
A rhabdomyosarcoma hydrogel model to unveil cell-extracellular matrix interactions†
Mattia
Saggioro
ab,
Stefania
D'Agostino
ab,
Anna
Gallo
ac,
Sara
Crotti
 d,
Sara
D'Aronco
d,
Diana
Corallo
e,
Giulia
Veltri
bf,
Gabriele
Martinez
c,
Antonella
Grigoletto
c,
Anna Maria
Tolomeo
bg,
Giovanni
Tafuro
c,
Marco
Agostini
hdg,
Sanja
Aveic
ei,
Valentina
Serafin
bf,
Alessandra
Semenzato
c,
Gianfranco
Pasut
*c and
Michela
Pozzobon
*ab
d,
Sara
D'Aronco
d,
Diana
Corallo
e,
Giulia
Veltri
bf,
Gabriele
Martinez
c,
Antonella
Grigoletto
c,
Anna Maria
Tolomeo
bg,
Giovanni
Tafuro
c,
Marco
Agostini
hdg,
Sanja
Aveic
ei,
Valentina
Serafin
bf,
Alessandra
Semenzato
c,
Gianfranco
Pasut
*c and
Michela
Pozzobon
*ab
aStem Cells and Regenerative Medicine Lab, Institute of Pediatric Research Città della Speranza, 35129 Padova, Italy. E-mail: michela.pozzobon@unipd.it; Tel: +390499640126
bDepartment of Women and Children Health, University of Padova, 35127 Padova, Italy
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, 35131 Padova, Italy. E-mail: gianfranco.pasut@unipd.it; Tel: +390498275694
dNIB Lab Institute of Pediatric Research Città della Speranza, 35129 Padova, Italy
eLaboratory of Target Discovery and Biology of Neuroblastoma, Institute of Pediatric Research Città della Speranza, 35129 Padova, Italy
fOncohematology Laboratory, Institute of Pediatric Research Città della Speranza, 35129 Padova, Italy
gL.i.f.e.L.a.b. Program, Consorzio per la Ricerca Sanitaria (CORIS), 35129 Padova, Italy
hFirst Surgical Clinic Section, Department of Surgical, Oncological and Gastroenterological Sciences, Padova University, 35128 Padova, Italy
iDepartment of Dental Materials and Biomaterials Research, RWTH Aachen University Hospital, Aachen, Germany
First published on 8th November 2021
Abstract
Three-dimensional (3D) culture systems have progressively attracted attention given their potential to overcome limitations of classical 2D in vitro systems. Among different supports for 3D cell culture, hydrogels (HGs) offer important advantages such as tunable mechanical and biological properties. Here, a biocompatible hyaluronic acid-polyethylene glycol HG was developed to explore the pro-migratory behavior of alveolar rhabdomyosarcoma (ARMS) cells. Proteomic analysis of ARMS xenografts unveiled the composition of the extracellular matrix (ECM) elucidating the most representative proteins. In parallel, HGs were obtained by the combination of a thiol-containing hyaluronic acid derivative and different polyethylene glycol (PEG) dimaleimide polymers. The selection of the optimal HG for ARMS cell growth was made based on degradation time, swelling, and cell distribution. Rheology measures and mechanical properties were assessed in the presence or absence of ECM proteins (collagen type I and fibronectin), as well as viability tests and cell distribution analysis. The role of ITGA5, the receptor of fibronectin, in determining ARMS cell migration was validated in vitro upon ITGA5 silencing. In vivo, cell dissemination and the capacity for engrafting were validated after injecting ARMS cell populations enriched for the level of ITGA5 in zebrafish embryos. To study the interactions with ARMS-specific ECM proteins (HG + P), the key players from the Rho and heat-shock pathways were investigated by reverse phase protein array (RPPA). Our data suggest that the developed 3D ARMS model is useful for identifying potential physical hallmarks that allow cancer cells to resist therapy, escape from the immune-system and increase dissemination.
1. Introduction
Two-dimensional (2D) cell culture systems have been so far the conventional approach for the study of cancer biology and drug screening. Although, there is an increasing awareness of their limited ability to reflect the three-dimensional (3D) tumor architecture, the extracellular matrix protein (ECM) composition, and the surrounding cell types that comprise the in vivo tumor milieu. Consequently, 3D cell culture systems are progressively getting consideration among translational research investigations to fill the gaps between the traditional 2D in vitro studies and the in vivo cancer models.1–4 Even though the distance between the cell-based studies and the animal models remains considerable, significant improvements have been made to closely resemble the complexity and heterogeneity of the in vivo tumor microenvironment (TME).5–7 An elegant example is given by the recent work of Chen and co-workers8 where a decellularized colon tissue has been used as a 3D scaffold that recapitulates colon-rectal cancer progression. Although natural scaffolds of biological origin well mimic the multifaced classes of ECM proteins and cytokines present in tissue samples, the intrinsic sample-to-sample variability and the lack of a comprehensive characterization of protein composition still represent an issue for a general and standardized use.9,10 Conversely, synthetic scaffolds might offer a simplified platform that can be engineered in a more controlled, highly reproducible manner and also on a high scale. A polymeric backbone can be modified to finely tune parameters such as porosity, stiffness, and degradation.11,12 Among the most widespread and easy-to-use synthetic scaffolds, hydrogels (HGs) offer tunable and constant chemical, physical and biological conditions to encapsulated cells. In this perspective, 3D HGs represent scaffolds in which cells are suspended within or adhered to the polymeric backbone, thus recapitulating the three-dimensionality of TME.11,13,14 So far, the complexity of proteins included in HGs is generally limited, making these models less representative of a tissue-like environment.15–17 Recently, the great advances of analytical technology allowed the analysis of the ECM protein composition of whole tissues, thus shedding light on the differences between healthy and diseased ECMs.18–20 The combination of the information of proteomic analysis of a given tissue with the development of a functionalized synthetic scaffold is particularly relevant for establishing specific and highly reproducible tissue-like 3D models.In this study we focused on the development of a hyaluronic acid (HA)-based hydrogel that resembles pediatric rhabdomyosarcoma (RMS), in particular the most aggressive alveolar (ARMS) subtype. Sarcomas are the most common cancers in childhood and adolescence. These tumors comprehend a large and heterogeneous group of mesenchymal malignancies. Among them, RMS is the most aggressive and with the lowest overall survival rates. Although therapy modifications improved the overall survival of patients affected by pediatric RMS, further efforts are needed to ameliorate the outcome for young patients, specifically those affected by ARMS. RMSs are classified as rare diseases21,22 and even if the genetic background of the cancer cells has been deeply investigated, a tremendous lack of information still persists for what concerns the composition of the TME. Consequently, to address this unmet medical need we adopted an integrated approach taking into consideration the development of a 3D HG model based on the proteomic characterization of the TME of ARMS. The proposed in vitro 3D model incorporates the most represented proteins in ARMS ECM, as identified by the proteomic analysis of xenogeneic tumor masses. The cell distribution, viability, and key proteins of the Rho pathway (responsible for actin cytoskeleton assembling and contraction23–25) in RH30 cell-cultured HGs were investigated.
The model can be further employed with cells of patient origin for drug testing, with a special focus on metastatic migration inhibition.
2. Experimental section
2.1 Cell culture and spheroid formation
The ARMS RH30 cell line was kindly provided by Solid Tumors Lab (Prof. Bisogno, Padova, Italy). GFP+ RH30 were kindly provided by Dr Rampazzo.26 Integrin alpha 5-negative RH30 cells (ITGA5−RH30) were obtained using ITGA5 siRNA (Santa Cruz Biotechnology, Dallas, Texas, USA) and Mirus TransIT-X2 Transfection Reagent (Mirus Bio, Wisconsin, USA) following the manufacturer's instructions. As a control a siRNA scramble sequence (siNEG, Santa Cruz Biotechnology, Dallas, Texas, USA) was used. Cells were grown in culture medium composed of high glucose DMEM (Gibco, Dublin, Ireland) supplemented with 10% fetal bovine serum (FBS, Gibco, Dublin, Ireland), 1% 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 penicillin/10000 μg mL−1 streptomycin (Gibco, Dublin, Ireland), and 1% 200 mM L-glutamine (Gibco, Dublin, Ireland) in tissue culture flasks (Sarstedt, Nümbrecht, Germany) at 37 °C, 5% CO2 and 95% relative humidity.
000 U mL−1 penicillin/10000 μg mL−1 streptomycin (Gibco, Dublin, Ireland), and 1% 200 mM L-glutamine (Gibco, Dublin, Ireland) in tissue culture flasks (Sarstedt, Nümbrecht, Germany) at 37 °C, 5% CO2 and 95% relative humidity.
For the injection in zebrafish embryos, RH30 cells were sorted for the gated expression of ITGA5 (high vs. low). ITGA5high and ITGA5low cell populations were selected and sorted. In particular, 3–5 × 106 cells per mL (N = 3) were stained with the anti-human ITGA5-PE directly conjugated antibody (Table 1) and analyzed on a MoFlo High Performance Cell Sorter (Beckman Coulter). Relative percentages of the two different subpopulations were calculated based on live gated cells (as indicated by physical parameters, side scatter and forward scatter, and 7AAD negative cells – Invitrogen, Carlsbad, USA).
| Target protein | Producer | Product code | Host | Application |
|---|---|---|---|---|
| ITGA5 PE | R&D Systems | FAB1864P | Mouse | Flow cytometry |
| ITGA5 | R&D Systems | FAB1864 | Mouse | IF |
| CD44 FITC | Thermo Fisher Scientific | 11-0441-82 | Mouse | Flow cytometry |
| 7-AAD | Thermo Fisher Scientific | 00-6993-50 | None | Flow cytometry |
| MYOG | Santa Cruz Biotechnology | sc-576 | Rabbit | IF |
| HuNu | Millipore | MAB1281 | Mouse | IF |
| aMo-594 Alexa Fluor | Invitrogen | A-11005 | Goat | II Ab/IF |
| aRb-594 Alexa Fluor | Invitrogen | A-21442 | Chicken | II Ab/IF |
| ARP2 N1c3 | Genetex | GTX103311 | Rabbit | RPPA |
| HSP70 | Enzo Life Sciences | ADI-SPA-810 | Mouse | RPPA |
| HSP90 | Enzo Life Sciences | SPA-830 | Mouse | RPPA |
| Integrin β1 | Cell Signaling Technology | 9699 | Rabbit | RPPA |
| n-WASP (Ser484/485) | Millipore | AB1964 | Rabbit | RPPA |
| FAK | Cell Signaling Technology | 3285S | Rabbit | RPPA |
| ROCK1 | Abnova | H00006093-M01 | Mouse | RPPA |
| ROCK2 | Abnova | H00009475-M02 | Mouse | RPPA |
| GAPDH | Genetex | GTX 8627408 | Mouse | RPPA |
For spheroid formation, GFP+ RH30 were detached from tissue culture flasks with 0.05% trypsin-EDTA (Merk, Darmstadt, Germany), resuspended in low glucose DMEM (Gibco, Dublin, Ireland) supplemented with 1:50 B27 (Gibco, Dublin, Ireland), 20 ng mL−1 EGF (ORF Genetics, Kopavogur, Iceland), 10 ng mL−1 bFGF (ORF Genetics, Kopavogur, Iceland) and 1% 10000 U mL−1 penicillin/10000 μg mL−1 streptomycin. Cells were seeded in ultra-low adhesion 96 well plates (Corning, New York, USA) at 10 cells per μL cell density and incubated at 37 °C, 5% CO2 and 95% relative humidity for 5 days.
2.2 Xenografts
2.3 Sample staining
For tissue sectioning, samples were fixed in 4% paraformaldehyde (PFA, Merk, Darmstadt, Germany) for 1 h, washed for 10 minutes in PBS on an orbital shaker, included in OCT embedding medium (Kaltek, Padua, Italy) using isopentane (Merk, Darmstadt, Germany) and liquid nitrogen and stored at −80 °C. 10 μm-thick slices (Leica CM1520 cryostat-Leica Biosystems, Wetzlar, Germany) were processed for immunohistochemistry and immunofluorescence. Haematoxylin and eosin staining was performed using a Hematoxylin and Eosin Rapid Frozen Sections Kit, (Bio-Optica, Milan, Italy). For immunofluorescence analyses, fixed cells or frozen sections were permeabilized for 15 min with 0.5% Triton X-100 (Bio-Rad, Hercules, USA), blocked for 15 minutes with 10% horse serum (Gibco, Dublin, Ireland) and incubated with primary antibodies overnight at 4 °C, (Table 1). After washing, slides were incubated 1 h at room temperature with secondary antibodies that were Alexa Fluor-conjugated. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Merk, Darmstadt, Germany) on glass slides or with 1:10000 Hoechst solution (Life Technologies, Carlsbad, USA) on multiwell plates (Corning, New York, USA). Phalloidin (Pha-488, Abcam, Cambridge, UK) staining was performed on whole-mount hydrogel samples that were fixed in 4% PFA for 2 h and washed for 30 minutes in PBS on an orbital shaker. Pha-488 was diluted in a solution of 0.5% Triton X-100 in PBS in ratio 1:2000 (v/v); incubation was maintained at room temperature for 2 h in the dark and under constant agitation on an orbital shaker. Z-Stacks images were acquired using a Zeiss LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Cell viability assays “(Live & Dead assay”-Thermo Fisher, Waltham, Massachusetts, USA; PrestoBlue ™ HS Cell Viability Reagent-Thermo Fisher, Carlsbad, USA) were performed. The first was applied on whole-mount samples of HG embedded cells according to the manufacturer's instructions at 7 days after seeding. Z-Stack images were acquired using a Zeiss LSM 800 confocal microscope. Cell density, roundness and maximum projection were evaluated using Fiji software.29 The second was used on HG + cells, HG + P + cells and Matrigel + cells and following the manufacturer's instruction fluorescence was read at 540–570 nm, at 24 h, 48 h, 96 h and 7 days.
2.4 Proteomic analysis
For proteomic analysis fresh ARMS tissue was processed as indicated in the protocol published by Naba and colleagues.18 Briefly, the decellularization of 6 xenogeneic samples occurred using the “Compartment Protein Extraction Kit” (Millipore, Burlington, Massachusetts, USA). After homogenization, a series of washing and centrifugation steps followed, in which the cytosolic, nuclear and cytoskeletal fractions were collected and separated from the extracellular components. The ECM-enriched fraction was then reduced with DTT (10 mM, Merk, Darmstadt, Germany) in 8 M urea (Merk, Darmstadt, Germany) and alkylated iodoacetamide (25 mM, Merk, Darmstadt, Germany) followed by the deglycosylation with PNGaseF (500000 U mL−1, New England Biolabs). To digest ECM proteins, Lys-C (0.1 μg μL−1, New England Biolabs) and trypsin (0.5 μg μL−1, Sequencing Grade Modified Tyrpsin, PROMEGA) were used until digestion was inactivated by acidification. Samples were finally desalted using Solid-Phase Extraction (SPE) cartridges (SupelTM-Select HLB SPE tubes, Supelco). An Ultimate 3000 HPLC system coupled to a Q Exactive (Thermo Fisher, Waltham, Massachusetts, USA) mass spectrometer was employed. The peptide mixtures were separated with a Biobasic C18 column, 1 mm ID × 150 mm, 5 μm, using a 3–45% linear gradient of acetonitrile (Suprapur grade, Romil) + 0.1% TFA (mobile phase B) in H2O + 0.1% TFA (mobile phase A) over 110 minutes of analysis. Mass spec data were acquired in data-dependent mode in the 300–1500 m/z mass range (this range is sufficient for the detection of very large peptides, having up to 5 or 6 charges). Instrumental parameters were set as follows: source: ESI (+); precursor charge (z) selection: from 2 to 5; precursor resolution: 10000; fragment resolution: 60000. LC-MS/MS data were processed by Proteome Discoverer 2.2 (Thermo Fisher, Waltham, Massachusetts, USA) using the Sequest HT algorithm for protein identification. Search parameters were set as follows: database, SwissProt (v2017-10-25); enzyme, Trypsin (max 2 missed cleavages); taxonomy, homo sapiens or Mus musculus; precursor mass tolerance, 10 ppm, fragment mass tolerance, 0.02 Da. Fixed modifications: carbamidomethyl (C). Dynamic modifications: oxidation (M, P, K); deamidation (N, Q), and phosphorylation (S, T, Y). An acceptable protein false discovery rate (FDR) was set <0.01 and a minimum of 2 non-redundant peptides was used to obtain protein identification.
2.5 Cell migration and invasion test
Migration assay was performed using 8 μm-pore Transwell inserts (Sarstedt, Nümbrecht, Germany) in a 24-well plate, alone or with fibronectin (from Merk, Darmstadt, Germany, 66 mg ml−1 (diluted) was used to coat the inner and outer surface of the Transwell membrane. Trans-wells with coating solution were kept in the incubator for 1 hour) as described elsewhere.26 Briefly, 1 × 105 cancer cells were seeded in the upper chamber, and the lower chamber was filled with serum-free medium. After 24 h, membranes were fixed in 4% PFA and stained with 1:10000 Hoechst solution (Life Technologies, Carlsbad, California, USA) for 15 minutes. Pictures were taken using a Zeiss Axio Observer (Carl Zeiss, Oberkochen, Germany) microscope before and after cleaning the upper side of the membranes with cotton wool. Migrated cells were counted on 4× magnification images using Fiji software.29 For invasion assay, Transwell inserts were pre-coated with 50 μL of Matrigel (Corning, New York, USA); staining and counting of the migrated cells were performed after 48 h, as previously described for the migration test.
2.6 Hydrogel production
For the development of the HA-based hydrogel, a thiol derivative of HA was synthesized as follows: 250 mg (0.618 mmol HA repetitive units) of 200 kDa HA (Fidia Farmaceutici – Abano Terme, Italy) were dissolved in 20 mL of anhydrous dimethyl sulfoxide (DMSO) together with 210 μL (3.09 mmol) of methanesulphonic acid (Merk, Darmstadt, Germany). After dissolution, 502 mg (3.09 mmol) of 1,1-carbonyldiimidazole (Merk, Darmstadt, Germany) were added. The solution was left under stirring for 1 h at room temperature, and then 206 mg (0.927 mmol) of 2-(2-pyridyldithio)ethylamine hydrochloride (SPDC) were added; triethylamine was poured dropwise until the solution turned basic. The reaction was let to proceed overnight under agitation at 40 °C to yield the HA-SPDC intermediate. Then, 2.5 mL of NaCl saturated solution were added and after 15 minutes the solution was added dropwise in cold ethanol, and the precipitate was filtered and washed by using a Gooch filter. The excess of ethanol was removed by drying the precipitate under nitrogen gas flow prior to solubilization in 0.5 M NaOH, and subsequent neutralization with HCl 0.5 M. The product was dialyzed using a membrane with a molecular weight cut-off of 14 kDa against 0.1 M acetate buffer at pH 5 for 48 h and then against water for 24 h. The dialyzed solution was then lyophilized. The product was characterized by (1) TNBS assay, to verify the absence of non-reacted SPDC, and (2) pyridine-2-thione absorbance measurement after its release from the HA intermediate by DTT reduction was performed to quantify the amount of SPDC bound to HA.The final step was the reduction of the disulphide bond in the SPDC molecule: 230 mg (0.55 mmol) of HA-SPDC were reacted with 195 mg (0.55 mmol) of DTT in 10 mL of 50 mM phosphate buffer with 2 mM EDTA at pH 7 for 1 h. The solution was dialyzed against the same buffer for 24 h and then against 1 mM EDTA for 48 h. The dialysis was kept under nitrogen flow. The product was then lyophilized, and the amount of sulfhydryl groups was determined by Ellman's assay.30
HA-SH was resuspended in the culture medium at final concentration 1% [w/v] by gentle pipetting. Pellets of 2 × 105 cells or 3 spheroids were resuspended in culture medium/HA-SH solution and transferred on a glass flat bottom 96 well plate or on a glass slide mounted with flexiPERM silicon chamber slide (Merk, Darmstadt, Germany). This solution mixture was finally crosslinked with 2, 10 or 25 kDa PEG-dimaleimide (Iris Biotech GMBH, Marktredwitz, Germany) to obtain the hydrogel. The ratio of maleimide and thiol groups was stoichiometrically kept at 1:1. Hydrogels were enriched with 100 μg mL−1 recombinant fibronectin (Merk, Darmstadt, Germany) and 600 μg mL−1 collagen I (Enzo, Farmingdale, USA), added to the HA solution before resuspending cells or spheroids.
2.7 Swelling
The swelling capacity of hydrogels was evaluated over time at 1 h, 3 h, 24 h, 48 h, 7 d, 14 d, 21 d and 30 d after gel formation. HGs were incubated in 5 mL of PBS at 37 °C and weighed at the corresponding time-points to obtain the swelled weight (Ws). After drying with absorbent paper, HGs were weighed again to obtain the dry weight post swelling (Wd(t)). Wd(0) is the dry weight at time zero, before the swelling.The equilibrium of swelling (Qs) and the percentual degradation (%deg) are expressed as follows:


2.8 Rheological analyses
Hydrogel blocks of 100 μL (4 mm long and 2 mm thick) with the addition of cells and with or without proteins (collagen I and fibronectin) underwent rheological analysis. Modular Compact Rheometer MCR 92 (Anton Paar, Graz, Austria), with a 25 mm parallel plate geometry with serrated surfaces was used. A stable axial force was achieved (0.03–0.05 N) and a temperature of 23 ± 0.05 °C was maintained. The rheological tests were conducted in triplicate.An amplitude sweep test was done and the storage (G′) and the loss (G′′) moduli were measured at a fixed frequency value of 1 rad per s and increasing the strain (γ) values from 0.2 to 2% in the linear viscoelastic region (LVR), in which the material does not undergo irreversible changes. This test was performed to highlight the hydrogels’ viscoelastic properties and physical stability. The testing parameters settled to outline the rheological characterization of the hydrogels and are listed in Table 2.
| Parameters | Values |
|---|---|
| Diameter of the geometry | 25 mm |
| Temperature | 23 ± 0.05 °C |
| Axial force | 0.03–0.05 N |
| Oscillation frequency | 1 rad per s |
| Oscillation strain | 0.2–2.0% |
| Data points per decade | 15 |
| Conditioning time | 2.0 s |
| Sampling time | 3.0 s |
2.9 Texture analysis
A compression test was performed on blocks of 200 μl hydrogel with cells and proteins, using a Texture Analyzer TMS-Pro, from Food Technology Corporation (Sterling, Virginia, USA). The instrument was equipped with a load cell of 10N and a cylindrical stainless-steel probe with a diameter of 1 cm. The hydrogel blocks were placed on a clean flat aluminum base. The analyses were conducted at room temperature. Through vertical displacement, the probe compressed each sample to a depth of 1 mm at a rate of 60 mm min−1 and then returned to the point it first came in contact with the sample. The pre-test speed was set at 30 mm min−1, while the test speed and the post-test speed were set at 60 mm min−1. The software Texture Lab Pro was used to collect and display the data. The force required to compress the sample was recorded as a function of the distance traveled by the probe and, from the curve plotted as load (N) vs. displacement (mm), the following parameters were obtained: hardness, consistency, and stiffness. Hardness is the positive peak and refers to the maximum value of force, expressed as Newton (N), needed to compress the sample. Consistency is the area under the positive curve (N*mm) and represents the total work of the sample against the deformation. Stiffness is the slope of the curve during the compression phase (N/mm2) and is a further indication of the firmness and deformability of the sample. The results obtained from the texture analysis were expressed as the mean of three repeated analysis ± standard deviation.2.10 Scanning electron microscopy
For scanning electron microscopy (SEM), samples were lyophilized prior to imaging with a CamScan MX3000 scanning electron microscope.2.11 Reverse phase protein array
The Reverse Phase Protein Assay (RPPA) was performed as previously described.31–34 In detail, protein extraction and quantification have been carried out as follows: 6 ARMS fresh tissue (∼20 mg), and a pool of 4 hydrogels with and 4 without ECM proteins were resuspended in 200 μL of “Lysis buffer” composed as follows: (for 1 mL) (915 μl of Tissue Protein Extraction Reagent (TPER) (Pierce, Rockford, IL), 60 μL of NaCl 5 M, 10 μL of sodium orthovanadate 100 mM, 10 μL of Pefabloc 200 mM (Roche Diagnostics, Basel, Switzerland) 1 μL of Aprotinin 5 mg ml−1 (Sigma-Aldrich, St Louis, MO, USA), 5 μl of pepstatin A 1 mg ml−1 (Sigma-Aldrich, St Louis, MO, USA), and 1 μl of leupeptin 5 mg ml−1 (Sigma-Aldrich, St Louis, MO, USA). Of note, to efficiently recover the proteins pellets were resuspended vigorously by pipetting or using disposable micropestles in the case of hydrogels and fresh ARMS tissue. The solution was then vortexed for 30 seconds and incubated on ice for 30 minutes repeating these passages 3 times. Finally, the suspension was centrifuged at 600g for 10 minutes at 4 °C and the supernatant was recovered and stored at −80 °C. “Pierce BCA Protein Assay Kit” (Thermo Fisher, Waltham, Massachusetts, USA) was used to determine the protein concentration using a BSA standard curve, as specified by the supplier. After incubation at 37 °C for 30 minutes, absorbance was measured with a spectrophotometer at λ = 562 nm. Lysates were loaded into a 384-well plate and serially diluted with lysis buffer into four-point dilution curves ranging from undiluted to 1:8 to have a more precise signal quantification and to avoid signal intensity saturation. Protein lysates were printed in quadruplicate in each array set onto nitrocellulose-coated slides (ONCYTE Nitrocellulose Film Slides, Grace Bio-Labs, Oregon, USA) with the 2470 Arrayer (Aushon BioSystems, Massachusetts, USA). Printed slides were stored desiccated at −20 °C until use. For RPPA analysis, one slide was stained for the total GAPDH protein amount to normalize the signal intensity of the other antibodies. This normalization was performed to avoid signal background contamination from the proteins derived from the hydrogel digestion. The other slides were stained with primary antibodies (Table 1), previously validated by western blot (WB) for single band specificity, on an automated slide stainer (Dako Autostainer Plus, Dako-Cytomation, Denmark).
The signal amplification stage was performed through the Amplification System Kit and then signal was revealed using diaminobenzidine/hydrogen peroxide (DAB) as a chromogen-substrate for 5 min (Dako-Cytomation, Denmark). TIF images of the antibody and total protein were analyzed using the Microvigene software (VigeneTech Inc, Massachusetts, USA) to extract numeric intensity protein values from the array images.
2.12 Statistical analysis
Data are expressed as means ± SD. For all experiments (qPCR and tissue analysis), statistical significance was determined using an equal-variance Student's t test or Mann–Whitney U test to compare the two groups. Kruskal–Wallis test was applied to compare all groups. GraphPad software 8 was used, and a p value below 0.05 was considered to be statistically significant. Legend: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.3. Results and discussion
3.1 Xenograft characterization and proteomic analysis of ARMS ECM for the selection of distinctive proteins to be loaded in HGs
In this work we created and characterized the first hydrogel-based 3D model of ARMS, showing that it is possible to shape the cell dissemination process in a tunable scaffold. Our aim was to define the composition of synthetic scaffolds that more closely recapitulate the features of ARMS native tissue. We decided to dissect the protein ECM composition of tumor in order to unveil the composition of this unexplored and complex environment. Our final idea was to engineer the synthetic scaffold that was enriched with the tumor-specific proteins. Indeed, besides cancer cells, the TME is composed of highly cross-linked proteins that play a paramount role in controlling cancer cell growth and dissemination.18,35A xenogeneic model of ARMS26 has been developed in order to characterize the TME. The contribution of human cells in ARMS xenogeneic tumor masses was assessed with human specific Human Nuclei antigen (HuNu) staining that showed an almost exclusive presence of human cells in the xenografts (HuNu+ DAPI+ 87.21% ± 1.23%) over mouse cell infiltrates (HuNu− DAPI+ 12.79% ± 1.23%) (Fig. 1B and C). The tumor mass preserved myogenic lineage as confirmed by a diffuse expression of the RMS pathological marker MyoG (Fig. 1D). From these evidence, we established that the xenogeneic ARMS tumor recapitulates the features of human ARMS.
 | ||
| Fig. 1 Proteomic analysis of the ARMS ECM: (A) haematoxylin and eosin staining of the ARMS xenograft obtained 3 weeks after from subcutaneous injection in Rag2−/−γc−/− mice of the human RH30 cell line. Scale bar 100 μm. (B) Immunofluorescence staining of the ARMS xenograft with Human Nuclei (HuNu) antigen (in red) and DAPI (in blue). Scale bar 50 μm. (C) Percentage of murine infiltrate and stromal cells (DAPI+HuNu−) and human cells (DAPI+HuNu+) per field in xenograft tissue sections (N = 5). (D) Immunofluorescence staining of ARMS molecular marker myogenin (MyoG) in red, DAPI in blue. Scale bar 200 μm. € Results of the proteomic analysis. Number of proteins identified per category. (F) Number of peptides identified for each protein belonging to the categories. (G) Relative abundance of each peptide (peptide abundance) according to the protein's categories. (H) Histogram showing peptide abundance for each protein identified in the category “core ECM proteins”. These are divided into: collagens, glycoproteins and glycosaminoglycans. Red bars are proteins of mouse origin and in blue proteins of human origin (N = 3). | ||
Xenogeneic samples were processed, as already described,18 to enrich samples with ECM proteins. ECM enriched samples were then prepared for MS analysis. The identified proteins were classified as “Core ECM”, “ECM associated” and “Others”.19 Among all the 555 identified proteins (Fig. 1E), residual not-ECM proteins were still present in the samples, 425 from human and 14 from mouse, respectively. The category of “core ECM proteins” was represented by 60 and 43 proteins of human and murine origin, respectively, while 8 human and 5 murine proteins were classified as “ECM associated proteins”. The number of single peptides (Fig. 1F) for the category of “core ECM proteins” was revealed to be 2085 human and 1352 murine peptides. Remarkably, peptide abundance (Fig. 1G), which accounted for the signal intensity of each peptide (correlated with the abundance of the protein) showed that 57.98% and 28.27% (86.25% in total) of the signal was generated by peptides of “core ECM proteins” from mouse and human source, respectively. Taken together these data indicated that the analyzed samples were enriched in ECM proteins.
In panel H of Fig. 1, each bar represents the total signal generated from the contribution (peptide abundance) of all the unique peptides belonging to each “core ECM protein”. This signal correlates with the relative abundance of the protein in the tissue, previously described.26,27 From this analysis we could conclude that the most represented “core ECM proteins” were collagens, mainly collagen 1, 2 and 3 isoforms (55.34% of the total peptide abundance signal); among glycoproteins the most represented were fibrillin, periostin and fibronectin (29.07% of the total peptide abundance signal). The latest is the transmembrane receptor of ITGA5, a key mediator of cell motility and metastasis.36,37 The contribution of glycosaminoglycans was marginal.
3.2 In vitro and in vivo ITGA5 dependent migration of ARMS cells
Among the identified core ECM proteins, we focused on the presence of fibronectin in ARMS TME for its known role in tumor progression, ECM organization and metastasis in many other types of cancer.38 To test if RH30 cancer cells can benefit from the presence of fibronectin in the tumor stroma, cells were transfected with small interfering RNAs targeting integrin a5 (ITGA5) (Table 3), which together with ITGB1 assembles in the classical fibronectin-binding heterodimer ITGA5B1.| Exp. no. | % 7AAD negative cells (alive cells) | % ITGA5+ cells after siRNA |
|---|---|---|
| #1 | 98.74 | 2.12 |
| #2 | 98.13 | 1.63 |
| #3 | 98.14 | 0.53 |
The in vitro migration assay showed how silencing of the ITGA5 subunit is sufficient to decrease cell migration through the membrane of a Boyden chamber (Fig. 2A). The same result was demonstrated when the migration test was performed using fibronectin coating (Fig. S1†). This result underlined the importance of ITGA5 as triggering molecule to bind fibronectin for enhancing cell migration.
 | ||
| Fig. 2 In vitro and in vivo ITGA5 dependent migration of ARMS cells: (A) Transwell migration assay of siNEG (top) RH30 cells and treated with siRNA ITGA5 (bottom). Hoechst (blue) immunofluorescence staining of migrated cells on the bottom surface of the Boyden chamber. The histogram shows the percentage of migrated cells after 24 h for siNEG (black) and siRNA ITGA5 (grey) (N = 3). (B) Transwell invasion of Matrigel coating: siNEG (top) RH30 cells and treated with siRNA ITGA5 (bottom). Hoechst (blue) immunofluorescence staining of cells invading Matrigel. Histogram showing cluster dimension (surface area) of siNEG (black) and siRNA ITGA5 (grey) treated cells. (C) Cytogram of parental RH30 stained with ITGA5-PE antibody and sorted in 2 populations: ITGA5high and ITGA5low based on the expression of ITGA5. (D) Injection of the sorted population in the duct of Cuvier of Tg(fli1:GFP) zebrafish embryos. (E) Immunofluorescence images show GFP vasculature of the zebrafish (green) and RH30 ITGA5high or ITGA5low marked with lipophilic dye Dil (red). Close up of the inter-somitic vasculature, scale bar: 50 μm. Graph: percentage of zebrafish embryos showing extravasation events was reported for the two sorted cell populations. Number of embryos analyzed: N = 62 for ITGA5high and N = 59 for ITGA5low. Dpi: days post injection. ** p < 0.01. | ||
In the invasion test, control and ITGA5 silenced cells seeded on Matrigel coated membranes, presented two completely different organizations: the former proliferated in small spherical cell clusters, while the latter organized in larger and elongated aggregates (Fig. 2B).
To compare in vivo the invasive potential of ITGA5B1 expressing RH30, two populations of RH30 cells were sorted according to ITGA5 expression levels: one with a higher expression of ITGA5 (called ITGA5high, 13.5% of the original population) and a second population expressing ITGA5 at lower levels (called ITGA5low, 27.2% of the original population, Fig. 2C). We demonstrated that the intracellular ITGA5 does not change in the two fractions, indicating the lack of influence of the internal amount of protein expression on motility (Fig. S2†). The obtained populations were fluorescently labeled and injected into the duct of Cuvier of two days post-fertilization Tg(fli1:GFP) zebrafish embryos39 (Fig. 2D). The expression of GFP by zebrafish endothelial cells highlights embryo's vasculature and allows the identification of extravasation events in vivo. At one-day post injection, 30.6 ± 3.68% of the zebrafish injected with ITGA5high cell population showed extravasation events in the trunk region of the transplanted animals. Moreover, those cells displayed the capability to migrate within the surrounding somites, the precursors of skeletal muscle cells. Only 8.5 ± 8.47% of the embryos injected with the RH30 ITGA5low population showed this pro-invasive phenotype (Fig. 2E). To prove that the different expression of ITGA5 is stable over time, and ITGA5high and ITGA5low was investigated by flow cytometry after 4 in vitro passages of the sorted populations (Fig. S3†). Approximately 20% of the cells retained the high-level expression of ITGA5 in the ITGA5high population while 9.1% of cells in the ITGA5low population regained the expression of ITGA5 at high levels (Fig. S3†).
Taken together, these results confirmed the crucial role of ITGA5 in defining RH30 ARMS cell migratory behavior both in vitro and in vivo. The connection between ECM proteins, integrins and cells is a key point for the anchorage and dissemination processes of cells,40 as here demonstrated with the in vivo injection experiment in zebrafish in which only the highly positive ITGA5 cell fraction was able to extravasate from blood circulation and reach the nearby muscle precursor cells.
3.3 Synthesis and characterization of the hyaluronic acid (HA) and polyethylene glycol (PEG) scaffold
The choice to recreate the ARMS ECM using the hydrogel as a mold was mainly triggered by the intrinsic properties of the hydrogel itself. The hydrophilic networks of polymers, cross-linked either by physical or chemical means, are ideal for recapitulating the ECM of tissues and organs.41 Hydrogels can be manipulated and engineered to have a range of characteristics that can fit the biochemical and biophysical properties of a tissue specific ECM as well as adequate porosity for the diffusion of nutrients to foster cell growth. Our hydrogels were based on a HA polymer (200 kDa) partially functionalized with a thiol bearing molecule, which was exploited as a crosslinking point with di-maleimide PEGs of different molecular weights (2–25 kDa). As is known, HA is a linear glycosaminoglycan composed of disaccharide repeating units of D-glucuronic acid (GlcA) and N-acetyl-D-glucosamine (GlcNAc), and is one of the main components in the ECM of living organisms. HA is not a simple structural component of the ECM but it has several physiological roles, also in function of its molecular weight, and greatly influences tumor progression.42 In this regard, the function of its physiological receptor CD44 is relevant, also highly expressed in RMS cells. Through the binding to CD44, HA triggers signaling pathways involved in tumor invasiveness and progression.43,44 On the other hand, thanks to its high biocompatibility, PEG is a widely exploited polymer for biological applications, going from protein conjugation to surface modification.45,46 PEG's high flexibility, confined functionalization only at the polymer end-chains, and its availability in different molecular weights are suitable features for the role as cross-linking agents for HA-based hydrogels. Furthermore, both HA and PEG are highly hydrophilic polymers thus ensuring the best milieu not only for the cells and for their growth but also for the proteins that can be loaded and retained into the hydrogels. From the development of these HA-based hydrogels, we selected PEG 10 kDa as the best cross-linking agent in terms of hydrogels’ stability and rheological properties.We aimed to recreate in vitro a reproducible and tunable platform for testing the effect of ECM proteins on ARMS cell migration. An HA-based hydrogel (HG) was developed considering that HA is the receptor of CD44, a surface antigen greatly expressed in ARMS (Fig. S4A†). PEG of different molecular weights (2 kDa, 10 kDa and 25 kDa,) was studied as the crosslinking agent (Fig. S5†). All tested PEGs formed compact hydrogels, almost instantaneously, with appreciable transparency and consistency (HA-SH functionalization range: 10–16%) (Fig. 3A). The time course of swelling/degradation of the different scaffolds was analyzed by incubation in PBS (Fig. 3B). After 24 h, the HG with PEG 2 kDa was completely degraded making any further characterization not possible. Conversely, HGs with PEG 10 and 25 kDa showed reduced degradation over 30 days (70.7% for PEG 10 kDa and 96.7% for PEG 25 kDa retained weight with respect to the initial values). The swelling behavior of the 10 and 25 kDa PEG HGs over time, shown in Fig. 3C, remained relatively constant and comparable among the two crosslinking agents (swelling coefficient ∼20%). Additional information regarding the topological morphology of the HGs comes from imaging with a scanning electron microscope (Fig. 3D and E). The HG with PEG 25 kDa showed a denser latex, in a filamentous network of fibers. Similarly, the HG prepared with PEG 10 kDa presented structured filamentous fibers but organized in a less firm matrix. The latter appeared more compatible to harbor cells.
 | ||
| Fig. 3 HA-based hydrogel characterization: (A) Gross-appearance of the hydrogel (HGs) obtained from the combination of HA-SH and Mal-PEG-Mal of three molecular weights: 2 kDa (purple), 10 kDa (blue) and 25 kDa (green). (B) Degradation over 30 days of the three different hydrogels calculated as a percentage of the ratio: dry weigh tx/dry weight t0 (N = 5). (C) Swelling ratio over 30 days of the HA-PEG 10 kDa and HA-PEG 25 kDa hydrogels calculated as a ratio of: wet weigh tx/dry weight tx (N = 5). (D) SEM images of HA-PEG 10 kDa. On the left scale bar 200 μm, magnification scale bar 50 μm. (E) SEM images of HA-PEG 25 kDa. On the left scale bar 200 μm, magnification scale bar 50 μm. (F) Rheological characterization of HA-PEG 10 kDa HG with and without collagen I and fibronectin (FN1). On the left storage and loss moduli for HG with (orange) and without (blue) ECM proteins. On the right histograms of hardness, softness and consistency obtained from compression tests (N = 5). n.s. = not significant. | ||
In this regard, the rheological and mechanical properties of HG with PEG 10 kDa in the absence or presence of ECM proteins (HG + P) (collagen I and fibronectin at concentrations of 600 μg mL−1 and 100 μg mL−1, respectively) were thoroughly studied. In the amplitude sweep test both HGs with and without ECM proteins displayed typical gel characteristics, with storage modulus higher than loss modulus (Fig. 3F left) in the range of the shear stress tested. Compression tests measured hardness (0.24 N and 0.27 N for HGs without proteins and with proteins, respectively), stiffness (0.14 N mm−2 and 0.15 N mm−2) and consistency (0.20 N mm and 0.21 N mm), showing that these parameters were not significantly affected by the addition of ECM proteins (Fig. 3F right). The samples possess similar resistance to wear, shrinkage, and degradation. When xenogeneic samples were tested, as expected the mechanical properties appeared to be very different compared to the ones of HG and HG + P (Fig. S6†). Indeed, the tissues possess multiple cell types and proteins with respect to our developed model. However, considering our hydrogels, the analyses confirmed that the HA-based hydrogel prepared with 10 kDa PEG formed a consistent and stable HG over a long period of time (30 days). Moreover, the addition of ECM proteins, for mimicking ARMS TME, did not impair the HG formation nor the physical properties of the scaffold. These results encouraged us to use the HG with PEG 10 kDa for the in vitro culture of RH30 cells and investigate their migratory ability in a 3D ARMS TME-mimicking environment.
3.4 Interaction of ARMS cells in the HA-based hydrogels with and without ECM proteins
Next, we aimed at determining the biocompatibility of the fabricated HA-based hydrogels for in vitro applications. ARMS cells (RH30) were resuspended in HA-SH, just before the reticulation step with the PEG counterpart, and stained with live and dead assay after 7 days of culture. The Z-stack of 3D volumes (638 μm × 638 μm × 500 μm) of cells in HGs were acquired with a confocal microscope. Only marginal levels of cell death were reported, thus showing an overall good viability of ARMS cells (Fig. 4A left panel). Object segmentation determined cell density and cell distribution on the z-axis. Volumes have been divided in segments of 100 μm in height and number of cells normalized on an area of 1 mm2 (Fig. 4A middle panel and histogram). Cell distribution across the intervals showed a density that was higher in the segment between 201 and 300 μm (4109 cells per mm2) and slightly less dense in the lowest and highest segments (2715 cell per mm2 and 2568 cells per mm2, respectively). | ||
| Fig. 4 RH30 cell distribution and viability in HG with PEG 10 kDa HG: (A) on the left. 3D volume view of RH30 cells seeded in the HG and stained with live and dead assay displaying alive cells in green and apoptotic cells in red. Middle. 3D reconstruction of cells seeded in HG where color is assigned using the thermal Look Up table (LUT) according to the position of each cell in the z-axis. On the right. Histogram showing cell distribution calculated as the number of cells per volume of 10 mm3 across the z-axis. (B) Immunofluorescence staining of phalloidin (green) and Hoechst (blue) in RH30 cells seeded in HG, HG with the addition of ECM proteins (HG + P) and Matrigel. On the right: histogram of the cell shape roundness factor calculated for cell shape embedded in HG (ocher), HG + P (light blue) and Matrigel (grey). (C) On the left, graphic representation of RH30-GFP spheroid invasion in HG at different timepoints. On the right, representative images of the analyzed spheroids. (D) Increase in the spheroid invasion area for the three different 3D supports: HG (ocher), HG + P (light blue) and Matrigel (grey), normalized to the spheroid area at t0. Day 12: positive tendence. ** p < 0.01; *** p < 0.001; **** p < 0.0001. | ||
The morphology of RH30 cells resuspended in the HG without ECM proteins (termed HG + cells) appeared more spherical-shaped, and the cells remained isolated in the HG matrix or grew in spherical clusters. Cells embedded in the HG with the structural collagen I and the glycoprotein fibronectin (HG + P) displayed a more elongated shape and were organized in multicellular strands resembling the morphology of cells in Matrigel (Fig. 4B). They were alive (Fig. S7†) and measurement of the morphological parameter “roundness” at 7 days after seeding confirmed that cells cultured on HG showed a more spherical shape (roundness: 0.761 ± 0.029) compared to RH30 grown in HG + P and Matrigel (roundness: 0.608 ± 0.033 and 0.446 ± 0.025, respectively). This shape resembled the one of the mesenchymal migratory phenotype.47,48
Under the HG + P condition, the cells were evenly distributed, alive and, most importantly, were interacting with each other and with the surrounding microenvironment. The important role of fibronectin in RMS toward ECM remodeling has been already proved49 and in a recent work of Keller's group, the presence of collagens in RMS has been investigated as elements that favor metastatization.50
Spheroids of RH30 cells were seeded under the three previously mentioned conditions (HG, HG + P and Matrigel) and cell migration at the edge of the spheroids was followed over time (1 day, 6 days and 12 days) by measuring the area covered from the maximum projection of the acquired z-stack (Fig. 4C). The strong GFP presence underlined the viability of spheroids and for each timepoint, areas were normalized to the area at day 1 for each condition separately. At 1 and 6 days after seeding, no evident differences could be seen among the 3 conditions. On day 12 however, cells growing in HG + P and Matrigel showed a higher grade of invasion when compared to the HG without proteins and to previous timepoints (Fig. 4D). Altogether these data suggest that ECM proteins in HG support the migration of RH30 cells.
The challenge of tuning cell migration and the interaction between the cytoskeleton and ECM has profound implications for studying the disease development and diagnosis. The observed cell elongation in the protein embedded hydrogel prompts us to investigate the molecular pathway characteristic of the mesenchymal mode of migration.
To shed light on how the actin cytoskeleton of cells is differentially regulated in HG and HG + P, a Reverse Phase Protein Array (RPPA) analysis on HG, HG + P and tissue samples from ARMS xenografts was performed (Fig. 5A). This sensitive and precise high-throughput technology for the quantitative measurement of signaling proteins in biological samples,51 allowed us to investigate the dissemination via the Rho pathway, starting from the ITGB1 through CDC42 and ARP2. ITGB1, which forms contact structures with the ECM, and the Focal Adhesion Kinase phosphorylation (FAK)52 proteins were not differentially expressed between the two HG conditions, while both conditions clearly differed from cells deriving from the xenograft (Fig. 5A). We can speculate that, as already proved for some epithelial cancers, in our HA-based scaffold, the cell surface CD44–hyaluronan interactions provide an integrin-independent mechanism of cell migration.53,54 To prove this hypothesis, a preliminary experiment of cell migration with sorted CD44+ and CD44− cells has been performed. We demonstrated that CD44+ cells with respect to CD44− possess higher migration ability, albeit comparable ITGA5 expression (Fig. S4B and C†). Rho-associated protein kinase (ROCK) 1 and 2, involved in actin organization, showed different behavior. ROCK1 was significantly more expressed in the HG + P + cells condition with respect to HG + cells, although ROCK2 was equally present in both HG states (Fig. S8†).
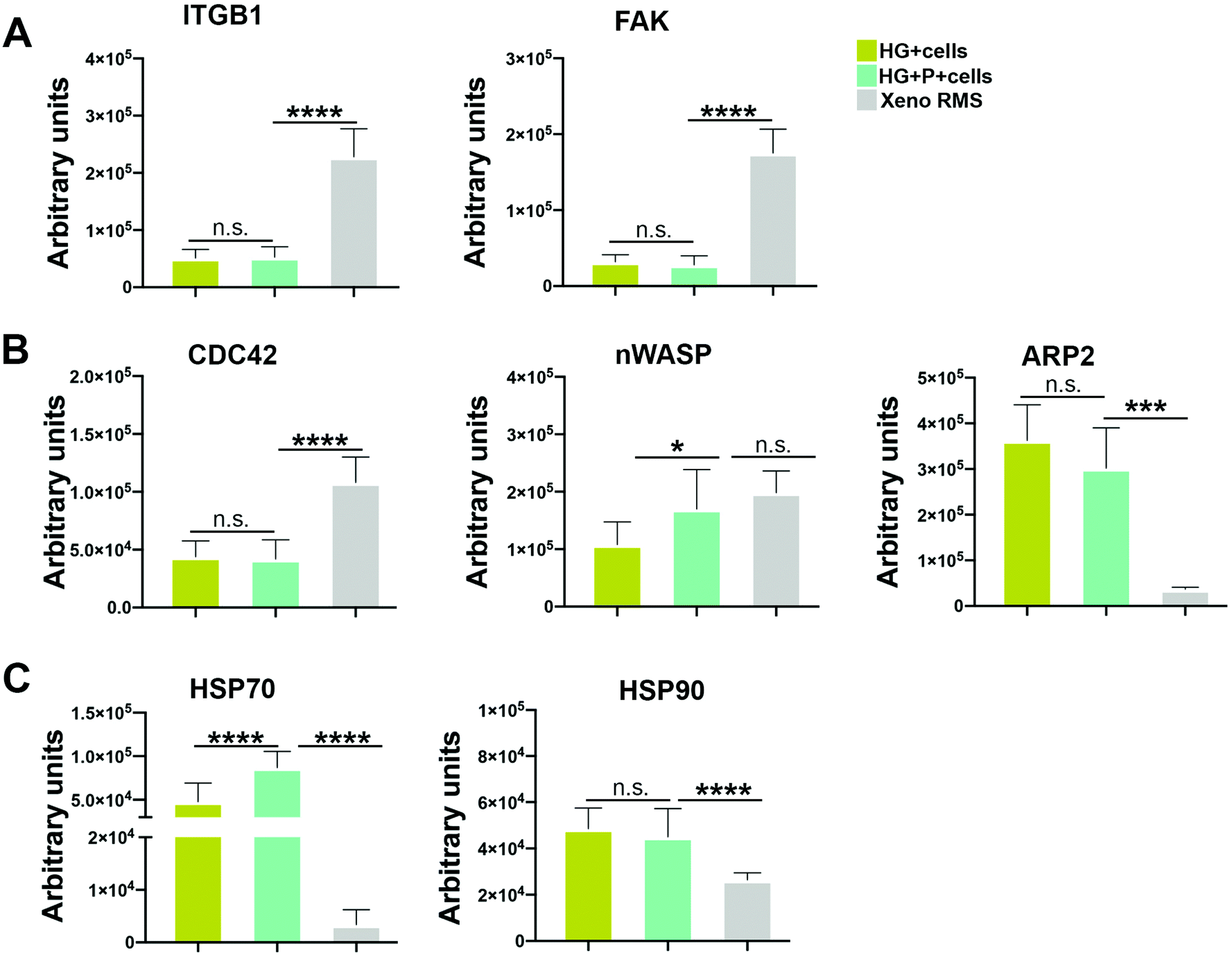 | ||
| Fig. 5 RPPA analysis of RH30 cells grown in HG, HG + P and xenograft models: total protein levels in HG (ocher), HG + P (light blue) and xenograft (light grey) are represented in the histograms. RPPA values are directly normalized by MicroVigene 5.6.0.8 software on GAPDH signals used as the loading control. (A) RPPA analysis of the upper branch of the pathway involving ITGb1 and FAK. (B) RPPA analysis of the lower branch of the pathway involving CDC42, n-WASP, ARP2 (C) RPPA analysis of total protein levels of HSP70 and HSP90, involved in anti-apoptotic and pro-migratory signaling. n.s. = not significant. *** p < 0.001; **** p < 0.0001. | ||
Among the analyzed proteins regulating actin polymerization (CDC42, ARP2/3 and n-WASP), n-WASP levels were higher in RH30 cells grown in HG + P compared to HG and similar to the expression levels of cells extracted from the xenograft (Fig. 5B). ARP2 was also highly expressed under both HG conditions (Fig. 5B). Regarding the heat-shock proteins, HSP70 and HSP90 proteins, sharing anti-apoptotic features, the strongest expression of HSP70 was seen in the HG + P, with the expression in HG being also higher when compared to the xenogeneic controls. The levels of HSP90 were similar for the HG and HG + P but significantly higher than that in the xenograft (Fig. 5C). The expression of the multifunctional HSP70 and HSP90 proteins, which suppress apoptosis, bypass cellular senescence and support metastasis,55–57 was an additional confirmation that cells were active and well preserved in the scaffold.
These data taken together suggest that morphological differences in RH30 elongation, when cultured in HG and HG + P, might not be due to differences in cell–ECM adhesions and formation of focal adhesions. Our preliminary results on CD44 will be more deeply investigated. The levels of actin polymerizing proteins are not dramatically changed between HG and HG + P, only levels of n-WASP differed across the two conditions. The high levels of HSP70 and HSP90 underlined the high cell vitality in the 3D model.
Other groups already demonstrated that the Wiskott–Aldrich syndrome protein (WASP) and the neural WASP (n-WASP) interact with actin filaments through the bound with the Arp2/3 complex and are regulated in turn by binding to GTP-bound Cdc42.58 Cells in our 3D model, and specifically under the condition with ECM proteins, significantly expressed n-WASP and phosphorylated Arp2, confirming that the phosphorylation of Arp2 protein increases cell motility.59 Further experiments are needed to clarify if n-WASP is the only effector for the morphological difference in cell elongation or there might be another mechanism regulating cell shape in the presence of ECM proteins.
4. Conclusions
In summary, RMS is a rare cancer of mesenchymal origin that has not been subject to systematic studies related to the microenvironment that surround the cells.In particular, the need to improve pediatric patient outcome affected by ARMS led us to the conclusion that the development and the testing of novel selective therapies directed toward suitable molecular targets, not only in tumor cells but also in the TME, are of paramount relevance. This approach requires an in vitro testing strategy that recapitulates the natural features of the tumors.
Our work created the first 3D model of ARMS with tumor-enriched ECM proteins. We are planning to perform proteomic analysis on patient specimens not only to validate our results but also to add more ECM proteins characteristics of ARMS TME. Although, in sarcoma, cancer cells are the most represented in TME and are self-sufficient in ECM remodeling,26 the heterogeneous cells isolated by patient biopsies will better exemplify the cell compartment present in ARMS.
With this work, we start defining, for the first time, the protein ECM composition of RMS. The incorporation of these ECM cues into 3D tumor models opens new avenues for the investigation of biological mechanisms of cancer cell migration and disease progression.
Author contributions
MS: conceptualization, collection and assembling of the data, writing. SD: 3D model assessment, collection and assembling of the data. AG: hydrogel synthesis, 3D model assessment. GM, AGr: hydrogel synthesis. SC, SD: proteomics. DC: zebrafish experiments. GV, VS: RPPA experiments. GT: rheological analysis. AMT, MA, SA, AS: collection and assembling of the data. MP, GP: conceptualization, data interpretation and curation, writing, funding acquisition. All the authors revised the manuscript and approved it in the present form.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We thank Elena Carbonin for her skillful support in hydrogel synthesis. Anna Maria Tolomeo is supported by the ‘Consorzio per la Ricerca Sanitaria’ (CORIS) of the Veneto Region, Italy (L.i.f.e.L.a.b. Program), grant number DGR1017, July 17, 2018. V. S. has received funding from Fondazione Associazione Italiana per la Ricerca sul Cancro (AIRC) under MFAG 2018 – ID. 21771 project – P.I. Serafin Valentina. GP's Lab has been supported from AIRC (IG 2017 – ID. 20244 project – P.I. Pasut Gianfranco) and by the Italian Ministry of Health (“Ricerca Finalizzata” GR-2011-02351128 – P.I. Pasut Gianfranco). This research was supported by the Department of Women and Children Health and of Pharmaceutical and Pharmacological Sciences, University of Padova.References
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CAS.
- S. A. Langhans, Front. Pharmacol., 2018, 9, 1–14 CrossRef PubMed.
- C. Fischbach, R. Chen, T. Matsumoto, T. Schmelzle, J. S. Brugge, P. J. Polverini and D. J. Mooney, Nat. Methods, 2007, 4, 855–860 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- N. Vidavsky, J. A. Kunitake, A. E. Chiou, P. A. Northrup, T. J. Porri, L. Ling, C. Fischbach and L. A. Estroff, Biomaterials, 2018, 179, 71–82 CrossRef CAS PubMed.
- C. Ringuette Goulet, G. Bernard, S. Chabaud, A. Couture, A. Langlois, B. Neveu, F. Pouliot and S. Bolduc, Biomaterials, 2017, 145, 233–241 CrossRef CAS PubMed.
- E. L. S. Fong, S.-E. Lamhamedi-Cherradi, E. Burdett, V. Ramamoorthy, A. J. Lazar, F. K. Kasper, M. C. Farach-Carson, D. Vishwamitra, E. G. Demicco, B. A. Menegaz, H. M. Amin, A. G. Mikos and J. A. Ludwig, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6500–6505 CrossRef CAS PubMed.
- H. J. Chen, Z. Wei, J. Sun, A. Bhattacharya, D. J. Savage, R. Serda, Y. Mackeyev, S. A. Curley, P. Bu, L. Wang, S. Chen, L. Cohen-Gould, E. Huang, X. Shen, S. M. Lipkin, N. G. Copeland, N. A. Jenkins and M. L. Shuler, Nat. Biotechnol., 2016, 1–9 Search PubMed.
- G. S. Hussey, J. L. Dziki and S. F. Badylak, Nat. Rev. Mater., 2018, 3, 159–173 CrossRef CAS.
- G. G. Giobbe, C. Crowley, C. Luni, S. Campinoti, M. Khedr, K. Kretzschmar, M. M. De Santis, E. Zambaiti, F. Michielin, L. Meran, Q. Hu, G. van Son, L. Urbani, A. Manfredi, M. Giomo, S. Eaton, D. Cacchiarelli, V. S. W. Li, H. Clevers, P. Bonfanti, N. Elvassore and P. De Coppi, Nat. Commun., 2019, 10, 5658 CrossRef PubMed.
- S. Nam, R. Stowers, J. Lou, Y. Xia and O. Chaudhuri, Biomaterials, 2019, 200, 15–24 CrossRef CAS PubMed.
- J. S. Miller, C. J. Shen, W. R. Legant, J. D. Baranski, B. L. Blakely and C. S. Chen, Biomaterials, 2010, 31, 3736–3743 CrossRef CAS PubMed.
- C. Fischbach, H. J. Kong, S. X. Hsiong, M. B. Evangelista, W. Yuen and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 399–404 CrossRef CAS PubMed.
- O. Hasturk, K. E. Jordan, J. Choi and D. L. Kaplan, Biomaterials, 2020, 232, 119720 CrossRef CAS PubMed.
- S. K. Seidlits, C. T. Drinnan, R. R. Petersen, J. B. Shear, L. J. Suggs and C. E. Schmidt, Acta Biomater., 2011, 7, 2401–2409 CrossRef CAS PubMed.
- E. Türker, Ü. H. Yildiz and A. Arslan Yildiz, Int. J. Biol. Macromol., 2019, 139, 1054–1062 CrossRef PubMed.
- J. Arulmoli, H. J. Wright, D. T. T. Phan, U. Sheth, R. A. Que, G. A. Botten, M. Keating, E. L. Botvinick, M. M. Pathak, T. I. Zarembinski, D. S. Yanni, O. V. Razorenova, C. C. W. Hughes and L. A. Flanagan, Acta Biomater., 2016, 43, 122–138 CrossRef CAS PubMed.
- A. Naba, K. R. Clauser and R. O. Hynes, J. Vis. Exp., 2015, 101, 1–9 Search PubMed.
- A. Naba, K. R. Clauser, S. Hoersch, H. Liu, S. A. Carr and R. O. Hynes, Mol. Cell. Proteomics, 2012, 11, 1–18 CrossRef PubMed.
- A. Naba, O. M. T. Pearce, A. Del Rosario, D. Ma, H. Ding, V. Rajeeve, P. R. Cutillas, F. R. Balkwill and R. O. Hynes, J. Proteome Res., 2017, 16, 3083–3091 CrossRef CAS PubMed.
- S. Ognjanovic, A. M. Linabery, B. Charbonneau and J. A. Ross, Cancer, 2009, 115, 4218–4226 CrossRef PubMed.
- E. R. Rudzinski, J. R. Anderson, D. S. Hawkins, S. X. Skapek, D. M. Parham and L. A. Teot, Arch. Pathol. Lab. Med., 2015, 139, 1281–1287 CrossRef PubMed.
- M. N. Hayes, K. McCarthy, A. Jin, M. L. Oliveira, S. Iyer, S. P. Garcia, S. Sindiri, B. Gryder, Z. Motala, G. P. Nielsen, J. P. Borg, M. van de Rijn, D. Malkin, J. Khan, M. S. Ignatius and D. M. Langenau, Cell Stem Cell, 2018, 22, 414–427 CrossRef CAS PubMed.
- S. Thuault, F. Comunale, J. Hasna, M. Fortier, D. Planchon, N. Elarouci, A. De Reynies, S. Bodin, A. Blangy and C. Gauthier-Rouvière, Mol. Biol. Cell, 2016, 27, 2653–2661 CrossRef CAS PubMed.
- F. Ramadan, A. Fahs, S. E. Ghayad and R. Saab, Cancer Metastasis Rev., 2020, 39, 287–301 CrossRef PubMed.
- S. D’Agostino, L. Tombolan, M. Saggioro, C. Frasson, E. Rampazzo, S. Pellegrini, F. Favaretto, C. Biz, P. Ruggieri, P. Gamba, P. Bonvini, S. Aveic, R. Giovannoni and M. Pozzobon, Front. Oncol., 2021, 1–14 Search PubMed.
- M. Pozzobon, M. Saggioro, S. D'Agostino, G. Bisogno, M. Muraca and P. Gamba, Methods Mol. Biol., 2017, 257–284 Search PubMed.
- K. Stoletov, V. Montel, R. D. Lester, S. L. Gonias and R. Klemke, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17406–17411 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carrera, E. Frise, K. Verena, L. Mark, P. Tobias, P. Stephan, R. Curtis, S. Stephan, S. Benjamin, T. Jean-Yves, J. W. Daniel, H. Volker, E. Kevin, T. Pavel and C. Albert, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- F. Cimmino, L. Pezone, M. Avitabile, L. Persano, M. Vitale, M. Sassi, S. Bresolin, V. Serafin, N. Zambrano, A. Scaloni, G. Basso, A. Iolascon and M. Capasso, J. Proteome Res., 2016, 15, 3643–3655 CrossRef CAS PubMed.
- V. Serafin, V. Lissandron, B. Buldini, S. Bresolin, M. Paganin, F. Grillo, N. Andriano, C. Palmi, G. Cazzaniga, S. Marmiroli, V. Conter, G. Basso and B. Accordi, Leukemia, 2017, 31, 1007–1011 CrossRef CAS PubMed.
- L. A. Liotta, V. Espina, A. L. Mehta, V. Calvert, K. Rosenblatt, D. Geho, P. J. Munson, L. Young, J. Wulfkuhle and E. F. Petricoin, Cancer Cell, 2003, 3, 317–325 CrossRef CAS PubMed.
- M. T. Zampini, C. Tregnago, V. Bisio, L. Simula, G. Borella, E. Manara, C. Zanon, F. Zonta, V. Serafin, B. Accordi, S. Campello, B. Buldini, A. Pession, F. Locatelli, G. Basso and M. Pigazzi, Leukemia, 2018, 32, 1124–1134 CrossRef CAS PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed.
- F. Pantano, M. Croset, K. Driouch, N. Bednarz-Knoll, M. Iuliani, G. Ribelli, E. Bonnelye, H. Wikman, S. Geraci, F. Bonin, S. Simonetti, B. Vincenzi, S. S. Hong, S. Sousa, K. Pantel, G. Tonini, D. Santini and P. Clézardin, Oncogene, 2021, 40, 1284–1299 CrossRef CAS PubMed.
- H. Jin and J. Varner, Br. J. Cancer, 2004, 90, 561–565 CrossRef CAS PubMed.
- G. Efthymiou, A. Saint, M. Ruff, Z. Rekad, D. Ciais and E. Van Obberghen-Schilling, Front. Oncol., 2020, 1–18 Search PubMed.
- D. Corallo, M. Donadon, M. Pantile, V. Sidarovich, S. Cocchi, M. Ori, M. De Sarlo, S. Candiani, C. Frasson, M. Distel, A. Quattrone, C. Zanon, G. Basso, G. P. Tonini and S. Aveic, Cell Death Differ., 2020, 27, 1225–1242 CrossRef PubMed.
- G. Sökeland and U. Schumacher, Mol. Cancer, 2019, 18, 12 CrossRef PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- J. Necas, L. Bartosikova, P. Brauner and J. Kolar, Vet. Med., 2008, 53, 397–411 CrossRef CAS.
- L. Y. W. Bourguignon, M. Shiina and J.-J. Li, Advances in Cancer Research, Academic Press Inc., 2014, vol. 123, pp. 255–275 Search PubMed.
- G. Tzircotis, R. F. Thorne and C. M. Isacke, J. Cell Sci., 2005, 118, 5119–5128 CrossRef CAS PubMed.
- S. Zalipsky and G. Pasut, Polymer-Protein Conjugates, Elsevier, 2020, pp. 3–22 Search PubMed.
- J. E. Leslie-Barbick, J. E. Saik, D. J. Gould, M. E. Dickinson and J. L. West, Biomaterials, 2011, 32, 5782–5789 CrossRef CAS PubMed.
- T. Uynuk-Ool, M. Rothdiener, B. Walters, M. Hegemann, J. Palm, P. Nguyen, T. Seeger, U. Stöckle, J. P. Stegemann, W. K. Aicher, B. Kurz, M. L. Hart, G. Klein and B. Rolauffs, J. Tissue Eng. Regener. Med., 2017, 11, 3508–3522 CrossRef CAS PubMed.
- K. Paňková, D. Rösel, M. Novotný and J. Brábek, Cell. Mol. Life Sci., 2010, 67, 63–71 CrossRef PubMed.
- H. Ito, M. Duxbury, E. Benoit, R. S. Farivar, J. Gardner-Thorpe, M. J. Zinner, S. W. Ashley and E. E. Whang, Biochem. Biophys. Res. Commun., 2004, 318, 594–600 CrossRef CAS.
- X. Lian, J. S. Bond, N. Bharathy, S. P. Boudko, E. Pokidysheva, J. F. Shern, M. Lathara, T. Sasaki, T. Settelmeyer, M. M. Cleary, A. Bajwa, G. Srinivasa, C. P. Hartley, H. P. Bächinger, A. Mansoor, S. H. Gultekin, N. E. Berlow and C. Keller, Front. Oncol., 2021, 11, 38 Search PubMed.
- S. Boellner and K.-F. Becker, Microarrays, 2015, 4, 98–114 CrossRef CAS PubMed.
- K. Polyak and R. A. Weinberg, Nat. Rev. Cancer, 2009, 9, 265–273 CrossRef CAS PubMed.
- R. C. Casey and A. P. N. Skubitz, Clin. Exp. Metastasis, 2000, 18, 67–75 CrossRef CAS PubMed.
- L. T. Senbanjo and M. A. Chellaiah, Front. Cell Dev. Biol., 2017, 1–6 Search PubMed.
- D. T. H. Dimas, C. D. Perlepe, T. N. Sergentanis, I. Misitzis, K. Kontzoglou, E. Patsouris, G. Kouraklis, T. Psaltopoulou and A. Nonni, Anticancer Res., 2018, 38, 1551–1562 CAS.
- E. Lesko, J. Gozdzik, J. Kijowski, B. Jenner, O. Wiecha and M. Majka, Anticancer Drugs, 2007, 18, 1173–1181 CrossRef CAS PubMed.
- E. Lukasiewicz, K. Miekus, J. Kijowski, J. Gozdzik, M. Wilusz, S. Bobis-Wozowicz, O. Wiecha and M. Majka, J. Physiol. Pharmacol., 2009, 60, 161–166 CAS.
- R. Rohatgi, P. Nollau, H.-Y. H. Ho, M. W. Kirschner and B. J. Mayer, J. Biol. Chem., 2001, 276, 26448–26452 CrossRef CAS PubMed.
- L. L. LeClaire, M. Baumgartner, J. H. Iwasa, R. D. Mullins and D. L. Barber, J. Cell Biol., 2008, 182, 647–654 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm00929j |
| This journal is © The Royal Society of Chemistry 2022 |