
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiochemical and structural characterization of Haemophilus influenzae nitroreductase in metabolizing nitroimidazoles†
Dake
Liu‡
a,
Thisuri N.
Wanniarachchi‡
b,
Guangde
Jiang
a,
Gustavo
Seabra
a,
Shugeng
Cao
 c,
Steven D.
Bruner
*b and
Yousong
Ding
*a
c,
Steven D.
Bruner
*b and
Yousong
Ding
*a
aDepartment of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), University of Florida, Gainesville, Florida 32610, USA. E-mail: yding@cop.ufl.edu
bDepartment of Chemistry, University of Florida, Gainesville, Florida 32611, USA. E-mail: bruner@chem.ufl.edu
cDepartment of Pharmaceutical Sciences, University of Hawai'i at Hilo, Hilo, Hawaii 96720, USA
First published on 16th February 2022
Abstract
Nitroheterocycle antibiotics, particularly 5-nitroimidazoles, are frequently used for treating anaerobic infections. The antimicrobial activities of these drugs heavily rely on the in vivo bioactivation, mainly mediated by widely distributed bacterial nitroreductases (NTRs). However, the bioactivation can also lead to severe toxicities and drug resistance. Mechanistic understanding of NTR-mediated 5-nitroimidazole metabolism can potentially aid addressing these issues. Here, we report the metabolism of structurally diverse nitroimidazole drug molecules by a NTR from a human pathogen Haemophilus influenzae (HiNfsB). Our detailed bioinformatic analysis uncovered that HiNfsB represents a group of unexplored oxygen-insensitive NTRs. Biochemical characterization of the recombinant enzyme revealed that HiNfsB effectively metabolizes ten clinically used nitroimidazoles. Furthermore, HiNfsB generated not only canonical nitroreduction metabolites but also stable, novel dimeric products from three nitroimidazoles, whose structures were proposed based on the results of high resolution MS and tandem MS analysis. X-ray structural analysis of the enzyme coupled with site-directed mutagenesis identified four active site residues important to its catalysis and broad substrate scope. Finally, transient expression of HiNfsB sensitized an E. coli mutant strain to 5-nitroimidazoles under anaerobic conditions. Together, these results advance our understanding of the metabolism of nitroimidazole antibiotics mediated by a new NTR group and reinforce the research on the natural antibiotic resistome for addressing the antibiotic resistance crisis.
Introduction
Antibiotics have brought revolutionary impacts to modern medicine and have been widely used for the treatment of a variety of infections and surgical procedures, cancer therapy and organ transplantation.1 These natural or synthetic compounds primarily target several major biological processes in microbes, including cell wall synthesis, protein synthesis, nucleic acid synthesis, and selected metabolic pathways.2,3 However, antibiotic resistance has become a global threat to human health and is constraining the clinical usage of antibiotics.4 This crisis is exacerbated, at least partially, by the misuse and overuse of antibiotics in the healthcare system and the agricultural sector,5 as pathogens and environmental microbes have the potential to evolve and/or acquire resistance mechanisms when antibiotics are present even at the sublethal level.6–8 In this regard, an advanced understanding of the evolution, dissemination and mechanisms of antibiotic resistance is critical for tackling the global antibiotic resistance crisis.9Nitroaromatics are an important family of antibiotics, e.g., chloramphenicol (1) and many 5-nitroimidazoles (Fig. 1). Chloramphenicol is the clinical choice for the treatment of many bacterial infections,10e.g., meningitis caused by Haemophilus influenzae and Neisseria meningitides. On the other hand, 5-nitroimidazoles have been used to treat infections caused by a variety of anaerobic bacteria and parasites, e.g., Trichomonas vaginalis, Helicobacter pylori, and Clostridium difficile.11 The representative 5-nitroimidazole metronidazole (MTZ, 2) is an essential medicine by the World Health Organization and had 5.8 million prescriptions in the USA alone in 2018.12 All 5-nitroimidazole antibiotics share the same 5-NO2-imidazole core and are structurally different primarily with substitutions on N1 and C2 (Fig. 1). The antimicrobial actions of 5-nitroimidazoles rely on the enzymatic one- or two-electron reduction of the 5-nitro group in microbial cells to generate toxic, short-lived intermediates (e.g., radical, 5-nitroso and 5-hydroxylamine species) and/or non-reactive metabolites (e.g., 5-aminoimidazoles) (Fig. S1, ESI†).13 The highly reactive species can modify microbial proteins, DNA and lipids, leading to the microbial killing, and reduction to 5-aminoimidazoles is one important mechanism of nitroimidazole resistance.14 Different types of microbial enzymes with a low redox potential can reduce the nitro group, such as ferredoxin, flavodoxin, thioredoxin reductases, and nitroreductases (NTRs).13 However, the generation of different reactive and non-reactive nitroimidazole reduction species in the enzyme reactions remains poorly characterized partly due to their chemical instability.15 Nonetheless, discovery and characterization of enzymes catalyzing the 5-nitroimidazole reduction provide opportunities to control drug activation and address the resistance of this important family of antibiotics.
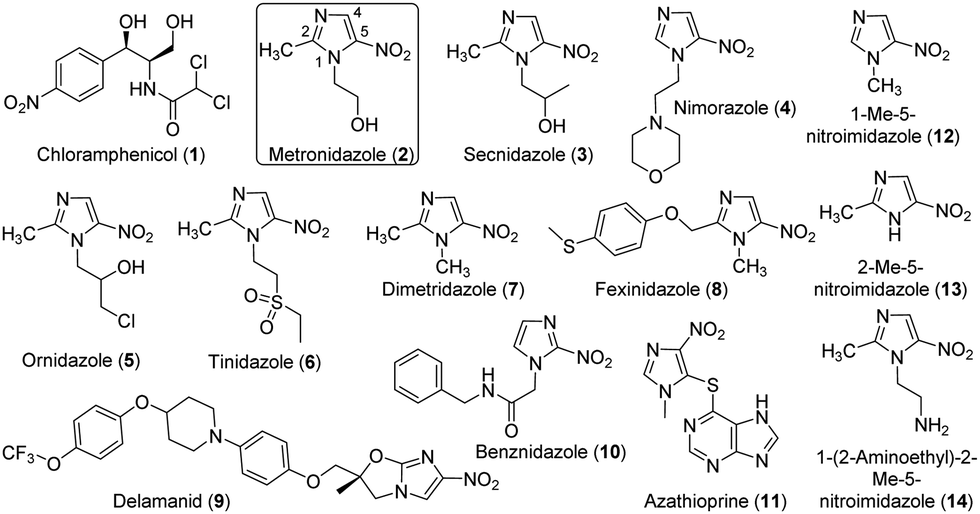 | ||
| Fig. 1 Chemical structures of chloramphenicol and selected nitro-imidazoles tested in this work. | ||
NTRs can be classified as type I oxygen-insensitive and type II oxygen-sensitive enzymes, depending on their oxygen sensitivity. The type II enzymes catalyze one-electron transfer in the nitro reduction and produce superoxide anion from molecular oxygen. The majority of well-characterized NTRs, however, belong to the type I, which can be further separated into the major and minor protein groups. The major group (e.g., NfsA from Escherichia coli) accepts only nicotinamide adenine dinucleotide phosphate (NADPH) as the electron donor, while those of the minor group (e.g., NfsB from E. coli) can use either nicotinamide adenine dinucleotide (NADH) or NADPH as the cofactor.16 The type I enzymes are generally homodimers with two flavin mononucleotide (FMN)-binding sites at the dimeric interface.17 Monomers adopt an α + β fold and their dimerization is required for noncovalent binding of FMNs and therefore the enzyme reactions.18,19 The enzymes typically follow a ping-pong bi–bi redox mechanism using a nicotinamide cofactor to supply electrons to the bound FMN,20 which subsequently reduces the nitroaromatics. Given the broad use of nitroaromatics,21 many wild type and engineered NTRs have been explored for a range of biotechnological and biomedical applications, such as bioremediation, cancer treatment and hypoxia imaging.14,22–24
The antimicrobial effects of 5-nitroimidazoles can be affected by the functions of various NTRs available in pathogens and other organisms. For example, the type I NTR RdxA of the human gastric pathogen H. pylori effectively activates MTZ (2),25 and its expression in E. coli leads to the host susceptibility to MTZ (2).26 Furthermore, multiple RdxA mutants are directly correlated to MTZ resistance in H. pylori.13 However, there are over 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 NTRs in public databases and only a small number have been functionally characterized.27,28 The vast unstudied NTRs can therefore be a valuable resource to advance the understanding of activation, metabolism and resistance of 5-nitroimidazoles and to develop a range of new applications.22,29 Herein, we report the detailed characterization of the type I NTR NfsB (HiNfsB) from Haemophilus influenzae in metabolizing clinically used 5-nitroimidazoles and its structural features. H. influenzae is a human pathogen in the proteobacteria phylum that can cause localized and invasive infections and is also a human commensal bacterium. In the present work, our bioinformatics studies discovered HiNfsB represents a group of 120 bacterial homologs, the vast majority of which have not been characterized yet. The recombinant enzyme showed varying activity toward 11 nitro-containing drugs, including chloramphenicol (1), 4- and 5-nitroimidazole antibiotics and the immunosuppressant azathioprine (11) (Fig. 1). In addition to known nitroimidazole reduction intermediates, we also observed stable, dimeric metabolites produced from three nitroimidazoles in high-resolution MS (HRMS) and tandem MS analyses. We further revealed the structural basis of the enzyme reaction and characterized the roles of four key active site residues through mutagenesis studies. Finally, the expression of HinfsB rendered the susceptibility of an E. coli mutant strain toward multiple 5-nitroimidazoles. Overall, this work provides a comprehensive characterization of HiNfsB in metabolizing nitroimidazole drugs and aids the development of this enzyme for biomedical and biotechnological applications.
000 NTRs in public databases and only a small number have been functionally characterized.27,28 The vast unstudied NTRs can therefore be a valuable resource to advance the understanding of activation, metabolism and resistance of 5-nitroimidazoles and to develop a range of new applications.22,29 Herein, we report the detailed characterization of the type I NTR NfsB (HiNfsB) from Haemophilus influenzae in metabolizing clinically used 5-nitroimidazoles and its structural features. H. influenzae is a human pathogen in the proteobacteria phylum that can cause localized and invasive infections and is also a human commensal bacterium. In the present work, our bioinformatics studies discovered HiNfsB represents a group of 120 bacterial homologs, the vast majority of which have not been characterized yet. The recombinant enzyme showed varying activity toward 11 nitro-containing drugs, including chloramphenicol (1), 4- and 5-nitroimidazole antibiotics and the immunosuppressant azathioprine (11) (Fig. 1). In addition to known nitroimidazole reduction intermediates, we also observed stable, dimeric metabolites produced from three nitroimidazoles in high-resolution MS (HRMS) and tandem MS analyses. We further revealed the structural basis of the enzyme reaction and characterized the roles of four key active site residues through mutagenesis studies. Finally, the expression of HinfsB rendered the susceptibility of an E. coli mutant strain toward multiple 5-nitroimidazoles. Overall, this work provides a comprehensive characterization of HiNfsB in metabolizing nitroimidazole drugs and aids the development of this enzyme for biomedical and biotechnological applications.
Results and discussion
HiNfsB reduces structurally diverse 5-nitroimidazoles in vitro
We first used HiNfsB as a query to mine its homologs in the non-redundant protein sequence database of NCBI by BLAST analysis. HiNfsB is selected because it has previously been shown to reduce chloramphenicol (1),30,31 leading the drug resistance. Furthermore, this enzyme can metabolize MTZ (2), nitrofurantoin, and 4-nitrobenzene derivatives,30,32 demonstrating substrate flexibility. However, MTZ metabolites generated by HiNfsB have not been reported,30 and the activity of this enzyme toward other nitroimidazoles is unknown. Our bioinformatic analysis identified 1046 HiNfsB homologs with 30–95% amino acid identity (as of August, 2021). The majority of these homologs (890) are from Firmicutes (e.g., Staphylococcus, Streptococcus and Bacillus genus), followed by Proteobacteria (114), Bacteroidetes (35), and Actinobacteria (7). We further analyzed these homologs by sequence similarity network (SSN) analysis to gain information on their functional relationship.33 With the cutoff of sequence identity at >60% (e value = 10−80), we generated 19 distinct clusters and 16 singletons (Fig. 2). In one cluster, HiNfsB is grouped with 113 homologs from the Proteobacteria phylum (87 from the γ-proteobacteria class and 27 from the β-proteobacteria class, e.g., Neisseria genus) and 7 from the Actinobacteria phylum (e.g., Rothia genus) (Table S1, ESI†). All of these HiNfsB homologs in the cluster have not been studied, except for only structure information of the homologs from Neisseria meningitidis ATCC 13091 (PDB: 6WT2) and four H. influenzae subspecies (e.g., PDB: 7LDQ). Interestingly, NfsB from the γ-proteobacterium E. coli (EcNfsB), which is one of the best-studied NfsBs,34 becomes a singleton in the SSN analysis. These results suggested that HiNfsB is functionally distinct from EcNfsB and belongs to a group of NTRs that have been poorly characterized so far.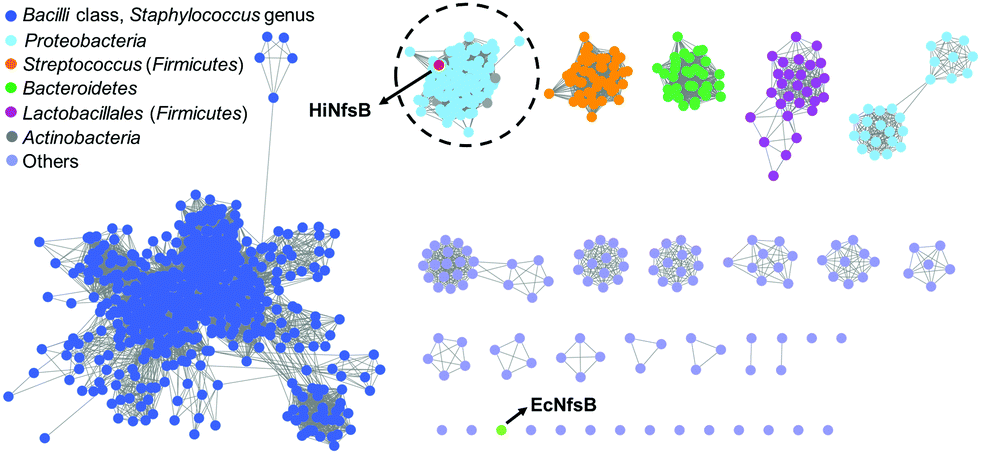 | ||
| Fig. 2 Sequence similarity network (SSN) analysis of HiNfsB homologs generated 19 distinct clusters and 16 singletons with a sequence identity of >60% (e value = 10−80). E. coli NfsB (EcNfsB) and H. influenzae NfsB (HiNfsB) are shown as light green and red, respectively. The HiNfsB cluster is highlighted. Different colours were used to represent homologs from various bacterial classes, genera and phyla as shown in the figure. | ||
We prepared recombinant HiNfsB through a single affinity column chromatography step after expressing a codon-optimized gene in E. coli (Fig. S2A, ESI†). The FMN content of HiNfsB was determined to be sub-stoichiometric at 0.52 ± 0.03 FMN per protein (Fig. S2B, ESI†). Nonetheless, as-purified recombinant HiNfsB was highly active toward MTZ (2) and chloramphenicol (1). With 1 μM enzyme, 2 mM MTZ (2) were completely metabolized in 3 hours (Fig. 3A) and 2 mM chloramphenicol (1) in 30 min (Fig. S3, ESI†).30 Furthermore, 0.1 μM HiNfsB consumed 21% of 2 (2 mM) in 10 min and the consumption was improved to about 29% after supplementing 1 μM FMN to the reaction (Fig. 3B). No MTZ (2) consumption was observed in negative controls lacking enzyme or an electron donor (Fig. 3A). We further determined that both NADH and NADPH equally supported the enzyme reaction (Fig. S4A, ESI†) and the enzyme showed a similar activity under both aerobic and anaerobic conditions (Fig. 3A), confirming HiNfsB to be a type I NTR. In addition, HiNfsB showed the highest activity toward MTZ at pH 7.4 and 25 °C (Fig. S4B, ESI†), which were used in the following enzyme reactions.
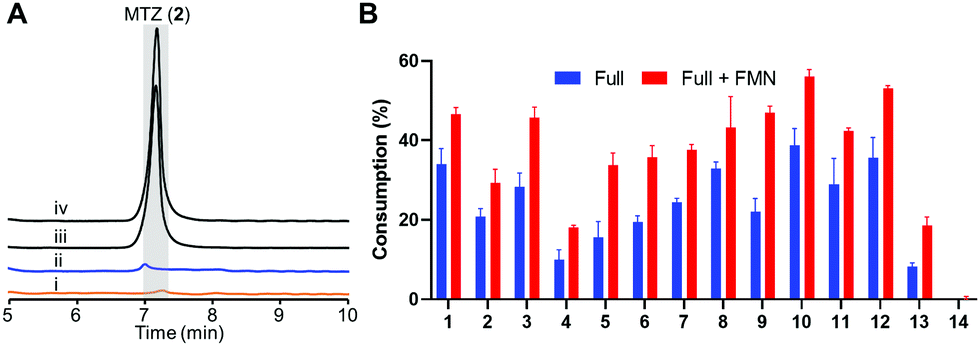 | ||
| Fig. 3 Recombinant HiNfsB metabolizes selected nitroimidazoles. (A) LC traces of the HiNfsB reactions with metronidazole (2) as substrate. The reactions contained 2 mM substrate, 1 μM enzyme, 1 mM NADP+, 20 mM glucose, and 16 μM glucose dehydrogenase (GDH) in 100 mM phosphate buffer (pH 7.4) and were incubated at room temperature for 3 hours under aerobic (i) or anaerobic (ii) condition. Negative controls lacked NADP+ (iii) or enzyme (iv). MTZ peak is highlighted in grey. (B) Consumption of 14 nitro-containing compounds by HiNfsB with or without 1 μM supplemented FMN. The reaction conditions were the same as (A) expect for the use of 0.1 μM enzyme and incubation for 10 min. The substrate was detected at 320 nm or 276 nm (for azathioprine). Consumptions (%) were determined as substrate peak areas at 10 min per those at 0 min × 100. The data represent means ± s.d. of three independent experiments. | ||
We next assessed the substrate scope of HiNfsB with nine additional clinically used nitroimidazoles (Fig. 1), including secnidazole (3), nimorazole (4), ornidazole (5), tinidazole (6), dimetridazole (7), fexinidazole (8), delamanid (9), benznidazole (10), and azathioprine (11). Similar to MTZ (2), compounds 3–6 have been used as antibiotics and antiprotozoal agents. Dimetridazole (7) has previously been used as a livestock feed additive due to its antiprotozoal activity. Fexinidazole (8) was recently approved for the treatment of African trypanosomiasis, while compound 10 is another antiparasitic agent for the treatment of Chagas disease. Delamanid (9) is a recently approved anti-TB agent that is used along with other drugs for the treatment of active drug-resistant tuberculosis. Different from other selected nitroimidazoles, azathioprine (11) is an immunosuppressant. All of these clinically used nitroimidazoles carry a 5-nitroimidazole moiety except for 4-nitroimidazole-containing delamanid (9) and azathioprine (11). These compounds are further different from one another by structurally diverse substituents on N1 and/or C2 of the nitroimidazole core (Fig. 1). For example, N1-substituents range from the proton and the methyl to the long bulky group in 9. Remarkably, HiNfsB demonstrated notable activity toward all substrates and the supplementation of FMN further enhanced the substrate consumption by an average of 1.6 times (Fig. 3B). After 10 min, HiNfsB showed the highest activity toward benznidazole (10, 38.8%) among all substrates, followed by fexnidazole (8, 32.9%). The three least active enzyme substrates were nimorazole (4), ornidazole (5), and tinidazole (6), but HiNfsB (0.1 μM) still consumed 10% to 19.5% substrates (2 mM) in 10 min (Fig. 3B). The effective consumption of compounds 8, 9, 10, and 11 by HiNfsB indicated the enzyme is tolerant to bulky substitutions on N1 and C2 of the nitroimidazole core and the nitro group at C4 and 5 of the imidazole (Fig. 3B). To gain additional insights into the substrate scope of HiNfsB, we tested 1-methyl-5-nitroimidazole (12) and 2-methyl-5-nitroimidazole (13) as dimetridazole (7) analogs and 1-(2-aminoethyl)-2-methyl-5-nitroimidazole (14) as MTZ (2) structural analog (Fig. 1). Strikingly, compound 12 turned to be the enzyme's second-best substrate as 35.6% of the substrate (2 mM) was consumed by 0.1 μM enzyme in 10 min, which was further improved to 53% in the presence of 1 μM FMN (Fig. 3B). On the other hand, the consumption of 13 by HiNFsB was about 2 to 3 times lower than 7, suggesting that the N1-methylation is favored by the enzyme. Compared with MTZ (2), compound 14 carries a 2-aminoethyl group on N1 but was barely consumed even with FMN supplementation (0.3%) (Fig. 3B). These results probably indicated that the physicochemical properties of the N1-substitution influence the enzyme nitro reduction reaction. We further computationally calculated the reduction potentials of all tested substrates. However, we found no correlation between substrate reduction potentials with their consumptions by HiNfsB (data not shown), illustrating the challenge in interpreting potential impacts of different substituents on nitroimidazole consumption by HiNfsB. Collectively, this work demonstrated that HiNfsB effectively metabolizes structurally diverse nitroimidazoles, particularly 5-nitroimidazoles. As a comparison, we prepared recombinant EcNfsB that contained 0.58 ± 0.06 FMN/protein, similar to HiNfsB (Fig. S2, ESI†). As-purified EcNfsB accepted compounds 1 to 11 as substrates but its activity was 1.2 to 9.2 times (in an average of 3.6 times) lower than HiNfsB under the same conditions (Fig. S5, ESI†), further demonstrating that HiNFsB represents a group of less-explored type I NfsB (Fig. 2).
The HiNfsB reaction generates both known and novel metabolites from nitroimidazoles
Despite effective substrate consumption, our HPLC analysis detected no notable products of almost all HiNfsB reactions, e.g., those with MTZ as substrate (Fig. S6, ESI†), agreeing with the inherent chemical instability of nitroimidazole reduction intermediates and metabolites.15 Indeed, extracted ion chromatogram (EIC) mass spectrometry (MS) also failed to detect nitroso, hydroxylamine, and amino species from the enzyme reactions except those containing compounds 8, 10 and 11 (Fig. 4A–C). For fexinidazole (8), we observed a putative hydroxylamine product (m/z = 266.1, Fig. 4A and Fig. S7, ESI†), while EIC analysis further discovered putative hydroxylamine and amine metabolites in the reactions with benznidazole (10) and azathioprine (11) as substrates according to the corresponding m/z values of these metabolites (Fig. 4B, C and Fig. S7, ESI†). To identify putative short-lived reactive species, we performed the enzyme reactions for 5 min and then incubated with benzyl chloroformate (CBZ) to derivatize putative hydroxylamine and amine species. EIC analysis of the derivatization mixtures identified a major new product generated from nimorazole (4) by HiNfsB (m/z = 329.1, Fig. 4D). This product was proposed to be dehydrated CBZ-hydroxylamino-nimorazole based on its high-resolution MS (observed [M + H]+: 329.1602; calculated [M + H]+: 329.1608) and MS/MS fragmentation (Fig. S8A, ESI†). EIC analysis also identified a small new peak whose content showed the expected molecular weight of CBZ-hydroxylamino-nimorazole (observed [M + H]+: 347.1694; calculated [M + H]+: 347.1714) and a similar MS/MS fragmentation to dehydrated CBZ-hydroxylamino-nimorazole (Fig. S8B, ESI†). Importantly, these two peaks had the same retention time, suggesting that the dehydrated product can be generated during the MS analysis and be more stable than CBZ-hydroxylamino-nimorazole. We further detected CBZ-hydroxylamine species in the HiNfsB reaction with substrates ornidazole (5) and dimetridazole (7) according to their corresponding m/z values and the results of HRMS/MS analysis supported their structure assignment (Fig. 4E, F and Fig. S8C, D, ESI†). On the other hand, this derivatization approach failed to detect any new product in all other reactions. Nonetheless, these results demonstrated that HiNfsB follows the classical two-electron chemistry of type I NTRs to generate multiple reactive and non-reactive metabolites from 5-nitroimidazoles17 and the chemical stability of these metabolites is significantly affected by original substrates themselves.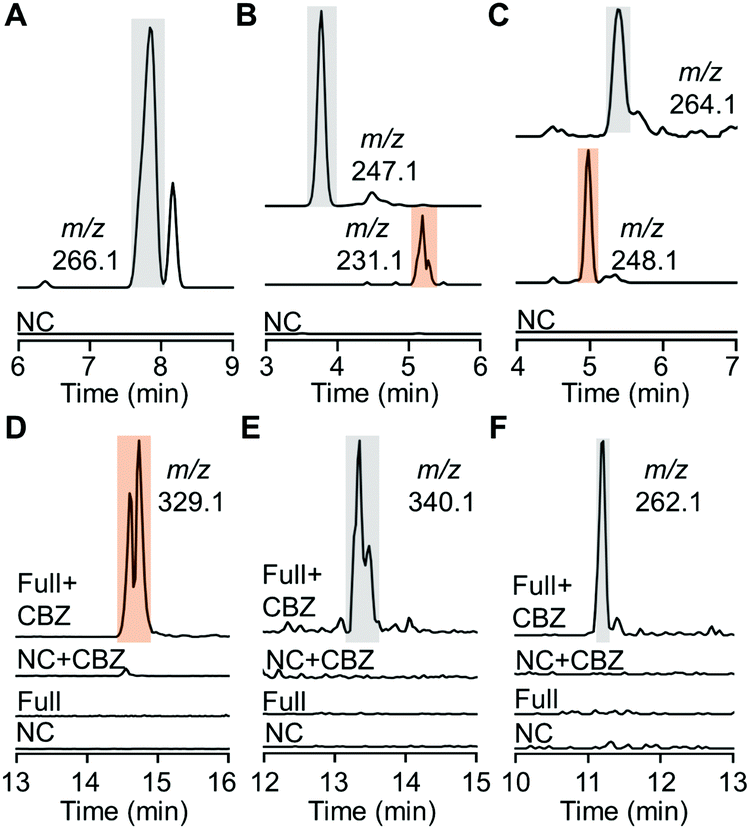 | ||
| Fig. 4 Metabolites produced from nitroimidazoles by HiNfsB. (A–C) EIC analysis of hydroxylamine and amine metabolites in the HiNfsB reactions with compounds 8, 10 and 11 as substrates, respectively. The full reactions contained 2 mM substrate, 1 μM enzyme, 1 mM NADP+, 20 mM glucose, and 16 μM GDH in 100 mM phosphate buffer (pH 7.4) and were incubated at room temperature for 3 hours. Corresponding m/z values were shown. (D–F) EIC analysis of CBZ derivatives in the HiNfsB reactions with compounds 4, 5 and 7 as substrates, respectively. The reaction conditions are the same as above, except for the use of 2 μM enzyme and incubation for 5 min. Negative control (NC) lacked enzyme. Corresponding m/z values were shown. The peaks of hydroxylamine metabolites and their CBZ derivatives are highlighted in grey, while those of amine metabolites and their CBZ derivatives are highlighted in orange. | ||
Careful inspection of the LC traces of the HiNfsB reactions with ornidazole (5), tinidazole (6) and dimetridazole (7) led to the identification of a new peak that has a higher retention time than the corresponding substrate (Fig. 5A). The molecular weights of all these peak contents are significantly higher than their corresponding substrates in the LC-HRMS analysis (Fig. 5A). Of note, the peak content generated from ornidazole (5) showed the characteristic isotope pattern of two chlorine atoms in the HRMS analysis. HRMS/MS analysis provided additional structural information of these peak contents and suggested dimeric metabolites formed from one molecule of reactive hydroxylamine species and one molecule of amine metabolite of ornidazole (5), tinidazole (6) and dimetridazole (7) generated by HiNfsB (Fig. S9–S11, ESI†). Potential pathways for the formation of these metabolites were proposed (Fig. S9–S11, ESI†) and await future validation. To assess if these new metabolites can be produced in living bacterial cells, we incubated tinidazole (6, 100 μM) with E. coli cells expressing HinfsB in the LB medium overnight. EIC analysis detected the same dimeric metabolite in the culture, and the induced expression of HiNfsB enhanced the amount of this metabolite (Fig. 5B). The metabolite showed a similar fragmentation pattern to the one from the enzyme reaction (Fig. S10B and C, ESI†). We also incubated EcNfsB expressing E. coli cells with compounds 5, 6, and 7 but did not observe these new metabolites (Fig. 5B), providing supportive evidence to the difference between EcNfsB and HiNfsB and the involvement of reactive intermediates in the production of these new metabolites. Of note, dimeric metabolites have been observed in the enzymatic metabolism of nitrobenzene and polynitroaromatics,35,36 likely through the condensation of two reactive nitroreduction species.
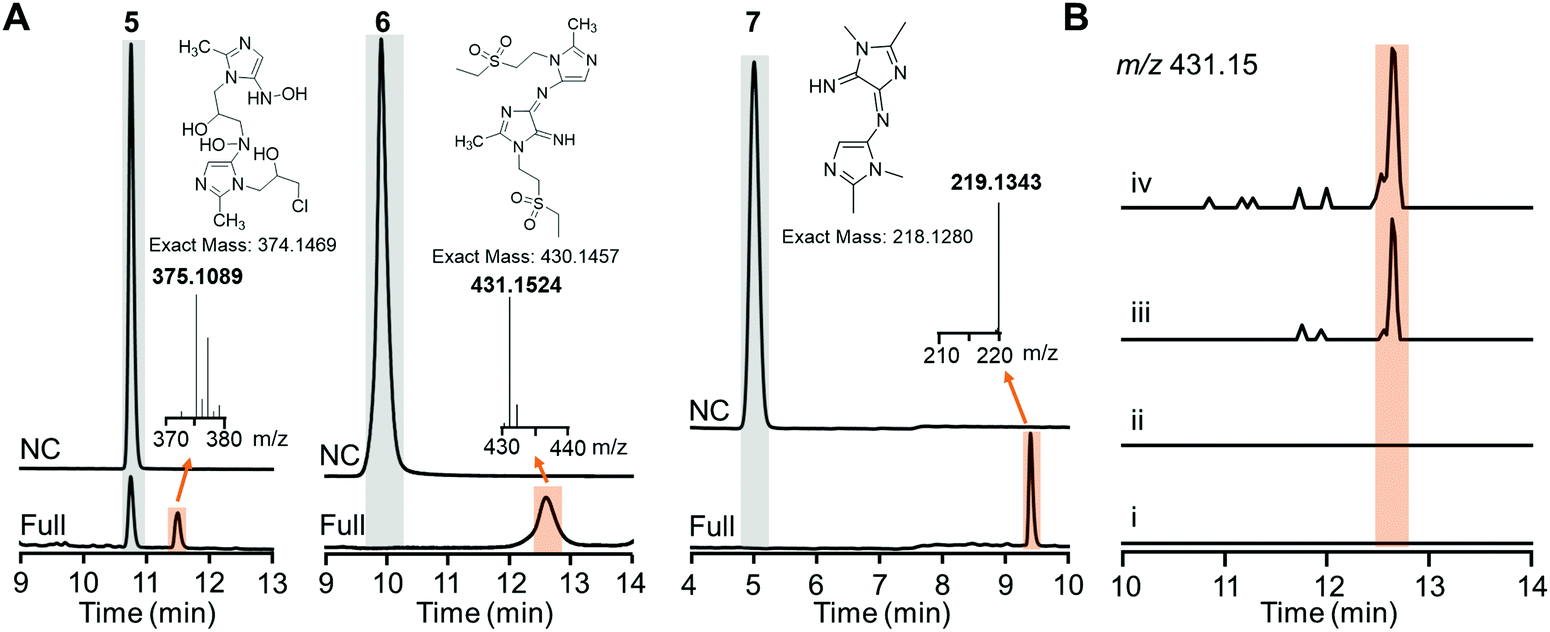 | ||
| Fig. 5 Proposed dimeric metabolites generated from nitroimidazoles by HiNfsB. (A) LC traces of the HiNfsB reactions with compounds 5, 6 and 7 as substrates. The reactions contained 2 mM substrate, 1 μM enzyme, 1 mM NADP+, 20 mM glucose, and 16 μM GDH in 100 mM phosphate buffer (pH 7.4) and were incubated at room temperature for 3 hours. Negative control (NC) lacked enzyme. The peaks of substrates and new metabolites are highlighted in grey and orange, respectively. HRMS spectra of these metabolites were included along with their putative chemical structures. (B) EIC analysis (m/z = 431.15) identified the putative dimeric metabolite from the crude extract of HiNfsB expressing E. coli culture fed with 6 (100 μM). (i) E. coli with the empty vector pET22b; (ii) EcNfsB expressing E. coli; (iii) HiNfsB expressing E. coli; (iv): HiNfsB expressing E. coli induced with 0.1 μM IPTG. All cultures were fed with 100 μM 6 and then incubated at 250 rpm, 37 °C overnight under aerobic conditions. Putative dimeric product was highlighted in orange. | ||
Overall X-ray structure of HiNfsB
To gain insight into the structural basis for HiNfsB catalyzed reduction of nitroimidazoles, we solved the X-ray crystal structure of HiNfsB soaked with substrate 1-methyl-5-nitroimidazole (12). Overall the structure showed a homodimer, a characteristic quaternary structure for members of the type I NTR family (Fig. 6A). Native-PAGE gel electrophoresis confirmed a dimeric HiNfsB complex in solution, consistent with our structural data (Fig. S12, ESI†). Each monomer contains a four-stranded, twisted β-sheet surrounded by seven α-helices. The β-sheet also makes up the core region of each monomer. The C-terminus of each monomer extends into the partner monomer as a β-strand, stabilizing the dimer. The overall structure closely resembles the characteristic α + β fold of other structurally known NTR homologs.19,37 EcNfsB is among the best-studied NTRs and its ligand-bound and unbound structures have been reported.19 Despite only ∼29% of sequence identity, HiNfsB and EcNfsB share overall similar secondary structural features (Fig. S13A and B, ESI†).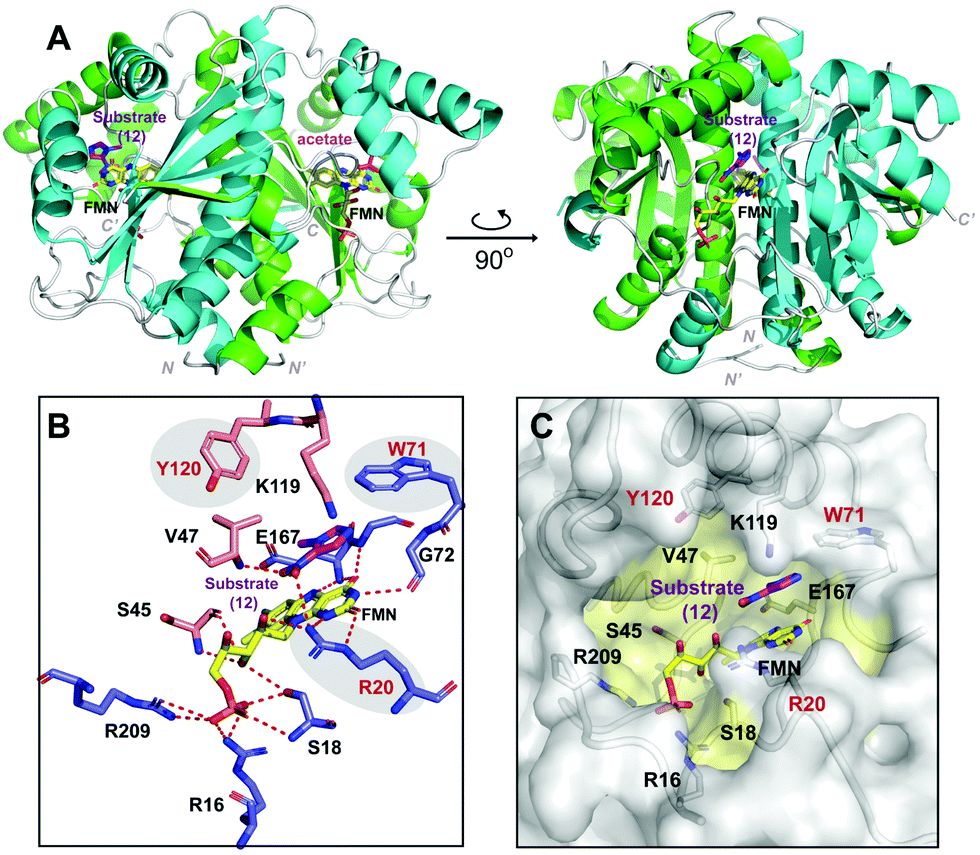 | ||
| Fig. 6 Overall protein structure of HiNfsB. (A) The two monomer chains of the active dimer are colored in green and cyan. FMN and ligands are bound in the dimer interface and are represented as colored sticks. (B) The interactions of HiNfsB/FMN/1-methyl-5-nitroimidazole (12). Residues from one monomer are colored in blue and those from the other are in light gold. Polar/hydrogen bonding interactions are shown as dash lines. (C) Accessible surface representation of the active site. 12 is shown in the pocket and residues lining the substrate/FMN binding pocket are highlighted in yellow. | ||
The cofactor FMN and the substrate bind at the dimer interface, resulting in an expected two active sites per homodimer. Well-defined electron density that correlates to FMN bound in both active sites shows clear non-covalent binding of the cofactor. The final refined protein model depicts an oxidized form of FMN, bound in a classical butterfly conformation.19,37 The re-face of FMN faces the hydrophobic substrate-binding cavity while the si-face is solvent exposed. The cofactor is bound to HiNfsB by polar interactions from both protein chains at the dimer interface (Fig. 6B). The phosphate group of FMN is stabilized by two arginine residues (Arg16 and Arg209) and one serine residue (Ser18) all belonging to a single monomer. In contrast, the FMN isoalloxazine ring and ribityl moieties make protein interactions with residues of both dimer subunits involving polar contacts with residues Ser18, Arg20, Ser45, Gly72 and Glu167.
The two HiNfsB monomers are highly similar (RMSD 0.802 Å, all atoms) and have only minor positional changes evident in flexible loop regions. Clear electron density for the substrate 1-methyl-5-nitroimidazole (12) is present in one of the two active sites of the dimer and electron density in the second active site is consistent with acetate (Fig. S14, ESI†). The asymmetric dimer with a single active site occupied has been previously observed in other flavoprotein structures.38,39 In addition, small molecules such as acetate are known competitive NTR inhibitors.17 Consistent with our results, five crystal structures of NfsBs from four H. influenzae subspecies have very recently been deposited into the protein data bank, each with a homodimer and three containing two acetates bound (e.g., PDB: 7LDQ). However, residues in the HiNsfB/12 active site are more ordered in the ligand-bound active site compared to HiNfsB/acetate, as shown by lower B-factors (Fig. S15, ESI†). Of note, our HiNfsB structure highly resembles the recently deposited ones. For example, the calculated RMSD between HiNfsB and PDB structure 7LDQ is 0.958 Å on all atoms (Fig. S16, ESI†). To investigate potential structural changes upon substrate binding, we sought to determine additional protein structures. We crystallized HiNfsB protein in the absence of any ligands. However, extra electron density was observed in the binding pocket adjacent to FMN. As it is not possible to accurately identify the bound ligand, the extra electron density was eventually modeled with nicotinate (Fig. S17, ESI†). Nicotinic acid is a cellular metabolite of E. coli and is likely copurified with recombinant HiNfsB. In addition, it was previously used as a substrate analog in the structural characterization of EcNfsB.19 The structure of HiNsfB/nicotinate is highly similar to HiNsfB/12 (RMSD = 0.742 Å, all atoms) and shows no apparent conformational change in the active site, indicating rigidity of the overall structure of the enzyme and a fixed solvent exposed active site. Further comparison of HiNsfB/nicotinate to the EcNsfB/nicotinate structure shows similar binding orientation of the nicotinate where the nicotinate ring stacks against the isoalloxazine ring of FMN (Fig. S17, ESI†). In addition, the active site pocket of all structures is of sufficient size to accommodate binding of structurally diverse substrates (Fig. 6C).
Key residues of HiNfsB relevant to the reduction of nitroimidazoles
1-Methyl-5-nitroimidazole (12) forms π-stacking and hydrophobic interactions with the re-face of FMN in the active site (Fig. 6B and C). The substrate 12 lies rotated relative to the nicotinate binding orientation however, the nitro groups are aligned in a similar position compared to EcNfsB/nicotinate structure (Fig. S13, ESI†),19 possibly as a result of the steric bulk of the methyl group at the C1 position. The nitro group of 12 is at a distance of 4.5 Å from the N5 of FMN isoalloxazine ring, an orientation that is ∼1 Å from an optimal favorable spacing for reduction (Fig. S13C, ESI†). In addition, 12 forms hydrogen bonding interactions with the Val47 backbone of chain A and Arg20 of chain B (Fig. 6B). The modest number of protein/ligand interactions is consistent with the high substrate promiscuity of HiNfsB (Fig. 3). Similarly, the carboxylate group of nicotinate interacts with the same residues (Val 47 and Arg20) in the nicotinate bound structure of HiNfsB. The side chain of Arg20 interacts with the nitro group of the substrate (12) along with hydrogen bonds to isoalloxazine ring and ribityl moiety of FMN, suggesting a general role for this residue in ligand binding and catalysis. Interestingly, Arg20 is conserved in only two of the closest structural homologs of HiNfsB (PDB: 6WT2 and 2HAY) but is a lysine residue in others, including EcNfsB. We further aligned the amino acid sequences of all 1046 HiNfsB homologs in Fig. 2. Indeed, Lys is found in the corresponding position of 860 homologs, while all 120 homologs in the HiNfsB cluster and the cluster in orange (homologs from Streptococcus, Fig. 2) carry Arg in this position (Fig. S18, ESI†). The conservation of Lys or Arg suggests the functional role of this position in type I NfsBs and potentially reflects the difference in chemical reactivity of HiNfsB and EcNfsB towards specific nitroimidazole substrates. To evaluate the role of Arg20, we prepared recombinant HiNfsB R20A and R20K mutants (Fig. S2, ESI†). The R20A mutant contained only 0.09 ± 0.02 FMN per protein, and the FMN content in the R20K mutant was similar to the wild type. These results confirm the involvement of Arg20 in the cofactor binding. We further tested the two mutants against all nitro-containing substrates, except for the least active substrate 14 (Fig. 3B). The R20A mutation showed no effect on the consumption of compounds 3 and 13 by HiNfsB but significantly decreased the consumption of all other tested substrates by up to 5 times (Fig. 7 and Fig. S19, ESI†). FMN supplementation enhanced the enzyme consumption of compounds 3, 4, 6, 7, 10, 12, and 13 by 1.3 to 6 times, but did not affect the rest substrates. Of note, the R20A mutant was inactive toward compound 8 but FMN supplementation led to the consumption of about 21% of this substrate. The R20K mutant retained significant activities toward all tested substrates, ranging from 7.6% (compound 12) to 21.9% (compound 10) (Fig. 7). FMN supplementation further improved the average substrate consumption from 13.0% to 28.4%. However, the catalytic activity of the R20K mutant was still significantly lower than HiNsfB (e.g., 13.0% vs. 22.0% average substrate consumption with as-purified enzymes), except for compound 13 for which as-purified R20K mutant was three times more active. Furthermore, the R20A mutant showed a 2.5-time higher activity than the R20K mutant toward compounds 3 and 11. Compared with EcNfsB, the R20K mutant showed a higher activity in consuming almost all other substrates (e.g., 6.3 times improvement toward chloramphenicol), except for compound 11 (Fig. S5B, ESI†). Collectively, these results demonstrated that R20 is a key residue in substrate binding and cofactor coordination.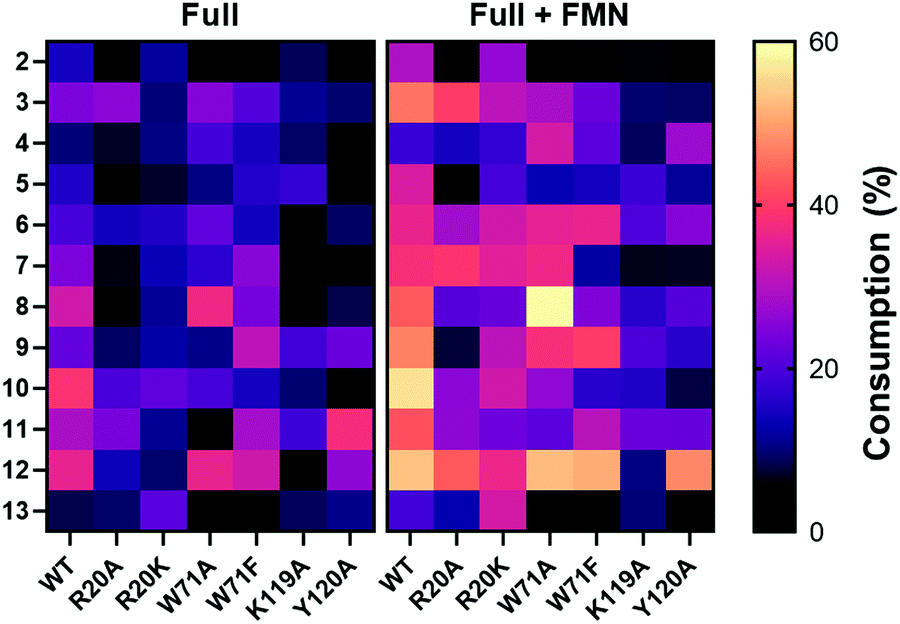 | ||
| Fig. 7 Heat map of the consumption of compounds 2–13 by WT HiNfsB and its R20A, R20K, W71A, W71A, Y120A and K119A mutants. The reactions contained 2 mM substrate, 0.1 μM enzyme, 1 mM NADP+, 20 mM glucose, and 16 μM GDH in 100 mM phosphate buffer (pH 7.4) and were incubated at room temperature for 10 min. The data represent means ± s.d. of three independent experiments. | ||
Previous studies on EcNsfB demonstrated that Phe70 and Phe124 regulate access of the anticancer prodrug CB1954 into the active site along with the regioselectivity in reducing its two nitro groups.40 Of note, nicotinic acid is stacked between the Phe124 and FMN (Fig. S13A, ESI†), and the F124K mutant improves the sensitivity of E. coli toward CB1954 by five times. Superposition of HiNfsB with EcNfsB (PDB 1ICU) shows that in HiNfsB these residues are replaced with Trp71 and Lys119, respectively (Fig. S13A, ESI†). Trp71 is in proximity to the ligand and may participate in substrate binding through hydrophobic and/or π interactions. Indeed, our molecular docking with different nitroimidazoles in the active site of HiNfsB revealed that Arg20, Trp71, Lys119 and Tyr120 all may influence substrate binding (e.g., ornidazole shown in Fig. S20, ESI†). To characterize the roles of Trp71, Lys119 and Tyr120, we generated W71A, W71F, K119A, and Y120A mutants (Fig. S2, ESI†). Compared with the wild type, the W71A mutant, with or without FMN supplement, showed the increased consumption of nimorazole (4) and fexinidazole (8) and lowered its activity toward almost all other substrates, particularly no consumption of azathioprine (11) without FMN supplement (Fig. 7). Similarly, except for nimorazole (4), the W71F mutant showed decreased activity toward almost all substrates. These mutants contained the same level of FMN as the WT (Fig. S2, ESI†). How the replacement of Trp71 with Ala and Phe influences HiNfsB's activity toward different nitroimidazoles awaits future studies. K119A and Y120A mutants also had the same level of FMN as the wild type (Fig. S2, ESI†) but both showed decreased activity toward almost all substrates (Fig. 7 and Fig. S19, ESI†). In particular, the K119A mutant lost its activity toward dimetridazole (7), fexinidazole (8), and 1-methyl-5-nitroimidazole (12) without FMN supplement. Collectively, these mutagenesis results revealed that the binding of nitroimidazoles to the enzyme active site involves multiple residues and different modifications on the nitroimidazole scaffold can influence enzyme activity.
E. coli cells expressing HinfsB become susceptible to 5-nitroimidazoles under anaerobic conditions
We next determined the minimal inhibition concentrations (MICs) of E. coli cells transformed with EcnfsB or HinfsB toward 11 nitroimidazoles, including metronidazole (2), secnidazole (3), nimorazole (4), ornidazole (5), tinidazole (6), dimetridazole (7), fexinidazole (8), benznidazole (10), 1-methyl-5-nitroimidazole (12), 2-methyl-5-nitroimidazole (13), and 1-(2-aminoethyl)-2-methyl-5-nitroimidazole (14). Compounds 12–14 are nitroimidazole structural analogs, while the rest have proven antibiotic activities (Fig. 1). Anti-TB agent delamanid (9) was not selected as its bulkier structure could lead to unequal penetration efficiency into E. coli cells with others. To eliminate basal metabolism of nitroimidazoles by NfsA and NfsB, both genes were deleted in E. coli MG1655 cells in a previous work41 and the resultant E. coli Δnfsa/b strain (DA65117 provided by Prof. Dan I. Andersson) was used here. The cells with or without IPTG induction were incubated with serial concentrations of these compounds under both aerobic and anaerobic conditions. Except for benznidazole (10), the MIC values of all tested compounds were 0.5 mM or higher with E. coli Δnfsa/b::EcnfsB and E. coli Δnfsa/b::HinfsB under aerobic conditions (Table 1). IPTG induction did not affect the susceptibility of E. coli Δnfsa/b::EcnfsB but reduced the MIC values of 3, 5, 6, 7 and 12 toward E. coli Δnfsa/b::HinfsB. Compounds 8, 13 and 14 possessed no antibacterial activity under all tested conditions. The lowest MIC was observed with secnidazole (3) at 62.5 μM. Benznidazole (10) is an antiparasitic drug and possesses potent antimicrobial activities toward both multiple aerobes and anaerobes.42 IPTG induction further reduced the MIC values of 10 toward both transformed E. coli strains by 8 times (Table 1). This result suggested that EcNfsB and HiNfsB can catalyze the nitroreduction of benznidazole in E. coli cells, agreeing with the in vitro consumption of this compound by these two enzymes (Fig. 3B and Fig. S5, ESI†). On the other hand, HiNfsB effectively consumed other tested nitroimidazoles (except for 14) in vitro, and it remains unclear how E. coli Δnfsa/b::HinfsB cells are more resistant to other compounds under aerobic conditions. We further evaluated the susceptibility of transformed E. coli cells toward nitroimidazoles under anaerobic conditions. Compounds 8, 13 and 14 remained inactive. On the other hand, we observed that the MIC values of almost all other compounds were decreased (Table 1), while benznidazole (10) was still among the most active compounds. Even without IPTG induction, the anaerobic conditions already led E. coli Δnfsa/b::HinfsB to be sensitive toward the majority of compounds with the lowest MIC value of 15.6 μM (10 and 12). As the T7 promoter is known to be leaky,43 a low level of HiNfsB may cause the increased susceptibility. In comparison, the susceptibility of E. coli Δnfsa/b::EcnfsB was improved by 2 times only toward compounds 5, 6, 7 and 10 under anaerobic conditions. IPTG induction further reduced the MIC values of all compounds except for 8, 13 and 14 by 2 to 16 times. The most active compounds (MIC = 7.8 μM) toward E. coli Δnfsa/b::HinfsB included 2, 6, 10 and 12. E. coli Δnfsa/b::EcnfsB showed the highest susceptibility toward 10 (MIC = 3.9 μM), followed by 12 with the MIC value of 31.3 μM. Interestingly, nimorazole (4) showed antibacterial activity (MIC = 31.3 μM) only when E. coli Δnfsa/b::HinfsB was induced by IPTG under anaerobic conditions. Overall, these results suggested that when expressed in E. coli, HiNfsB effectively reduces a range of nitroimidazoles to cytotoxic species under anaerobic conditions, making them bactericidal agents. This work further lays the basis toward the investigation of other factors than the bioactivation of nitroimidazoles in influencing their bactericidal activities.| Compound | EcNfsB | EcNfsB + IPTG | HiNfsB | HiNfsB + IPTG | ||||
|---|---|---|---|---|---|---|---|---|
| +O2 | −O2 | +O2 | −O2 | +O2 | −O2 | +O2 | −O2 | |
| a The concentration unit is μM. | ||||||||
| 2 | >500 | 500 | >500 | 125 | >500 | 62.5 | >500 | 7.8 |
| 3 | >500 | 500 | >500 | 250 | >500 | >500 | 62.5 | 62.5 |
| 4 | >500 | >500 | >500 | 500 | >500 | 500 | >500 | 31.3 |
| 5 | >500 | 250 | >500 | 62.5 | >500 | 62.5 | 250 | 15.6 |
| 6 | 500 | 250 | 500 | 250 | >500 | 62.5 | 250 | 7.8 |
| 7 | 500 | 250 | 500 | 62.5 | 500 | 31.3 | 125 | 15.6 |
| 8 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| 10 | 31.3 | 15.6 | 3.9 | 3.9 | 62.5 | 15.6 | 7.8 | 7.8 |
| 12 | 500 | 500 | 500 | 31.3 | 500 | 15.6 | 125 | 7.8 |
| 13 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| 14 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
Conclusions
In the present study, we focused on the biochemical and structural characterization of a less-explored, type I group B NTR from a human pathogen H. influenzae (HiNfsB). HiNfsB exhibited nitroreduction activity toward a series of structurally diverse nitroimidazoles (Fig. 3B), including the anti-TB agent delamanid (9) and the immunosuppressant azathioprine (11) carrying a 4-nitroimidazole moiety. The broad substrate scope of HiNfsB indicates that HiNfsB and its unexplored homologs (Fig. 2) can contribute to the activation and resistance of many nitro-containing drug molecules and candidates and can find a range of biotechnological and biomedical applications. In addition to canonical reactive and nonreactive species, we detected a new type of stable dimeric metabolites generated from compounds 5, 6 and 7 by HiNfsB (Fig. 4 and 5). The dimeric metabolite of tinidazole (6) was also detected in HiNfsB expressing E. coli culture. Future studies are needed to unambiguously determine the structures of these metabolites, examine if they can be produced from other nitro-containing compounds by other enzymes in different biological samples, and characterize the potential biological functions of these metabolites. Our X-ray structural analysis of the enzyme coupled with site-directed mutagenesis further identified structural elements and residues important to the enzyme catalysis and broad substrate scope (Fig. 6 and 7). R20 is critical to the coordination of FMN cofactor and substrate binding, while W71, K119 and Y120 may affect the substrate scope of the enzyme. Finally, HiNfsB rendered its heterologous host E. coli Δnfsa/b to be significantly more susceptible toward nitroimidazoles, particularly under anaerobic conditions (Table 1). Collectively, this work uncovered the role and mechanistic base of HiNfsB in metabolizing 5-nitroimidazoles and other nitro-containing drugs. The results advance our fundamental understanding of the metabolism, resistance and toxicity of an important family of antibiotics, guiding their future uses and development. Furthermore, our work suggests the exploration of the vast majority of unstudied NTRs for biotechnological and biomedical studies.Experimental procedures
General methods
Reagents and chemicals were purchased from major vendors including Thermo Scientific, New England Biolabs, Fisher Scientific and Sigma-Aldrich. Plasmid preparation and DNA purification were performed with GeneJET Plasmid Miniprep Kit and GeneJETGel Extraction Kit (Thermo Scientific), respectively. Primers were ordered from Sigma-Aldrich and Genewiz provided DNA sequencing service. E. coli DH5α was used for routine cloning studies and E. coli BL21-GOLD(DE3) (Agilent) was used for protein expression in Luria-Bertani broth or Terrific broth. E. coli Δnfsa/b was provided by Prof. Dan I. Andersson (Uppsala University). A Shimadzu Prominence UHPLC system (Kyoto, Japan) coupled with a PDA detector was used for HPLC analysis. HRMS data were generated on a Thermo Fisher Q Exactive Focus mass spectrometer equipped with an electrospray probe on Universal Ion Max API source.Bioinformatic analysis
To generate the SSN of HiNfsB homologs, HiNfsB sequence from Haemophilus influenzae Rd KW20 (NCBI Reference Sequence: NC_000907.1) was utilized as a query for NCBI Basic Local Alignment Search Tool (BLAST).44 Generated sequence set with 25–95% identity was then subject to redundancy removal in CD-HIT with a threshold at 95%.45 The resulted 1064 sequences were then used to generate the SSN utilizing EFI-Enzyme Similarity Tool with 60% (e value = 10−80) threshold46 and visualized in Cytoscape.47Mutagenesis, cloning and transformation of Nfsb
Codon optimized HinfsB gene (accession number: NC_000907.1) was synthesized with Eurofins (Fig. S21, ESI†) and EcnfsB was amplified from isolated genomic DNA (Gene ID: 945778) using primers shown in Table S2 (ESI†). Standard molecular biology protocols were followed to clone the gene products into pET22b through NdeI and HindIII digestion sites for transforming E. coli BL21-GOLD competent cells.2 This cloning method leads to recombinant proteins with a C-terminal extension of 13 amino acids, with a His6-tag. Site-directed mutagenesis was performed via overlapping PCR (Table S2, ESI†).Protein expression and purification
TB medium supplemented with 100 μg mL−1 ampicillin was inoculated with 0.1% overnight culture of transformed E. coli cells. The culture was incubated at 37 °C, 250 rpm until OD600 reached 0.6. Cell culture was then cooled down to room temperature and protein expression was induced by isopropyl-β-D-thiogalactopyranoside (IPTG, 0.1 mM) at 16 °C, 250 rpm overnight. Cells were harvested by centrifugation and the pellet was resuspended in lysis buffer (0.5 g mL−1). The cells were lysed by sonication for 2 min and collected by centrifugation at 18000 rpm at 4°C for 20 min. Protein purification was carried out with Ni-NTA resin (Thermo) following our established protocols.2 Purification buffers were as follows: lysis buffer: 25 mM Tris-Cl, pH 8.0, 100 mM NaCl, 20 mM imidazole, 3 mM βME and 10% glycerol; washing buffer: 25 mM Tris-Cl, pH 8.0, 100 mM NaCl, 30 mM imidazole, 3 mM βME and 10% glycerol; elution buffer: 25 mM Tris-Cl, pH 8.0, 100 mM NaCl, 50–300 mM imidazole, 3 mM βME, 10% glycerol; desalting buffer: 25 mM Tris-Cl, pH 8.0, 100 mM NaCl, 3 mM βME, 10% glycerol. Purified proteins were desalted using a PD-10 column and exchanged into the desalting buffer. Protein concentrations were determined by NanoDrop and the solutions were then aliquoted and stored at −80°C until use.
Flavin content quantification
Purified enzyme was denatured by boiling. Flavin released in the supernatant was then analyzed by HPLC with an Agilent Poroshell 120 EC-C18 column (2.7 μm, 4.6 × 50 mm). The analyst was first eluted with 1% solvent B (methanol) for 1 min and followed by a linear gradient of 5 mM ammonium acetate (pH 6.0) to 100% methanol in 20 min. The flow rate was set as 1 mL min−1 and the flavin was detected at 451 nm. Commercially available FMN was used to generate a standard curve for quantification based on the area under the peak.NfsB reactions
The enzyme reaction (100 μL) in 100 mM phosphate buffer (pH = 7.4) typically contained 0.1 or 1 μM enzyme, 2 mM substrate, 20 mM glucose, 16 μM GDH, and 1 mM NADP+. Substrate stock solutions (100 mM) were prepared in DMSO. FMN (1 μM) was supplemented in the reactions with 0.1 μM enzyme as indicated. The reaction was aerobically incubated at room temperature, 400 rpm on thermostat (Eppendorf). The anaerobic reactions were performed in an anaerobic chamber (Whitley DG250). To determine the optimal pH for the HiNfsB reaction, the above reactions with 0.1 μM HiNfsB were performed in acetic acid–sodium acetate buffer, phosphate buffer or sodium carbonate–sodium bicarbonate buffer, which together covered a pH range of 5 to 10. The reactions were performed for 10 min under aerobic conditions. To determine the optimal temperature, the reactions with 0.1 μM enzyme were incubated at 4, 16, 25, 30, 37, and 45 °C for 10 min. At the indicated time, acetonitrile (200 μL) was used to terminate the reaction. After mixing well, the solution was centrifuged at 18000 rpm, 4 °C for 20 min, and 10 μL supernatants were used for LC-MS and HPLC analysis. All experiments were performed in triplicate. To derivatize the amine and hydroxylamine intermediates, 150 μL supernatants of acetonitrile-quenched reactions were chilled at −20 °C for 10 min and then incubated with 12 μL of CBZ solution (97 wt%). The mixture was then incubated at room temperature for 30 min. The reactions were then terminated by 20 μL of 0.1 M 1-aminoadamantane and the solutions were further incubated at room temperature for 30 min. Supernatants (10 μL) were used for LC-MS and HPLC analysis.
HPLC analysis
The products were analyzed by a Shimadzu Prominence UHPLC system (Kyoto, Japan) fitted with a YMC-Pack Ph column (5 μm, 250 × 4.6 mm), coupled with a PDA detector. Analytes were eluted first with 1% solvent B (acetonitrile with 0.1% formic acid) for 2 min and then with a linear gradient of 1–20% solvent B in 14 min, followed by another linear gradient of 20–99% solvent B in 1 min. The solvent A was water with 0.1% formic acid. The column was further cleaned with 99% solvent B for 2 min and then re-equilibrated with 1% solvent B for 2 min. The flow rate was set as 0.8 mL min−1, and the products were detected at 320 nm or 276 nm (for azathioprine).LC-MS analysis
Samples were analyzed by LC-MS using a YMC-Pack Ph column (5 μm, 250 × 4.6 mm). Analytes were eluted first with 1% solvent B (acetonitrile with 0.1% formic acid) for 2 min and then with a linear gradient of 1–20% solvent B in 14 min, followed by another linear gradient of 20–99% solvent B in 1 min. The column was further cleaned with 99% solvent B for 2 min and then re-equilibrated with 1% solvent B for 2 min. The flow rate was set as 0.8 mL min−1. Electrospray ionization was used and ion counts for a particular m/z peak were determined by peak height. LC-HRMS and HRMS/MS experiments were conducted on Thermo Scientific™ Q Exactive Focus mass spectrometer with Dionex™ Ultimate™ RSLC 3000 uHPLC system, equipped with H-ESI II probe on Ion Max API Source. Analytes were separated on an Agilent Poroshell 120 EC-C18 column (2.7 μm, 3.0 × 50 mm) by solvent A (water with 0.1% formic acid) and B (acetonitrile containing 0.1% formic acid) with a flow rate of 0.5 mL min−1. The column was run with 10% B for 2 min, 10–95% B in 8.5 min, 95% B for 2.5 min and 95 to 10% B in 0.5 min, and then re-equilibrated in 2% B for 2 min. MS1 signals were acquired under the Full MS positive ion mode covering a mass range of m/z 100–500, with a resolution at 70000 and an AGC target at 1 × 106. Precursor ions were selected in the orbitrap typically with an isolation width of 3.0 m/z and fragmented in the HCD cell with step-wise collision energies (CE) of 20, 25, and 30.
Crystallization and structure determination
Purified HiNfsB was concentrated to 12 mg mL−1 and incubated with a 10-fold molar excess of 1-methyl-5-nitroimidazole (12) overnight before setting up crystal trays. The protein was screened for crystallization using commercial screens and the sitting drop vapor diffusion method at 4 °C. A single yellow, rod-shaped protein crystal was observed in 8% v/v tacsimateTM pH 5.0 and 20% w/v polyethylene glycol 3350 after ∼1 month of incubation. Additional protein crystals were obtained using the hanging drop vapor diffusion method with 0.2 M potassium sodium tartrate tetrahydrate, 0.1 M sodium citrate tribasic dihydrate pH 5.6 and 2.0 M ammonium sulfate at 4 °C without substrate. Crystals were cryoprotected in 20% glycerol and 80% original crystallization condition and X-ray diffraction data were collected at the synchrotron source at Argonne National Laboratory, APS beamline 23-ID-D, using a wavelength of 1.033 Å. Collected diffraction data were integrated, merged and scaled using the program XDS.48 Data quality was accessed with Xtriage. An initial phase solution was obtained using molecular replacement with PDB entry 6WT2 (∼56% sequence identity) as the search model. The final refined atomic model was completed by several rebuilding and refinement cycles using PHENIX.REFINE49 and COOT.50 Water molecules were placed in the structure by manual inspection of the 2Fo−Fc map. The refinement statistics for the structure of HiNfsB in a complex with 1-methyl-5-nitroimidazole (12) and nicotinic acid are shown in Table S3 (ESI†). Figures were prepared with PyMOL. The atomic coordinates and structure factors of HiNfsB/12 and HiNfsB/nicotinate have been deposited in the PDB under the ID codes 7T2Z and 7T33, respectively.Molecular docking
Molecular docking experiments were performed with AutoDock Vina using the crystal structure of HiNfsB solved in this work.51 The structure was prepared for docking using MGLTool, including the removal of crystallographic waters. FMN was kept in the structure. By centering on the ribityl moiety of FMN, the grid box was defined with parameters: center_x = 16.736, center_y = 11.088, center_z = −0.590, size x = y = z = 40 Å, energy_range = 4, exhaustiveness = 8. The results were analyzed with PyMOL. The binding mode used to elucidate protein–ligand interactions in the active site was chosen considering the orientation of the substrate 12 and nicotinic acid in the elucidated crystal structures, distance from the nitro group to N5 of FMN, and the binding energies.Susceptibility of E. coli Δnfsa/b with Hinfsb or Ecnfsb gene against nitroimidazoles
The susceptibility test followed an established broth dilution method.52 The construct of pET22b with Hinfsb or Ecnfsb was first transformed into E. coli Δnfsa/b competent cells to generate E. coli Δnfsa/b::HinfsB and E. coli Δnfsa/b::EcnfsB. A single colony was then inoculated in LB with ampicillin for growing overnight. The culture was then suspended in fresh LB to achieve 1 × 108 CFU mL−1. The suspension was then diluted for 100 folds to give around 106 CFU mL−1 inoculum in 96-well plates with 100 μL LB with ampicillin/well. Two-fold dilutions of selected nitroimidazole antibiotics were then inoculated in triplicate. Positive controls well (inoculated) and negative controls (non-inoculated) were included. The microplates were incubated at 37 °C for 20 hours and cell growth was quantitated by a microplate reader at 600 nm to determine MIC (minimum inhibitory concentration) values. For the anaerobic susceptibility test, all reagents, experimental materials and media were pre-incubated in an anaerobic chamber overnight before experiments. Bacterial strains were grown in LB broth in the anaerobic chamber overnight at 37 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by startup funds provided by the University of Florida (Y. D.) and NIH (R35 GM128742 to Y. D.). We thank Jim Rocca and Garret Rubin for their technical support and the staff of LS-CAT, ANL for help with data collection. We thank Prof. Dan I. Andersson (Uppsala University) for sharing the E. coli Δnfsa/b strain.References
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- P. Zhang, B. S. MacTavish, G. Yang, M. Chen, J. Roh, K. R. Newsome, S. D. Bruner and Y. Ding, ACS Chem. Biol., 2020, 15, 2281–2288 CrossRef CAS PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- Y. Abouelhassan, A. T. Garrison, H. Yang, A. Chavez-Riveros, G. M. Burch and R. W. Huigens, 3rd, J. Med. Chem., 2019, 62, 7618–7642 CrossRef CAS PubMed.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- M. A. Kohanski, M. A. DePristo and J. J. Collins, Mol. Cell, 2010, 37, 311–320 CrossRef CAS PubMed.
- N. Czekalski, E. Gascón Díez and H. Bürgmann, ISME J., 2014, 8, 1381–1390 CrossRef CAS PubMed.
- J. H. Bethke, A. Davidovich, L. Cheng, A. J. Lopatkin, W. Song, J. T. Thaden, V. G. Fowler, Jr., M. Xiao and L. You, Sci. Adv., 2020, 6, eaax3173 CrossRef CAS PubMed.
- J. Park, A. J. Gasparrini, M. R. Reck, C. T. Symister, J. L. Elliott, J. P. Vogel, T. A. Wencewicz, G. Dantas and N. H. Tolia, Nat. Chem. Biol., 2017, 13, 730–736 CrossRef CAS PubMed.
- G. P. Dinos, C. M. Athanassopoulos, D. A. Missiri, P. C. Giannopoulou, I. A. Vlachogiannis, G. E. Papadopoulos, D. Papaioannou and D. L. Kalpaxis, Antibiotics, 2016, 5, 20 CrossRef PubMed.
- I. Brook, Expert Opin. Pharmacother., 2011, 12, 1691–1707 CrossRef CAS PubMed.
- https://clincalc.com/DrugStats/Drugs/Metronidazole .
- S. A. Dingsdag and N. Hunter, J. Antimicrob. Chemother., 2018, 73, 265–279 CrossRef CAS PubMed.
- C. Thomas and C. D. Gwenin, Biology, 2021, 10, 388 CrossRef CAS PubMed.
- H. K. Leiros, S. Kozielski-Stuhrmann, U. Kapp, L. Terradot, G. A. Leonard and S. M. McSweeney, J. Biol. Chem., 2004, 279, 55840–55849 CrossRef CAS PubMed.
- J. Whiteway, P. Koziarz, J. Veall, N. Sandhu, P. Kumar, B. Hoecher and I. B. Lambert, J. Bacteriol., 1998, 180, 5529–5539 CrossRef CAS PubMed.
- P. R. Race, A. L. Lovering, R. M. Green, A. Ossor, S. A. White, P. F. Searle, C. J. Wrighton and E. I. Hyde, J. Biol. Chem., 2005, 280, 13256–13264 CrossRef CAS PubMed.
- P. Macheroux, B. Kappes and S. E. Ealick, FEBS J., 2011, 278, 2625–2634 CrossRef CAS PubMed.
- A. L. Lovering, E. I. Hyde, P. F. Searle and S. A. White, J. Mol. Biol., 2001, 309, 203–213 CrossRef CAS PubMed.
- W. Pitsawong, J. P. Hoben and A. F. Miller, J. Biol. Chem., 2014, 289, 15203–15214 CrossRef CAS PubMed.
- K. S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- J. N. Copp, A. M. Mowday, E. M. Williams, C. P. Guise, A. Ashoorzadeh, A. V. Sharrock, J. U. Flanagan, J. B. Smaill, A. V. Patterson and D. F. Ackerley, Cell Chem. Biol., 2017, 24, 391–403 CrossRef CAS PubMed.
- M. D. Roldan, E. Perez-Reinado, F. Castillo and C. Moreno-Vivian, FEMS Microbiol. Rev., 2008, 32, 474–500 CrossRef CAS PubMed.
- Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407–6416 CrossRef CAS PubMed.
- M. Martinez-Julvez, A. L. Rojas, I. Olekhnovich, V. Espinosa Angarica, P. S. Hoffman and J. Sancho, FEBS J., 2012, 279, 4306–4317 CrossRef CAS PubMed.
- G. Sisson, J. Y. Jeong, A. Goodwin, L. Bryden, N. Rossler, S. Lim-Morrison, A. Raudonikiene, D. E. Berg and P. S. Hoffman, J. Bacteriol., 2000, 182, 5091–5096 CrossRef CAS PubMed.
- E. Akiva, J. N. Copp, N. Tokuriki and P. C. Babbitt, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E9549–E9558 CrossRef CAS PubMed.
- J. N. Copp, E. Akiva, P. C. Babbitt and N. Tokuriki, Biochemistry, 2018, 57, 4651–4662 CrossRef CAS PubMed.
- R. S. Boddu, O. Perumal and D. K, Biotechnol. Appl. Biochem., 2021, 68, 1518–1530 CAS.
- T. S. Crofts, P. Sontha, A. O. King, B. Wang, B. A. Biddy, N. Zanolli, J. Gaumnitz and G. Dantas, Cell Chem. Biol., 2019, 26, 559–570 CrossRef CAS PubMed e556.
- A. L. Smith, A. L. Erwin, T. Kline, W. C. T. Unrath, K. Nelson, A. Weber and W. N. Howald, Antimicrob. Agents Chemother., 2007, 51, 2820–2829 CrossRef CAS PubMed.
- K. D. Green, M. Y. Fosso, A. S. Mayhoub and S. Garneau-Tsodikova, Bioorg. Med. Chem. Lett., 2019, 29, 1127–1132 CrossRef CAS PubMed.
- H. J. Atkinson, J. H. Morris, T. E. Ferrin and P. C. Babbitt, PLoS One, 2009, 4, e4345 CrossRef PubMed.
- E. M. Williams, M. H. Rich, A. M. Mowday, A. Ashoorzadeh, J. N. Copp, C. P. Guise, R. F. Anderson, J. U. Flanagan, J. B. Smaill, A. V. Patterson and D. F. Ackerley, Biochemistry, 2019, 58, 3700–3710 CrossRef CAS PubMed.
- P. van Dillewijn, R. M. Wittich, A. Caballero and J. L. Ramos, Appl. Environ. Microbiol., 2008, 74, 6703–6708 CrossRef CAS PubMed.
- P. van Dillewijn, R. M. Wittich, A. Caballero and J. L. Ramos, Appl. Environ. Microbiol., 2008, 74, 6820–6823 CrossRef CAS PubMed.
- C. A. Haynes, R. L. Koder, A. F. Miller and D. W. Rodgers, J. Biol. Chem., 2002, 277, 11513–11520 CrossRef CAS PubMed.
- E. Johansson, G. N. Parkinson, W. A. Denny and S. Neidle, J. Med. Chem., 2003, 46, 4009–4020 CrossRef CAS PubMed.
- A. G. O’Neill, B. A. Beaupre, Y. Zheng, D. Liu and G. R. Moran, Appl. Environ. Microbiol., 2020, 86, e01758-20 CrossRef PubMed.
- J. I. Grove, A. L. Lovering, C. Guise, P. R. Race, C. J. Wrighton, S. A. White, E. I. Hyde and P. F. Searle, Cancer Res., 2003, 63, 5532–5537 CAS.
- R. Roemhild, M. Linkevicius and D. I. Andersson, PLoS Biol., 2020, 18, e3000612 CrossRef PubMed.
- H. Hof, Antimicrob. Agents Chemother., 1989, 33, 404–405 CrossRef CAS PubMed.
- F. Baneyx, Curr. Opin. Biotechnol, 1999, 10, 411–421 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed.
- Y. Huang, B. Niu, Y. Gao, L. Fu and W. Li, Bioinformatics, 2010, 26, 680–682 CrossRef CAS PubMed.
- R. Zallot, N. Oberg and J. A. Gerlt, Biochemistry, 2019, 58, 4169–4182 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00238d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |