
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural impact of thioamide incorporation into a β-hairpin†
Kristen E.
Fiore
 a,
Martijn J.
Patist
a,
Sam
Giannakoulias
a,
Cheng-Hsin
Huang
b,
Hitesh
Verma
c,
Bhavesh
Khatri
c,
Richard P.
Cheng
b,
Jayanta
Chatterjee
c and
E. James
Petersson
*a
a,
Martijn J.
Patist
a,
Sam
Giannakoulias
a,
Cheng-Hsin
Huang
b,
Hitesh
Verma
c,
Bhavesh
Khatri
c,
Richard P.
Cheng
b,
Jayanta
Chatterjee
c and
E. James
Petersson
*a
aDepartment of Chemistry, University of Pennsylvania, 231 S. 34th Street, Philadelphia, 19104, USA. E-mail: ejpetersson@sas.upenn.edu
bDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei 10617, Taiwan
cMolecular Biophysics Unit, Indian Institute of Science, Bangalore 560012, India
First published on 5th April 2022
Abstract
The thioamide is a naturally-occurring single atom substitution of the canonical amide bond. The exchange of oxygen to sulfur alters the amide's physical and chemical characteristics, thereby expanding its functionality. Incorporation of thioamides in prevalent secondary structures has demonstrated that they can either have stabilizing, destabilizing, or neutral effects. We performed a systematic investigation of the structural impact of thioamide incorporation in a β-hairpin scaffold with nuclear magnetic resonance (NMR). Thioamides as hydrogen bond donors did not increase the foldedness of the more stable “YKL” variant of this scaffold. In the less stable “HPT” variant of the scaffold, the thioamide could be stabilizing as a hydrogen bond donor and destabilizing as a hydrogen bond acceptor, but the extent of the perturbation depended upon the position of incorporation. To better understand these effects we performed structural modelling of the macrocyclic folded HPT variants. Finally, we compare the thioamide effects that we observe to previous studies of both side-chain and backbone perturbations to this β-hairpin scaffold to provide context for our observations.
Introduction
The thioamide is an intriguing isostere of the canonical amide bond. Although it differs by only a single atom, the thioamide has unique chemical and physical properties that have been employed by chemists and biophysicists. For example, the thioamide has a lower oxidation potential.1 Consequently, thioamides can quench fluorescence in a distance dependent manner: Förster resonance energy transfer (FRET)-based quenching of UV fluorophores and photoinduced electron transfer (PeT)-based quenching of visible fluorophores.2 Therefore, fluorophore/thioamide pairs can be utilized as minimal biophysical probes to study protein folding or dynamics. This has been done to monitor peptide–protein binding,3–5 to monitor protease activity in real time,6 and to monitor protein conformational changes during refolding,3 unfolding,7,8 or misfolding.5Another unique property of the thioamide is that is has a red-shifted π-to-π* absorption,9 giving it a unique circular dichroism (CD) signature.10 This red-shifted absorption also lowers the excitation energy required for photoisomerization.11 Therefore, upon irradiation the thioamide can selectivity photoisomerize from trans-to-cis, enabling its use as a photoswitch in peptides.12–15
Nature installs thioamides in ribosomally synthesized and post-translationally modified peptides (RiPPs),16 as well as in at least two proteins: methyl coenzyme M reductase (MCR)17,18 and the uL16 protein of the E. coli 70S ribosome.19 Although the method of installation is well-studied, the effect of thioamidation on protein function is relatively unknown.
To develop a systematic understanding of how the thioamide can affect biological activity, as well as to promote its utility as a biophysical probe, a more fundamental understanding of the structural impact of thioamide incorporation is needed. Previously, small molecule studies have suggested that the lower electronegativity of the thioamide sulfur results in it being a weaker hydrogen bond acceptor.20–22 Conversely, the lower N–H pKa (12 versus 14)23 should result in the thioamide being a stronger hydrogen bond donor (Fig. 1).24,25 Additionally the larger van der Waals radius of sulfur26 results in a longer C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond27,28 which could be perturbative depending upon the environment. To determine the impact on protein thermodynamic stability, we incorporated thioamides into native proteins of different secondary structures: calmodulin (α-helical), the B1 domain of protein G (GB1, β-sheet), and collagen (PPII triple-helix).29 In some cases, reasonable explanations could be made as to why incorporation at some positions was more destabilizing than others based on the existing structures of the native proteins. However, many seemingly similar thioamide substitutions resulted in very different effects on protein stability. We were particularly intrigued by the effects on GB1, where substitution in the same β-strand had destabilizing effects differing by 2 kcal mol−1. The same variation was observed in another β-sheet structure, the Pin1 WW domain (three β-strands), where the thermostability (ΔTM(thio–oxo)) varied from −0.9 to 14.8 °C depending upon the microenvironment of the position.30 To further investigate the effects of thioamides in β-sheets, we turned to model peptide systems, which have proven to be valuable for rigorous investigation of protein modifications.
S double bond27,28 which could be perturbative depending upon the environment. To determine the impact on protein thermodynamic stability, we incorporated thioamides into native proteins of different secondary structures: calmodulin (α-helical), the B1 domain of protein G (GB1, β-sheet), and collagen (PPII triple-helix).29 In some cases, reasonable explanations could be made as to why incorporation at some positions was more destabilizing than others based on the existing structures of the native proteins. However, many seemingly similar thioamide substitutions resulted in very different effects on protein stability. We were particularly intrigued by the effects on GB1, where substitution in the same β-strand had destabilizing effects differing by 2 kcal mol−1. The same variation was observed in another β-sheet structure, the Pin1 WW domain (three β-strands), where the thermostability (ΔTM(thio–oxo)) varied from −0.9 to 14.8 °C depending upon the microenvironment of the position.30 To further investigate the effects of thioamides in β-sheets, we turned to model peptide systems, which have proven to be valuable for rigorous investigation of protein modifications.
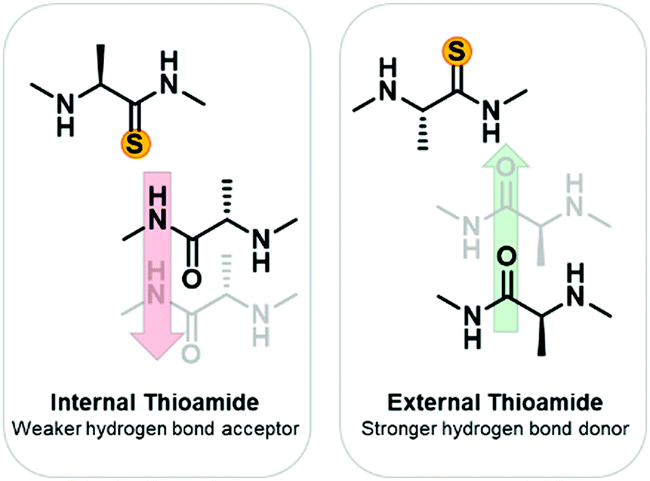 | ||
| Fig. 1 Expected thioamide effects on hydrogen bonding. Based on previous small molecule studies, it is expected that an internal thioamide will be disruptive to β-hairpin structure due to the thioamide being a weaker hydrogen bond acceptor and having a larger van der Waals radius. An external thioamide will be stabilizing since it is a stronger hydrogen bond donor than an amide. | ||
Thioamides have been previously investigated in α-helical31,32 and poly-proline II (PPII) helical model systems.33 The structural impacts were scaffold and position specific. In contrast, there has been very limited study of thioamides in β-sheet model systems with only four examples to our knowledge. In two of the studies, thioamide substitution occurred in the turn, and therefore is not informative on how the chemical and physical properties of the thioamide affect cross-strand β-sheet interactions.34,35 In a well-studied β-hairpin, the tryptophan zipper (Trpzip), thioamides were incorporated as hydrogen bond acceptors and the thermodynamic stability measured using CD spectroscopy.36 Overall, the thioamides within the strands were destabilizing by about 1 kcal mol−1, with the terminal position being the least perturbative. Thioamides were also incorporated into a TrpZip with an azobenzene derivative at the β-turn.37 This allowed for control of the folding state of the peptide by photo-initiated cis/trans isomerization (cis = folded, trans = unfolded). Folding was observed with time-resolved IR spectroscopy and CD. For these TrpZips, two thioamides were incorporated to observe site-specific coupling spectroscopically. Thioamides on the same strand serving as hydrogen bond donors were minimally perturbative with unfolding rates similar to the all-amide reference. Thioamides on opposite strands serving as hydrogen bond donors stabilized the β-hairpin relative to the reference. Thioamides on opposite strands as hydrogen bond acceptors strongly destabilized the β-hairpin.
In these studies, the impact of thioamides on β-sheets are largely interpreted in terms of differing hydrogen bonding properties. However, such a simple interpretation is inconsistent with our observations in GB1, where local structure significantly altered the impact of thioamide substitution. Moreover, although these two studies include elegant kinetic and thermodynamic studies, they lack any direct structural information. Although TrpZips have been well-characterized, we were interested in studying a less-folded β-hairpin scaffold that might be more sensitive to effects of the thioamide and one for which an extensive body of literature on other non-covalent interactions is available for comparison. Therefore, we have designed a systematic investigation of thioamide incorporation using a model β-hairpin system that meets these requirements and performed structural analysis with NMR. This experimental data is supplemented with structural modelling of the macrocyclic folded variants.
Results and discussion
Scaffold design
In choosing a host scaffold for our thioamide guests, we analyzed β-hairpins that are water soluble, monomeric, and have significant β-sheet character. A well-established construct that meets these characteristics is the Gellman “YKL” β-hairpin.38,39 This scaffold has been utilized to study the β-hairpin stabilization of cross-strand interactions38 and strand length,40 as well as the β-sheet propensity of charged derivatives of β-branched-amino acids41 and aza-amino acids.42 This extensively studied scaffold is also a good starting point for structural characterization as several previous studies have reported structural models based on NMR data.38–40,43 This β-hairpin is enforced with a stabilized two-residue β-turn, proGly (where pro is D-proline). Examination of 2 residue β-hairpin loops in proteins determined that there is a preference for type I′ and type II′ β-turns44 and pro promotes the right-handed twist needed for this biologically relevant β-turn.45 Conversely, ProGly incorporation results in a left-handed turn that eradicates the β-sheet structure, which can be utilized for synthesis of an unfolded control peptide.39 This 12 residue anti-parallel “YKL” β-sheet is stabilized by a diagonal cation–π interaction (Tyr2 and Lys9). To avoid electrostatic associations from the termini, the N-terminus is acetylated, and the C-terminus is a carboxamide (Fig. 2). We hypothesized that the β-hairpin would be stabilized when the thioamide is positioned as a hydrogen bond donor.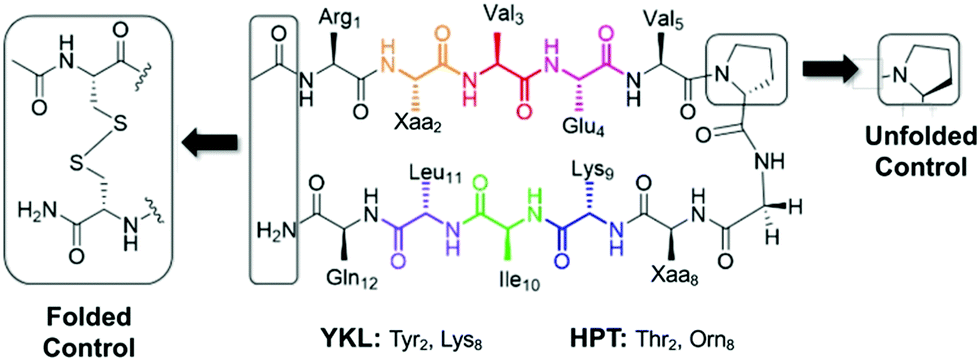 | ||
| Fig. 2 YKL and HPT test β-hairpin scaffolds. The unfolded control has a ProGly β-turn. The folded control has terminal cysteines that are oxidized to form a disulfide linked cyclic peptide. The folded control was only synthesized for the HPT scaffold. | ||
YKL scaffold CSD analysis
We initially chose to incorporate thioamides at the following hydrogen bond donor positions: Glu4, Lys9 and Leu11. These constructs are denoted YKL-GluS4, YKL-LysS9, and YKL-LeuS11-OH, using the superscript S convention for naming thioamides from Mahanta et al.16 The LeuS11 peptide was synthesized with a C-terminal carboxylate (indicated as –OH) due to the propensity for thioamides at the penultimate position to cause hydrolysis and epimerization at C-terminal amides (Fig. S4, ESI†).3 For each thioamide position of interest as well as the all-amide reference peptide denoted “YKL”, two peptides were synthesized, the test peptide (with proGly at the turn) and the corresponding unfolded control peptide. The unfolded control has the same sequence as the test peptide but has a ProGly β-turn so it does not have any β-sheet secondary structure.Except for GluS4, the test peptides displayed significant anti-parallel β-sheet character (minimum at 215 nm) in the far-UV region of their CD spectra (Fig. S6A, ESI†). This indicated that the level of folding in the thioamide peptides was comparable to the YKL reference peptide. For YKL-GluS4, the π-to-π* absorbance of the thioamide has a greater intensity, which complicates the spectra and could explain the lack of a minimum at 215 nm. Since the contribution of a thioamide residue to the CD spectrum is not well-defined, we sought other measurements to quantify the relative stabilities of the thioamide variants. CD thermal melts measured at the 215 nm signature were linear, and therefore could not be fit to two-state models to derive folding energetics (Fig. S6B, ESI†). Therefore, we turned to 2D NMR measurements instead. Peptides were dissolved in sodium deuteroacetate buffer (Table S3, ESI†)‡ and TOCSY and ROESY were collected at 10 °C. Lateral NOEs between Tyr2 and Leu11, as well as diagonal NOEs between Tyr2 and Lys9 were observed for all test β-hairpins (Fig. S7, ESI†). This suggests that all thioamide-containing variants are in a β-hairpin conformation. Although the connectivities of the observed NOEs differ slightly, the similarity of the overall patterns is enough to deem the structures alike.
As expected, the unfolded controls lack cross-strand NOEs (Fig. S8, ESI†). Inclusion of the ProGly β-hairpin allows calculation of the chemical shift deviation (CSD or Δδ = test δ − random coil δ) with the ProGly unfolded control (Δδ = test δ − unfolded control δ). β-Hairpins are dynamic structures, and CSD analysis is reflective of the global average, providing a quantitative measure indicative of secondary structure. A ΔδHα of greater than 0.1 ppm for three consecutive residues is considered to be evidence of a β-sheet.46 The ΔδHα data suggest that all tested thioamide-peptides have β-sheet character comparable to the YKL reference peptide (Fig. 3). At each thioamide position, the ΔδHα for the n + 1 residue is significantly different in comparison to other peptides. However, this is merely indicative of a local perturbation of the electronic environment since ΔδHα is not increased for other residues. Although YKL-GluS4 has β-sheet character according to ΔδHα, the values are lower than the other positions tested. This could be due to additional conformational rigidity from the nearby β-turn, which results in a less favorable β-hairpin structure with a thioamide at Glu4. Thus, we conclude from these data as well as variable temperature NMR experiments that the thioamide hydrogen bond donor substitutions do not significantly increase the stability of the β-hairpin (Table S4, ESI†).
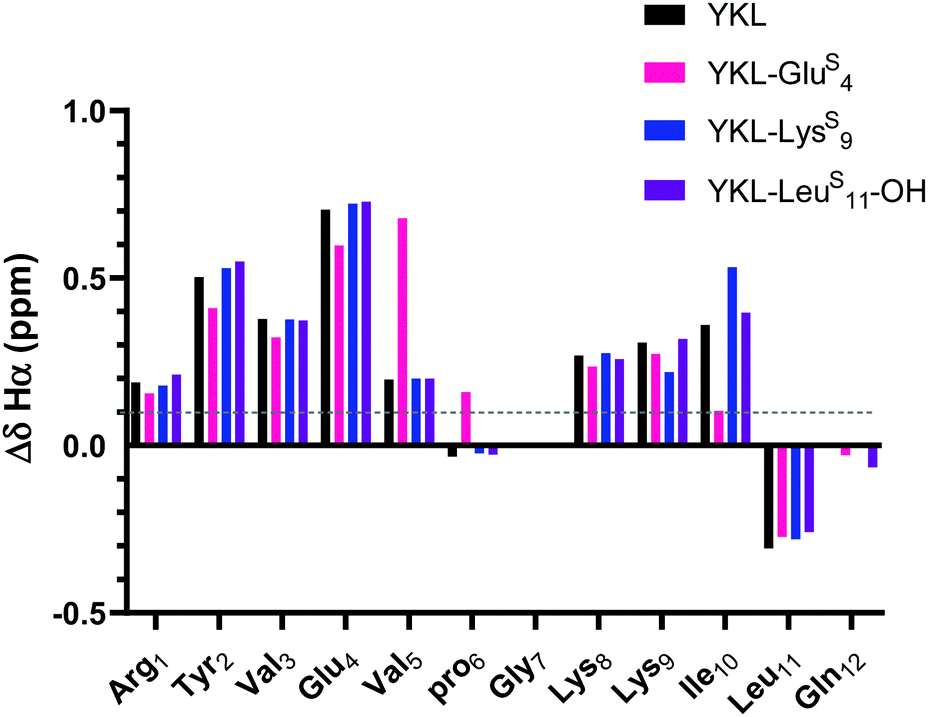 | ||
| Fig. 3 ΔδHα (test δHα − unfolded control δHα) for YKL-GluS4, YKL-LysS9, and YKL-LeuS11-OH in comparison to YKL. Three consecutive ΔδHα values of greater than 0.1 ppm (shown in figure) is indicative of β-sheet structure. Besides slight variations around where the thioamide is incorporated, the ΔδHα values are like YKL for all positions tested suggesting that the thioamide is not increasing foldedness. ΔδNH analysis also demonstrates the same trends (Fig. S9, ESI†). | ||
HPT scaffold CSD analysis
In light of the unexpected failure of the thioamide hydrogen bond donor to increase foldedness, we questioned whether the YKL scaffold was too stable to observe potential perturbations to structure due to thioamide incorporation. Removal of the cation–π interaction would decrease the stability of the scaffold, making it more sensitive to perturbation. Indeed, the Cheng lab previously replaced Tyr2 with Thr to achieve this purpose and used Orn8 instead of Lys8 to help with chemical shift assignment.47–49 Also, the internal Tyr side-chain causes ring-currents that result in upfield shifts of other internal protons. Therefore, removal of Tyr additionally leaves the internal chemical shifts unaffected by the ring-currents and allows for more accurate determination of the effect of incorporation of an internal thioamide. This variant of the YKL scaffold is referred to as HPT (HairPins with Thr at position 2) (Fig. 2). The hypothesis remained that incorporation of a thioamide as a hydrogen bond donor would increase foldedness, whereas a thioamide as a hydrogen bond acceptor would decrease foldedness.Thioamides were incorporated at Thr2, Val3, Ile10, and Leu11 in both test (proGly) and unfolded (ProGly) forms: HPT-ThrS2, HPT-ValS3, HPT-IleS10, and HPT-LeuS11-OH. Again, a carboxylate was included at the C-terminus of the LeuS11 peptide, in this case due to hydrolysis of the amide form (Fig. S4, S5 and Tables S6, S7, ESI†). The test β-hairpins were first examined by CD and have a significant thioamide π-to-π* absorption band (Fig. S10, ESI†). However, the less stable test HPT peptides do not have as strong a β-sheet signature at 218 nm, which is further complicated by the strong thioamide absorbance. Therefore, the effect of thioamide incorporation on global secondary structure could not be determined using CD. As a result, we again relied on NMR for information.
HPT, HPT-ThrS2, HPT-ValS3, HPT-IleS10, and HPT-LeuS11-OH were dissolved in sodium deuteroacetate or phosphate buffer to 1–10 mM concentration (Table S3, ESI†).2 TOCSY and ROESY spectra were collected at 25 °C. The HPT NMR data was obtained from a previous publication.49
For the test peptide, the HPT β-hairpin has both lateral NOEs between Thr2 and Leu11, as well as diagonal NOEs between Thr2 and Lys9. All thioamide test β-hairpins have mainly lateral NOEs between Glu4 and Lys9, with HPT-ThrS2 having a second cross-strand NOE between Thr2 and Leu11 (Fig. S11, ESI†). The presence of cross-strand NOEs for all positions tested is suggestive of β-hairpin conformation. As expected, all unfolded controls lack cross-strand NOEs (Fig. S12, ESI†). For a more quantitative measure of β-hairpin structure, we again used CSD analysis.
The ΔδHα values for HPT are generally lower than those for YKL, indicating that it has less β-sheet structure, in agreement with the CD data and expectations based on prior literature.49 The ΔδHα values for the HPT-ThrS2, HPT-IleS10 and HPT-LeuS11-OH peptides demonstrate that all have β-sheet structure, whereas HPT-ValS3 does not (Fig. 4). Besides HPT-IleS10, these observations agree with our YKL work that a thioamide as a hydrogen bond donor is neutral, and additionally agrees with our hypothesis that an internal thioamide is destabilizing. We did not expect that an internal thioamide (IleS10) could be minimally perturbing. The difference observed for both internal thioamide positions (ValS3 and IleS10) could be due to the right-handed twist of the β-hairpin,50 which would better accept the additional steric bulk at Ile10, but not Val3.
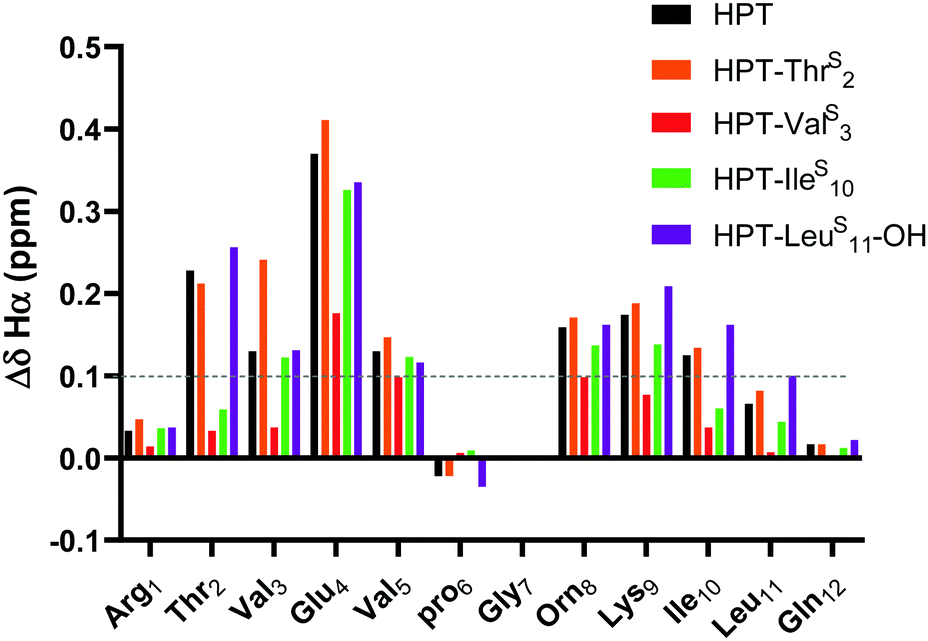 | ||
| Fig. 4 ΔδHα (test δHα − unfolded control δHα) for HPT-ThrS2, HPT-ValS3, HPT-IleS10, and HPT-LeuS11-OH in comparison to HPT. Three consecutive ΔδHα values of greater than 0.1 ppm (shown in figure) is indicative of β-sheet structure. The ΔδHα data demonstrates that HPT-ThrS2, HPT-IleS10, and HPT-LeuS11-OH have β-sheet character, whereas HPT-ValS3 does not. Discussion of ΔδNH is in the ESI† (Fig. S13). | ||
HPT scaffold ΔΔG analysis
To quantitatively compare the effect of thioamide incorporation, folded controls were synthesized to allow for calculation of fraction folded (%) and ΔΔGFolding. For the folded control, terminal cysteines were added and oxidized to produce a disulfide-linked cyclic peptide following a strategy previously employed by the Cheng laboratory (Fig. 2).43,47–49 However, this analysis is only valid if the folded controls display sufficient chemical shift dispersion. As the β-hairpin is more folded, both δHα and δNH should be shifted downfield. This was observed when the thioamide is externally facing (i.e., positioned as a hydrogen bond donor). However, the folded controls with an internal thioamide did not display enough dispersion to be a true folded control (Fig. 5). This could be due to puckering as the internal electron-rich thioamide is forced towards the opposing strand due to steric constraints. Since we observed abundant cross-strand NOEs for the HPT-ValS3 and HPT-IleS10 folded controls (Fig. S14, ESI†), we performed fraction folded analysis despite the lack of chemical shift dispersion. We note that in Fig. 5 ΔδHα is dramatically affected for the residue following the thioamide, consistent with previous studies of the steric and electronic impact of thioamide incorporation on neighboring residues.51,52 The ΔδHα value is increased relative to the HPT value for more stable peptides with externally facing thioamides and decreased for less stable peptides with internally facing thioamides.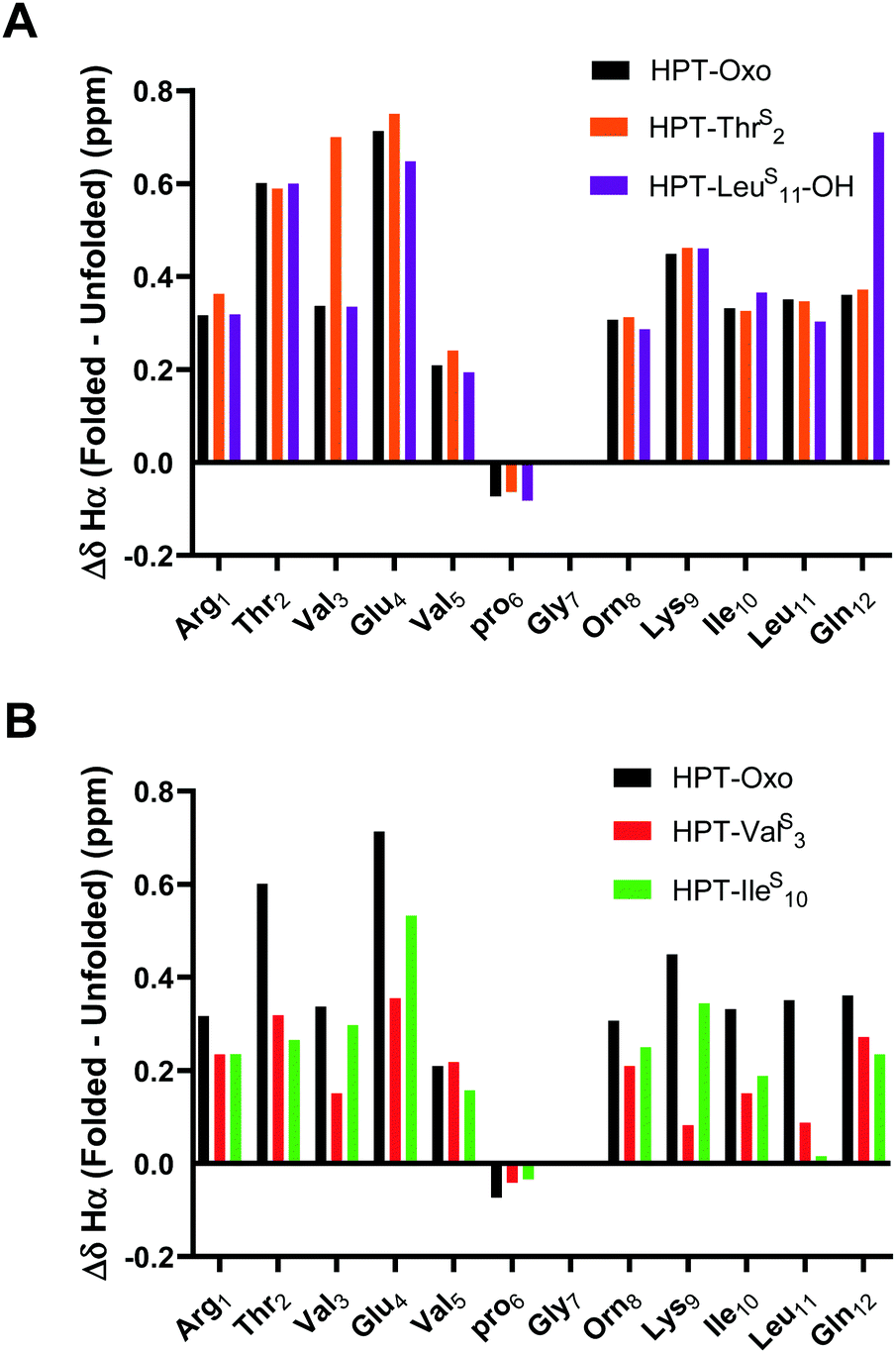 | ||
| Fig. 5 ΔδHα (folded δHα − unfolded control δHα) for external thioamides (A, HPT-ThrS2 and HPT-LeuS11-OH) and internal thioamides (B, HPT-ValS3 and HPT-IleS10) in comparison to HPT. Folded controls with internal thioamides result in a decreased chemical shift dispersion. The dispersion can be quantified by comparing the ΔδHα between the folded and unfolded controls. For the external thioamides (A), there is a similar dispersion observed for all the peptides. This is not observed for the internal thioamides (B), where the values greatly differ in comparison to HPT. | ||
Fraction folded was calculated for each residue of a test β-hairpin using eqn (1). The final fraction folded value reported is an average of position 3 and 10. These positions were chosen because Val3 and Ile10 are in the middle of the β-strands and therefore not affected by the flexible termini or the constrained β-turn. Secondly, for Val3 and Ile10, Hα is externally facing and therefore δHα is not affected by the internal micro-environment of the β-hairpin which may be perturbed by the thioamide. Thus, δHα for these positions is a direct indicator of foldedness. Similar to ΔδHα analysis, HPT-ThrS2 has the same fraction folded value as HPT (Table 1). HPT-LeuS11-OH has a higher fraction folded, HPT-IleS10 is slightly less folded, and HPT-ValS3 is the least folded. ΔΔGFolding was calculated with eqn (2) and (3). The ΔΔGFolding provide a general metric that can be compared to other β-hairpin studies to place the magnitude of the observed effects in context.
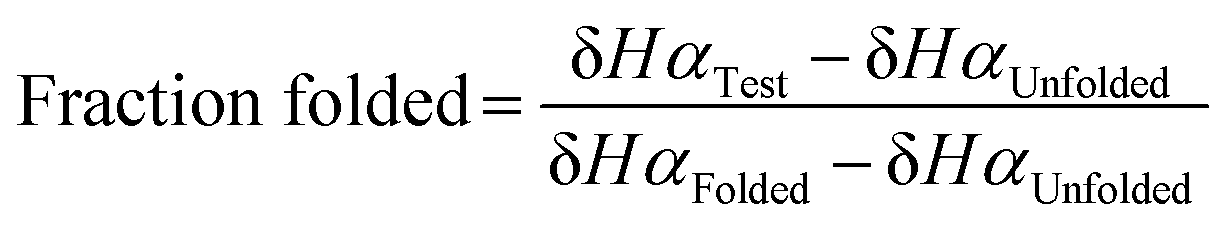 | (1) |
 | (2) |
| ΔΔG = ΔGThio − ΔGoxo | (3) |
| Peptide | Fraction folded (%) | ΔΔGFolding (kcal mol−1) |
|---|---|---|
| Fraction folded calculated using eqn (1), reported as an average of positions 3 and 10. Eqn (2) and (3) were used to calculate ΔΔGFolding. Both methods of analysis are suggestive that HPT-ThrS2 and HPT-IleS10 have a similar energy of folding and fraction folded profile to HPT. HPT-LeuS11-OH is more folded and has a more favorable free energy of folding than HPT. Thioamide incorporation at ValS3 is disruptive to β-hairpin structure. See Table S8 (ESI) for all values used in calculating fraction folded and ΔΔG. | ||
| HPT | 38 ± 1 | — |
| HPT-ThrS2 | 38 ± 3 | 0.01 ± 0.1 |
| HPT-ValS3 | 25 ± 1 | 0.38 ± 0.01 |
| HPT-IleS10 | 37 ± 5 | 0.04 ± 0.1 |
| HPT-LeuS11-OH | 42 ± 3 | −0.09 ± 0.1 |
These data clearly show that even in a short β-hairpin the context of the thioamide substitution is very important. While the thioamide as hydrogen bond acceptor can indeed be destabilizing (HPT-ValS3), it can also be a neutral modification (HPT-IleS10). Likewise, while the thioamide as hydrogen bond donor can be stabilizing in the more sensitive HPT scaffold (HPT-LeuS11-OH), it too can be neutral (HPT-ThrS2), depending on local context.
Structural modelling
To elucidate the mechanistic basis of the thioamide effects we observed with NMR, we utilized structural modelling with PyRosetta.53 Since the folded HPT peptides exhibited significantly stronger NOEs (Fig. S11 and S14, ESI†) due to their macrocyclic constraint, we took advantage of this stability to avoid difficulty in modelling and analysis because of the flexibility in the test peptides. As a starting point, we used the average NMR structure (PDB ID 1jy9) previously solved for a YKL derivative with four additional Thr residues at the termini (see Steric interactions sub-section below).40 The structure was modified to convert it to the HPT folded control by removing the two terminal Thr residues, converting the penultimate Thr residues to Cys, forming the disulfide bond, acetylating the N-terminus, converting the C-terminus to a carboxamide, and converting Tyr2 to Thr and Lys8 to Orn. Following generation in PyRosetta, the initial HPT structure is similar to the starting 1jy9 PDB structure (Fig. S16, ESI†). Next, we performed a constrained relax in Rosetta using the NOE-derived distances for the HPT folded control to generate our final HPT structure (Fig. S17 and S18, ESI†). Previously, the Petersson laboratory developed Rosetta patches for the thioamide based on ab initio calculations and experimental data for thioamides in protease substrates.54–56 With the thioamide patches and the corresponding distance constraints, the thioamide folded control peptides: HPT-ThrS2, HPT-ValS3, HPT-IleS10, and HPT-LeuS10 were simulated in PyRosetta. On average, 20 constraints were used per structure and only three distance pairs per β-hairpin had a deviation of greater than 0.15 Å between the experimentally-derived and computed distances (Table S9, ESI†).The HPT-ValS3 folded control structure deviates greatly from the HPT folded control with a backbone root mean squared deviation (RMSD) of 2.15 Å (Fig. 6 and Fig. S20, Table S10, ESI†). Although the structure near the turn overlays well with the HPT folded control, accommodation of the internal thioamide at Val3 results in a dramatic twist in the hairpin at Thr2/Leu11. For the non-perturbing HPT-ThrS2 and HPT-IleS10 folded macrocycles, the structures of both are more like the HPT folded peptides (Fig. 6 and Fig. S19, S21, Table S10, ESI†). The C-terminal strand for HPT-ThrS2 is closer to the N-terminal strand (hydrogen bonds among the four terminal residues are 0.5 Å shorter), potentially a result of the thioamide acting as a stronger hydrogen bond donor to the carbonyl of Ile10 as well as a flip of the Thr2 side-chain due to breaking of a hydrogen bond with the Thr2 carbonyl (Fig. S19, ESI†). In agreement with our proposed hypothesis, the internal thioamide at Ile10 is better accepted due to the right-handed twist of this scaffold, allowing for a longer hydrogen bond with the Val3 N–H without altering the hairpin shape (RMSD = 0.63 Å). For the slightly stabilized HPT-LeuS11, the overall structure is like the HPT folded control (RMSD = 1.23 Å), with a nearly identical structure near the hairpin turn, but with an additional twist at the terminus near the thioamide (Fig. 6 and Fig. S22, Table S10, ESI†). Although the Leu11–N–H/Arg1CO hydrogen bond distance does not change, the angle does, leading to a rotation of the Leu11 side-chain and movement relative to the CysC/CysN disulfide bond. Experimentally, this can be seen from an increase in the Leu11 and CysC/CysN NOE distances (Table S10, ESI†).
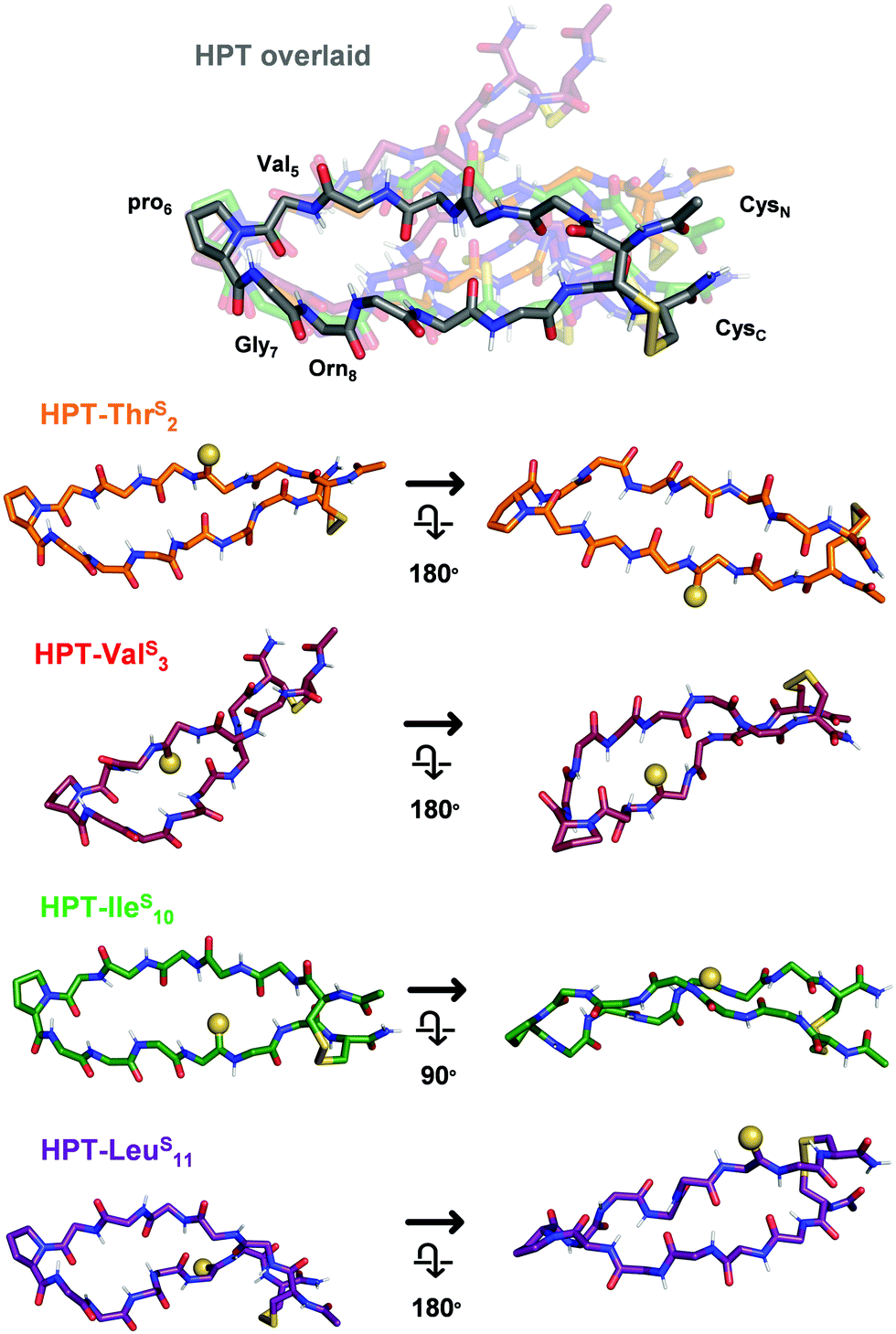 | ||
| Fig. 6 Structural models of the HPT folded control peptides. Only the backbone is displayed, except for pro6 and the terminal Cys residues. In HPT overlaid, all of the thioamide HPT peptides are aligned to HPT (grey, other peptides in indicated colors) based on the coordinates of Val5, pro6, Gly7, and Orn8 to enable comparison (RMSDs in Table S10, ESI†). Each thioamide peptide structure from the overlay is displayed individually from two angles, with the thioamide shown as a sphere. HPT-ThrS2 and HPT-IleS10 are similar in structure to HPT. As a result of the right-hand twist of the β-hairpin, the thioamide for HPT-IleS10 is more solvent-exposed. HPT-ValS3 has a dramatic twist, with differences in backbone arrangement around the internal thioamide. HPT-LeuS11 has a more pronounced twist at the terminus near the thioamide. Additional views and discussion of structures are in Fig. S18–S22 (ESI†). | ||
To further analyze these macrocyclic peptide structures, we used a Backrub57 protocol to generate ensembles. With slightly higher average deviations from the experimentally-derived distances (Table S11, ESI†), these ensembles (except for HPT-IleS10) demonstrate low backbone RMSDs (<1 Å) for the 10 lowest energy structures (Fig. S23–S26 (ESI†), links to coordinate files in pdb format are also provided), and align well with the constrained relax structures. For the HPT-IleS10 folded macrocycle, there is increased rotation at the β-turn and the strand opposite the thioamide. This provides a potential mechanism for how the corresponding test peptide can accommodate the internal thioamide (Fig. S25, ESI†).
We note that these mechanistic explanations must be taken with some caution as a relatively small number of NOEs were available for modelling constraints. Additionally, the chemical shift dispersion is very small for HPT-ValS3, where the folded control structure deviates significantly from HPT (backbone RMSD of 2.15 Å), raising some concern over whether it is a true folded control. While these structures provide snapshots of the folded control structures, simulation of the unfolded and test peptides would be required for direct comparison of energetics to the experimental results. However, by simulating these structures we were able to provide plausible mechanistic explanations for how IleS10 is well-accepted because of the right-hand twist, whereas ValS3 is destabilizing and results in a different configuration than HPT.
Discussion
As noted, we chose the YKL/HPT scaffold because multiple studies have used it as a host system to investigate the β-sheet propensity of various amino acids and their derivatives. These studies have included a variety of non-covalent interactions such as ion-pairing, π-system interactions, and steric constraints. A review of these findings which utilize the amino acid derivatives shown in Fig. 7 can be found in the ESI.† To enable accurate comparisons, all stability measurements are reported as ΔΔGFolding with the parent peptide as a reference.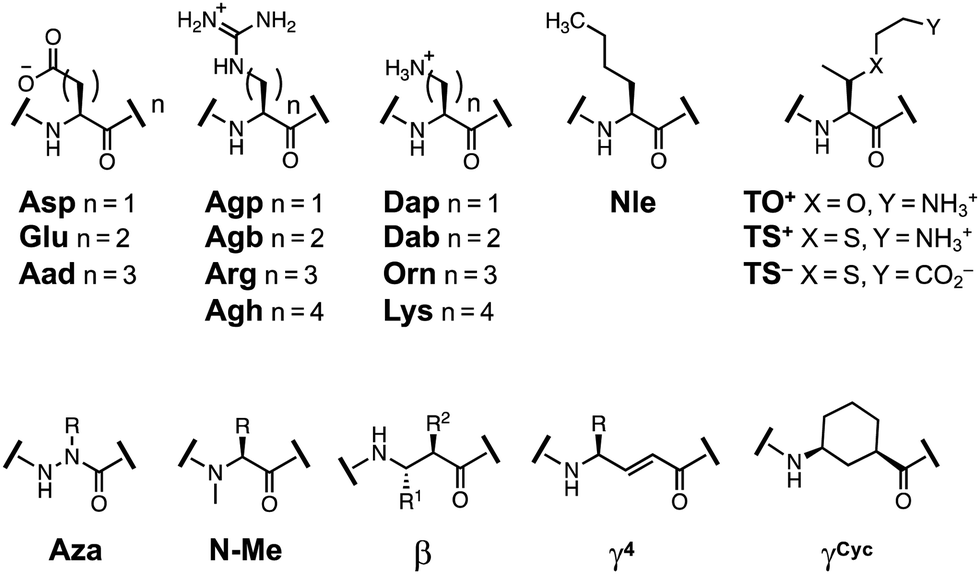 | ||
| Fig. 7 Structures of amino acids derivatives previously studied in β-hairpin scaffolds. | ||
To place the thioamide modification in the context of the field, we found the following interactions important to mention. Strengthening cation–π interactions by methylation of Lys9 or Arg9 side-chains stabilized the hairpin by about −0.2 kcal mol−1 per methylation.58–61 Sulfur–arene interactions62 increased the stability by −0.3 to −0.5 kcal mol−1.63 Side-chain phosphorylation demonstrated that anion–π interactions were destabilizing by ∼1 kcal mol−1.64,65 Introduction of charged β-branched derivatives (TS4− and TS9+) was highly stabilizing (−0.5 kcal mol−1 and −0.6 kcal mol−1), whereas TO+ was slightly destabilizing (+0.1 kcal mol−1).41 Addition of β-branched residues to the termini increased stability by −0.3 kcal mol−1.40 For backbone derivatives, Aza-Val3 incorporation was disruptive to foldedness (1.26 kcal mol−1), whereas aza-Gly3 was better accepted (0.75 kcal mol−1), but still less stable than YKL.42 β-Amino acid or linear (E)-vinylogous γ4-residues substitution was moderately destabilizing (0.5–0.6 kcal mol−1).66,67 Whereas a cyclically constrained γ-residue was stabilizing (−0.3 to −0.6 kcal mol−1).67,68 These perturbation studies are summarized in Table 2.
| Interaction | Perturbationref. | Effect on ΔΔGFolding |
|---|---|---|
| Cation–π | Methylation of Lys9 across from Trp258 | −0.2 kcal mol−1 per methylation |
| Replacement of Lys9 with Arg across from Trp261 | −0.3 kcal mol−1 | |
| Methylation of Arg9 across from Trp259 | −0.6 kcal mol−1 for first methylation | |
| Sulfur–arene | Replacement of Lys9 with Met across from Trp2 or Phe263 | −0.3 to −0.5 kcal mol−1 |
| Anion–π | Phosphorylated Ser9, Thr9, or Tyr9 across from Trp264,65 | +∼1 kcal mol−1 |
| Steric | 2 Thr added to each terminus40 | −0.3 kcal mol−1 |
| TS4− substitution41 | −0.5 kcal mol−1 | |
| TS9+ substitution41 | −0.6 kcal mol−1 | |
| TO9+ substitution41 | +0.1 kcal mol−1 | |
| Backbone | γCyc substitution67,68 | −0.3 to −0.6 kcal mol−1 |
| γ4 substitution67 | +0.5 kcal mol−1 | |
| β-Amino acid replacement of two α-amino acids66 | +0.5 to 0.6 kcal mol−1 per αα substitution | |
| Aza-Gly3 substitution42 | +0.8 kcal mol−1 | |
| Aza-Val3 substitution42 | +1.3 kcal mol−1 |
Thioamide effects are comparable in scale to these previous modifications. HPT-ThrS2, HPT-IleS10, and HPT-LeuS11-OH demonstrate a similar energy of folding to HPT (−0.09 to +0.04 kcal mol−1) where thioamide incorporation is less stabilizing than a cation–π or ion-pairing interaction. The internal thioamide at HPT-ValS3 is the most disruptive (0.38 kcal mol−1), but is still not as disruptive as β-amino acid incorporation (0.5–0.6 kcal mol−1) or phosphorylation (1 kcal mol−1).
There are differing opinions as to the relative importance of backbone hydrogen bonds, side-chain electrostatic and/or hydrophobic interactions on β-hairpin stability, and their importance can change depending on the β-hairpin construct.69–71 For the scaffolds we have discussed, it appears as though hydrophobic interactions such as aromatic stacking or addition of the β-branched derivatives are more stabilizing than electrostatic interactions (ion-pairing, cation–π). The favorable hydrophobic interaction suggests that desolvation could play a major role in stability for the β-hairpin. Since the thioamide is less polar than the canonical amide bond, an internal thioamide could further stabilize a hydrophobic interaction. Indeed, a recent study by Chatterjee indicates that the altered desolvation of the thioamide contributes to stability in the Pin1 WW β-sheet system.30 Stabilization by desolvation of the thioamide would be most prominent for internal thioamides. However, in the structural modelling of the HPT folded controls with internal thioamides, the thioamides are within hydrogen bonding distance of the opposing strand and do not appear to be engaging with a hydrophobic pocket (Fig. S20 and S21, ESI†). It is important to note that the previously discussed hydrophobic interactions are between side-chains (or thioamide and a side-chain), therefore the same might not be true for the backbone. Also, these β-hairpins, particularly the less stable HPT scaffold, are flexible substrates lacking tertiary structure so the ability to stabilize via backbone desolvation is very limited.
Conformational rigidity and hydrogen bonding are both backbone properties that can influence the stability of this β-hairpin scaffold. The combination of these properties, as well as differences in the micro-environment of each residue result in a complex system that does not behave as predicted based on small molecule studies. Our results show that an external thioamide can be slightly stabilizing (HPT-LeuS11-OH), whereas an internal thioamide can be destabilizing (HPT-ValS3), as predicted. However, they also show that an internal thioamide can be neutral (HPT-IleS10) without significantly altering the peptide structure. The fact that these trends do not match simple interpretations of the hydrogen bonding properties of the thioamide demonstrates that the effect of incorporation is position specific.
The results also reflect the importance of certain interactions at a position in the β-hairpin. The increased stability of HPT-LeuS11-OH where the thioamide is positioned as a hydrogen bond donor suggests that backbone hydrogen bonding is important at the terminus. The lack of change in stability for HPT-ThrS2 and all thioamide-containing YKL β-hairpins suggests that backbone hydrogen bonds are less impactful at this position in HPT and in the YKL scaffold. However, interactions cannot always be neatly parsed into backbone and side-chain effects. For example, in our model of HPT-ThrS2 we observe that breaking of a backbone CO side-chain OH hydrogen bond upon thionation leads to a twist that contributes to overall stability (Fig. S19, ESI†). Observations such as these highlight the importance of structural data attained using the macrocyclized folded peptides in understanding the impact of thioamide modification in model peptides and the growing number of thioamide-containing natural products.
Previous experimental work has suggested that introduction of a thioamide reduces conformational flexibility,72,73 and theoretical studies demonstrate increased steric constraints for the n + 1 residue.51,52,74,75 Thioamide incorporation at a residue closer to the β-turn (GluS4) in the YKL scaffold, has β-sheet character based on ΔδHα analysis, but it is less prevalent than the other positions tested. The decreased stability of YKL-GluS4 could be due to an inability of Val5 to accommodate the additional conformational restraint since it is already constrained by pro6 and the β-turn. This would also explain the dramatic twist observed in our structural modelling work for the HPT-ValS3 folded control. Since this macrocyclic construct is also sterically constrained by the disulfide, the twist occurs to relieve the rigidity imposed by the thioamide.
Even in the macrocyclic folded peptides, the overall stability derives from an interplay of interactions that vary by position, making it difficult to define a single causative feature for thioamide stability effects. However, the NMR-derived models of these macrocyclic systems enable one to apply more sophisticated electronic structure calculations51 to help to explain stability effects as well as observations such as the effect on the ΔδHα value for the n + 1 residue. We will pursue such computational analysis in conjunction with additional structure determination efforts for constrained systems.
Conclusions
The collection and analysis of 1H–1H NMR data for thioamide incorporation into two β-hairpin scaffolds, as well as structural modelling of the macrocyclic folded controls, suggests structural trends which deviate from expectations based on previous thioamide small molecule studies. For a stable scaffold, the YKL β-hairpin, incorporation of thioamides as hydrogen bond donors did not increase foldedness. Instead, all positions of incorporation demonstrated a similar structure to that of the YKL parent peptide. In a less stable scaffold, the HPT β-hairpin, thioamide incorporation had different structural impacts depending on position. Incorporation of a thioamide as a hydrogen bond donor was either minimally stabilizing (HPT-LeuS11-OH) or neutral (HPT-ThrS2). Conversely, incorporation as a hydrogen bond acceptor was either destabilizing (HPT-ValS3) or neutral (HPT-IleS10). To elucidate why these two positions were different we performed structural modelling of the folded controls. The conformation of HPT-ValS3 is highly unlike the others as a result of structural alterations to accommodate the destabilizing internal thioamide. Conversely, the folded control of HPT-IleS10 is similar in structure to HPT. In this position, the internal thioamide is more solvent exposed due to the right-handed twist of the β-hairpin, and therefore the thioamide steric bulk is better accommodated. This deviation from expectation based on the environment of the thioamide residue follows our previous observations with protein secondary structures.29Our results reinforce the idea that it is difficult to develop simple rules regarding how thioamide modifications will impact β-sheet structure since specific details such as twists, conformational rigidity or the relative importance of those hydrogen bonding interactions will play a major role. Currently, for utilization of the thioamide as a non-perturbing biophysical probe in fluorescence quenching or CD experiments, we recommend consulting the wild-type protein structure and incorporating the thioamide at β-sheet locations where the residue does not engage as a hydrogen bond acceptor. Although we have observed here that hydrogen bond acceptor positions can be tolerated, they are best avoided until criteria for identifying tolerated positions are determined. The increases in stability observed to date for incorporation as a hydrogen bond donor are minimal and should not significantly alter protein folding. To realize computational models that are predictive of the structural impact of thioamide incorporation at a position in a protein, we will use macrocyclic peptides like those shown to be useful in structure determination here, as well as host–guest studies of peptide/protein complexes, to gather sufficient data for machine learning models similar to those that have been successful in our protease studies.54 Ultimately, we hope to be able to rationally design peptides containing single or even multiple thioamide substitutions, as well as full-sized proteins synthesized through SPPS and/or native chemical ligation.3,5,30,33,76–80
Author contributions
K. E. F. synthesized and purified the HPT-ThrS2 and HPT-ValS3 peptides, collected CD data for all HPT peptides, and acquired NMR data for the HPT-IleS10 and HPT-LeuS11-OH peptides. M. P. synthesized and purified the HPT-IleS10, HPT-LeuS10-OH, and HPT peptides. C.-H. H. acquired the NMR data for the HPT-ThrS2 and HPT-Vals3 peptide and initially made assignments. H. V. and B. K. synthesized all YKL peptides, performed CD analysis, collected NMR data (including variable temperature studies), and made initial assignments. K. E. F. processed, assigned, and analyzed all NMR data (using the initial assignments for assistance). S. G. G. performed the structural modelling. K. E. F. and E. J. P. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Pennsylvania (Penn) and the National Science Foundation (NSF CHE-1708759 to E. J. P.). K. E. F. thanks the National Institutes of Health (NIH) for funding through the Structural Biology & Molecular Biophysics Training Program (T32-GM-008275). S. G. thanks the NSF for funding through the NSF Graduate Research Fellowship Program (DGE-1845298). The University of Pennsylvania Bruker AVANCE NEO 600 MHz NMR spectrometer was supported by NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, and the Vagelos Institute for Energy Science and Technology. At Penn, the 600 MHz spectrometer is managed by Dr Jun Gu and Dr Chad Lawrence. J. C. acknowledges IISc (MHRD), DST-FIST, UGC-CAS, and DBT-IISc partnership program for funding and infrastructural support. R. C. acknowledges support from the Ministry of Science and Technology in Taiwan (MOST-110-2113-M-002-010). Bruker AVIII 800 MHz spectrometer in the Instrumentation Center at National Taiwan University was supported by the Ministry of Science and Technology in Taiwan (MOST-110-2731-M-002-001). At NTU, the 800 MHz spectrometer is managed by Dr Shing-Jong Huang. K. E. F. acknowledges Dr Chad Lawrence for his assistance with acquiring the NMR data at Penn.Notes and references
- F. G. Bordwell, D. J. Algrim and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 5903–5904 CrossRef CAS.
- E. J. Petersson, J. M. Goldberg and R. F. Wissner, Phys. Chem. Chem. Phys., 2014, 16, 6827–6837 RSC.
- R. F. Wissner, S. Batjargal, C. M. Fadzen and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 6529–6540 CrossRef CAS PubMed.
- J. M. Goldberg, R. F. Wissner, A. M. Klein and E. J. Petersson, Chem. Commun., 2012, 48, 1550–1552 RSC.
- S. Batjargal, Y. J. Wang, J. M. Goldberg, R. F. Wissner and E. J. Petersson, J. Am. Chem. Soc., 2012, 134, 9172–9182 CrossRef CAS PubMed.
- J. M. Goldberg, X. Chen, N. Meinhardt, D. C. Greenbaum and E. J. Petersson, J. Am. Chem. Soc., 2014, 136, 2086–2093 CrossRef CAS PubMed.
- J. M. Goldberg, S. Batjargal and E. J. Petersson, J. Am. Chem. Soc., 2010, 132, 14718–14720 CrossRef CAS PubMed.
- J. M. Goldberg, L. C. Speight, M. W. Fegley and E. J. Petersson, J. Am. Chem. Soc., 2012, 134, 6088–6091 CrossRef CAS PubMed.
- M. Hollósi, E. Kollát, J. Kajtár, M. Kajtár and G. D. Fasman, Biopolymers, 1990, 30, 1061–1072 CrossRef.
- T. Sifferlen, M. Rueping, K. Gademann, B. Jaun and D. Seebach, Helv. Chim. Acta, 1999, 82, 2067–2093 CrossRef CAS.
- J. Zhao, D. Wildemann, M. Jakob, C. Vargas and C. Schiene-Fischer, Chem. Commun., 2003, 2810–2811 RSC.
- J. Helbing, H. Bregy, J. Bredenbeck, R. Pfister, P. Hamm, R. Huber, J. Wachtveitl, L. De Vico and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 8823–8834 CrossRef CAS PubMed.
- H. Satzger, C. Root, P. Gilch, W. Zinth, D. Wildemann and G. Fischer, J. Phys. Chem. B, 2005, 109, 4770–4775 CrossRef CAS PubMed.
- H. Bregy, H. Heimgartner and J. Helbing, J. Phys. Chem. B, 2009, 113, 1756–1762 CrossRef CAS PubMed.
- Y. Huang, Z. Cong, L. Yang and S. Dong, J. Pept. Sci., 2008, 14, 1062–1068 CrossRef CAS PubMed.
- N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142–163 CrossRef CAS PubMed.
- W. Grabarse, F. Mahlert, S. Shima, R. K. Thauer and U. Ermler, J. Mol. Biol., 2000, 303, 329–344 CrossRef CAS PubMed.
- D. D. Nayak, N. Mahanta, D. A. Mitchell and W. W. Metcalf, eLife, 2017, 6, 1–18 CrossRef PubMed.
- Z. L. Watson, F. R. Ward, R. Méheust, O. Ad, A. Schepartz, J. F. Banfield and J. H. D. Cate, eLife, 2020, 9, e60482 CrossRef CAS PubMed.
- C. Alemán, J. Phys. Chem. A, 2001, 105, 6717–6723 CrossRef.
- H.-J. Lee, Y.-S. Choi, K.-B. Lee, J. Park and C.-J. Yoon, J. Phys. Chem. A, 2002, 106, 7010–7017 CrossRef CAS.
- M. Hollósi, M. Zewdu, E. Kollát, Z. Majer, M. Kajtár, G. Batta, K. Kövér and P. Sándor, Int. J. Pept. Protein Res., 1990, 36, 173–181 CrossRef PubMed.
- K. A. Jensen, Arch. Pharm. Chem. Sci. Ed., 1981, 9, 93–116 CAS.
- E. P. Dudek and G. O. Dudek, J. Org. Chem., 1967, 32, 823–824 CrossRef CAS.
- M. Hollósi, Z. Majer, M. Zewdu, F. Ruff, M. Kajtár and K. E. Kövér, Tetrahedron, 1988, 44, 195–202 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. R. Truter, J. Chem. Soc., 1960, 0, 997–1007 RSC.
- R. Bardi, A. M. Piazzesi, C. Toniolo, O. E. Jensen, R. S. Omar and A. Senning, Biopolymers, 1988, 27, 747–761 CrossRef CAS.
- C. R. Walters, D. M. Szantai-Kis, Y. Zhang, Z. E. Reinert, W. S. Horne, D. M. Chenoweth and E. J. Petersson, Chem. Sci., 2017, 8, 2868–2877 RSC.
- B. Khatri, S. Raghunathan, S. Chakraborti, R. Rahisuddin, S. Kumaran, R. Tadala, P. Wagh, U. D. Priyakumar and J. Chatterjee, Angew. Chem., Int. Ed., 2021, 60, 24870–24874 CrossRef CAS PubMed.
- A. Reiner, D. Wildemann, G. Fischer and T. Kiefhaber, J. Am. Chem. Soc., 2008, 130, 8079–8084 CrossRef CAS PubMed.
- J. H. Miwa, L. Pallivathucal, S. Gowda and K. E. Lee, Org. Lett., 2002, 4, 4655–4657 CrossRef CAS PubMed.
- R. W. Newberry, B. VanVeller and R. T. Raines, Chem. Commun., 2015, 51, 9624–9627 RSC.
- J. H. Miwa, A. K. Patel, N. Vivatrat, S. M. Popek and A. M. Meyer, Org. Lett., 2001, 3, 3373–3375 CrossRef CAS PubMed.
- B. Khatri, P. Majumder, J. Nagesh, A. Penmatsa and J. Chatterjee, Chem. Sci., 2020, 11, 9480–9487 RSC.
- R. M. Culik, H. Jo, W. F. Degrado and F. Gai, J. Am. Chem. Soc., 2012, 134, 8026–8029 CrossRef CAS PubMed.
- J. Spekowius, R. Pfister and J. Helbing, J. Phys. Chem. B, 2021, 125, 7662–7670 CrossRef CAS PubMed.
- F. A. Syud, H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 8667–8677 CrossRef CAS PubMed.
- H. E. Stanger and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 4236–4237 CrossRef CAS.
- H. E. Stanger, F. A. Syud, J. F. Espinosa, I. Giriat, T. Muir and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12015 CrossRef CAS PubMed.
- S. J. Maynard, A. M. Almeida, Y. Yoshimi and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 16683–16688 CrossRef CAS PubMed.
- M. A. McMechen, E. L. Willis, P. C. Gourville and C. Proulx, Molecules, 2019, 24, 1999 CrossRef PubMed.
- F. A. Syud, J. F. Espinosa and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 11577–11578 CrossRef CAS.
- B. L. Sibanda and J. M. Thornton, Nature, 1985, 316, 170–174 CrossRef CAS PubMed.
- C. Chothia, J. Mol. Biol., 1973, 75, 295–302 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS PubMed.
- L.-H. Kuo, J.-H. Li, H.-T. Kuo, C.-Y. Hung, H.-Y. Tsai, W.-C. Chiu, C.-H. Wu, W.-R. Wang, P.-A. Yang, Y.-C. Yao, T. W. Wong, S.-J. Huang, S.-L. Huang and R. P. Cheng, Biochemistry, 2013, 52, 7785–7797 CrossRef CAS PubMed.
- H.-T. Kuo, S.-L. Liu, W.-C. Chiu, C.-J. Fang, H.-C. Chang, W.-R. Wang, P.-A. Yang, J.-H. Li, S.-J. Huang, S.-L. Huang and R. P. Cheng, Amino Acids, 2015, 47, 885–898 CrossRef CAS PubMed.
- H.-T. Kuo, C.-J. Fang, H.-Y. Tsai, M.-F. Yang, H.-C. Chang, S.-L. Liu, L.-H. Kuo, W.-R. Wang, P.-A. Yang, S.-J. Huang, S.-L. Huang and R. P. Cheng, Biochemistry, 2013, 52, 9212–9222 CrossRef CAS PubMed.
- C.-H. Huang, T. W. Wong, C.-H. Yu, J.-Y. Chang, S.-J. Huang, S.-L. Huang and R. P. Cheng, Molecules, 2021, 26, 1346 CrossRef CAS PubMed.
- B. J. Lampkin and B. VanVeller, J. Org. Chem., 2021, 86, 18287–18291 CrossRef CAS PubMed.
- D. R. Artis and M. A. Lipton, J. Am. Chem. Soc., 1998, 120, 12200–12206 CrossRef CAS.
- S. Chaudhury, S. Lyskov and J. J. Gray, Bioinformatics, 2010, 26, 689–691 CrossRef CAS PubMed.
- S. Giannakoulias, S. R. Shringari, C. Liu, H. A. T. Phan, T. M. Barrett, J. J. Ferrie and E. J. Petersson, J. Phys. Chem. B, 2020, 124, 8032–8041 CrossRef CAS PubMed.
- J. Yoon, J. J. Ferrie and E. J. Petersson, J. Phys. Chem. B, 2020, 124, 10653–10662 CrossRef CAS PubMed.
- H. A. T. Phan, S. G. Giannakoulias, T. M. Barrett, C. Liu and E. J. Petersson, Chem. Sci., 2021, 12, 10825–10835 RSC.
- C. A. Smith and T. Kortemme, J. Mol. Biol., 2008, 380, 742–756 CrossRef CAS PubMed.
- R. M. Hughes, M. L. Benshoff and M. L. Waters, Chem. – Eur. J., 2007, 13, 5753–5764 CrossRef CAS PubMed.
- R. M. Hughes and M. L. Waters, J. Am. Chem. Soc., 2006, 128, 12735–12742 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, Protein Sci., 2003, 12, 2443–2452 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, Protein Sci., 2004, 13, 2515–2522 CrossRef CAS PubMed.
- A. J. Riemen and M. L. Waters, J. Am. Chem. Soc., 2009, 131, 14081–14087 CrossRef CAS PubMed.
- A. J. Riemen and M. L. Waters, Org. Biomol. Chem., 2010, 8, 5411–5417 RSC.
- G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2012, 134, 15906–15913 CrossRef CAS PubMed.
- G. A. Lengyel, Z. E. Reinert, B. D. Griffith and W. S. Horne, Org. Biomol. Chem., 2014, 12, 5375–5381 RSC.
- G. A. Lengyel, G. A. Eddinger and W. S. Horne, Org. Lett., 2013, 15, 944–947 CrossRef CAS PubMed.
- A. J. Maynard, G. J. Sharman and M. S. Searle, J. Am. Chem. Soc., 1998, 120, 1996–2007 CrossRef CAS.
- C. S. Colley, S. R. Griffiths-Jones, M. W. George and M. S. Searle, Chem. Commun., 2000, 593–594 RSC.
- S. R. Griffiths-Jones, A. J. Maynard and M. S. Searle, J. Mol. Biol., 1999, 292, 1051–1069 CrossRef CAS PubMed.
- T. T. Tran, J. Zeng, H. Treutlein and A. W. Burgess, J. Am. Chem. Soc., 2002, 124, 5222–5230 CrossRef CAS PubMed.
- H. Verma, B. Khatri, S. Chakraborti and J. Chatterjee, Chem. Sci., 2018, 9, 2443–2451 RSC.
- T. T. Tran, A. W. Burgess, H. Treutlein and J. Zeng, J. Mol. Graphics Modell., 2001, 20, 245–256 CrossRef CAS PubMed.
- T. T. Tran, H. Treutlein and A. W. Burgess, J. Comput. Chem., 2001, 22, 1026–1037 CrossRef CAS.
- Y. Huang, J. J. Ferrie, X. Chen, Y. Zhang, D. M. Szantai-Kis, D. M. Chenoweth and E. J. Petersson, Chem. Commun., 2016, 52, 7798–7801 RSC.
- C. R. Walters, J. J. Ferrie and E. J. Petersson, Chem. Commun., 2018, 54, 1766–1769 RSC.
- S. Batjargal, Y. Huang, Y. J. Wang and E. J. Petersson, J. Pept. Sci., 2014, 20, 87–91 CrossRef CAS PubMed.
- Y. J. Wang, D. M. Szantai-Kis and E. J. Petersson, Org. Biomol. Chem., 2015, 13, 5074–5081 RSC.
- B. Khatri, P. Bhat and J. Chatterjee, J. Pept. Sci., 2020, 26, e3248 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Peptide synthesis and characterization data, and details of NMR-based computational modeling. Links to coordinates for structures are provided. See DOI: 10.1039/d1cb00229e |
‡ For NMR experiments, the YKL peptides were dissolved in 100 mM sodium deuteroacetate buffer pH 3.8 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v H2O/D2O). The HPT peptides were dissolved in 50 mM sodium deuteroacetate pH 5.5 (9:1 v/v H2O/D2O) or 50 mM NaH2PO4 pH 5.5 (9:1 v/v H2O/D2O). Variability in solubility based on construct and experimental time required for data collection is the reason different buffers and pH values were used. Since the buffer remained consistent for the test, unfolded control, and folded control peptides of each HPT thioamide position, the difference in salt should have minimal to no effect on fraction folded and ΔΔG analysis. Discussion of NMR collection across the different universities can be found in Table S3 and Fig. S15 (ESI†). :1 v/v H2O/D2O). The HPT peptides were dissolved in 50 mM sodium deuteroacetate pH 5.5 (9:1 v/v H2O/D2O) or 50 mM NaH2PO4 pH 5.5 (9:1 v/v H2O/D2O). Variability in solubility based on construct and experimental time required for data collection is the reason different buffers and pH values were used. Since the buffer remained consistent for the test, unfolded control, and folded control peptides of each HPT thioamide position, the difference in salt should have minimal to no effect on fraction folded and ΔΔG analysis. Discussion of NMR collection across the different universities can be found in Table S3 and Fig. S15 (ESI†). |
| This journal is © The Royal Society of Chemistry 2022 |