
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide/protein-based macrocycles: from biological synthesis to biomedical applications
Wen-Hao
Wu†
a,
Jianwen
Guo†
bc,
Longshuai
Zhang
bc,
Wen-Bin
Zhang
 *a and
Weiping
Gao
*bc
*a and
Weiping
Gao
*bc
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry & Physics of Ministry of Education, Center for Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, P. R. China. E-mail: wenbin@pku.edu.cn
bDepartment of Geriatric Dentistry, Beijing Laboratory of Biomedical Materials, Peking University School and Hospital of Stomatology, Beijing, 100081, P. R. China. E-mail: gaoweiping@hsc.pku.edu.cn
cBiomedical Engineering Department, Peking University, Beijing, 100191, P. R. China
First published on 9th June 2022
Abstract
Living organisms have evolved cyclic or multicyclic peptides and proteins with enhanced stability and high bioactivity superior to their linear counterparts for diverse purposes. Herein, we review recent progress in applying this concept to artificial peptides and proteins to exploit the functional benefits of these macrocycles. Not only have simple cyclic forms been prepared, numerous macrocycle variants, such as knots and links, have also been developed. The chemical tools and synthetic strategies are summarized for the biological synthesis of these macrocycles, demonstrating it as a powerful alternative to chemical synthesis. Its further application to therapeutic peptides/proteins has led to biomedicines with profoundly improved pharmaceutical performances. Finally, we present our perspectives on the field and its future developments.
Introduction
Cyclization is a simple modification of a linear polymer chain by intramolecular chemical ligation, forming macrocycles with certain changes in properties.1–4 Depending on the sites of ring closure (head-to-tail vs. side-chain ligation), macrocycles may exist in different types. The categorization and variation of macrocycles is a subject of study in chemical topology which concerns the invariant properties of a molecule during continuous deformations such as bending, twisting, and stretching, or simply put, the connectivity and spatial relationship of a molecule.5–12 To illustrate this concept, we take the molecular graphs of three macrocycle variants shown in Fig. 1A as an example.8,13 Graph I has a connectivity completely different from the other two. Although Graph II and III have identical connectivity, the latter has an additional entwined spatial relationship in 3D space. As a result, all of them have distinct topology from each other. To provide a broad coverage of complex cyclic structures, the concept of macrocycles is stretched in this review to cover a broad range of cyclic variants with different topologies as shown in Fig. 1B. Specifically, when cyclization occurs on non-entangling polymer chains, (multi)cyclic and cyclic-branched macrocycle variants are obtained; when cyclization occurs on knotted or entangled precursors, macrocyclic knots and links with nonplanar molecular graphs (molecular graphs which could only be embedded in 3D space) are usually obtained where the molecular segments are mechanically interlocked (i.e., there is an entanglement between molecular entities such that they cannot be separated without breaking or distorting chemical bonds). In both cases, cyclization plays a vital role in defining connectivity and preventing the disentanglement of polymer chains, which could help reshape the conformational space of the polymer and endow properties distinct from its linear counterpart. In peptides and proteins, cyclization exerts significantly more conformation restriction on the unfolded state rather than the folded state, thereby increasing the stability of the folded state.14–16 Eliminating the free termini of polypeptide chains also reduces the possibility of enzymatic degradation by exopeptidases.17–19 The effects are perhaps even more significant for those with nonplanar molecular graphs, where tight coupling between molecular segments and dynamic transition between different states are anticipated.20,21 Therefore, over the past decades, peptide/protein-based macrocycles have emerged as a unique class of macromolecular therapeutics.22–30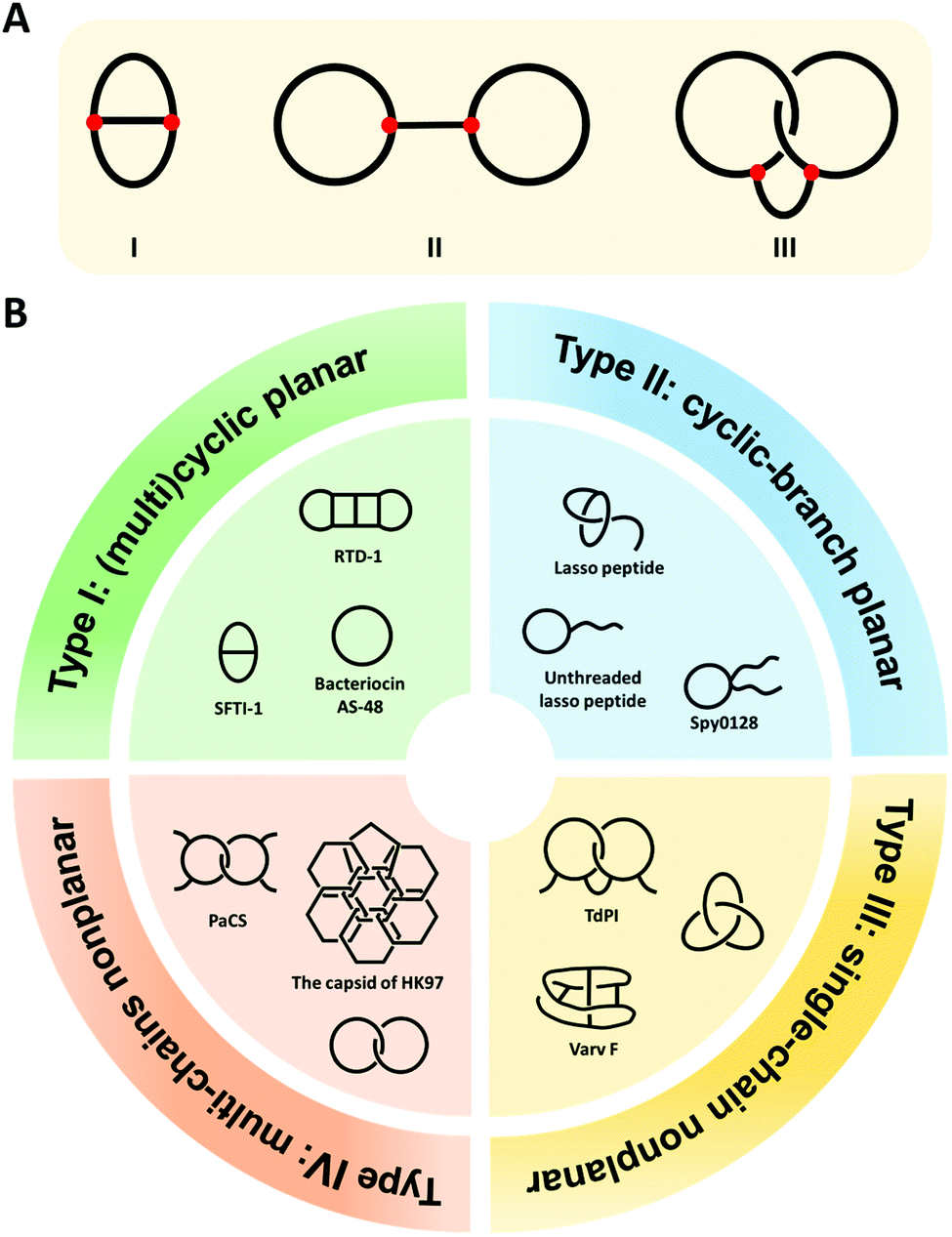 | ||
| Fig. 1 (A) The molecular graphs of three macrocycles with different chemical topologies. The line represents a polymer chain with a red vertex being a branch point. (B) General illustration and typical examples of the four categories of the naturally occurring peptide/protein-based macrocycles with distinct topologies. Type I: sunflower trypsin inhibitor 1 (SFTI-1) with planar θ-curve topology, rhesus-θ defensin-1 (RTD-1) with multicyclic topology,51 and bacteriocin AS-48 with pure cyclic topology; Type II: lasso peptide and Spy0128 domain with cyclic-branch topologies; Type III: cyclotide varv F with K3,3 graph topology, tick-derived protease inhibitor (TdPI) with interchain linked topology,52 and trefoil knot; Type IV: Pyrobaculum aerophilum citrate synthase (PaCS) with [2]catenane or Hopf link topology and the capsid of HK97 with “chain-mail” topology. | ||
Peptide/protein macrocycles are not uncommon in nature. Although nascent proteins are strictly linear due to the template polymerization mechanism of ribosomal synthesis,31,32 living organisms have evolved diverse cyclic peptides/proteins via post-translational processing to combat diseases or as part of their defense mechanisms. The most common form is simple macrocycles formed by seamlessly linking the N- and C-termini after expression.33 Up to now, more than 1400 sequences of naturally occurring simple cyclic peptides/proteins have been documented.34,35 The largest group of cyclic peptides were discovered from plants, named cyclotides.36–38 Lasso peptides are perhaps the second largest group with a cyclic peptide threaded by its tail (Fig. 1B).39,40 The disulfide bond formation leads to even more complex topologies, such as cysteine knots and ladders (embedded rings that are threaded by the disulfide bonds)33,41 and mechanically interlocked, multicyclic protein links (Fig. 1B).20 Occasionally, the cyclic structures can also be fixed by other covalent linkages, such as thioester, ester, or isopeptide bonds. The beautiful molecular ‘chain-mail’ structure of bacteriophage HK97 capsid is composed of mechanically interlocked protein rings that are covalently closed by isopeptide bonds (Fig. 1B).42–44 While their cyclization mechanism differs from case to case, it seems that folding and assembly are all critical in pre-organizing the protein precursors for ring closure.35,40,43,45,46 Cyclization confines the conformational space of peptides/proteins and thus, imparts diverse functions while enhancing their stability.33,35 For example, cyclotide Kalata B1 exhibits cytotoxicity, anti-microbial, and anti-virus activities, as well as excellent proteolytic resistance.33,47,48 A larger cyclic bacteriocin AS-48, isolated from Enterococcus faecalis S-48, has a remarkably high melting temperature (Tm) of 93 °C (Fig. 1B).49,50 The protein catenane Pyrobaculum aerophilum citrate synthase (PaCS) displays a Tm about 10 °C higher than the linear control without disulfide bonds (Fig. 1B).20 The HK97 capsid can even tolerate up to 5 M of guanidinium hydrochloride.42,43
Inspired by the functional benefits observed in these naturally occurring peptide/protein-based macrocycles, much effort has been directed to synthesizing artificial peptide/protein-based macrocycles as well as their variants. While nature evolves these compounds for optimal fitness, artificial systems can fully unleash the power of cyclization in engineering their properties. To date, there has been considerable success in their syntheses, with a rapidly expanded toolbox of broad ranges of efficient technologies for chemical cyclization.53–60 In this review, we shall focus on their biological synthesis and biomedical applications. Unlike organic synthesis, biosynthesis relies on the cellular machinery and is often genetically encoded to allow programming of the products’ structures. It is usually highly efficient, specific, and capable of generating considerable complexity with high bioactivity both in vitro and in vivo.59,60 By relating genotype to phenotype, biosynthesis also provides a facile approach for the rapid discovery of bioactive leads.61–63 The chemical space could be even further expanded by incorporating non-canonical amino acids.64–66 The biosynthesis of diverse peptide/protein-based macrocycles and their variants thus provides a rich resource for exploring their biomedical applications.
Overview of peptide/protein-based macrocycles
As shown in Fig. 1B, we categorize peptide/protein-based macrocycles into four types. On one hand, (multi)cyclic and cyclic-branched macrocycles with planar molecular graphs (molecular graphs which enable to be embedded in 2D space) are classified as Type I and Type II. The former results from main-chain cyclization (such as the protein bacteriocin AS-48) and the latter has at least one terminal of the chain remaining (such as the Spy0128 domain originated from Streptococcus pyogenes67). For example, sunflower trypsin inhibitor 1 (SFTI-1) belongs to Type I and possesses a θ-curve topology which is an embedding of the Greek letter θ in the plane.68 Lasso peptides and proteins usually belong to Type II. On the other hand, macrocycles with nonplanar molecular graphs are classified either as Type III (single-chain) or Type IV (multi-chain). Their formation could arise from multiple covalent linkages.41 For example, cyclotide varv F has been found to possess a K3,3 graph (a prototypical nonplanar graph that cannot be embedded in the plane in such a way that its edges intersect only at their end points).69 When folding and assembly are involved, highly complex structures, such as knots and links, emerge. Knots are the embeddings of a circle in 3D space. Although proteins are typically linear, knotted proteins do exist with entanglements upon folding. In nature, the topologies of such macrocycles are often further reinforced by disulfide bonds from cysteine residues. Sulkowska and coworkers70,71 have systematically looked into this problem and identified many nonplanar topologies in the protein data bank. Catenanes or links comprise two or more mechanically interlocked rings, while [2]catenane or Hopf link consists of two rings linked together exactly once. The existence of link-form macrocycles is rare in nature. Pyrobaculum aerophilum citrate synthase (PaCS) is a branched catenane since the disulfide bond only closes a relatively small ring. The capsid of HK97 possesses a “chain-mail” topology which is composed of multiple mechanically interlocked rings.42–44 In view of all these macrocycle variants, it is apparent that chemical ligation tools are of primary importance owing to their determinant role in ring closure, whereas folding and assembly further increase the overall structural complexity. Below, we summarize various ligation tools for cyclization and then present “assembly-reaction” synergy as an effective strategy for creating diverse peptide/protein-based macrocycle variants.Chemical tools for main-chain cyclization
Main-chain cyclization in biological synthesis relies either on genetically encoded protein ligation chemistry or enzymatic ligation chemistry with high sequence specificity. The former includes intein-mediated protein splicing or split-intein-mediated trans-splicing reactions. The latter involves many enzymes such as Sortase A, Butelase 1, OaAEP, etc.Expressed protein ligation is a strategy for protein ligation or cyclization based on an intein-mediated splicing reaction.72 It was first reported independently by Muir73 and Xu74 groups in 1998. The intein-bearing protein precursor generates a C-terminal α-thioester group via S/O acyl transfer, which then undergoes reversible trans(thio)esterification with the N-terminal residue (Cys, Ser, or Thr) of another protein precursor to form a branched oxy(thio)ester intermediate. After succinimide formation and subsequent S/O to N shift, the intermediate converts to a conjugate ligated with a native peptide bond.75 The process is similar to that of native chemical ligation,76 but both precursors are produced by recombinant technique, not chemical synthesis. It has been successfully employed for protein backbone cyclization where the reactive Cys and intein are appended at the N- and C-termini of the target protein, respectively (Fig. 2A). The first case was reported by Camarero and Muir77 on the cyclization of Src homology 3 domain from the c-Crk adaptor protein which increases its ligand binding affinity by 6-folds. Afterward, a series of peptides (such as a brain-binding peptide, complementarity determining region H3/C2)78 and proteins (such as β-lactamase (BLA),79 thioredoxin, maltose binding protein74) were cyclized by the same method. More interestingly, it could also be applied in living cells.80
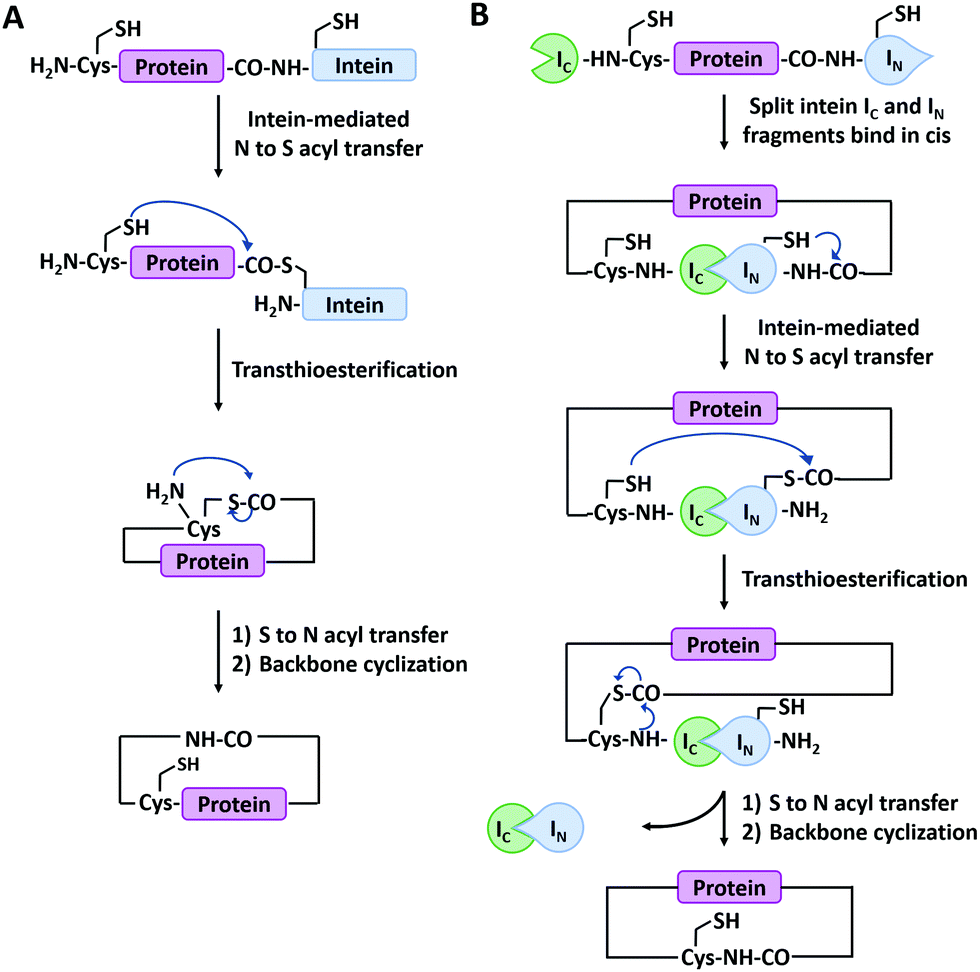 | ||
| Fig. 2 Schematic illustration of protein head-to-tail cyclization by expressed protein ligation (A), and split-intein mediated reaction (B). | ||
Split-intein-mediated ligation takes advantage of the fact that intein domains may be split into two parts, named N-intein (IN) and C-intein (IC), which can spontaneously reconstitute to form a native peptide bond linkage between their fusion proteins while excising themselves off the fusion.81,82 This protein splicing event can be used for protein cyclization (Fig. 2B). Benkovic and co-workers83 first used split Ssp inteins to produce cyclic peptides and proteins by fusing IN and IC at the C- and N-termini of target protein, respectively. The obtained circular dihydrofolate reductase (DHFR) exhibited improved thermal and proteolytic stability over its linear counterpart. It has later been used on other proteins as well, including green fluorescence protein (GFP),84 xylanase,85 and a peptide HIV inhibitor.86 Notably, this strategy is also applicable in cultured cells. For example, Camarero and co-workers87 synthesized cyclotide MCoTI-I by both expressed protein ligation and split-intein-mediated ligation, the latter of which outperformed the former in the in vivo expression. Besides Ssp inteins, a lot of split inteins with high reactive activity and robust extein tolerance have been developed by genomic sequencing and rational engineering (e.g., gp41-1, gp41-8, NrdJ-1, and IMPDH-1).72 Among them, a split intein derived from Nostoc puntiforme PCC73102 (Npu), which possesses robust trans-splicing activity with an efficiency of 98%, has been widely used.88 By varying the positions of split sites in Npu intein, two mutually orthogonal split intein pairs (102 residues for IN1 and 36 residues for IC1; 15 residues for IN2 and 123 residues for IC2) have been developed, which greatly enhances our capability to synthesize complex protein macrocycle variants.89,90
Sortase A is a kind of transpeptidase enzyme that could anchor proteins onto the cell wall surface. It was first reported by Schneewind and co-workers91 in 1999 and has received wide applications since then. It usually recognizes a pentapeptide sequence of LPXTG (X represents any amino acid) at the C-terminus and an oligoglycine sequence at the N-terminus of the substrate and catalyzes the cleavage of the amide bond between T and G to generate an acyl-enzyme thioester intermediate, which was subsequently attacked by the amine group at the N-terminal oligoglycines for ligation or cyclization (Fig. 3A).92 Boder and co-workers first used Sortase A to cyclize a bifunctional precursor protein, Gly3-GFP-LPETG-His6.93 The final products were characterized as a mixture of both monomer and dimer. The more specific cyclization of GFP was then realized by Ploegh group94 and Gao group23 and further extended to biomedical applications. Since then, a series of cyclic proteins, such as histatin-195 and interferon-α (IFN-α),22,24,25 were produced by this method. These cyclic peptides and proteins showed enhanced stability and improved biological activity compared with their linear counterparts. Nowadays, Sortase A has been engineered to be independent of Ca2+ and capable of recognizing different pentapeptide sequences, greatly enriching this toolbox.75 A Ca2+-independent Sortase A discovered from Streptococcus pyogenes has been successfully applied to cyclize GFP in vivo.96,97 Compared with intein-mediated cyclization, Sortase A requires fewer recognition sequences in the precursor protein. However, its application in vivo is quite limited due to the complication of intracellular nucleophiles such as the ε-amino group of lysine.98 Sortase A also suffers from low turnover rates (the molar ratio of the enzyme to protein substrate is usually from 0.1 to 1) and undesirable reverse reaction.94 It thus requires a lot of enzymes and a large excess of one substrate to promote the reaction, which is not ideal for protein cyclization.
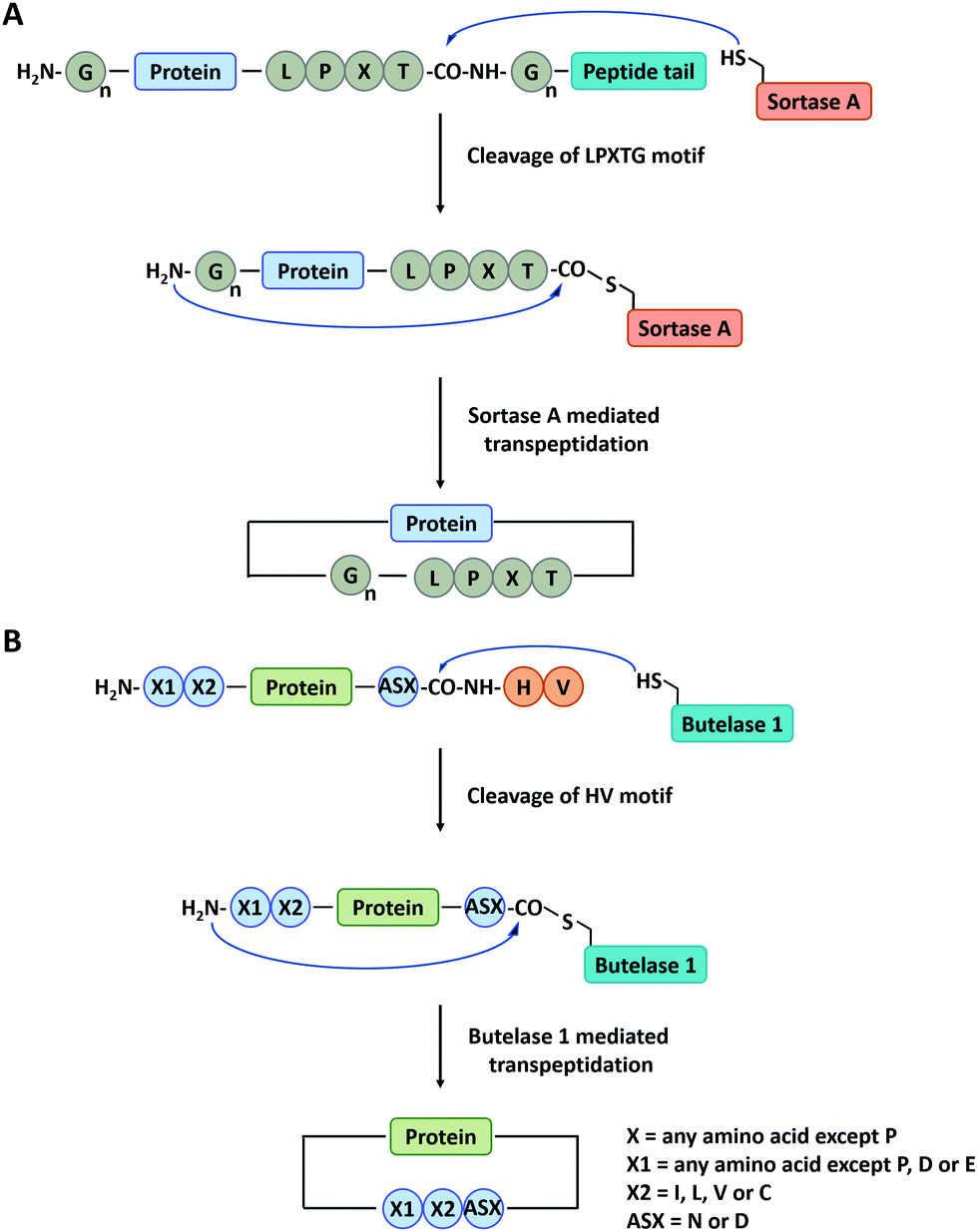 | ||
| Fig. 3 Schematic illustration of protein head-to-tail cyclization by Sortase A mediated reaction (A), and Butelase 1 mediated reaction (B). | ||
Apart from Sortase A, other enzymes have also been developed to produce peptide/protein-based macrocycles. The most representative case is a class of peptide ligases in the asparaginyl endoproteases (AEP) family, which requires very short recognition motifs to ligate a range of targets.99 Butelase 1 is the first AEP peptide ligase purified from the cyclotide-producing plant Clitoria ternatea.100,101 This enzyme recognizes the C-terminal D/N-HV sequence of polypeptide precursor and catalyzes the cleavage of the amide bond between the D/N and H to generate an acyl-enzyme intermediate. Then, target cyclization proceeds via ligation of the D/N residue to the N-terminal amino acid residue (Fig. 3B).102,103 Tam and co-workers102 have constructed cyclic GFP, human growth hormone (hGH), and interleukin-1 receptor antagonist via Butelase 1 mediated ligation. Compared with Sortase A, Butelase 1 shows exceptionally high efficiency (>90% yield, low enzyme to substrate molar ratio: 0.001 to 0.01, and short reaction time) and better tolerance to recognition amino acids (almost all amino acids at the N-terminus).102,104 However, until now, Butelase 1 still cannot be recombinantly expressed in an active form on a large scale, which limits its availability and wide use.104
OaAEP1b is another kind of AEP peptide ligase isolated from the plant Oldenlandia affinis with a similar catalytic mechanism to that of Butelase 1 but different recognition sequences at the C-terminus of protein precursor (NGL, NAL, or NCL).105,106 Despite usually lower catalytic efficiencies than Butelase 1, OaAEP1b could be expressed in E. coli and activated under acidic conditions.107 Based on the structure-based mutagenesis of OaAEP1b, Wu and co-workers107 engineered an OaAEP1b variant which shows hundreds of times faster catalytic kinetics than the wild-type. This variant was demonstrated to be highly efficient for the ligation and cyclization of peptides and proteins. By now, a lot of peptide/protein-based macrocycles have been synthesized by OaAEP1b, including GFP and its variants,108 intrinsically disordered malarial vaccine candidate Plasmodium falciparum merozoite surface protein 2,109 and several cyclotides (MCoTI-II, Kalata B1, a kallikrein-related peptidase 5 inhibitor based on the SFTI-1 scaffold and a potent α-conotoxin from Conus victoriae).105,110,111
The natural translation process in ribosome is limited to 20 canonical amino acid building blocks, however, genetic code reprogramming (GCR) provides a new opportunity to incorporate hundreds of non-canonical amino acids, especially side chain-substituted amino acids, into the sequence of peptides/proteins. In general, GCR is based on the “mis-acylation” of tRNA molecules with non-canonical amino acids for subsequent incorporation into polypeptide chains during translation.112 Suga and co-workers113 have shown that GCR could generate backbone-cyclized polypeptides during ribosomal synthesis using recombinant elements for peptides synthesis (Fig. 4). They introduced a Cys-Pro-glycolic acid (C-P-HOG) sequence into the peptide chain via GCR, which could self-rearrange to form a diketopiperazine–thioester intermediate, and then react with an N-terminal Cys residue in the same way as native chemical ligation. This elegant strategy not only enables the synthesis of several naturally occurring backbone-cyclized peptides, such as eptidemnamide, scleramide, RTD-1, and SFTI-1 but also provided another option for the generation of cyclic peptide libraries and screening of such libraries in vitro to identify potential lead drugs. The involvement of non-canonical amino acids often leads to lower production yields. Once a target is identified, the short cyclic peptide may also be synthesized using solid-phase peptide synthesis, obviating the problem of scale-up in the recombinant ribosomal synthesis. It should be noted that since GCR typically needs flexizyme-mediated tRNA acylation,114 it is challenging to use this system in cells for making large-sized protein molecules.
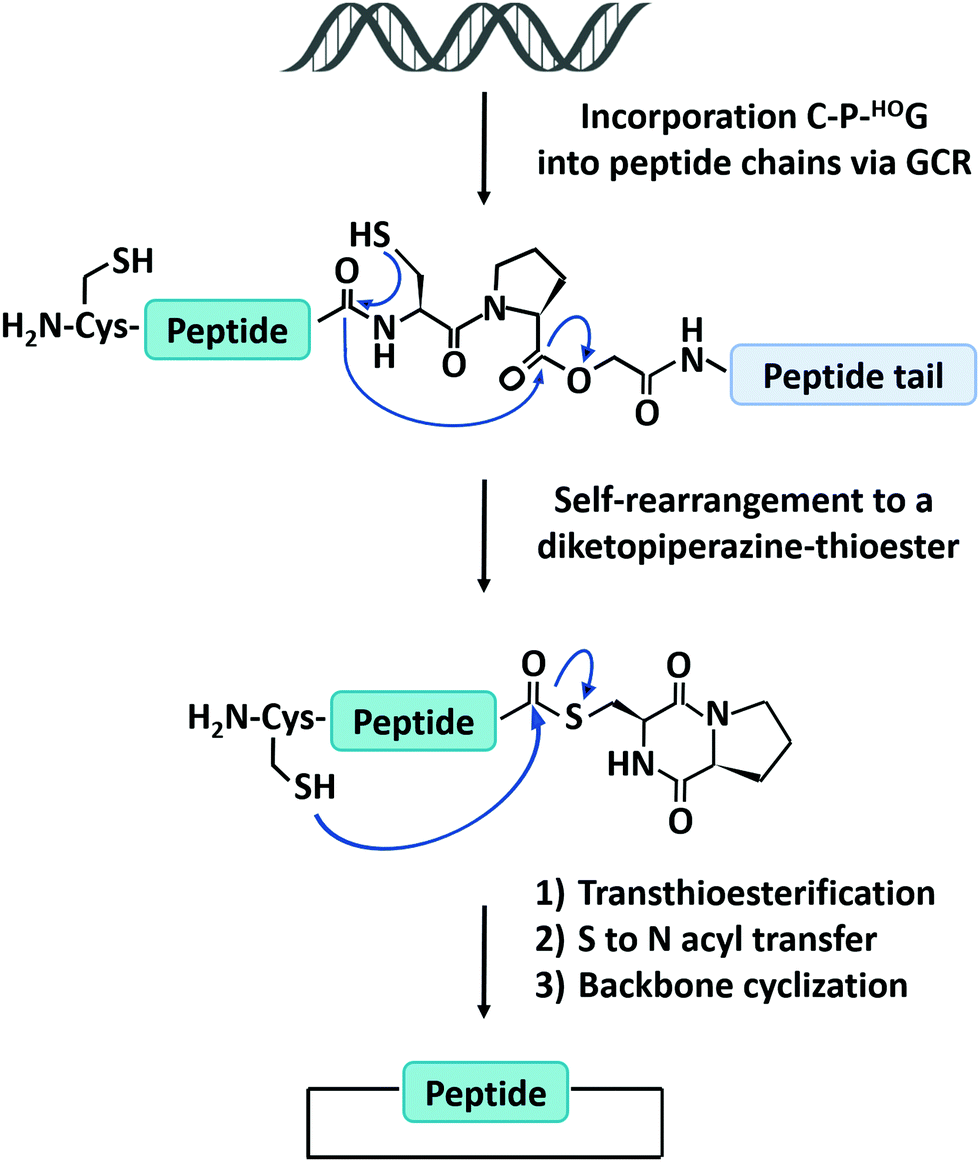 | ||
| Fig. 4 Schematic illustration of the head-to-tail cyclization of peptides by genetic code reprogramming. | ||
Chemical tools for side-chain cyclization
In nature, there are many types of side-chain covalent linkages, such as disulfide bond, isopeptide bond, or ester/thioester bond.67,115–118 Despite wide existence in natural proteins, the disulfide bond is unstable under reducing conditions. Thus, we mainly focus on other more stable covalent linkages, such as isopeptide bonds which is an amide bond formed between the side chains of amino acids like Lys and Asn/Asp.115,119 It has commonly been found in microbial protein domains, such as the Spy0128 and fibronectin-binding protein (FbaB) from Streptococcus pyogenes.67,115,119 Notably, these protein domains can be split to develop chemical reaction tools and have been widely used.118,120–124Howarth and co-workers125 first dissected Spy0128 into two types of peptide/protein reactive pairs, termed as pilin-C (residues 18–229)/isopeptag (16 amino acids at the C-terminal) and pilin-N/isopeptag-N. The two parts of each reactive pair can reconstitute and spontaneously form an isopeptide bond irreversibly between Lys and Asn via an autocatalysis process. However, the inefficient reaction (60% yield) and relatively large molecular weight (∼35 kDa) limit their application. Howarth and co-workers126 then divided the CnaB2 domain (the second immunoglobulin-like collagen adhesin domain)119 derived from FbaB into SpyTag (the C-terminal β-strand composed of 13 amino acid residues) and SpyCatcher (138 residues at the N-terminal), giving rise to another peptide/protein reactive pair. The SpyTag/SpyCatcher chemistry shows high reactivity with a second-order rate constant (k) of ∼1.4 × 103 M−1 s−1. It was further optimized into the second and third generations with exceptional reactivity via rational engineering and directed evolution (k ∼ 2.0 × 105 M−1 s−1 for SpyCatcher002 and k ∼ 5.5 × 105 M−1 s−1 for SpyCatcher003).127,128 Meanwhile, the reaction is broadly applicable both in vitro and in cellulo. As a supplement, the SnoopTag/SnoopCatcher pair was also created by splitting the D4 domain of RrgA adhesin from Streptococcus pneumoniae, which shows comparable efficiency but mutually orthogonal reactivity to SpyTag/SpyCatcher ligation.129 Up to now, many peptide/protein reactive pairs have been developed, constituting a rich toolbox for side-chain ligation.118
Compared to main-chain ligation, the genetically encoded peptide/protein reactive pairs, such as SpyTag/SpyCatcher chemistry, are advantageous in producing cyclic proteins and their variants. First, the reaction is highly efficient and robust. Second, since tag and catcher react not only at the termini but also inside the protein chain, they can enable domain-selective cyclization with intact N-and C-termini for further modification.130 For example, by genetically fusing SpyTag and SpyCatcher to the N- and C-termini of the target protein, Howarth and co-workers131,132 successfully realized the side-chain cyclization of a series of enzymes, including BLA, DHFR, and phytase, leading to the cyclic-branched structures as we categorized in Type II (Fig. 5). These enzymes exhibit remarkably enhanced thermal stability in many regards: (1) improved anti-aggregation behavior; (2) excellent thermal resilience, tending to refold and restore the catalytic activity better. These functional benefits are presumably attributed to the cyclic-branched topology as well as the extraordinary stability of the SpyTag/SpyCatcher complex. Besides, other proteins such as firefly luciferase133 and lichenase,134 have also been cyclized by SpyTag/SpyCatcher chemistry to improve their performance in bioactivity and stability.
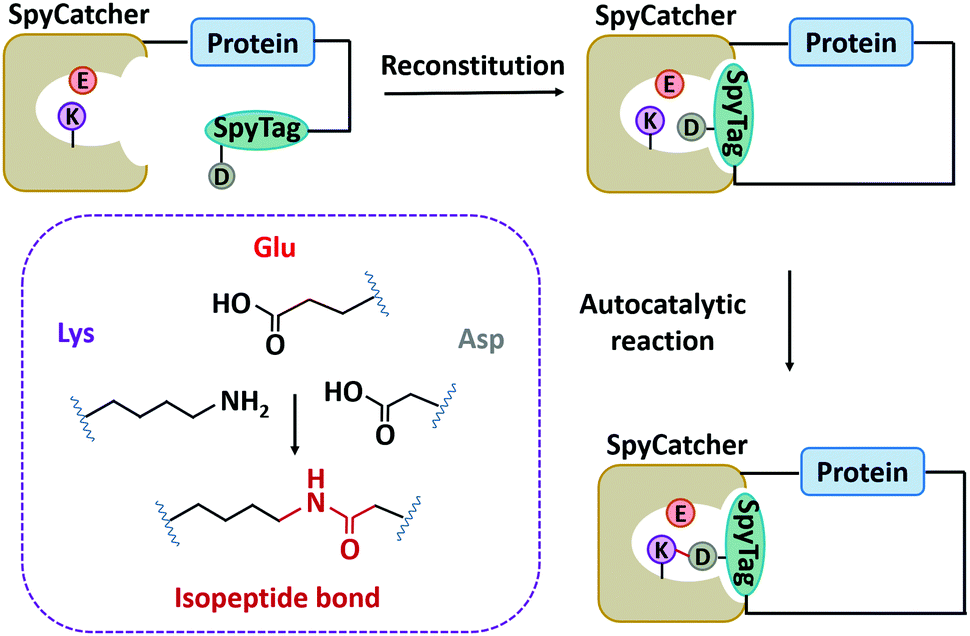 | ||
| Fig. 5 Schematic illustration of protein side-chain cyclization by SpyTag/SpyCatcher chemistry. | ||
Although SpyTag/SpyCatcher chemistry has been successful for biologically inspired synthesis of protein macrocycles, especially those with complex topological structures,130 it is not traceless. The large SpyTag/SpyCatcher complex (∼15 kDa) after cyclization might unexpectedly alter the structure and function of the original protein due to the increased steric hindrance and potential immunogenicity. To reduce the size of the ligation scar, Howarth and co-workers135,136 have developed “SpyTag/KTag/SpyLigase” and “SnoopTagJr/DogTag/SnoopLigase” systems by further splitting the SpyTag/SpyCatcher and SnoopTag/SnoopCatcher pairs into three parts. The SpyLigase and SnoopLigase catalyze the reaction of SpyTag/KTag and SnoopTagJr/DogTag, respectively, forming an isopeptide bond with a scar of only 5–6 kDa molecular weight. Recently, Zhang and co-workers137 have found an alternative way to split SpyCatcher, leading to BDTag (25 amino acids), and SpyStapler (∼8 kDa). SpyStapler can efficiently catalyze the isopeptide bonding between SpyTag and BDTag. This tool has later been adapted to develop an active-template synthesis of protein catenanes.138 The small size of fused tags and higher coupling efficiency make these tools attractive for researchers in the field of chemical biology, synthetic biology, protein engineering, and biomaterials.
“Assembly-reaction” synergy
Compared with Type I and Type II macrocycles, Type III or Type IV macrocycles possess nonplanar molecular graphs and potentially remarkable functional benefits, such as enhanced thermal/chemical stability and proteolytic resistance. To address the challenge in their syntheses, the “assembly-reaction” synergy has been developed, which utilizes entwined protein domains as a template to guide the chain entanglement and combines with genetically encoded ligation chemistry to provide a module way for producing diverse topological peptide/protein macrocycles (knots, links, branched links, etc.).139–141Recently, Iwaï and co-workers142 have reported the production of a mathematical protein trefoil knot with closed ends. The trefoil knot is the simplest knot that could not be embedded in the plane. A deeply trefoil-knotted YibK from Pseudomonas aeruginosa was cyclized by split intein and Sortase A, respectively, to generate the trefoil knot exhibiting increased thermal stability and reduced aggregation propensity (Fig. 6). In comparison, protein catenanes contain two or more mechanically interlocked rings with higher structural complexity. Based on the “assembly-reaction” synergy, Zhang and co-workers143,144 have synthesized a series of protein catenanes using the entwined p53dim homodimerization domain57 and SpyTag/SpyCatcher chemistry in cellulo. The expressed nascent protein precursors first pre-organize into an entangled intermediate assembly as guided by p53dim followed by ring closure through SpyTag/SpyCatcher reaction to lock in the catenane topology (Fig. 7A).143
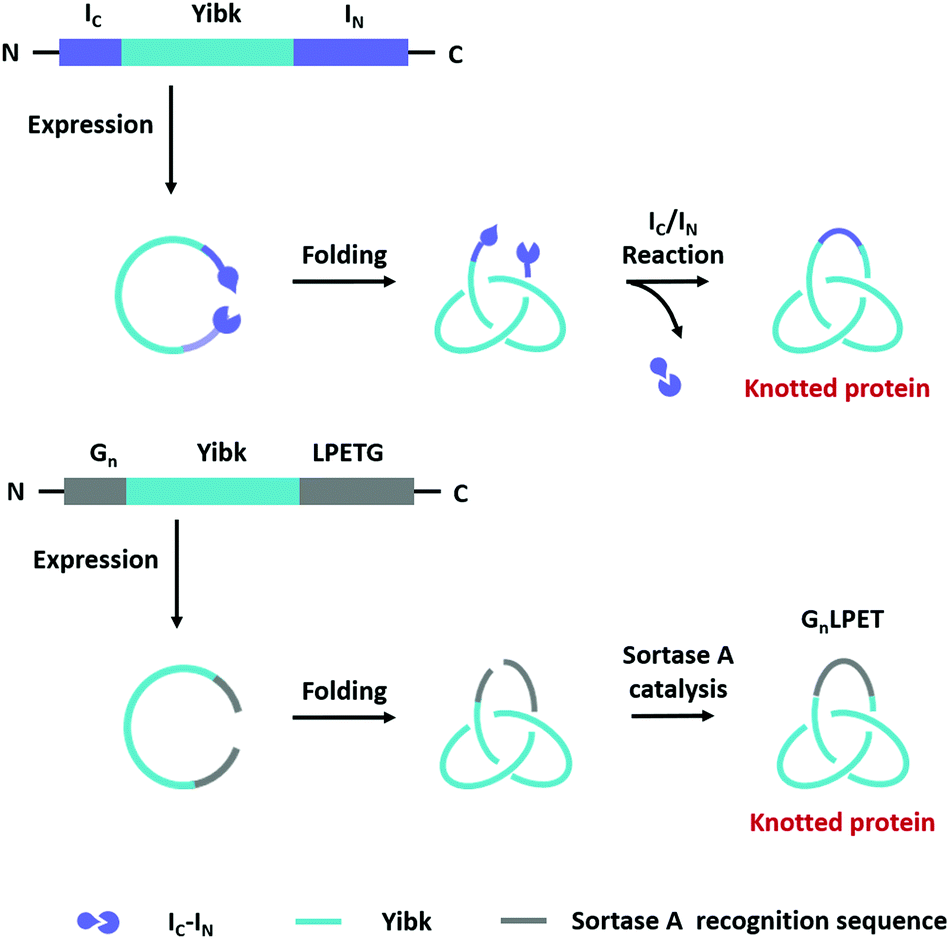 | ||
| Fig. 6 Schematic illustration of biosynthesis processes of protein knots. | ||
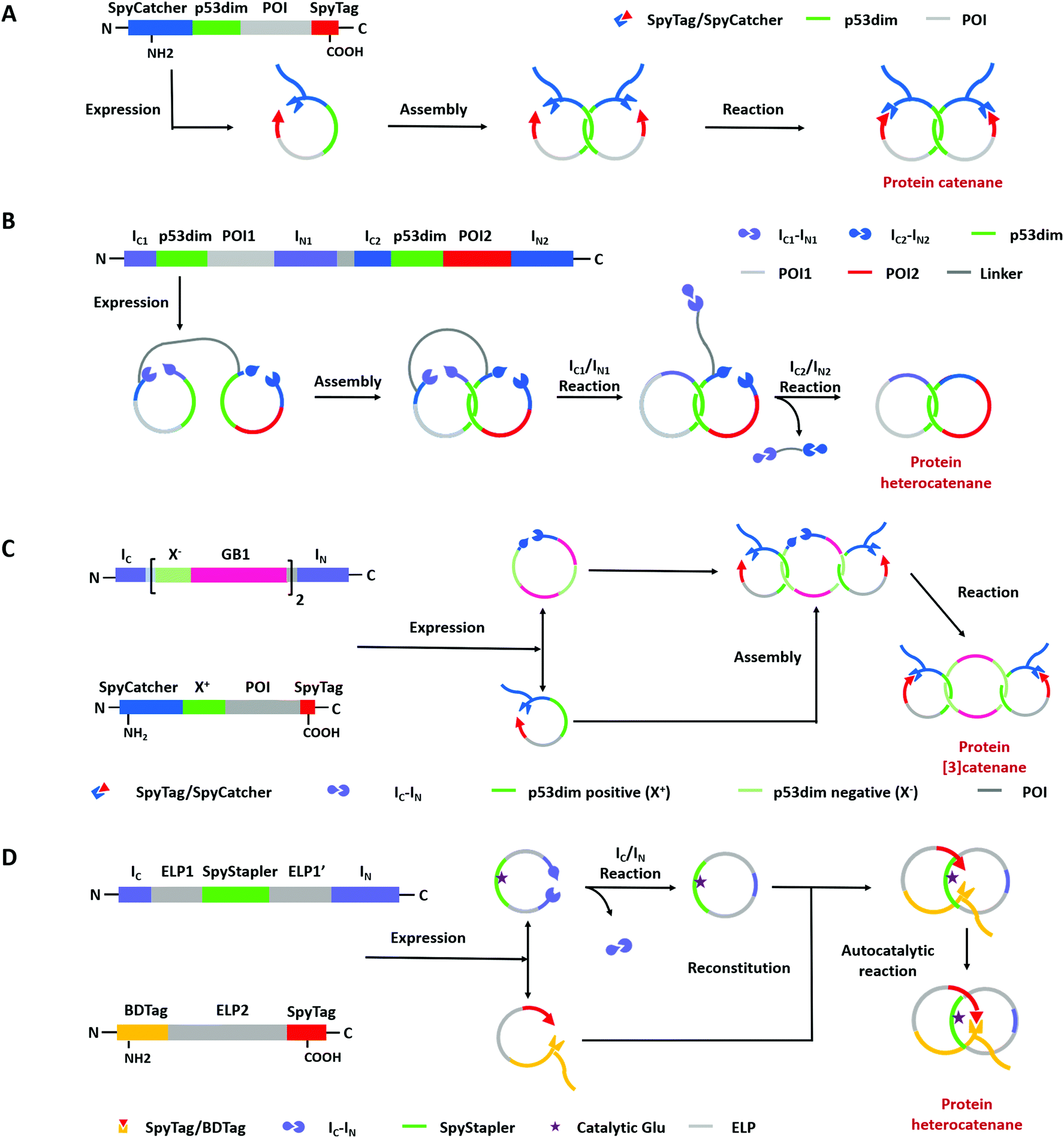 | ||
| Fig. 7 Schematic illustration of biosynthesis processes of protein links via “assembly-reaction” synergy: (A) protein homocatenanes; (B) main-chain cyclized protein heterocatenane by streamlined synthesis approach; (C) protein [3]catenane; (D) branched protein heterocatenane based on the active template strategy (POI is short for the protein of interest). | ||
A lot of proteins, including unstructured ELP and well-folded GFP, BLA, or DHFR have been concatenated by this modular approach.143,144 Due to the domain-selective feature and intact termini of SpyTag/SpyCatcher, this strategy could be extended to synthesize star-like protein catenanes and protein pretzelanes.145,146 Moreover, lasso proteins, though not included in the nonplanar molecular graphs, could also be produced by a similar strategy. The precursor protein SpyCatcher-p53dim-SpyTag-p53dim would form the cyclic-branched structure with the tail threaded in the ring due to “assembly-reaction” synergy.147 By replacing the peptide/protein reactive pair with orthogonal split intein chemistry, Zhang and co-workers148 recently developed an autonomous streamlined synthesis of traceless protein heterocatenanes with two distinct rings. With a sequence of multiple post-translational processes including p53dim guided chain interlacing and orthogonal intein-mediated ring closure, the nascent linear chain could be transformed into a main-chain heterocatenane directly in cells (Fig. 7B). The topological diversity has been further expanded by using an engineered p53dim heterodimer. Protein [3]catenane and [4]catenane composed of three or four interlocked rings were successfully synthesized with the combination of p53dim heterodimer for controlled multi-chain intertwining and mutually orthogonal split-intein-mediated ligation and SpyCatcher/SpyTag reaction for closing the primary and secondary rings, respectively (Fig. 7C).149 Recently, by rewiring the connectivity of the SpyTag/BDTag/SpyStapler complex in 3D space, an active template strategy was developed to enable selective protein heterocatenane synthesis. The chain entanglement was introduced by folding and the reconstituted complex served to catalyze the isopeptide bond formation for ring closure, leading to the heterocatenane topology (Fig. 7D).138,150 Although still in its infancy, the biosynthesis of non-planar protein macrocycles, such as protein knots and links, is promoted by the development of genetically encoded protein entangling motifs and ligation tools. These macrocycles with nonplanar graphs often exhibit extra stability and other functional benefits, which holds great promise in protein therapeutics.
Biomedical applications of peptide macrocycles
Cyclization has been demonstrated as a simple yet effective strategy to develop therapeutic peptides, particularly as inhibitors for a wide range of protein targets.28–30,151–153 The framework of cyclic peptides imparts defined and pre-organized conformations and enables highly specific target engagement through reducing the entropic loss upon binding.154,155 Meanwhile, the absence of termini in cyclic peptides often leads to enhanced proteolytic resistance, which allows them to exert functions in harsh conditions.33,35,37 Recently, the genotype–phenotype linkage of peptide macrocycles has facilitated drug discovery via rapid preparation and high-throughput screening of libraries consisted of hundreds of millions of peptide macrocycles with low cost.61,62,156–158 Examples include phage display,159 split-intein circular ligations of peptides and proteins (SICLOPPS),160 and mRNA display.161 In phage display, the cyclization is usually achieved by disulfide bond formation.62 Here, we focus on the progress in the other two directions which utilizes more stable covalent linkages for cyclization.SICLOPPS is a method for the high-throughput screening of macrocyclic peptide variants produced by intracellular expression and split intein mediated cyclization (Fig. 8A).160,162 It facilitates the construction of libraries comprised of up to about 108 sequences and the screening process can be performed both in prokaryotes and in eukaryotes.163 For example, in order to evolve peptide macrocycles to suppress the toxicity of human α-synuclein, a presynaptic neuronal protein relevant to Parkinson's disease,164,165 Lindquist and co-workers26 constructed a library containing more than 30 million cyclic peptide octamers via intein-mediated protein trans-splicing. After selection on a series of filtering assays in a yeast synucleinopathy model, two peptide macrocycles were picked and confirmed to possess reproducible and specific activities with suppressed toxicity. These selected peptide macrocycles were introduced into C. elegans Parkinson's disease model and found to significantly reduce dopaminergic neurodegeneration. Another example aimed to engineer peptide macrocycles to regulate the interactions between the heterodimeric hypoxia-inducible factor (HIF)-1α and -1β, which is crucial for cancer therapy.166,167 Tavassoli and co-workers27 constructed a library containing more than 3.2 million cyclic hexapeptides by SICLOPPS and screened out active peptide macrocycles for the inhibition of HIF-1 heterodimerization. One of the selected peptide macrocycles, named cyclo-CLLFVY, was observed as a good inhibitor to bind with the PAS-B domain (amino acid residues 235–350) of HIF-1α. These examples demonstrate the potential of SICLOPPS in developing highly active macrocyclic peptide drugs for disease treatments.
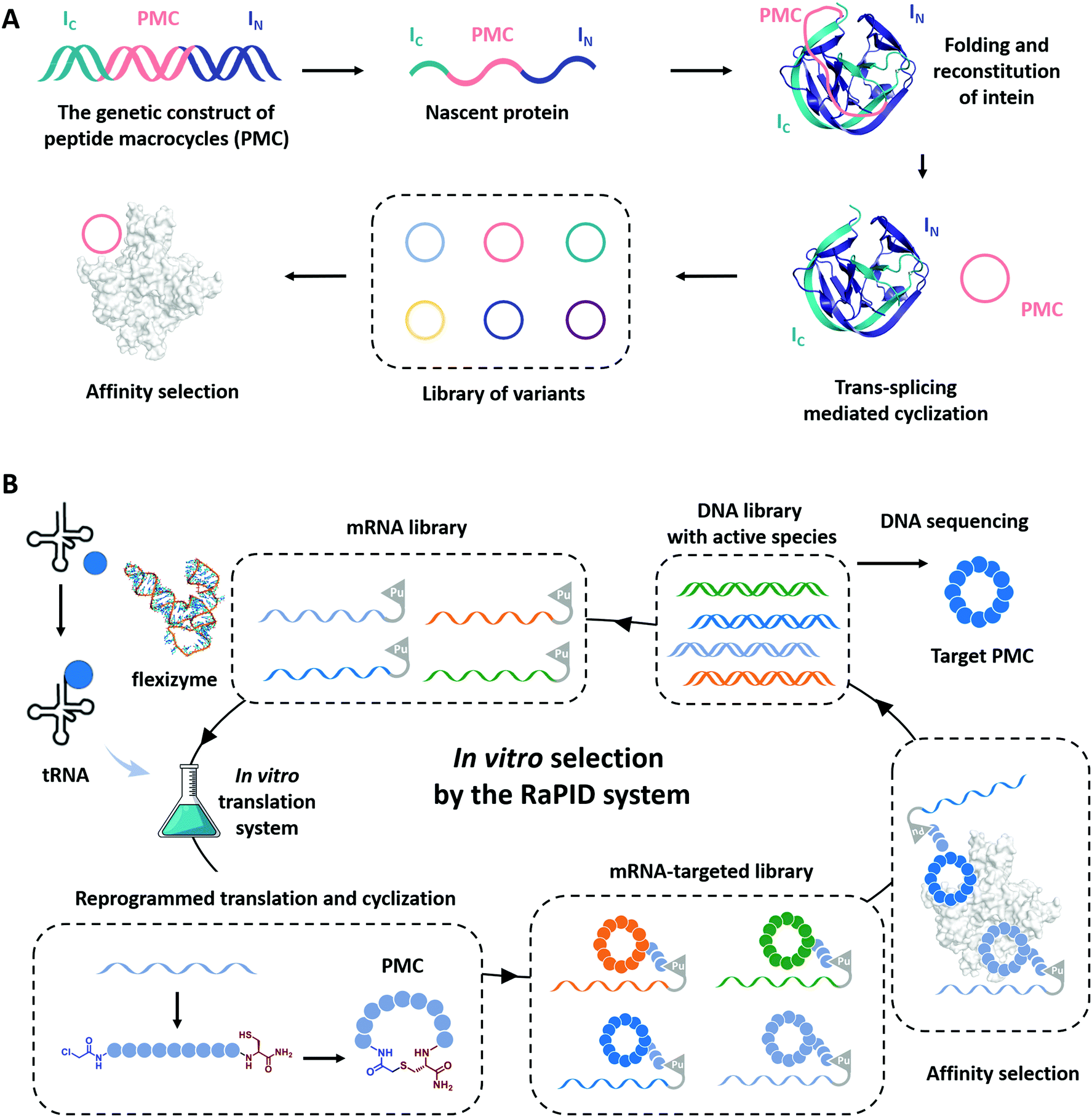 | ||
| Fig. 8 Schematic illustration of the evolution processes of SICLOPPS (A) and mRNA display (B). | ||
Unlike phage display or SICLOPPS, the in vitro transcription and translation process and the ability to conveniently incorporate noncanonical amino acids notably enlarge the library size of mRNA display to up to 1012–1014.63,157,168 Moreover, with flexizymes,66,169 a class of artificially evolved ribozymes capable of charging tRNA with various non-standard amino acids, Suga and co-workers170,171 have developed an attractive mRNA display methodology, termed random nonstandard peptide integrated discovery (RaPID) platform. As shown in Fig. 8B, the RaPID system allows the synthesis of cyclized peptide libraries by genetic code reprogramming and spontaneous posttranslational cyclization. Iterative rounds of affinity-based selection and gradual enrichment of the active species give rise to the desired peptide macrocycles for targeted proteins. Using this technology, they successfully identified a series of peptide macrocycle-based inhibitors to factor XIIa (an initiator of the contact system),172–174 calcium and integrin-binding protein 1 (an intracellular protein implicated in the survival and proliferation of triple-negative breast cancer),175 influenza viral envelope protein hemagglutinin,176 and human epidermal growth factor receptor (EGFR),177,178 which provide potent drug candidates in antithrombotics, pneumonia prevention, and cancer therapy. Through effective cyclization and incorporating noncanonical amino acids, such as β-amino acids, D-amino acids, and L-carboranylalanine, these peptide macrocycles showed not only high inhibitory activity but also protease resistance and/or good cell permeability. Other exciting examples include the discovery of low nanomolar inhibitors of prolyl hydroxylase isoform 2,179 isoform-selective Akt kinase180 or NAD-dependent deacetylase sirtuin 2,181 and the evolvement of cyclic peptides with the high binding ability to the interleukin-6 receptor.182 The RaPID also exhibited its commercial promise to develop the potential macrocyclic inhibitor of programmed death 1/programmed death ligand 1(PD-1/PD-L1).183
With these advances, the identification of peptide macrocycles with target biological functions is no longer considered a major obstacle in drug discovery and development. More significant challenges now may lie in the poor membrane permeability, low oral bioavailability, and metabolic instability of these drug candidates.157 Cyclization is expected to play vital roles in optimizing, at least partially, these crucial pharmacological properties for clinical translation.
Biomedical applications of protein macrocycles
Compared with small-molecule drugs, protein-based therapeutics exhibit unique functional benefits, such as high specificity and activity and fewer side effects. However, they also suffer some disadvantages, including inherent instability and poor pharmacokinetics.184,185 To solve these problems, a lot of works on protein-macrocycle-based therapeutics have emerged, which can not only improve the stability of protein drugs but also lead to other functional benefits, such as optimized tissue penetration or binding affinity.24,25,186 Moreover, these benefits could be further improved by combining with other strategies, such as PEGylation,187 which remarkably optimize the pharmacokinetic behaviors of protein drugs. Herein, we would like to discuss the cyclization of important protein-based therapeutics, such as cytokines, enzymes, and single-chain variable fragment (scFv) antibodies, to illustrate how cyclization influences their behaviors.Cytokines are a class of small-molecule proteins regulating cell growth, differentiation, and immune responses.188 As an important section of protein-based therapeutics, many cytokines, such as IFN, granulocyte colony stimulating factor (GCSF), interleukins, and hGH, have been approved by FDA for the treatment of various diseases. As an elegant example, Ploegh and co-workers22 constructed a cyclic-branched IFN-α coupled with 10 kDa PEG through two sequential transacylation procedures catalyzed by two Sortase A variants189 recognizing different sequences (LPETA for SrtAstrep and LPETG for SrtAStaph) (Fig. 9A). The engineered cyclic-branched IFN-α showed not only enhanced in vivo stability and thermal stability over its linear counterpart (Fig. 9B and C), but also improved pharmacokinetics over its non-PEGylated counterpart (Fig. 9D). However, considering the multiple-step synthesis approach and the low efficiency of PEGylation, this strategy is complicated by a low overall yield (∼10%). Alternatively, Lu24 and Gao25 groups reported two methods to construct long-circulating cyclic IFN-α, respectively. Compared with cyclic-branched IFN-α, these cyclic IFN-α exhibited better ex vivo and in vivo tumor penetration, which should be explained by the compact structures. These results suggest that cyclization may be combined with other strategies, such as PEGylation22,190 and ABD fusion,25 to further improve its performance.
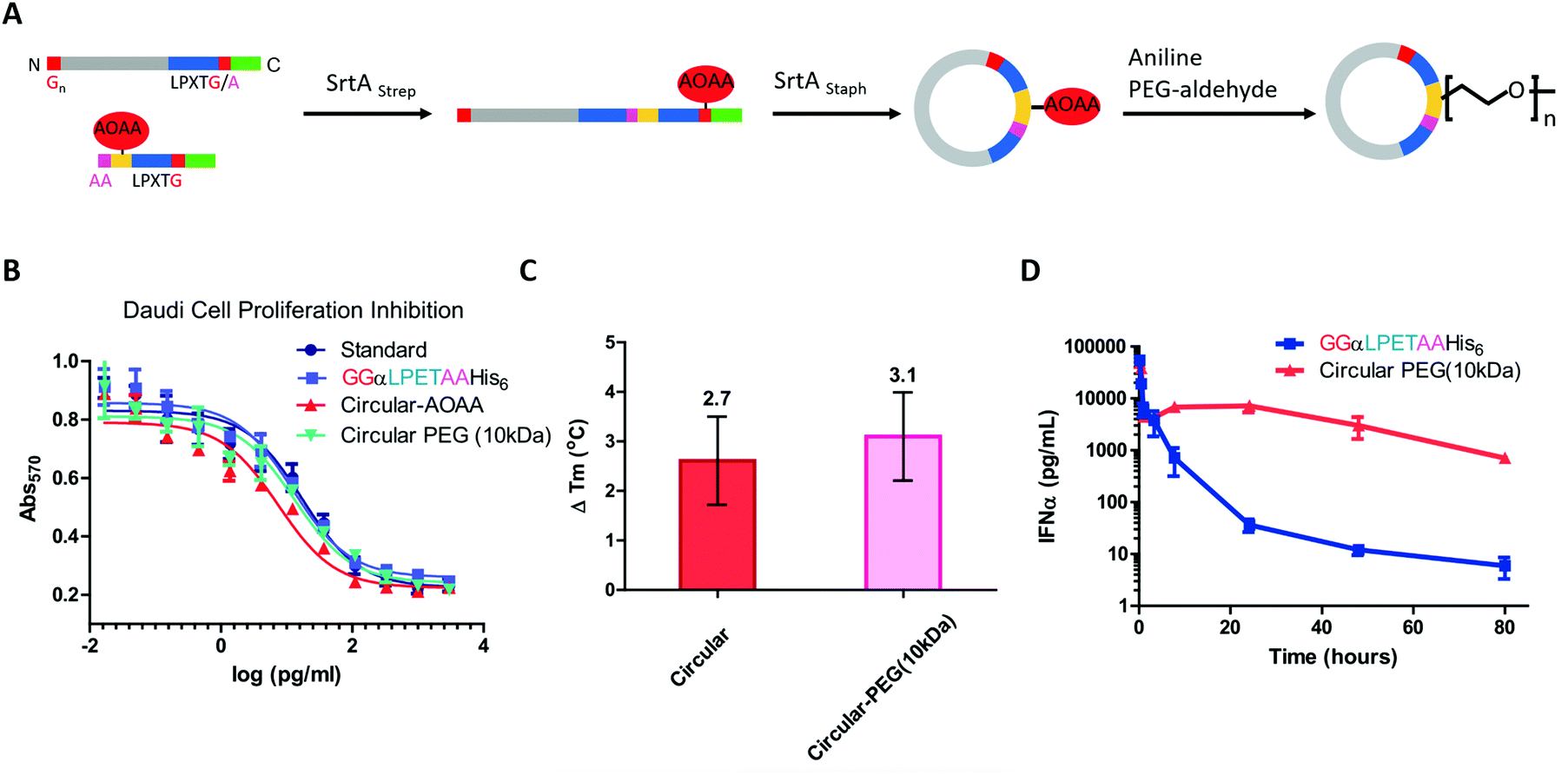 | ||
| Fig. 9 Synthesis of cyclic-branched IFN-α and PEG conjugate via two sequential transacylation procedures mediated by Sortase A (A). The cyclic-branched IFN-α and PEG conjugate shows optimized bioactivity (B), improved thermal stability (C), and prolonged circulation time (D).22 | ||
Apart from IFN-α, GCSF was also cyclized by Sortase A22 or split-intein191-mediated ligation. Honda and co-workers191 constructed a series of connecting loops to develop different cyclic variants, and one of them showed better thermal resistance with a 13 °C increase in Tm. hGH192 and interleukin-1 receptor antagonist102 were also cyclized to show the enhanced stability against thermal denaturation and comparable biological activities. However, it is difficult to evaluate their therapeutic potential due to the lack of in vivo data.
Enzymes are also a kind of potential therapeutics. Nine recombinant enzymes have been approved for clinical use from January 2014 to July 2018.194 However, unlike the cyclization of cytokines, studies on the cyclization of enzymes have mainly focused on model proteins. As a classical model, BLA has been cyclized either by intein-mediated peptide ligation79 or SpyTag/SpyCatcher chemistry132 as mentioned above. All these cyclic BLA and cyclic-branched BLA showed better stability against thermal denaturation than their linear counterparts.
Monoclonal antibodies seem to be the most important section of protein-based therapeutics considering their dominance in terms of product approvals or market value.194 They usually show high affinity and specificity but need to be produced in mammalian cells with a high cost. Thus, scFv antibodies, whose heavy and light variable fragments are usually connected with a short peptide linker, have emerged to be an alternative option. However, scFv antibodies suffer from their instability. The inter-domain interactions would lead to aggregate formation. To suppress scFv oligomerization, Morioka and co-workers193 constructed a kind of cyclic scFv antibody by Sortase A-mediated peptide ligation (Fig. 10A). By doing so, aggregation was markedly suppressed without disturbing the binding activities and thermal stability of the scFv antibody (Fig. 10B). This work is a useful attempt to produce cyclic scFv antibodies, holding great promise for further use in biomedical fields.
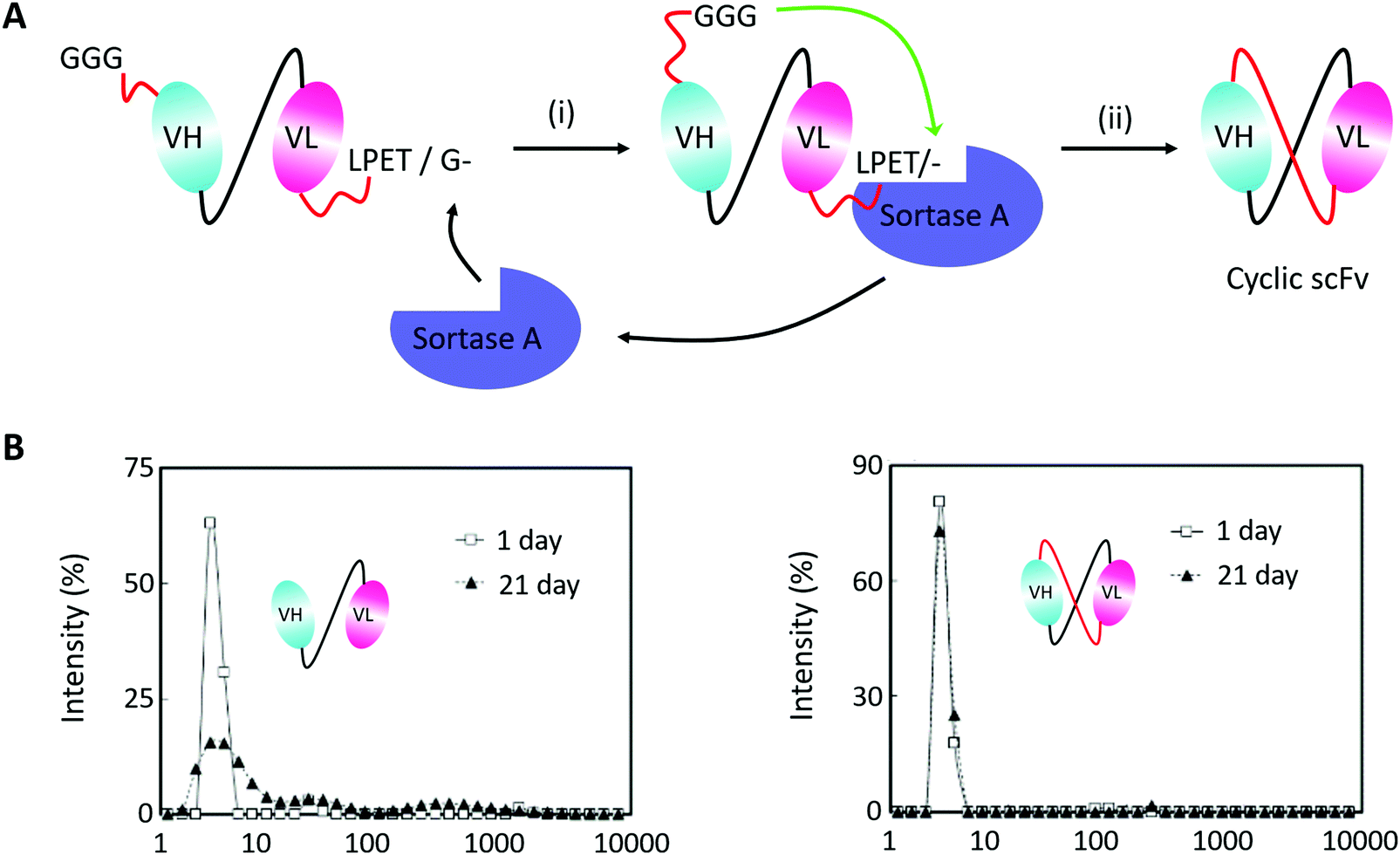 | ||
| Fig. 10 (A) Scheme for Sortase A-mediated scFv cyclization and (B) analysis of molecular size distributions by dynamic light scattering: linear scFv (left) and cyclic scFv (right) (B).193 | ||
Besides simple cyclization, a more complicated, higher order protein [n]catenane based artificial antibody has been developed by Zhang and co-workers.149 Based on the synthetic strategy mentioned above, a human epidermal growth factor receptor 2 (HER2)-specific affibody (AffiHER2 that targets the HER2 receptor specifically) has been genetically encoded into the scaffolds of protein [3]catenane or [4]catenane, giving rise to bivalent and trivalent artificial antibodies termed [3]catbody and [4]catbody. Due to the multivalent effect and stability enhancement of the [n]catenane scaffold, both [3]catbody and [4]catbody exhibit increased binding affinities to HER2 receptors, significantly prolonged circulation time, and optimized tumor accumulation. This indicates that protein macrocycles and their variants can not only increase the stability of target protein drugs but also bring in other functional benefits, such as multi-valent effects.
Conclusions
Inspired by naturally occurring peptide/protein macrocycles and their variants with enhanced stability and biological activity, extensive efforts have been directed to explore the biological syntheses and biomedical applications of such macrocycles over the past two decades. There are many advantages in biological synthesis, including high efficiency and selectivity, fully genetically programmable, and compatible with various functional proteins, making it a convenient way to produce peptide/protein-based macrocycles with tailor-designed topologies and functional benefits. In particular, significant advances have been gained in discovering new cyclic peptide-based drugs and in improving the pharmacokinetic properties of protein drugs.Despite these progresses, much work is still urgently needed. The extensive presence of peptide/protein-based macrocycles in nature should be understood in detail, particularly their formation mechanism and structural diversity. The detailed study on the structure–property relationship requests a moderate diversity of artificial peptide/protein-based macrocycles. Although the “assembly-reaction” synergy is powerful, there is still limited toolkits of genetically encoded protein entangling and ligation motifs. This toolbox should be largely expanded and optimized in terms of their reactivity and specificity to facilitate the synthesis of diverse macrocycles. Computer-aided methodology and direct evolution have been considered powerful to optimize these protein toolkits. In addition, it is also important to consider the scale-up synthesis of these macrocycles in industry. With their rapid development, we believe that more and more peptides/protein-based macrocycles will be successfully developed with increasing structural complexity, evolving from simple circle to multicycles (Type I), cyclic-branch (Type II), and further to knots and links (Type III and IV). Functional benefits will be gained in this endeavor, which shall open a new avenue for peptide/protein-based drug discovery and enable advanced therapeutics for biomedical applications.
Author contributions
The review was written, and the literature analyzed, through contributions by all authors. Each of the authors gave approval to the final version of the manuscript. W. W. H. and J. G. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R&D Program of China (No. 2020YFA0908100), the National Natural Science Foundation of China (No. 21534006, 21925102, 21991132, 81991505, 92056118), and Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202006). W. B. Z. is sponsored by Bayer Investigator Award. W. W. H. acknowledges Boya Postdoc Fellowship from Peking University and a fellowship from China Postdoctoral Science Foundation (No. 2020TQ0005).Notes and references
- T. Yamamoto and Y. Tezuka, Polym. Chem., 2011, 2, 1930–1941 RSC.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791–796 CrossRef CAS PubMed.
- Y. Tezuka, Acc. Chem. Res., 2017, 50, 2661–2672 CrossRef CAS PubMed.
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS.
- Y. Tezuka, Topological polymer chemistry: progress of cyclic polymers in syntheses, properties, and functions, World Scientific, 2013 Search PubMed.
- E. Flapan, When topology meets chemistry: a topological look at molecular chirality, Cambridge University Press, 2000 Search PubMed.
- K. Shimokawa, K. Ishihara and Y. Tezuka, Topology of Polymers, Springer Nature, 2019 Search PubMed.
- Y. Tezuka, Isr. J. Chem., 2020, 60, 67–74 CrossRef CAS.
- X.-W. Wang and W.-B. Zhang, Trends Biochem. Sci., 2018, 43, 806–817 CrossRef CAS PubMed.
- Z. Qu, S. Z. Cheng and W.-B. Zhang, Trends Chem., 2021, 3, 402–415 CrossRef CAS.
- W.-B. Zhang and S. Z. Cheng, Giant, 2020, 1, 100011 CrossRef.
- Y. Tezuka and T. Deguchi, Topological Polymer Chemistry, Springer Nature, 2022 Search PubMed.
- H.-X. Zhou, Acc. Chem. Res., 2004, 35, 123–130 CrossRef PubMed.
- H.-X. Zhou, J. Mol. Biol., 2003, 332, 257–264 CrossRef CAS PubMed.
- H.-X. Zhou, J. Am. Chem. Soc., 2003, 125, 9280–9281 CrossRef CAS PubMed.
- T. L. Aboye and J. A. Camarero, J. Biol. Chem., 2012, 287, 27026–27032 CrossRef CAS PubMed.
- R. J. Clark, M. Akcan, Q. Kaas, N. L. Daly and D. J. Craik, Toxicon, 2012, 59, 446–455 CrossRef CAS PubMed.
- N. K. Williams, E. Liepinsh, S. J. Watt, P. Prosselkov, J. M. Matthews, P. Attard, J. L. Beck, N. E. Dixon and G. Otting, J. Mol. Biol., 2005, 346, 1095–1108 CrossRef CAS PubMed.
- D. R. Boutz, D. Cascio, J. Whitelegge, L. J. Perry and T. O. Yeates, J. Mol. Biol., 2007, 368, 1332–1344 CrossRef CAS PubMed.
- C. Zong, M. J. Wu, J. Z. Qin and A. J. Link, J. Am. Chem. Soc., 2017, 139, 10403–10409 CrossRef CAS PubMed.
- M. W. Popp, S. K. Dougan, T. Y. Chuang, E. Spooner and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3169–3174 CrossRef CAS PubMed.
- J. Hu, W. Zhao, Y. Gao, M. Sun, Y. Wei, H. Deng and W. Gao, Biomaterials, 2015, 47, 13–19 CrossRef CAS PubMed.
- Y. Hou, Y. Zhou, H. Wang, R. Wang, J. Yuan, Y. Hu, K. Sheng, J. Feng, S. Yang and H. Lu, J. Am. Chem. Soc., 2018, 140, 1170–1178 CrossRef CAS PubMed.
- J. Guo, J. Sun, X. Liu, Z. Wang and W. Gao, Biomaterials, 2020, 250, 120073 CrossRef CAS PubMed.
- J. A. Kritzer, S. Hamamichi, J. M. Mccaffery, S. Santagata, T. A. Naumann, K. A. Caldwell, G. A. Caldwell and S. Lindquist, Nat. Chem. Biol., 2009, 5, 655–663 CrossRef CAS PubMed.
- E. Miranda, I. K. Norderen, A. L. Male, C. E. Lawrence, F. Hoakwie, F. Cuda, W. Court, K. R. Fox, P. A. Townsend and G. K. P. Ac Kham, J. Am. Chem. Soc., 2013, 135, 10418–10425 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961–2004 CrossRef CAS PubMed.
- D. Whitford, Proteins: Structure and Function, J. Wiley & Sons, Hoboken, NJ, 2005 Search PubMed.
- G. A. Brar and J. S. Weissman, Nat. Rev. Mol. Cell Biol., 2015, 16, 651–664 CrossRef CAS PubMed.
- M. Trabi and D. J. Craik, Trends Biochem. Sci., 2002, 27, 132–138 CrossRef CAS PubMed.
- C. K. Wang, Q. Kaas, L. Chiche and D. J. Craik, Nucleic Acids Res., 2008, 36, D206–D210 CrossRef CAS PubMed.
- L. Cascales and D. J. Craik, Org. Biomol. Chem., 2010, 8, 5035–5047 RSC.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, J. Mol. Biol., 1999, 294, 1327–1336 CrossRef CAS PubMed.
- D. C. Ireland, R. J. Clark, N. L. Daly and D. J. Craik, J. Nat. Prod., 2010, 73, 1610–1622 CrossRef CAS PubMed.
- R. Burman, S. Gunasekera, A. A. Strömstedt and U. Göransson, J. Nat. Prod., 2014, 77, 724–736 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed.
- M. O. Maksimov, S. J. Pan and L. A. James, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- N. L. Daly and D. J. Craik, Curr. Opin. Chem. Biol., 2011, 15, 362–368 CrossRef CAS PubMed.
- R. L. Duda, Cell, 1998, 94, 55–60 CrossRef CAS PubMed.
- W. R. Wikoff, L. Liljas, R. L. Duda, H. Tsuruta, R. W. Hendrix and J. E. Johnson, Science, 2000, 289, 2129–2133 CrossRef CAS PubMed.
- C. Helgstrand, W. R. Wikoff, R. L. Duda, R. W. Hendrix, J. E. Johnson and L. Liljas, J. Mol. Biol., 2003, 334, 885–899 CrossRef CAS PubMed.
- A. D. Gillon, I. Saska, C. V. Jennings, R. F. Guarino, D. J. Craik and M. A. Anderson, Plant J., 2008, 53, 505–515 CrossRef CAS PubMed.
- I. Saska, A. D. Gillon, N. Hatsugai, R. G. Dietzgen, I. Hara-Nishimura, M. A. Anderson and D. J. Craik, J. Biol. Chem., 2007, 282, 29721–29728 CrossRef CAS PubMed.
- O. Saether, D. J. Craik, I. D. Campbell, K. Sletten, J. Juul and D. G. Norman, Biochemistry, 1995, 34, 4147–4158 CrossRef CAS PubMed.
- N. L. Daly and D. J. Craik, J. Biol. Chem., 2000, 275, 19068–19075 CrossRef CAS PubMed.
- M. Martínez-Bueno, M. Maqueda, A. Gálvez, B. Samyn, J. V. Beeumen, J. Coyette and E. Valdivia, J. Bacteriol., 1994, 176, 6334–6339 CrossRef PubMed.
- C. González, G. M. Langdon, M. Bruix, A. Gálvez and M. Rico, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11221–11226 CrossRef PubMed.
- P. Tongaonkar, P. Tran, K. Roberts, J. Schaal, G. Ösapay, D. Tran, A. J. Ouellette and M. Selsted, J. Leukocyte Biol., 2011, 89, 283–290 CrossRef CAS PubMed.
- G. C. Paesen, C. Siebold, K. Harlos, M. F. Peacey, P. A. Nuttall and D. Stuart, J. Mol. Biol., 2007, 368, 1172–1186 CrossRef CAS PubMed.
- L. S. Zhang and J. P. Tam, J. Am. Chem. Soc., 1997, 119, 2363–2370 CrossRef CAS.
- Y.-Q. Chen, C. Chen, Y. He, M. Yu, L. Xu, C.-L. Tian, Q.-X. Guo, J. Shi, M. Zhang and Y. M. Li, Tetrahedron Lett., 2014, 55, 2883–2886 CrossRef CAS.
- M. Chen, S. Wang and X. Yu, Chem. Commun., 2019, 55, 3323–3326 RSC.
- L. Z. Yan and P. E. Dawson, Angew. Chem., Int. Ed., 2001, 40, 3625–3627 CrossRef CAS PubMed.
- J. W. Blankenship and P. E. Dawson, J. Mol. Biol., 2003, 327, 537–548 CrossRef CAS PubMed.
- J. W. Blankenship and P. E. Dawson, Protein Sci., 2010, 16, 1249–1256 CrossRef PubMed.
- H. C. Hayes, L. Y. Luk and Y.-H. Tsai, Org. Biomol. Chem., 2021, 19, 3983–4001 RSC.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- A. D. Foster, J. D. Ingram, E. K. Leitch, K. R. Lennard, E. L. Osher and A. Tavassoli, J. Biomol. Screening, 2015, 20, 563–576 CrossRef CAS PubMed.
- C. Sohrabi, A. Foster and A. Tavassoli, Nat. Rev. Chem., 2020, 4, 90–101 CrossRef CAS.
- Y. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
- K. Josephson, M. C. Hartman and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 11727–11735 CrossRef CAS PubMed.
- T. G. Heckler, L. H. Chang, Y. Zama, T. Naka, M. S. Chorghade and S. M. Hecht, Biochemistry, 1984, 23, 1468–1473 CrossRef CAS PubMed.
- M. Ohuchi, H. Murakami and H. Suga, Curr. Opin. Chem. Biol., 2007, 11, 537–542 CrossRef CAS PubMed.
- H. J. Kang, F. Coulibaly, F. Clow, T. Proft and E. N. Baker, Science, 2007, 318, 1625–1628 CrossRef CAS PubMed.
- S. J. de Veer, A. M. White and D. Craik, Angew. Chem., Int. Ed., 2021, 60, 8050–8071 CrossRef CAS PubMed.
- C. K. Wang, S. H. Hu, J. L. Martin, T. Sjogren, J. Hajdu, L. Bohlin, P. Claeson, U. Goransson, K. J. Rosengren and J. Tang, J. Biol. Chem., 2009, 284, 10672–10683 CrossRef CAS PubMed.
- P. Dabrowski-Tumanski and J. I. Sulkowska, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3415–3420 CrossRef CAS PubMed.
- J. I. Sulkowska, Curr. Opin. Struct. Biol., 2020, 60, 131–141 CrossRef CAS PubMed.
- N. H. Shah and T. W. Muir, Chem. Sci., 2014, 5, 446–461 RSC.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 6075–6710 Search PubMed.
- T. C. Evans, J. Biol. Chem., 1999, 274, 3923–3926 CrossRef CAS PubMed.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2019, 120, 3051–3126 CrossRef PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- J. A. Camarero and T. W. Muir, J. Am. Chem. Soc., 1999, 121, 5597–5598 CrossRef CAS.
- M. Q. Xu and T. C. Evans, Jr., Methods, 2001, 24, 257–277 CrossRef CAS PubMed.
- H. Iwai and A. Plückthun, FEBS Lett., 1999, 459, 166–172 CrossRef CAS PubMed.
- J. A. Camarero, D. Fushman, D. Cowburn and T. W. Muir, Biorg. Med. Chem., 2001, 9, 2479–2484 CrossRef CAS.
- G. Volkmann and H. Iwai, Mol. BioSyst., 2010, 6, 2110–2121 RSC.
- Y. Li, Biotechnol. Lett., 2015, 37, 2121–2137 CrossRef CAS PubMed.
- C. P. Scott, E. Abel-Santos, M. Wall, D. C. Wahnon and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13638–13643 CrossRef CAS PubMed.
- H. Iwai, A. Lingel and A. Pluckthun, J. Biol. Chem., 2001, 276, 16548–16554 CrossRef CAS PubMed.
- M. C. Waldhauer, S. N. Schmitz, C. Ahlmanneltze, J. G. Gleixner, C. C. Schmelas, A. G. Huhn, C. Bunne, M. Buscher, M. Horn, N. Klughammer, J. Kreft, E. Schäfer, P. A. Bayer, S. G. Krämer, J. Neugebauer, P. Wehler, M. P. Mayer, R. Eils and B. D. Ventura, Mol. BioSyst., 2015, 11, 3231–3243 RSC.
- T. S. Young, D. D. Young, I. Ahmad, J. M. Louis, S. J. Benkovic and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11052–11056 CrossRef CAS PubMed.
- K. Jagadish, R. Borra, V. Lacey, S. Majumder, A. Shekhtman, L. Wang and J. A. Camarero, Angew. Chem., Int. Ed., 2013, 52, 3126–3131 CrossRef CAS PubMed.
- H. Iwai, S. Züger, J. Jin and P. H. Tam, FEBS Lett., 2006, 580, 1853–1858 CrossRef CAS PubMed.
- J. Zettler, V. Schütz and H. D. Mootz, FEBS Lett., 2009, 583, 909–914 CrossRef CAS PubMed.
- A. A. Sesilja, Z. Sara, B. Edith, I. Hideo and L. Hilal, PLoS One, 2009, 4, e5185 CrossRef PubMed.
- S. K. Mazmanian, G. Liu and H. Ton-That, Science, 1999, 285, 760 CrossRef CAS PubMed.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed.
- R. Parthasarathy, S. Subramanian and E. T. Boder, Bioconjugate Chem., 2007, 18, 469–476 CrossRef CAS PubMed.
- J. M. Antos, W. L. Popp, R. Ernst, G. L. Chew, E. Spooner and H. L. Ploegh, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed.
- J. G. Bolscher, M. J. Oudhoff, K. Nazmi, J. M. Antos, C. P. Guimaraes, E. Spooner, E. F. Haney, J. J. Garcia Vallejo, H. J. Vogel, W. van’t Hof, H. L. Ploegh and E. C. Veerman, FASEB J., 2011, 25, 2650–2658 CrossRef CAS PubMed.
- K. Strijbis, E. Spooner and H. L. Ploegh, Traffic, 2012, 13, 780–789 CrossRef CAS PubMed.
- J. Zhang, S. Yamaguchi, H. Hirakawa and T. Nagamune, J. Biosci. Bioeng., 2013, 116, 298–301 CrossRef CAS PubMed.
- J. J. Bellucci, J. Bhattacharyya and A. Chilkoti, Angew. Chem., Int. Ed., 2015, 54, 441–445 CAS.
- M. A. Jackson, E. K. Gilding, T. Shafee, K. S. Harris and D. J. Craik, Nat. Commun., 2018, 9, 2411 CrossRef CAS PubMed.
- N. L. Daly, K. J. Rosengren and D. J. Craik, Adv. Drug Delivery Rev., 2009, 61, 918–930 CrossRef CAS PubMed.
- G. K. T. Nguyen, W. H. Lim, P. Q. T. Nguyen and J. P. Tam, J. Biol. Chem., 2012, 287, 17598–17607 CrossRef CAS PubMed.
- G. K. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, J. Am. Chem. Soc., 2015, 137, 15398–15401 CrossRef CAS PubMed.
- G. K. Nguyen, X. Hemu, J. P. Quek and J. P. Tam, Angew. Chem., Int. Ed., 2016, 55, 12802–12806 CrossRef CAS PubMed.
- M. Schmidt, A. Toplak, P. Quaedflieg, J. Maarseveen and T. Nuijens, Drug Discovery Today, 2017, 26, 11–16 CrossRef PubMed.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, D. Van and D. J. Craik, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed.
- Q. Wu, H. L. Ploegh and M. C. Truttmann, ACS Chem. Biol., 2017, 12, 664–673 CrossRef CAS PubMed.
- R. Yang, Y. H. Wong, G. Nguyen, J. P. Tam, J. Lescar and B. Wu, J. Am. Chem. Soc., 2017, 139, 5351–5358 CrossRef CAS PubMed.
- K. M. Mikula, I. Tascón, J. J. Tommila and H. Iwa, FEBS Lett., 2017, 591, 1285–1294 CrossRef CAS PubMed.
- K. S. Harris, R. F. Guarino, R. S. Dissanayake, P. Quimbar, O. C. McCorkelle, S. Poon, Q. Kaas, T. Durek, E. K. Gilding, M. A. Jackson, D. J. Craik, N. L. van der Weerden, R. F. Anders and M. A. Anderson, Sci. Rep., 2019, 9, 10820 CrossRef PubMed.
- B. J. Smithies, Y.-H. Huang, M. A. Jackson, K. Yap, E. K. Gilding, K. S. Harris, M. A. Anderson and D. J. Craik, ACS Chem. Biol., 2020, 15, 962–969 CrossRef CAS PubMed.
- K. Yap, J. Du, F. B. H. Rehm, S. R. Tang, Y. Zhou, J. Xie, C. K. Wang, S. J. de Veer, L. H. L. Lua, T. Durek and D. J. Craik, Nat. Protoc., 2021, 16, 1740–1760 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Trends Biochem. Sci., 2014, 39, 400–408 CrossRef CAS PubMed.
- T. Kawakami, A. Ohta, M. Ohuchi, H. Ashigai, H. Murakami and H. Suga, Nat. Chem. Biol., 2009, 5, 888–890 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. – Eur. J., 2013, 19, 6530–6536 CrossRef CAS PubMed.
- H. Kwon, C. J. Squire, P. G. Young and E. N. Baker, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1367–1372 CrossRef CAS PubMed.
- M. Walden, J. M. Edwards, A. M. Dziewulska, R. Bergmann, G. Saalbach, S. Y. Kan, O. K. Miller, M. Weckener, R. J. Jackson and S. L. Shirran, eLife, 2015, 4, e06638 CrossRef PubMed.
- J. A. Pointon, W. D. Smith, G. Saalbach, A. Crow, M. A. Kehoe and M. J. Banfield, J. Biol. Chem., 2010, 285, 33858–33866 CrossRef CAS PubMed.
- F. Zhang and W. B. Zhang, Chin. J. Chem., 2020, 38, 864–878 CrossRef CAS.
- R. M. Hagan, R. Björnsson, S. A. Mcmahon, B. Schomburg and U. Schwarz-Linek, Angew. Chem., Int. Ed., 2010, 49, 8421–8425 CrossRef CAS PubMed.
- G. Yin, J. Wei, Y. Shao, W.-H. Wu, L. Xu and W.-B. Zhang, Chin. Chem. Lett., 2021, 32, 353–356 CrossRef CAS.
- F. Sun and W. B. Zhang, Chin. J. Chem., 2020, 38, 894–896 CrossRef CAS.
- L. Xu and W.-B. Zhang, Polymer, 2018, 155, 235–247 CrossRef CAS.
- J. Fang and W.-b Zhang, Acta Polym. Sin., 2018, 429–444 Search PubMed.
- J. Wei, L. Xu, W.-H. Wu, F. Sun and W.-B. Zhang, Sci. China: Chem., 2022, 1–11 Search PubMed.
- B. Zakeri and M. Howarth, J. Am. Chem. Soc., 2010, 132, 4526–4537 CrossRef CAS PubMed.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS PubMed.
- A. H. Keeble, A. Banerjee, M. P. Ferla, S. C. Reddington, I. Anuar and M. Howarth, Angew. Chem., Int. Ed., 2017, 56, 16521–16525 CrossRef CAS PubMed.
- A. H. Keeble, P. Turkki, S. Stokes, I. N. A. Khairil Anuar, R. Rahikainen, V. P. Hytonen and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26523–26533 CrossRef CAS PubMed.
- G. Veggiani, T. Nakamura, M. D. Brenner, R. V. Gayet, J. Yan, C. V. Robinson and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1202–1207 CrossRef CAS PubMed.
- W.-B. Zhang, F. Sun, D. A. Tirrell and F. H. Arnold, J. Am. Chem. Soc., 2013, 135, 13988–13997 CrossRef CAS PubMed.
- C. Schoene, S. P. Bennett and M. Howarth, Sci. Rep., 2016, 6, 21151 CrossRef CAS PubMed.
- C. Schoene, J. O. Fierer, S. P. Bennett and M. Howarth, Angew. Chem., Int. Ed., 2014, 53, 6101–6104 CrossRef CAS PubMed.
- M. Si, Q. Xu, L. Jiang and H. Huang, PLoS One, 2016, 11, e0162318 CrossRef PubMed.
- J. Wang, Y. Wang, X. Wang, D. Zhang, S. Wu and G. Zhang, Biotechnol. Biofuels, 2016, 9, 79 CrossRef PubMed.
- J. O. Fierer, G. Veggiani and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1176–E1181 CrossRef CAS PubMed.
- C. M. Buldun, J. X. Jean, M. R. Bedford and M. Howarth, J. Am. Chem. Soc., 2018, 140, 3008–3018 CrossRef CAS PubMed.
- X. L. Wu, Y. Liu, D. Liu, F. Sun and W.-B. Zhang, J. Am. Chem. Soc., 2018, 140, 17474–17483 CrossRef CAS PubMed.
- X.-D. Da and W.-B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 11097–11104 CrossRef CAS PubMed.
- X. W. Wang and W.-B. Zhang, Trends Biochem. Sci., 2018, 43, 806–817 CrossRef CAS PubMed.
- T.-t Yang, X.-D. Da and W.-B. Zhang, Acta Polym. Sin., 2020, 51, 804–816 CAS.
- L. Xu and W.-B. Zhang, Sci. China: Chem., 2018, 61, 3–16 CrossRef CAS.
- S. T. D. Hsu, Y. T. C. Lee, K. M. Mikula, S. M. Backlund and H. Iwa, Front. Chem., 2021, 9, 663241 CrossRef CAS PubMed.
- X. W. Wang and W.-B. Zhang, Angew. Chem., Int. Ed., 2016, 55, 3442–3446 CrossRef CAS PubMed.
- X. W. Wang and W.-B. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13985–13989 CrossRef CAS PubMed.
- D. Liu, W.-H. Wu, Y. J. Liu, X. L. Wu, Y. Cao, B. Song, X. Li and W.-B. Zhang, ACS Cent. Sci., 2017, 3, 473–481 CrossRef CAS PubMed.
- X. Bai, Y. Liu, J. Lee, J. Fang, W.-H. Wu, J. Seo and W.-B. Zhang, Giant, 2022, 10, 100092 CrossRef CAS.
- Y. Liu, W.-H. Wu, S. Hong, J. Fang, F. Zhang, G. X. Liu, J. Seo and W. B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19153–19161 CrossRef CAS PubMed.
- Y. Liu, Z. Duan, J. Fang, F. Zhang, J. Xiao and W.-B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 16122–16127 CrossRef CAS PubMed.
- W.-H. Wu, X. Bai, Y. Shao, C. Yang, J. J. Wei, W. Wei and W.-B. Zhang, J. Am. Chem. Soc., 2021, 143, 18029–18040 CrossRef CAS PubMed.
- Q.-H. Guo, Y. Jiao, Y. Feng and J. F. Stoddart, CCS Chem., 2021, 3, 1542–1572 CrossRef CAS.
- R. M. Freidinger, D. F. Veber, D. S. Perlow, J. R. Brooks and R. Saperstein, Science, 1980, 210, 656–658 CrossRef CAS PubMed.
- D. F. Veber, R. G. Strachan, S. J. Bergstrand, F. W. Holly, C. F. Homnick, R. Hirschmann, M. L. Torchiana and R. Saperstein, J. Am. Chem. Soc., 1976, 98, 2367–2369 CrossRef CAS.
- E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef CAS PubMed.
- P. Edman, Annu. Rev. Biochem., 1959, 28, 69–96 CrossRef CAS PubMed.
- D. A. Horton, G. T. Bourne and M. L. Smythe, J. Comput.-Aided Mol. Des., 2002, 16, 415–431 CrossRef CAS PubMed.
- K. Jin, Future Med. Chem., 2020, 12, 1687–1690 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- N. K. Bashiruddin and H. Suga, Curr. Opin. Chem. Biol., 2015, 24, 131–138 CrossRef CAS PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed.
- C. P. Scott, E. Abel-Santos, M. Wall, D. C. Wahnon and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13638–13643 CrossRef CAS PubMed.
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297–12302 CrossRef CAS PubMed.
- A. Tavassoli, Curr. Opin. Chem. Biol., 2017, 38, 30–35 CrossRef CAS PubMed.
- A. Tavassoli and S. Benkovic, Nat. Protoc., 2007, 2, 1126–1133 CrossRef CAS PubMed.
- T. F. Outeiro and S. Lindquist, Science, 2003, 302, 1772–1775 CrossRef CAS PubMed.
- A. A. Cooper, A. D. Gitler, A. Cashikar, C. M. Haynes, K. J. Hill, B. Bhullar, K. Liu, K. Xu, K. E. Strathearn and F. Liu, Science, 2006, 313, 324–328 CrossRef CAS PubMed.
- B. Keith, R. S. Johnson and M. C. Simon, Nat. Rev. Cancer, 2012, 12, 9–22 CrossRef CAS PubMed.
- A. Giaccia, B. G. Siim and R. S. Johnson, Nat. Rev. Drug Discovery, 2003, 2, 803–811 CrossRef CAS PubMed.
- K. Josephson, A. Ricardo and J. W. Szostak, Drug Discovery Today, 2014, 19, 388–399 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. – Eur. J., 2013, 19, 6530–6536 CrossRef CAS PubMed.
- T. Passioura, T. Katoh, Y. Goto and H. Suga, Annu. Rev. Biochem., 2014, 83, 727–752 CrossRef CAS PubMed.
- Y. Goto and H. Suga, Acc. Chem. Res., 2021, 54, 3604–3617 CrossRef CAS PubMed.
- T. Katoh and H. Suga, J. Am. Chem. Soc., 2022, 144, 2069–2072 CrossRef CAS PubMed.
- W. Liu, S. J. de Veer, Y.-H. Huang, T. Sengoku, C. Okada, K. Ogata, C. N. Zdenek, B. G. Fry, J. E. Swedberg and T. Passioura, J. Am. Chem. Soc., 2021, 143, 18481–18489 CrossRef CAS PubMed.
- D. J. Ford, N. M. Duggan, S. E. Fry, J. Ripoll-Rozada, S. M. Agten, W. Liu, L. Corcilius, T. M. Hackeng, R. van Oerle and H. M. Spronk, J. Med. Chem., 2021, 64, 7853–7876 CrossRef CAS PubMed.
- V. A. Haberman, S. R. Fleming, T. M. Leisner, A. C. Puhl, E. Feng, L. Xie, X. Chen, Y. Goto, H. Suga, L. V. Parise, D. Kireev, K. H. Pearce and A. A. Bowers, ACS Med. Chem. Lett., 2021, 12, 1832–1839 CrossRef CAS PubMed.
- M. Saito, Y. Itoh, F. Yasui, T. Munakata, D. Yamane, M. Ozawa, R. Ito, T. Katoh, H. Ishigaki and M. Nakayama, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- S. Imanishi, T. Katoh, Y. Yin, M. Yamada, M. Kawai and H. Suga, J. Am. Chem. Soc., 2021, 143, 5680–5684 CrossRef CAS PubMed.
- Y. Yin, N. Ochi, T. W. Craven, D. Baker, N. Takigawa and H. Suga, J. Am. Chem. Soc., 2019, 141, 19193–19197 CrossRef CAS PubMed.
- T. McAllister, T.-L. Yeh, M. Abboud, I. Leung, E. Hookway, O. King, B. Bhushan, S. Williams, R. Hopkinson and M. Münzel, Chem. Sci., 2018, 9, 4569–4578 RSC.
- Y. Hayashi, J. Morimoto and H. Suga, ACS Chem. Biol., 2012, 7, 607–613 CrossRef CAS PubMed.
- J. Morimoto, Y. Hayashi and H. Suga, Angew. Chem., Int. Ed., 2012, 124, 3479–3483 CrossRef.
- T. Passioura, W. Liu, D. Dunkelmann, T. Higuchi and H. Suga, J. Am. Chem. Soc., 2018, 140, 11551–11555 CrossRef CAS PubMed.
- T. Zarganes-Tzitzikas, M. Konstantinidou, Y. Gao, D. Krzemien, K. Zak, G. Dubin, T. A. Holak and A. Dömling, Expert Opin. Ther. Pat., 2016, 26, 973–977 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef CAS PubMed.
- A. Purkayastha and T. J. Kang, Biotechnol. Bioprocess Eng., 2019, 24, 702–712 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- J. Oppenheim, Int. J. Hematol., 2001, 74, 3–8 CrossRef CAS PubMed.
- J. M. Antos, G. L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, W. L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- J. Zhou, X.-Y. Zhang and Z.-Q. Su, Chin. J. Polym. Sci., 2021, 39, 1093–1109 CrossRef CAS.
- T. Miyafusa, R. Shibuya, W. Nishima, R. Ohara, C. Yoshida and S. Honda, ACS Chem. Biol., 2017, 12, 2690–2696 CrossRef CAS PubMed.
- N. Rasche, J. Tonillo, M. Rieker, S. Becker, B. Dorr, D. Ter-Ovanesyan, U. A. Betz, B. r Hock and H. Kolmar, Bioconjugate Chem., 2016, 27, 1341–1347 CrossRef CAS PubMed.
- S. Yamauchi, Y. Kobashigawa, N. Fukuda, M. Teramoto, Y. Toyota, C. Liu, Y. Ikeguchi, T. Sato, Y. Sato and H. Kimura, Molecules, 2019, 24, 2620 CrossRef CAS PubMed.
- G. Walsh, Nat. Biotechnol., 2018, 36, 1136–1145 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |