
Physical mixture of a cyclic lipopeptide vaccine induced high titres of opsonic IgG antibodies against group A streptococcus†
Harrison Y. R.
Madge‡
 a,
Wenbin
Huang‡
a,
Lachlan
Gilmartin
a,
Berta
Rigau-Planella
a,
Waleed M.
Hussein
a,
Zeinab G.
Khalil
b,
Prashamsa
Koirala
a,
Viviene S.
Santiago
b,
Robert J.
Capon
b,
Istvan
Toth
abc and
Rachel J.
Stephenson
*a
a,
Wenbin
Huang‡
a,
Lachlan
Gilmartin
a,
Berta
Rigau-Planella
a,
Waleed M.
Hussein
a,
Zeinab G.
Khalil
b,
Prashamsa
Koirala
a,
Viviene S.
Santiago
b,
Robert J.
Capon
b,
Istvan
Toth
abc and
Rachel J.
Stephenson
*a
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane 4072, Australia. E-mail: r.stephenson@uq.edu.au
bInstitute for Molecular Bioscience, The University of Queensland, St Lucia, Queensland 4072, Australia
cSchool of Pharmacy, The University of Queensland, Brisbane 4072, Australia
First published on 26th November 2021
Abstract
Untreated or reoccurring group A Streptococcus (GAS) infection can lead to a number of post-infection complications, including rheumatic heart disease. There is no licenced vaccine for the treatment or prevention of GAS infection. We identified that a cyclic decapeptide plays a significant positive influence on the adjuvant activity of several lipid-antigen mixtures. Here, three synthetic vaccine components were synthesised: (1) J8-PADRE represents the GAS B cell antigen (J8) conjugated to the universal T helper epitope (PADRE); (2) a synthetic toll like receptor 2 (TLR2) ligand based on a C16 alkyl chain lipid moiety; and (3) a cyclic carrier deca-peptide. Previously, through structure-immune activity investigations, it was observed that a physical mixture of these three components had significantly higher IgG immune responses when compared to a fully conjugated vaccine construct. Expanding the scope of this structure–activity investigation, we show that the presence of the cyclic peptide is required for the induction of a strong, balanced Th1/Th2 immune response when compared with lipid and antigen only, and cyclic lipopeptide plus B/T cell antigen physical mixtures.
Introduction
The progress of modern vaccine research, specifically the development of peptide-based vaccine technology, is predominantly governed by the lack of safe and effective adjuvants.1,2 Many modern vaccine designs, including subunit synthetic-peptide or protein vaccines, have been developed to circumvent issues with traditional whole organism vaccines.3 Whole organism vaccines are based on either live attenuated or killed pathogens, and are still the most common form of vaccination employed today.3 However, these vaccines suffer from several disadvantages, ranging from possible autoimmune reactions from the inclusion of unnecessary deleterious biological components, to more practical issues, including a higher cost of production, and the need for cold chain transportation and live pathogen culturing.3 Further, at the forefront of vaccine research are several pathogens in which whole organism vaccines are unsuitable due to both a lack of efficacy and safety (e.g. GAS, cancer and human immune deficiency virus).3–5Subunit vaccines, specifically synthetic peptide vaccines, contain only the minimal antigenic components from the pathogenic microorganism. Subunit vaccines can be chemically synthesised thereby they are completely characterisable compounds. Application of subunit vaccines reduces potential side effects and not only allows protection against organisms which contain potentially toxic components, but also allows for the targeting of highly neutralising or conserved epitopes avoiding induction of cursory non-protective immune responses.6 However, these short peptide epitopes (10–50 amino acids in length) do not possess intrinsic immunogenic properties and therefore alone do not induce suitable immune responses.7 As such, current research has focused on the development of delivery systems and next generation adjuvants to improve the immunogenicity of small antigenic epitopes. The current array of adjuvants approved for human use is generally dominated by aluminium salts, including aluminium hydroxide, aluminium phosphate, and amorphous aluminium hydroxy phosphate sulphate.8 These aluminium-based adjuvants were the first to be used in humans, after the discovery of their adjuvant properties in 1926, and in 1932 they were included in a commercial vaccine cosctruct.9 They would then continue to be the only adjuvants used in licensed vaccines for approximately 70 years.9
The adjuvant concept was first proposed at the beginning of the 20th century when vaccines comprised of diphtheria and tetanus purified toxoids failed to produce an effective immune response.8 More recent adjuvants approved for human use include, oil-in-water emulsions (e.g. MF59 and AS03), AS04 (3′-O-deacylated monophosphoryl lipid A [MPLA] adsorbed onto aluminium salts), CpG oligodeoxynucleotides (ODN) and AS01, MPLA, and saponin QS-21 formulated into liposomes.10 However, all these examples are either approved for only certain vaccines and formulations, or are only suitable for certain antigen classes.5 To date, most commercially available adjuvants, specifically aluminium-based adjuvants, are particularly ineffective at stimulating immune responses to peptide-based antigens.5,11,12
An elegant solution for the stimulation of an immune response against short peptides has been the display of peptide epitopes on self-adjuvant delivery systems incorporating synthetic toll like receptor ligands.13 These systems mimic naturally occurring pathogen associated molecular patterns, allowing the vaccine to target antigen presenting cells.13 The use of lipoamino acid-containing delivery systems chemically conjugated to minimum antigenic B cell epitopes combined with universal T helper epitopes (necessary to achieve a balanced Th1/Th2 response and for T cell activation of B cells) has been well established. These lipid-based delivery systems are demonstrated to induce high antibody titres and protection against disease challenge for a number of pathogenic organisms (e.g. GAS, malaria and hookworm).14–17 The latter contention has been that chemical conjugation between all vaccine components (B cell epitope, T helper epitope and lipid adjuvant) was necessary to induce the desired immune response.3,18 However, during our recent study on the structure–activity relationship of self-adjuvant cyclic lipopeptides for the delivery of conserved GAS peptide epitopes, we observed high levels of opsonic IgG induced by a physical mixture of vaccine components, significantly higher than the best performing conjugated vaccine groups.19
In the present study we aimed to investigate the potential of a cyclic lipopeptide adjuvant by performing immunological analysis of varying physical mixture compositions in order to identify effective adjuvants co-administered with peptide antigens. Adjuvants capable of performing suitable immune activation and inducing immune responses to co-administered antigens, without chemical conjugation, present several benefits over systems requiring conjugation. For example, the conjugation of certain peptide antigens to the delivery system could potentially alter the secondary structure of the epitope, which in some cases (e.g. GAS J8 epitope) are essential for the induction of antibodies which recognise the natural pathogen. Further, certain synthetic processes used for the production of these conjugated systems often require sophisticated multiple conjugation strategies, leading to by-products and/or low reaction yields.20 The benefits of physically-mixed adjuvants, include their ability to be used across a wide range of vaccine products and antigens, reducing the number of synthetic steps affording an easier and cheaper synthesis. Further, the capacity to alter the adjuvant antigen ratio has been shown to have a significant effect on the induced immune response.21,22
Here, we focused on GAS as a model organism, with no vaccine currently approved. Data published in 2017 reported the global incidence of GAS infection at approximately 2–4 people per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in developed countries.23 This number was shown to rise as high as 20 times (12–80 people per 100000) within some indigenous populations in both the USA and Australia.24,25 Worldwide, as of 2015, there were over 300000 deaths attributed to GAS infection primarily through post-infectious complications, such as rheumatic fever and rheumatic heart disease.26
000 in developed countries.23 This number was shown to rise as high as 20 times (12–80 people per 100000) within some indigenous populations in both the USA and Australia.24,25 Worldwide, as of 2015, there were over 300000 deaths attributed to GAS infection primarily through post-infectious complications, such as rheumatic fever and rheumatic heart disease.26
We examined the use of the same building blocks from our previous work,19 the combined conserved GAS B cell antigen and universal T helper epitope J8-PADRE, a known toll like receptor 2 (TLR2) ligand (C16 lipoamino acid),27 and cyclic decapeptide carrier. These vaccine building blocks were synthesised and conjugated using standard solid phase peptide chemistry, copper catalysed click reactions and orthogonal protecting strategies. These compounds displaying differing degrees of conjugation were then physically mixed to afford the vaccine libraries (Fig. 1) which were assessed in vivo for their ability to produce GAS antigen-specific protective immune responses by examining the levels of induced opsonic IgG against several clinical GAS isolates. Two separate immunological assessments were carried out under the same experimental conditions (Study 1 and Study 2; Fig. 1) with Study 1 assessing the structure–activity relationship of lipid and antigen conjugation to the cyclic decapeptide, and Study 2 investigating the role of lipid conjugation and cyclic decapeptide.
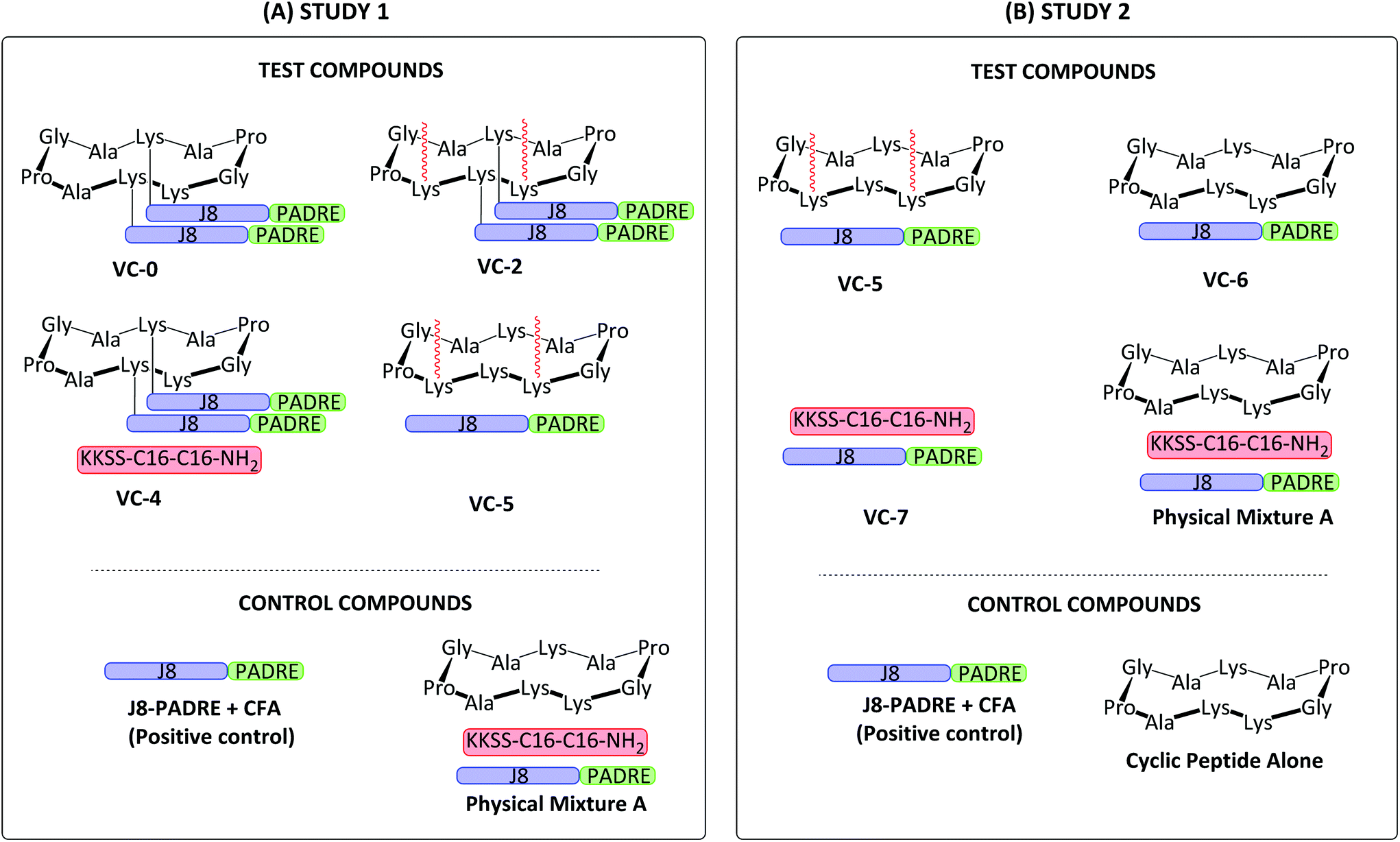 | ||
| Fig. 1 Schematic structure of Study 1 (A) and Study 2 (B) vaccine candidates assessed in vivo. Study 1 investigated the structure–activity relationship of lipid and antigen conjugation to the cyclic decapeptide. Study 2 investigated the role of lipid conjugation and cyclic decapeptide. GAS J8 B cell antigen (QAEDKVKQSREAKKQVEKALKQLEDKVQ; blue); universal T helper epitope PADRE (AKFVAAWTLKAAA; green); synthetic lipid (KKSS-C16-C16-NH2; red).19 | ||
Materials & methods
General procedures
Peptide synthesis grade N,N′-dimethylformamide (DMF), trifluoracetic acid (TFA), piperidine and high performance liquid chromatography (HPLC) grade acetonitrile (MeCN) were purchased from Merck Biosciences (Kilsyth, VIC, Australia). Fluorenylmethyloxycarbonyl (Fmoc)-protected L-amino acids, Rink amide MBHA resin (100–200 mesh, 0.52 mmol g−1 loading) and 2-chlorotrityl chloride (2-CTC) resin (100–200 mesh, 1% DVB) were purchased from Novabiochem (Melbourne, VIC, Australia). HATU was purchased from Mimotopes (Mulgrave, VIC, Australia). 4-Pentynoic acid was purchased from Flurochem Ltd (Graphite Way, Hadfield, UK). Goat anti-mouse IgG (H + L)-horseradish peroxidase (HRP) conjugated was purchased from Bio-Rad Laboratories (Gladesville, NSW, Australia). PBS tablets were purchased from Medicago (Uppsala, Sweden). Tween 20 (polyoxyethylene(20)sorbitan monolaurate) was purchased from Ajax Finechem (Brisbane, QLD, Australia). O-Phenylenediamine dihydrochloride (OPD) peroxidase substrate (SIGMAFAST OPD tablet) was purchased from Sigma-Aldrich (St. Louis, USA). Skim milk powder was purchased from Merck Biosciences (Kilsyth, VIC, Australia). Horse blood Todd–Hewitt broth, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dulbecco's modified Eagle's medium (DMEM), sodium dodecyl sulphate (SDS), L-glutamine, penicillin, streptomycin and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Scoresby, VIC, Australia). All remaining reagents were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). Dry DCM was prepared by drying over 3 Å molecular sieves overnight.Purification was performed using reverse phase (RP)-HPLC on either a Shimadzu semi-preparative (flow rate: 5 mL min−1) system equipped with a CBM-20A controller, LC-20AT pump, SIL-10A auto injector, SPD-20A UV/Vis detector set to a wavelength of 214 nm, FRC-10A fraction collector with a Vydac C4 column (Altima 5 μm, 22 × 250 mm); or a Shimadzu preparative (flow rate: 20 mL min−1) system equipped with a CBM-20A controller, LC-20AP pumps, SPD-20A UV/Vis detector set to a wavelength of 214 nm and a Vydac C18 (Altima 5 μm, 22 × 250 mm) or C4 (Protein 214TP1022) column. A gradient of solvent A (TFA:H2O; 100:0.1) and solvent B (MeCN:H2O:TFA; 90:10:0.1) was used. Purity was >95% for final vaccine components as measured by analytical RP-HPLC. Analytical RP-HPLC was performed using a Shimadzu RP-HPLC with an LC-20AB pump, a SIL-20AC HT autosampler and an SPD-M10A detector set to a wavelength of 214 nm with a Vydac C18 (218TP; 5 μm, 4.6 × 250 mm) or C4 (214TP; 5 μM, 4.6 × 250 mm) column. A 1 mL min−1 gradient of solvent A and solvent B was used. Electrospray ionisation mass spectrometry (ESI-MS) was performed on a PerkinElmer-Sciex API3000 quadrupole mass spectrometer with Analyst 1.4 software (Applied Biosystems/MDS Sciex, Toronto, Canada), operating with a constant flow (1:1) of solvent C (0.1% acetic acid in water) and solvent D (MeCN:H2O:acetic acid; 90:10:0.1) at a rate of 0.5 mL min−1. NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer. Dynamic light scattering (DLS) measurements were taken using a Zetasizer Nano ZP instrument (Malvern Instrument, UK) with Malvern Zetasizer Analyser 6.2 software. Transmission electron microscopy (TEM) images were captured on a JEM-1010 TEM (HT7700 Exalens, HITACHI Ltd, JEOL Ltd, Japan) operated at 80 kV, using negative staining. Circular dichroism spectra were obtained on a Jasco J-710 CD spectrometer (Jasco Corp., Japan). Experimental procedures were approved by the University of Queensland Animal Ethics Committee (AEC number: SCMB/AIBN/069/17). Mice were obtained from the Animal Resource Centre (Perth, Western Australia) and housed at the AIBN UQBR facility (Brisbane, QLD, Australia).
Synthesis of vaccine building blocks
The J8-PADRE GAS antigen, BB1–BB4 building blocks and conjugated vaccine compounds (VC-0 and VC-2, Fig. 1) were synthesised as previously described.19
Azido-functionalised J8-PADRE (N3-QAEDKVKQSREAKKQVEKALKQLEDKVQAKFVAAWTLKAAA) was synthesized by standard Fmoc SPPS on Rink amide MBHA resin using standard 20% piperidine in DMF deprotection (2 × 10 min, RT) and 4.2 eq. HATU/6.2 eq. DIPEA (2 × 30 min, RT) mediated coupling steps. Azido acetic acid (4.2 eq.) was coupled at the conclusion of the J8-PADRE sequence. Following addition of azido acetic acid, the peptide was worked up using the standard protocol: washing of the resin thoroughly with DMF, MeOH and DCM and transferred to a desiccator overnight. The dried resin-peptide was treated with a cleavage solution of TFA:TIPS:H2O (95:2.5:2.5) and stirred at RT for 3 h. The crude peptide was precipitated with ice-cold diethyl ether and isolated by centrifugation. The crude peptide was dissolved in MeCN:H2O (30:70) and lyophilised before being purified using preparative RP-HPLC. Pure fractions (as determined by ESI-MS and analytical RP-HPLC) were collated and lyophilised. Characterisation was performed by analytical RP-HPLC and ESI-MS (ESI, Fig. S1†).
Azido-functionalised J8-PADRE R t = 18.8 min (C18 column, 0–100% solvent B, 30 min). Molecular weight (C207H347N63O61): 4694.43 g mol−1. ESI-MS: [M + 3H]+3m/z 1566.4 (calcd: 1565.8), [M + 4H]+4m/z 1175.2 (calcd: 1174.6), [M + 5H]+5m/z 939.9 (calcd: 939.9), [M + 6H]+6m/z 784.0 (calcd: 784.0), [M + 7H]+7m/z 671.5 (calcd: 671.5). Yield: 165.7 mg, 33%.
BB1 (KKSS-C16-C16-NH2, Fig. S2†) was synthesised by the same standard Fmoc SPPS on Rink amide MBHA resin as described for Azido-functionalised J8-PADRE, differing only in incorporation of C16 lipoamino acids. Here, 2 (Dde protected C16 lipomanio acid)28 was preactivated and coupled to the resin using HATU and DIPEA as described previously. Dde removal was completed by treatment of the resin with a 2% hydrazine in DMF solution (3 × 10 min). Following cleavage and work up, the dried crude lipo-peptide was purified by preparative RP-HPLC and was determined to be >95% pure by analytical HPLC (ESI, Fig. S2†).
BB1 (KKSS-C16-C16-NH2) Rt = 20.9 min (C4 column, 0–100% solvent B, 30 min). Molecular weight (C50H99N9O8): 954.4 g mol−1. ESI-MS: [M + 1H]+1m/z 955.0 (calcd: 955.4). Yield: 10.6 mg, 48%.
Cyclic peptide alkynes BB2 and BB3 (Scheme 1) were synthesised following our previously published protocol.19 Briefly, BB2 and BB3 were first synthesised in their linear confirmations (BB2-L: NH2-K1APGA1APG-OH and BB3-L: NH2-K1KPGA1APG-OH, respectively) by Fmoc SPPS on 2-CTC resin (0.5 mmol scale). Following 2-CTC resin pre-activation with the first amino acid of the sequence (Fmoc-Gly-OH) and capping of unreacted chloride groups, the remaining sequence was coupled identically to those on rink amide resin. As described in our previous publication,19 the cyclic peptides were alkyne-functionalised by the addition of Fmoc-lysine-[N-4-pentynoic acid]-OH19 (1, Scheme 1) during SPPS of the peptide backbone. As per peptides synthesised on Rink amide resin, completed 2-CTC resin-bound peptides were washed thoroughly with DMF, MeOH, and DCM, followed by drying in vacuo. Cleavage of 2-CTC bound peptides were achieved by treatment of the dried resin-peptides with acetic acid/trifluoroethanol (TFE)/DCM (1:1:4, 10 mL) for 30 min at RT. The resin was isolated by filtration and treated a second time with the same cleavage solution. Following isolation of the resin by filtration, both filtrates were combined, and the solvent was evaporated in vacuo, leaving the crude peptide, which was lyophilised to afford a white solid, which was used without further purification.
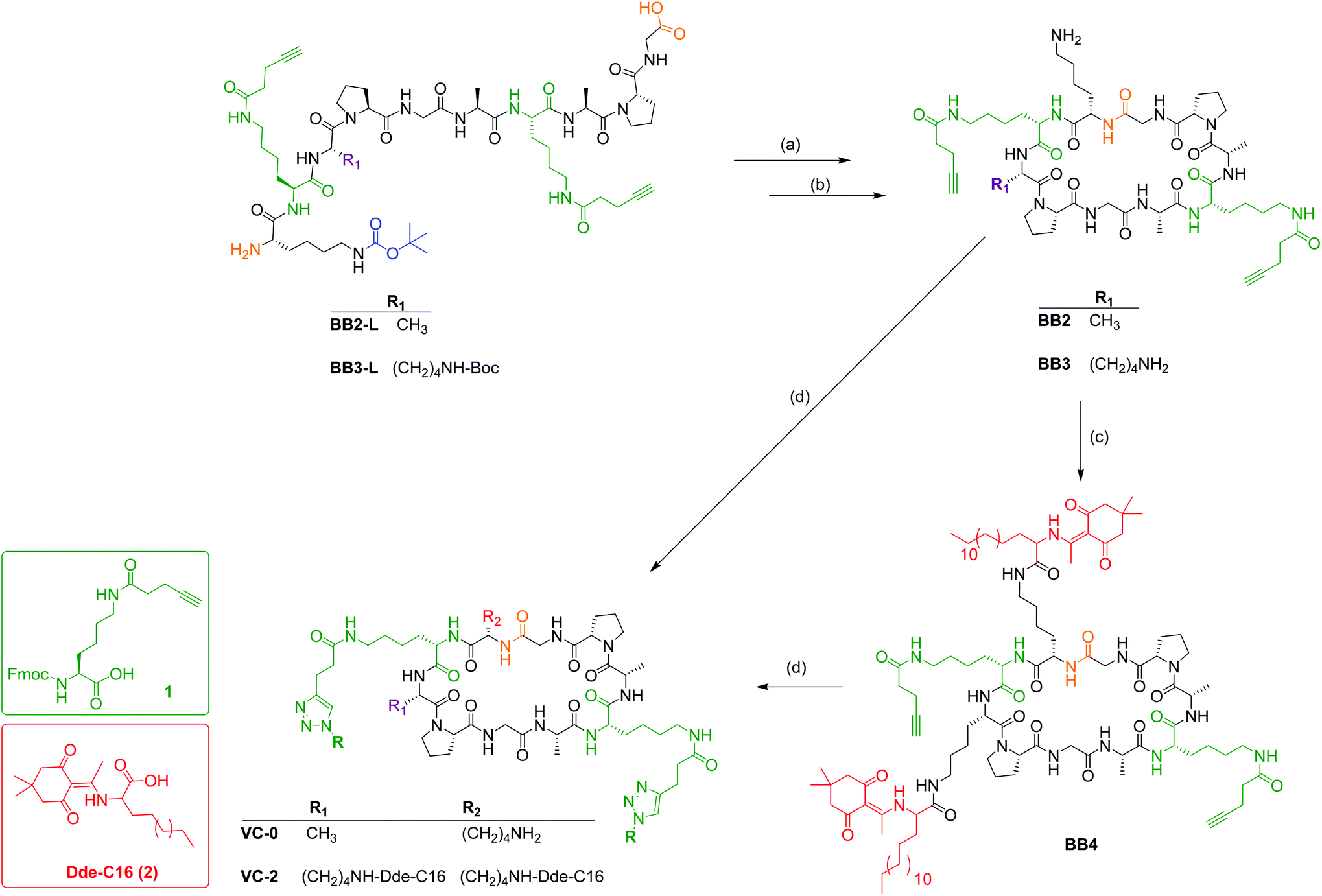 | ||
| Scheme 1 Synthetic strategy for the synthesis of BB2-BB4, VC-0 and VC-2. (a) BB2-L or BB3-L (0.5 mg mL−1) in DMF, HATU (2 eq.), DIPEA (3 eq.), RT, 4 h. (b) Trifluoroacetic acid (TFA)/H2O (95:5), RT, 3 h. (c) (2, 4.2 eq.; red), HATU (4.0 eq.), DIPEA (5.0 eq.), RT, overnight. (d) N3-J8-PADRE, pentanol:MeOH (30:70), CuSO4 (8.8 eq.), NaAsc (17.6 eq.), 36 °C, 2 h. | ||
Each linear peptide was then cyclised, firstly by dissolving in DMF (0.5 mg mL−1) followed by the addition of HATU (4 eq.) and DIPEA (5 eq.) and left to stir overnight at RT. Solvent was removed in vacuo before sidechain protecting groups were removed by treatment of the crude cyclic peptides with 95% TFA. Following removal of TFA in vacuo, the crude cyclic peptides were dissolved in MeCN:H2O (30:70) and lyophilised. BB2 was purified by preparative RP-HPLC and was determined to be >95% pure by analytical RP-HPLC (ESI, Fig. S3†). BB3 was used as crude in the following step.
BB2 R t = 19.0 min (C18 column, 0–100% solvent B, 30 min). Molecular weight (C51H79N13O12): 1066.27 g mol−1. ESI-MS: [M + 1H]+1m/z 1066.7 (calcd: 1067.27), [M + 2H]+2m/z 533.8 (calcd: 534.1). Yield: 6.4 mg, 37%.
Cyclic lipopeptide BB4 (Scheme 1) was synthesised by coupling Dde-C16 (synthesised as previously reported28) to the two unprotected sidechain amines of the previously Boc-protected lysine moieties. This was achieved by the standard amino acid coupling protocol outlined for Fmoc SPPS. Differing from our previously published synthesises, following solvent removal, the crude lipopeptide was dissolved in MeCN:H2O and purified by preparative RP-HPLC to afford pure BB4 (>95% as determined by analytical RP-HPLC) (ESI, Fig. S4†).
BB4 R t = 27.3 min (C4 column, 0–100% solvent B, 30 min). Molecular weight (C106H172N16O18): 1958.64 g mol−1. ESI-MS: [M + 1H]+1m/z 1960.2 (calcd: 1959.64), [M + 2H]+2m/z 980.0 (calcd: 980.32), [M + 3H]+3m/z 654.2 (calcd: 653.9) Yield: 4.8 mg, 16%.
Conjugated vaccine constructs (VC-0 and VC-2; Fig. 1) were synthesised following our previously reported protocols.19 Briefly, VC-0 and VC-2 were synthesised by copper catalysed alkyne–azide 1,3-dipolar cycloaddition between azido-functionalised antigen (J8-PADRE) and alkyne-functionalised cyclic peptide (BB1, VC-0 and BB4, VC-2). BB1 (or BB4) (1 eq.) was dissolved in a minimal volume of MeOH (BB1) (or 1-pentanol for BB4) and added to a solution of pure azido-J8-PADRE (2.5 eq.) in MeOH. Following addition of CuSO4 (8.8 eq.) and sodium ascorbate (17.6 eq.), the reaction was stirred at 36 °C and monitored by analytical RP-HPLC. After completion of the reaction (approx. 2 h), the reaction was diluted with Milli Q water (20 mL) and lyophilised. Crude vaccine constructs were purified using semi-preparative RP-HPLC to afford pure VC-0 (or VC-2), respectively. Pure vaccine constructs were characterised by analytical RP-HPLC and ESI-MS (See ESI, Fig. S5 and S6†).
VC-2 R t = 21.0 min (C4 column, 0–100% solvent B, 30 min). Molecular weight (C520H866N142O140): 11347.50 g mol−1. ESI-MS: [M + 6H]+6m/z 1892.7 (calcd: 1892.3), [M + 7H]+7m/z 1622.0 (calcd: 1622.1), [M + 8H]+8m/z 1419.4 (calcd: 1419.4), [M + 9H]+9m/z 1261.9 (calcd: 1261.8), [M + 10H]+10m/z 1136.1 (calcd: 1135.8), [M + 11H]+11m/z 1032.5 (calcd: 1032.6). Yield: 2.3 mg, 14%.
VC-0 R t = 19.6 min (C18 column, 0–100% solvent B, 30 min). Molecular weight (C471H787N141O134): 10569.33 g mol−1. ESI-MS: [M + 8 H]+8m/z 1324.4 (calcd: 1322.2), [M + 9H]+9m/z 1175.7 (calcd: 1175.4), [M + 10H]+10m/z 1058.0 (calcd: 1057.9), [M + 11H]+11m/z 962.1 (calcd: 961.8), [M + 12H]+12m/z 881.8 (calcd: 881.8), [M + 13H]+13m/z 813.7 (calcd: 814.0). Yield: 3.5 mg, 19%.
Immunological assessment in mice
In vivo assessment was conducted in two separate studies. Study 1 (A, Fig. 1) was undertaken initially to corroborate the results observed in our initial cyclic peptide investigation and to assess the structure–activity relationship of lipid and antigen conjugation to cyclic peptide. Once these trends were confirmed, Study 2 (B, Fig. 1) was commenced to further explore the role of lipid conjugation and cyclic peptide presence. All animal protocols used were approved by the Institute's ethics committee (University of Queensland Animal Ethics Committee: SCMB/AIBN/069/17) and performed in accordance with National Health and Medical Research Council (NHMRC) of Australia guidelines.
Study 1: 4–6-week old female C57BL/6 mice (Animal Resource Centre, Perth, Western Australia, n = 5 mice/group) were immunised subcutaneously at the tail base on day 0 with 30 μg of VC-2 or an antigen normalised dose of varying composition physical mixture (VC-0, VC-4, VC-5; Physical Mixture A) compositions shown in ESI (Table S1†) dissolved in 50 μl of phosphate-buffered saline (PBS), followed by three boosts on days 21, 38 and 41 post primary immunisation. A negative control group was administered 50 μl of PBS. The positive control group received a primary immunisation of 30 μg of J8-PADRE emulsified with CFA in PBS (1:1; 50 μL) followed by thee boosts of J8-PADRE (30 μg in 50 μL PBS). Serum was collected one day prior to each immunisation and on day 41, seven days following the final immunisation. Blood was collected from mice tail tip and allowed to clot for at least 30 min at 37 °C. Serum was collected after centrifugation for 10 min at 1000g and stored at −80 °C.
Study 2: was undertaken with identical mice, timeline, positive and negative controls, and serum collection protocols. Mice were administered in this study with either Physical Mixture A, VC-5, VC-6, VC-7 or BB2 (cyclic peptide alone) in an antigen-normalised dose to VC-2.
Determination of total J8-Specific IgG antibody titre
J8-specific serum IgG titre was determined by a previously described ELISA protocol.29 Briefly, plates were coated with J8 peptide (previously synthesised19) (50 μg per plate) in a volume of 100 μL per well overnight at 4 °C. The peptide coating solution was removed, and the wells blocked with 5% skim milk/PBS-Tween-20 for 1.5 h at 37 °C. The plates were then washed (3× with PBS-Tween-20 buffer). Mouse sera were two-fold serially diluted in 0.5% skim milk/PBS-Tween-20 buffer, starting at an initial dilution of 1:100 to a final dilution of 1:12800 and incubated for 1.5 h at 37 °C. The plates were washed (5x) before horseradish-conjugated goat anti-mouse IgG (H + L) was added at a dilution of 1:3000 in 0.5% skim milk/PBS-Tween-20 for 1.5 h at 37 °C. After washing, 100 μl of OPD substrate was prepared and added according to the manufacturer's instructions and incubated at RT for 20 min in the dark. Absorbance was measured at 450 nm. Antibody titre was defined as the lowest concentration to give an absorbance (at 450 nm) of more than 3 standard deviations above the average of the negative control (serum from mice immunised with PBS). Statistical significance (P < 0.05) was determined using One-way ANOVA with Tukey's multiple comparisons test.
Opsonisation assay
The opsonisation assay was completed as described previously.30 Briefly, clinical isolates were provided by Princess Alexandra hospital (Brisbane, Australia) and included: 2727; ACM-2002 (human abscess – lymph gland); GC2 203 (wound swab); D3840 (nasopharynx swab); and D2612 (nasopharynx swab). Bacterial isolates were streaked on Todd–Hewitt broth supplemented with 5% yeast extract agar plates, and incubated at 37 °C for 24 h. Single colonies were transferred to Todd-Hewitt broth (5 mL) supplemented with 5% yeast extract and incubated for 24 h at 37 °C to give approximately 4.6 × 106 colony forming units (CFU) ml−1. The culture was serially diluted to 102 in PBS and an aliquot (10 μL) was mixed with heat-inactivated sera (10 μL) and horse blood (80 μL). The inactivated sera were prepared by heating in a water bath at 50 °C for 15 min. Bacteria were incubated in the presence of sera and incubated in a 96-well plate at 37 °C for 3 h. The bacterial survival was analysed by withdrawing an aliquot (10 μL) from the culture material, which was then plated on Todd-Hewitt agar plates supplemented with 5% yeast extract and 5% horse blood. Plates were incubated at 37 °C for 24 h and colonies enumerated to colony forming units (CFU). Opsonic activity of the antibodies (anti-peptide) sera (percentage reduction in mean CFU) was calculated as (1 − [CFU in the presence of anti-peptide sera]/[mean CFU in the presence of un-treated wells]) × 100. The assay was performed in duplicate from two independent cultures.Cytotoxicity (MTT) assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cytotoxicity assay was performed using NCI-H46 and HEK-293 cell lines. NCI-H46 and HEK-293 were grown in RPMI 1640 medium and Dulbecco's modified Eagle's medium (DMEM), respectively, as adherent monolayers in supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, penicillin (100 U ml−1), and streptomycin (100 μg ml−1) in a humidified 37 °C incubator (5% CO2). Cells were harvested with trypsin and dispensed into 96-well assay plates at 2000 cells per well and incubated for 18 h at 37 °C with 5% CO2. Physical Mixture A and VC-5 were dissolved in PBS, and aliquots (10 μl) were tested over two final concentrations ranging from 0.6 mg mL−1 to 0.3 mg mL−1. Negative control wells were treated with 1% aqueous DMSO. Positive control wells were treated with sodium dodecyl sulphate (SDS). After a 48 h incubation at 37 °C, 10 μL MTT in PBS (5 mg ml−1) was added to each well (final concentration 0.5 mg ml−1), and the microtiter plates incubated for a further 4 h at 37 °C with 5% CO2. After this final incubation, the medium was aspirated, and precipitated formazan crystals were dissolved in DMSO (100 μl per well). The absorbance of each well was then measured at 600 nm. All experiments were performed in triplicate.Dynamic light scattering
Individual compounds J8-PADRE, BB1 (KKSS-C16-C16-NH2) and BB2 (0.05 mg mL−1 in PBS) and Physical Mixture A, VC-5, VC-6 and VC-7 (0.05 mg mL−1 in PBS) were analysed using dynamic light scattering (DLS) to measure the particle size (z average) and poly dispersity index (PDI). The samples were treated with ultrasonication for 5 min and then transferred into disposable cuvettes. Measurements were taken at 25 °C and 173° light scattering.Transmission electron microscopy
Physical Mixture A, VC-6 and VC-5 (0.05 mg mL−1 in PBS) were individually applied to glow-discharged carbon-coated copper 200 mesh grids (Ted Pella) and negative-stained with 2% uranyl acetate.Secondary structure analysis
Individual compounds (J8-PADRE, BB1 and BB2 and Physical Mixture A, VC-6 and VC-7) were placed in a 1 mm quartz cuvette and CD spectra was measured at 23 °C. Data was collected between 195 and 275 nm with a continuous scanning speed of 50 nm min−1.Results and discussion
Vaccine synthesis
This study aimed to extend previously published findings around the adjuvant effects of a physically mixed vaccine containing a GAS B cell antigen conjugated to a T helper epitope, lipid moiety and cyclic decapeptide (Physical Mixture A; Fig. (1A)) that was the most immunogenic vaccine candidate following structure-immune activity analysis of antibody titres and bacterial opsonisation potential.19Vaccine building blocks and conjugated vaccine compounds were synthesised using standard Fmoc SPPS chemistry, and solution phase coupling and copper catalysed alkyne azide cycloadditions.19 The building blocks BB1-BB4 and the conjugated vaccines (VC-0 and VC-2) were synthesised as previously published.19 The combined immunological epitope (J8-PADRE; J8 shown to be conserved between multiple GAS strains31) was synthesised on Rink amide MBHA resin.
Cyclic peptides were synthesised in their linear confirmations on 2-CTC resin which generated carboxyl C-terminal peptides which were then cyclised in the solution phase (Scheme 1). We reported the use of crude cyclic lipopeptide alkyne for the synthesis of VC-2 due to the high hydrophobicity (and hence poor solubility) of compound BB4 and inability to purify by RP-HPLC, however, in this study we required pure cyclic lipopeptide BB4 for use in the VC-5 physical mixture. To allow for BB4 purification by preparative RP-HPLC, following coupling of the Dde-C16 lipoamino acid, the crude BB4 reaction mixture was washed with hexane to remove excess Dde-C16 lipoamino acid. This reaction mixture was then solubilised in HPLC-compatible solvent (acetonitrile/water) for efficient purification. Apart from these two alterations, all compounds were synthesised as previously reported.19
In vivo immunological studies
In vivo immunological assessment was conducted in two separate studies. Study 1 (A, Fig. 1) was undertaken to examine the effect of conjugation between the antigens (GAS B cell and universal T helper) and carrier decapeptide following the promising response from Physical Mixture A in previous investigations. Study 2 (B, Fig. 1) was commenced to further explore the role of the cyclic decapeptide, and the effect of conjugation of lipid TLR2 ligands to this cyclic decapeptide carrier. In both studies, female C57BL/6 mice were immunised subcutaneously at the tail base on days 0, 21, 28, and 35 with; Study 1–30 μg VC-2 and an antigen normalised dose of VC-0, VC-4, VC-5 or Physical Mixture A; and Study 2 – VC-2 normalised doses of Physical Mixture A, VC-5, VC-6, VC-7, and Cyclic Peptide alone (BB2) (Fig. 1). Both studies shared negative control (PBS) and positive control (30 μg of J8-PADRE emulsified in 50:50 PBS/CFA primary immunisation; and 30 μg J8-PADRE in PBS for boosts). In both studies, blood was collected from the tail tip 7 days following the last injection (day 41). Serum was analysed for total antigen-specific serum IgG antibodies by enzyme-linked immunosorbent assay (ELISA).
We observed the high potency of Physical Mixture A, which was able to elicit levels of anti-J8 antibodies with no significant difference to the positive control across both studies (Fig. 2). This is a highly promising result considering that CFA is seen as the gold standard in vaccine development, and is a highly efficacious adjuvant, but unfortunately, it is restricted from human use.32 The toxicity of this CFA adjuvant has been illustrated by the death of two animals from this group during the study (Study 1). By comparing the compounds in Study 1 with the CFA-adjuvanted control (Fig. 2) we observed no significant difference between Physical Mixture A and the J8-PADRE + CFA positive control (P < 0.0639), and significant differences for the J8-PADRE + CFA positive control with VC-5 (P < 0.0003), VC-4 (P < 0.0001), and VC-2 (P < 0.0015) indicating the conjugation of any of the vaccine components to the cyclic core reduces the adjuvant activity of the mixture (Fig. 2).
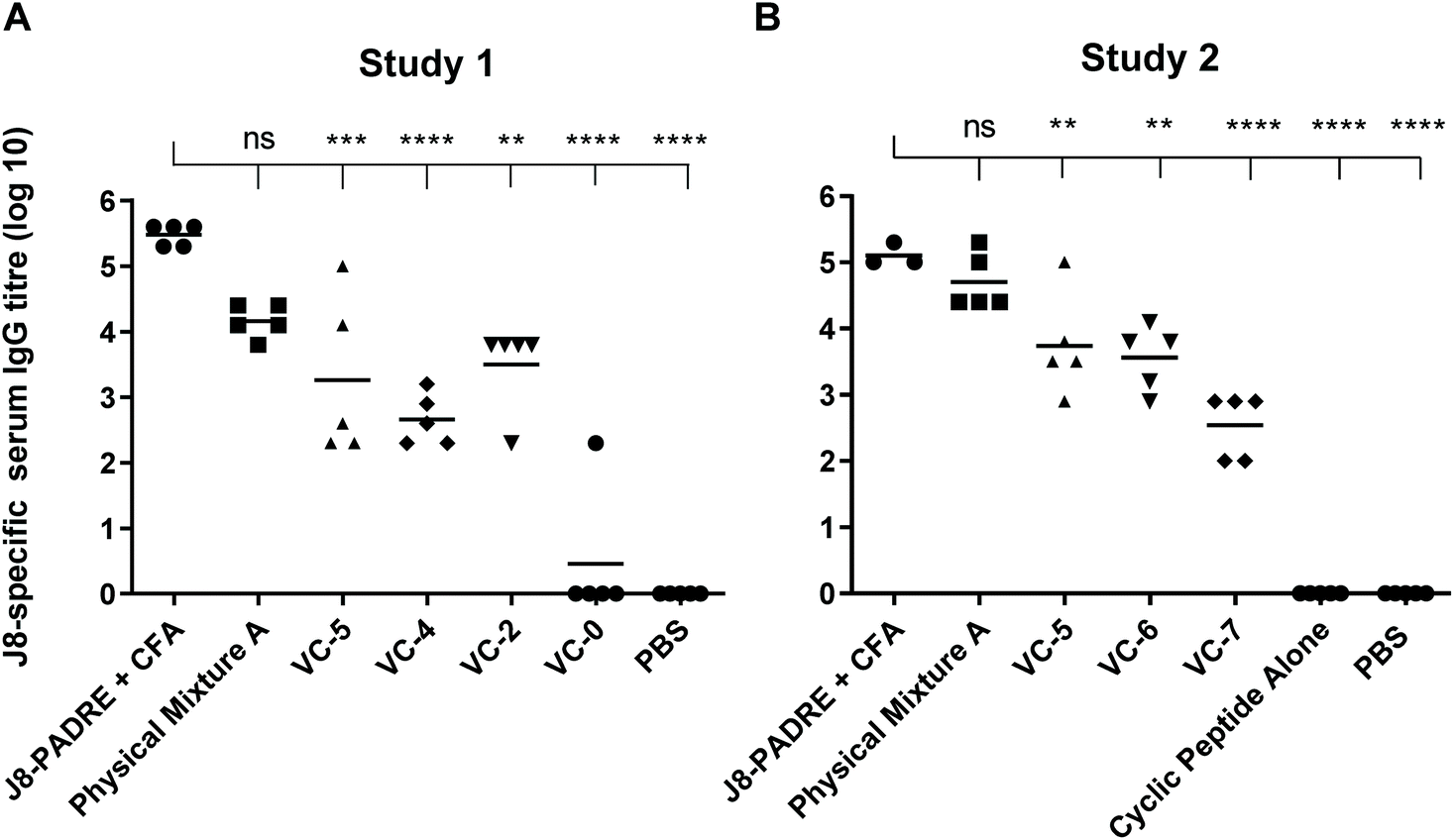 | ||
| Fig. 2 J8-specific serum IgG antibody titres (log 10) at day 41 following primary immunisation and three boosts induced in response to subcutaneous immunisation of C57BL/6 mice (n = 5 per group) with the vaccine candidates (A and B, Fig. 1), the negative control (PBS) and positive control (J8-PADRE + CFA), as determined by ELISA. Statistical analysis was performed using a one-way ANOVA followed by Tukey post-hoc test. Antibody titres against J8 are shown for individual mice with the average titre indicated by a bar. (ns, P > 0.05; *, P < 0.05; ** P < 0.01; *** P < 0.001; ****, P < 0.0001). | ||
In Study 2 we again observed no significant difference between Physical Mixture A and the positive control (J8-PADRE + CFA; Fig. 2). Comparing Physical Mixture A, VC-6 and VC-7 (Study 2) we observed statistically significant differences between Physical Mixture A and both the two-component mixtures, VC-6 and VC-7 (Fig. 2). The results indicate that for most effective adjuvant activity, the presence of all three vaccine components (lipid, antigen and cyclic peptide) are essential. Lipid or the cyclic decapeptide physically mixed with antigen (VC-6 and VC-7) show some activity, however, this is significantly lower than that of the tri-component mixture (Physical Mixture A).
In vitro opsonisation assay
Protection against GAS in a murine challenge model and reduction in airway and skin colony counts has been correlated with high titres of opsonic antibodies.33 Following the final bleed (day 41), serum from each vaccinated mouse was pooled into vaccine groups and assessed for its ability to effectively opsonize five clinically relevant strains of GAS bacteria (Fig. 3). Across Studies 1 and 2 we again saw the high opsonic potential of antibodies produced by immunisation with Physical Mixture A (average 87% across Study 1 and 77% Study 2) with similar levels of colony reduction to mice immunised with J8-PADRE + CFA (average 90% across Study 1 and 83% Study 2). VC-5 also showed high opsonic potential with an average of 75% across the five strains in Study 1, and 58% across the five strains in Study 2. This result is likely due to the two high responding mice (IgG titres) observed in the individual mouse ELISA results (Fig. 2). In Study 2, VC-5, VC-6 and VC-7 follow the trends of the antibody titres observed from ELISA.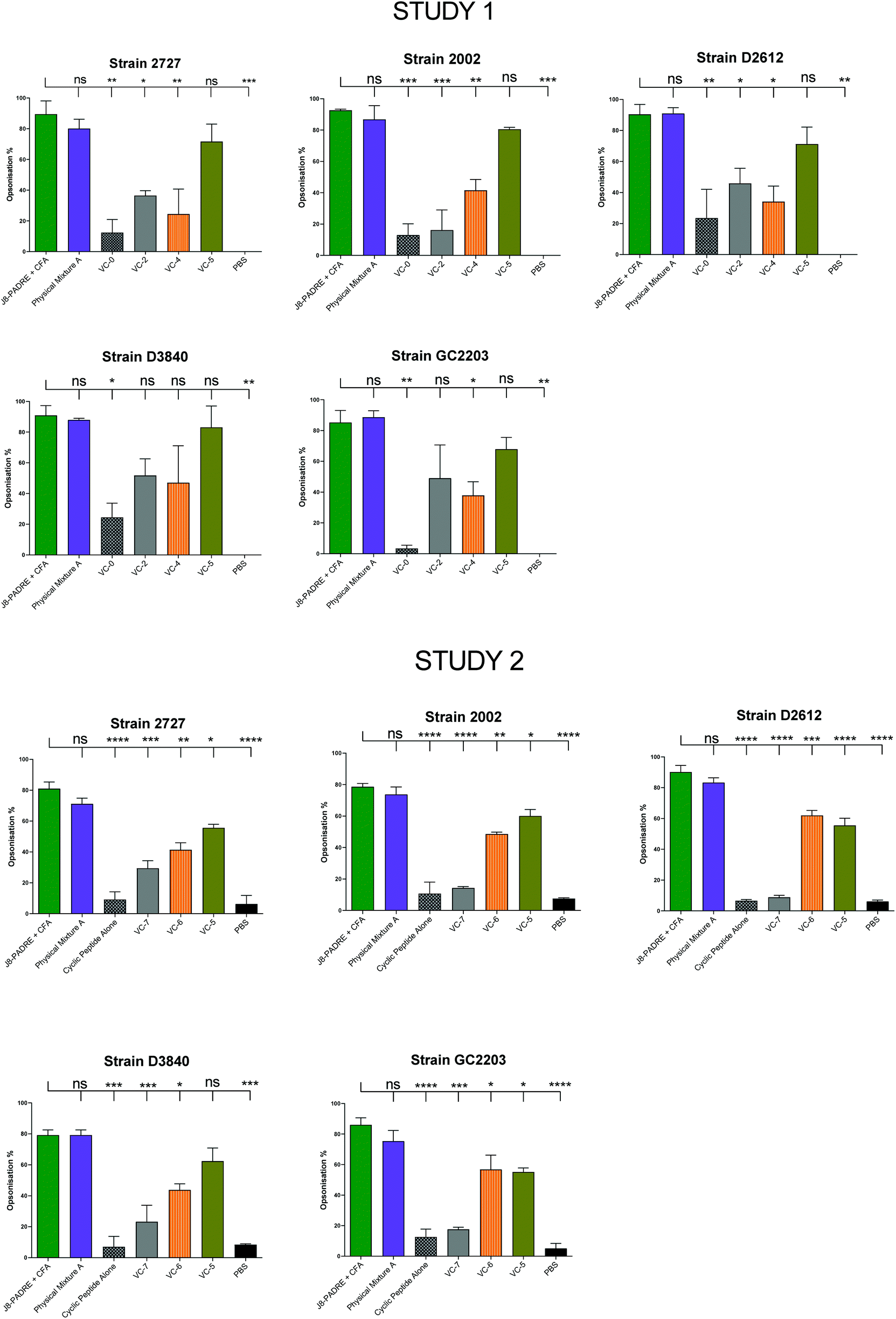 | ||
| Fig. 3 Average opsonisation percentage of different GAS strains by serum collected after the final bleed (day 41 following primary immunisation and three boosts) from mice immunised with the vaccine candidates (VC-0, VC-2, VC-4, VC-5, VC-6, VC-7, Cyclic Peptide Alone, and Physical Mixture A;Fig. 1), and the negative (PBS) and positive (J8-PADRE + CFA) controls from Study 1 (Top) and Study 2 (bottom). Results are represented as opsonisation percentage compared to reference untreated wells, and error is represented as standard error of the mean (SEM). | ||
To gain further insight into the varied responses observed between vaccine groups, we analysed the antibody subclasses for primary vaccine groups of interest (J8-PADRE + CFA, Physical Mixture A, VC-5 and VC-6). C57BL/6 mice produce four subclasses of IgG (IgG1, IgG2b, IgG2c and IgG3).34 Of particular interest to vaccine development is the activation of a Th1 verse a Th2 response by the vaccine candidate. A balanced Th1/Th2 (humoral verse cellular) response is necessary for an effective protective immune response for vaccines (e.g. bacteria or virus).35 In the subclasses produced by the C57BL/6 mice, IgG1 is predominantly induced by the Th2 pathway, and IgG2c by the Th1 pathway.36 Assessing the IgG2c/IgG1 (Th1/Th2) ratio provided an indication of the state of the Th1/Th2 bias induced by the vaccine candidates. Here, both the CFA-adjuvanted positive control (J8-PADRE + CFA) and Physical Mixture A induced the most balanced immune response with a Th1/Th2 ratio of <1.5 (Fig. 4). Whereas, VC-5 and VC-6 (which performed poorer than Physical Mixture A), showed a stronger Th1 biased immune response with an IgG2c/IgG1 ratio significantly higher than Physical Mixture A (Physical Mixture A verse VC-5, P < 0.002; Physical Mixture A verse VC-6, P < 0.0001). We can rationally accept these results as we expect a slight Th1 bias due to the use of a predominantly Th1-inducing T-helper epitope (PADRE). Further, the similarity of the Th1/Th2 ratio observed for the CFA adjuvanted group along with previously reported values for this Th1-inducing adjuvant.37,38
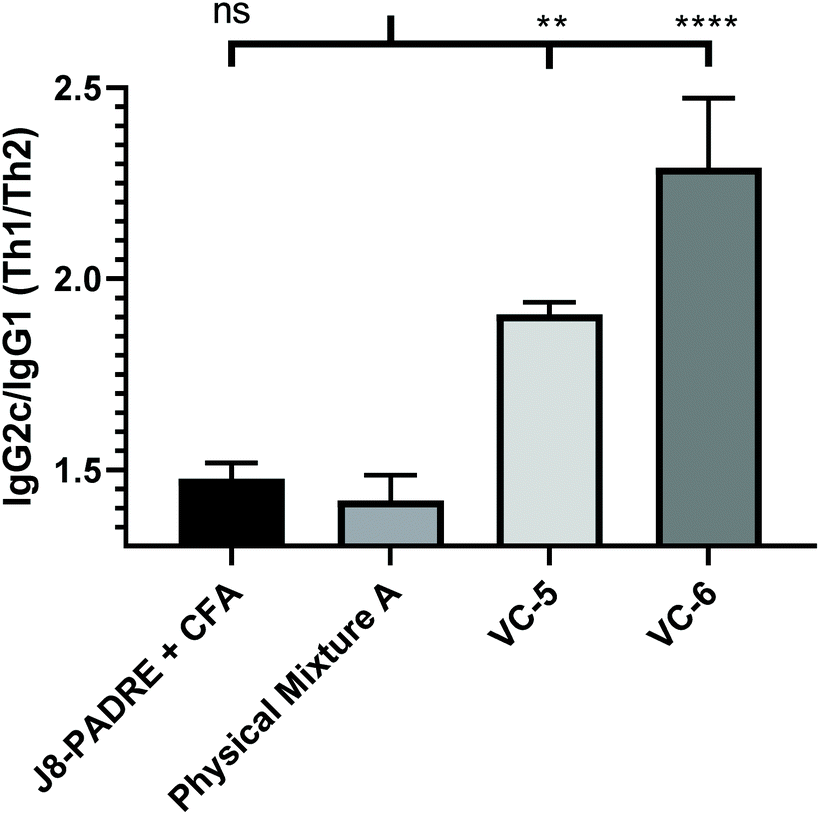 | ||
| Fig. 4 IgG2c/IgG1 ratio for pooled serum at day 41 (following the primary immunisation and three boosts) induced in response to subcutaneous immunisation of C57BL/6 mice (n = 5 per group) with the highest performing vaccine candidates (Physical Mixture A, VC-5 and VC-6), and positive control (J8-PADRE + CFA), as determined by ELISA. Statistical analysis was performed using a one-way ANOVA followed by Tukey post-hoc test. IgG2c/IgG1 ratios are shown calculated from IgG1 and IgG2c titres for pooled serum against J8. (ns, P > 0.05; *, P < 0.05; ** P < 0.01; *** P < 0.001; ****, P < 0.0001). | ||
Biophysical characterisation
Some synthetic peptide-based vaccines, particularly lipid adjuvanted vaccines, benefit from their ability to self-assemble into nanoparticles in an aqueous environment.39 Nanoparticulate vaccines have been investigated for all manner of infectious diseases and cancer, and have shown preferential trafficking to lymph nodes and uptake by antigen presenting cells.40 Not only can nanoparticle-based vaccines show increased immunogenicity, their size can also effect the activation of Th1 verse Th2 immune responses.41We initially examined each component of the physical mixtures separately by DLS (J8-PADRE, BB1, cyclic peptide alone [BB2] and BB4). Each of these compounds displayed markedly different particle sizes and distributions in solution (Table S2†). J8-PADRE, BB2 and BB4 showed two distinct polydisperse populations of particles with significant differences in size (J8-PADRE: 310.5 ± 29.9 nm, 92.2 ± 9.2 nm; BB2: 242.2 ± 14.7 nm, 5138 ± 198.9 nm and Cyclic Peptide Alone [BB2]: 184.1 ± 16.1, 5324 ± 196.2 nm). The third vaccine component BB1 (KKSS-C16-C16-NH2) formed a single particle size (1713 ± 210.7 nm). Following formulation of the three vaccine components to yield the vaccine candidate groups, Physical Mixture A, VC-5, VC-6 and VC-7, these were also analysed by DLS. Each of the final vaccine mixtures formed single particle distributions with narrower PDI indicating some form of interaction between all three separate compounds (Fig. 5). Interestingly, VC-5, VC-6 and VC-7 formed much smaller particles (249.3 ± 8.1 nm, 228.6 ± 17.3 nm and 200.3 ± 8.4 nm) respectively, when compared to Physical Mixture A (857.1 ± 56.9 nm).
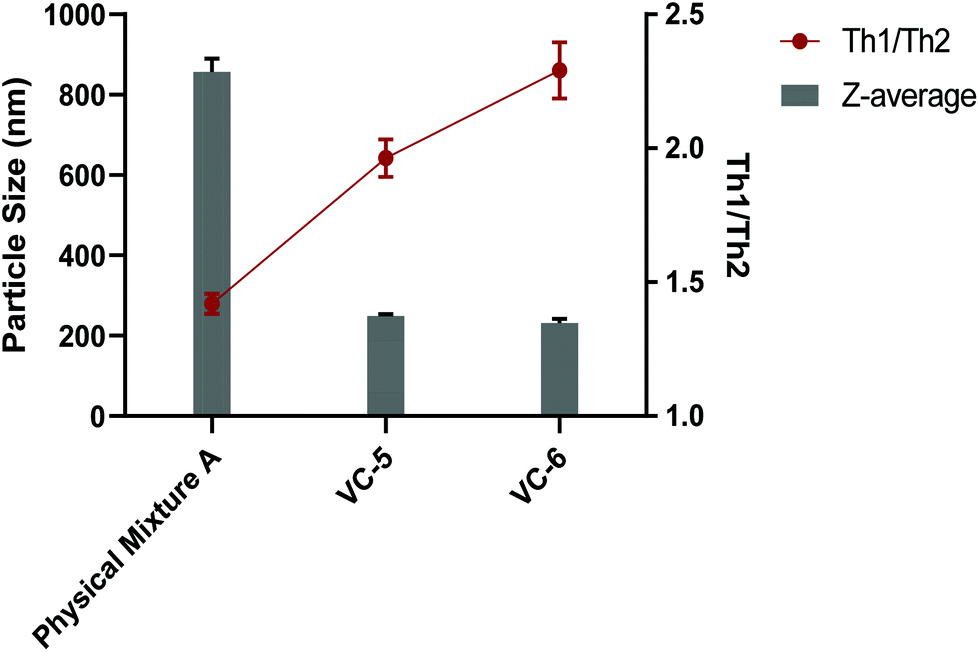 | ||
| Fig. 5 Vaccine particle sizes (as determined by DLS intensity at a concentration of 0.05 mg mL−1) and Th1/Th2 ratio (of pooled serum as determined by ELISA) for vaccine candidates which induced high titres of >50% opsonising IgG in immunisation study 2 (Physical Mixture A, VC-5 and VC-6). | ||
In theory, this difference in vaccine candidate particle size can explain the difference in Th1/Th2 ratio observed between the Physical Mixture A, VC-5 and VC-6 vaccine groups. It is widely accepted that particulate vaccines (or antigens) in the small (<500 nm) range preferentially induce Th1 (or cellular) immune responses, whereas larger particulate antigens and vaccines (>500 nm) preferentially induce Th2 (or humoral) immune responses.42 This phenomenon is most simplistically described as small particle sizes (virus sized particles) inducing cellular immunity with larger particles (bacterial sized particles) inducing humoral antibody responses. This immune response modulation is understood to be associated with the different mechanisms by which differently sized particles are up taken into APCs. With small particles (<500 nm) being up taken by endocytosis compared with large particles (>500 nm) being up taken by phagocytosis.42 This skewing of the immune response, either Th1 or Th2, is the bodies attempt at mounting the most effective response against a particular pathogen (Th1 or cellular response against intracellular pathogens and a Th2 or humoral response against extracellular pathogens).43
To confirm the particle sizes observed from DLS analysis, we examined Physical Mixture A, VC-5 and VC-6 by transmission electron microscopy (TEM). In the images shown in Fig. 6, Physical Mixture A generates small spherical particles (∼250 nm in diameter) which aggregate to form larger (800–900 nm) particles. This TEM confirms the particle size result from DLS and explains the large poly dispersity index observed for Physical Mixture A. VC-5 and VC-6 generates very small, non-uniform (100–200 nm) particles, consistent with the particle size result from DLS and explains the small poly dispersity index observed in both mixtures.
 | ||
| Fig. 6 Transmission electron microscopy images of Physical Mixture A (top), VC-5 (middle) and VC-6 (bottom) at 0.1 mg mL−1. White bars indicate (2.0 μm, 1.0 μm, 500 nm, 200 nm), negative staining 2% uranyl acetate. | ||
Circular dichroism was also performed to analyse the secondary structure of the vaccine components and the Physical Mixtures (Fig. S7†). We observed expected beta sheet like confirmation of the cyclic peptide, BB2 and the presence of an alpha-helix like structure in J8-PADRE which has previously been shown to be critical for the recognition of the native GAS protein.44 Of all analysed mixtures, Physical Mixture A was observed to have the highest alpha helix content which provides evidence to explain both the high antibody titres (antibody specificity) and the high opsonisation potential of serum elicited by this compound.
Cytotoxicity (MTT) assay
To further evaluate the suitability of the lead compounds from this study (Physical Mixture A and VC-5) as potential vaccine candidates/adjuvants, their toxicity was assessed across two common cell lines at identical concentrations to those used in the in vivo study (0.6 mg mL−1) and half in vivo concentration (0.3 mg mL−1) (Fig. 7). No statistically significant toxicity was observed across either cell line for both compounds assessed. This result highlights the safety profile of peptide-based vaccines and peptide-based vaccine delivery systems.45 Further, the low level of non-significant toxicity observed for Physical Mixture A (in vivo concentration) could be a contributing factor to the adjuvant effect of this vaccine.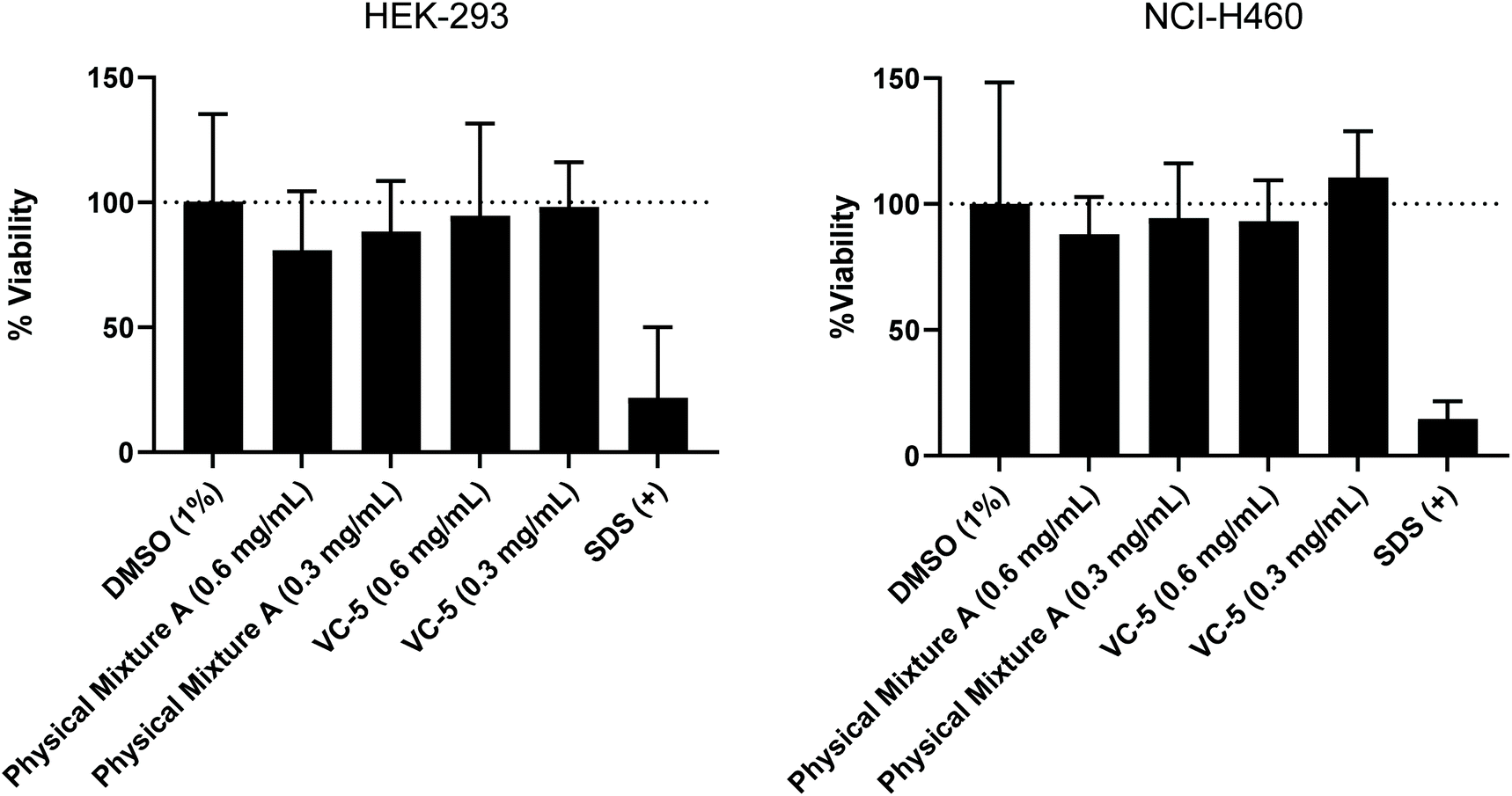 | ||
| Fig. 7 MTT cell viability assay was performed (in triplicate) on HEK-293 and NCI-H460 cell lines with two concentrations of compounds (0.6 and 0.3 mg mL−1). | ||
Conclusion
We have proven that a physical mixture of vaccine components is superior in inducing opsonic immune responses against multiple strains of GAS bacteria. This structure–activity analysis investigated the degree of conjugation of these components concluding that all three vaccine components were necessary for a strong and balanced immune response. Conjugation of the lipid moiety to the cyclic decapeptide was not necessary for a strong IgG response, however, the presence of the cyclic decapeptide significantly increased the adjuvant capability of the vaccine components by inducing an enhanced balanced Th1/Th2 immune activation. The combination of vaccine components had a significant effect on the particle size, with the addition of the cyclic decapeptide producing large (∼850 nm) nanoparticles when formulated in aqueous solution. Interestingly, the cyclic lipo-peptide with antigen vaccine and the cyclic peptide with antigen vaccine produced significantly smaller particles compared with the tri-vaccine formulation (cyclic peptide plus antigen plus lipid). The results support further investigation of these building blocks as adjuvants for GAS peptide antigens.Author contributions
R.J.S and I.T designed the experimental study. H.Y.R.M, W.H, L.G and B.R contributed to compound synthesis and purification. H.Y.R.M and W.H performed the in vivo animal experiments, ELISA analysis and biophysical characterisation. W.M.H, Z.G.K, V.S.S and R.J.C performed the in vitro opsonisation and MTT toxicity assays. P.K performed the transmission electron microscopy. H.Y.R.M drafted the manuscript which was critically reviewed by R.J.S and I.T. All authors contributed to editing of the manuscript and approved its final version.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by National Health and Medical Research Council (NHMRC) program grants APP1132975 and APP1158748. The authors acknowledge the UQBR AIBN animal house facilities. We also acknowledge A. D. Paterson and P. Harris (Centre for Clinical Research, The University of Queensland) for providing GAS isolates 2727, ACM-2002, GC2 203, D3840, and D2612. We thank Susan E. Bates and Robert W. Robey (NCE, NIH, Bethesda, MD, USA) for providing the NCI-H460 cells, and Dr Frank Fontaine (IMB, UQ) for providing the HEK293 cells.References
- R. J. Malonis, J. R. Lai and O. Vergnolle, Peptide-Based Vaccines: Current Progress and Future Challenges, Chem. Rev., 2020, 120(6), 3210–3229 CrossRef CAS.
- B. Pulendran, S. Arunachalam, P. O’Hagan and D. T. O’Hagan, Emerging concepts in the science of vaccine adjuvants, Nat. Rev. Drug Discovery, 2021, 20(6), 454–475 CrossRef CAS PubMed.
- M. Skwarczynski and I. Toth, Peptide-based synthetic vaccines, Chem. Sci., 2016, 7(2), 842–854 RSC.
- W. C. Madeleine, Pathogenesis of Group A Streptococcal Infections, Clin. Microbiol. Rev., 2000, 13(3), 470 CrossRef PubMed.
- F. Azmi, A. A. Ahmad Fuaad, M. Skwarczynski and I. Toth, Recent progress in adjuvant discovery for peptide-based subunit vaccines, Hum. Vaccines Immunother., 2014, 10(3), 778–796 CrossRef CAS PubMed.
- L. M. Mayr, B. Su and C. Moog, Non-Neutralizing Antibodies Directed against HIV and Their Functions, Front. Immunol., 2017, 8, 1590–1590 CrossRef PubMed.
- W. Li, M. D. Joshi, S. Singhania, K. H. Ramsey and A. K. Murthy, Peptide Vaccine: Progress and Challenges, Vaccines, 2014, 2(3), 515–536 CrossRef CAS.
- S. Shi, H. Zhu, X. Xia, Z. Liang, X. Ma and B. Sun, Vaccine adjuvants: Understanding the Structure and Mechanism of Adjuvanticity, Vaccine, 2019, 37(24), 3167–3178 CrossRef CAS PubMed.
- A. Di Pasquale, S. Preiss, F. Tavares Da Silva and N. Garçon, Vaccine Adjuvants: from 1920 to 2015 and Beyond, Vaccines, 2015, 3(2), 320–343 CrossRef CAS.
- G. Del Giudice, R. Rappuoli and A. M. Didierlaurent, Correlates of Adjuvanticity: A Review on Adjuvants in Licensed Vaccines, Elsevier, 2018, pp. 14–21 Search PubMed.
- N. Garçon and A. Di Pasquale, From Discovery to Licensure, the Adjuvant System Story, Hum. Vaccines Immunother., 2017, 13(1), 19–33 CrossRef PubMed.
- E. Mata, M. Igartua, R. M. Hernández, J. E. Rosas, M. E. Patarroyo and J. L. Pedraz, Comparison of the Adjuvanticity of Two Different Delivery Systems on the Induction of Humoral and Cellular Responses to Synthetic Peptides, Drug Deliv., 2010, 17(7), 490–499 CrossRef CAS.
- M. Skwarczynski and I. Toth, Lipid-Core-Peptide System for Self-Adjuvanting Synthetic Vaccine Delivery, Bioconj. Prot., 2011, 751, 297–308 CAS.
- S. Bartlett, R. M. Eichenberger, R. J. Nevagi, K. A. Ghaffar, N. Marasini, Y. Dai, A. Loukas, I. Toth and M. Skwarczynski, Lipopeptide-Based Oral Vaccine Against Hookworm Infection, J. Infect. Dis., 2019, 221(6), 934–942 CrossRef.
- S. H. Apte, P. L. Groves, M. Skwarczynski, Y. Fujita, C. Chang, I. Toth and D. L. Doolan, Vaccination with Lipid Core Peptides Fails to Induce Epitope-Specific T Cell Responses but Confers Non-Specific Protective Immunity in a Malaria Model, PLoS One, 2012, 7(8), e40928 CrossRef CAS.
- C. Olive, M. Batzloff, A. Horváth, T. Clair, P. Yarwood, I. Toth and M. F. Good, Potential of Lipid Core Peptide Technology as a Novel Self-Adjuvanting Vaccine Delivery System for Multiple Different Synthetic Peptide Immunogens, Infect. Immun., 2003, 71(5), 2373 CrossRef CAS.
- P. M. Moyle, A. Horvath, C. Olive, M. F. Good and I. Toth, Development of Lipid-Core-Peptide (LCP) Based Vaccines for the Prevention of Group A Streptococcal (GAS) Infection, Lett. Pept. Sci., 2003, 10(5–6), 605–613 CrossRef CAS.
- A. De Titta, M. Ballester, Z. Julier, C. Nembrini, L. Jeanbart, A. J. Van Der Vlies, M. A. Swartz and J. A. Hubbell, Nanoparticle Conjugation of CpG Enhances Adjuvancy for Cellular Immunity and Memory Recall at Low Dose, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(49), 19902–19907 CrossRef CAS.
- H. Y. R. Madge, H. Sharma, W. M. Hussein, Z. G. Khalil, R. J. Capon, I. Toth and R. J. Stephenson, Structure–Activity Analysis of Cyclic Multicomponent Lipopeptide Self-Adjuvanting Vaccine Candidates Presenting Group A Streptococcus Antigens, J. Med. Chem., 2020, 63(10), 5387–5397 CrossRef CAS.
- W. M. Hussein, T.-Y. Liu, P. Maruthayanar, S. Mukaida, P. M. Moyle, J. W. Wells, I. Toth and M. Skwarczynski, Double Conjugation Strategy to Incorporate Lipid Adjuvants into Multiantigenic Vaccines, Chem. Sci., 2016, 7(3), 2308–2321 RSC.
- K. L. Bengtsson and A. Sjölander, Adjuvant Activity of Iscoms: Effect of Ratio and Co-Incorporation of Antigen and Adjuvant, Vaccine, 1996, 14(8), 753–760 CrossRef CAS PubMed.
- C. B. Fox, R. M. Kramer, V.,L. Barnes, Q. M. Dowling and T. S. Vedvick, Working Together: Interactions Between Vaccine Antigens and Adjuvants, Ther. Adv. Vaccines, 2013, 1(1), 7–20 CrossRef PubMed.
- J. Oliver, M. Wilmot, J. Strachan, S. St George, C. R. Lane, S. A. Ballard, M. Sait, K. Gibney, B. P. Howden and D. A. Williamson, Recent trends in invasive group A Streptococcus disease in Victoria, Commun. Dis. Intell., 2019, 43, 43 Search PubMed.
- J. J. Ferretti, D. L. Stevens and V. A. Fischetti, ed. Streptococcus pyogenes: Basic Biology to Clinical Manifestations [Internet], University of Oklahoma Health Sciences Center, Oklahoma City (OK), 2016 Search PubMed.
- P. J. May, A. C. Bowen and J. R. Carapetis, The Inequitable Burden of Group A Streptococcal Diseases in Indigenous Australians, Med. J. Aust., 2016, 205(5), 201–203 CrossRef PubMed.
- D. A. Watkins, C. O. Johnson, S. M. Colquhoun, G. Karthikeyan, A. Beaton, G. Bukhman, M. H. Forouzanfar, C. T. Longenecker, B. M. Mayosi and G. A. Mensah, Global, regional, and national burden of rheumatic heart disease, 1990–2015, N. Engl. J. Med., 2017, 377(8), 713–722 CrossRef.
- M. Zaman, A.-B. M. Abdel-Aal, K. S. M. Phillipps, Y. Fujita, M. F. Good and I. Toth, Structure–Activity Relationship of Lipopeptide Group A Streptococcus (GAS) Vaccine Candidates on Toll-Like Receptor 2, Vaccine, 2010, 28(10), 2243–2248 CrossRef CAS PubMed.
- P. B. Ross, A. R. Falconer and I. Thot, N-1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl (N-Dde) Lipoamino Acids, Molbank, 2008, 2008(2), M566 CrossRef.
- K. A. Ghaffar, N. Marasini, A. K. Giddam, M. R. Batzloff, M. F. Good, M. Skwarczynski and I. Toth, Liposome-Based Intranasal Delivery of Lipopeptide Vaccine Candidates Against Group A Streptococcus, Acta Biomater., 2016, 41, 161–168 CrossRef.
- N. Marasini, A. K. Giddam, Z. G. Khalil, W. M. Hussein, R. J. Capon, M. R. Batzloff, M. F. Good, I. Toth and M. Skwarczynski, Double Adjuvanting Strategy for Peptide-Pased Vaccines: Trimethyl Chitosan Nanoparticles for Lipopeptide Delivery, Nanomedicine, 2016, 11(24), 3223–3235 CrossRef CAS.
- C. Dai, Z. G. Khalil, W. M. Hussein, J. Yang, X. Wang, L. Zhao, R. J. Capon, I. Toth and R. J. Stephenson, Opsonic Activity of Conservative Versus Variable Regions of the Group A Streptococcus M Protein, Vaccines, 2020, 8(2), 210 CrossRef CAS.
- J. de S. Apostólico, V. A. S. Lunardelli, F. C. Coirada, S. B. Boscardin and D. S. Rosa, Adjuvants: Classification, Modus Operandi, and Licensing, J. Immunol. Res., 2016, 2016, 1459394–1459394 Search PubMed.
- M. Skwarczynski, G. Zhao, J. C. Boer, V. Ozberk, A. Azuar, J. G. Cruz, A. K. Giddam, Z. G. Khalil, M. Pandey, M. A. Shibu, W. M. Hussein, R. J. Nevagi, M. R. Batzloff, J. W. Wells, R. J. Capon, M. Plebanski, M. F. Good and I. Toth, Poly(Amino Acids) as a Potent Self-Adjuvanting Delivery System for Peptide-Based Nanovaccines, Sci. Adv., 2020, 6(5), eaax2285 CrossRef CAS.
- S. S. Weber, J. Ducry and A. Oxenius, Dissecting the Contribution of IgG Subclasses in Restricting Airway Infection with Legionella Pneumophila, J. Immunol., 2014, 193(8), 4053–4059 CrossRef CAS.
- P. Kidd, Th1/Th2 balance: The Hypothesis, Its Limitations, and Implications for Health and Disease, Altern. Med. Rev., 2003, 8(3), 223–246 Search PubMed.
- R. M. Martin, J. L. Brady and A. M. Lew, The Need for IgG2c Specific Antiserum When Isotyping Antibodies from C57BL/6 and NOD Mice, J. Immunol. Methods, 1998, 212(2), 187–192 CrossRef CAS.
- L. L. de Oliveira, K. C. Coltri, C. R. B. Cardoso, M.-C. Roque-Barreira and A. Panunto-Castelo, T Helper 1−Inducing Adjuvant Protects against Experimental Paracoccidioidomycosis, PLoS Neglected Trop. Dis., 2008, 2(3), e183 CrossRef.
- D. Kim, A. Monie, L. He, Y. C. Tsai, C. F. Hung and T. C. Wu, Role of IL-2 secreted by PADRE-specific CD4 + T cells in enhancing E7-specific CD8 + T-cell immune responses, Gene Ther., 2008, 15(9), 677–687 CrossRef CAS.
- G. Zhao, S. Chandrudu, M. Skwarczynski and I. Toth, The application of self-assembled nanostructures in peptide-based subunit vaccine development, Eur. Polym. J., 2017, 93, 670–681 CrossRef CAS.
- K. T. Gause, A. K. Wheatley, J. Cui, Y. Yan, S. J. Kent and F. Caruso, Immunological Principles Guiding the Rational Design of Particles for Vaccine Delivery, ACS Nano, 2017, 11(1), 54–68 CrossRef CAS.
- H. Y. R. Madge, R. J. Stephenson and I. Toth, Chapter 20 - Nanocarrier-based vaccine delivery systems for synthetic peptide vaccines, in Handbook of Nanotechnology Applications, ed. W. J. Lau, K. Faungnawakij, K. Piyachomkwan and U. R. Ruktanonchai, Elsevier, 2021, pp. 509–535 Search PubMed.
- M. V. Baranov, M. Kumar, S. Sacanna, S. Thutupalli and G. van den Bogaart, Modulation of Immune Responses by Particle Size and Shape, Front. Immunol., 2021, 11, 3854 Search PubMed.
- S. D. Xiang, A. Scholzen, G. Minigo, C. David, V. Apostolopoulos, P. L. Mottram and M. Plebanski, Pathogen recognition and development of particulate vaccines: does size matter?, Methods, 2006, 40(1), 1–9 CrossRef CAS PubMed.
- V. Ozberk, M. Pandey and M. F. Good, Contribution of cryptic epitopes in designing a group A streptococcal vaccine, Hum. Vaccines Immunother., 2018, 14(8), 2034–2052 CrossRef PubMed.
- S. Reynolds, M. Pandey, J. Dooley, A. Calcutt, M. Batzloff, V. Ozberk, J.-L. Mills and M. Good, Preclinical Safety and Immunogenicity of Streptococcus Pyogenes (Strep A) Peptide Vaccines, Sci. Rep., 2021, 11(1), 127 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01333e |
| ‡ Authors contributed equally to this manuscript |
| This journal is © The Royal Society of Chemistry 2022 |