and fibril traces exhibit isotope peaks in the 1570–1590 cm−1 region, consistent with oligomeric polymorphs73 or separate β-sheet populations.43
The last set of double labels are V32G33, whose spectra are shown in Fig. 1S–X. 2-State kinetics are observed in solution, indicating a transition from monomer to fibril with little to no regular secondary structure present during the lag phase when oligomers are present.
Data in Fig. 2A–X is analogous to that presented in Fig. 1A–X except experiments in Fig. 2 were performed in the presence of DOPC vesicles (see Methods).
Fig. 2A–F display results for L12A13-labeled hIAPP. The diagonal slices and matrix decomposition analysis in Fig. 2D and E, respectively, reveals 2-state behavior. Interestingly, this is not the behavior without vesicles, for which 3-state kinetics were observed (Fig. 1D and E). Based on the frequencies these states appear at, the data presented here suggests that while aggregating in the presence of vesicles, L12A13 sample either α-helix or fibrillar states, but we see no evidence of the short β-sheets that were observed in solution.
Fig. 2G–L display results for L16V17 labeled hIAPP in the presence of vesicles. For this set of labels, 2-state kinetics are observed, as was observed in buffer. Thus, the vesicles do not induce a structural change in the L16V17 region of the oligomer.
Fig. 2M–R display results for isotope-labeled hIAPP at G24A25, residing in the FGAIL region that forms a parallel β-sheet in oligomers in buffer. These results show that G24A25 proceeds in a 3-state kinetic model with 2D IR spectra that exhibit a clearly resolved isotope labeled peak at 1580 cm−1; the spectra and kinetics of G24A25 with vesicles is very similar to what was seen in buffer. Therefore, the FGAIL oligomer, as monitoring by G24A25, is still an intermediate in the aggregation pathway of hIAPP whether or not amyloid formation occurs in the presence of DOPC vesicles.
Data for the final hIAPP label, V32G33, is shown in Fig. 2S–X. It undergoes 2-state kinetics, similar to the data taken without vesicles, although some subtle differences exist in the spectra (such as the intensity of the random coil peak).
We also note that aggregation in the presence of DOPC vesicles is more rapid. The lag time, as measured by the half-rise time of the sigmoidal kinetics in Fig. 1D and 2D, is about twice as short in the presence of vesicles than without, taking about 200 minutes without vesicles and 100 minutes with vesicles. Because aggregation is faster, data was only collected for 300 minutes in Fig. 2 instead of 500 minutes, as in Fig. 1. The kinetics in Fig. 1 and 2 are plotted on the same x-axis to illustrate the increase in aggregation rate. Reproducibility of kinetics is difficult in amyloid aggregation experiments, but the trend holds for all data collected at each set of sites, and is consistent with membrane enhanced aggregation kinetics reported previously.74,75 Due to convection currents when hIAPP is mixed with the vesicle solution, all hIAPP will come into contact with a vesicle during the time that it takes to place the sample in the spectrometer. Thus, even the earliest measured 2D IR spectra will already be impacted by interactions with vesicles.76 That being said, we cannot determine if hIAPP remains bound to the membranes during the course of the experiment, but the change in structure at L16V17 and the increase in aggregation rate (decreased lag time) proves that the membranes does alter the hIAPP structure and the kinetics.
Discussion
In the Results above, we presented data from 4 sites of hIAPP over the course of aggregation both in the absence- and presence-of vesicles. The primary question that we are trying to address is whether the FGAIL oligomer that we have extensively studied and modeled in buffer is also involved in hIAPP aggregation in the presence of vesicles. In buffer, the proteins evolve from monomers to oligomers and then to amyloid fibrils.22,42 The ability to spectroscopically resolve each of the 3 states depends on the couplings between the isotope labeled residues.43 We have previously established that all residues studied here have sizeable inter-molecular couplings once the fibrils have formed because of the close proximity between β-strands.42,43,65 Even residues G24A25 that are at the edge of a disordered loop still couple to the adjacent hIAPP polypeptides above and below it in the fibril.34,43 Thus, the spectra for all 4 sets of amino acids here are expected to exhibit a distinct spectrum when fibrils are formed. That being said, not all amino acids will necessarily report on the formation of the oligomers. If a residue has a disordered structure in both the monomer and the oligomer, then its spectrum will be unchanged during the lag phase and new spectral features will not be observed until fibrils are formed. We expected G24A25 to exhibit a distinct oligomer spectrum when aggregation proceeded in buffer, because we knew from our extensive prior work that the FGAIL region adopts a parallel β-sheet in the lag phase prior to fibrils formation.42 Indeed, G24A25 in buffer did indeed exhibit 3-state kinetics. Thus, some residues will exhibit 2-state kinetics and others 3-state kinetics, depending on whether those particular residues have a unique secondary/quaternary structure in the oligomer.Shown in Fig. 3 is a graphical summary of the structural conclusions drawn from the 2D IR spectra and kinetics presented above. We break the aggregation kinetics into monomer, oligomer and fibril, reflecting their respective timepoints during the kinetic traces. The amino acids measured in this study are labeled by the secondary structure drawn from the interpretation of the 2D IR spectra. Secondary structure assignments are also given to other residues as measured in prior studies. Shown in Fig. 4 is a graphical cartoon of the 3-state aggregation pathway, highlighting the similarities and differences in the aggregation mechanism in buffer and in the presence of DOPC vesicles.
 | ||
| Fig. 3 Double – label structural information for hIAPP monomers, oligomers, and fibrils. The hIAPP primary sequence (top) displays the sites of each double-label, colored cyan. *, +, and ^ denote structural assignments given in ref. 43, 42, and 34, respectively. | ||
 | ||
| Fig. 4 Proposed mechanism of assembly of amyloid fibrils in solution and in solution with vesicles consistent with the results of the double labels used in this study and previous work.34,42,43 | ||
The most important result of this study is that, when hIAPP aggregates in solution with DOPC vesicles, oligomers with a parallel β-sheet at G24A25 are observed like they are in buffer. As indicated in Fig. 3, in buffer the parallel β-sheet in the oligomer extends from at least F23 to L27, spanning the FGAIL sequence.42 Therefore, at least a portion of the oligomeric β-sheet is present when aggregation is impacted by DOPC vesicles.
This finding is of interest because, it is thought, that the FGAIL β-sheet is the reason that the oligomers have a large and stable population during the lag phase. Amyloid aggregation is typically thought to be a nucleation event with the formation of the oligomers acting as the rate limiting step. That traditional nucleation model is at odds with the observations here and our previous work in buffer,41 because if the oligomers are the rate limiting step to fibril formation, then their population should be very low since they are unlikely to form and once they do form, they nucleate fibrillization. But, instead, oligomers are readily observed during the lag phase. In buffer, we have concluded that the rate limiting step in the free energy landscape of aggregation is the unstructuring of the FGAIL β-sheet in the oligomer in order to adopt the fold of the fibril.41 The need to unstructure the FGAIL β-sheet creates a barrier that kinetically traps the oligomers, which accounts for why they can be easily observed experimentally. The results presented in this article indicate that the FGAIL β-sheet of the oligomers still forms even when aggregation occurs with DOPC vesicles. Thus, the oligomers are a meta-stable species regardless of whether they form in buffer or in the presence of DOPC vesicles. Fig. 4 provides a cartoon rendition of aggregation in buffer (top panel) and in buffer with DOPC vesicles present (bottom panel) consistent with our results presented here and those presented in previous work.34,42,43
We cannot determine from these experiments whether the oligomer assembles on the vesicles and remains bound or whether the vesicles serve to nucleate the oligomer with subsequent aggregation occurring in solution. hIAPP is positively charged and binds strongly to negative vesicles, including DOPC.8,10,77 The N-terminus of hIAPP is amphipathic and forms α-helices on anionic lipids as established by CD spectroscopy30 and site-specific EPR labeling.31 Interactions between hIAPP and zwitterionic vesicles like DOPC have previously been observed although there is limited high resolution structural data available.61 One postulate is that the N-terminal helices coalesce into helical bundles on the membranes, enabling the β-sheets of the amyloid fibrils.30,31,74,78,79 Helical bundles may serve as Ca2+ ion channels that initiate an apoptotic cascade.16,74,78–80 The atomic structure is only known in SDS micelles by NMR28 and on POPS vesicles by EPR31 as mentioned above. An additional structure solved by Ramamoorthy and co-workers found residues A5-V17 and S20-L23 of rat IAPP in helical conformations on DPC micelles.81 Thus, the helical structure that we observe in the oligomer might be caused by hIAPP binding to the vesicles, albeit creating a shorter helix than seen previously by NMR or EPR (because we observe helicity at L12A13 but not L16V17).30,31
Alternatively, rather than the polypeptides binding to the membrane and then organizing into oligomers, the membrane might instead be nucleating oligomer formation similar to the way that many small molecules alter amyloid aggregation.42,82–85 It is well-known that helical inducing agents catalyze hIAPP aggregation.86,87 For example, a few percent of hexafluoroisopropanol or trifluoroethanol added to buffer causes faster amyloid aggregation.75,86 Helical polypeptides added to hIAPP also induce amyloid fiber formation.87 Thus, the vesicles might increase the proportion of alpha-helices, which then serve to nucleate oligomers. Indeed, we have previously pointed out the similarity of leucine rich repeat proteins to the known secondary structure of hIAPP, and hypothesized that the tertiary structure of hIAPP has a similar fold.43 Leucine rich repeat proteins have helices that stabilize short, 3–4 residue β-sheets.43,88 If the oligomers have a structure similar to the Leucine rich repeat proteins, then inducing helicity might stabilize the FGAIL β-sheet, whether or not the oligomer remains bound to the vesicles.
Conclusions
In this manuscript we reported the structure throughout aggregation of hIAPP at four regions. Two sets of labels were placed near the N-terminus, one set of labels was placed in the FGAIL region, and one set of labels was placed in the C-terminus. Experiments were performed in vitro, either in buffer or when mixed with vesicles made of DOPC, which is a zwitterionic lipid. In buffer, residues L12A13 and G24A25 exhibited 3-state kinetics, reflecting an α-helix to short β-sheet to fibrillar β-sheet for L12A13 and reflecting a random coil to short β-sheet to fibrillar β-sheet for G24A25. In buffer with DOPC vesicles, the parallel FGAIL oligomer sheet is still observed, but the L12A13 β-sheet is not. Our experiments cannot determine if the oligomers are bound to the vesicles or if the vesicles are nucleating/stabilizing oligomer formation by inducing helices, for example, but either way, they are impacting the kinetics, the structure at L12A13, and retaining the G24A25 β-sheet. Since this oligomer has a well-defined parallel β-sheet, it is hopeful that it might be targeted by compounds that recognize its structure. Indeed, in buffer, artificial macrocycles pre-programmed with the FGAIL sequence altered the aggregation kinetics by stabilizing the oligomer's β-sheet.42 In the future, lipids of more complex structural composition including negatively-charged lipids,89–92 could be studied using 2D IR spectroscopy to further examine the effects of sterics and electrostatics on hIAPP aggregation kinetics.Author contributions
S. S. D., M. M., and C. R. F. conducted data curation, formal analysis, investigation, and validation. M. M. coded software and performed non-negative matrix factorization for spectral decomposition. M. T. Z. provided project administration, supervision, and visualization. S. S. D. and M. T. Z. wrote the paper.Funding sources
This work was supported by the National Institutes of Health (R01DK079895).Conflicts of interest
To ameliorate conflicts of interest, the University of Wisconsin-Madison requires us to disclose that M. T. Z. is part owner of PhaseTech Spectroscopy, Inc, a company that manufactures 2D spectrometers such as those used in this report.References
- D. L. Hay, S. Chen, T. A. Lutz, D. G. Parkes and J. D. Roth, Pharmacol. Rev., 2015, 67, 564–600 CrossRef CAS PubMed.
- W. A. Scherbaum, Exp. Clin. Endocrinol. Diabetes, 1998, 106, 97–102 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- A. Clark, G. J. Cooper, C. E. Lewis, J. F. Morris, A. C. Willis, K. B. Reid and R. C. Turner, Lancet, 1987, 330, 231–234 CrossRef.
- G. J. Cooper, A. C. Willis, A. Clark, R. C. Turner, R. B. Sim and K. B. M. Reid, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8628–8632 CrossRef CAS PubMed.
- R. L. Hull, G. T. Westermark, P. Westermark and S. E. Kahn, J. Clin. Endocrinol. Metab., 2004, 89, 3629–3643 CrossRef CAS PubMed.
- A. Lorenzo, B. Razzaboni, G. C. Weir and B. A. Yankner, Nature, 1994, 368, 756–760 CrossRef CAS PubMed.
- K. Sasahara, Biophys. Rev., 2018, 10, 453–462 CrossRef CAS PubMed.
- K. Sasahara, K. Morigaki and K. Shinya, FEBS J., 2014, 281, 2597–2612 CrossRef CAS PubMed.
- K. Sasahara, K. Morigaki, T. Okazaki and D. Hamada, Biochemistry, 2012, 51, 6908–6919 CrossRef CAS PubMed.
- L. Caillon, A. R. F. Hoffmann, A. Botz and L. Khemtémourian, J. Diabetes Res., 2016, 2016, 5639875 Search PubMed.
- L. Khemtémourian, J. A. Killian, J. W. M. Höppener and M. F. Engel, Exp. Diabetes Res., 2008, 2008, 421287 CrossRef PubMed.
- M. F. M. Engel, L. Khemtémourian, C. C. Kleijer, H. J. D. Meeldijk, J. Jacobs, A. J. Verkleij, B. de Kruijff, J. A. Killian and J. W. M. Höppener, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6033–6038 CrossRef CAS PubMed.
- Y. Hirakura, W. W. Yiu, A. Yamamoto and B. L. Kagan, Amyloid, 2000, 7, 194–199 CrossRef CAS PubMed.
- A. Quist, I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10427–10432 CrossRef CAS PubMed.
- T. A. Mirzabekov, M. Lin and B. Kagan, J. Biol. Chem., 1996, 271, 1988–1992 CrossRef CAS PubMed.
- M. Anguiano, R. J. Nowak and P. T. Lansbury Jr, Biochemistry, 2002, 41, 11338–11343 CrossRef CAS PubMed.
- J. R. Brender, S. Salamekh and A. Ramamoorthy, Acc. Chem. Res., 2012, 45, 454–462 CrossRef CAS PubMed.
- J. D. Green, L. Kreplak, C. Goldsbury, X. L. Blatter, M. Stolz, G. S. Cooper, A. Seelig, J. Kistler and U. Aebi, J. Mol. Biol., 2004, 342, 877–887 CrossRef CAS PubMed.
- S. A. Jayasinghe and R. Langen, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2002–2009 CrossRef CAS PubMed.
- M. F. M. Sciacca, S. A. Kotler, J. R. Brender, J. Chen, D. Lee and A. Ramamoorthy, Biophys. J., 2012, 103, 702–710 CrossRef CAS PubMed.
- A. Abedini, A. Plesner, P. Cao, Z. Ridgway, J. Zhang, L. H. Tu, C. T. Middleton, B. Chao, D. J. Sartori, F. Meng, H. Wang, A. G. Wong, M. T. Zanni, C. B. Verchere, D. P. Raleigh and A. M. Schmidt, eLife, 2016, 5, e12977 CrossRef PubMed.
- J. Janson, R. H. Ashley, D. Harrison, S. McIntyre and P. C. Butler, Diabetes, 1999, 48, 491–498 CrossRef CAS PubMed.
- L. Milanesi, T. Sheynis, W. Xue, E. V. Orlova, A. L. Hellewell, R. Jelinek, E. W. Hewitt, S. E. Radford and H. R. Saibil, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20455–20460 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2011, 133, 7240 CrossRef CAS PubMed.
- C. Wu and J. E. Shea, PLoS Comput. Biol., 2013, 9, e1003211 CrossRef CAS PubMed.
- M. B. Erlach, H. R. Kalbitzer, R. Winter and W. Kremer, Biophys. Chem., 2019, 254, 106239 CrossRef PubMed.
- R. P. R. Nanga, J. R. Brender, S. Vivekanandan and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2337–2342 CrossRef CAS PubMed.
- J. Cort, Z. Liu, G. Lee, S. M. Harris, K. S. Prickett, L. S. Gaeta and N. H. Andersen, Biochem. Biophys. Res. Commun., 1994, 204, 1088–1095 CrossRef CAS PubMed.
- J. D. Knight, J. A. Hebda and A. D. Miranker, Biochemistry, 2006, 45, 9496–9508 CrossRef CAS PubMed.
- M. Apostolidou, S. A. Jayasinghe and R. Langen, J. Biol. Chem., 2008, 283, 17205–17210 CrossRef CAS PubMed.
- S. Kumar and A. D. Miranker, Chem. Commun., 2013, 49, 4749–4751 RSC.
- D. C. Rodriguez Camargo, K. J. Korshavn, A. Jussupow, K. Raltchev, D. Goricanec, M. Fleisch, R. Sarkar, K. Xue, M. Aichler, G. Mettenleiter, A. K. Walch, C. Camilloni, F. Hagn, B. Reif and A. Ramamoorthy, eLife, 2017, 6, e31226 CrossRef PubMed.
- S. Luca, W. Yau, R. Leapman and R. Tycko, Biochemistry, 2007, 46, 13505–13522 CrossRef CAS PubMed.
- C. Röder, T. Kupreichyk, L. Gremer, L. U. Schäfer, K. R. Pothula, R. B. G. Ravelli, D. Willbold, W. Hoyer and G. F. Schröder, Nat. Struct. Mol. Biol., 2020, 27(7), 660–667 CrossRef PubMed.
- Q. Cao, D. R. Boyer, M. R. Sawaya, P. Ge and D. S. Eisenberg, Nat. Struct. Mol. Biol., 2020, 27, 653–659 CrossRef CAS PubMed.
- F. Weirich, L. Gremer, E. A. Mirecka, S. Schiefer, W. Hoyer and H. Heise, PLoS One, 2016, 11, e0161243 CrossRef PubMed.
- P. Arosio, T. P. J. Knowles and S. Linse, Phys. Chem. Chem. Phys., 2015, 17, 7606–7618 RSC.
- D. C. Rodriguez Camargo, S. Chia, J. Menzies, B. Mannini, G. Meisl, M. Lundqvist, C. Pohl, K. Bernfur, V. Lattanzi, J. Habchi, S. I. Cohen, T. P. J. Knowles, M. Vendruscolo and S. Linse, Front. Mol. Biosci., 2021, 8, 757425 CrossRef PubMed.
- L. Zhang and J. D. Schmit, Isr. J. Chem., 2017, 57, 738–749 CrossRef CAS PubMed.
- A. L. Serrano, J. P. Lomont, L. H. Tu, D. P. Raleigh and M. T. Zanni, J. Am. Chem. Soc., 2017, 139, 16748–16758 CrossRef CAS PubMed.
- L. Buchanan, E. Dunkelberger, H. Tran, P. Cheng, C. Chiu, P. Cao, D. Raleigh, J. J. De Pablo, J. Nowick and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19285–19290 CrossRef CAS PubMed.
- M. Maj, J. P. Lomont, K. L. Rich, A. M. Alperstein and M. T. Zanni, Chem. Sci., 2018, 9, 463–474 RSC.
- C. Betsholtz, L. Christmansson, U. Engström, F. Rorsman, V. Svensson, K. H. Johnson and P. Westermark, FEBS Lett., 1989, 251, 261–264 CrossRef CAS PubMed.
- P. Westermark, U. Engstrom, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS PubMed.
- K. Tenidis, M. Waldner, J. Bernhagen, W. Fischle, M. Bergmann, M. Weber, M. L. Merkle, W. Voelter, H. Brunner and A. Kapurniotu, J. Mol. Biol., 2000, 295, 1055–1071 CrossRef CAS PubMed.
- T. T. Ashburn, M. Auger and P. T. Lansbury Jr., J. Am. Chem. Soc., 1991, 114, 790–791 CrossRef.
- G. G. Glenner, E. D. Eanes and C. A. Wiley, Biochem. Biophys. Res. Commun., 1988, 155, 608–614 CrossRef CAS PubMed.
- Y. Kiriyama and H. Nochi, Cells, 2018, 7, 95 CrossRef PubMed.
- P. Hamm and M. Zanni, Concepts and methods of 2D infrared spectroscopy, Cambridge University Press, 2011 Search PubMed.
- C. T. Middleton, A. M. Woys, S. S. Mukherjee and M. T. Zanni, Methods, 2010, 52, 12–22 CrossRef CAS PubMed.
- L. E. Buchanan, E. B. Dunkelberger and M. T. Zanni, in Protein Folding and Misfolding, ed. H. Fabien and D. Naumann, Springer, Berlin, Heidelberg, 1st edn, 2012, ch. 9, vol. 1, pp. 217–237 Search PubMed.
- M. K. Petti, J. P. Lomont, M. Maj and M. T. Zanni, J. Phys. Chem. B, 2018, 122, 1771–1780 CrossRef CAS PubMed.
- S.-H. Shim and M. T. Zanni, Phys. Chem. Chem. Phys., 2009, 11, 748–761 RSC.
- D. B. Strasfeld, Y. L. Ling, R. Gupta, D. P. Raleigh and M. T. Zanni, J. Phys. Chem. B, 2009, 113, 15679–15691 CrossRef CAS PubMed.
- S.-H. Shim, R. Gupta, Y. L. Ling, D. B. Strasfeld, D. P. Raleigh and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6614–6619 CrossRef CAS PubMed.
- T. O. Zhang, M. Grechko, S. D. Moran and M. T. Zanni, Methods Mol. Biol., 2016, 1345, 21–41 CrossRef CAS PubMed.
- M. W. Berry, M. Browne, A. N. Langville, V. P. Pauca and R. J. Plemmons, Comput. Stat. Data Anal., 2007, 52, 155–173 CrossRef.
- W. Rock, Y.-L. Li, P. Pagano and C. M. Cheatum, J. Phys. Chem. A, 2013, 117, 6073–6083 CrossRef CAS PubMed.
- S. K. Karthick Kumar, A. Tamimi and M. D. Fayer, J. Chem. Phys., 2012, 137, 184201 CrossRef CAS PubMed.
- X. Zhang, J. R. St. Clair, E. London and D. P. Raleigh, Biochemistry, 2017, 56, 376–390 CrossRef CAS PubMed.
- T. Skotland and K. Sandvig, Nat. Commun., 2019, 10, 2752 CrossRef PubMed.
- R. Akter, P. Cao, H. Noor, Z. Ridgway, L. H. Tu, H. Wang, A. G. Wong, X. Zhang, A. Abedini, A. M. Schmidt and D. P. Raleigh, J. Diabetes Res., 2016, 2016, 2798269 Search PubMed.
- S. S. Dicke, A. M. Alperstein, K. L. Schueler, D. S. Stapleton, S. P. Simonett, C. R. Fields, F. Chalyavi, M. P. Keller, A. D. Attie and M. T. Zanni, J. Phys. Chem. B, 2021, 125, 9517–9525 CrossRef CAS PubMed.
- C. R. Fields, S. S. Dicke, M. K. Petti, M. T. Zanni and J. P. Lomont, J. Phys. Chem. Lett., 2020, 11, 6382–6388 CrossRef CAS PubMed.
- M. Grechko and M. T. Zanni, J. Chem. Phys., 2012, 137, 184202 CrossRef PubMed.
- E. B. Dunkelberger, M. Grechko and M. T. Zanni, J. Phys. Chem. B, 2015, 119, 14065–14075 CrossRef CAS PubMed.
- C. T. Middleton, P. Marek, P. Cao, C. C. Chiu, S. Singh, A. M. Woys, J. J. De Pablo, D. P. Raleigh and M. T. Zanni, Nat. Chem., 2012, 4, 355–360 CrossRef CAS PubMed.
- A. Abedini and D. P. Raleigh, J. Mol. Biol., 2006, 355, 274–281 CrossRef CAS PubMed.
- Z. Ridgway, K. H. Lee, A. Zhyvoloup, A. Wong, C. Eldrid, E. Hannaberry, K. Thalassinos, A. Abedini and D. P. Raleigh, Biophys. J., 2020, 118, 1142–1151 CrossRef CAS PubMed.
- J. S. Fortin and M. Benoit-Biancamano, Amyloid, 2015, 22, 194–202 CrossRef CAS PubMed.
- J. P. Lomont, K. L. Rich, M. Maj, J. J. Ho, J. S. Ostrander and M. T. Zanni, J. Phys. Chem. B, 2018, 122, 144–153 CrossRef CAS PubMed.
- L. E. Buchanan, M. Maj, E. B. Dunkelberger, P. N. Cheng, J. S. Nowick and M. T. Zanni, Biochemistry, 2018, 57, 6470–6478 CrossRef CAS PubMed.
- D. Raleigh, X. Zhang, B. Hastoy and A. Clark, J. Mol. Endocrinol., 2017, 59, R121–R140 CAS.
- J. A. Williamson, J. P. Loria and A. D. Miranker, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- Y. L. Ling, D. B. Strasfeld, S. H. Shim, D. P. Raleigh and M. T. Zanni, J. Phys. Chem. B, 2009, 113, 2498–2505 CrossRef CAS PubMed.
- J. Lesma, F. Bizet, C. Berardet, N. Tonali, S. Pellegrino, M. Taverna, L. Khemtémourian, J.-L. Soulier, C. van Heijenoort, F. Halgand, T. Ha-Duong, J. Kaffy and S. Ongeri, Front. Cell Dev. Biol., 2021, 9, 2531 Search PubMed.
- M. Pannuzzo, A. Raudino, D. Milardi, C. La Rosa and M. Karttunen, Sci. Rep., 2013, 3, 2781 CrossRef PubMed.
- X. Dong, Q. Qiao, Z. Qian and G. Wei, Biochim. Biophys. Acta, Biomembr., 2018, 9, 1826–1839 CrossRef PubMed.
- S. Casas, A. Novials, F. Reimann, R. Gomis and F. M. Gribble, Diabetologia, 2008, 51, 2252–2262 CrossRef CAS PubMed.
- R. P. R. Nanga, J. R. Brender, J. Xu, K. Hartman, V. Subramanian and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 8252–8261 CrossRef CAS PubMed.
- Y. Xu, R. Maya-Martinez, N. Guthertz, G. R. Heath, I. W. Manfield, A. L. Breeze, F. Sobott, R. Foster and S. E. Radford, Nat. Commun., 2022, 13, 1040 CrossRef CAS PubMed.
- L. M. Young, J. C. Saunders, R. A. Mahood, C. H. Revill, R. J. Foster, L. H. Tu, D. P. Raleigh, S. E. Radford and A. E. Ashcroft, Nat. Chem., 2015, 7, 73–81 CrossRef CAS PubMed.
- A. Nath, D. E. Schlamadinger, E. Rhoades and A. D. Miranker, Biochemistry, 2015, 54, 3555–3564 CrossRef CAS PubMed.
- S. Kumar, D. E. Schlamadinger, M. A. Brown, J. M. Dunn, B. Mercado, J. A. Hebda, I. Saraogi, E. Rhoades, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2015, 22, 369–378 CrossRef CAS PubMed.
- K. Yanagi, M. Ashizaki, H. Yagi, K. Sakurai, Y. H. Lee and Y. Goto, J. Biol. Chem., 2011, 286, 23959–23966 CrossRef CAS PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736–739 CrossRef CAS PubMed.
- J. Bella, K. L. Hindle, P. A. Mcewan and S. C. Lovell, Cell. Mol. Life Sci., 2008, 65, 2307–2333 CrossRef CAS PubMed.
- S. A. Jayasinghe and R. Langen, Biochemistry, 2005, 44, 12113–12119 CrossRef CAS PubMed.
- J. D. Knight and A. D. Miranker, J. Mol. Biol., 2004, 341, 1175–1187 CrossRef CAS PubMed.
- P. E. S. Smith, J. R. Brender and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 4470–4478 CrossRef CAS PubMed.
- D. H. J. Lopes, A. Meister, A. Gohlke, A. Hauser, A. Blume and R. Winter, Biophys. J., 2007, 93, 3132–3141 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally. |
| ‡ Michał Maj completed his postdoctoral research at University of Wisconsin-Madison. He is currently a Professor at the University of Gothenburg, Box 462, 405 30 Gothenburg, Sweden. |
| This journal is © The Royal Society of Chemistry 2022 |