
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLandscaping macrocyclic peptides: stapling hDM2-binding peptides for helicity, protein affinity, proteolytic stability and cell uptake†
Aline D.
de Araujo
 ,
Junxian
Lim
,
Kai-Chen
Wu
,
Huy N.
Hoang
,
Huy T.
Nguyen
and
David P.
Fairlie
*
,
Junxian
Lim
,
Kai-Chen
Wu
,
Huy N.
Hoang
,
Huy T.
Nguyen
and
David P.
Fairlie
*
Division of Chemistry and Structural Biology, ARC Centre of Excellence for Innovations in Peptide and Protein Science, Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: d.fairlie@uq.edu.au
First published on 31st May 2022
Abstract
Cyclic peptides that modulate protein–protein interactions can be valuable therapeutic candidates if they can be delivered intact to their target proteins in cells. Here we systematically compare the effects of different helix-inducing cyclization constraints on the capacity of a macrocyclic peptide component to confer α-helicity, protein-binding affinity, resistance to degradative proteases and cell uptake to a 12-residue peptide fragment of tumor suppressor protein p53. We varied the helix-inducing constraint (hydrocarbon, lactam, aliphatic or aromatic thioether, etc.) and the position of the cyclization linker (i to i + 4 or i to i + 7 bridges) in order to sculpt the macrocyclic size, stabilize its structure, and promote cell uptake. We find that rigidifying the macrocycle leads to higher alpha helicity, target affinity and proteolytic stability to different extents, whereas cell uptake of compounds shown here is mostly driven by hydrophobicity and aromaticity of the macrocycle.
Introduction
Protein–protein interactions (PPIs) control most cellular processes, but their dysregulation can lead to disease. Finding ways to activate or inhibit PPIs is important for design of future therapeutics.1 Most PPIs involve large, shallow, and highly polar interacting surfaces (1500–3000 Å2).2 These present significant challenges for binding conventional small molecule drugs that require small hydrophobic binding pockets in proteins.1 PPIs are often mediated by helical motifs, comprising 1–4 α-helical turns (4–15 amino acids).3–6 Synthetic peptides corresponding to such helical regions of proteins are attractive leads for drug design.3–6 However, in water outside proteins these peptides are not thermodynamically stable α-helices, raising the entropic penalty for target binding. They are also usually too polar to enter cells where most PPIs are located. To address these limitations, two residues on the same face of a putative peptide helix may be covalently crosslinked (‘stapled’), for example by lactamization,3 olefin ring-closing metathesis (RCM),5,6 cysteine thiolation7 or 1,3-dipolar cycloaddition8 (Fig. 1), to form a macrocyclic component that may stabilize a peptide helix. Incorporating such crosslinks can also potentially alter aqueous versus membrane solubility, enhance chemical and metabolic stability, and increase cell uptake and bioavailability.3–9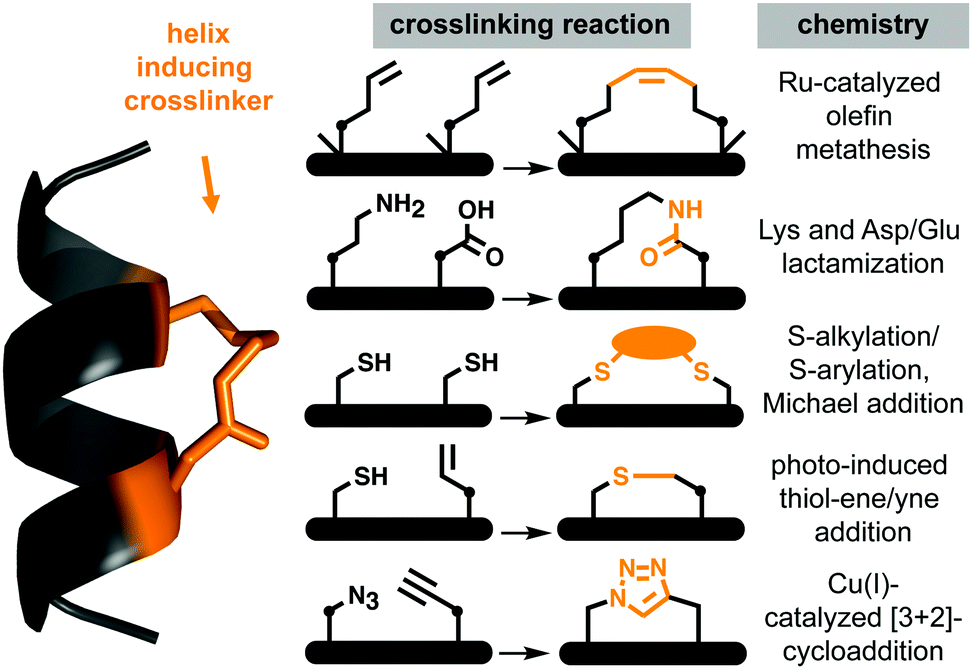 | ||
| Fig. 1 Chemical macrocyclization strategies to stabilize short peptide sequences into helices via side-chain crosslinking. The stapling constraints are usually imposed between (i → i + 4)- (pictured) or (i → i + 7)-positions within the peptide. • means (CH2)n, n = 0, 1, 2,…. | ||
A remarkable feature of some peptides with hydrocarbon-linked macrocyclic components is their ability to enter cells and bind to intracellular proteins.6 Their membrane permeability may be due to extended hydrophobicity introduced by the linker, facilitating interaction with phospholipid membranes. However, increasing hydrophobicity too much may lead to aggregation or cell lysis. Moreover, hydrophobicity is arguably not the sole contributor to cell uptake, with amphipathicity, charge, dipole and helicity also potentially contributing to endocytosis and release into the cytosol.10,11
Systematic studies show that a specific lactam bridge across five residues (i → i + 4) is more α-helix inducing than hydrocarbon, cysteinyl and triazole linkers in cyclic pentapeptides alone12 or in 10–20 residue peptides.12,13 Others found that lactam and hydrocarbon linkers at the centre of a specific 12-mer peptide induced comparable helicity.14 Positioning an (i → i + 4) hydrocarbon linker in a BCL-2 binding peptide with positive charges led to increased helicity and cell uptake only when positioned at the amphipathic boundary.10 Fewer (i → i + 7) crosslinkers15,16 have been comparatively investigated for their effects on biophysical properties of peptides.
Here we systematically evaluate the impact of (i → i + 4) and (i → i + 7) crosslinkers of varying composition, hydrophobicity and flexibility, on structural and biophysical properties of a bioactive peptide, Ac-LTFEHYWAQLTS-CONH2 (1).17 Discovered by phage display, this peptide inhibits interaction of tumor suppressor protein p53 with hDM2, a protein that prevents p53 action and transports it from nucleus to cytosol. hDM2 is also a ligase that attaches ubiquitin covalently to p53, marking it for proteasomal degradation. Compounds that bind to hDM2 can reactivate the p53 pathway for cell-cycle arrest and apoptosis of tumor cells.18 However, this peptide is prone to proteolytic degradation and unable to cross cell membranes, prompting studies on increasing its stability and cell uptake.19–21 Here we focus on comparing hydrocarbon, thioether, triazole and lactam linkers in 1 for their ability to increase α-helicity, hDM2 affinity, resistance to proteases, and cell uptake without cell lysis.
Results
To incorporate helix-inducing cyclic constraints into the peptide, we were careful to retain essential hDM2-binding residues (Phe3, Trp7, Leu10) but replaced His5 with Glu5 to improve solubility without affecting binding to hDM2.19 To measure cell uptake, the N-terminal acetyl group was replaced by β-alanine linked to fluorescein (FITC). We investigated effects of different cyclic components on α-helicity (measured by circular dichroism (CD) spectra in aqueous media), binding affinity for hDM2 (measured by competition for hDM2 binding of FITC-labelled 1 using fluorescence polarization (FP) assays), and proteolytic stability (using LCMS after incubation with chymotrypsin, pepsin or proteinase K). Later we describe their hydrophobicity and propensity for cell uptake and cell lysis.(i → i + 4)-crosslinkers
Positions 4 and 8 in 1 were stapled (Fig. 2A) with a hydrocarbon linker5 (2a), a Lys-Asp lactam bridge3 (2b), a para-benzyl bis-thioether linker22 (2c), a perfluorophenyl bis-thioether23 (2d) or its homocysteine variant (2e).24 Since the lactam bridge is known to be the most effective helix inducer from the C-terminus,13 we included analogue 2f (Fig. 2A). We also synthesized bicyclic 2g and 2h, since two bridges (two lactams or one hydrocarbon/one lactam) can strongly enforce α-helicity,13,25 the hydrocarbon creating a larger hydrophobic surface to assist membrane interaction.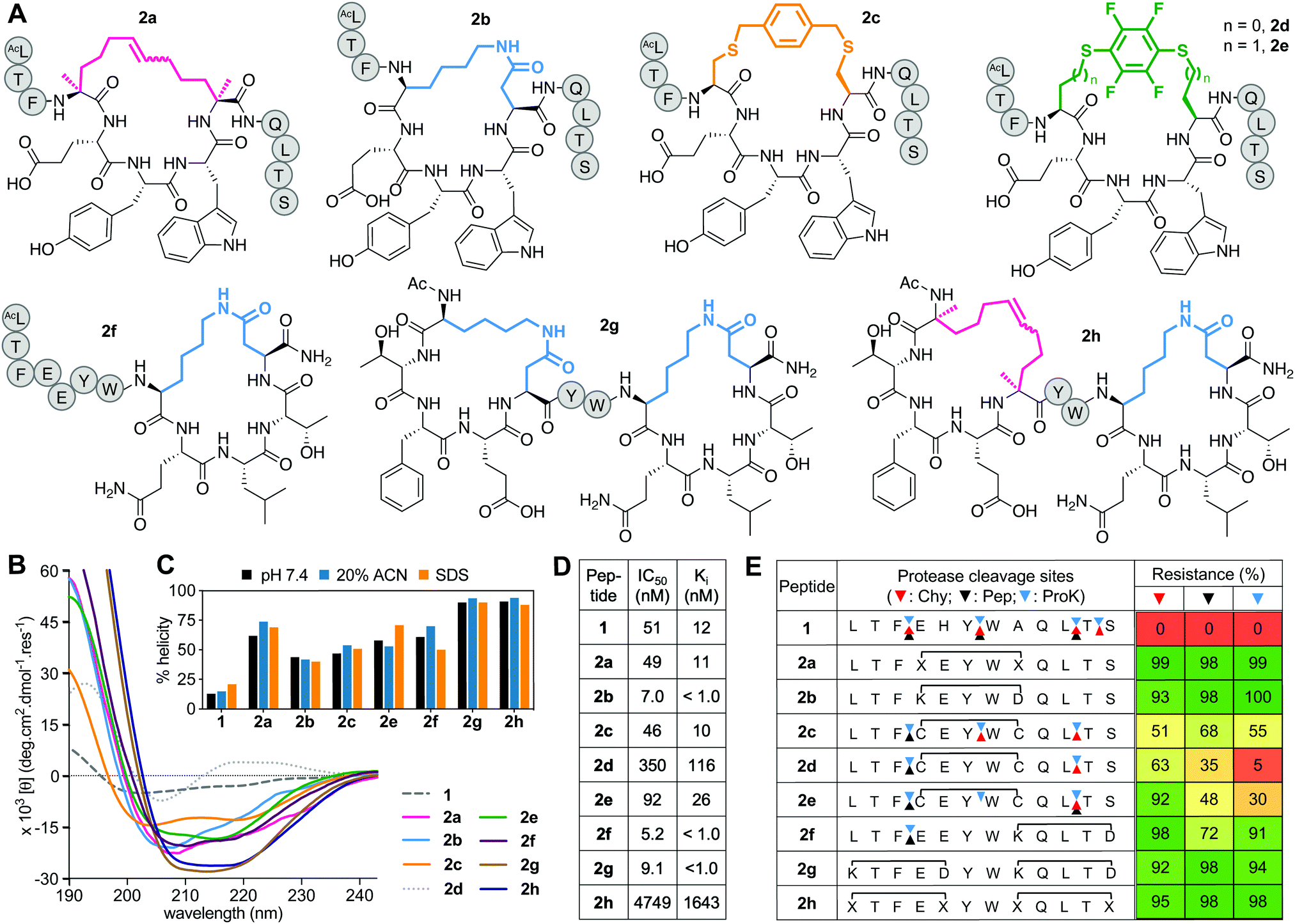 | ||
| Fig. 2 A. Structures of (i → i + 4)-crosslinked peptides 2a–h. B. CD spectra for 2a–hvs.1 (50 μM) in 10 mM phosphate buffer (pH 7.4) at 22 °C. C. Percent helicity calculated from CD spectra (θ220) in 10 mM phosphate buffer (pH 7.4), 20% ACN or 20 mM SDS buffer. D. IC50 = mean peptide concentration for inhibiting 50% binding of 1-FITC (10 nM) to hDM2 (25 nM) measured by FP. pIC50 ± SEM values are reported in Fig. S1 (ESI†). Binding affinity (Ki) calculated from IC50 values. E. Heatmap indicating percent peptide intact after 2 h incubation with chymotrypsin (Chy), pepsin (Pep) or proteinase K (ProK) at 22 °C at pH 8 or 2 (Pep only), measured by LCMS. Arrows point to respective cleavage sites as revealed by LCMS analysis of the cleaved fragments. | ||
Peptides 2a and 2e induced ∼60% helicity in aqueous phosphate buffer (10 mM, pH 7.4) (Fig. 2B), with the lactam linkage conferring greater helicity from the C-terminus (2f). Thioether 2c had much less helicity than 2a. Compound 2d gave a distorted CD curve, possibly obscured by strong absorbance from the fluoroaryl moiety,23,24 but not for 2e that had some helix-like ellipticity despite a perfluoroaryl crosslink. Adding a second helix-inducing constraint into 2f, either a second lactam (2g) or hydrocarbon (2h), enhanced helicity (∼90%) independent of solvent (Fig. 2C), consistent with other findings.3a,13,26 Only small variations in helicity (±12%) were observed in CD spectra for 1 or 2a–h in 10 mM phosphate buffer, 20% acetonitrile in water or 20 mM SDS buffer (sodium dodecyl sulfate, 10 mM sodium phosphate, 0.15 M NaF buffer pH 7.4) (Fig. 2C). Enhanced α-helicity did not correlate with binding affinity for hDM2, as determined by competitive binding with 1-FITC to hDM2 (Fig. 2D). Only lactam peptides 2b, 2f, 2g had higher affinity than 1. The N-terminal hydrocarbon cycle in 2h had reduced binding affinity for hDM2.
Stapling protected against proteolysis (Fig. 2E) compared to unstapled 1 that was cleaved within 1 h by chymotrypsin, pepsin or proteinase K at positions 3–4, 6–7, 10–11, 11–12. Hydrocarbon or lactam stapling (2a, 2b, 2g, 2h) maintained peptides intact, cleavage only occurring in 2f outside the cycle at the distant 3–4 position, whereas thioethers 2c–e were not fully protected (Fig. 2E), with positions outside and inside the cycle vulnerable to cleavage. Results are consistent with constrained α-helices26a,27d,27d not being flexible enough to present the extended β-strand conformation required for substrate cleavage by proteases.27
Aliphatic (i → i + 7)-crosslinkers
The prototype of this series has an α-methylated 11-membered alkene tether5 between positions 4 and 11 (3a, Fig. 3A) and was developed into an anticancer drug candidate.19 Alternatively, cysteines may be linked with aliphatic crosslinkers using thiol–ene photochemistry,15 thiolation under high temperature and base28 or with Zn2+ salts,29 or via selenocysteine stapling.16 Here we use Zn(OAc)2-assisted thiolation with dibromooctane29 to make (i → i + 7) thioethers 3b–e (Fig. 3A), with cysteines (L-Cys4/L-Cys11, 3b; D-Cys4/L-Cys11, 3c) or α-methyl cysteines (L-(αMe) Cys4/L-(αMe)Cys11, 3d; D-(αMe)Cys4/L-(αMe)Cys11, 3e).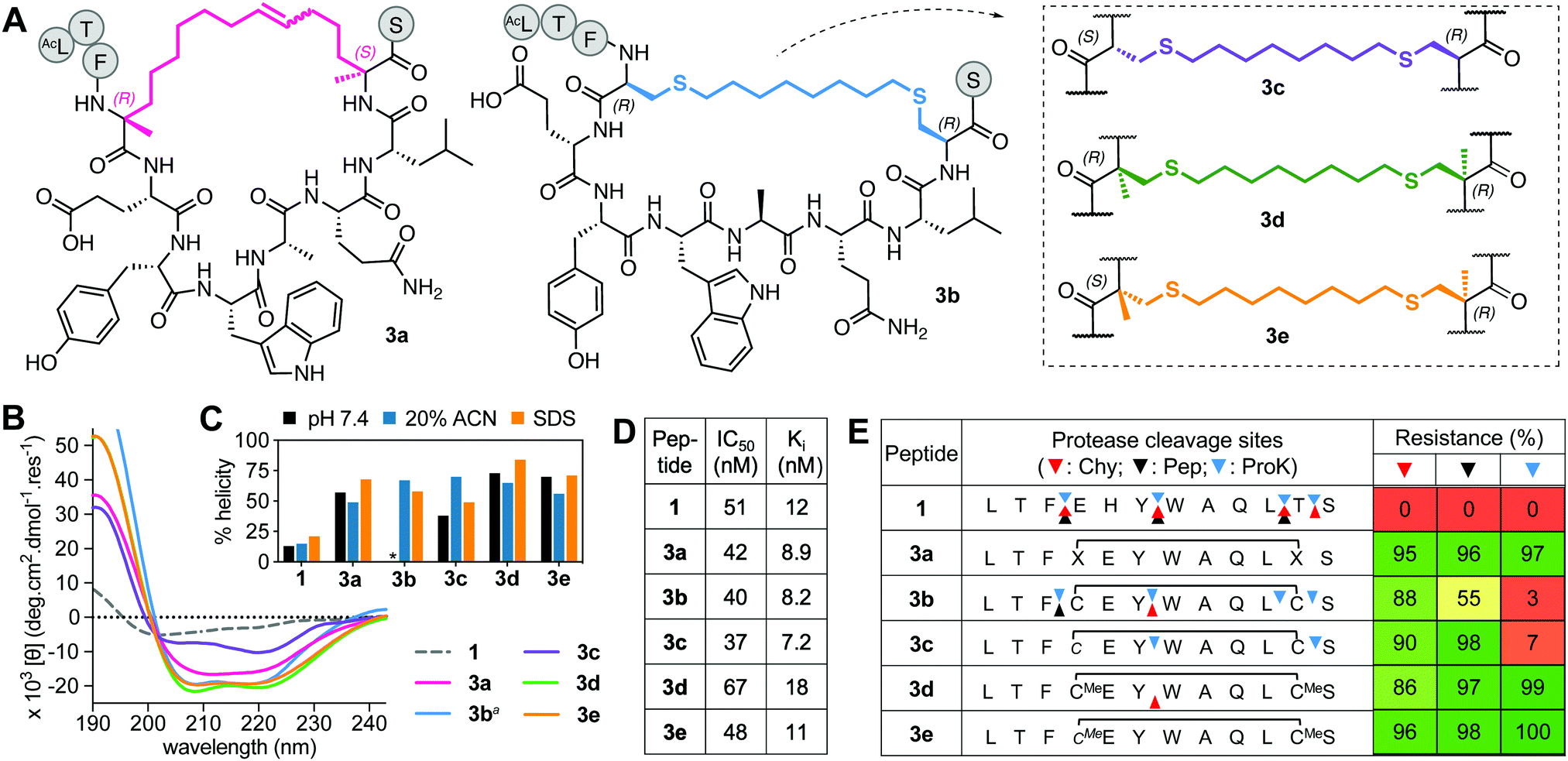 | ||
| Fig. 3 A. Structures of aliphatic (i → i + 7)-crosslinked peptides 3a–e. B. CD spectra for 3a–evs.1 (50 μM) in 10 mM phosphate buffer (pH 7.4, 22 °C) except 3ba in 20% ACN. C. Percent helicity calculated from CD spectra (θ220) in 10 mM phosphate buffer, 20% ACN or 20 mM SDS buffer (* not helical). D. IC50 is mean peptide concentration to inhibit 50% binding of 1-FITC (10 nM) to hDM2 (25 nM) measured by FP. pIC50 ± SEM values are reported in Fig. S1 (ESI†). Binding affinity (Ki) calculated from IC50 values. E. Heatmap indicating percent peptide intact after 2 h incubation with chymotrypsin (Chy), pepsin (Pep) or proteinase K (ProK) at 22 °C and pH 8 or 2 (Pep only), measured by LCMS. Arrows point to respective cleavage sites as revealed by LCMS analysis of the cleaved fragments. | ||
In aqueous buffer, 3b did not show a CD spectrum typical of an α-helix (Fig. S2, ESI†), possibly due to aggregation, while 3c showed some helicity (38%, Fig. 3B). The α-methyl groups in 3d and 3e increased helicity to 70% (Fig. 3B). The α-methylated 3a also had higher helicity (57%) than non-αMe analogues. Helicity was greater in the presence of SDS micelles (Fig. 3C). However, helicity did not correlate with binding affinity for hDM2 (Fig. 3C and D), suggesting that some degree of induced fit occurs on binding to this protein, with all five analogues having high affinity but α-methylation offering no enhanced binding. Further, altering the length of the aliphatic linker in 3b by one methylene or introducing an E- or Z-alkene at a central position in the linker, did not significantly impact helicity (in 20% acetonitrile) or hDM2 affinity of the thioether.29
The D- or L-configuration and α-methyl modifications had more impact on stability to proteases (Fig. 3E). Upon cyclization, positions 6–7 and 10–11 inside the macrocycle became significantly protected against cleavage, but not entirely in the case of 3b, perhaps because of its non-helical structure in aqueous buffer (Fig. 3E and Fig. S2, ESI†). Cyclization did shelter position 11–12 from degradation except to proteinase K. D-Cys at position 4 prevented proteolysis (3c), whereas α-methylation fully protected neighbouring positions from cleavage by all enzymes.
Aromatic (i → i + 7)-crosslinkers
This series includes the benzyl bistriazole-linked peptide 4a, via a two-component construction based on a triazole-stapling approach,8 and four variants of a biphenyl-crosslinked bisthioether macrocycle 4b (Fig. 4A), with a (i → i + 7)-cysteine staple previously reported21 with altered L- and D-Cys or αMeCys residues at the crosslinking site (4b–e). Peptide 4a showed similar binding to hDM2 as previous aliphatic (i → i + 7)-crosslinked counterparts, but had reduced helix induction (27%, Fig. 4B and C). Despite unnatural amino acids at stapling sites, 4a was partially vulnerable to cleavage by proteinase K and completely cleaved by pepsin (acidic pH required for pepsin activity led to triazole protonation and destabilization) (Fig. 4E). Compound 4b was highly helical (72%) in water but did not have higher hDM2 affinity. Incorporating α-methylation into 4b at the Cys stapling site to form 4d did not increase helicity or affinity but did confer strong resistance to protease degradation (Fig. 4E). Compound 4c was the least α-helical (24%), as D-residues disrupt helix propensity, but helicity was increased by α-methylation at cysteine (64% in 4e) without much impact on hDM2 affinity, and protected positions 3–4 and 11–12 against proteolysis (Fig. 4E), as for the previous series.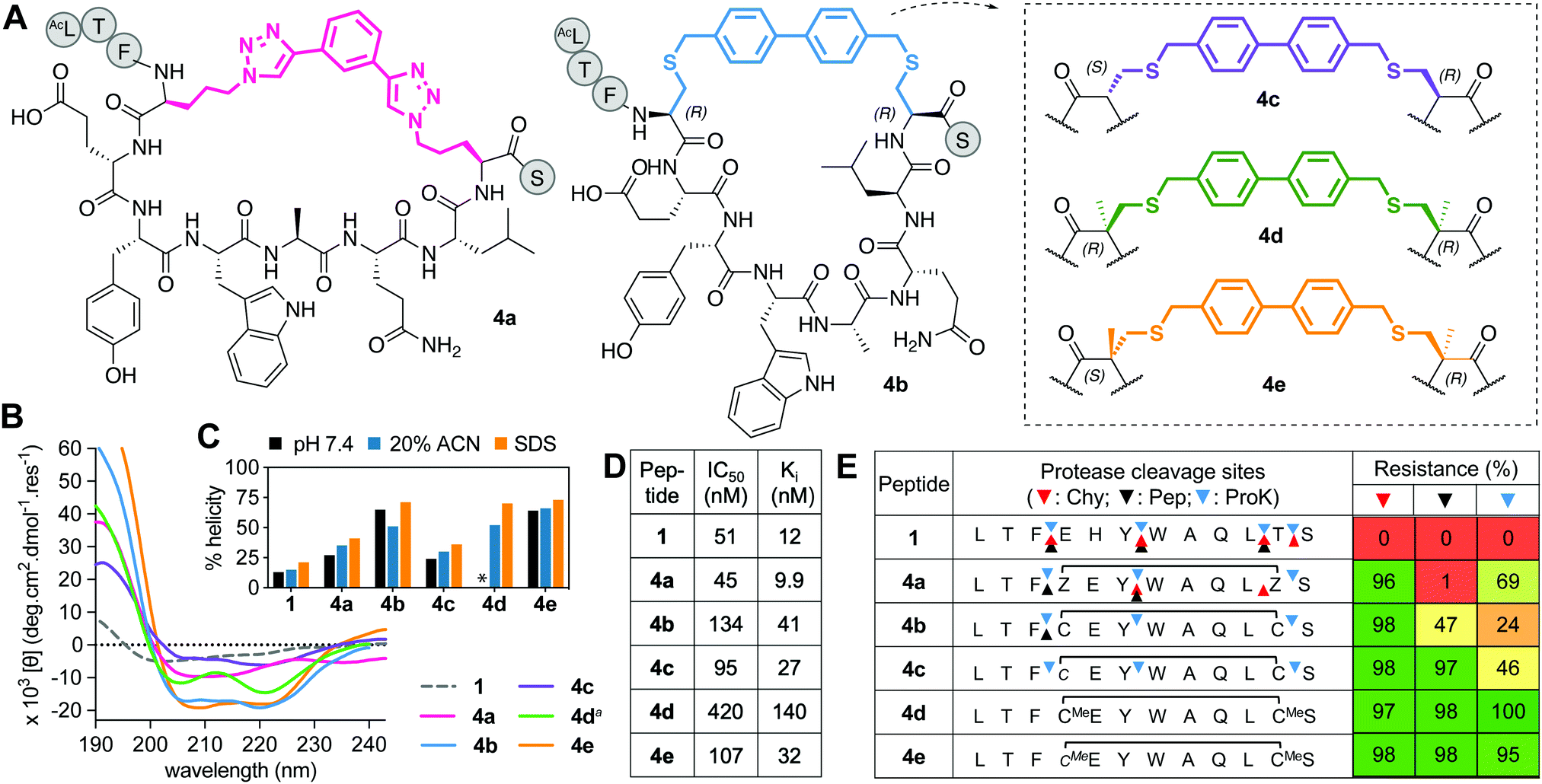 | ||
| Fig. 4 A. Structures of aromatic (i → i + 7)-crosslinked peptides 4a–e. B. CD spectra for 4a–evs.1 (50 μM) in 10 mM phosphate buffer (pH 7.4, 22 °C), except 4da in 20% ACN. C. Percent helicity calculated from CD spectra (θ220) in 10 mM phosphate buffer, 20% ACN or 20 mM SDS buffer (* not helical, Fig. S2, ESI†). D. IC50 = mean peptide concentration to inhibit 50% binding of 1-FITC (10 nM) to hDM2 (25 nM) measured by FP. pIC50 ± SEM values are reported in Fig. S1, ESI.† Binding affinity (Ki) calculated from IC50 values. E. Heatmap indicating percent peptide intact after 2 h incubation with chymotrypsin (Chy), pepsin (Pep) or proteinase K (ProK) at 22 °C at pH 8 or 2 (Pep only), measured by LCMS. Arrows show cleavage sites revealed by LCMS of the cleaved fragments. | ||
Perfluorobiphenyl (i → i + 7)-crosslinkers
Incorporating perfluorobiaryl linkers to form cyclic peptides was first demonstrated by SN-arylation of cysteine sidechains with a decafluorobiphenyl linker23 that enhanced cell penetration of peptides.23,30 However, is this linker an effective helix-inducing constraint? In the one report of helicity,23 (i → i + 4)-stapling of a 14-mer peptide with perfluorobiphenyl only modestly altered α-helicity. We created a Cys4-Cys11 (i → i + 7)-crosslink with decafluorobenzyl, but the macrocycle (5a, Fig. 5A) had little helicity (Fig. 5B and C), weak hDM2 binding (Fig. 5D), and was degraded by proteases (Fig. 5E).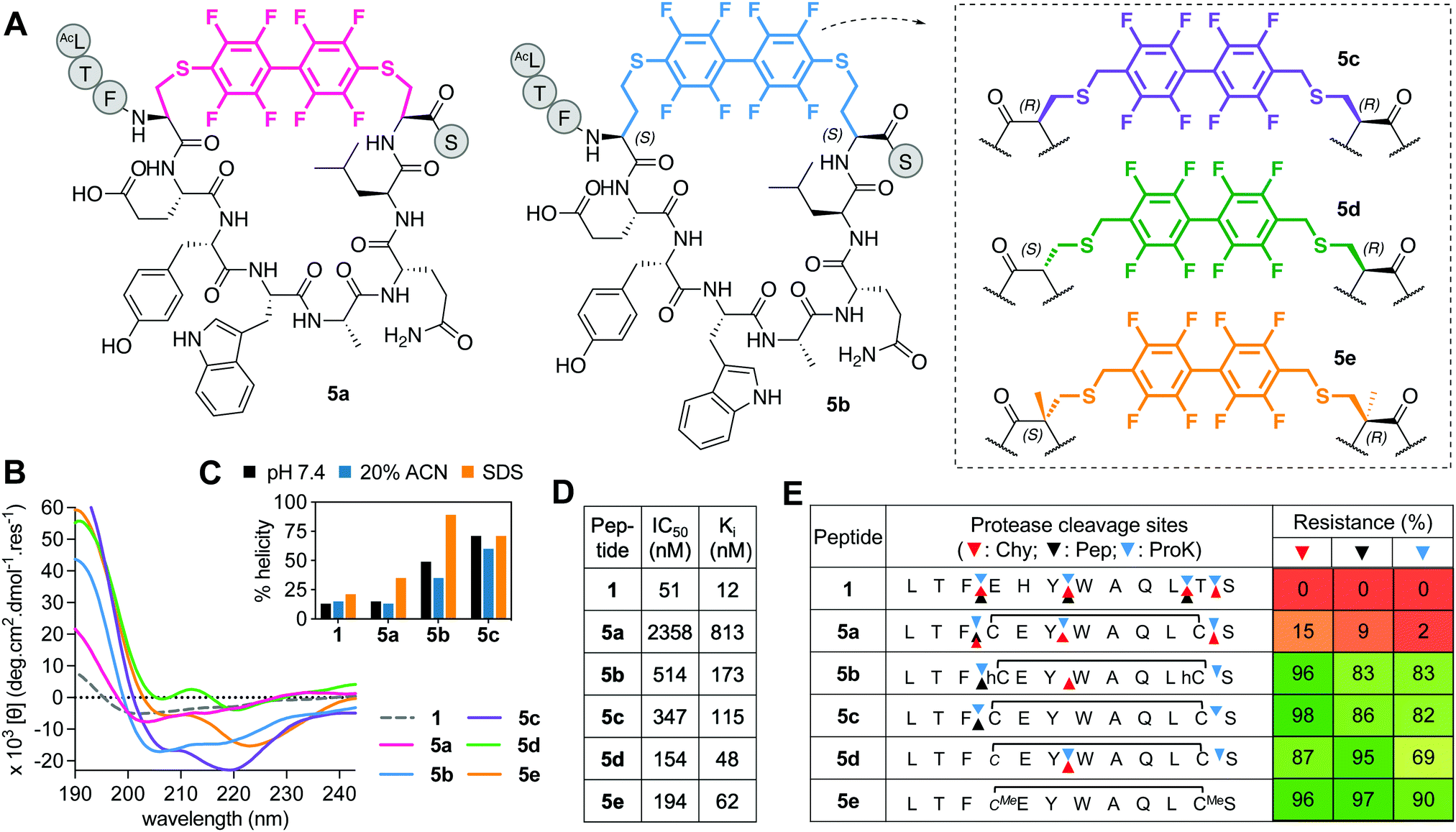 | ||
| Fig. 5 A. Structures of perfluorobiphenyl (i → i + 7)-crosslinked peptides 5a-e. B. CD spectra for 5a–evs.1 (50 μM) in 10 mM phosphate buffer (pH 7.4, 22 °C). C. Percent helicity calculated from CD spectra (θ220) in 10 mM phosphate buffer, 20% ACN or 20 mM SDS buffer (5d,e not helical). D. IC50 = mean peptide concentration to inhibit 50% binding of fluorescent 1-FITC (10 nM) to hDM2 (25 nM) measured by FP. pIC50 ± SEM values are reported in Fig. S1 (ESI†). Binding affinity (Ki) calculated from IC50 values. E. Heatmap indicating percent peptide intact after 2 h incubation with chymotrypsin (Chy), pepsin (Pep) or proteinase K (ProK) at 22 °C, pH 8 or 2 (Pep only), measured by LCMS. Arrows point to the respective cleavage sites as revealed by LCMS analysis of the cleaved fragments. | ||
Towards a better (i → i + 7) linker, we constructed new perfluorobiphenyl analogues 5b–e that expanded ring size, altered D-, L-, α-methylated Cys, and altered the position of the connecting S–C bond. Increasing macrocycle size by two methylene units (5b) enhanced helicity, hDM2 binding, and protease resistance (Fig. 5). D- or α-methyl Cys substitution made positions 3–4 and 11–12 more protease resistant (Fig. 5E). Compounds 5d and 5e had unidentified CD curves (Fig. 5B), but increased hDM2 affinity vs. other perfluorobiphenyl analogues. Overall, perfluorination increased protease stability, but led to a ≥3-fold decrease in binding affinity for hDM2 versus1.
Hydrophobicity
Differences in hydrophobicity between compounds were compared by HPLC retention times (Fig. 6A), calculated octanol/water partition coefficients (clogP), and calculated total hydrophobic surface area (tHSA). HPLC retention time (Fig. 6A) varied considerably with cyclization linker (lactams eluting faster), correlating with both clogP over 9 log units (Fig. 6B and C) and calculated tHSA over 200 Å2 (Fig. 6B and D). HPLC retention time correlated better with clogP (ChemDraw 20.0, R2 = 0.84, Fig. 6C) than other calculations (e.g. AlogP, Fig. S3, ESI†). Using energy-minimized α-helical models (Maestro, Schrödinger), we estimated tHSA and total surface area for each peptide and found a significant correlation between tHSA and HPLC retention time (Fig. 6D).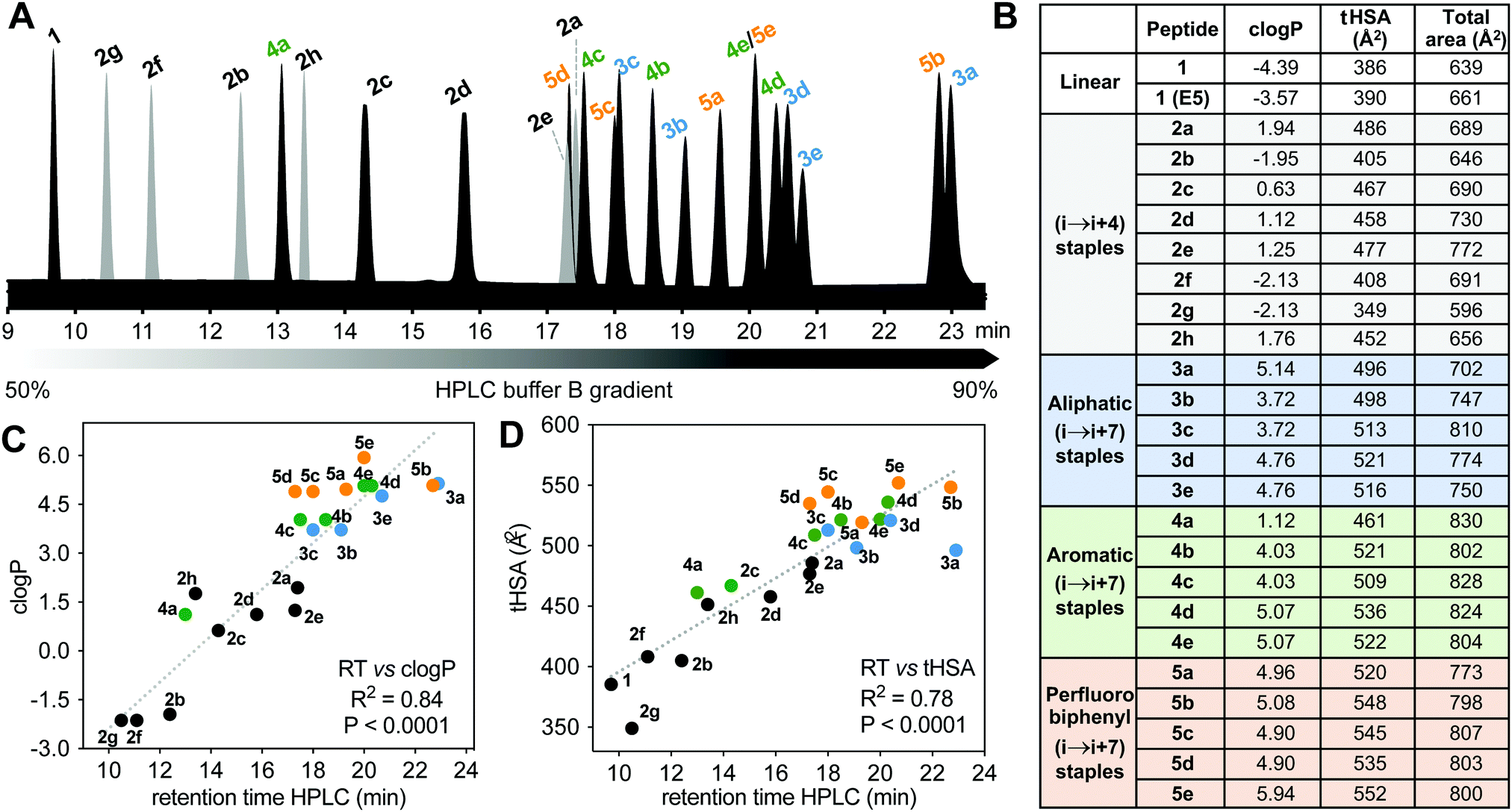 | ||
| Fig. 6 Hydrophobicity of 1, 2a–h, 3a–e, 4a–e, 5a–e. A. HPLC analysis showing retention time for each peptide using a C18 column (Phenomenex Luna), at 1 mL min−1 flow and 2.5 buffer B (0.1% TFA, 90% ACN) gradient per minute. B. Calculated parameters: logP (clogP), total hydrophobic surface area (tHSA) and total surface area for each peptide. C and D. Correlation between clogP (2D hydrophobicity) and HSA (3D hydrophobic surface area) calculated for each peptide vs. experimental HPLC retention. | ||
Lactam bridges hardly altered surface area or volume relative to acyclic 1, whereas hydrocarbon and perfluoroaryl-linked homocysteine staples conferred the greatest hydrophobicity in each series. As expected, hydrophobicity substantially increased (28–40%) with hydrophobic (i → i + 7)-crosslinkers. Two α-methyl groups increased clogP by one long unit, but did not change tHSA much (0.5–4.5%) (Fig. 6B). Noticeably, perfluorobiphenyl analogues 5b and 5c had substantially different HPLC retention times despite similar clogP and tHSA, suggesting an impact of different geometries adjacent to the biphenyl ring. Outlier 3a had intermediate tHSA contrasting with its high HPLC retention time and clogP.
Cell uptake
Cell uptake of the FITC-labelled peptides into HeLa cells was quantified by flow cytometry under serum-free conditions (Fig. 7A), with cell-penetrating peptide TAT as control. The fluorescent label did not significantly change hydrophobicity relative to N-acetylated peptides, according to HPLC retention time (Fig. S4, ESI†). clogP and total hydrophobic surface area (tHSA) calculated for N-acetylated peptides was correlated with cell uptake (Fig. 7B and C). Only peptides with high hydrophobicity (clogP > 3.5, tHSA > 475 Å2) entered HeLa cells (Fig. 7). Most FITC-labelled peptides were α-helical in membrane-simulating SDS buffer (Fig. S5, ESI†), but there was no overall correlation between helicity and cell uptake, even among peptides with high clogP values.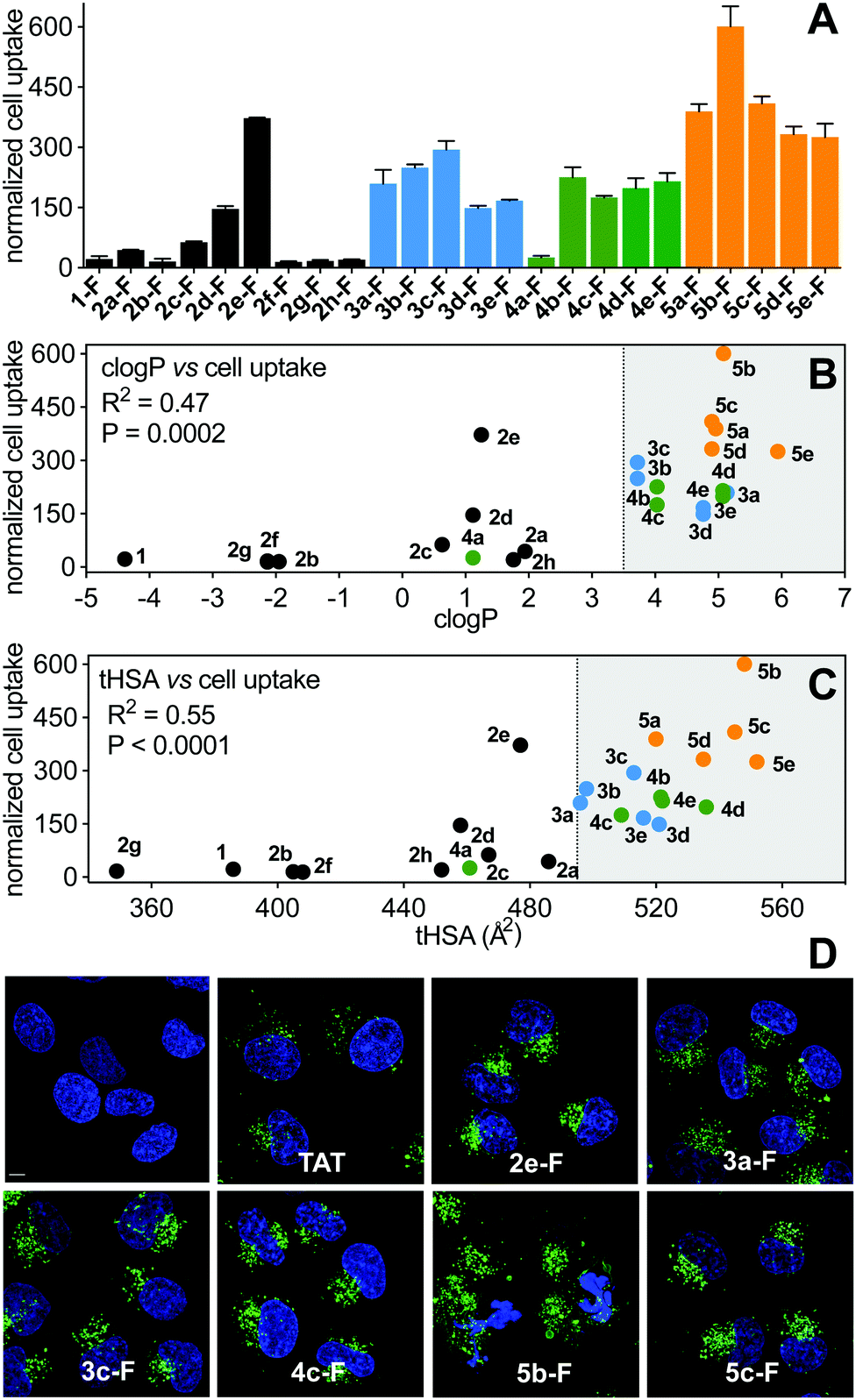 | ||
| Fig. 7 A. Cell uptake of FITC-labelled peptides (5 μM) in HeLa cells measured by flow cytometry. Median fluorescence intensity measured after treatment with trypan blue (to eliminate membrane-bound peptide fluorescence) and normalized to TAT. Data = mean ± SEM. B and C. Correlation of cell uptake with clogP (B) or tHSA (C). D. Live confocal microscopy shows internalization of FITC-labelled peptides (5 μM) into HeLa cells after 3 h. Nuclei stained with Hoechst (blue), peptide green (FITC channel). Scale bar represents 5 μm. | ||
Of the (i → i + 4)-series, only 2e-FITC has substantially entered the cells, despite similar properties to 2a-FITC that has only marginally more uptake than 1-FITC. The shorter perfluorophenyl-stapled compound 2d-FITC also exhibited moderate internalization despite its unruled structure and reduced lipophilicity, suggesting that the perfluorophenyl thioether promotes cell uptake. Lactam linkages were too polar to promote cell uptake, even when combined with a second hydrocarbon bridge (2h-FITC, clogP < 3.5; HSA < 475 Å2). Of the (i → i + 7)-series, cell uptake correlated somewhat with peptide hydrophobicity (R2 = 0.55); aliphatic and non-fluorinated aromatic staples displaying comparable activity. The very hydrophobic perfluorobiphenyl compounds were the most internalized into HeLa cells, with 5b-FITC the most cell uptaken peptide. α-Methylation stabilized helical structure, but surprisingly did not increase cell uptake.
Live cell confocal microscopy was used to visualize internalization of non-lytic FITC-labelled peptides (2e, 3a, 3c, 4c, 5c; see next section under cell lysis) into live HeLa cells, avoiding artefacts of cell fixation. All peptides (5 μM) exhibited significantly more cell uptake than cell-penetrating peptide TAT (Fig. 7D), in agreement with flow cytometry results (Fig. 7A). Peptides showed a punctate intracellular fluorescence distribution, typical of endocytosis and sequestration.31 Similar experiments with lytic peptide 5b-FITC showed even more peptide in the membrane and some endocytotic internalization early during incubation, followed by significant cell damage and cell death after 3 h (Fig. 7D). None of the stapling configurations studied here led to fast endosomal release to the cytosol in HeLa cells. We note that hydrocarbon-stapled peptides such as 3a have been reported to reach the cytosol in sufficient amounts to activate p53-apoptotic pathways in other cancer cell lines.19 One such peptide is ALRN-6924, a promising p53/hDM2 inhibitor peptide drug currently undergoing clinical trials.32
Cell lysis
Non-specific lysis of HeLa cells by the N-acetylated peptides (12 μM, 1 h, Fig. 8) was measured colorimetrically by lactate dehydrogenase (LDH) liberated from the cytosol. HeLa cells were used as they do not respond quickly to activation of the p53 pathway, and early onset of cell damage suggests non-specific membrane lysis induced by the peptides and not by triggering apoptosis. LDH release after 1 h (Fig. 8A) indicated that only the most hydrophobic peptides (3a, 5a, 5b, 5e) promoted cell lysis (clogP ≥ 5, Fig. 8B) in a concentration-dependent manner (Fig. S6, ESI†). At lower concentrations (<6 μM), 3a did not cause significant lysis, whereas 5b was cytotoxic even at 1 μM (Fig. S6, ESI†). Peptides 3c, 4c and 5c with lower hydrophobicity (clogP 3.5–5), internalized into HeLa cells with no lysis even at 50 μM (Fig. S6, ESI†).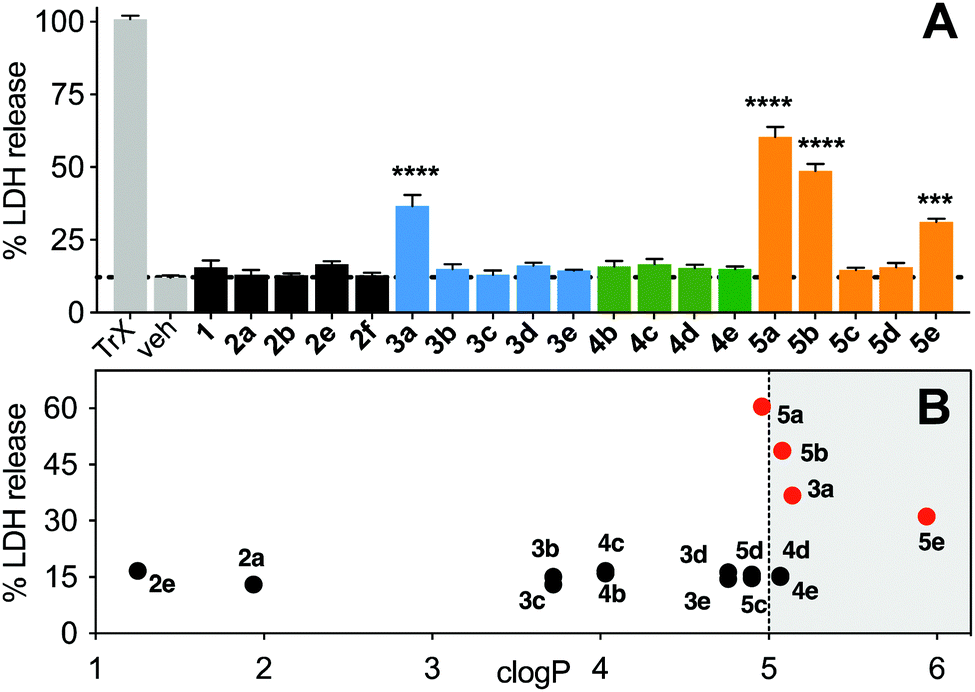 | ||
| Fig. 8 A. LDH release after incubation of stapled peptides (12 μM) in HeLa cells for 1 h. Data = mean ± SEM. One-way ANOVA, *** (P < 0.001), **** (P < 0.0001). B. LDH release versus calculated hydrophobicity (clogP). | ||
Discussion
We have systematically compared the impact of diverse helix-inducing cyclisation constraints on the structure, affinity (hDM2), proteolytic resistance and cell uptake properties of a 12-mer peptide inhibitor of oncogenic p53/hDM2). The linear sequence (1) has little α-helicity, is rapidly and completely degraded by proteases, and its sequence is not sufficiently hydrophobic or amphipathic to permeate cell membranes. Crosslinking two amino acid sidechains at (i, i + 4) or (i, i + 7) positions to constrain one- or two-turns of a helix, respectively, can be optimised to form macrocyclic components that can mitigate these problems.The field of peptide stapling has expanded considerably over the last decade, with different chemical approaches to constrain peptides into helices. However, macrocyclization is limited by chemoselective reactions that work in solution with unprotected peptides (thiolation, click reaction) or are high-yielding conversions on solid-phase (RCM, lactamization). In particular, stapling across longer distances (i → i + 7) with retention of helicity can be more challenging, with fewer literature reports despite yielding the most clinically advanced stapled peptide to date (ALRN-6924).32 Here, we used RCM, S-alkylation and S-arylation chemistry to insert diverse (i → i + 7)-crosslinkers into 1 and compared their properties to (i → i + 4)-crosslinkers.
Enhancing helicity of 1
All (i, i + 4) staples substantially increased α-helicity in water relative to 1 (Fig. 2). Maximum helicity in water for one (i → i + 4) staple was 60% (2a (hydrocarbon), 2f (lactam)) and one (i → i + 7) staple was 70% (3d, 3e (α-methyl octanyl thioether), while ≥90% helicity was achieved with two (i → i + 4) crosslinks (2g, 2h). α-Methyl substitution increased helicity in aliphatic or aromatic thioether stapled compounds, particularly after the switch to D-Cys at positions 4 (3e, 4e, 5e), mirroring the strategy used to develop the (i → i + 7)-hydrocarbon6a in 3a.Solvent-exposed lactam staples showed greater hDM2 affinity
The N-terminus of p53 is intrinsically disordered but folds into an amphipathic helix upon binding to hDM2. A short sequence corresponding to this p53 region has negligible helicity and binds weakly to hDM2, but high affinity can be obtained by insertion of a helix-inducing constraint33 that maintains residues corresponding to Phe19, Trp23, Leu26 of the p53 on the same helical face (Fig. 9). Surprisingly, we found no correlation between % helicity promoted by the various PDI-staples and peptide binding affinity for hDM2, indicating that the induced-fit for 1 is a powerful driver even without preorganized helicity in the peptide.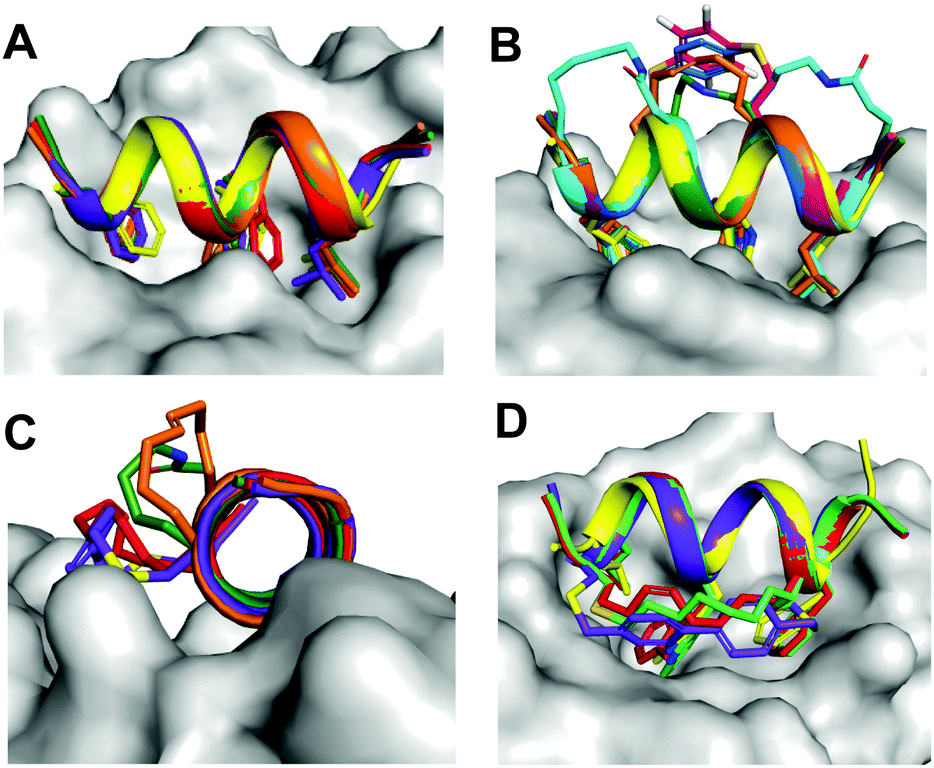 | ||
| Fig. 9 Molecular modelling showing superimposition of stapled peptides bound to hDM2. A. Alignment of Phe3-Trp7-Leu10 at the same hDM2-binding site. B–D. The (i, i + 4)-crosslinkers of (2a (orange), 2b (green), 2c (blue), 2e (magenta), 2f (cyan) project into solvent, whereas (i, i + 7)-staples of 3a (red), 3c (green), 4c (purple) face a hydrophobic pocket located at the boundary of the PPI interface. | ||
Molecular modelling was used to investigate conformational limitations to adopting the hDM2-binding helix in the presence of the crosslinking staples. The (i → i + 4) crosslinkers project into solvent and so do not interfere with hDM2 interactions. However, (i → i + 7) staples face a hydrophobic pocket at the hDM2 surface (Fig. 9). The small size of lactam bridges together with superior helix induction conferred the highest hDM2 affinity to peptides 2b and 2f, enabling them to project hydrophobic Phe3, Trp7 and Leu10 sidechains deep into hDM2 without steric interference from the stapled linker. For the (i → i + 7) series, bulkier aromatic crosslinkers were less favourable than aliphatic linkers for hDM2 affinity, possibly due to unfavourable contacts with the rim of the hDM2 binding site. Such contacts were observed in the crystal structure of hDM2 bound to a (i → i + 7) hydrocarbon-crosslinked p53 peptide,34 and were proposed to assist binding. Controversially, we found here that hydrophilic (i → i + 4) staples that did not interact with the hDM2 protein surface bound even more avidly.
Staples affect proteolytic resistance
Most proteases recognize an extended or β-strand like ligand conformation in their active sites.27 Peptides constrained to, or flexible enough to adopt, that structure are cleaved faster than peptides constrained to helical or turn structures.27d Insertion of the crosslinking staples can slow peptide degradation, by reinforcing α-helical structure and reducing propensity to form an extended conformation recognized and processed by proteases, or by creating a protective shield that denies protease access to amino acid residues adjacent to the staple. All stapling constructs investigated here increased resistance to three common digestive proteases that rapidly cleave unstapled 1. Whereas most staples conferred strong resistance to chymotrypsin, some left the peptide vulnerable to the action of proteinase K and pepsin, emphasizing the importance of measuring degradation against multiple proteases to gauge stability. Pepsin digestion is valuable for assessing peptide proteolysis in acidic environments such as found in the digestive tract.Cysteine-linked peptides showed the lowest protection, allowing cleavage at positions inside and outside the cycle, but this was mitigated by a D-Cys (partially) or α-methyl substituent. Implementing both modifications led to thioether peptides with higher proteolytic resistance, comparable to hydrocarbon-stapled peptides with α-methyl substitution (2a, 3avs.3e, 4e, 5e). An unstapled analogue of peptide 2a strongly resisted the action of proteinase K, implying that the α-methyl alkane residue per se offers strong proteolytic protection to the 12-mer peptide (Fig. S7, ESI†). Interestingly, lactam staples that contained no D- or α-substituted amino acids were exceptionally stable to degradation, suggesting a robust helical configuration that was protective. Moreover, single lactam or hydrocarbon (i → i + 4)-stapling across positions 4–8 was sufficient to shelter the peptide bonds from the proteases, including blocking hydrolysis of residues located outside the macrocycle but held in a helical conformation. Similar protection was only observed in the (i → i + 7)-series when α-methylation was also implemented. Double stapling (2g, 2h) was not required to secure full proteolytic resistance to the 12-mer peptide, but this may be necessary for longer sequences.35
A hydrophobic balance for cell uptake without lysis
Unlike many studies of cell penetrating peptides, the sequence used here lacks positively charged amino acids. Indeed, incorporating Glu5 introduced a negative charge, yet the peptides still interacted with membranes and were taken up into cells, dictated primarily by the hydrophobic linker. Peptides with hydrophilic linkers (lactams, triazoles) did not internalize readily, regardless of how well they stabilized helical structure. Only chemical modifications that enlarged the solvent-exposed hydrophobic surface area of the molecule led to higher cell uptake, independently of helicity. α-Methyl substitution incrementally increased lipophilicity of the peptide, but it did not significantly increase the hydrophobic surface area and did not enhance cell uptake.Overall, (i → i + 7)-crosslinked peptides were more taken up into cells than (i → i + 4)-counterparts because they considerably expanded the hydrophobic surface of the peptide. Of the (i → i + 4) series, the increase in cell uptake was small but noticeable, with lactam ≪ hydrocarbon < benzyl ≪ perfluorophenyl, a trend also observed for staples in a peptide that binds to estrogen receptor coactivator.14 Perfluoro-phenyl analogue 2e was an exception. Of the (i → i + 7)-series, aliphatic and biphenyl crosslinkers gave comparable cell uptake, but perfluorination of the aromatic ring triggered greater entry. Compounds with greater hydrophobicity (clogP > 5) led to cell lysis.
In summary, a compromise between helicity, protein affinity, protease resistance, hydrophobic surface area and cell membrane association are required in these kinds of peptides to achieve greater entry to cancer cells, not get trapped in endosomes, and interact strongly with target intracellular proteins without causing cell lysis.
Author contributions
A. D. A. synthesized peptides, measured CD spectra, protease stability and binding affinity. J. L. and K.-C. W. performed cell assays. H. N. H. calculated structural and biophysical properties. H. T. N. synthesized crosslinkers and α-methyl-cysteine units. D. P. F. obtained funding. A. D. A. and D. P. F. wrote the paper with input from all authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank ARC (DP180103244, DP160104442, CE200100012) and NHMRC (SPRF1117017, 1128908 1145372 and 2009551) for funding.References
- (a) D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS PubMed; (b) H. Lu, Q. Zhou, J. He, Z. Jiang, C. Peng, R. Tong and J. Shi, Signal Transduction Targeted Ther., 2020, 5, 213 CrossRef PubMed; (c) C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502–508 CrossRef CAS PubMed.
- S. Jones and J. M. Thornton, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13–20 CrossRef CAS PubMed.
- (a) N. E. Shepherd, G. Abbenante and D. P. Fairlie, Angew. Chem., Int. Ed., 2004, 43, 2687–2690 CrossRef CAS PubMed; (b) N. E. Shepherd, H. N. Hoang, G. Abbenante and D. P. Fairlie, J. Am. Chem. Soc., 2005, 127, 2974–2983 CrossRef CAS PubMed; (c) R. S. Harrison, N. E. Shepherd, H. N. Hoang, G. Ruiz-Gómez, T. A. Hill, R. W. Driver, V. S. Desai, P. R. Young, G. Abbenante and D. P. Fairlie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11686–11691 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- (a) H. Wang, R. S. Dawber, P. Zhang, M. Walko, A. J. Wilson and X. Wang, Chem. Sci., 2021, 12, 5977–5993 RSC; (b) N. S. Robertson and D. R. Spring, Molecules, 2018, 23, 959 CrossRef PubMed; (c) M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 CrossRef CAS PubMed.
- (a) C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS; (b) L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed; (c) P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed.
- D. P. Fairlie and A. Dantas de Araujo, Biopolymers, 2016, 106, 843–852 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, S.-T. Quah, M. Rossmann, L. Laraia, N. Skold, T. J. Sum, P. J.-E. Rowling, T. L. Joseph, C. Verma, M. Hyvonen, L. S. Itzhaki, A. R. Venkitaraman, C. J. Brown, D. P. Lane and D. R. Spring, Chem. Sci., 2014, 5, 1804–1809 RSC.
- Peptide stapling reviews: (a) L. McDougall and A. G. Jamieson, eLS, John Wiley & Sons, Ltd, 2022, https://doi.org/10.1002/9780470015902.a002840 Search PubMed; (b) X. Li, S. Chen, W.-D. Zhang and H.-G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed; (c) M. Moiola, M. G. Memeo and P. Quadrelli, Molecules, 2019, 24, 3654 CrossRef CAS PubMed; (d) Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2014, 44, 91–102 RSC.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS PubMed.
- A. D. Kalafatovic and E. Giralt, Molecules, 2017, 22, 1929 CrossRef PubMed.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS PubMed.
- H. N. Hoang, R. W. Driver, R. L. Beyer, T. A. Hill, A. D. de Araujo, F. Plisson, R. S. Harrison, L. Goedecke, N. E. Shepherd and D. P. Fairlie, Angew. Chem., Int. Ed., 2016, 55, 8275–8279 CrossRef CAS PubMed.
- Y. Tian, Y. Jiang, J. Li, D. Wang, H. Zhao and Z. Li, ChemBioChem, 2017, 18, 2087–2093 CrossRef CAS PubMed.
- Y. Wang and D. H. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- A. Dantas de Araujo, S. R. Perry and D. P. Fairlie, Org. Lett., 2018, 20, 1453–1456 CrossRef CAS PubMed.
- B. Hu, D. M. Gilkes and J. Chen, Cancer Res., 2007, 67, 8810–8817 CrossRef CAS PubMed.
- G. Sanz, M. Singh, S. Peuget and G. Selivanova, J. Mol. Cell Biol., 2019, 11, 586–599 CrossRef CAS PubMed.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–3454 CAS.
- (a) K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, F. Jiang, T. Wang, Y.-D. Wu and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 8013–8017 CrossRef CAS PubMed; (b) G. Philippe, Y.-H. Huang, O. Cheneval, N. Lawrence, Z. Zhang, D. P. Fairlie, D. J. Craik, A. D. de Araujo and S. T. Henriques, J. Pept. Sci., 2016, 106, 853–863 CrossRef CAS PubMed; (c) G. Philippe, A. Mittermeier, N. Lawrence, Y.-H. Huang, N. D. Condon, A. Loewer, D. J. Craik and S. T. Henriques, ACS Chem. Biol., 2021, 16, 414–428 CrossRef CAS PubMed; (d) P. G. Dougherty, J. Wen, X. Pan, A. Koley, J.-G. Ren, A. Sahni, R. Basu, H. Salim, G. Appiah Kubi, Z. Qian and D. Pei, J. Med. Chem., 2019, 62, 10098–10107 CrossRef CAS PubMed; (e) Y. H. Lau, Y. Wu, M. Rossmann, B. X. Tan, P. de Andrade, Y. S. Tan, C. Verma, G. J. McKenzie, A. R. Venkitaraman, M. Hyvönen and D. R. Spring, Angew. Chem., Int. Ed., 2015, 54, 15410–15413 CrossRef CAS PubMed.
- Z. Muppidi, X. Wang, J. Li, J. Chen and Q. Lin, Chem. Commun., 2011, 47, 9396–9398 RSC.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- S. J.-M. Verhoork, C. E. Jennings, N. Rozatian, J. Reeks, J. Meng, E. K. Corlett, F. Bunglawala, M. E.-M. Noble, A. G. Leach and C. R. Coxon, Chem. – Eur. J., 2019, 25, 177–182 CrossRef CAS PubMed.
- A. D. de Araujo, J. Lim, K. C. Wu, Y. Xiang, A. C. Good, R. Skerlj and D. P. Fairlie, J. Med. Chem., 2018, 61, 2962–2972 CrossRef PubMed.
- (a) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020–13041 CrossRef CAS PubMed; (b) M. J. Kelso, R. L. Beyer, H. N. Hoang, A. S. Lakdawala, J. P. Snyder, W. P. Oliver, T. A. Robertson, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2004, 126, 4828–4842 CrossRef CAS PubMed; (c) R. L. Beyer, H. N. Hoang, T. G. Appleton and D. P. Fairlie, J. Am. Chem. Soc., 2004, 126, 15096–15105 CrossRef CAS PubMed.
- (a) J. D.-A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2005, 105, 973–1000 CrossRef CAS PubMed; (b) P. K. Madala, J. D.-A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2010, 110(6), PR1–PR31 CrossRef CAS PubMed; (c) J. D.-A. Tyndall and D. P. Fairlie, J. Mol. Recognit., 1999, 12, 363–370 CrossRef CAS PubMed; (d) D. P. Fairlie, J. D.-A. Tyndall, R. C. Reid, A. K. Wong, G. Abbenante, M. J. Scanlon, D. R. March, D. A. Bergman, C. L.-L. Chai and B. A. Burkett, J. Med. Chem., 2000, 43, 1271–1281 CrossRef CAS PubMed.
- (a) G. T. Perell, R. L. Staebell, M. Hairani, A. Cembran and W. C.-K. Pomerantz, ChemBioChem, 2017, 18, 1836–1844 CrossRef CAS PubMed; (b) G. Zhang, F. Barragan, K. Wilson, N. Levy, A. Herskovits, M. Sapozhnikov, Y. Rodríguez, L. Kelmendi, H. Alkasimi, H. Korsmo, M. Chowdhury and G. Gerona-Navarro, Angew. Chem., Int. Ed., 2018, 57, 17073–17078 CrossRef CAS PubMed.
- A. D. de Araujo, H. T. Nguyen and D. P. Fairlie, ChemBioChem, 2021, 22, 1784–1789 CrossRef CAS PubMed.
- C. M. Fadzen, J. M. Wolfe, C.-F. Cho, E. A. Chiocca, S. E. Lawler and B. L. Pentelute, J. Am. Chem. Soc., 2017, 139, 15628–15631 CrossRef CAS PubMed.
- D. Y. Yoo, S. A. Barros, G. C. Brown, C. Rabot, D. Bar-Sagi and P. S. Arora, J. Am. Chem. Soc., 2020, 142, 14461–14471 CrossRef CAS PubMed.
- (a) M. N. Saleh, M. R. Patel, T. M. Bauer, S. Goel, G. S. Falchook, G. I. Shapiro, K. Y. Chung, J. R. Infante, R. M. Conry, G. Rabinowits, D. S. Hong, J. S. Wang, U. Steidl, G. Naik, V. Guerlavais, V. Vukovic, D. A. Annis, M. Aivado and F. Meric-Bernstam, Clin. Cancer Res., 2021, 27, 5236–5247 CrossRef CAS PubMed; (b) C. Morrison, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS PubMed.
- F. Bernal, A. F. Tyler, S. J. Korsmeyer, L. D. Walensky and G. L. Verdine, J. Am. Chem. Soc., 2007, 129, 2456–2457 CrossRef CAS PubMed.
- S. Baek, P. S. Kutchukian, G. L. Verdine, R. Huber, T. A. Holak, K. W. Lee and G. M. Popowicz, J. Am. Chem. Soc., 2011, 134, 103–106 CrossRef PubMed.
- G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, chemical, structural and biological characterisation data. Experimental procedures for compound syntheses, characterization by HPLC and MS analysis, hDM2 binding assays, proteolysis analysis and cell-based assays (PDF). See DOI: https://doi.org/10.1039/d1cb00231g |
| This journal is © The Royal Society of Chemistry 2022 |