
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelical sulfono-γ-AApeptides with predictable functions in protein recognition
Peng
Sang
,
Yan
Shi
,
Lulu
Wei
and
Jianfeng
Cai
 *
*
Department of Chemistry, University of South Florida, 4202 E. Fowler Ave., Tampa, FL 33620, USA. E-mail: jianfengcai@usf.edu
First published on 20th May 2022
Abstract
Sulfono-γ-AApeptides are a subset of possible sequence-specific foldamers that might be considered for the design of biomimetic drug molecular structures. Although they have been studied for a relatively short period of time, a number of structures and functions have been designed or discovered within this class of unnatural peptides. Examples of utilizing these sulfono-γ-AApeptides have demonstrated the potential that sulfono-γ-AApeptides can offer, however, to date, their application in biomedical sciences yet remains unexplored. This review mainly summarizes the helical folding conformations of sulfono-γ-AApeptides and their biological application as helical mimetics in medicinally relevant protein–protein interactions (PPIs) and assesses their potential for the mimicry of other α-helices for protein recognition in the future.
 Peng Sang | Peng Sang is a Research Associate at the University of South Florida. His research interests include the exploration of folding structures of peptidomimetics and the development of novel peptide foldamers potentially targeting viruses, cancer, diabetes, etc. |
 Yan Shi | Yan Shi is a Research Associate at the University of South Florida. Her current research interests focus on AApeptide folding, combinatorial screening of γ-AApeptides and structure-based drug design, synthesis and biological evaluation. |
 Lulu Wei | Lulu Wei is a Graduate Student at the University of South Florida. Her research interest is focused on the synthesis and combinatorial screening of γ-AApeptides. |
 Jianfeng Cai | Jianfeng Cai is a Professor in the Department of Chemistry at the University of South Florida. His research group focuses on the development and application of AApeptide-based peptidomimetics. |
Introduction
In the past two decades, our understanding of protein structures and functions has advanced rapidly, providing an in-depth understanding of the mechanisms of various biological processes. As an outgrowth of this understanding, researchers designed a variety of important unnatural oligomers (foldamers) to mimic the secondary and tertiary structures of natural peptides and proteins1–7 and achieved impressive results.8–12 To date, these foldamer systems have been used to mimic various aspects of the folding and organization of natural peptides and proteins. A number of outstanding studies based on unnatural foldamers that have been developed involve β-peptides,13,14 peptoids,15–17 β-peptoids,18,19 oligoureas,20 azapeptides,21–23 and others.γ-AApeptides (γ-substituted-N-acylated-N-aminoethyl amino acids) are a class of peptidomimetics developed in recent years by our group.24,25 Their backbone structure is derived from γ-chiral PNA (peptide nucleic acid), and each repeating unit is compared with a conventional dipeptide residue and contains the same number of side chains as α-peptides of the same lengths (Fig. 1). As a subset of γ-AApeptides, sulfono-γ-AApeptides have emerged as a new class of foldameric helical mimetics in recent years (Fig. 1)26 and have been employed to tackle some continuing challenges in modulating α-helix-mediated PPIs.27–33 As a class of proteolytically stable peptidomimetics, sulfono-γ-AApeptides and their related peptide hybrids exhibit unique folding stability by adopting a series of robust helical structures with well-defined hydrogen bond patterns.34–39 This type of unnatural peptidomimetics has the following advantages: (1) half of the side chains are derived from the chiral groups of canonical amino acids, whereas the other half of the side chains are introduced by sulfonyl chlorides, providing an enormous chemical diversity. (2) They can form well-defined and stable secondary structures analogous to that of an α-helix, enabling the mimicry of the structures of natural peptides or proteins and maintaining or even improving the functions of their natural counterparts. (3) Their inherent resistance to degradation and enhanced cell permeability make this class of peptidomimetics ideal lead compounds or drug candidates.
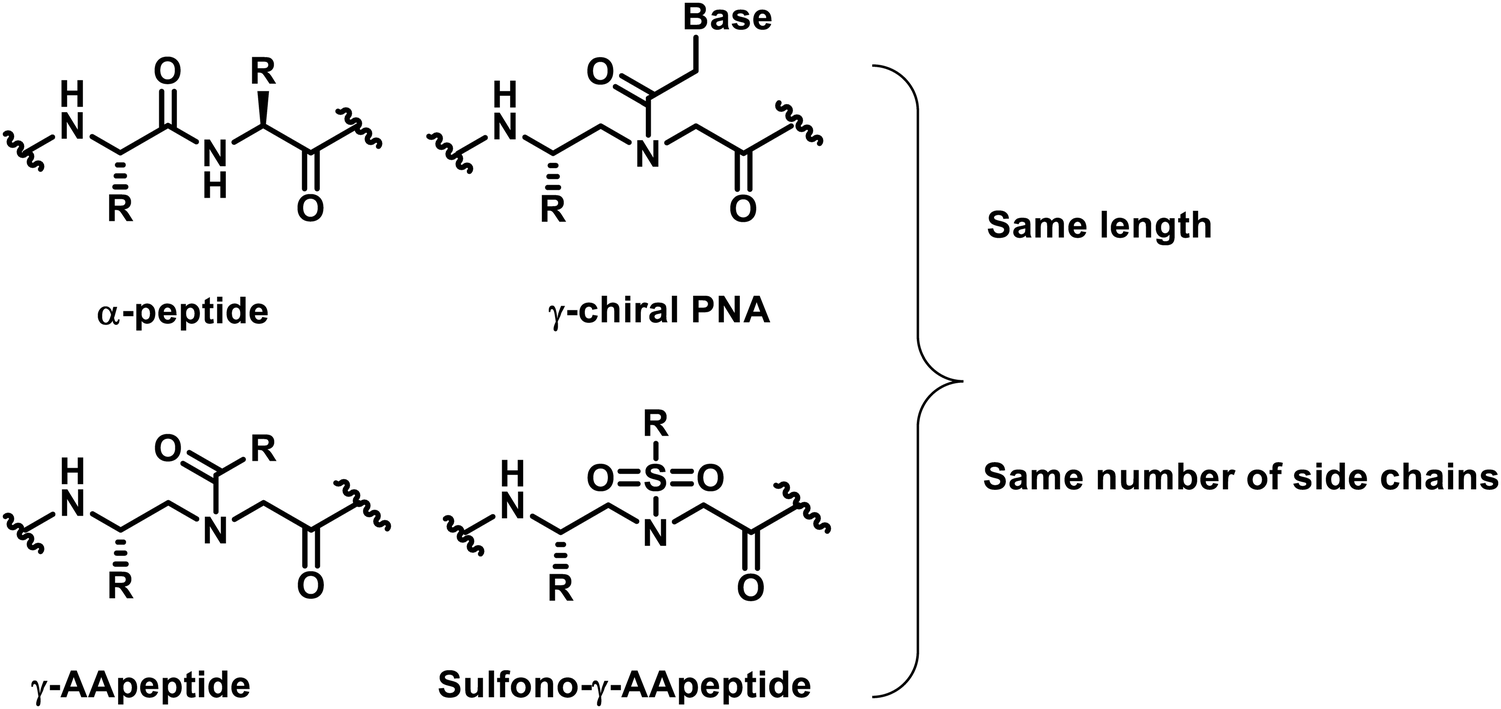 | ||
| Fig. 1 Chemical structures of α-peptide, γ-chiral PNA, γ-AApeptide and sulfono-γ-AApeptide. | ||
To date, we have conducted the research on the structure and functional applications of sulfono-γ-AApeptides in chemical biology and biomedical sciences and have obtained some promising results. Our exploration on sulfono-γ-AApeptides further expands our understanding of the structure, stability and internal mechanism of peptides and proteins. In this review, we focused on the review of the helical folding conformations of sulfono-γ-AApeptides and their biological applications as helical mimetics in medicinally relevant PPIs and provide our perspective on their potential in the mimicry of other biological molecules in the future.
Helical conformations
Polyamide sequences composed of both α-amino acid residues and sulfono-γ-AA residues with different ratios will adopt different types of helical conformations. The naming of the helical forms of sulfono-γ-AApeptides and their peptide hybrids adopts a convention based on the number of atoms in the pseudo-hydrogen-bonded ring.40,41Left-handed 414-helices
The 2D NMR spectroscopy investigation of homogeneous L-sulfono-γ-AApeptides provided an early indication that sulfono-γ-AApeptides are able to form helical structures.42 However, the structure generated by the NOE-constrained molecular dynamics was elusive, because the helical chirality could not be derived, and the hydrogen bonding mode was uncertain owing to dynamic solution structures. Recently, we have finally clarified the helicity and hydrogen bonding mode of the helix based on the atomic-level structural information of homogeneous L-sulfono-γ-AApeptides through their crystal structures (Fig. 2A).37 Peptides formed from homogeneous sulfono-γ-AApeptide building blocks derived from L-amino acids adopt left-handed 14-helices (Fig. 2B).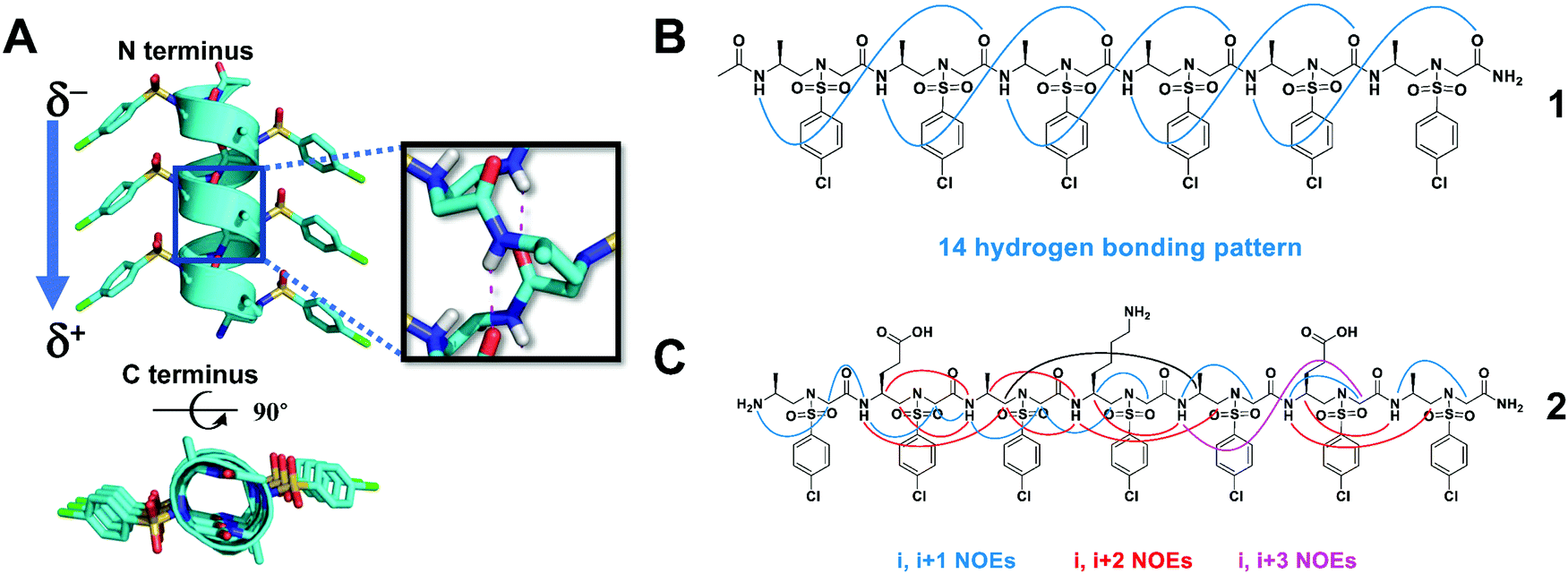 | ||
| Fig. 2 (A) Side and top views of the crystal structure of the representative oligomer 1. Hydrogen bonding is shown in red. (B) The intramolecular 14-hydrogen-bonding pattern of 1 detected in the crystal structure. (C) Summary of the detected NOESY cross-peaks of 5 mM oligomer 2 between hydrogen atoms on non-adjacent residues in CD3OH (10 °C). Three types of NOEs are displayed in different colors. Reproduced with permission from ref. 37. Copyright 2018 by John Wiley & Sons, Inc. | ||
The 14-helix is stabilized by hydrogen bonding between an amide proton at position i and a main chain carbonyl at position i + 3, forming a series of highly consistent 14-membered rings. This hydrogen bonding mode results in the formation of a macrodipole with a partial positive charge at the C-terminal and a partial negative charge at the N-terminal, which is the opposite of the macrodipole formed in a canonical α-helix. Due to its unique hydrogen bond pattern and side chain arrangement, it is named a 414-helix, meaning that each turn of the helix contains 4 side chains (three residues), and 14 atoms are in the pseudo-loop formed by intramolecular hydrogen bonds. The helical pitch of these L-sulfono-γ-AApeptides is 5.1 Å, which is very close to that of the α-helix (5.4 Å). The side chains are perfectly located on top of each other along the spiral axis, implying their ability to mimic the side chains of the α-helix on multiple helical faces. The existence of this secondary structure in the solution is also supported by circular dichroism (CD), 2D NMR spectroscopy (Fig. 2C) and molecular dynamics simulations in various solvents.37 The preference for left-handed helix formation is further rationalized by molecular dynamics simulations in methanol.37
Right-handed and left-handed 413-helices
Systematic conformational searches and crystal structures of the α/L-sulfono-γ-AA hybrid peptides have revealed inherent preferences for different helical conformations. Sequences with alternating L-sulfo-γ-AA structural units and α-amino acids in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 repeating pattern can form a right-handed helical conformation with a virtually identical helical pitch of 5.34 Å and a radius of 3.05 Å (Fig. 3A and B).34,36,38 The side chains of this type of peptide were almost perpendicular to the helical axis and point away from the peptide axis. The series of crystal structures we have obtained clearly reveal the neat and uniform 13-hydrogen bond pattern between the backbone carbonyl group of each residue and the amide N–H of the fourth residue with a distance of 1.95–2.11 Å. Therefore, the 413-helix is designated for this type of helical foldamer. The CD spectra show strong positive Cotton effects between 205 and 215 nm, also suggesting that 1:1 α/sulfono-γ-AApeptides adopt right-handed helical conformations. 2D NMR spectroscopy was further used to investigate the atomic-scale details of intramolecular interactions in solution, and the results are consistent with the single crystal data. It is also noted that the helical handedness of α/sulfono-γ-AA hybrid peptides is completely dependent on their chiral side chains, as α/D-sulfono-γ-AA hybrid peptides adopt the conformation of the left-handed 413-helix (Fig. 3C and D).34
:1 repeating pattern can form a right-handed helical conformation with a virtually identical helical pitch of 5.34 Å and a radius of 3.05 Å (Fig. 3A and B).34,36,38 The side chains of this type of peptide were almost perpendicular to the helical axis and point away from the peptide axis. The series of crystal structures we have obtained clearly reveal the neat and uniform 13-hydrogen bond pattern between the backbone carbonyl group of each residue and the amide N–H of the fourth residue with a distance of 1.95–2.11 Å. Therefore, the 413-helix is designated for this type of helical foldamer. The CD spectra show strong positive Cotton effects between 205 and 215 nm, also suggesting that 1:1 α/sulfono-γ-AApeptides adopt right-handed helical conformations. 2D NMR spectroscopy was further used to investigate the atomic-scale details of intramolecular interactions in solution, and the results are consistent with the single crystal data. It is also noted that the helical handedness of α/sulfono-γ-AA hybrid peptides is completely dependent on their chiral side chains, as α/D-sulfono-γ-AA hybrid peptides adopt the conformation of the left-handed 413-helix (Fig. 3C and D).34
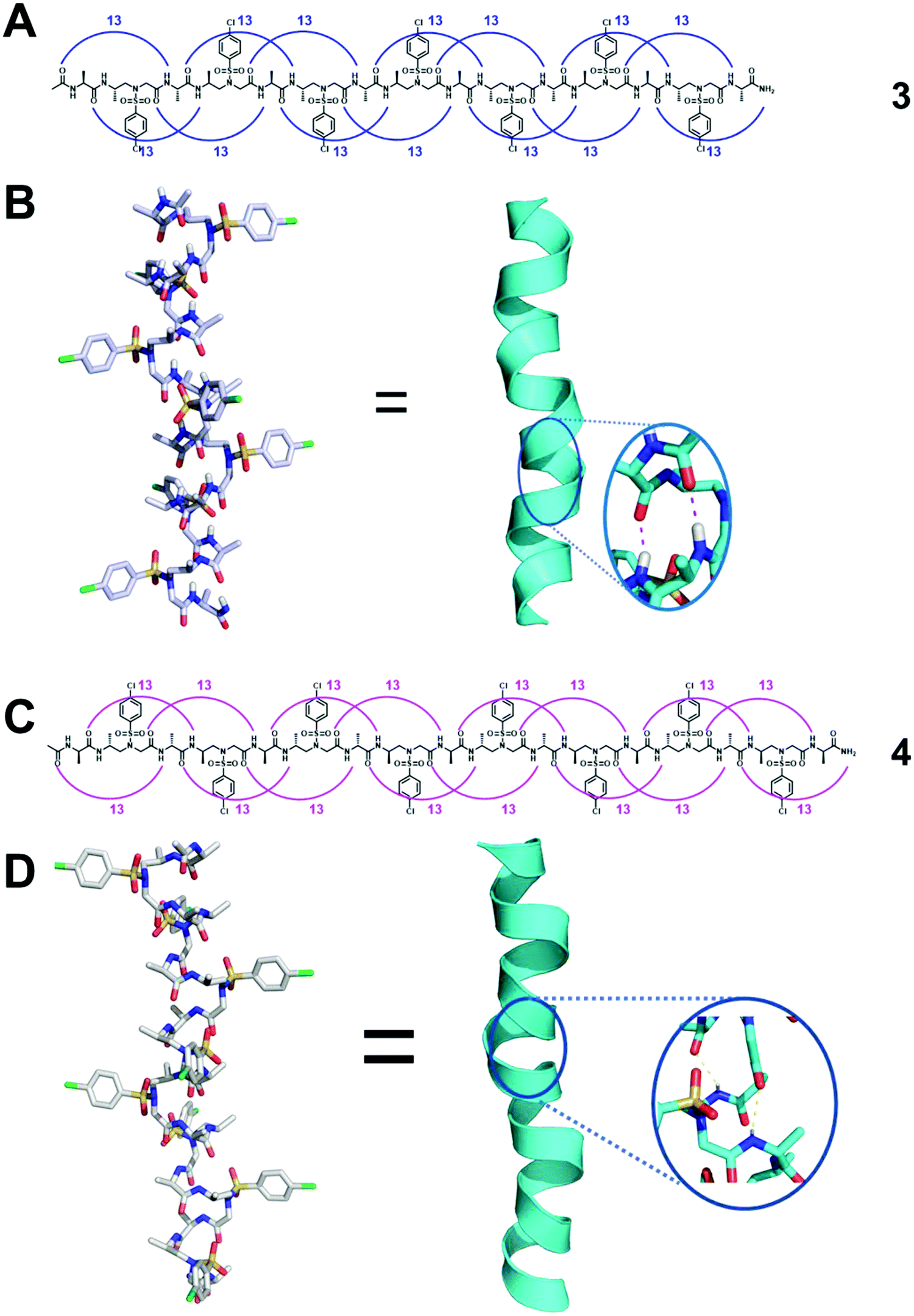 | ||
| Fig. 3 (A) Hydrogen bonding pattern of the representative oligomer 3 detected in crystal structures. (B) Crystal structure of 3 stabilized by intramolecular hydrogen bonds (magenta dashed line in the inset). (C) Hydrogen bonding pattern of the representative left-handed oligomer 4 detected in crystal structures. (D) Crystal structure of 4 stabilized by intramolecular hydrogen bonds (yellow dashed line in the inset). Reproduced with permission from ref. 38. Copyright 2018 American Chemical Society. | ||
Right-handed 4.516–14 helices
Heterogeneous oligomers consisting of a 2:1 pattern of L-α/D-sulfono-γ-AA amino acids can adopt a 16/14-helix conformation.39 The characteristic feature of this helix is an intertwined network of 16- and 14-membered hydrogen-bonded rings (Fig. 4). The discovery of this secondary structure further strengthens our confidence in using unnatural peptidomimetics and proteomimetics to mimic natural peptides and proteins.
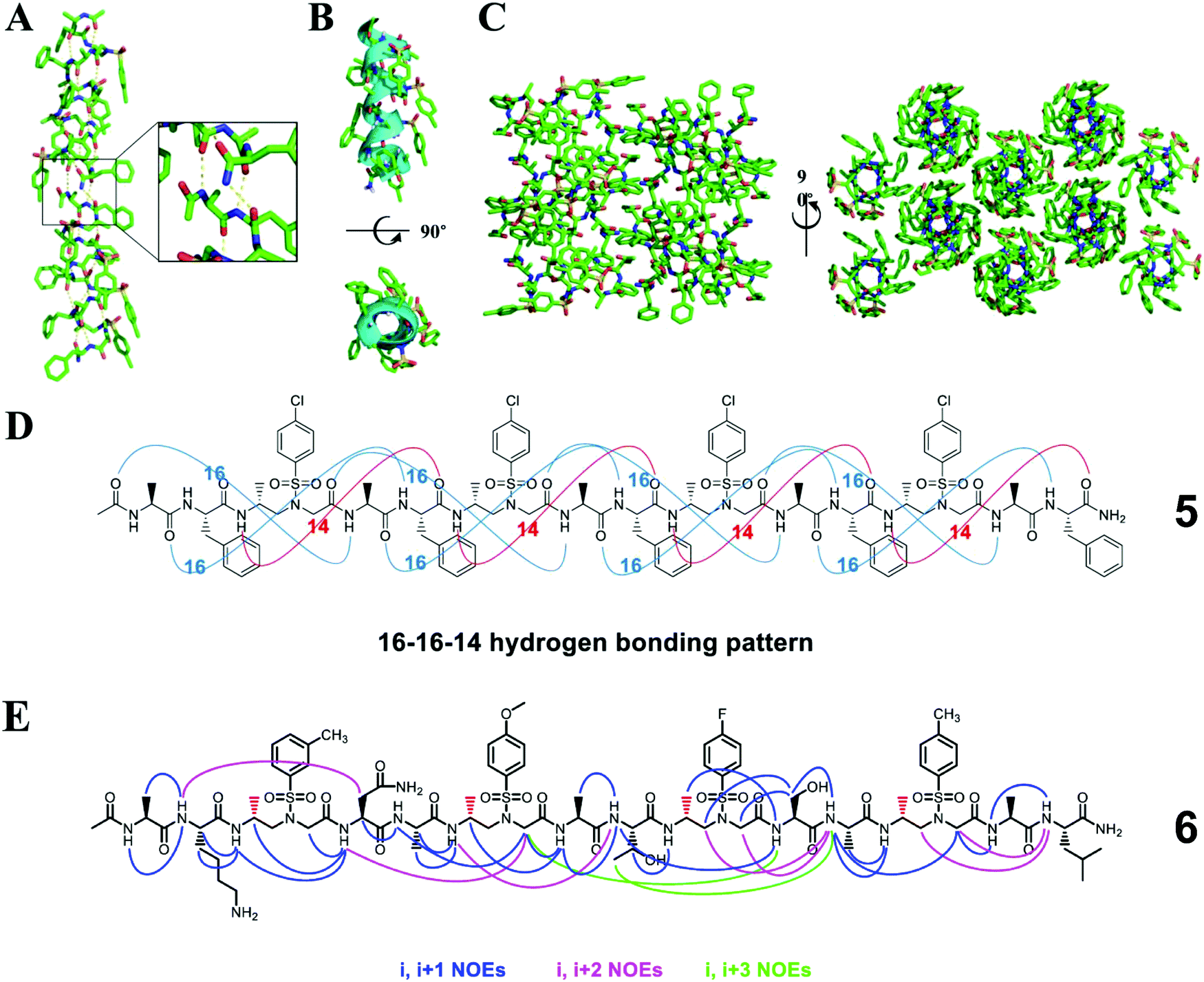 | ||
| Fig. 4 (A) Helical structure of a representative crystal 5 packing along the peptide axis; the intermolecular hydrogen-bonding pattern was shown in the inset for clarity. (B) Cartoon representation of 5 shown in oval to further clarify the helix. (C) Crystal packing of oligomer 5 viewed perpendicular and then down to the helical axis. (D) 16–16–14-Hydrogen-bonding pattern detected in the crystal structure of 5. (E) Summary of the detected NOESY cross-peaks of 5 mM oligomer 6 between protons on nonadjacent residues in CD3OH (10 °C). Three types of NOEs are displayed in different colors. Each D-sulfono-γ-AApeptide unit is considered as one residue. Reproduced with permission from ref. 39. Copyright 2017 by American Chemical Society. | ||
The 16/14-helix has been elucidated at the atomic level by single-crystal X-ray crystallography analysis and studied in organic solvents by CD and 2D-NMR.39 All the single crystals reveal a right-handed helical structure and an almost uniform helix radius of 2.6 Å, a pitch of 5.1 Å, and 4.5 residues per turn and show the same 16–16–14 hydrogen bond pattern. This helical mode formed between the N–H group of the α-amino acid residue and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the α-amino acid four residues earlier (type A 16-hydrogen bonding) or between the N–H group of the α-amino acid residue and the CO group of the D-sulfono-γ-AA four residues earlier (type B 16-hydrogen bonding) or between the N–H group of the D-sulfono-γ-AA residue and the CO group of the D-sulfono-γ-AA four residues later (14-hydrogen bonding) i + 4 → i hydrogen bonding with a distance of 2.1 Å (H⋯O distance). In contrast to the uniform arrangement of the amide bonds and the helix axis of the 14- and 13-helices, there are two types of amide bond orientations in the 16/14 helix. Therefore, this helical conformation was named the 4.516–14 helix, which represents the number of residues in each helical turn, and 16 or 14 atoms participate in the ring formed by intramolecular hydrogen bonds. Compared with the 310 helix and the α-helix, the 4.516–14 helix is not packed tightly, but is slightly tighter than the π spiral.39
O group of the α-amino acid four residues earlier (type A 16-hydrogen bonding) or between the N–H group of the α-amino acid residue and the CO group of the D-sulfono-γ-AA four residues earlier (type B 16-hydrogen bonding) or between the N–H group of the D-sulfono-γ-AA residue and the CO group of the D-sulfono-γ-AA four residues later (14-hydrogen bonding) i + 4 → i hydrogen bonding with a distance of 2.1 Å (H⋯O distance). In contrast to the uniform arrangement of the amide bonds and the helix axis of the 14- and 13-helices, there are two types of amide bond orientations in the 16/14 helix. Therefore, this helical conformation was named the 4.516–14 helix, which represents the number of residues in each helical turn, and 16 or 14 atoms participate in the ring formed by intramolecular hydrogen bonds. Compared with the 310 helix and the α-helix, the 4.516–14 helix is not packed tightly, but is slightly tighter than the π spiral.39
Biologically active sulfono-γ-AApeptides
Inhibitors of p53-MDM2/MDMX PPIs
p53 is a tumor suppressor, which plays an important role in the process of carcinogenic transformation, protecting higher organisms from cancer.43 MDM2 and MDMX have considerable structural homology; binding to the N-terminus of p53 causes the inhibition of transcriptional activity and degradation. Studies have shown that the overexpression of MDM2 and MDMX is a key factor leading to a variety of human cancers, making them promising targets for the development of anti-tumor drugs.44 The interaction of p53-MDM2 has been deeply studied, which provides a good platform for verifying the effectiveness of helical sulfono-γ-AApeptide mimics.Homogeneous L-sulfono-γ-AApeptides adopt a left-handed helix structure with a defined 14-atom hydrogen bond pattern, and its pitch is 5.1 Å, which is similar to α-peptide (5.4 Å).37 We speculated that the α-helical structure of p53 can be mimicked by the homogeneous L-sulfono-γ-AApeptides to inhibit the p53-MDM2/MDMX interaction. Based on this assumption, we designed and synthesized a series of L-sulfono-γ-AApeptides. The lead compound 7 obtained from this is one of the most effective unnatural peptidomimetic inhibitors for this interaction (Fig. 5).
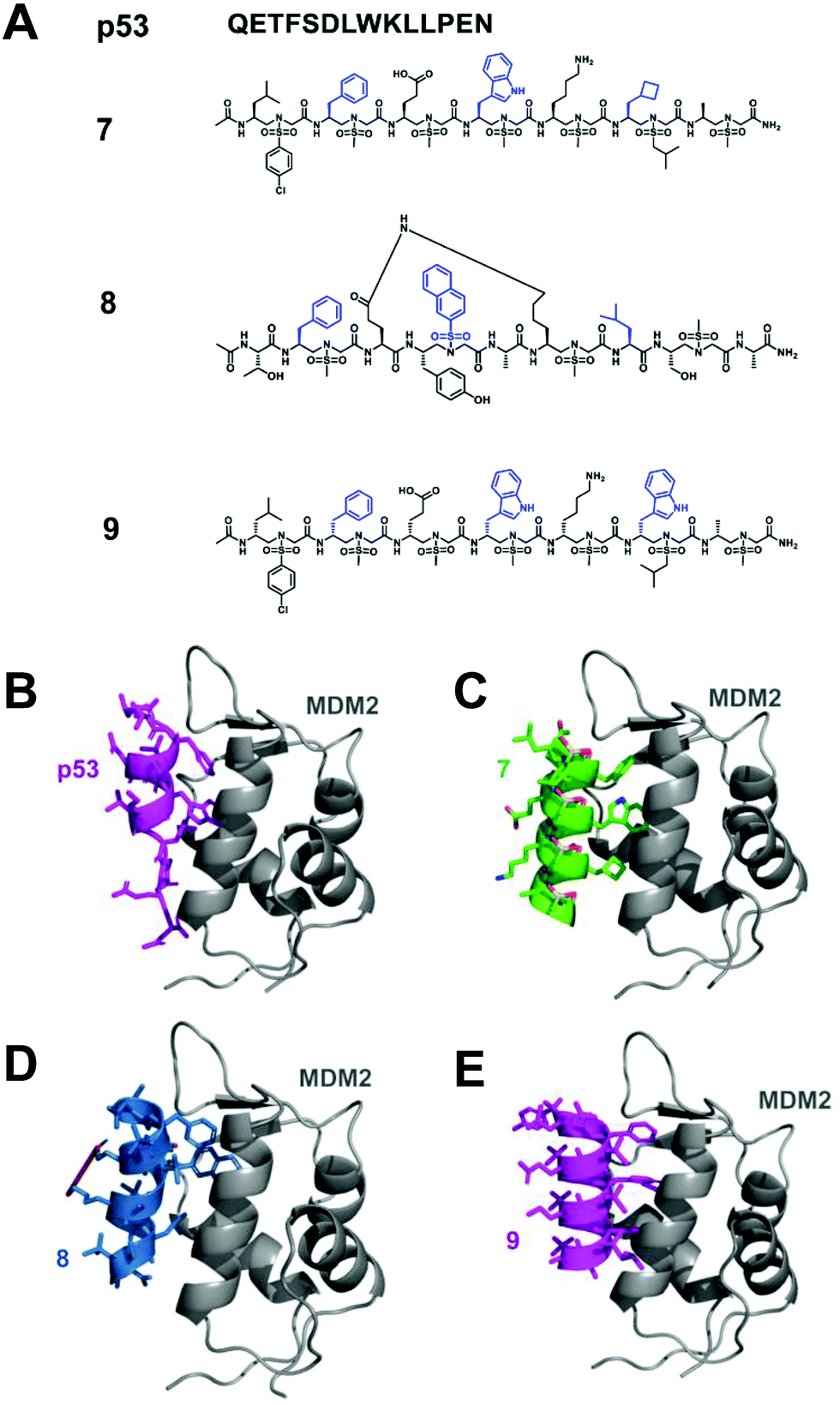 | ||
| Fig. 5 (A) Structures of p53, lead linear homogeneous L-sulfono-γ-AApeptide 7, lead stapled 1:1 α/sulfono-γ-AApeptide 8 and lead linear homogeneous D-sulfono-γ-AApeptide 9 investigated for the disruption of the p53-MDM2 interaction. The side chains mimicking Phe19, Trp23, and Leu26 in p53 are shown in blue. (B) Crystal structure of the interaction of p53 with MDM2 (PDB: 1YCR). (C) Modeling of the lead homogeneous L-sulfono-γAApeptide 7.32 (D) Modeling of the stapled 1:1 α/sulfono-γ-AApeptide 8.29 (E) Designed lead homogeneous D-sulfono-γ-AApeptide 9 interaction with MDM2.30 Reproduced with permission from ref. 30. Copyright 2020 by American Chemical Society. | ||
Although homogeneous L-sulfono-γ-AApeptides can be successfully applied to inhibit α-helix-mediated p53-MDM2/MDMX PPIs, there may be some limitations in mimicking the right-handed α-helix structure using the left-handed backbone. Our previous studies have shown that 1:1 α/L-sulfono-γ-AApeptides have a stable right-handed 413 pinwheel-like helical structure with a pitch of 5.34 Å, which is similar to the pitch of an α-helix (5.4 Å).38 Therefore, we envision that this type of peptidomimetic may be used to inhibit/modulate protein–protein interactions. To test this hypothesis, we took the p53–MDM2/MDMX interaction as an example, and the results showed that the lead compound 8 can bind tightly to MDM2/MDMX and block the p53–MDM2/MDMX interaction (Fig. 5).29
As the enantiomer of homogeneous L-sulfono-γ-AApeptides, homogeneous D-sulfono-γ-AApeptides should have a right-handed conformation like the α-helical peptide. We speculated that it might make the design of the α-helical simulation easier and more straightforward. Therefore, we also assessed whether it is suitable for the mimicry of helical domains in protein–protein interactions.30 The results show that the lead compound 9 can bind well to MDM2 and competitively block the p53-MDM2 interaction at the binding site (Fig. 5).
Inhibitors of BCL9-β-catenin PPIs
The Wnt/β-catenin signaling pathway plays an important role in embryonic development, tissue homeostasis, and several types of human cancers.45 As the central medium of signal transduction, β-catenin controls the expression of several key genes that regulate the cell cycle and apoptosis. The transcriptional activation of the Wnt/β-catenin signaling pathway depends on the formation of the β-catenin-BCL9 (or B9L) or β-catenin/TCF/LEF super complex.46,47 Therefore, the Wnt/β-catenin signal transduction can be inhibited by blocking the interaction of β-catenin-BCL9, so as to develop new anticancer drugs. Although there have been reports of using small molecule and peptide inhibitors to block the β-catenin-BCL9 interaction, there are few examples of unnatural peptidomimetic inhibitors.48–57 It is still a big challenge to design effective peptidomimetic inhibitors to enter cells and block the β-catenin–BCL9 interaction.We designed and synthesized a series of left-handed homogeneous sulfono-γ-AApeptides to mimic the helical structure of natural BCL9 (Fig. 6).33 The results show that the left-handed homogeneous sulfono-γ-AApeptides can structurally and functionally mimic the α-helical domain of BCL9 and selectively and more efficiently block the cancer-related β-catenin–BCL9 interaction in cells. The enzymatic stability of peptide drugs is critical to their biological activity, while sulfono-γ-AApeptides show no detectable degradation and exhibit extremely high stability, which increases their therapeutic application potential. This work can expand the use of sulfono-γ-AApeptides in the preparation of potent and cell-permeable peptidomimetic drugs and will have many applications in biomedicine and chemical biology.
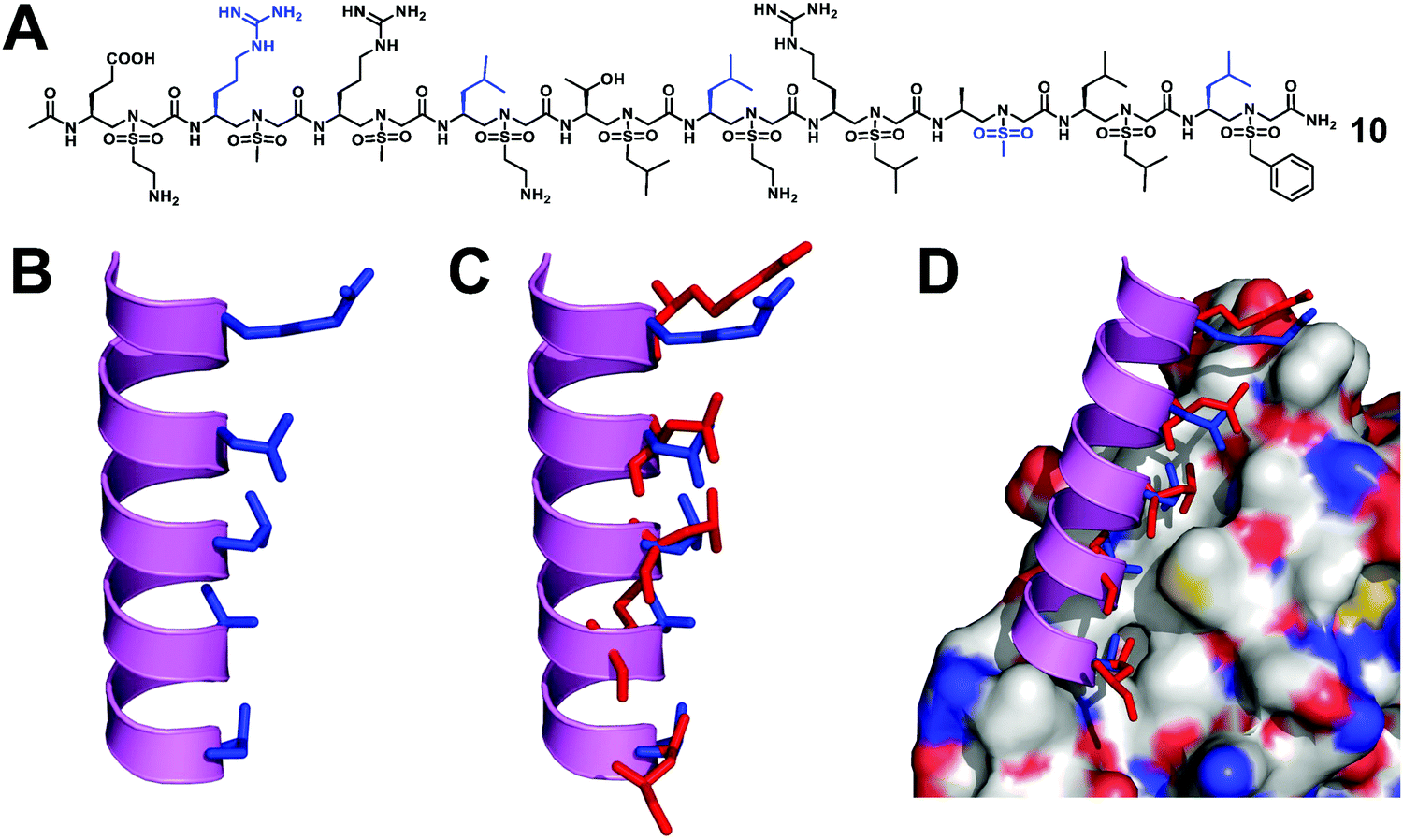 | ||
| Fig. 6 (A) Structure of a representative lead compound 10 investigated for the disruption of the β-catenin–BCL9 interaction. (B) Proposed structure of sulfono-γ-AApeptide 10. (C) Overlay of 10 with the critical residues of the BCL9 helical peptide. (D) Overlay of 10 with the critical residues of BCL9 on the binding surface of β-catenin (PDB: 2GL7). Reproduced with permission from ref. 33. Copyright 2019 by Proceedings of the National Academy of Sciences. | ||
Agonists of GLP-1R
The glucagon-like peptide-1 receptor (GLP-1R)58 belongs to the B subfamily of G protein-coupled receptors (GPCR), and its glucagon helical peptide ligand GLP-1 analogue is expected to be a therapeutic drug candidate for type 2 diabetes and obesity.59 However, the half-life of natural GLP-1 is very short; it will be rapidly degraded by proteases and excreted by the kidneys.60 Therefore, improving the stability of GLP-1 is essential for the development of diabetes drugs. The use of helical mimics is an effective strategy for the development of proteolytically stable GLP-1R agonists. However, the existing GLP-1 mimics need to retain most of the natural amino acid residues in order to maintain their biological activity.61–64 The reports of the unnatural backbone to mimic GLP-1, which may be due to the difference in the helicity between the α-helix and helical peptidomimetic, resulting in the use of completely unnatural peptidomimetics to mimic a long and complex α like GLP-1 helix is extremely challenging. Since the peptidomimetics with a complete unnatural backbone are more resistant to proteolysis than hybrid peptides, the development of GLP-1 helical peptide mimics with the entire unnatural backbone is more practical.We have conducted an in-depth analysis of the GLP-1/GLP-1R interaction mechanism and the single crystal structure of sulfono-γ-AApeptides and designed and synthesized a series of L-sulfono-γ-AApeptides which may be able to mimic the natural GLP-1 (Fig. 7).31 The results show that these peptidomimetics can mimic the residues on multiple faces of the GLP-1 α-helical domain in terms of structure and function. Among them, the lead compound obtained showed potent GLP-1R agonistic activity in both cell-based experiments and oral glucose tolerance tests in animals. More importantly, the degradation of sulfono-γ-AApeptides was not observed in the enzymatic stability and human serum stability experiments.
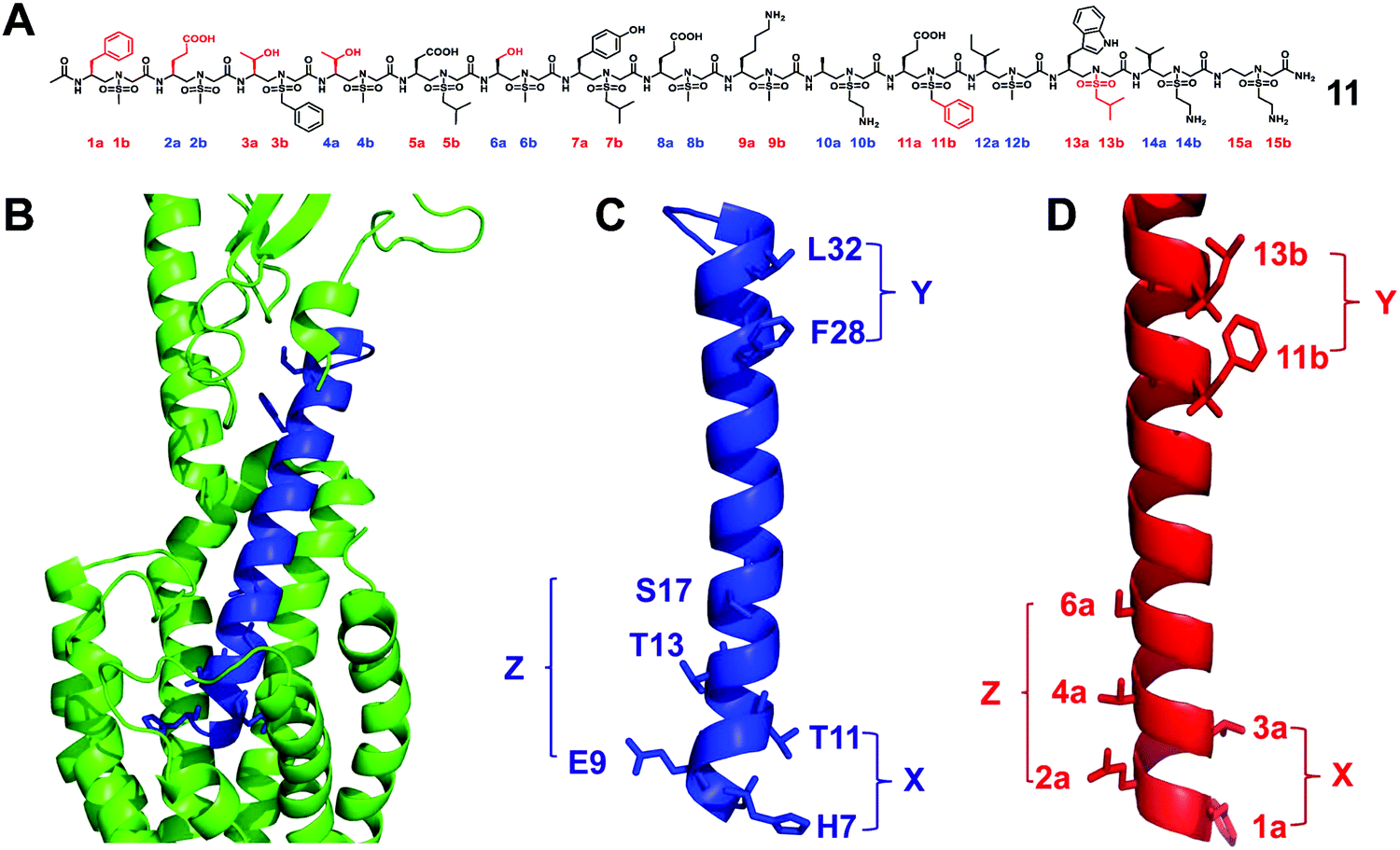 | ||
| Fig. 7 (A) Structure of the lead sulfono-γ-AApeptide 11. (B) GLP-1 binds to GLP-1R (PDB: 5VAI). GLP-1 (7-36) is shown in blue and GLP-1R is represented in green cartoon. (C) The helical domain of GLP-1 with critical residues shown as sticks. (D) Design of sulfono-γ-AApeptide 11, with side chains mimicking the important residues in C. (X, Y, and Z denote the faces of the residues on the helix, respectively.) Reproduced with permission from ref. 31. Copyright 2020 by the American Association for the Advancement of Science. | ||
The α/sulfono-γ-AApeptide hybrid analogues of glucagon
Glucagon is an essential hormone for regulating glucose homeostasis, and it is also a counter-regulatory hormone for insulin.65 Unlike GLP-1, which stimulates glucose uptake, the role of glucagon is to bind to the glucagon receptor and increase the concentration of glucose in the blood stream. Glucagon is considered to be an effective treatment for severe hypoglycemia; however, its plasma half-life is only a few minutes, which limits its widespread clinical application.66 Interestingly, although significant efforts to develop GLP-1 analogues with improved stability, there are few reports on the development of stable glucagon analogues, despite the fact that the development of proteolytically stable glucagon analogues can be quite important in biomedical sciences.67In order to explore the potential applications of α/sulfono-γ-AApeptide hybrids by the use of minimal insertion of sulfono-γ-AA residues to stabilize short-acting biologically active peptides, we chose this long and complex glucagon peptide as the target.28 We tried to exchange α amino acids with sulfono-γ-AA and other unnatural residues and combined with extension strategies, including functionalized peptides with polyethylene glycol (PEG) and long fatty acid chains to promote the binding of albumin and extend the in vivo activity, expecting to develop glucagon analogues with enhanced stability and prolonged biological activity (Fig. 8).
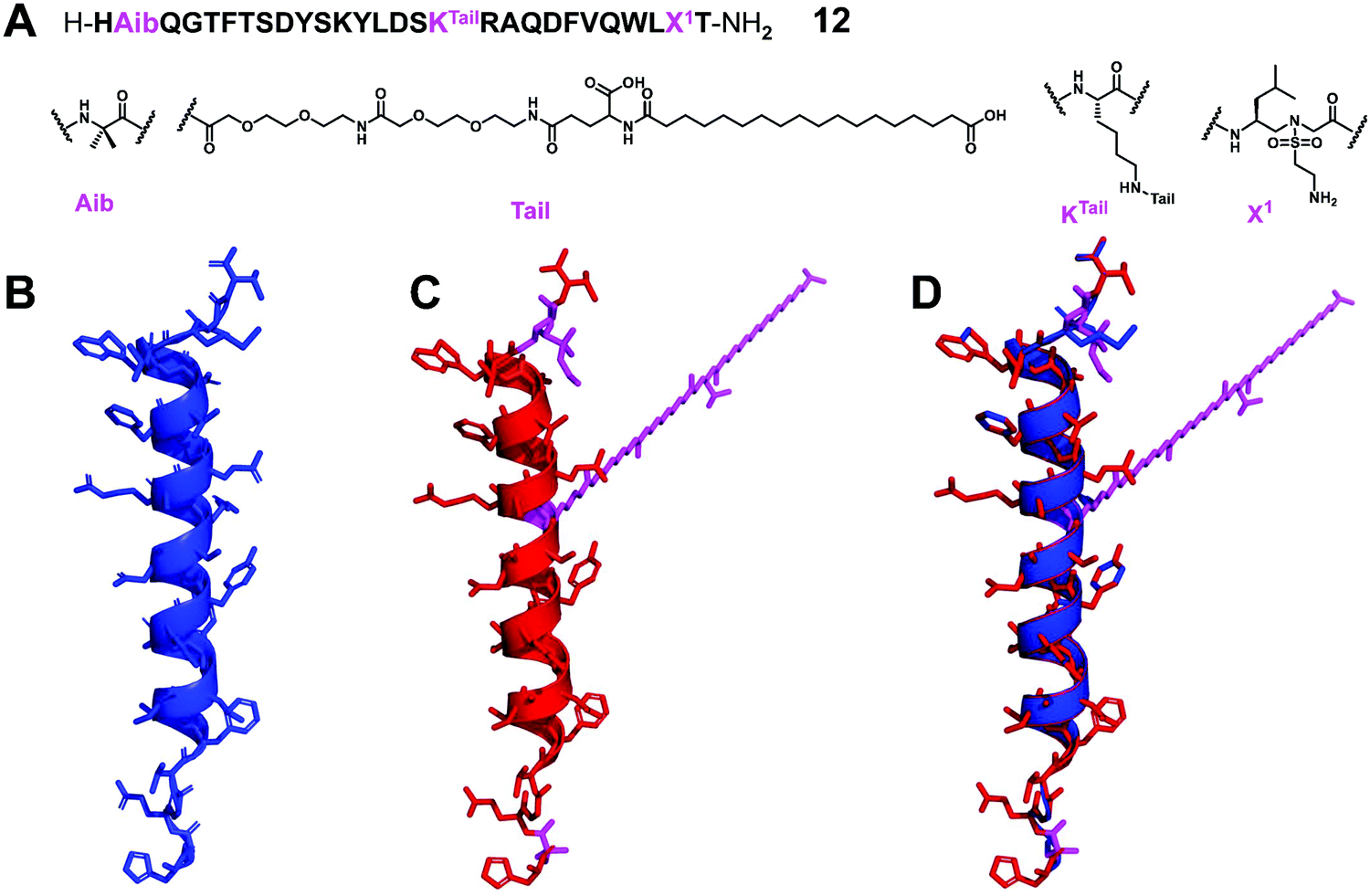 | ||
| Fig. 8 (A) Structure of the lead α/sulfono-γ-AApeptide hybrid glucagon analogue 12. (B–D) Crystal structure of glucagon (B) (PDB: 1GCN), modeling of the lead α/sulfono-γ-AApeptide hybrid 12 (C), and superimposition of glucagon with the lead α/sulfono-γ-AApeptide hybrid 12 (D). Reproduced with permission from ref. 28. Copyright 2021 by American Chemical Society. | ||
From the result point of view, this strategy shows enhanced stability and prolonged in vivo activity, while retaining natural glucagon-like activity. This work represents the first successful example of using sulfono-γ-AA residues to stabilize glucagon. This new backbone modification method may help to develop peptides with therapeutic potential. This strategy can be used to explore and develop stabilized analogues of other short-acting bioactive peptides.
Modulating angiogenesis by mimicking the VEGF
The regulation of key biological processes through peptide mimetics provides a huge opportunity to regulate processes that lead to serious diseases. One of the processes is angiogenesis, the formation of new blood vessels, which is a hallmark of cancer cells. The upregulation of angiogenesis leads to the proliferation of the vasculature network and subsequent tumor progression and metastasis. The down-regulation of angiogenesis is related to diseases such as ischemic heart disease, organ repair and wound healing.68–70By the use of the helical sulfono-γ-AApeptide scaffold, we designed unnatural peptidic foldamers that mimic the key helix binding domain on VEGF-A (helix α1) to inhibit the interaction of VEGF-A with VEGFR-1 and VEGFR-2.27 Another significant finding we reported is that we found that with only one residue difference (replace Phe with Trp), our sulfono-γ-AApeptide based helical mimetics 13 and 14 can activate or inhibit angiogenesis, respectively (Fig. 9). Our subsequent findings suggested that this distinct regulation of angiogenesis may be due to the selective binding of 13 to VEGFR-1 and 14 to VEGFR-2, respectively. The computational modeling and biological studies suggested that the binding selectivity is due to the slightly different binding interfaces of VEGFR-1 and VEGFR-2. This is also the first time that this hypothesis has been deduced by small unnatural peptidomimetics rather than large proteins. Overall, these molecules thus provide us a key to switch the angiogenic signaling, a biological process that balances the effects of proangiogenic and antiangiogenic factors, where imbalances lead to several diseases including cancer.
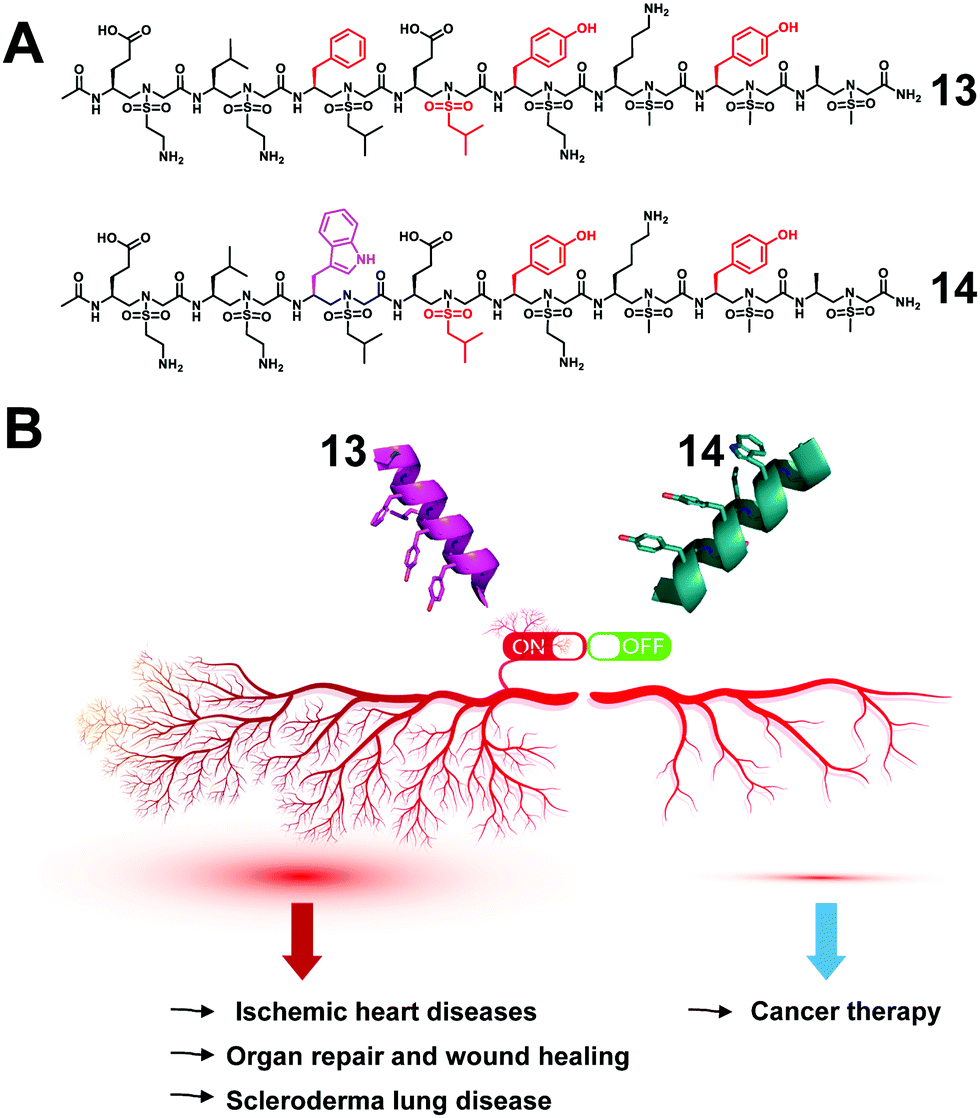 | ||
| Fig. 9 (A) Structures of the lead sulfono-γ-AApeptides 13 and 14. (B) Modulation of the angiogenic switch with the sulfono-γ-AApeptide mimics of VEGF-A. Reproduced with permission from ref. 27. Copyright 2022 by American Chemical Society. | ||
The effectiveness of angiogenic therapies, mostly targeting one element of the VEGF pathway, has been limited. This is believed to be due to the complexity of the pathway and ambiguity of the role of VEGF receptors at different stages of diseases. We believe that our success in effectively targeting VEGFR-1 and VEGR-2 selectively represents a major development in a holistic approach for targeting cancer and other diseases by modulating angiogenic receptors, and in addition, this also presents an extraordinary chemical biology tool to interrogate angiogenesis signaling.
Conclusions
Sulfono-γ-AApeptides represent a small subset of possible sequence-specific oligomers that researchers may consider when designing biomimetic structures. Although these types of unnatural peptidomimetics have been studied for just a relatively short period, a number of lead compounds with promising biological activity have been discovered through the design based on their crystal structures. Applying this structure-based drug design method to the field of protein–protein interactions should lead to the discovery of ligands for various drug-related targets, just like the case of β-peptides, oligoureas, and peptoids. So far, some progress has been made in the use of the secondary structures of sulfono-γ-AApeptides to design new peptidomimetic lead compounds, and the use of the well-defined tertiary structure of this type of peptidomimetic to mimic more complex natural peptides represents an additional challenge. If good progress can be made in the mimicry of the tertiary and quaternary structure of sulfono-γ-AApeptides in future research, it will have more fundamental significance for understanding protein folding and the design of drugs and functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the NIH RO1AI152416 (J. C.) and the NIH RO1 AG056569 (J. C.).Notes and references
- T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- J. S. Laursen, J. Engel-Andreasen and C. A. Olsen, Acc. Chem. Res., 2015, 48, 2696–2704 CrossRef CAS PubMed.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS PubMed.
- E. A. John, C. J. Massena and O. B. Berryman, Chem. Rev., 2020, 120, 2759–2782 CrossRef CAS PubMed.
- P. Milbeo, J. Martinez, M. Amblard, M. Calmès and B. Legrand, Acc. Chem. Res., 2021, 54, 685–696 CrossRef CAS PubMed.
- D. A. Guarracino, J. A. Riordan, G. M. Barreto, A. L. Oldfield, C. M. Kouba and D. Agrinsoni, Chem. Rev., 2019, 119, 9915–9949 CrossRef CAS PubMed.
- D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366–1375 CrossRef CAS PubMed.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed.
- M. Pasco, C. Dolain and G. Guichard, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, pp. 89–125 DOI:10.1016/B978-0-12-409547-2.12565-X.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- R. Gibadullin, C. J. Randall, J. Sidney, A. Sette and S. H. Gellman, J. Am. Chem. Soc., 2021, 143, 6470–6481 CrossRef CAS PubMed.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS PubMed.
- K. J. Lee, G. Bang, Y. W. Kim, M. H. Shin and H.-S. Lim, Bioorg. Med. Chem., 2021, 48, 116423 CrossRef CAS PubMed.
- Y. Fukuda, M. Yokomine, D. Kuroda, K. Tsumoto, J. Morimoto and S. Sando, Chem. Sci., 2021, 12, 13292–13300 RSC.
- J. S. Laursen, P. Harris, P. Fristrup and C. A. Olsen, Nat. Commun., 2015, 6, 7013 CrossRef CAS PubMed.
- M. Vestergaard, B. Skive, I. Domraceva, H. Ingmer and H. Franzyk, Int. J. Mol. Sci., 2021, 22, 5617 CrossRef CAS PubMed.
- L. Cussol, L. Mauran-Ambrosino, J. Buratto, A. Y. Belorusova, M. Neuville, J. Osz, S. Fribourg, J. Fremaux, C. Dolain, S. R. Goudreau, N. Rochel and G. Guichard, Angew. Chem., Int. Ed., 2021, 60, 2296–2303 CrossRef CAS PubMed.
- D. Sabatino, C. Proulx, P. Pohankova, H. Ong and W. D. Lubell, J. Am. Chem. Soc., 2011, 133, 12493–12506 CrossRef CAS PubMed.
- J. Gante, Synthesis, 1989, 405–413 CrossRef CAS.
- R. G. Ohm, M. Mulumba, R. M. Chingle, Ahsanullah, J. Zhang, S. Chemtob, H. Ong and W. D. Lubell, J. Med. Chem., 2021, 64, 9365–9380 CrossRef CAS PubMed.
- Y. Shi, P. Teng, P. Sang, F. She, L. Wei and J. Cai, Acc. Chem. Res., 2016, 49, 428–441 CrossRef CAS PubMed.
- P. Teng, Y. Shi, P. Sang and J. Cai, Chem. – Eur. J., 2016, 22, 5458–5466 CrossRef CAS PubMed.
- P. Sang, Y. Shi, B. Huang, S. Xue, T. Odom and J. Cai, Acc. Chem. Res., 2020, 53, 2425–2442 CrossRef CAS PubMed.
- S. Abdulkadir, C. Li, W. Jiang, X. Zhao, P. Sang, L. Wei, Y. Hu, Q. Li and J. Cai, J. Am. Chem. Soc., 2022, 144, 270–281 CrossRef CAS PubMed.
- P. Sang, H. Zeng, C. Lee, Y. Shi, M. Wang, C. Pan, L. Wei, C. Huang, M. Wu, W. Shen, X. Li and J. Cai, J. Med. Chem., 2021, 64, 13893–13901 CrossRef CAS PubMed.
- Y. Shi, P. Sang, J. Lu, P. Higbee, L. Chen, L. Yang, T. Odom, G. Daughdrill, J. Chen and J. Cai, J. Med. Chem., 2020, 63, 13187–13196 CrossRef CAS PubMed.
- P. Sang, Y. Shi, P. Higbee, M. Wang, S. Abdulkadir, J. Lu, G. Daughdrill, J. Chen and J. Cai, J. Org. Chem., 2020, 85, 10552–10560 CrossRef CAS PubMed.
- P. Sang, Z. Zhou, Y. Shi, C. Lee, Z. Amso, D. Huang, T. Odom, V. T.-B. Nguyen-Tran, W. Shen and J. Cai, Sci. Adv., 2020, 6, eaaz4988 CrossRef CAS PubMed.
- P. Sang, Y. Shi, J. Lu, L. Chen, L. Yang, W. Borcherds, S. Abdulkadir, Q. Li, G. Daughdrill, J. Chen and J. Cai, J. Med. Chem., 2020, 63, 975–986 CrossRef CAS PubMed.
- P. Sang, M. Zhang, Y. Shi, C. Li, S. Abdulkadir, Q. Li, H. Ji and J. Cai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10757–10762 CrossRef CAS PubMed.
- P. Teng, M. Zheng, D. C. Cerrato, Y. Shi, M. Zhou, S. Xue, W. Jiang, L. Wojtas, L.-J. Ming, Y. Hu and J. Cai, Commun. Chem., 2021, 4, 58 CrossRef CAS.
- Y. Shi, G. Yin, Z. Yan, P. Sang, M. Wang, R. Brzozowski, P. Eswara, L. Wojtas, Y. Zheng, X. Li and J. Cai, J. Am. Chem. Soc., 2019, 141, 12697–12706 CrossRef CAS PubMed.
- P. Teng, G. M. Gray, M. Zheng, S. Singh, X. Li, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2019, 58, 7778–7782 CrossRef CAS PubMed.
- F. She, P. Teng, A. Peguero-Tejada, M. Wang, N. Ma, T. Odom, M. Zhou, E. Gjonaj, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2018, 57, 9916–9920 CrossRef CAS PubMed.
- P. Teng, Z. Niu, F. She, M. Zhou, P. Sang, G. M. Gray, G. Verma, L. Wojtas, A. van der Vaart, S. Ma and J. Cai, J. Am. Chem. Soc., 2018, 140, 5661–5665 CrossRef CAS PubMed.
- P. Teng, N. Ma, D. C. Cerrato, F. She, T. Odom, X. Wang, L.-J. Ming, A. van der Vaart, L. Wojtas, H. Xu and J. Cai, J. Am. Chem. Soc., 2017, 139, 7363–7369 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071–13072 CrossRef CAS.
- D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi and S. H. Gellman, Nature, 1997, 387, 381–384 CrossRef CAS PubMed.
- H. Wu, Q. Qiao, Y. Hu, P. Teng, W. Gao, X. Zuo, L. Wojtas, R. W. Larsen, S. Ma and J. Cai, Chem. – Eur. J., 2015, 21, 2501–2507 CrossRef CAS PubMed.
- L. Chen, W. Borcherds, S. Wu, A. Becker, E. Schonbrunn, G. W. Daughdrill and J. Chen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4624–4629 CrossRef CAS PubMed.
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS PubMed.
- R. Nusse and H. Clevers, Cell, 2017, 169, 985–999 CrossRef CAS PubMed.
- J. Sampietro, C. L. Dahlberg, U. S. Cho, T. R. Hinds, D. Kimelman and W. Xu, Mol. Cell, 2006, 24, 293–300 CrossRef CAS PubMed.
- L. M. van Tienen, J. Mieszczanek, M. Fiedler, T. J. Rutherford and M. Bienz, eLife, 2017, 6, e20882 CrossRef PubMed.
- M. Feng, J. Q. Jin, L. Xia, T. Xiao, S. Mei, X. Wang, X. Huang, J. Chen, M. Liu, C. Chen, S. Rafi, A. X. Zhu, Y.-X. Feng and D. Zhu, Sci. Adv., 2019, 5, eaau5240 CrossRef CAS PubMed.
- M. Zhang, Z. Wang, Y. Zhang, W. Guo and H. Ji, J. Med. Chem., 2018, 61, 2989–3007 CrossRef CAS PubMed.
- K. B. Teuscher, M. Zhang and H. Ji, J. Med. Chem., 2017, 60, 157–169 CrossRef CAS PubMed.
- J. A. Wisniewski, J. Yin, K. B. Teuscher, M. Zhang and H. Ji, ACS Med. Chem. Lett., 2016, 7, 508–513 CrossRef CAS PubMed.
- M. Zhang, J. A. Wisniewski and H. Ji, Anal. Biochem., 2015, 469, 43–53 CrossRef CAS PubMed.
- L. R. Hoggard, Y. Zhang, M. Zhang, V. Panic, J. A. Wisniewski and H. Ji, J. Am. Chem. Soc., 2015, 137, 12249–12260 CrossRef CAS PubMed.
- M. de la Roche, T. J. Rutherford, D. Gupta, D. B. Veprintsev, B. Saxty, S. M. Freund and M. Bienz, Nat. Commun., 2012, 3, 680 CrossRef PubMed.
- Z. Wang, M. Zhang, V. Quereda, S. M. Frydman, Q. Ming, V. C. Luca, D. R. Duckett and H. Ji, J. Med. Chem., 2021, 64, 12109–12131 CrossRef CAS PubMed.
- Z. Li, M. Zhang, K. B. Teuscher and H. Ji, J. Med. Chem., 2021, 64, 11195–11218 CrossRef CAS PubMed.
- Z. Wang, M. Zhang, W. Luo, Y. Zhang and H. Ji, J. Med. Chem., 2021, 64, 5886–5904 CrossRef CAS PubMed.
- Y. Zhang, B. Sun, D. Feng, H. Hu, M. Chu, Q. Qu, J. T. Tarrasch, S. Li, T. Sun Kobilka, B. K. Kobilka and G. Skiniotis, Nature, 2017, 546, 248–253 CrossRef CAS PubMed.
- D. J. Drucker, Cell Metab., 2018, 27, 740–756 CrossRef CAS PubMed.
- K. Hupe-Sodmann, G. P. McGregor, R. Bridenbaugh, R. Göke, B. Göke, H. Thole, B. Zimmermann and K. Voigt, Regul. Pept., 1995, 58, 149–156 CrossRef CAS PubMed.
- L. M. Johnson, S. Barrick, M. V. Hager, A. McFedries, E. A. Homan, M. E. Rabaglia, M. P. Keller, A. D. Attie, A. Saghatelian, A. Bisello and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 12848–12851 CrossRef CAS PubMed.
- M. V. Hager, L. M. Johnson, D. Wootten, P. M. Sexton and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 14970–14979 CrossRef CAS PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. H. Zimmer, G. Guichard and S. R. Goudreau, Nat. Commun., 2019, 10, 924 CrossRef PubMed.
- P. M. Levine, A. T. Balana, E. Sturchler, C. Koole, H. Noda, B. Zarzycka, E. J. Daley, T. T. Truong, V. Katritch, T. J. Gardella, D. Wootten, P. M. Sexton, P. McDonald and M. R. Pratt, J. Am. Chem. Soc., 2019, 141, 14210–14219 CrossRef CAS PubMed.
- M. J. Pereira, K. Thombare, A. Sarsenbayeva, P. G. Kamble, K. Almby, M. Lundqvist and J. W. Eriksson, Mol. Cell. Endocrinol., 2020, 503, 110696 CrossRef CAS PubMed.
- I. Banerjee, M. Salomon-Estebanez, P. Shah, J. Nicholson, K. E. Cosgrove and M. J. Dunne, Diabetic Med., 2019, 36, 9–21 CrossRef CAS PubMed.
- J. R. Chabenne, P. A. Mroz, J. P. Mayer and R. D. DiMarchi, J. Med. Chem., 2020, 63, 3447–3460 CrossRef CAS PubMed.
- P. Shubik, J. Cancer Res. Clin. Oncol., 1982, 103, 211–226 CrossRef CAS PubMed.
- K. Gupta and J. Zhang, Postgraduate Med. J., 2005, 81, 236–242 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |