
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrole–protein interactions in H-NOX and HasA†
Christopher M.
Lemon
 *abc,
Amos J.
Nissley
d,
Naomi R.
Latorraca
ac,
Elizabeth C.
Wittenborn
b and
Michael A.
Marletta
*abd
*abc,
Amos J.
Nissley
d,
Naomi R.
Latorraca
ac,
Elizabeth C.
Wittenborn
b and
Michael A.
Marletta
*abd
aDepartment of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA. E-mail: clemon@berkeley.edu; marletta@berkeley.edu
bCalifornia Institute for Quantitative Biosciences (QB3), University of California, Berkeley, CA 94720, USA
cMiller Institute for Basic Research in Science, University of California, Berkeley, CA 94720, USA
dDepartment of Chemistry, University of California, Berkeley, CA 94720, USA
First published on 21st March 2022
Abstract
Replacing the native porphyrin cofactor in haem proteins has led to the development of novel designer proteins for a variety of applications. In most cases, haem analogues bind in a way that is comparable to the iron porphyrin, but this is not necessarily the case for complexes bearing non-exchangeable ligands. This study probes how a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O corrole binds to functionally disparate hemoproteins: a haem-dependent oxygen sensor (H-NOX) and a haem-scavenging protein (HasA). The results demonstrate that the protein–cofactor interactions are distinct from the native, haem-bound holoprotein. In H-NOX, the PO unit primarily hydrogen bonds with the haem-ligating histidine (H102), rather than the hydrogen-bonding network that stabilises the Fe(II)–O2 complex in the native protein. In the absence of H102, the protein is still able to bind the corrole, albeit at reduced levels. Molecular dynamics simulations were utilised to determine the flexibility of apo H-NOX and revealed the coupled motion of key residues necessary for corrole binding. In the case of HasA, the PO unit does not primarily interact with either the haem-ligating histidine (H32) or tyrosine (Y75). Instead, histidine 83, the hydrogen-bonding partner for Y75, is critical for PO corrole binding. The conformation of HasA is interrogated by site-specifically labelling the protein and exploiting Förster resonance energy transfer (FRET) to determine the dye–cofactor distance. HasA reconstituted with the PO corrole exhibits an extended, apo-like conformation. Together, these results demonstrate that non-natural cofactors can bind to proteins in unexpected ways and highlight the need to uncover these interactions for the further development of designer haem proteins.
O corrole binds to functionally disparate hemoproteins: a haem-dependent oxygen sensor (H-NOX) and a haem-scavenging protein (HasA). The results demonstrate that the protein–cofactor interactions are distinct from the native, haem-bound holoprotein. In H-NOX, the PO unit primarily hydrogen bonds with the haem-ligating histidine (H102), rather than the hydrogen-bonding network that stabilises the Fe(II)–O2 complex in the native protein. In the absence of H102, the protein is still able to bind the corrole, albeit at reduced levels. Molecular dynamics simulations were utilised to determine the flexibility of apo H-NOX and revealed the coupled motion of key residues necessary for corrole binding. In the case of HasA, the PO unit does not primarily interact with either the haem-ligating histidine (H32) or tyrosine (Y75). Instead, histidine 83, the hydrogen-bonding partner for Y75, is critical for PO corrole binding. The conformation of HasA is interrogated by site-specifically labelling the protein and exploiting Förster resonance energy transfer (FRET) to determine the dye–cofactor distance. HasA reconstituted with the PO corrole exhibits an extended, apo-like conformation. Together, these results demonstrate that non-natural cofactors can bind to proteins in unexpected ways and highlight the need to uncover these interactions for the further development of designer haem proteins.
Introduction
Haem proteins represent a versatile platform for the development of designer proteins with novel functionality,1–3 most prominently as artificial metalloenzymes. Often, haem proteins are directly used with the native iron porphyrin cofactor.4 Directed evolution can be utilised to reconfigure active and/or allosteric sites to engender novel reactivity.5 Alternatively, substitution of the iron centre with a different transition metal provides another inroad to novel chemical reactivity.6–9 Together, these approaches have greatly expanded the scope of chemical transformations that haem proteins can mediate.10–13 In addition to artificial metalloenzymes, the incorporation of non-iron metalloporphyrins in haem proteins has enabled the development of designer proteins as optical oxygen sensors,14,15 MRI contrast agents,16,17 spectroscopic probes,18–20 and tools to interrogate protein function.21–23A less explored approach is substitution of the porphyrin cofactor with an alternative tetrapyrrole macrocycle or a related ligand.24,25 These include corrole,26–28 porphycene,29–31 phthalocyanine,32,33 salophen,32,34–37 and tetradehydrocorrin38,39 (Fig. 1). In general, these ligands exhibit chemical properties (e.g., optical, redox) and reactivity that are distinct from porphyrins. This approach has been utilised to develop artificial metalloenzymes,26,29–31,35–39 antibiotics,32–34 and fluorescent proteins.28 Expansion of this approach could explore uncharted chemical space for a variety of applications that range from energy conversion to biomedical sensing and imaging.
 | ||
| Fig. 1 Tetrapyrrole macrocycles and related ligands that have been incorporated into haem proteins. For each ligand, the listed elements represent the specific complexes that have been utilised and those highlighted in red have been structurally characterised. | ||
Another approach to develop proteins with novel functionality is de novo protein design;40 this has been utilised for a variety of applications that range from fluorescent sensors41,42 to vaccine development.43 Computational protein design goes beyond the confines of extant proteins and can leverage specific protein–cofactor interactions to enhance cofactor binding. This is accomplished by modulating the residues that interact with the cofactor to tailor the binding pocket. In the case of a metallocofactor, this could include the protein-based ligands that bind to the metal centre. However, there are few examples where de novo protein design has been exploited to bind small-molecule cofactors, namely haem or non-biological iron porphyrins.44–48 This methodology has been refined and extended to design proteins that are selective for specific zinc porphyrins, enabling discrimination of both the metal centre and peripheral substituents.49–51 Examples of proteins specifically designed to bind non-porphyrin cofactors are exceedingly rare.52,53 The lack of studies that have determined the molecular interactions between proteins and abiological cofactors is one obstacle that hampers the broad application of this approach. After identifying critical and/or preferred contacts, this chemical information can then be utilised to design proteins that bind unnatural synthetic cofactors.
For the non-porphyrin complexes in Fig. 1, it is generally assumed that the cofactor directly replaces haem, resulting in a similar fold with protein–cofactor interactions that are analogous to the native holoprotein. This has been confirmed by X-ray crystal structures of haem proteins that have been substituted with a variety of complexes.30–33,36,38 However, this may not always be the case, particularly for cofactors that have distinctive coordination preferences or non-exchangeable axial ligands. Of the examples listed in Fig. 1, the PO corrole (P-1) would not be expected to bind to the protein in a way that is analogous to the native haem cofactor. Given that this corrole does not bind histidine (i.e., no P–NHis bond) and remains a five-coordinate complex in solution,28 it is unclear how this molecule would interact with haem proteins.
To address this question, this study sought to uncover the molecular details of P-1 binding in the haem nitric oxide/oxygen binding (H-NOX) protein from Caldanaerobacter subterraneus (Cs) and the haem acquisition system protein A (HasA) from Pseudomonas aeruginosa (Pa). Key residues in the haem binding pocket of these proteins were modulated. Corrole binding in these variants was analysed by absorption and emission spectroscopy. Molecular dynamics (MD) simulations were utilised to assess the flexibility and accessibility of key residues in apo H-NOX. The conformation of P-1-bound HasA was determined using Förster resonance energy transfer (FRET) with fluorescein-labelled cysteine variants. Together, these experiments demonstrate that P-1 binding in Cs H-NOX and Pa HasA is distinct from haem binding in the native holoprotein.
Results
P-1 binding in H-NOX variants
To investigate how corrole P-1 binds to Cs H-NOX, several mutations were made around the haem binding pocket (Fig. 2a). Haem binding in these variants is presented in the ESI† (Fig. S1 and S2). The out-of-plane coordination of the PO unit could place the oxygen atom in a position to interact with the hydrogen bonding network (W9, N74, Y140) that stabilises the native Fe(II)–O2 haem complex. The W9F and Y140F variants both exhibit P-1 binding that is nearly identical to the WT H-NOX (Table 1 and Fig. 2b). Similarly, the emission of these variants is unperturbed and comparable to the WT protein (Fig. 2c and Table 1). These results suggest that the distal hydrogen bonding network is not directly involved in corrole binding.
 | ||
Fig. 2 (a) Structure of the Fe(II)–O2 complex of WT Cs H-NOX (PDB ID: 3TF0), highlighting key residues in cyan: H102, P115, and the hydrogen bonding network W9, Y140, and N74. (b) Normalised (280 nm) absorption spectra of H-NOX variants reconstituted with P-1: WT ( ), W9F ( ), W9F ( ), H102G ( ), H102G ( ), H102S ( ), H102S ( ), P115A ( ), P115A ( ), and Y140F ( ), and Y140F ( ). (c) Emission spectra of absorbance-matched samples (A(520) = 0.0421 ± 0.0004) of H-NOX variants reconstituted with P-1: WT ( ). (c) Emission spectra of absorbance-matched samples (A(520) = 0.0421 ± 0.0004) of H-NOX variants reconstituted with P-1: WT ( ), W9F ( ), W9F ( ), H102G ( ), H102G ( ), H102S ( ), H102S ( ), P115A ( ), P115A ( ), and Y140F ( ), and Y140F ( ). ). | ||
| Protein | Ratioa | P-1 incorp.b (%) | Rel. Em.c |
ϕ
f × 102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
|---|---|---|---|---|
| a Soret to 280 nm absorbance ratio for the representative dataset in Fig. 2a. b Average P-1 incorporation relative to WT H-NOX, accounting for differences in ε280 for protein variants; errors reflect one standard deviation. c Integrated emission intensity relative to WT H-NOX. d Fluorescence quantum yield relative to WT H-NOX. | ||||
| WT | 5.06 | 100 | 1.00 | 2.5 |
| W9F | 5.42 | 97 ± 2 | 0.99 | 2.5 |
| H102G | 3.10 | 59 ± 5 | 1.02 | 2.6 |
| H102S | 1.86 | 37 ± 4 | 1.07 | 2.7 |
| P115A | 2.19 | 42 ± 3 | 0.83 | 2.1 |
| Y140F | 4.97 | 96 ± 2 | 0.99 | 2.5 |
The low fluorescence quantum yield of P-1 bound to H-NOX relative to HasA (2.5 vs. 7.3%) might be due to quenching by H102. P-1 does not bind histidine, and DFT calculations confirm that imidazole binding is destabilising.28 The H102G variant exhibited impaired corrole incorporation (59% relative to WT H-NOX). To rescue corrole binding with a hydrogen bonding partner, H102S was prepared. Surprisingly, this variant incorporates less corrole (37% relative to WT H-NOX). Since the absorption spectra are nearly identical for these three variants (Fig. S3, ESI†), the histidine side chain does not directly bind to the phosphorus centre (i.e., no P–NHis bond). These results suggest that, although H102 is not absolutely required, it does play a key role in P-1 binding.
Interestingly, corrole emission in H102G and H102S is approximately the same as WT H-NOX, indicating that H102 does not quench corrole fluorescence. However, there are a few subtle differences in the emission spectra of these variants. The full width at half maximum (FWHM) of the main emission transition for the H102 variants is nominally wider than the WT protein: 15 nm for WT, 16 nm for H102G, and 17 nm for H102S. Moreover, the Q(0,1) emission transition (∼620 nm) is more pronounced for the H102 variants, resulting in a decrease of the Q(0,0) to Q(0,1) intensity ratio: 7.74, 7.21, and 6.17 for WT, H102G, and H102S, respectively. Together, these results indicate that the corrole exhibits greater conformational flexibility in the H102 variants compared to the WT protein.
In an effort to enhance corrole fluorescence in H-NOX, the P115A variant was expressed. It has been previously demonstrated that P115 contributes to the highly distorted porphyrin macrocycle in the O2-bound WT protein.54 A protein environment that engenders distortion of the corrole macrocycle could result in lower fluorescence. The P115A variant exhibits low corrole binding (42%) that is similar to H102S (Table 1). In contrast to the other variants, P115A does not exhibit a split Soret band, indicating that the protein environment surrounding the corrole is distinct. This is also evident from the emission spectrum, which demonstrates that P-1 fluorescence is lower than the other variants (Fig. 2c).
Given the impaired binding of P-1 to the H102G, H102S, and P115A variants, additional reconstitution experiments were performed using other cofactors to ensure that these apoproteins are properly folded and competent to bind exogenous cofactors. Pd(II) mesoporphyrin IX (MPIX) was selected to test binding in H102G and H102S because this cofactor does not require a histidine residue, since Pd(II) porphyrins do not bind axial ligands. WT, H102G, and H102S bound Pd(MPIX) to the same extent (Fig. S4, ESI†). Similarly, P115A reconstituted with Zn protoporphyrin IX (PPIX) exhibits cofactor binding that is identical to the WT H-NOX (Fig. S5a, ESI†). Interestingly, Zn(PPIX) fluorescence for the P115A variant is lower than the WT protein (Fig. S5b, ESI†), albeit not to the same extent as observed for P-1. This suggests that there is a subtle difference in the protein fold that quenches cofactor fluorescence relative to WT H-NOX. Together, these results indicate that apo H102G, H102S, and P115A are appropriately folded and enable cofactor binding. Decreased binding of P-1 in these variants reflects the absence of specific protein–corrole interactions that are uniquely required for this cofactor.
Apo H-NOX molecular dynamics simulations
Since P-1 incorporation is impaired for P115A, H102S, and H102G, and the P115A mutation changes the chemical environment of the corrole, these point mutations may alter the conformational dynamics of H-NOX. To test this, molecular dynamics (MD) simulations were performed to survey the flexibility of the protein and determine the solvent accessibility of key residues. Since P-1 incorporation is accomplished by reconstitution, apo structures are more likely to reveal determinants of corrole binding than holoprotein structures. In the absence of a crystal structure of apo H-NOX, MD simulations of H-NOX were initiated after removing the haem and O2 from the WT structure (PDB ID: 3TF0). In simulation, the αF helix containing H102, as well as the loop connecting αF and αE, quickly (<100 ns) moves to partially occupy the space previously filled by haem (Fig. S6, ESI†).In these simulations, H102 rapidly reorients to a ‘flipped’ conformation (Fig. 3a), occupying the space previously filled by P115. Additionally, the distance between the αF helix and the neighbouring β1 strand, containing P115, increases significantly. Importantly, H102 can spontaneously switch between the ‘flipped’ conformation and an ‘upright’ (crystallographic) conformation. When H102 transitions back to the ‘upright’ conformation, P115 also assumes its crystallographic position, indicating that these two residues are tightly coupled. There are two key metrics that describe this interaction: the χ1 torsion angle for H102 and the Cα–Cα distance between P115 and H102 (Fig. 3b). Over six 1 μs simulations of WT apo H-NOX (Fig. 3c and Fig. S7a, ESI†), H102 more frequently adopts the ‘upright’ conformation (χ1 ∼ −70°) rather than the ‘flipped’ conformation (χ1 ∼ −180°), resulting in (on average) a shorter Cα–Cα distance (Fig. 3d). These simulations suggest that P115 constrains the movement of H102.
 | ||
| Fig. 3 (a) In simulation, H102 spontaneously transitions between the crystallographic ‘upright’ conformation (χ1 ∼ −70°) and a ‘flipped’ conformation (χ1 ∼ −180°). (b) Metrics used to characterise conformational changes in the simulations: distance between H102 and P115 Cα atoms (turquoise), and the H102 χ1 torsion angle (grey). (c) Representative simulations of WT and P115A, depicting the metrics illustrated in (b) over the course of the 1 μs simulation. Stars indicate the times that representative snapshots were extracted from the simulation, illustrated in (a). (d) Histograms constructed by aggregating values for each metric for the six WT simulations. (e) Histograms for the six P115A simulations. | ||
To test the hypothesis that the essential role of P115 arises due to its ability to control the conformation of H102, simulations using the P115A structure (PDB ID: 3EEE) after removing haem and O2 were also performed. It should be noted that 3EEE is nearly identical to 3TF0; alignment of the two structures yields a Cα root-mean-square deviation of 0.623 Å. In simulations of apo P115A H-NOX (Fig. S7b, ESI†), the αF helix exhibits a similar deviation as observed in the WT simulations. H102 also quickly transitions to the ‘flipped’ conformation, but unlike the WT simulations, it rarely transitions back to an ‘upright’ conformation (Fig. 3e). Moreover, transitioning back to the ‘upright’ conformation is not coupled to the repositioning of the A115 backbone and the β1 strand. Taken together, P115 imposes a constraint on the conformational dynamics of H102 by increasing its propensity to adopt a haem-engageable orientation.
P-1 binding in HasA variants
To evaluate P-1 binding in HasA, variants were prepared that modify residues on either the H32 loop or the Y75 loop (Fig. 4a). Haem binding in these variants is presented in the ESI† (Fig. S8–S10). For variants on the H32 loop (H32S, H32C, and V38C), corrole binding is nearly identical to the WT protein (Table 2 and Fig. 4b, c). If H32 were sufficiently close to P-1, then it would be expected that the lone pair of electrons on the imine nitrogen of the side chain could quench fluorescence. If this were the case, the emission of the H32S variant should be enhanced relative to the WT protein. Similarly, if residue 32 were sufficiently close to the corrole, the H32C variant would exhibit lower fluorescence than the H32S via heavy atom quenching.55 The similar emission intensity from these variants indicates that H32 is not sufficiently close to P-1 to cause fluorescence quenching, suggesting that the H32 loop does not engage with the corrole. | ||
Fig. 4 (a) Structure of WT Pa HasA (PDB ID: 3ELL), highlighting key residues in cyan: H32, Y75, and H83. (b) Normalised (280 nm) absorption spectra of HasA variants reconstituted with P-1: WT ( ), H32C ( ), H32C ( ), H32S ( ), H32S ( ), V38C ( ), V38C ( ), and Y75F ( ), and Y75F ( ). (c) Emission spectra of absorbance-matched samples (A(520) = 0.0411 ± 0.0004) of HasA variants reconstituted with P-1: WT ( ). (c) Emission spectra of absorbance-matched samples (A(520) = 0.0411 ± 0.0004) of HasA variants reconstituted with P-1: WT ( ), H32C ( ), H32C ( ), H32S ( ), H32S ( ), V38C ( ), V38C ( ), and Y75F ( ), and Y75F ( ). ). | ||
| Protein | Ratioa | P-1 incorp.b (%) | Rel. Em.c |
ϕ
f × 102d |
|---|---|---|---|---|
| a Soret to 280 nm absorbance ratio for the representative dataset in Fig. 4a. b Average P-1 incorporation relative to WT HasA, accounting for differences in ε280 for protein variants; errors reflect one standard deviation. c Integrated emission intensity relative to WT HasA. d Fluorescence quantum yield relative to WT HasA. | ||||
| WT | 5.76 | 100 | 1.00 | 7.3 |
| H32C | 5.30 | 93 ± 5 | 1.02 | 7.4 |
| H32S | 5.65 | 96 ± 3 | 0.99 | 7.2 |
| V38C | 6.06 | 101 ± 5 | 1.03 | 7.5 |
| Y75F | 2.80 | 45 ± 3 | 0.47 | 3.5 |
| H83A | 0 | 0 | — | — |
| H83D | 0 | 0 | — | — |
| H83S | 0 | 0 | — | — |
Conversely, mutations on the Y75 loop drastically affect P-1 binding. In an attempt to enhance corrole emission, variants of H83 were prepared. Since this residue is not essential for haem binding,56 it may not be necessary for P-1 binding. Surprisingly, the H83A variant did not bind any corrole; only apoprotein was recovered after reconstitution. Next, the H83D variant was prepared to provide a hydrogen bonding partner for Y75 that could also increase the phenolate character of the tyrosine, but no corrole binding was observed. Finally, H83S was prepared to provide a small, flexible hydrogen bonding partner, but this variant did not bind corrole. These results indicate that H83 is absolutely required to bind P-1.
Given the direct interaction between H83 and P-1, it is expected that the lone pair of electrons on the imine nitrogen of the side chain would quench emission. To see if protonation of H83 enhances corrole fluorescence, pH-dependent studies were performed. Pa HasA is remarkably stable over a wide pH range, exhibiting minimal protein precipitation between pH 3 and 11 (Fig. S11, ESI†). This is consistent with the previously reported stability of HasA over the pH 4 to 10 range.57 At pH 5, the extent of reconstitution of WT apo HasA with P-1 was identical to the standard conditions at pH 6.5. Moreover, the emission intensity is identical at pH 5 and 6.5 (Fig. S12, ESI†), suggesting that H83 is protonated at pH 6.5.
To determine if the haem-ligating tyrosine is necessary for corrole binding, the Y75F variant was prepared. In this case, P-1 binding is diminished (45% relative to WT HasA). Interestingly, the absorption spectrum of this variant is blue-shifted relative to the WT and H32 loop variants; this is particularly apparent for the Soret band. This shift likely reflects a change in the dielectric constant of the medium surrounding the corrole. Additionally, the emission intensity of the Y75F variant is significantly quenched relative to the WT protein. These results demonstrate that, although Y75 is not absolutely required for corrole binding, it does directly interact with the cofactor.
Determining HasA conformation using FRET
Since the H32 loop does not appear to engage with P-1, FRET was used to interrogate the conformation of corrole-bound HasA. The protein was labelled with a fluorescent dye, utilising FRET to measure the dye–cofactor distance. Since Pa HasA does not have any native cysteine residues, site-specific labelling can be accomplished without interference from non-target residues. To this end, the H32C and V38C HasA variants were prepared because these residues move significantly upon haem binding, as evidenced by the apo (PDB ID: 3MOK) and holo (PDB ID: 3ELL) crystal structures (Fig. 5a and b). Fluorescein-5-maleimide was selected as the dye because its emission profile overlaps well with the absorption profile of P-1 bound to WT HasA (Fig. S13, ESI†). Results for haem-bound, fluorescein-labelled HasA variants are presented in the ESI† (Fig. S14–S16 and Table S1). | ||
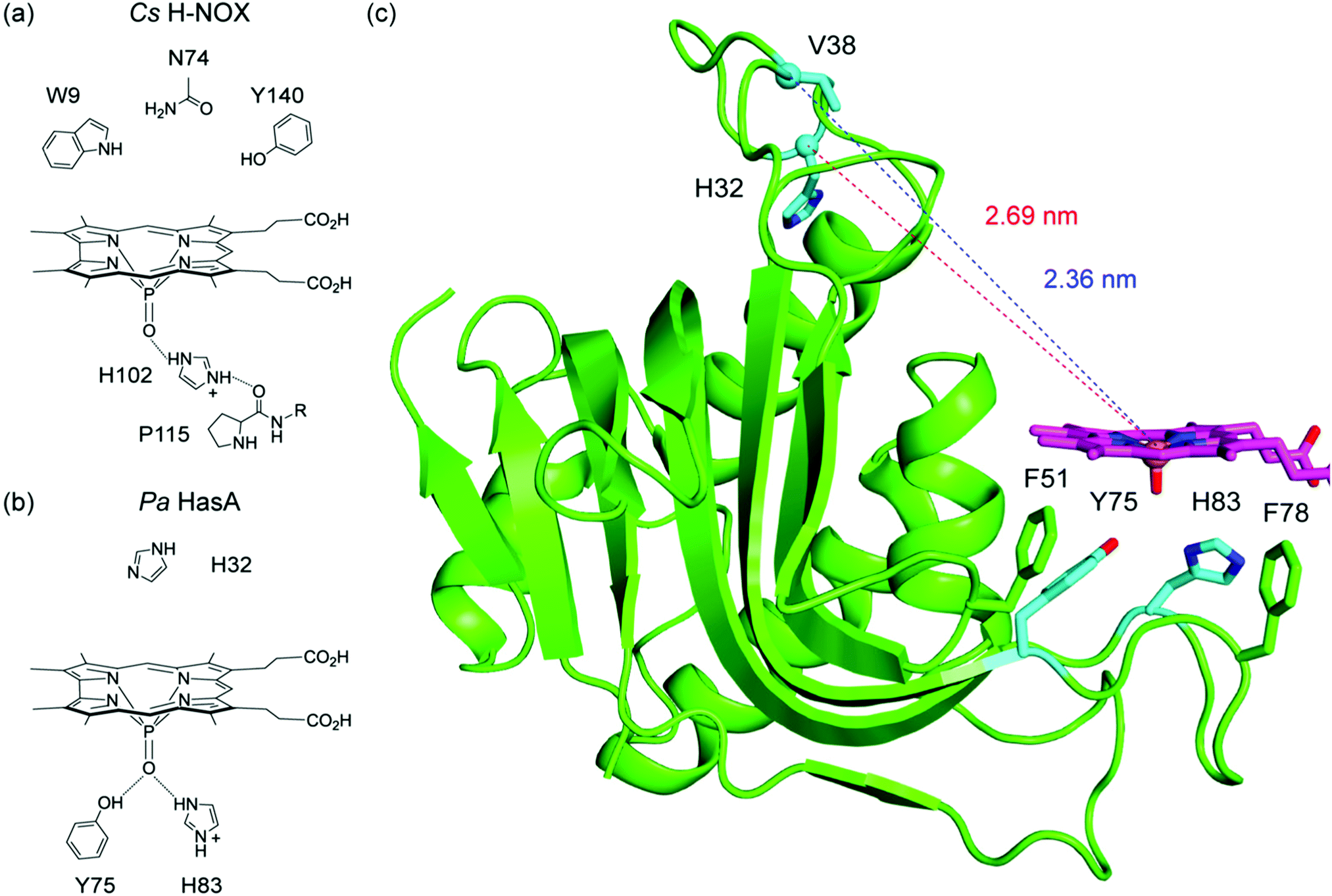
Fig. 5 Structures of (a) holo (PDB ID: 3ELL) and (b) apo Pa HasA (PDB ID: 3MOK), illustrating the large-scale conformational change upon haem binding. Key residues are highlighted in cyan. (c) Absorption (solid lines) and emission spectra (λex = 470 nm, dashed lines) of fluorescein-labelled apo ( ) and P-1 (—) V38C HasA in TEA buffer at pH 6.5. Emission spectra are for absorbance-matched samples: A(470) = 0.0481 ± 0.0002. ) and P-1 (—) V38C HasA in TEA buffer at pH 6.5. Emission spectra are for absorbance-matched samples: A(470) = 0.0481 ± 0.0002. | ||
Since attempts to label V38C HasA reconstituted with P-1 led to extensive corrole loss, reconstitution of fluorescein-labelled protein was preferred (Fig. 5c). P-1 incorporation, as judged by the Soret to 280 nm ratio, is not as high as it is for the unlabelled sample (4.22 vs. 6.06), indicating that P-1 incorporation is ∼70%. Based on the steady-state data, the apparent FRET efficiency is 63%. Correcting for the presence of apoprotein, the estimated FRET efficiency is 75%. Since the presence of apoprotein complicates the steady-state data, time-resolved methods were utilised to determine a more accurate FRET efficiency (Fig. S16, ESI†). Fluorescein-labelled apo V38C cleanly fits to a monoexponential decay with a lifetime of 4.11 ns. The P-1 reconstituted sample exhibits biexponential kinetics; the long component is consistent with the apoprotein, while the short component has a lifetime of 0.91 ns. The time-resolved data yields a FRET efficiency of 78%, which is consistent with the corrected steady-state data (75%). This corresponds to a donor–acceptor distance of 3.16 nm.
This distance suggests that the protein exhibits an open, apo-like conformation. Based on the holo HasA structure, the Fe to Cα of V38 distance is 1.03 nm (Fig. 5a). At most, the dye, linker, and side chain would add 1.36 nm (Fig. S17, ESI†), to give an expected maximum distance of 2.39 nm. This is substantially shorter than the FRET-determined distance of 3.16 nm. Instead, the FRET distance is similar to the O atom of the Y75 side chain to Cα of V38 distance of 2.54 nm observed in the apo HasA structure (Fig. 5b). The difference between the FRET and crystal structure distances is 0.62 nm, which reasonably accounts for the cysteine side chain, the maleimide, and the dye.
To corroborate this result, the H32C variant was also studied. As with V38C, reconstitution of fluorescein-labelled H32C with P-1 is incomplete. After accounting for the presence of apoprotein, the FRET efficiency is 68% (Fig. S18, ESI†). This is lower than that for V38C (75%), which is expected given that H32 is farther away from the putative corrole binding site. This result is corroborated with time-resolved data. The fluorescein lifetime of 1.06 ns gives a FRET efficiency of 74%, which corresponds to a 3.28 nm distance. This is consistent with the 2.80 nm distance between the O atom of the Y75 side chain and Cα of H32 (Fig. 5b). Together, these results indicate that HasA adopts an open, apo-like conformation when P-1 is bound.
Discussion
Data interpretation and structural model for P-1 binding in H-NOX
The results of corrole binding to the protein variants, in conjunction with MD simulations, enable a model of P-1 binding in Cs H-NOX. Variants that modify the distal hydrogen bonding network (W9F, Y140F) exhibit P-1 binding that is the same as the WT protein. It has previously been demonstrated that the distal subdomain, containing the hydrogen bonding network, moves ∼4.5 Å upon O2 binding.58 Given the lack of direct contacts between P-1 and the H-bonding network, the distal subdomain is likely shifted away from the haem binding pocket, analogous to the unliganded haem structure. The increased space in the haem binding pocket would enable greater conformational flexibility of the corrole.While it could be the case that the absence of one H-bonding residue can be compensated by another, it is more likely that the PO unit primarily interacts with H102. This is evident from the significant decrease in corrole binding for the H102 variants. In the absence of H102, hydrogen bonding of the propionate chains on the corrole to the Y–S–R motif serves as the only point of contact. However, this interaction alone is insufficient for full corrole incorporation. The broader emission profiles and pronounced Q(0,1) emission band for the H102 variants suggest that the corrole exhibits greater conformational flexibility. This is consistent with the idea that the distal subdomain has moved away from the haem binding pocket. The diminished rigidity of the binding pocket is likely one reason for the decrease in corrole incorporation.
The other residue that plays a key role in P-1 binding is P115. The absence of P115 leads to a significant decrease in corrole binding and the absorption spectrum is distinct from the other H-NOX variants, displaying a single Soret band. Additionally, corrole emission is lower than the other H-NOX variants. These spectral differences suggest that the corrole resides in a more hydrophilic environment such that P-1 is more solvent-exposed in the P115A variant. Given the strong coupling between H102 and P115 observed in the MD simulations, perhaps the absence of P115 does not enable the proper movements of H102, precluding proper cofactor recognition. Indeed, MD simulations of the apo P115A variant indicate that H102 is primarily in the ‘flipped’ conformation. This rotamer is likely less amenable to bind P-1 given that the corrole cannot simultaneously engage H102 and the Y–S–R motif.
Given the potential similarity between the P-1 bound H-NOX and the Fe(II) unliganded structure (PDB ID: 5JRU), we looked for interactions that could further stabilise corrole binding. In the unliganded structure, the N–H proton of the H102 side chain could hydrogen bond with the backbone carbonyl of P115 (Fig. S19a, ESI†). Therefore, if H102 were protonated when P-1 is bound, this residue could hydrogen bond with both the PO unit and the backbone carbonyl of P115. Given the necessity of P115 for corrole binding, this is an intriguing possibility. The structure of the P115A variant (PDB ID: 3EEE) exhibits a different conformation of the protein backbone at A115 (Fig. S19b, ESI†), which likely precludes this contact from forming in the variant. It was previously demonstrated that P-1 incorporation decreases with increasing pH,28 suggesting that protonation of this residue occurs at lower pH to enhance corrole binding. Moreover, protonation of H102 explains why the WT protein and H102 variants exhibit the same emission intensity. If H102 were not protonated, then the lone pair of electrons on the imine nitrogen would be sufficiently close to the corrole to observe significant fluorescence quenching. Together, these observations suggest that H102 is protonated, which can then form an additional hydrogen bond with P115 to rigidify the haem binding pocket and enhance corrole binding.
Fig. 6a illustrates a proposed model for P-1 binding in Cs H-NOX. Rather than interacting with the distal hydrogen bonding network, the PO unit of the corrole hydrogen bonds to H102. This residue is protonated, forming an additional H-bond to the backbone carbonyl of P115. Together, these interactions rigidify the haem binding pocket and stabilise corrole binding.
 | ||
| Fig. 6 (a) Proposed model for P-1 binding in Cs H-NOX. The PO unit hydrogen bonds with H102. At pH 6.5, H102 is protonated and also hydrogen bonds with the backbone carbonyl of P115, rigidifying the corrole in the haem binding pocket. (b) Proposed model for P-1 binding in Pa HasA. The PO unit hydrogen bonds with both Y75 and protonated H83 to form a small hydrogen bonding network. The H32 loop does not engage with the corrole, and FRET studies confirm that the protein adopts an apo-like structure. (c) Docking model between P-1 and the apo HasA structure (PDB ID: 3MOK). The cofactor is flanked by F51 and F78, which may serve as hydrophobic gatekeeping residues for cofactor recognition. Adapted with permission from ref. 3. Copyright 2021 American Chemical Society. | ||
Data interpretation and structural model for P-1 binding in HasA
In the case of HasA, modulation of residues on the H32 loop has no effect on corrole binding. Since corrole absorption does not change in the presence or absence of H32, the histidine side chain does not directly coordinate to the phosphorus centre. Moreover, corrole emission in the two H32 variants is not enhanced relative to the WT protein. This suggests that H32 is sufficiently far away from the corrole (>10 Å)55 such that no quenching is observed.Alternatively, the absence of quenching by H32 could be due to protonation. In a previous study, the pKa values for histidine residues in HasA from Serratia marcescens were determined using NMR spectroscopy. The pKa of H32 in the apoprotein is 7.3 and this value significantly decreases to <4.8 when Ga(PPIX) is bound.57 Since H32 does not directly bind to P-1, this residue likely has a pKa that is similar to the apoprotein when the corrole is bound. Therefore, H32 is largely protonated in these experiments at pH 6.5.
To determine the conformation of the H32 loop when P-1 is bound, FRET experiments were utilised. The V38C and H32C variants were labelled with fluorescein maleimide. Based on the FRET analysis, the fluorescein label at V38C and H32C with P-1 exhibit distances of 3.16 nm and 3.28 nm, respectively. These values are consistent with the apo conformation of HasA, indicating that the H32 loop exhibits an extended conformation when P-1 is bound. This observation corroborates previous studies, which suggest the H32 loop does not randomly sample conformations. Instead, movement of the H32 loop is triggered by the formation of a high-spin haem intermediate, implying an induced fit mechanism.59 Since P-1 is unable to recapitulate haem binding, the H32 loop remains in an apo conformation.
Since the H32 loop is not involved in corrole binding, modulation of the Y75 loop has a significant effect on P-1 binding. The Y75F variant exhibits lower corrole binding. Although this residue is not necessary, it provides a contact for P-1 that enables complete corrole loading. The Y75F variant exhibits decreased fluorescence, suggesting that P-1 resides in a more hydrophilic environment. Alternatively, the loss of Y75 could pull P-1 closer to H83, thereby quenching fluorescence (via the lone pair on the imine nitrogen). Based on the S. marcescens study, the pKa of H83 in the apoprotein is 5.6 and increases to 9.7 when Ga(PPIX) is bound.57 Therefore, H83 is likely protonated at pH 6.5 when P-1 is bound to HasA. This is supported by the pH-dependent emission studies of WT HasA with P-1.
Surprisingly, no corrole binding is observed in the absence of H83. After initial experiments with H83A, we hypothesised that the lack of corrole binding may be due to the loss of a hydrogen bonding residue. We then attempted to recapitulate the proposed roles of H83 with other, non-histidine variants. H83 forms a hydrogen bond with Y75 and may facilitate deprotonation to enable haem ligation.56,60 The H83D variant was expressed because the aspartate side chain is an appropriate length to hydrogen bond to Y75 (Fig. S10, ESI†). However, no corrole binding was observed. The H83S variant was prepared in an attempt to provide a flexible, non-histidine H-bonding partner for P-1, but no corrole binding was observed. Together, these results indicate that H83, similar to H102 in H-NOX, is critical for P-1 binding. Unlike H-NOX, HasA lacks a Y–S–R motif, so there is no other point of contact in the protein for P-1 in the absence of H83.
Fig. 6b illustrates a proposed model for corrole binding in Pa HasA. The PO unit of P-1 hydrogen bonds with both Y75 and H83. This hydrogen bonding network may also include water and/or buffer molecules to further stabilise corrole binding. It was previously demonstrated that haem recognition initially occurs at the Y75 loop; hydrophobic patches on this loop stabilise cofactor binding.59,61 The H32 loop does not interact with P-1 and, based on the FRET experiments, adopts an apo-like structure. Given the similarity to apo HasA, a docking model was generated using the apo HasA structure (PDB ID: 3MOK) and the DFT-optimised geometry of P-1 (Fig. 6c).28 The corrole was positioned such that the PO unit forms suitable H-bonds to both Y75 and H83 while avoiding steric clashes with Y138 and M141. The macrocycle is flanked by two hydrophobic residues, F51 and F78, that may serve as gatekeeping residues to hold the cofactor in place. With this docking model, we can gauge how it compares to the FRET distances. The distances between the P atom and Cα of V38 and H32 are 2.36 nm and 2.69 nm, respectively (Fig. 6c). The corresponding FRET distances are 3.16 for V38C and 3.28 for H32C. The difference between these metrics is 0.80 nm for V38C and 0.59 nm for H32C, which reasonably accounts for the cysteine side chain, the maleimide, and the dye.
Insight into the biochemistry of Cs H-NOX and Pa HasA
Although this study involves an unnatural haem analogue, these experiments provide some insight into the native biochemistry of Cs H-NOX and Pa HasA. MD simulations demonstrate that H102 in Cs H-NOX is conformationally flexible, exhibiting a ∼90° rotation. This motion had only previously been observed for NO-sensing H-NOX proteins (e.g., Shewanella oneidensis). In the unliganded state of So H-NOX, the proximal histidine coordinates to the iron centre. Upon NO binding, the Fe–His bond breaks and the side chain rotates ∼90°.23 Conversely, Cs H-NOX, an O2 sensor, forms a six-coordinate NO complex, retaining the Fe–His bond.58 MD simulations demonstrate that H102 is flexible in Cs H-NOX, adopting conformations similar to those in NO-bound So H-NOX. This suggests that this flexibility is inherent to H-NOX proteins. Consequently, the formation of a six-coordinate NO complex in Cs H-NOX is not likely a constraint imposed by the protein fold. Instead, it may be a consequence of the electronic structure of the haem cofactor that is conferred by the secondary coordination sphere (i.e., the distal hydrogen bonding network).The coupling of P115 and H102 sheds some light on the role of this proline residue, which is highly conserved in H-NOX domains, from bacterial H-NOX to mammalian sGC. P115 modulates the conformation54 and redox potential62 of the haem cofactor. Mutation of the proline to an alanine also has a functional consequence. The P117A variant of the Shewanella woodyi H-NOX constitutively activates the cognate diguanylate cyclase.63 While these observations are consequences of the mutation, they do not necessarily describe the fundamental role of the proline or explain why it is so highly conserved. Since P115 forces H102 to preferentially adopt an ‘upright’ conformation, it could be that P115 poises H102 for haem binding in the native holoprotein. The influence of P115 likely extends to the conformation of the αF helix, which bears H102. The αF helix is involved in the binding interface between the H-NOX and histidine kinase for both Shewanella oneidensis64 and Vibrio cholerae.65 Perhaps P115 serves a fundamental role to modulate the αF helix conformation and regulate activity. This could explain why this proline residue is so highly conserved across H-NOX domains.
P-1 binding in Pa HasA corroborates previous observations and hypotheses regarding the native holoprotein. Corrole binding occurs on the Y75 loop, which is consistent with observations that haem binding first occurs at this site.59 However, hydrophobicity of the Y75 loop is not sufficient for cofactor binding. Attempts to reconstitute the apoprotein with Pd(MPIX) failed, resulting in no porphyrin binding. Since the Pd(II) porphyrin does not bind axial ligands, it is clear that an additional protein–cofactor interaction is necessary. This is accomplished by the formation of the Fe–Tyr bond for haem or the PO⋯His hydrogen bond for P-1. The observation that P-1-bound HasA adopts an apo-like structure supports the idea that closure of the H32 loop occurs by an induced-fit mechanism upon haem binding.59
Given the necessity of H83 for P-1 binding, this residue may play a critical role in the initial steps of haem acquisition. Additionally, it has been posited that H83 serves to increase the phenolate character of Y75.57,60 This contention seems to be further supported by the holo/haem H83D variant, which exhibits a UV-vis absorption spectrum nearly identical to the WT protein (Fig. S9, ESI†). This contrasts the other H83 variants, which exhibit distinct spectra. The aspartate side chain can seemingly fill the role of H83 by acting as a suitable hydrogen bonding partner with a comparable distance to the native residue (Fig. S10, ESI†). However, since the H83D variant does not bind P-1, it is likely that H83 serves additional roles through π-stacking and hydrophobic interactions that stabilise cofactor binding. Together, these experiments support previously reported hypotheses regarding H83 and underscore the multifaceted role of this conserved residue.56
Conclusions
Mutagenesis of key residues in the haem binding pocket of Cs H-NOX and Pa HasA has identified critical contacts between the protein and corrole P-1. In both cases, the corrole exhibits a binding mode that is distinct from haem. The primary interaction occurs between a histidine residue and the PO unit of P-1. In contrast to all previously characterised examples of non-porphyrin cofactors bound to haem proteins, this histidine interaction does not involve direct coordination to the central element of the macrocycle. MD simulations of apo H-NOX reveal that H102 and P115 are tightly coupled, thereby explaining the necessity of both residues for full corrole incorporation. FRET studies with site-specific labelling of HasA variants demonstrate that the protein adopts an extended, apo-like conformation when P-1 is bound. Together, this study illustrates that unnatural haem analogues can bind to proteins in unexpected ways and underscores the need to identify these interactions, particularly in cases of cofactor reconstitution.
The continued elucidation of key interactions between small molecules and proteins provides foundational research for a variety of applications, ranging from drug development to de novo protein design. The key interaction between a PO unit and a protonated histidine residue revealed in this study could be leveraged for drug design, targeting histidine side chains that are integral to protein active or allosteric sites. For example, bacterial histidine kinases are emerging targets for antibiotics.66 Drug molecules with a PO moiety could target the active-site histidine residue of these proteins as an inroad to novel antibiotics.
Given the ability of Pa HasA to bind a diverse array of haem analogues, this protein is an attractive platform for artificial metalloenzymes. One barrier to achieving this application is the necessity of two protein-based ligands, rendering the complex coordinatively saturated. Indeed, all previously reported examples of cofactors bound to HasA require both Y75 and H32. Consequently, one of these ligands must dissociate in order to bind a substrate at the metal centre. It is likely that the loss of one protein-based ligand would lower the affinity for the haem analogue, resulting in potential cofactor loss. Conversely, P-1 binding in Pa HasA only requires the Y75 loop, making this construct a suitable scaffold for catalysis, particularly because phosphorus corroles can serve as photocatalysts that mediate organic transformations.67 Additionally, phosphorus corroles are potent photosensitisers that can generate singlet oxygen and/or superoxide from molecular oxygen for photodynamic therapy.68 The H-NOX and HasA conjugates with corrole P-1 provide a facile means of imparting water solubility to utilise phosphorus corroles for advanced photochemical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CML and NRL acknowledge the Miller Institute at UC Berkeley for a Postdoctoral Fellowship. AJN was supported by an NIH training grant: T32 066698. This research was supported by a grant from the National Institutes of Health (NIH): R01GM127854. We thank Berkeley Research Computing for providing computational resources through the Savio cluster. We thank Sarah Chen for performing the cloning and initial expression of S82C HasA and Deirdre Hanley for assisting with experiments to complete this study. We thank Trevor Roberts for helping with time-resolved emission data, as well as Professor Naomi Ginsberg for allowing us to access the instrument. We thank Allie Batka for assisting with mass spectrometry, and we thank Prof. Matthew Francis for allowing us to access the instrument. Prof. Susan Marqusee, Dr. John Hangasky, and the members of the Marletta lab are graciously thanked for critically evaluating this manuscript.References
- M. O. Senge, S. A. MacGowan and J. M. O’Brien, Chem. Commun., 2015, 51, 17031–17063 RSC.
- E. Sitte and M. O. Senge, Eur. J. Org. Chem., 2020, 3171–3191 CrossRef CAS PubMed.
- C. M. Lemon and M. A. Marletta, Acc. Chem. Res., 2021, 54, 4565–4575 CrossRef CAS PubMed.
- J. A. McIntosh, C. C. Farwell and F. H. Arnold, Curr. Opin. Chem. Biol., 2014, 19, 126–134 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- M. Bordeaux, R. Singh and R. Fasan, Bioorg. Med. Chem., 2014, 22, 5697–5704 CrossRef CAS PubMed.
- M. W. Wolf, D. A. Vargas and N. Lehnert, Inorg. Chem., 2017, 56, 5623–5635 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- M. Jeschek, S. Panke and T. R. Ward, Trends Biotechnol., 2018, 36, 60–72 CrossRef CAS PubMed.
- K. Vong, I. Nasibullin and K. Tanaka, Bull. Chem. Soc. Jpn., 2021, 94, 382–396 CrossRef CAS.
- J. Nibbs, S. A. Vinogradov, J. M. Vanderkooi and B. Zelent, Photochem. Photobiol., 2004, 80, 36–40 CrossRef CAS PubMed.
- M. B. Winter, E. J. McLaurin, S. Y. Reece, C. Olea, D. G. Nocera and M. A. Marletta, J. Am. Chem. Soc., 2010, 132, 5582–5583 CrossRef CAS PubMed.
- V. S. Lelyveld, E. Brustad, F. H. Arnold and A. Jasanoff, J. Am. Chem. Soc., 2011, 133, 649–651 CrossRef CAS PubMed.
- M. B. Winter, P. J. Klemm, C. M. Phillips-Piro, K. N. Raymond and M. A. Marletta, Inorg. Chem., 2013, 52, 2277–2279 CrossRef CAS PubMed.
- M. C. Findlay and J. C. W. Chien, Eur. J. Biochem., 1977, 76, 79–83 CrossRef CAS PubMed.
- O. C. Choon and G. A. Rodley, J. Inorg. Biochem., 1983, 19, 189–202 CrossRef CAS.
- C. Deniau, J. Couprie, C. Simenel, V. Kumar, I. Stojiljkovic, C. Wandersman, M. Delepierre and A. Lecroisey, J. Biomol. NMR, 2001, 21, 189–199 CrossRef CAS PubMed.
- B. M. Hoffman and D. H. Petering, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 637–643 CrossRef CAS PubMed.
- E. A. Dierks, S. Hu, K. M. Vogel, A. E. Yu, T. G. Spiro and J. N. Burstyn, J. Am. Chem. Soc., 1997, 119, 7316–7323 CrossRef CAS.
- M. A. Herzik, R. Jonnalagadda, J. Kuriyan and M. A. Marletta, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4156–E4164 CrossRef CAS PubMed.
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945–954 CrossRef CAS PubMed.
- K. Oohora and T. Hayashi, Dalton Trans., 2021, 50, 1940–1949 RSC.
- T. Matsuo, A. Hayashi, M. Abe, T. Matsuda, Y. Hisaeda and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 15124–15125 CrossRef CAS PubMed.
- M. Bröring, F. Brégier, O. Burghaus and C. Kleeberg, Z. Anorg. Allg. Chem., 2010, 636, 1760–1766 CrossRef.
- C. M. Lemon and M. A. Marletta, Inorg. Chem., 2021, 60, 2716–2729 CrossRef CAS PubMed.
- K. Oohora, Y. Kihira, E. Mizohata, T. Inoue and T. Hayashi, J. Am. Chem. Soc., 2013, 135, 17282–17285 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, Y. Kihira, H. Sugimoto, Y. Shiro and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 18460–18463 CrossRef CAS PubMed.
- T. Hayashi, D. Murata, M. Makino, H. Sugimoto, T. Matsuo, H. Sato, Y. Shiro and Y. Hisaeda, Inorg. Chem., 2006, 45, 10530–10536 CrossRef CAS PubMed.
- C. Shirataki, O. Shoji, M. Terada, S. Ozaki, H. Sugimoto, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2014, 53, 2862–2866 CrossRef CAS PubMed.
- Y. Shisaka, Y. Iwai, S. Yamada, H. Uehara, T. Tosha, H. Sugimoto, Y. Shiro, J. K. Stanfield, K. Ogawa, Y. Watanabe and O. Shoji, ACS Chem. Biol., 2019, 14, 1637–1642 CrossRef CAS PubMed.
- G. Centola, D. J. Deredge, K. Hom, Y. Ai, A. T. Dent, F. Xue and A. Wilks, ACS Infect. Dis., 2020, 6, 2073–2085 CrossRef CAS PubMed.
- M. Ohashi, T. Koshiyama, T. Ueno, M. Yanase, H. Fujii and Y. Watanabe, Angew. Chem., Int. Ed., 2003, 42, 1005–1008 CrossRef CAS PubMed.
- T. Ueno, T. Koshiyama, M. Ohashi, K. Kondo, M. Kono, A. Suzuki, T. Yamane and Y. Watanabe, J. Am. Chem. Soc., 2005, 127, 6556–6562 CrossRef CAS PubMed.
- D. K. Garner, L. Liang, D. A. Barrios, J.-L. Zhang and Y. Lu, ACS Catal., 2011, 1, 1083–1089 CrossRef CAS PubMed.
- T. Hayashi, Y. Morita, E. Mizohata, K. Oohora, J. Ohbayashi, T. Inoue and Y. Hisaeda, Chem. Commun., 2014, 50, 12560–12563 RSC.
- K. Oohora, Y. Miyazaki and T. Hayashi, Angew. Chem., Int. Ed., 2019, 58, 13813–13817 CrossRef CAS PubMed.
- P.-S. Huang, S. E. Boyken and D. Baker, Nature, 2016, 537, 320–327 CrossRef CAS PubMed.
- J. C. Klima, L. A. Doyle, J. D. Lee, M. Rappleye, L. A. Gagnon, M. Y. Lee, E. P. Barros, A. A. Vorobieva, J. Dou, S. Bremmer, J. S. Quon, C. M. Chow, L. Carter, D. L. Mack, R. E. Amaro, J. C. Vaughan, A. Berndt, B. L. Stoddard and D. Baker, Nat. Commun., 2021, 12, 856 CrossRef CAS PubMed.
- A. Quijano-Rubio, H.-W. Yeh, J. Park, H. Lee, R. A. Langan, S. E. Boyken, M. J. Lajoie, L. Cao, C. M. Chow, M. C. Miranda, J. Wi, H. J. Hong, L. Stewart, B.-H. Oh and D. Baker, Nature, 2021, 591, 482–487 CrossRef CAS PubMed.
- S. Boyoglu-Barnum, D. Ellis, R. A. Gillespie, G. B. Hutchinson, Y.-J. Park, S. M. Moin, O. J. Acton, R. Ravichandran, M. Murphy, D. Pettie, N. Matheson, L. Carter, A. Creanga, M. J. Watson, S. Kephart, S. Ataca, J. R. Vaile, G. Ueda, M. C. Crank, L. Stewart, K. K. Lee, M. Guttman, D. Baker, J. R. Mascola, D. Veesler, B. S. Graham, N. P. King and M. Kanekiyo, Nature, 2021, 592, 623–628 CrossRef CAS PubMed.
- N. R. L. Rojas, S. Kamtekar, C. T. Simons, J. E. McLean, K. M. Vogel, T. G. Spiro, R. S. Farid and M. H. Hecht, Protein Sci., 1997, 6, 2512–2524 CAS.
- F. V. Cochran, S. P. Wu, W. Wang, V. Nanda, J. G. Saven, M. J. Therien and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 1346–1347 CrossRef CAS PubMed.
- I. V. Korendovych, A. Senes, Y. H. Kim, J. D. Lear, H. C. Fry, M. J. Therien, J. K. Blasie, F. A. Walker and W. F. DeGrado, J. Am. Chem. Soc., 2010, 132, 15516–15518 CrossRef CAS PubMed.
- F. Nastri, R. Bruni, O. Maglio and A. Lombardi, in Coordination Chemistry in Protein Cages: Principles, Design, and Applications, ed. T. Ueno and Y. Watanabe, John Wiley & Sons, Hoboken, NJ, 2013, pp. 45–85 Search PubMed.
- A. D’Souza and S. Bhattacharjya, Biochemistry, 2021, 60, 431–439 CrossRef PubMed.
- H. C. Fry, A. Lehmann, J. G. Saven, W. F. DeGrado and M. J. Therien, J. Am. Chem. Soc., 2010, 132, 3997–4005 CrossRef CAS PubMed.
- H. C. Fry, A. Lehmann, L. E. Sinks, I. Asselberghs, A. Tronin, V. Krishnan, J. K. Blasie, K. Clays, W. F. DeGrado, J. G. Saven and M. J. Therien, J. Am. Chem. Soc., 2013, 135, 13914–13926 CrossRef CAS PubMed.
- N. F. Polizzi, Y. Wu, T. Lemmin, A. M. Maxwell, S.-Q. Zhang, J. Rawson, D. N. Beratan, M. J. Therien and W. F. DeGrado, Nat. Chem., 2017, 9, 1157–1164 CrossRef CAS PubMed.
- P. Braun, E. Goldberg, C. Negron, M. von Jan, F. Xu, V. Nanda, R. L. Koder and D. Noy, Proteins, 2011, 79, 463–476 CrossRef CAS PubMed.
- M. M. Sheehan, M. S. Magaraci, I. A. Kuznetsov, J. A. Mancini, G. Kodali, C. C. Moser, P. L. Dutton and B. Y. Chow, Biochemistry, 2018, 57, 6752–6756 CrossRef CAS PubMed.
- C. Olea, E. M. Boon, P. Pellicena, J. Kuriyan and M. A. Marletta, ACS Chem. Biol., 2008, 3, 703–710 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- R. Kumar, H. Matsumura, S. Lovell, H. Yao, J. C. Rodríguez, K. P. Battaile, P. Moënne-Loccoz and M. Rivera, Biochemistry, 2014, 53, 2112–2125 CrossRef CAS PubMed.
- N. Wolff, C. Deniau, S. Létoffé, C. Simenel, V. Kumar, I. Stojiljkovic, C. Wandersman, M. Delepierre and A. Lecroisey, Protein Sci., 2002, 11, 757–765 CrossRef CAS PubMed.
- C. W. Hespen, J. J. Bruegger, C. M. Phillips-Piro and M. A. Marletta, ACS Chem. Biol., 2016, 11, 2337–2346 CrossRef CAS PubMed.
- E. T. Yukl, G. Jepkorir, A. Y. Alontaga, L. Pautsch, J. C. Rodriguez, M. Rivera and P. Moënne-Loccoz, Biochemistry, 2010, 49, 6646–6654 CrossRef CAS PubMed.
- A. Y. Alontaga, J. C. Rodriguez, E. Schönbrunn, A. Becker, T. Funke, E. T. Yukl, T. Hayashi, J. Stobaugh, P. Moënne-Loccoz and M. Rivera, Biochemistry, 2009, 48, 96–109 CrossRef CAS PubMed.
- G. Jepkorir, J. C. Rodríguez, H. Rui, W. Im, S. Lovell, K. P. Battaile, A. Y. Alontaga, E. T. Yukl, P. Moënne-Loccoz and M. Rivera, J. Am. Chem. Soc., 2010, 132, 9857–9872 CrossRef CAS PubMed.
- C. Olea, J. Kuriyan and M. A. Marletta, J. Am. Chem. Soc., 2010, 132, 12794–12795 CrossRef CAS PubMed.
- S. Muralidharan and E. M. Boon, J. Am. Chem. Soc., 2012, 134, 2044–2046 CrossRef CAS PubMed.
- M. Rao, M. A. Herzik, A. T. Iavarone and M. A. Marletta, Biochemistry, 2017, 56, 1274–1284 CrossRef CAS PubMed.
- Y. Guo, A. T. Iavarone, M. M. Cooper and M. A. Marletta, Biochemistry, 2018, 57, 1779–1789 CrossRef CAS PubMed.
- A. E. Bem, N. Velikova, M. T. Pellicer, P. van Baarlen, A. Marina and J. M. Wells, ACS Chem. Biol., 2015, 10, 213–224 CrossRef CAS PubMed.
- A. Mahammed, K. Chen, J. Vestfrid, J. Zhao and Z. Gross, Chem. Sci., 2019, 10, 7091–7103 RSC.
- C. M. Lemon, Pure Appl. Chem., 2020, 92, 1901–1919 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and additional results. See DOI: 10.1039/d2cb00004k |
| This journal is © The Royal Society of Chemistry 2022 |