
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalyzed Dowd–Beckwith radical-polar crossover reaction for the synthesis of medium-sized carbocyclic compounds†
Tushar
Singha
 ,
Ganesh Arjun
Kadam
and
Durga Prasad
Hari
*
,
Ganesh Arjun
Kadam
and
Durga Prasad
Hari
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore, 560012, India. E-mail: dphari@iisc.ac.in
First published on 25th May 2023
Abstract
The Dowd–Beckwith reaction, a ring-expansion of carbonyl compounds via alkoxy radicals, is a powerful approach for synthesizing medium to large-sized carbocyclic scaffolds, which takes advantage of existing ring structures and avoids entropic and enthalpic factors that arise from the end-to-end cyclization strategies. However, the Dowd–Beckwith ring-expansion followed by H-atom abstraction is still the dominating pathway, which hampers its synthetic applications, and there currently exist no reports on the functionalization of ring-expanded radicals using non-carbon based nucleophilic reagents. Herein, we report a redox-neutral decarboxylative Dowd–Beckwith/radical-polar crossover (RPC) sequence that delivers functionalized medium-sized carbocyclic compounds with broad functional group tolerance. The reaction allows one-carbon ring-expansion of 4-, 5-, 6-, 7-, and 8-membered ring substrates and can also be applied to three-carbon chain incorporation, enabling remote functionalization in medium-sized rings.
Introduction
Medium-sized carbocyclic compounds, in particular, oxygenated seven- and eight-membered rings, are important frameworks that can be found in a wide variety of naturally occurring bioactive molecules and pharmaceutical drugs (Scheme 1A).1 Furthermore, the introduction of medium-sized conformationally flexible rings enhances the secondary binding of an inhibitor without adding to the structure's stereochemical complexity.2 These salient features have witnessed an upsurge in both the synthesis and applications of these privileged motifs in medicinal chemistry discovery programmes within academia and the pharmaceutical industry.1 Nevertheless, unfavourable enthalpy and entropic factors and as well as transannular interactions make medium-sized ring synthesis very challenging and substrates with a fixed reaction site are required.3 As a result, varying the ring size usually requires a different substrate design, which is both time-consuming and expensive. In this context, ring-expansion strategies are highly attractive as they take advantage of the existing ring structures and avoid the kinetic and thermodynamic challenges associated with direct cyclization approaches.3b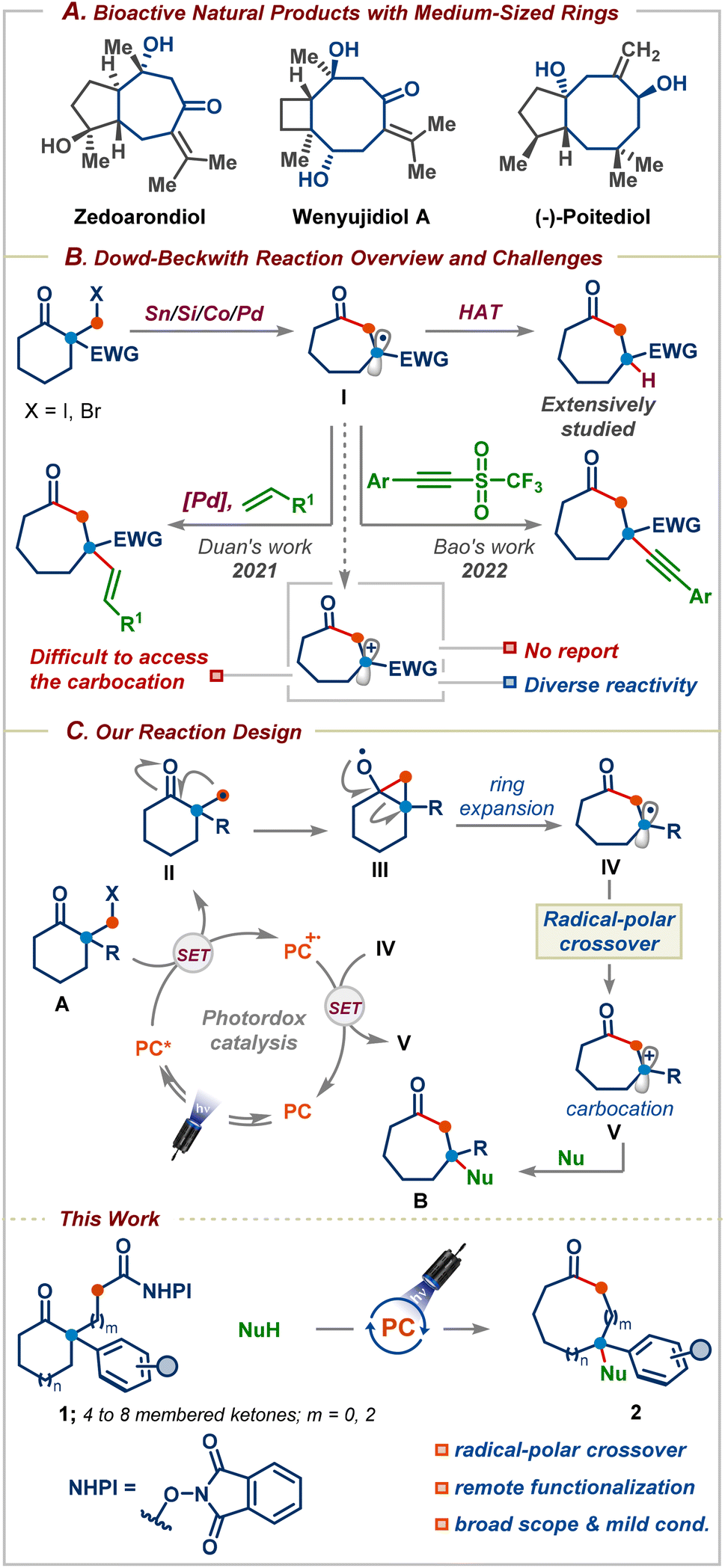 | ||
| Scheme 1 (A) Bioactive natural products containing medium-sized rings, (B) Dowd–Beckwith reaction overview and challenges, and (C) Dowd–Beckwith/radical-polar crossover (this work). | ||
Three decades ago, Dowd and Beckwith first reported an elegant approach for the synthesis of medium-sized rings based on the ring-expansion of ketones via alkoxy radicals (Scheme 1B).4 The efficacy of this reaction has unsurprisingly resulted in its application to the synthesis of several complex bioactive natural products and pharmaceuticals.5 Despite the emergence of the Dowd–Beckwith reaction,6 this reaction often requires elevated temperatures, environmentally unfriendly haloketone substrates, and stoichiometric amounts of tin-based reagents, which are unstable and it is tedious to remove the tin by-products whose disposal poses a hazardous problem.6a Furthermore, the ring-expansion followed by H-atom abstraction is still the dominating pathway, which hampers its application in the synthesis of functionalized medium-sized rings.6a A recent breakthrough has been realized based on light-induced palladium catalysis for the Dowd–Beckwith/Heck type coupling cascade for the synthesis of β-alkenylated cyclic ketones possessing a quaternary carbon center (Scheme 1B).7 Very recently, Bao and co-workers successfully trapped the ring-expanded radical with a triflone reagent to access medium-sized rings (Scheme 1B).8 Although these strategies have been successful, they are mainly limited to activated carbon-based radical-trapping reagents. To the best of our knowledge, there are no reports on the functionalization of the Dowd–Beckwith ring-expanded radicals using non-carbon-based nucleophilic reagents. Thus, the development of mechanistically distinct strategies for applying nucleophilic coupling partners could open up significant chemical space and lead to new opportunities in organic synthesis due to the vast abundance and diversity of nucleophilic reagents. Evidently, such a strategy requires oxidation of the radical I before reacting with its nucleophilic partner (Scheme 1B). In this context, we sought to consider the possibility of a net-neutral radical/polar crossover strategy9 to induce a Dowd–Beckwith/carbon–heteroatom bond formation cascade. We envisaged that the reduction of alkyl radical precursor A by an excited state photocatalyst would furnish the primary radical II that could add onto the neighbouring carbonyl group to provide cyclopropyloxy radical III (Scheme 1C). Subsequent ring-expansion would give radical intermediate IV. To achieve cationic radical-polar crossover (RPC), the radical IV should be electron-rich, unlike in the traditional Dowd–Beckwith reaction where the ring-expanded radical I is often electron deficient. Therefore, we believed that incorporating an aryl/alkyl group α to the carbonyl group is highly important, which not only facilitates the ring-expansion but also offers a means to divert the prototypical reactivity and provide carbocations upon single-electron oxidation. Finally, the carbocation V would subsequently be entrapped by a nucleophile. Herein, we demonstrated the successful realization of a redox-neutral decarboxylative Dowd–Beckwith ring-expansion followed by the RPC sequence for the synthesis of functionalized medium-sized carbocyclic compounds. The reaction proceeds under ambient conditions, displays excellent functional group tolerance, and is shown to allow one-carbon ring-expansion of 4-, 5-, 6-, 7-, and 8-membered ring substrates. Furthermore, we show that this process can also be applied to three-carbon chain incorporation, enabling remote functionalization in medium-sized rings. Finally, the obtained products could be easily transformed into valuable cyclic 1,3-diols and seven-membered ring-containing indoles and cyclobutane derivatives.
Results and discussion
We began our investigation by studying the reaction of ethyl 1-(iodomethyl)-2-oxocyclohexane-1-carboxylate (3), a commonly employed radical precursor for the Dowd–Beckwith reaction.6a Irradiation of 3 in MeOH using 2 mol% 4-CzIPN as a photocatalyst didn’t deliver the desired ring-expansion product 4, and the starting material was simply reisolated (Scheme 2). The lack of reactivity was attributed to the high positive reduction potential of electrophilic radical intermediate VI, which can be difficult to oxidize using the ground-state oxidized photocatalyst. To overcome this, we sought an alternative radical precursor that would enable the RPC step to afford the carbocation intermediate. Subjecting 2-(iodomethyl)-2-phenylcyclohexan-1-one (5) to previous conditions, although it didn't deliver the desired methanol addition product 2a, it gave the enone product 6via a possible intermediate VII, which could be formed through RPC followed by iodide addition (Scheme 2). However, analysis of the crude reaction mixture showed a very poor conversion of 5, perhaps arising from inefficient single-electron reduction of the C–I bond (E1/2 = −2.04 V vs. SCE of 5).10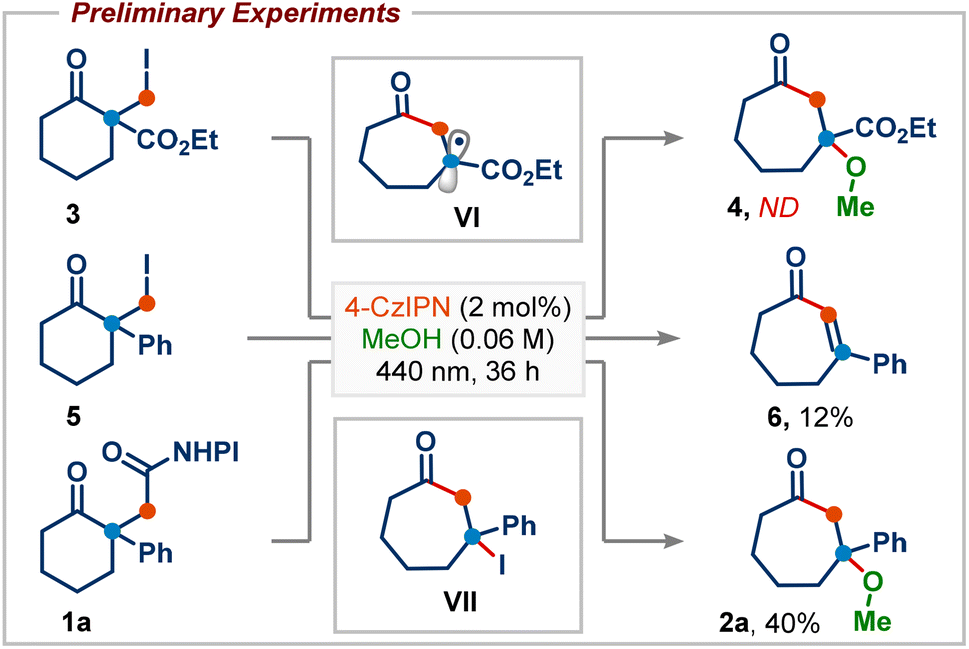 | ||
| Scheme 2 Preliminary experiments. ND = not detected. | ||
Recently, numerous research groups have found that redox-active esters are more reactive than alkyl iodides and provide alkyl radicals via single-electron reduction followed by decarboxylation with the release of a non-nucleophilic leaving group.11 Pleasingly, irradiation of 1a (E1/2 = −1.23 V vs. SCE of 1a)10 in MeOH provided the desired ring-expansion product 2a with 40% yield (Scheme 2).12 A substantial increase in the reaction yield was observed by employing more reducing photocatalysts, probably due to the more favourable single-electron transfer between the excited state IrIII and redox-active ester 1a (Table 1, entries 2–4). A common limitation in trapping carbocations with oxygen nucleophiles is that they are often required as solvents.13 This renders the process uneconomical, especially when expensive or non-commercially available alcohols are used. Next, we screened various solvents using methanol as the reagent (Table 1, entries 5–9). We were delighted to find that product 2a was obtained in 91% yield using 2.5 equiv. of MeOH in acetonitrile solvent (Table 1, entry 9). This shows that the use of methanol as a solvent is not required. Photocatalyst loading could be reduced to 1 mol% without affecting the reaction yield (Table 1, entry 10). Control experiments highlighted the importance of both light and the photocatalyst in promoting the reaction, as the product was not observed in their absence (Table 1, entries 11 and 12).
|
|
||||
|---|---|---|---|---|
| Entry | PC (2 mol%) | Solvent | MeOH (xx equiv.) | Yieldb |
| a General conditions: 1a (0.1 mmol), solvent (0.06 M), photocatalyst (2 mol%), 440 nm, 36 h. b NMR yields using dibromomethane as the internal standard. c 0.24 M instead 0.06 M. d Using 1 mol% of Ir(ppy)3. e No light. ND = not detected. | ||||
| 1 | 4-CzIPN | MeOH | — | 40 |
| 2 | Ir(ppy)3 | MeOH | — | 83 |
| 3 | [Ir]-1 | MeOH | — | 77 |
| 4 | [Ir]-2 | MeOH | — | 80 |
| 5 | Ir(ppy)3 | DCM | 5.0 | 66 |
| 6 | Ir(ppy)3 | Toluene | 5.0 | 16 |
| 7 | Ir(ppy)3 | DMF | 5.0 | ND |
| 8 | Ir(ppy)3 | MeCN | 5.0 | 85 |
| 9c | Ir(ppy)3 | MeCN | 2.5 | 91 |
| 10c,d | Ir(ppy)3 | MeCN | 2.5 | 92 |
| 11c,e | Ir(ppy)3 | MeCN | 2.5 | ND |
| 12c | — | MeCN | 2.5 | ND |
|
||||

Having established reaction conditions for the decarboxylative Dowd–Beckwith/RPC reaction, we explored the scope of the transformation (Scheme 3). A wide range of primary alcohols appended to acyclic chains or to various rings (cyclopropyl, cyclobutyl, and cyclohexyl) can serve as suitable nucleophiles, generating corresponding products in good to high yields (products 2b–2e). Benzylic, allylic, and propargylic alcohols were tolerated well in this reaction (products 2f–2h). Both cyclic and acyclic secondary alcohols furnished the corresponding seven-membered ring-containing congested ethers 2i–2l in moderate to good yields. Given the natural abundance of alcohols, we proceeded to use bio-compatible alcohols: the terpenes menthol and borneol underwent the desired transformation successfully, providing the ethers 2m and 2n in 42% (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and 49% (1:1 dr) yields, respectively. Tertiary alcohols did not give the desired products, presumably because of their low nucleophilicity. We were pleased to find that simply replacing the alcohol in our reaction with water cleanly afforded the desired product 2o in 83% yield. The reaction was not limited to oxygen nucleophiles, N- and C-centered nucleophiles were also competent. Pyrazole underwent addition to generate 3-aminocycloheptan-1-one derivative 2p in 53% yield. 1,3,5-Trimethoxybenzene delivered the product 2q with an all-carbon quaternary center. The structure of 2q was determined by X-ray analysis (CCDC 2263930).
:1 dr) and 49% (1:1 dr) yields, respectively. Tertiary alcohols did not give the desired products, presumably because of their low nucleophilicity. We were pleased to find that simply replacing the alcohol in our reaction with water cleanly afforded the desired product 2o in 83% yield. The reaction was not limited to oxygen nucleophiles, N- and C-centered nucleophiles were also competent. Pyrazole underwent addition to generate 3-aminocycloheptan-1-one derivative 2p in 53% yield. 1,3,5-Trimethoxybenzene delivered the product 2q with an all-carbon quaternary center. The structure of 2q was determined by X-ray analysis (CCDC 2263930).
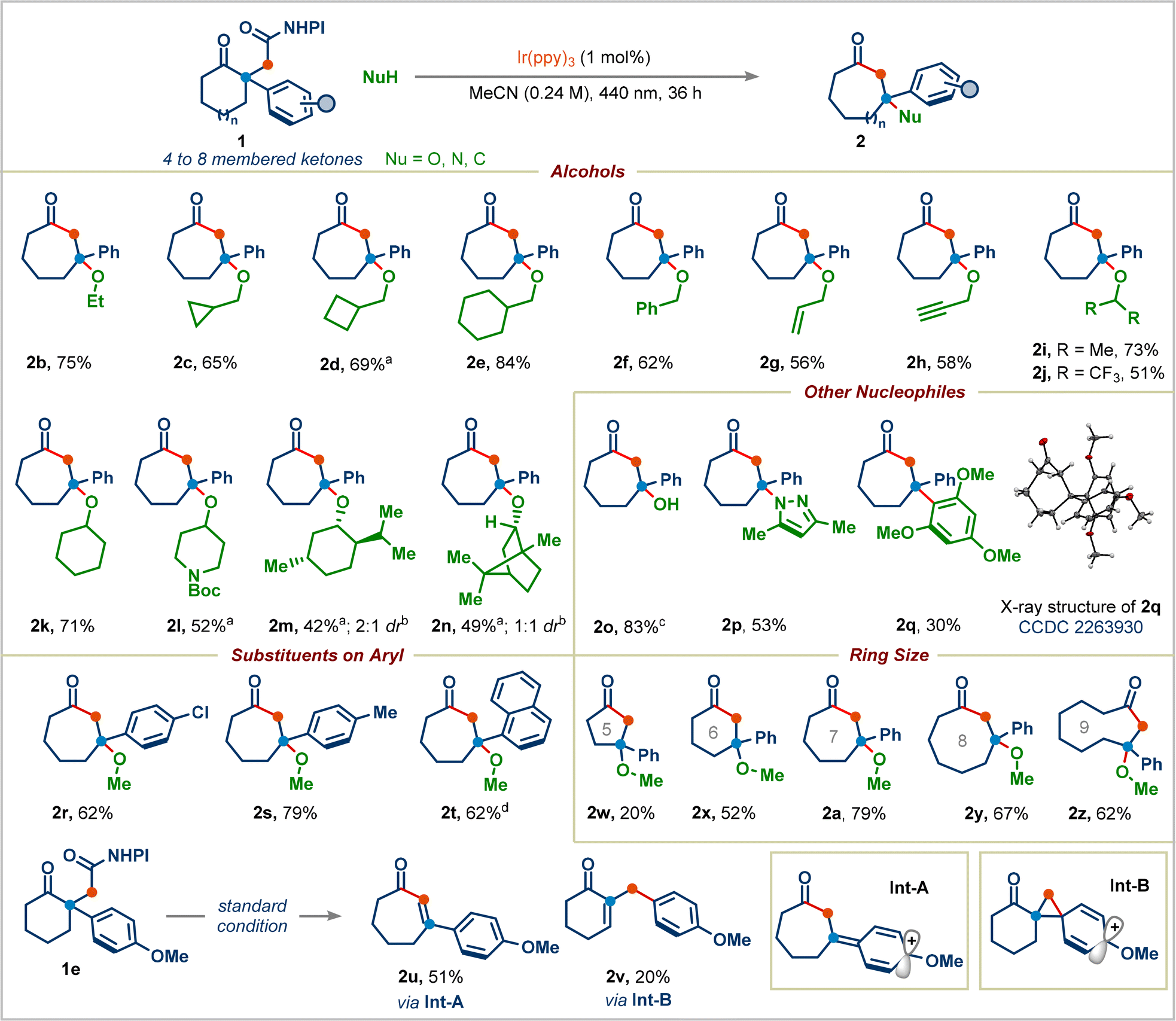 | ||
| Scheme 3 Scope of one-carbon ring-expansion. Standard conditions: 1 (0.3 mmol), alcohol (5.0 equiv.) and Ir(ppy)3 (1 mol%) in acetonitrile (0.24 M), 440 nm Kessil lamp, 36 h. Yields are of isolated products. aUsing 2.5 equiv. of the alcohol. bdr was determined by 1H-NMR. cUsing 0.1 mmol of 1a and 5.0 equiv. of water. dUsing 0.2 mmol of 1d. ND = not detected. | ||
We next proceeded to examine the effect of substituents on the aryl group in redox-active esters: electron-withdrawing or electron-donating substituted-aryl groups and naphthyl group furnished the corresponding ring-expansion products 2r–2t in good yields. Surprisingly, 4-methoxyphenyl substituted redox-active ester 1e didn't deliver the desired ring-expansion addition product; instead, it gave the enone product 2u and 1,2-aryl migrated product 2v in 51% and 20% yields viaInt-A and Int-B, respectively (see the ESI† for the complete mechanism). We were pleased to find that a range of redox-active esters bearing four-to eight-membered rings could react to deliver the one-carbon ring-expansion products 2w–2z. Notably, when substrate 7 containing a three-carbon side chain was subjected to standard conditions, the three-carbon ring-expansion product 8a was formed in 70% yield (Scheme 4). This example demonstrates the potential of this strategy in incorporation of functional groups in remote positions of medium-sized rings. Cyclobutylmethyl, benzyl, and cyclohexyl alcohols were also viable substrates in this three-carbon ring-expansion reaction (products 8b–8d). Notably, a simple fluoride (addition of Et3N·3HF) gave the corresponding tertiary fluoride 8e in 67% yield.
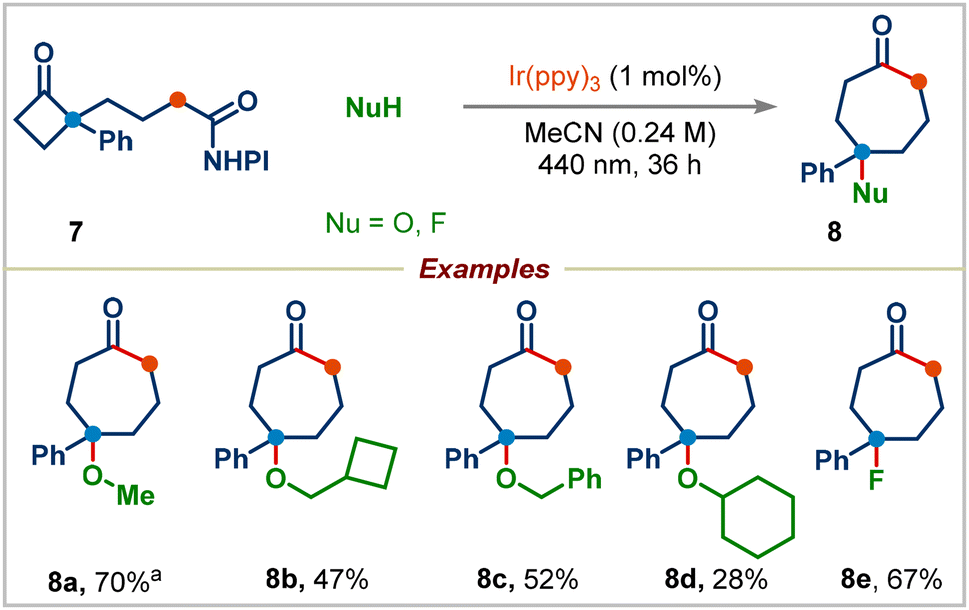 | ||
| Scheme 4 Scope of three-carbon ring-expansion. Standard conditions: 7 (0.25 mmol), alcohol (2.5 equiv.) and Ir(ppy)3 (1 mol%) in acetonitrile (0.24 M). aReaction was performed in 0.15 mmol scale using 5.0 equiv. of methanol. | ||
We next focused on establishing continuous flow conditions for this reaction since the batch reaction takes 36 h for the reaction to reach completion. We were delighted to find that the reaction time was reduced fifteen-fold compared to the batch condition without affecting the yield (Scheme 5A). The reaction was amenable to scale-up, with product 2a formed in 78% yield on a 1.0 mmol scale. Subsequently, we sought to highlight the synthetic utility of the obtained products by carrying out several downstream transformations (Scheme 5B). Reduction of 2a with NaBH4 afforded the cycloheptane-1,3-diol 9, a common core present in several natural products and pharmaceuticals (Scheme 1A), in excellent yield with 3:1 dr.14 The addition of PhMgBr to 2a delivered a more substituted cycloheptane-1,3-diol 10 in 71% yield with 15:1 dr.14 The reaction of 2a with phenylhydrazine in the presence of AcOH gave the corresponding seven-membered ring-containing indole derivative 11. Finally, treatment of 2a with p-TsOH followed by irradiation using thioxanthone as the photocatalyst furnished highly substituted cyclobutane derivative 12 (CCDC 2264117) in 36% yield with 10:1 dr over two steps.15
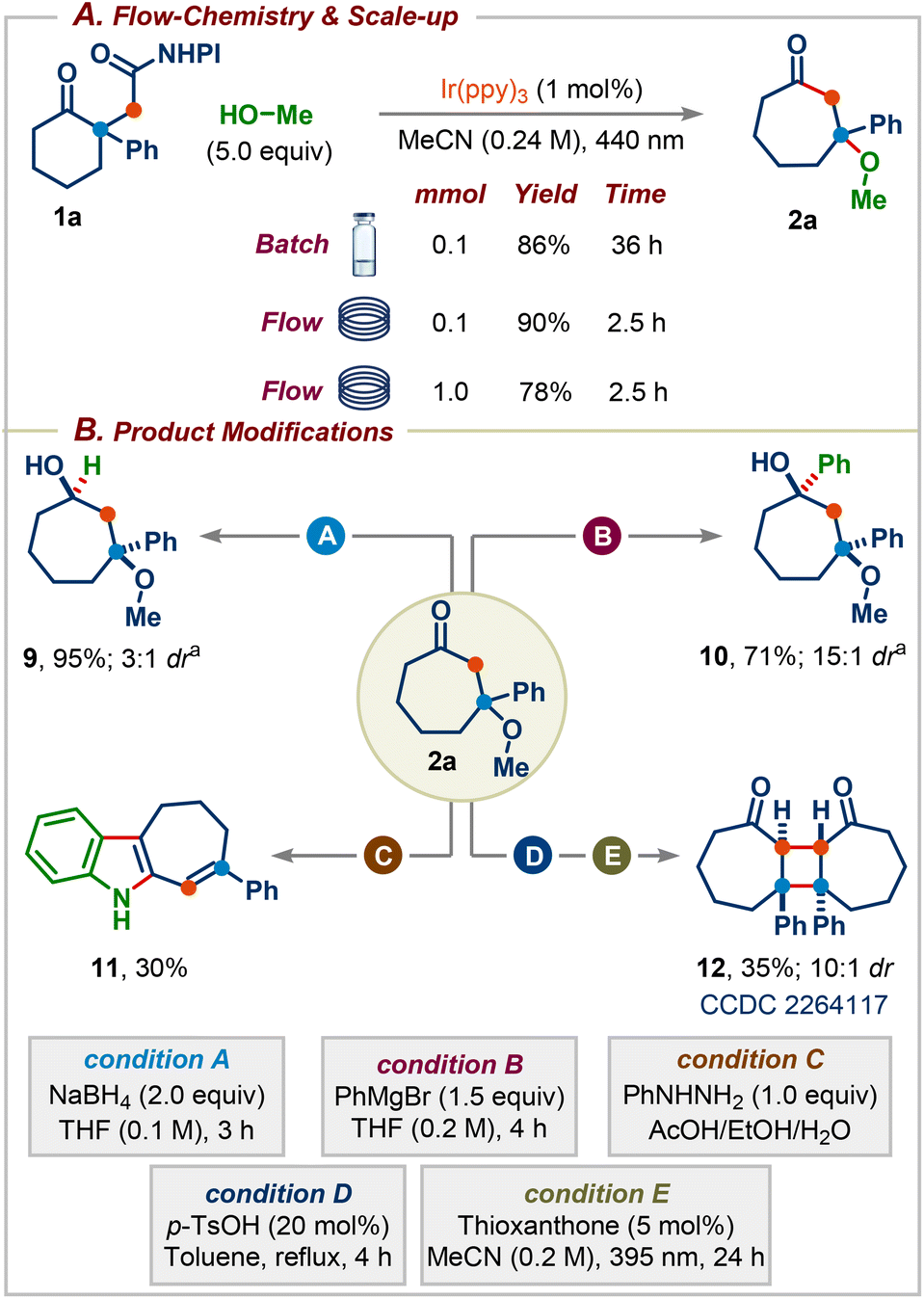 | ||
| Scheme 5 (A) Flow-chemistry and reaction scale-up. (B) Synthetic utility of the obtained products. adr was determined by 1H-NMR. | ||
Next, a series of experiments were performed to probe the key reaction intermediates involved in the reaction (Scheme 6A). The addition of 1.1 equiv. of TEMPO to the standard reaction conditions led to complete inhibition of the reaction, with the detection of TEMPO adducts 13 and/or 14, supporting the radical intermediacy during the reaction (Scheme 6B). When CD3OD was used, no α-deuterium incorporation was observed in the product, which reveals that the product 2ad was not formed via the enone intermediate 6 (Scheme 6C). Furthermore, irradiating the enone 6 in MeOH in the presence of the photocatalyst didn't deliver the methanol addition product 2a (Scheme 6D). These results further rule out a mechanism proceeding through the enone intermediate 6 and support the mechanism proposed in Scheme 1C.
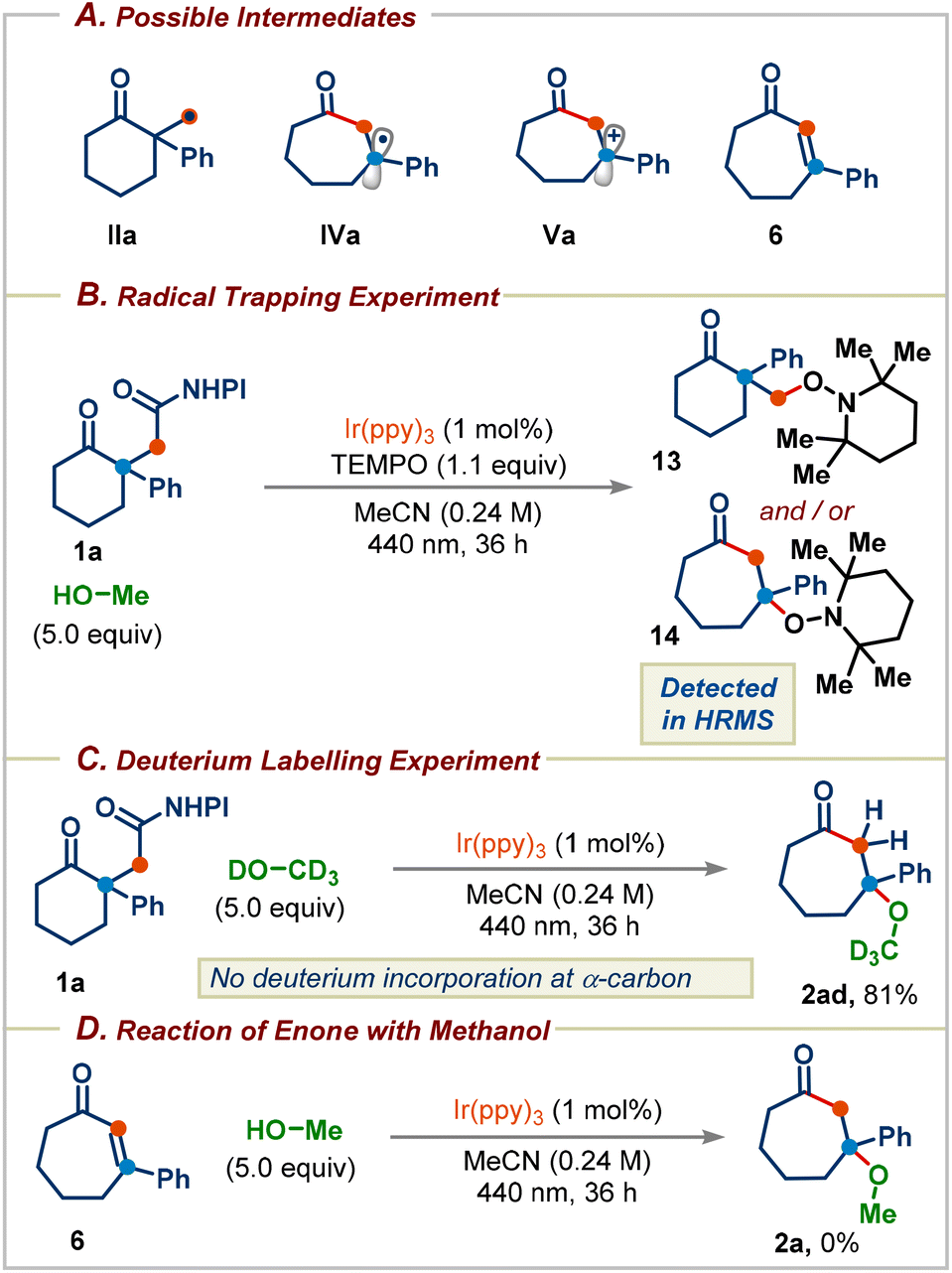 | ||
| Scheme 6 (A) Possible key intermediates. (B) Radical trapping experiment with TEMPO. (C) Deuterium labelling experiment using methanol-d4. (D) Reaction of enone with methanol. | ||
Conclusions
In summary, we have developed a novel redox-neutral decarboxylative Dowd–Beckwith/RPC strategy for the synthesis of functionalized medium-sized carbocyclic compounds using an Ir photocatalyst under visible light irradiation. The use of redox-active esters and the aryl group α to the carbonyl were key to the success of the reaction. A wide range of nucleophilic reagents are amenable to this transformation, including alcohols, water, C-, N-, and F-nucleophiles. The method shows broad applicability across various redox-active esters with different ring sizes and substituents and has been demonstrated to achieve the three-carbon ring-expansion, enabling remote functionalization successfully. Finally, the obtained products could be easily transformed into useful cyclic 1,3-diols and seven-membered ring-containing indoles and cyclobutane derivatives. We anticipate that this reaction will expand the utility of the Dowd–Beckwith reaction in organic synthesis due to the vast abundance and diversity of nucleophilic reagents.Data availability
General information, experimental procedures, characterization data for all new compounds, and NMR spectra are in the ESI.† Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number CCDC 2263930 and 2264117.Author contributions
T. S. designed the project and carried out optimization studies and substrate scope. G. A. K. helped in preparing starting materials for the substrate scope. T. S. and D. P. H. wrote the manuscript with suggestions from G. A. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. P. H. thanks the Indian Institute of Science (IISc) Bangalore for the start-up grant and infrastructure. Financial support from the Science and Engineering Research Board (SERB), Government of India (File CRG/2022/007372), is gratefully acknowledged. T. S. thanks the Ministry of Education, Government of India for PMRF, and G. A. K. thanks the University Grant Commission (U. G. C.) for doctoral fellowships. We thank Kishorkumar Sindogi for solving the X-ray crystal structures.Notes and references
- (a) J. Cossy, S. Arseniyadis, C. Meyer and R. H. Grubbs, Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts, Wiley, 2011, pp. 1–43 Search PubMed; (b) T. V. Nguyen, J. M. Hartmann and D. Enders, Synthesis, 2013, 45, 845–873 CrossRef CAS; (c) K. T. de Oliveira, B. M. Servilha, L. de C. Alves, A. L. Desiderá and T. J. Brocksom, in Stud. Nat. Prod. Chem., ed. R. Atta ur, Elsevier, 2014, vol. 42, pp. 421–463 Search PubMed; (d) Y.-J. Hu, L.-X. Li, J.-C. Han, L. Min and C.-C. Li, Chem. Rev., 2020, 120, 5910–5953 CrossRef CAS PubMed; (e) X.-C. Yu, C.-C. Zhang, L.-T. Wang, J.-Z. Li, T. Li and W.-T. Wei, Org. Chem. Front., 2022, 9, 4757–4781 RSC.
- K. R. Romines, K. D. Watenpaugh, P. K. Tomich, W. J. Howe, J. K. Morris, K. D. Lovasz, A. M. Mulichak, B. C. Finzel and J. C. Lynn, J. Med. Chem., 1995, 38, 1884–1891 CrossRef CAS PubMed.
- (a) J. R. Donald and W. P. Unsworth, Chem. – Eur. J., 2017, 23, 8780–8799 CrossRef CAS PubMed; (b) A. K. Clarke and W. P. Unsworth, Chem. Sci., 2020, 11, 2876–2881 RSC.
- (a) P. Dowd and S. C. Choi, J. Am. Chem. Soc., 1987, 109, 3493–3494 CrossRef CAS; (b) A. L. J. Beckwith, D. M. O'Shea, S. Gerba and S. W. Westwood, J. Chem. Soc., Chem. Commun., 1987, 666–667, 10.1039/C39870000666.
- Selected examples: (a) G. Mehta, N. Krishnamurthy and S. R. Karra, J. Am. Chem. Soc., 1991, 113, 5765–5775 CrossRef CAS; (b) M. T. Crimmins, Z. Wang and L. A. McKerlie, J. Am. Chem. Soc., 1998, 120, 1747–1756 CrossRef CAS; (c) E. Piers, M. Gilbert and K. L. Cook, Org. Lett., 2000, 2, 1407–1410 CrossRef CAS PubMed; (d) M. Takano, A. Umino and M. Nakada, Org. Lett., 2004, 6, 4897–4900 CrossRef CAS PubMed; (e) C. Fu, Y. Zhang, J. Xuan, C. Zhu, B. Wang and H. Ding, Org. Lett., 2014, 16, 3376–3379 CrossRef CAS PubMed; (f) K. Yu, Z.-N. Yang, C.-H. Liu, S.-Q. Wu, X. Hong, X.-L. Zhao and H. Ding, Angew. Chem., Int. Ed., 2019, 58, 8556–8560 CrossRef CAS PubMed; (g) F. L. Duecker, R. C. Heinze and P. Heretsch, J. Am. Chem. Soc., 2020, 142, 104–108 CrossRef CAS PubMed; (h) B. Yang, G. Wen, Q. Zhang, M. Hou, H. He and S. Gao, J. Am. Chem. Soc., 2021, 143, 6370–6375 CrossRef CAS PubMed; (i) Y. Zhao, J. Hu, R. Chen, F. Xiong, H. Xie and H. Ding, J. Am. Chem. Soc., 2022, 144, 2495–2500 CrossRef CAS PubMed.
- Review articles: (a) P. Dowd and W. Zhang, Chem. Rev., 1993, 93, 2091–2115 CrossRef CAS; (b) T. Singha, A. Rouf Samim Mondal, S. Midya and D. Prasad Hari, Chem. – Eur. J., 2022, 28, e202202025 CAS Selected examples:; (c) P. Dowd and W. Zhang, J. Am. Chem. Soc., 1991, 113, 9875–9876 CrossRef CAS; (d) S. Kim, G. H. Joe and J. Y. Do, J. Am. Chem. Soc., 1993, 115, 3328–3329 CrossRef CAS; (e) A. Studer and S. Amrein, Angew. Chem., Int. Ed., 2000, 39, 3080–3082 CrossRef CAS; (f) J. Hierold and D. W. Lupton, Org. Lett., 2012, 14, 3412–3415 CrossRef CAS PubMed; (g) Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100–15104 CrossRef CAS PubMed; (h) Y. Liu and Y.-Y. Yeung, Org. Lett., 2017, 19, 1422–1425 CrossRef CAS PubMed; (i) H. Xin, L.-N. Guo, M. Yang, C. Guan, Z.-H. Yuan and X.-H. Duan, Org. Chem. Front., 2023, 10, 1147–1152 RSC.
- L. Chen, L.-N. Guo, S. Liu, L. Liu and X.-H. Duan, Chem. Sci., 2021, 12, 1791–1795 RSC.
- Q. Zhang, M.-F. Chiou, C. Ye, X. Yuan, Y. Li and H. Bao, Chem. Sci., 2022, 13, 6836–6841 RSC.
- Review articles on the radical-polar crossover strategy: (a) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS PubMed; (b) S. Sharma, J. Singh and A. Sharma, Adv. Synth. Catal., 2021, 363, 3146–3169 CrossRef CAS.
- See the ESI† for cyclic voltammograms.
- Review articles: (a) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (b) P. Niu, J. Li, Y. Zhang and C. Huo, Eur. J. Org. Chem., 2020, 2020, 5801–5814 CrossRef CAS; (c) H. Chen, Y. A. Liu and X. Liao, Synthesis, 2020, 53, 1–29 Search PubMed Selected papers:; (d) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed; (e) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132–11135 CrossRef CAS PubMed; (f) J. Wang, T. Qin, T.-G. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 9676–9679 CrossRef CAS PubMed; (g) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed; (h) E. W. Webb, J. B. Park, E. L. Cole, D. J. Donnelly, S. J. Bonacorsi, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 9493–9500 CrossRef CAS PubMed; (i) D. C. Salgueiro, B. K. Chi, I. A. Guzei, P. García-Reynaga and D. J. Weix, Angew. Chem., Int. Ed., 2022, 61, e202205673 CrossRef CAS PubMed.
- We didn't observe the formation of the direct methoxylation product, which is expected to form via oxidation of the primary radical followed by trapping with methanol.
- (a) G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15, 4398–4401 CrossRef CAS PubMed; (b) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240–1243 CrossRef CAS PubMed.
- The stereochemistry of the major diastereomer was assigned based on a similar reaction reported in the literature: Y. Senda, H. Sakurai, S. Nakano and H. Itoh, Bull. Chem. Soc. Jpn., 1996, 69, 3297–3303 CrossRef CAS.
- K. Singh, W. Trinh and J. D. Weaver, Org. Biomol. Chem., 2019, 17, 1854–1861 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2263930 and 2264117. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01908j |
| This journal is © The Royal Society of Chemistry 2023 |