
Spherical α-helical polypeptide-mediated E2F1 silencing against myocardial ischemia-reperfusion injury (MIRI)†
Rongying
Zhu‡
a,
Mengying
Hou‡
b,
Yang
Zhou
b,
Huan
Ye
b,
Lei
Chen
a,
Chenglong
Ge
b,
Shanzhou
Duan
a,
Lichen
Yin
 *b and
Yongbing
Chen
*a
*b and
Yongbing
Chen
*a
aDepartment of Cardiothoracic Surgery, the Second Affiliated Hospital of Soochow University, Suzhou 215002, China. E-mail: chentongt@sina.com
bInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Soochow University, Suzhou 215123, China. E-mail: lcyin@suda.edu.cn
First published on 31st August 2022
Abstract
Apoptosis of cardiomyocytes is a critical outcome of myocardial ischemia-reperfusion injury (MIRI), which leads to the permanent impairment of cardiac function. Upregulated E2F1 is implicated in inducing cardiomyocyte apoptosis, and thus intervention of the E2F1 signaling pathway via RNA interference may hold promising potential for rescuing the myocardium from MIRI. To aid efficient E2F1 siRNA (siE2F1) delivery into cardiomyocytes that are normally hard to transfect, a spherical, α-helical polypeptide (SPP) with potent membrane activity was developed via dendrimer-initiated ring-opening polymerization of N-carboxyanhydride followed by side-chain functionalization with guanidines. Due to its multivalent structure, SPP outperformed its linear counterpart (LPP) to feature potent siRNA binding affinity and membrane activity. Thus, SPP effectively delivered siE2F1 into cardiomyocytes and suppressed E2F1 expression both in vitro and in vivo after intramyocardial injection. The E2F1–miR421–Pink1 signaling pathway was disrupted, thereby leading to the reduction of MIRI-induced mitochondrial damage, apoptosis, and inflammation of cardiomyocytes and ultimately recovering the systolic function of the myocardium. This study provides an example of membrane-penetrating nucleic acid delivery materials, and it also provides a promising approach for the genetic manipulation of cardiomyocyte apoptosis for the treatment of MIRI.
1. Introduction
Acute myocardial infarction is an ischemic heart disease caused by the rupture of atherosclerotic plaques in coronary arteries, accounting for the leading rate of mobility and mortality worldwide.1,2 Clinically, pharmacological thrombolysis or direct coronary intervention is commonly used to restore the coronary blood flow to the ischemic myocardium.3–5 However, a growing body of evidence suggests that reperfusion will also lead to myocardial ischemia-reperfusion injury (MIRI), impairing the myocardial tissues and causing cardiomyocyte apoptosis.6 MIRI involves a complex process related to multiple pathophysiological factors, such as elevated oxidative stress,7,8 inflammation,9,10 and mitochondrial dysfunction,11,12 which collectively lead to the apoptosis of cardiomyocytes. Therefore, rescuing cardiomyocytes from apoptosis during MIRI is crucial for the protection of cardiac function. Cell cycle-related proteins often take charge of regulating cell proliferation, differentiation, and apoptosis during cell cycle progression, among which E2F1 has been found to be unregulated and closely related to the development of inflammation during MIRI.13–15 In addition, upregulated E2F1 contributes to the damage of the myocardium by impairing the mitochondrial function through the E2F1–miR-421–Pink1 signaling pathway.16 Therefore, the inhibition of E2F1 expression to reduce cardiomyocyte inflammation and mitochondrial damage may be a promising strategy for the treatment of MIRI.Nowadays, RNA interference (RNAi) mediated by small interfering RNA (siRNA) has become a revolutionary therapeutic paradigm for various human diseases because of its high specificity and efficiency in silencing the expression of disease-related genes via degradation of mRNA.17–19 siRNA is membrane impermeable and prone to hydrolysis by nucleases, and therefore an effective delivery vector is highly necessary to mediate efficient intracellular siRNA delivery.20–24 Cationic polymers are an important category of gene vectors as exemplified by polyethyleneimine, polyamide dendrimers, poly(β-amino ester), and polypeptides, and they have been demonstrated to mediate effective siRNA delivery.25–33 These cationic materials form nanocomplexes (NCs) with negatively charged nucleic acids via the electrostatic interaction, thereby facilitating the cytosolic delivery often via the endocytosis pathway. However, efficient cardiomyocyte transfection is challenging because the myocardium has a compact ventricular muscle structure and cardiomyocytes are slowly dividing with poor endocytic capability. Thus, polycationic NCs that normally enter cells via endocytosis are poorly taken up by cardiomyocytes, and they tend to be trapped in the endolysosomes after internalization, ultimately leading to a weak transfection efficiency.34,35 Therefore, there is an urgent need to develop siRNA delivery vectors with potent membrane activity, which can enable highly efficient cytosolic transport and endolysosomal escape.
To overcome the membrane barrier against siRNA delivery, we have previously developed a cationic, α-helical polypeptide, which features high membrane penetrating capability that is closely related to its rigid, rod-like α-helical conformation. As such, the helical polypeptide mediates efficient gene transfection by facilitating the cellular uptake and endolysosomal escape of nucleic acid cargoes.36–39 To enhance the membrane activity as well as gene delivery efficiency, the polypeptide is tailored in terms of the cationic charges, hydrophobicity, and aromaticity.40–42 Besides these structural parameters, engineering the multivalency of polycations has been demonstrated as an effective strategy to strengthen the binding affinity with gene cargoes and reinforce the molecular interactions with cell membranes. To this end, a spherical α-helical polypeptide (SPP) was herein developed using amine-terminated, third-generation polyamidoamine (PAMAM, G3-NH2) as a macro-initiator to induce the polymerization of γ-(4-propyloxybenzyl)-L-glutamic acid-N-carboxyanhydride (POBLG-NCA), followed by conjugation of guanidine onto polypeptide side-chain terminals. Compared with the linear α-helical polypeptide (LPP) prepared with hexamethyldisilazane (HMDS) as the initiator, SPP had stronger siRNA binding affinity and membrane activity due to its multivalent topology that enhanced the positive charge density. Therefore, SPP mediated effective trans-membrane delivery of E2F1 siRNA (siE2F1) into cardiomyocytes and promoted efficient E2F1 silencing in the myocardium after intramyocardial administration, alleviating mitochondrial damage and inhibiting cardiomyocyte apoptosis to protect the myocardium from MIRI.
2. Results and discussion
2.1. Synthesis and characterization of SPP
POBLG–NCA was synthesized according to published procedures (Fig. S1†).40,41 Subsequently, the spherical polypeptide PAMAM–PPOBLG was synthesized via ring-opening polymerization (ROP) of POBLG–NCA as initiated by G3-NH2 at the monomer to initiator [M]/[I] molar ratio of 100/1. The characteristic peak of NCA in the 1H NMR spectrum (6.70 ppm) disappeared after ROP (Fig. S2†), indicating the successful synthesis of PAMAM–PPOBLG. The linear polypeptide PPOBLG was synthesized via HMDS-initiated ROP of POBLG–NCA according to published procedures.41 The degree of polymerization (DP) and the polydispersity index (Đ) of PAMAM–PPOBLG (DP = 87, Đ = 1.13) and POBLG (DP = 93, Đ = 1.18) were determined by gel permeation chromatography (Fig. S3†). Subsequently, the cationic, spherical α-helical polypeptide SPP and linear α-helical polypeptide LPP were prepared by conjugating 6-azide hexaguanidine onto the side chains of PAMAM–PPOBLG and PPOBLG by click chemistry (Fig. S4–S6†). 1H NMR analysis showed that the characteristic peaks of the triazole ring at 8.16 ppm and 6-azide hexaguanidine at 3.03 and 4.45 ppm appeared after click chemistry, indicating the successful synthesis of SPP (Fig. S7†). The circular dichroism (CD) spectra of SPP and LPP revealed two negative peaks at 208 nm and 222 nm, indicating that both of them adopted the α-helical structure with the calculated helicity of 61.7% and 68.1%, respectively (Fig. S8†).2.2. Preparation and characterization of SPP/siE2F1 NCs
The siRNA condensation abilities of SPP and LPP were first evaluated by agarose gel electrophoresis. As shown in Fig. 1A, when the SPP/siRNA weight ratio reached 4, siRNA migration was completely retarded, while a higher LPP/siRNA weight ratio of 6 was required to completely retard siRNA migration. It thus indicated that the spherical SPP had stronger siRNA binding affinity than the linear LPP, which was mainly ascribed to the multivalent structure and higher cationic charge density of SPP. Furthermore, at the SPP/siRNA weight ratio ≥ 4, SPP could effectively condense siRNA to form positively charged NCs with the hydrodynamic diameter of 150–200 nm (Fig. 1B). As the weight ratio of SPP/siRNA increased, the particle size of SPP/siRNA NCs gradually decreased while the positive zeta potential gradually increased. When the ratio reached over 10, neither the particle size nor the zeta potential significantly changed, and therefore the optimal SPP/siRNA weight ratio of 10 was identified and applied to the following experiments unless otherwise specified. Consistently, the transmission electron microscopy (TEM) image showed that the particle size of SPP/siRNA NCs was about 175 nm (Fig. 1C).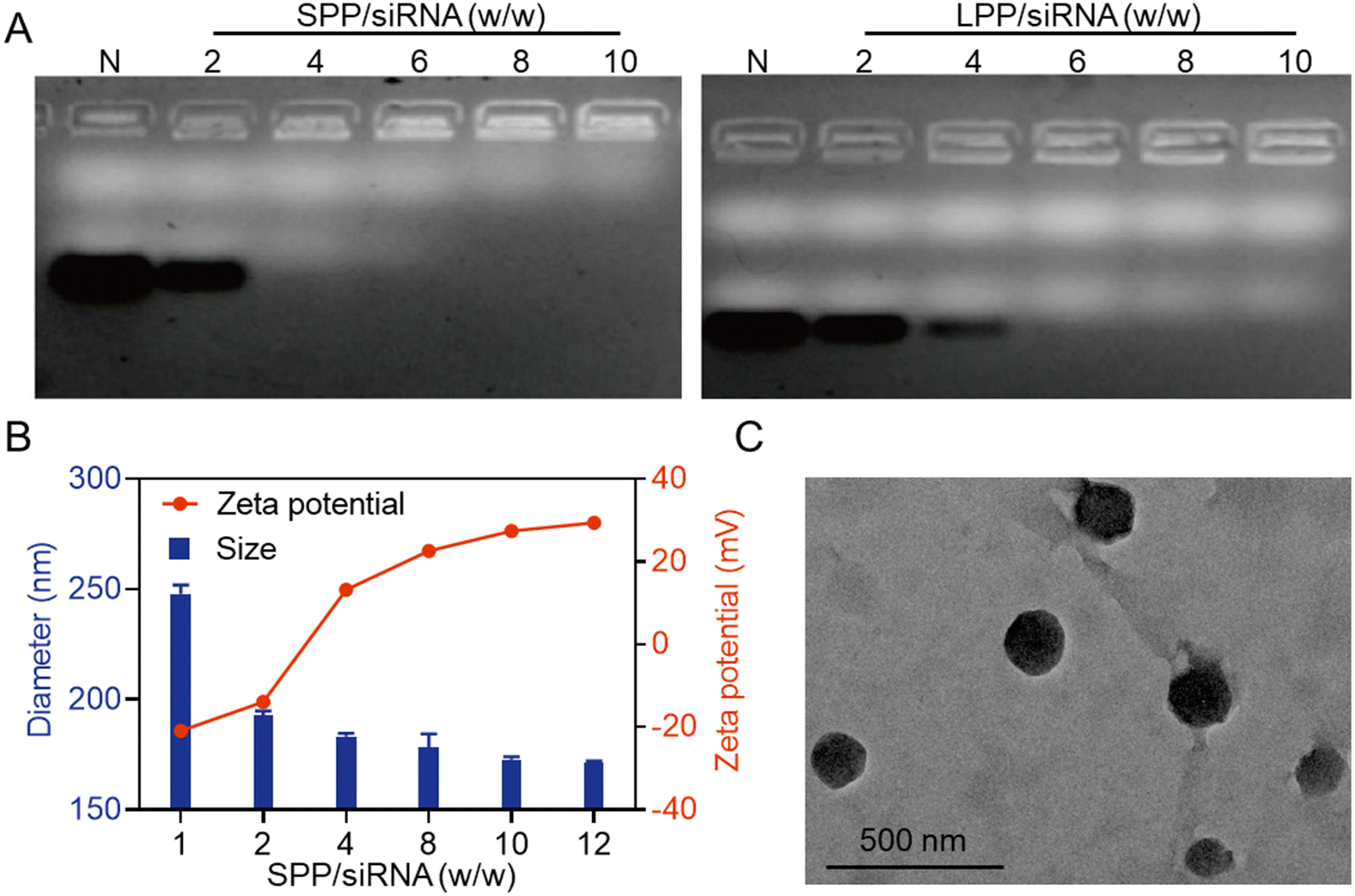 | ||
| Fig. 1 Characterization of SPP/siRNA NCs. (A) siRNA condensation by SPP and LPP at various polypeptide/siRNA weight ratios as evaluated by agarose gel electrophoresis. N represents naked siRNA. (B) Particle size and zeta potential of SPP/siRNA NCs at various SPP/siRNA weight ratios. (C) TEM image of SPP/siRNA NCs (SPP/siRNA = 10, w/w). | ||
Subsequently, the ability of SPP to protect siRNA from degradation by nucleases was investigated. As shown in Fig. S9,† siRNA in SPP/siRNA NCs remained intact after incubation with fresh rat serum (with plenty of nucleases) for 4 h, while in contrast, naked siRNA was mostly degraded. It thus indicated that SPP/siRNA NCs could effectively protect siRNA from hydrolytic degradation via stable complexation of the siRNA cargo.
2.3. In vitro cellular uptake and intracellular kinetics of SPP/FAM–siRNA NCs
The internalization level of polymer/FAM–siRNA NCs in H9C2 cells (rat cardiomyocyte) was first determined by flow cytometry, and SPP mediated a significantly higher cellular uptake level of FAM–siRNA than LPP and 25k PEI (Fig. 2A). Similar results were obtained when the cellular uptake level was determined by spectrofluorimetry (Fig. S10†). It thus indicated that the spherical SPP possessed higher membrane activity than the linear LPP and the commercial transfection reagent 25k PEI to mediate the trans-membrane delivery of siRNA. | ||
| Fig. 2 Cellular internalization and intracellular kinetics of SPP/FAM–siRNA NCs in H9C2 cells. (A) Flow cytometric histograms and the mean fluorescence intensity of H9C2 cells after 4 h incubation with naked FAM–siRNA or various FAM–siRNA-containing NCs (1 μg FAM–siRNA per mL, SPP/siRNA = 10, LPP/siRNA = 10, 25k PEI/siRNA = 5, w/w, n = 3). (B) CLSM images of H9C2 cells after incubation with SPP/FAM–siRNA NCs (SPP/FAM–siRNA = 10, w/w) for different times. Cell nuclei were stained with Hoechst 33258 (blue) and endolysosomes were stained with Lysotracker deep red (red). The co-localization ratios between FAM–siRNA and endolysosomes are listed (n = 10). | ||
Subsequently, the internalization pathway of SPP/FAM–siRNA NCs was studied by pre-treating H9C2 cells with various endocytic inhibitors. Chlorpromazine (CPZ) can inhibit clathrin-mediated endocytosis; genistein (GNT) and methyl-β-cyclodextrin (mβCD) can inhibit caveolae-mediated endocytosis; wortmannin (WTM) can inhibit macropinocytosis.43 As shown in Fig. S11,† only WTM treatment led to pronounced inhibition of the uptake level by ∼50%, while other inhibitors showed a negligible inhibitory effect. It thus indicated that part of the SPP/FAM–siRNA NCs entered the cells through macropinocytosis, while the remaining may enter cells via the non-endocytic mechanism. Upon effective internalization into cells, the SPP/FAM–siRNA NCs could effectively escape from the entrapment by endolysosomes, as evidenced by the obvious separation of green fluorescence (FAM–siRNA) from red fluorescence (endolysosomes stained by Lysotracker deep red) in H9C2 cells after 2 h and 4 h treatment with NCs (Fig. 2B).
2.4. In vitro gene silencing efficiency of SPP/siE2F1 NCs
Consistent with the above-mentioned cellular uptake level and endolysosomal escape capability, SPP/siE2F1 NCs promoted significantly higher gene silencing efficiency (72.8%) than LPP/siE2F1 NCs (50.9%) and 25k PEI/siE2F1 NCs (39.3%, Fig. 3A). It has been reported that the E2F1–miR421–Pink1 signaling pathway plays an important role in cardiomyocyte apoptosis, wherein overexpressed E2F1 activates miR421 expression and inhibits Pink1 transcription, subsequently impairing the mitochondrial function and inducing cardiomyocyte apoptosis.35 Therefore, the expression levels of miR421 and Pink1 were further determined. As expected, miR-421 was downregulated while Pink1 was upregulated after H9C2 cells were transfected with SPP/siE2F1 NCs (Fig. 3A). Furthermore, SPP/siRNA NCs exhibited lower cytotoxicity than LPP/siRNA NCs (Fig. S12†), which may be attributed to the shorter length of each single polypeptide on SPP that can reduce the damage to the cell membrane. | ||
| Fig. 3 SPP/siE2F1 NCs alleviated anoxia-induced mitochondrial damage, ROS burden, and the apoptosis of H9C2 cells. (A) Relative mRNA levels of E2F1, miR421, and Pink1 in H9C2 cells (n = 3). (B) Polarization of the mitochondrial membrane as detected via JC-1 staining. (C) Relative mitochondrial membrane potential (Ψm) as determined by spectrofluorimetry (n = 3). (D) Intracellular ROS level as detected by DCFH-DA staining. (E) Intracellular ATP level as determined by spectrofluorimetry (n = 3). (F) Flow cytometric analysis of H9C2 cells stained with annexin V-FITC/PI. The apoptotic ratio (early apoptosis + late apoptosis) of cells is listed. H9C2 cells were treated with various NCs (SPP/siE2F1 = 10, SPP/siSC = 10, LPP/siE2F1 = 10, 25k PEI/siE2F1 = 5, w/w) for 4 h, incubated in fresh medium for 20 h, and challenged with anoxia for 6 h before the above assessments. | ||
2.5. Inhibition of mitochondrial damage
MIRI causes severe damage to mitochondria in cardiomyocytes, which could be indicated by a decrease in the mitochondrial membrane potential (Ψm).44,45 JC-1 is a mitochondrial dye which aggregates to form a polymer and emits red fluorescence when the mitochondrial membrane potential is high. In contrast, when the mitochondria are damaged to afford a low membrane potential, JC-1 fails to aggregate and thus emits green fluorescence.46 As shown in Fig. 3B, a large amount of green fluorescence appeared in anoxia-treated H9C2 cells, while the green fluorescence was greatly diminished and the red fluorescence was enhanced after treatment with SPP/siE2F1 NCs. Consistently, quantitative analysis showed that SPP/siE2F1 NCs could remarkably inhibit the anoxia-induced mitochondrial membrane depolarization, as evidenced by the recovery of the mitochondrial membrane potential by ∼86% (Fig. 3C).Mitochondria are the main site of ROS and ATP production. Physiologically, ROS are balanced through the redox system in mitochondria. However, such a balance is disrupted upon MIRI-caused mitochondrial damage, thus resulting in ROS burden. In addition, impairment of the mitochondria leads to decreased ATP production.47–49 As shown in Fig. 3D, the intracellular ROS level (green fluorescence after staining with DCFH-DA) in H9C2 cells was increased after anoxia treatment, which however was significantly reduced after SPP/siE2F1 NC treatment. In contrast, the ATP level decreased in anoxia-treated H9C2 cells, while SPP/siE2F1 NCs significantly increased the ATP level (Fig. 3E). These results collectively suggested that SPP/siE2F1 NCs could effectively inhibit the mitochondrial damage caused by MIRI, as a result of the efficient disruption of the E2F1–miR421–Pink1 signaling pathway.
2.6. In vitro anti-apoptosis efficiency of SPP/siE2F1 NCs
Mitochondrial damage will often lead to cell apoptosis.50 Therefore, the anti-apoptosis ability of SPP/siE2F1 NCs in cardiomyocytes was further assessed by flow cytometry. As shown in Fig. 3F, the apoptotic ratio of anoxia-challenged cardiomyocytes decreased to ∼9% after treatment with SPP/siE2F1 NCs, significantly lower than that of anoxia-treated cells (∼45%) and close to that of un-challenged cardiomyocytes (∼5%). Combined with the above findings, it was substantiated that SPP/siE2F1 NCs could effectively decrease mitochondrial damage, reduce ROS production, and restore ATP production in cardiomyocytes after anoxia challenge, ultimately inhibiting cardiomyocyte apoptosis.2.7. In vivo gene silencing and anti-apoptosis efficiency of SPP/siE2F1 NCs
The in vivo gene silencing efficiency of intramyocardially injected SPP/siE2F1 NCs in the ischemic myocardium was first determined by real-time PCR. As shown in Fig. 4A, SPP/siE2F1 NCs effectively decreased the E2F1 mRNA level in the IR-injured myocardium by ∼66%, significantly outperforming LPP/siE2F1 NCs (∼38%) and 25k PEI/siE2F1 NCs (∼23%). Western blot analysis further demonstrated that SPP/siE2F1 NCs can effectively downregulate the E2F1 protein level (Fig. 4B). These results thus demonstrated that SPP could mediate efficient intracellular siE2F1 delivery after intramyocardial administration, thus promoting effective gene silencing in the ischemic myocardium. | ||
| Fig. 4 Intramyocardially injected SPP/siE2F1 NCs (250 μg siE2F1 per kg) mediated effective myocardial E2F1 silencing to alleviate cardiomyocyte apoptosis post MIRI. (A) Relative E2F1 mRNA level in the ischemic myocardium (n = 4). (B) Western blot analysis on the E2F1 protein level in the ischemic myocardium. (C) Representative images of ischemic myocardium sections stained with TUNEL. (D) Apoptosis ratio of the myocardium calculated from (C) (n = 4). | ||
The anti-apoptotic effect of SPP/siE2F1 NCs was explored by TUNEL staining. MIRI led to a large number of apoptotic cardiomyocytes, and the apoptotic ratio reached 38.2% in the ischemic myocardium (Fig. 4C and D). In contrast, the percentage of apoptotic cardiomyocytes (brown) in the myocardium of MIRI rats was significantly reduced after treatment with SPP/siE2F1 NCs, conferring the apoptotic ratio of 12.8%, significantly lower than that in the myocardium treated with LPP/siE2F1 NCs (21.9%) and 25k PEI/siE2F1 NCs (23.5%). It therefore demonstrated that SPP/siE2F1 NCs could effectively inhibit cardiomyocyte apoptosis after the efficient E2F1 silencing.
2.8. Alleviation of histopathological symptoms and reduction of infarct size
To study the recovery of the IR-injured myocardium after NC treatment, heart tissues were first analyzed by hematoxylin–eosin (H&E) staining. As shown in Fig. 5A, cardiomyocytes were disordered and deformed, and a large amount of inflammatory cells infiltrated into the myocardium of IR-injured rats. However, these symptoms were significantly alleviated after the administration of SPP/siE2F1 NCs. Consistently, the expression level of TNF-α, an important pro-inflammatory cytokine, was significantly downregulated in the myocardium sections of MIRI rats treated with SPP/siE2F1 NCs (Fig. S13 and S14†). Moreover, MIRI-induced myocardial fibrosis was notably diminished by SPP/siE2F1 NCs. As revealed by Masson's trichrome (MT) staining wherein the stained muscle fibers appear red while the stained collagen fibers appear blue,51 the fibrotic area of the myocardium in MIRI rats was significantly decreased from 39.1% (saline group) to 12.3% after treatment with SPP/siE2F1 NCs (Fig. 5B and D).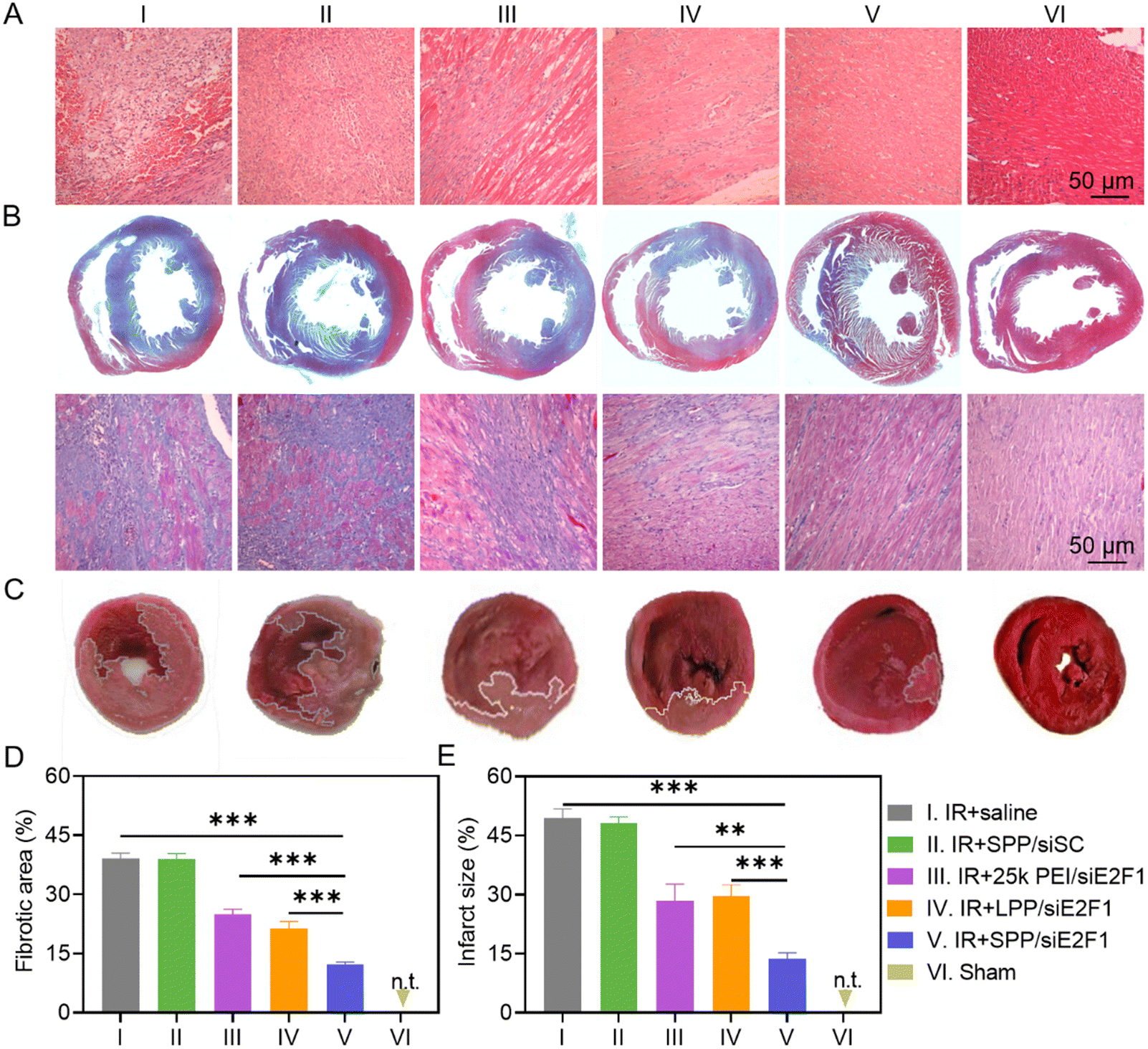 | ||
| Fig. 5 SPP/siE2F1 NCs (250 μg siE2F1 per kg) alleviated myocardial damage and reduced the infarct size in rats after MIRI. (A) Representative images of myocardium sections stained with H&E (A), MT (B), and TTC (C). (D) Fibrotic area calculated from MT staining images (n = 4). (E) Infarct size of the myocardium calculated from the TTC staining images (n = 4). Histopathology analysis and infarct size measurement were performed at 7 d post NC administration. Rats with no MIRI served as the sham group. | ||
Subsequently, the myocardial infarct size in MIRI rats was determined by 2,3,5-triphenyte-trazoliumchloride (TTC) staining. In normal cells, abundant dehydrogenase can reduce TTC into insoluble red triphenylformazan, whereas the dehydrogenase activity is decreased in dead or inactive cardiomyocytes that cannot be stained or can only be slightly stained.52 As shown in Fig. 5C and E, the myocardial infarct area of MIRI rats after treatment with SPP/siE2F1 NCs (13.6%, light red area) was significantly smaller than that of MIRI rats treated with 25k PEI/siE2F1 NCs (28.4%), LPP/siE2F1 NCs (29.7%), and saline (49.4%), indicating that SPP/siE2F1 NCs could effectively reduce the infarct size of MIRI rats, which could be attributed to the efficient silencing of E2F1, inhibition of mitochondrial damage, and reduction of cardiomyocyte apoptosis.
2.9. Recovery of cardiac function
The inhibition of inflammatory cell infiltration, myocardial fibrosis, and ischemia could improve the systolic function of the left ventricle, thus contributing to the recovery of cardiac function. Cardiac function of MIRI rats after treatment with SPP/siE2F1 NCs was assessed by echocardiography on day 3 after MIRI. As shown in Fig. 6A and S15, rats treated with SPP/siE2F1 NCs showed less change in the left ventricular function and insignificant ventricular dilatation when compared with sham rats without MIRI. The values of the ejection fraction (EF, %) and fraction shortening (FS, %) of rats treated with SPP/siE2F1 NCs were also close to those of sham rats yet significantly higher than those of MIRI rats treated with LPP/siE2F1 NCs or 25k PEI/siE2F1 NCs (Fig. 6B and C). It therefore demonstrated that via efficient inhibition of cardiomyocyte apoptosis and myocardium inflammation as a result of E2F1 silencing, SPP/siE2F1 NCs could effectively restore the systolic function of rat hearts post MIRI.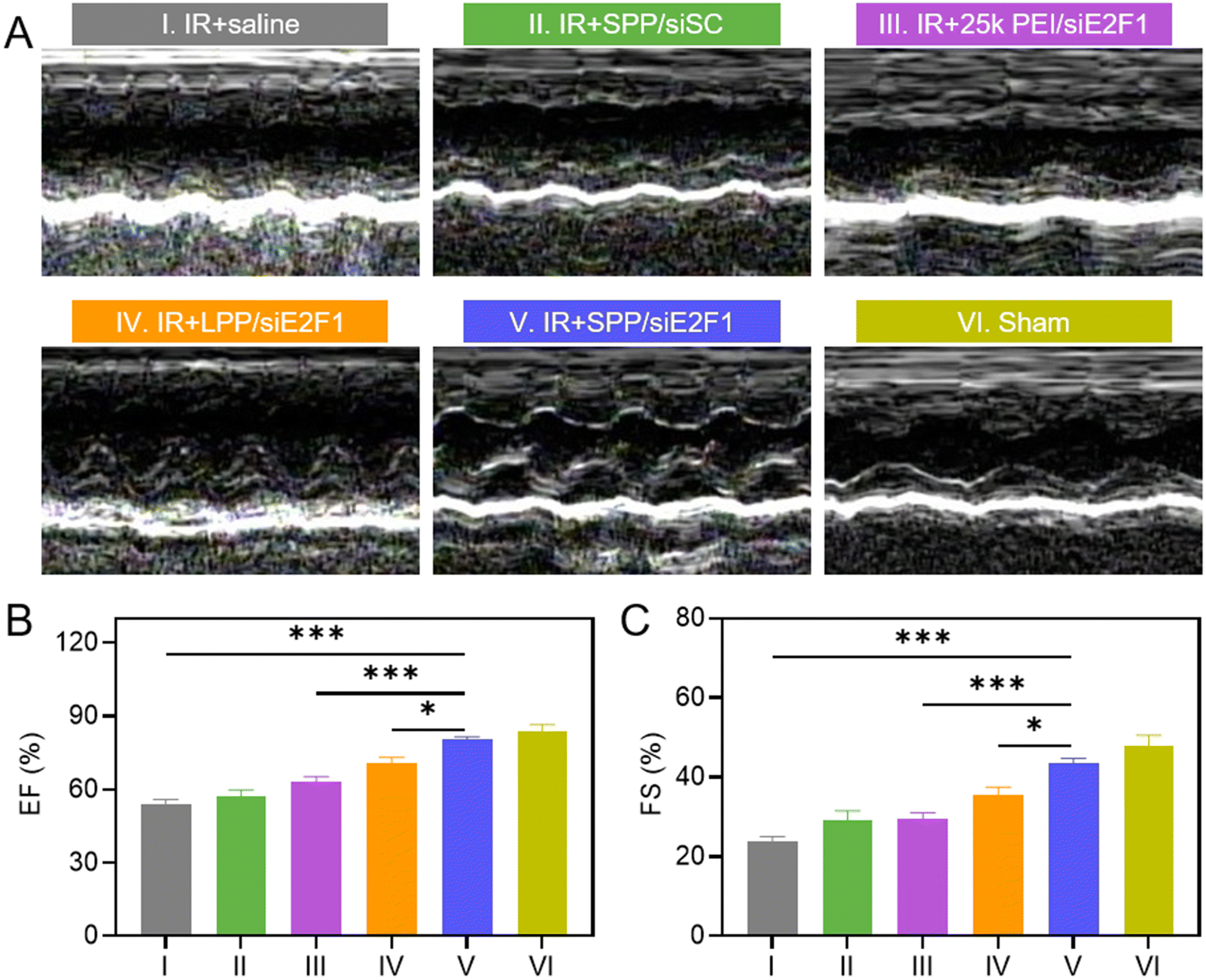 | ||
| Fig. 6 SPP/siE2F1 NCs (250 μg siE2F1 per kg) recovered cardiac function against MIRI. (A) Echocardiographs of MIRI rats post treatment with saline or various NCs. Ejection fraction (EF, B) and fraction shortening (FS, C) of the left ventricle as determined by echocardiography (n = 4). Rats with no MIRI served as the sham control. Echocardiographic analysis of rats was performed at 3 d post NC administration. | ||
2.10. Biocompatibility of SPP/siE2F1 NCs
The in vivo biocompatibility of SPP/siE2F1 NCs was examined after intramyocardial injection (250 μg siE2F1 per kg) into male Sprague-Dawley (SD) rats as described above for the efficacy study. Compared to saline-treated rats, SPP/siE2F1 NC-treated rats showed negligible abnormalities in the context of representative hematological parameters and biochemical parameters (Fig. S16†). In addition, in the H&E-stained cross-sections of the major organs (heart, liver, spleen, lungs, and kidneys), no necrosis, inflammation, edema, or other pathological symptoms were detected (Fig. S17†). These results suggested the desirable biocompatibility of SPP/siE2F1 NCs after intramyocardial injection.3. Conclusion
In this study, a spherical α-helical polypeptide SPP with potent membrane activity was developed as an effective vehicle for siRNA delivery into cardiomyocytes. SPP exhibited higher siRNA binding affinity, higher trans-membrane delivery efficiency, and lower cytotoxicity than linear LPP, mainly attributed to its spherical topology with multivalent presentation of the rod-like α-helix. After intramyocardial injection, SPP/siE2F1 NCs led to pronounced E2F1 silencing in the myocardium, thereby interrupting the E2F1–miR421–Pink1 signaling pathway to reduce mitochondrial damage, relieve ROS burden, and inhibit the apoptosis of cardiomyocytes, finally rescuing the myocardium from MIRI and restoring the cardiac function. This study provides an effective polymeric solution to overcome the membrane barrier against nucleic acid delivery, and it provides a promising genetic approach for the management of MIRI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (81903068, 82172076, and 51873142), the Jiangsu Key Research and Development Plan (Social Development) Project (BE2020653 and BE2021642), the Suzhou Science and Technology Development Project (SYS2019072), the Collaborative Innovation Center of Suzhou Nano Science & Technology, the 111 project, the National Tutorial System Training Project for Young Talents of the Suzhou Health System (Qngg2021010), the Suzhou Key Laboratory of Nanotechnology and Biomedicine, and the Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.References
- G. W. Reed, J. E. Rossi and C. P. Cannon, Lancet, 2017, 389, 197–210 CrossRef.
- D. Zhao, J. Liu, M. Wang, X. Zhang and M. Zhou, Nat. Rev. Cardiol., 2019, 16, 203–212 CrossRef PubMed.
- M. Mahmoudi, M. Yu, V. Serpooshan, J. C. Wu, R. Langer, R. T. Lee, J. M. Karp and O. C. Farokhzad, Nat. Nanotechnol., 2017, 12, 845–855 CrossRef CAS PubMed.
- G. Heusch and B. J. Gersh, Eur. Heart J., 2017, 38, 774–784 CAS.
- S. R. Mehta, D. A. Wood, R. F. Storey, R. Mehran, K. R. Bainey, H. Nguyen, B. Meeks, G. Di Pasquale, J. López-Sendón, D. P. Faxon, L. Mauri, S. V. Rao, L. Feldman, P. G. Steg, Á. Avezum, T. Sheth, N. Pinilla-Echeverri, R. Moreno, G. Campo, B. Wrigley, S. Kedev, A. Sutton, R. Oliver, J. Rodés-Cabau, G. Stanković, R. Welsh, S. Lavi, W. J. Cantor, J. Wang, J. Nakamya, S. I. Bangdiwala and J. A. Cairns, N. Engl. J. Med., 2019, 381, 1411–1421 CrossRef PubMed.
- D. M. Yellon and D. J. Hausenloy, N. Engl. J. Med., 2007, 357, 1121–1135 CrossRef CAS PubMed.
- J. Gao, Y. Song, Q. Wang, J. Chen, Q. Li, H. Tan, W. Yakufu, N. Zhang, S. Li, J. Zhang, H. Yang, Z. Wang, Z. Huang and J. Ge, Chem. Eng. J., 2022, 446, 136960 CrossRef CAS.
- K. Raedschelders, D. M. Ansley and D. D. Y. Chen, Pharmacol. Ther., 2012, 133, 230–255 CrossRef CAS PubMed.
- H. Tan, Y. Song, J. Chen, N. Zhang, Q. Wang, Q. Li, J. Gao, H. Yang, Z. Dong, X. Weng, Z. Wang, D. Sun, W. Yakufu, Z. Pang, Z. Huang and J. Ge, Adv. Sci., 2021, 8, 2100787 CrossRef CAS PubMed.
- J. Shi, Y. Yang, N. Yin, C. Liu, Y. Zhao, H. Cheng, T. Zhou, Z. Zhang and K. Zhang, Small Methods, 2022, 6, 2101158 CrossRef CAS.
- X. Sun, R. Gao, W. Li, Y. Zhao, H. Yang, H. Chen, H. Jiang, Z. Dong, J. Hu, J. Liu, Y. Zou, A. Sun and J. Ge, Bioact. Mater., 2021, 6, 2058–2069 CrossRef CAS PubMed.
- X. Han, R. Wang, X. Song, F. Yu, C. Lv and L. Chen, Biomaterials, 2018, 156, 134–146 CrossRef CAS PubMed.
- E. Angelis, P. Zhao, R. Zhang, J. I. Goldhaber and W. R. MacLellan, J. Mol. Cell. Cardiol., 2011, 51, 919–926 CrossRef CAS PubMed.
- M. G. Ertosun, F. Z. Hapil and O. Z. E. S. Osman Nidai, Cytokine Growth Factor Rev., 2016, 31, 17–25 CrossRef PubMed.
- K. Wang, T. An, L. Y. Zhou, C. Y. Liu, X. J. Zhang, C. Feng and P. F. Li, Cell Death Differ., 2015, 22, 743–754 CrossRef CAS.
- K. Wang, L. Y. Zhou, J. X. Wang, Y. Wang, T. Sun, B. Zhao, Y. J. Yang, T. An, B. Long, N. Li, C. Y. Liu, Y. Gong, J. N. Gao, Y. H. Dong, J. Zhang and P. F. Li, Nat. Commun., 2015, 6, 7619 CrossRef CAS.
- Y. Guo, J. Qin, Q. Zhao, J. Yang, X. Wei, Y. Huang, M. Xie, C. Zhang and Y. Li, Adv. Sci., 2022, 9, 2105875 CrossRef CAS PubMed.
- Y. Wang, M. Hou, S. Duan, Z. Zhao, X. Wu, Y. Chen and L. Yin, Bioact. Mater., 2022, 17, 320–333 CrossRef CAS PubMed.
- Y. Chen, S. Zhi, W. Liu, J. Wen, S. Hu, T. Cao, H. Sun, Y. Li, L. Huang, Y. Liu, P. Liang and J. Huang, Small Methods, 2020, 4, 2000309 CrossRef CAS.
- Y. Dong, D. J. Siegwart and D. G. Anderson, Adv. Drug Delivery Rev., 2019, 144, 133–147 CrossRef CAS.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS.
- M. P. Stewart, A. Sharei, X. Ding, G. Sahay, R. Langer and K. F. Jensen, Nature, 2016, 538, 183–192 CrossRef CAS PubMed.
- B. Kim, J. H. Park and M. J. Sailor, Adv. Mater., 2019, 31, 1903637 CrossRef CAS.
- J. Conde, A. Ambrosone, Y. Hernandez, F. Tian, M. McCully, C. C. Berry, P. V. Baptista, C. Tortiglione and J. M. De La Fuente, Nano Today, 2015, 10, 421–450 CrossRef CAS.
- X. L. Feng, F. T. Lü, L. B. Liu and S. Wang, Chin. Sci. Bull., 2013, 58, 2762–2766 CrossRef CAS.
- T. Zhang, Y. Huang, X. Ma, N. Gong, X. Liu, L. Liu, X. Ye, B. Hu, C. Li, J. H. Tian, A. Magrini, J. Zhang, W. Guo, J. F. Xing, M. Bottini and X. J. Liang, Nano Lett., 2018, 18, 6301–6311 CrossRef CAS PubMed.
- N. Feng, L. Liang, M. Fan, Y. Du, C. Chen, R. Jiang, D. Yu, Y. Yang, M. Zhang, L. Deng, X. Li, N. Geng, M. Xian, Q. Qin, X. Li, Q. Tan, F. Luo, F. Song, H. Qi, Y. Xie and F. Guo, ACS Nano, 2021, 15, 15874–15891 CrossRef CAS PubMed.
- C. A. Hong, A. A. Eltoukhy, H. Lee, R. Langer, D. G. Anderson and Y. S. Nam, Angew. Chem., 2015, 127, 6844–6848 CrossRef.
- H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef CAS PubMed.
- X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., 2014, 126, 12016–12021 CrossRef.
- Y. Dong, T. Yu, L. Ding, E. Laurini, Y. Huang, M. Zhang, Y. Weng, S. Lin, P. Chen, D. Marson, Y. Jiang, S. Giorgio, S. Pricl, X. Liu, P. Rocchi and L. Peng, J. Am. Chem. Soc., 2018, 140, 16264–16274 CrossRef CAS PubMed.
- J. Ren, P. Hu, E. Ma, X. Zhou, W. Wang, S. Zheng and H. Wang, Appl. Mater. Today, 2022, 27, 101445 CrossRef.
- W. Y. Tong, M. Alnakhli, R. Bhardwaj, S. Apostolou, S. Sinha, C. Fraser, T. Kuchel, B. Kuss and N. H. Voelcker, J. Nanobiotechnol., 2018, 16, 38 CrossRef PubMed.
- E. Xu, W. M. Saltzman and A. S. Piotrowski-Daspit, J. Controlled Release, 2021, 335, 465–480 CrossRef CAS PubMed.
- I. M. S. Degors, C. Wang, Z. U. Rehman and I. S. Zuhorn, Acc. Chem. Res., 2019, 52, 1750–1760 CrossRef CAS PubMed.
- H. He, N. Zheng, Z. Song, K. H. Kim, C. Yao, R. Zhang, C. Zhang, Y. Huang, F. M. Uckun, J. Cheng, Y. Zhang and L. Yin, ACS Nano, 2016, 10, 1859–1870 CrossRef CAS PubMed.
- J. Yan, X. Liu, F. Wu, C. Ge, H. Ye, X. Chen, Y. Wei, R. Zhou, S. Duan, R. Zhu, Y. Zheng and L. Yin, Adv. Mater., 2022, 34, 2109517 CrossRef CAS.
- Y. Deng, Y. Zhou, Q. Liang, C. Ge, J. Yang, B. Shan, Y. Liu, X. Zhou and L. Yin, Adv. Funct. Mater., 2021, 31, 2101033 CrossRef CAS.
- Y. Liu and L. Yin, Adv. Drug Delivery Rev., 2021, 171, 139–163 CrossRef CAS PubMed.
- C. Ge, J. Yang, S. Duan, Y. Liu, F. Meng and L. Yin, Nano Lett., 2020, 20, 1738–1746 CrossRef CAS.
- F. Li, Y. Li, Z. Zhou, S. Lv, Q. Deng, X. Xu and L. Yin, ACS Appl. Mater. Interfaces, 2017, 9, 23586–23601 CrossRef CAS PubMed.
- Q. Liang, F. Li, Y. Li, Y. Liu, M. Lan, S. Wu, X. Wu, Y. Ji, R. Zhang and L. Yin, Biomater. Sci., 2019, 7, 3717–3728 RSC.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- Y. Zhang, A. Khalique, X. Du, Z. Gao, J. Wu, X. Zhang, R. Zhang, Z. Sun, Q. Liu, Z. Xu, A. C. Midgley, L. Wang, X. Yan, J. Zhuang, D. Kong and X. Huang, Adv. Mater., 2021, 33, 2006570 CrossRef CAS.
- W. Marin, D. Marin, X. Ao and Y. Liu, Int. J. Mol. Med., 2021, 47, 485–499 CrossRef CAS PubMed.
- Y. Li, J. Dang, Q. Liang and L. Yin, Biomaterials, 2019, 209, 138–151 CrossRef CAS PubMed.
- G. S. Shadel and T. L. Horvath, Cell, 2015, 163, 560–569 CrossRef CAS PubMed.
- E. Bertero and C. Maack, Circ. Res., 2018, 122, 1460–1478 CrossRef CAS.
- J. Zhang, C. Li, Q. Xue, X. Yin, Y. Li, W. He, X. Chen, J. Zhang, R. L. Reis and Y. Wang, Small Methods, 2021, 5, 2100539 CrossRef CAS.
- X. Luo, X. Gong, L. Su, H. Lin, Z. Yang, X. Yan and J. Gao, Angew. Chem., Int. Ed., 2021, 60, 1403–1410 CrossRef CAS PubMed.
- A. H. Cohen, Am. J. Clin. Pathol., 1976, 65, 631–643 CrossRef CAS PubMed.
- M. Hou, X. Wu, Z. Zhao, Q. Deng, Y. Chen and L. Yin, Acta Biomater., 2022, 143, 344–355 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01075e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |