
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating fluorescence by photo-switched spin-state conversions in an iron(II)-based SCO-MOF†
Fei-Fei
Yan
 ,
Wen-Jing
Jiang
,
Nian-Tao
Yao
,
Pan-Dong
Mao
,
Liang
Zhao
,
Hui-Ying
Sun
,
Yin-Shan
Meng
and
Tao
Liu
*
,
Wen-Jing
Jiang
,
Nian-Tao
Yao
,
Pan-Dong
Mao
,
Liang
Zhao
,
Hui-Ying
Sun
,
Yin-Shan
Meng
and
Tao
Liu
*
State Key Laboratory of Fine Chemicals, Frontier Science Center for Smart Materials, Dalian University of Technology, 2 Linggong Road, Dalian 116024, China. E-mail: liutao@dlut.edu.cn
First published on 26th May 2023
Abstract
Manipulating fluorescence by photo-switched spin-state conversions is an attractive prospect for applications in smart magneto-optical materials and devices. The challenge is how to modulate the energy transfer paths of the singlet excited state by light-induced spin-state conversions. In this work, a spin crossover (SCO) FeII-based fluorophore was embedded into a metal–organic framework (MOF) to tune the energy transfer paths. Compound 1 {Fe(TPA-diPy)[Ag(CN)2]2}·2EtOH (1) has an interpenetrated Hofmann-type structure, wherein the FeII ion is coordinated by a bidentate fluorophore ligand (TPA-diPy) and four cyanide nitrogen atoms and acts as the fluorescent-SCO unit. Magnetic susceptibility measurements revealed that 1 underwent an incomplete and gradual spin crossover with T1/2 = 161 K. Photomagnetic studies confirmed photo-induced spin state conversions between the low-spin (LS) and high-spin (HS) states, where the irradiation of 532 and 808 nm laser lights converted the LS and HS states to the HS and LS states, respectively. Variable-temperature fluorescence spectra study revealed an anomalous decrease in emission intensity upon the HS → LS transition, confirming the synergetic coupling between the fluorophore and SCO units. Alternating irradiation of 532 and 808 nm laser lights resulted in reversible fluorescence intensity changes, confirming spin state-controlled fluorescence in the SCO-MOF. Photo-monitored structural analyses and UV-vis spectroscopic studies demonstrated that the photo-induced spin state conversions changed energy transfer paths from the TPA fluorophore to the metal-centered charge transfer bands, ultimately leading to the switching of fluorescence intensities. This work represents a new prototype compound showing bidirectional photo-switched fluorescence by manipulating the spin states of iron(II).
Introduction
Fluorophores that can exhibit switchable fluorescent emission in response to external stimuli such as light, heat, pressure, and magnetic or electric field are promisingly applicable in information storage,1–5 bio-sensing,6,7 fluorescence imaging,8–12 and molecular devices.13,14 It is highly desired to manipulate the fluorescent signal to fulfill the demands for advanced photoelectric technologies. Commonly, fluorescence switching mechanisms involve changes in the energy transfer path,15–17 excimer/exciplex formation,18–20 or HOMO–LUMO gap,21,22 which can be attained by chemical modifications and stacking modes of component molecules. In particular, controlling or switching the fluorescent signal via a light stimulus is more interesting owing to its noninvasive, supersensitive and accessible characteristics in comparison to other physical stimuli.23,24 Among the reported light-responsive molecular systems, photo-induced isomerization, such as cis/trans and ring opening/closing transformations, are extensively studied. These organic fluorescent molecular systems commonly undergo drastic molecular conformational transformation or bond breaking/formation in solution in switching processes.25 Moreover, fluorescence switching in these organic solids suffers from the drawbacks of low conversion and repetition efficiency, slow transformation rate and fatigue damage under UV light irradiation, which hamper their further applications for optoelectronic devices.26 It is, therefore, challenging to build molecular solids whose fluorescence properties can be manipulated without significant conformational changes, leading to high conversion efficiency and durability.Spin crossover (SCO) complexes are well-known switchable materials, in which spin states of transition metals with 3d4–3d7 electron configuration can be interconverted between high-spin (HS) and low-spin (LS) states under external stimuli such as temperature, light, pressure, and applied electric field.27–31 Recent studies have proven that the fluorescence intensity of SCO complexes can be effectively modulated by spin-state conversions.32–39 The fluorophore acts as a photosensitizer responsible for absorbing photons and emitting fluorescence, while the SCO unit is a better energy acceptor. When a coupling path is built between them, the energy transfer efficiency from the fluorophores to metal ions will be different for the HS and LS states due to the varied overlaps of emission bands of the ligand and absorption bands of the metal ion. It is noteworthy that an SCO molecule changes its coordination bond lengths, but no substantial conformational change occurs upon spin conversions, suggesting high durability against electronic state conversions. In addition, the HS and LS states of an SCO metal ion can be mutually manipulated by light irradiation, called light-induced excited spin state trapping (LIESST) and reverse LIESST (RE-LIESST) effects.40,41 The light-induced process usually occurs on a picosecond scale, enabling a rapid response upon light irradiation.42 The SCO units are, therefore, expected to provide a feasible approach to manipulate the fluorescence properties by the LIESST and RE-LIESST effects. To date, several fluorescence–SCO complexes showed synergy of spin crossover and emission behavior, among which only two SCO complexes exhibited the modulation of fluorescence upon light-induced LS → HS transition but the recovery of fluorescence requires time-consuming thermal treatment.43,44 In 2018, our group reported a mononuclear complex [FeL2(NCS)2], where L is a fluorophore ligand of (pyrene-1-yl)-N-(3,5-di(pyridin-2-yl)-4H-1,2,4-triazol-4-yl)methanimine, with the photoinduced spin crossover accompanied by a 20% change in the fluorescence emission intensity from the fluorophore.44 As far as we know, the SCO complex showing reversible modulation in fluorescence intensity upon light-induced spin-state conversions has not been reported. One reason is that the photo-responsive properties of the SCO unit are ultra-sensitive to subtle structural changes. In addition, the fluorophore is also easily quenched when embedded into paramagnetic metal complexes. Therefore, the state-of-the-art design of fluorescence–SCO molecules should be paid attention to preserve both the fluorescence and photo-induced SCO properties.45 The coupling of the fluorophore and SCO unit is key to managing an effective energy transfer for varying fluorescence intensities.
Hofmann-type FeII-based metal–organic frameworks (MOFs) are potential candidates to achieve the synergy of the SCO and fluorescence properties, because they have the advantages of flexible porous structures, high thermal stability, and multifunctional coupling properties, which can be pre-designed purposefully by the incorporation of appropriate functional ligands and guest molecules.46–51 In addition, a considerable number of Hofmann-type FeII-based SCO-MOFs were reported to show the LIESST effect,48,52 which makes us confident in realizing the bidirectional controls of the SCO and fluorescence properties by the LIESST and RE-LIESST effects. With these concerns, we designed a new fluorescent ligand N-phenyl-4-(4-pyridinyl)-N-[4-(4-pyridinyl)phenyl]aniline (TPA-diPy) with a strong fluorescence emission at 470 nm, and [Ag(CN)2]− was chosen as a building block to construct the fluorescent SCO-MOF. A newly designed Fe(II) complex of {Fe(TPA-diPy)[Ag(CN)2]2}·2EtOH (1) displays synergetic SCO-fluorescence properties, where the reversible and bidirectional modulations of fluorescence intensity upon spin-state conversions by alternating 532 and 808 nm light irradiation are realized. To our knowledge, 1 is the first example exhibiting reversibly switched fluorescence by light-induced spin-state conversions.
Results and discussion
The reaction of Fe(ClO4)2·6H2O and a bidentate ligand TPA-diPy with K[Ag(CN)2] in a mixed solvent of ethanol and DMF yielded {Fe(TPA-diPy)[Ag(CN)2]2}·2EtOH (1). Pale yellow crystals of 1 suitable for single crystal X-ray analyses were obtained by slow diffusion of component solutions using an H-shaped tube. The powder X-ray diffraction (XRD) measurement at room temperature confirmed the phase purity (Fig. S1†), and the thermogravimetric analysis (TGA) showed that 1 is stable below 360 K (Fig. S2†). The single crystal X-ray diffraction data were collected at 60, 120, and 270 K, and the crystallographic data are listed in Table S1–3.† Single crystal structure analyses show that 1 crystallizes in the monoclinic space group C2/c at the measured temperatures. The asymmetric unit contains one independent FeII ion, half of the TPA-diPy ligand, one [Ag(CN)2]− unit, and one guest EtOH molecule (Fig. S3†). The FeII ion adopts a slightly distorted octahedral coordination geometry, in which the equatorial positions are occupied by four cyanide nitrogen atoms from the [Ag(CN)2]− entities, and the apical positions are coordinated by two nitrogen atoms from the TPA-diPy ligands (Fig. 1a). The [Ag(CN)2]− units bridge the FeII ions to form a 3D Hoffmann-type framework, composed of [Fe6{Ag(CN)2}6] grids, in which the TPA-diPy ligands, running across the MOF frameworks, bridge the FeII ions (Fig. 1b and S4†). Uncoordinated ethanol molecules are located in the voids and form hydrogen bonds with the aniline groups. At 270 K, the Fe–NTPA-diPy and Fe–Ncyanide bond lengths are in the range of 2.195(5) and 2.131(7)-2.157(5) Å, respectively, characteristic of HS FeII ions. As the temperature was decreased to 120 K, the Fe–NTPA-diPy and Fe–Ncyanide bond lengths decreased to 2.092(6) and 2.035(7)-2.064(5) Å, respectively. Upon further cooling the sample to 60 K, 1 experienced subtle shortenings in the Fe–NTPA-diPy (0.011 Å) and Fe–Ncyanide (<0.016 Å) bond lengths. These variations in bond lengths suggested that 1 underwent an incomplete thermally induced SCO behavior.53–55 Based on the structural analyses, we conclude that the distorted octahedral geometry [FeIIN6] is a typical SCO coordination moiety, and the TPA-diPy ligand synthesized by grafting pyridyl groups onto the triphenylamine is the fluorophore. | ||
| Fig. 1 (a) Coordination environment of the FeII center for compound 1 at 120 K. (b) A 3D Hofmann-type MOF with [Fe6{Ag(CN)2}6] grids. The red dashed lines represent C–H⋯O interactions between the ligands and EtOH molecules. Hydrogen atoms and solvent molecules were omitted for clarity. Color codes: Fe, orange; Ag, pale green; N, violet; C, pale gray; H, blue; O, red. | ||
Magnetic susceptibility measurements were performed on the polycrystalline sample 1 in the temperature range from 2 to 300 K under a DC field of 1000 Oe (Fig. 2a). At 300 K, the χMT value is 3.82 cm3 mol−1 K, which is characteristic of an HS FeII ion (S = 2 and g = 2.26). As the temperature was lowered, the χMT products gradually decreased to 1.37 cm3 mol−1 K at 20 K, followed by a sudden decrease to 0.73 cm3 mol−1 K at 2 K. The results suggest an incomplete spin crossover to the LS state with T1/2 = 161 K. Considering the closest interatomic distance (Fe⋯Fe) of 10.207 Å, the antiferromagnetic contribution to the χMT values is negligibly small above 20 K, and the sudden decrease of the χMT values below 20 K is mainly attributed to the zero-field splitting of the remaining HS FeII species. The χMT values in the heating mode traced the same temperature profile as in the cooling mode, suggesting no apparent thermal hysteresis.
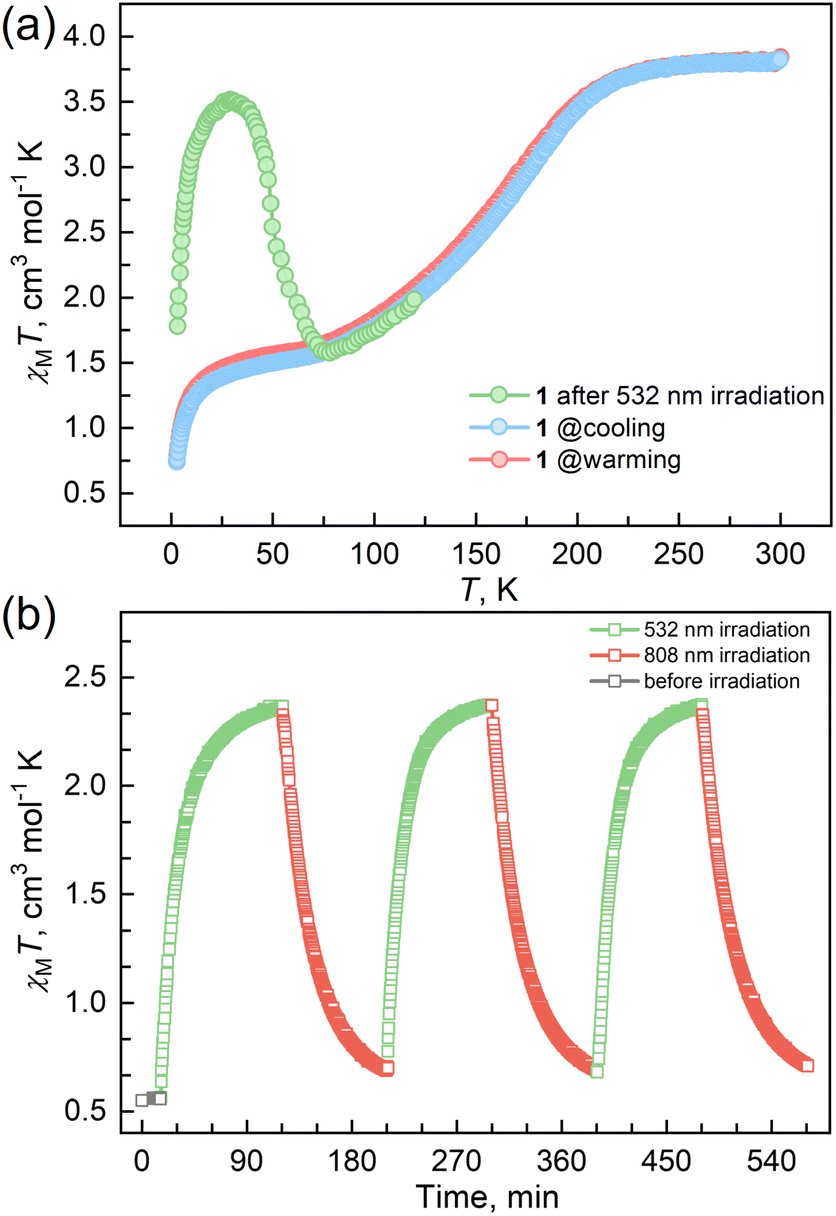 | ||
| Fig. 2 (a) Temperature-dependent susceptibilities of 1 under a DC field of 1000 Oe. (b) Plots of χMT vs. time under cycles of successive irradiation at 532 nm and 808 nm lasers at 10 K for 1. | ||
57Fe Mössbauer spectra of 1 were recorded at 275 and 40 K to characterize the spin states of the FeII centers (Fig. S5 and Table S5†). In the high-temperature phase at 275 K, a Mössbauer spectrum is composed of one quadrupole doublet, of which Mössbauer parameters are δ (isomer shift) = 0.96 mm s−1 and ΔEQ (quadrupole splitting) = 1.15 mm s−1, characteristic of the HS-FeII species. As the temperature was lowered to 40 K, the quadruple doublet (δ = 1.07 mm s−1 and ΔEQ = 2.01 mm s−1) assigned to the HS FeII component was decreased, and an additional quadrupole singlet (δ = 0.38 mm s−1 and ΔEQ = 0.17 mm s−1) was observed, assignable to the LS-FeII species. The peak area ratio for the HS and LS species is HS/LS = 0.39/0.61, consistent with the value (0.40/0.60) estimated from the magnetic susceptibility data at 40 K.
Variable temperature UV-vis absorption spectra were measured first because they can provide guidelines for choosing excitation light wavelengths suitable for photomagnetic experiments. According to UV-vis absorption spectra, we found that 1 in the high temperature (HT) phase at 300 K showed a broad absorption band at 840 nm, while in the low temperature (LT) phase at 100 K two absorption bands around 410–450 and 500–590 nm were observed (Fig. 3). Therefore, we decided to use 532 and 808 nm laser lights to investigate the photo-responsive properties of 1. Excitation around 532 nm, corresponding to MLCT or d–d (1A1→1T1) transitions in many LS-FeII materials, induces a dynamical sub-ps spin transition towards the metastable high spin (HS*) state,40,41 while excitation around 808 nm (d–d (5T2→5E)) in some HS-FeII materials induces a LS excited state (LS*), which kinetically decays to the LS* state within 40 ps.42 Photomagnetic measurements were then performed to study the LIESST and RE-LIESST effects (Fig. 2b). When 1 in the LT phase was irradiated using the 532 nm laser light at 10 K, the χMT values increased from 0.56 to 2.36 cm3 mol−1 K within 90 min. After turning off the laser light and raising the temperature to 30 K, the χMT values increased to a maximum of 3.50 cm3 mol−1 K. This suggests that most LS FeII species turned to a metastable HS*-FeII state in the metastable high temperature (HT*) phase by the LIESST effect. Upon heating the sample in the HT* phase, the χMT values decreased and merged with the values for the non-irradiated sample at 75 K, indicative of the thermal relaxation to the LT phase. In contrast, the 808 nm laser light irradiation on the HT* phase at 10 K for 90 min decreased the χMT values from 2.36 to 0.69 cm3 mol−1 K. These results clearly indicate that the HS*-FeII state (generated after irradiation with 532 nm light) can be reversibly transformed into the LS state using 808 nm light, thus confirming the RE-LIESST effect. It is worth noting that magnetic susceptibility switching by alternating the 532 and 808 nm laser lights can be well repeated three times, suggesting the repeatability and durable stability of 1 in the photo-switching. The reversible photo-response properties of 1 set the foundation for the manipulation of fluorescence properties by the LIESST and RE-LIESST effects.
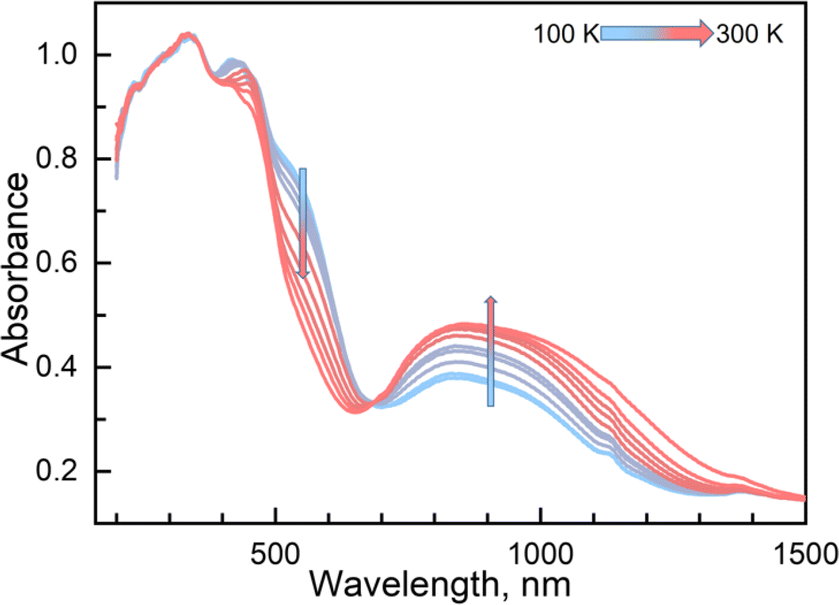 | ||
| Fig. 3 Temperature-dependent UV-vis spectra of 1 in a solid from 100 to 300 K in heating mode. | ||
To verify the synergy between fluorescence and SCO in compound 1, temperature-dependent fluorescence spectra for the solid samples of 1 and ligand TPA-diPy were recorded from 80 to 300 K in the heating mode, where the excitation light of λex = 398 nm was used for all measurements. The emission spectra of TPA-diPy exhibited a broad emission band with a peak maximum at λem = 471 nm. The peak intensity decreased monotonously upon heating due to the thermal quenching effect (Fig. S6†). In contrast to the ligand, 1 exhibited substantial temperature dependence in the fluorescence spectra (Fig. 4). The emission spectra of 1 at 80 K showed two bands with peak maxima at 450 and 480 nm. As the temperature was raised from 80 K, the emission intensities of the two bands became stronger and the bands merged into one and reached the maximum value at 220 K, followed by a decrease in intensity upon further temperature increases. Temperature dependences of the emission intensity showed a similar profile to that of the χMT values up to 225 K, above which the thermal quench became predominant. The anomalous fluorescence enhancement was observed only in the temperature range from 80 to 220 K, a similar temperature region in the spin crossover, confirming the synergetic coupling between the fluorescence emission and SCO.
 | ||
| Fig. 4 (a) Temperature-dependent fluorescence emission spectra for compound 1 in a solid state at λex = 398 nm with heating mode from 80 to 300 K. (b) χMT and the fluorescence intensity of the maximum emission as a function of temperature for 1. | ||
The LIESST and RE-LIESST effects on the fluorescence emission behavior were studied for 1, where laser light irradiation (532 and 808 nm) and the subsequent spectral measurements were performed at 10 K with the excitation light of λex = 398 nm (Fig. 5a and S7†). It should be noted that no LIESST effect was observed with the 398 nm light irradiation. Before the irradiation, 1 showed an emission peak at ca. 427 nm. Interestingly, the fluorescence intensity significantly increased after the 532 nm laser light irradiation and reached saturation after the irradiation for 30 minutes. Considering the photomagnetic data, the majority of LS FeII ions in 1 were converted to the metastable HS* state in the HT* phase by 532 nm light irradiation. It is also worth noting that the maximum fluorescence intensity in the HT* phase is about ca. 1.8 times larger than that of the unirradiated sample. When 1 in the HT* phase was exposed to the 808 nm laser light, the fluorescence intensity decreased and reached the same value as in the LT phases. It is also confirmed that the cyclic photo-irradiation experiments gave a reversible recovery of fluorescence intensities (Fig. 5b). The results demonstrate that the fluorescence intensity can be reversibly modulated by the LIESST and RE-LIESST processes. As far as we know, 1 is the first example in a family of SCO complexes to realize reversible photo-switched fluorescence by manipulating spin states.
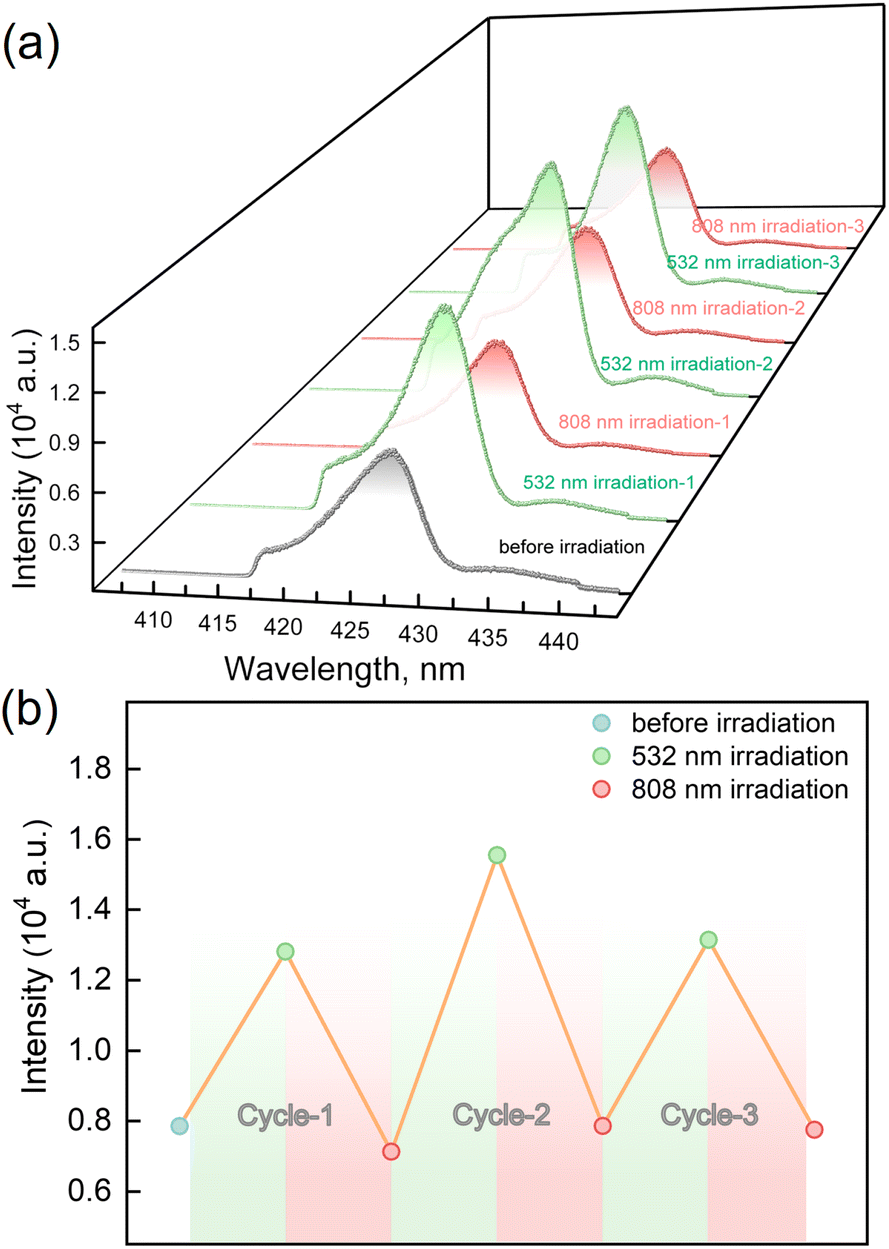 | ||
| Fig. 5 (a) Fluorescence emission spectra under cycles of alternative irradiation of 532 nm (green line) and 808 nm (red line) lights at 10 K under λex = 398 nm. (b) Fluorescence intensity of maximum emission for 1 under cycles of successive irradiation by 532 nm and 808 nm lights at 10 K (λex = 398 nm). | ||
Photo-monitored single crystal X-ray analyses provided the structural basis for the photo-switched luminescence upon light-induced spin state conversions.56,57 The single crystal of 1 was gradually cooled down to the LT phase at 30 K, which is a mixture of the HS and LS species (HS/LS = 0.40/0.60). The 532 nm laser light irradiation converted the LT phase to the HT* phase, confirming LIESST behavior. Note that 1 retains the same space group of C2/c throughout the photo-irradiation experiments. The cell volume in the HS* phase was increased by 2.61% from 3356.3(14) Å3 to 3433.8(3) Å3 (Table S1–3†), and the average Fe–N bond lengthened from 2.061(3) Å to 2.165(2) Å upon the irradiation (Table 1). These bond length changes indicate the occurrence of spin conversion from the LS to the HS* state. Afterward, the crystal in the HS* phase was irradiated with the 808 nm laser light. Upon irradiation, the average Fe–N bond lengths decreased to 2.009(3) Å and the unit cell volume shrank to 3359.5(4) Å3, suggesting the HS* to LS state conversion. It is worth mentioning that the average bond length (2.009(3) Å) in the LS state after the 808 nm laser irradiation is shorter than that of the pristine non-irradiated crystal (2.061(3) Å), suggesting the smaller HS/LS ratio in the LS phase generated by RE-LIESST. The temperature dependence of the crystal parameters for 1 in the metastable HS* phase showed a slight decrease in the unit cell volume from 3434(4) Å3 at 30 K to 3342.1(8) Å3 at 70 K (Table S4†). Further heating the crystal increased the unit cell volume to 3360.6(9) Å3 at 90 K. The result suggests that the metastable HS* phase relaxes back to the thermally stable LT phase at 90 K. The above findings confirm the alternating spin state switches of the FeII ion with 532 and 808 nm laser irradiation. Volume change and intermolecular reorganizations may also influence the Förster resonance energy transfer (FRET) process and thereby the fluorescence properties. To check this, we overlapped the crystal structures of 1 before and after light irradiation. The overlapped diagram indicated that no significant intermolecular reorganizations and structural rearrangements occurred (Fig. S8†). This suggests that the intermolecular structural rearrangement is not the main reason for the changes in fluorescence. The LIESST and RE-LIESST are the keys to manipulating the fluorescence by switching the spin states of FeII ions.
| 1 | 30 K | 30 K532 nm | 30 K808 nm |
|---|---|---|---|
| Fe1–NTPA-dipy | 2.094(6) | 2.191(4) | 2.027(8) |
| Fe1–Ncy | 2.056(6) | 2.155(4) | 2.010(8) |
| Fe1–Ncy | 2.033(7) | 2.149(5) | 1.991(8) |
| Fe1–Navg | 2.061(3) | 2.165(2) | 2.009(3) |
| V | 3356.3(14) | 3443.8(3) | 3359.0(4) |
Couplings between the SCO and fluorophore originate from the Förster resonance energy transfer (FRET), which requires effective spectral overlaps between the emission bands of the fluorophore and the absorption bands of the SCO unit (Fig. 6).32,45 Therefore, variable-temperature UV-vis absorption spectra of 1 were recorded to probe the spectral overlaps (Fig. 3). At 100 K, two absorption bands at around 410–450 and 500–590 nm were observed in the UV-vis spectrum, assignable to MLCT and d–d (1A1 → 1T1) bands of the LS FeII ion, respectively.40,41 As the temperature was raised to 300 K, the intensity of the two absorption bands gradually became smaller. At the same time, a broad absorption band at 700–1000 nm, corresponding to the d–d (5T2 → 5E) transition of the HS FeII ion, became stronger.42,47,58 It can be seen that the emission band of the fluorophore significantly overlapped with the absorption bands of the LS FeII species (Fig. 7). This suggests that the energy transfer occurs from the singlet excited state to the MLSLCT band in the LS FeII species, leading to fluorescence quenching via FRET. In contrast, the less overlapped emission and MHSLCT bands in the HS FeII state limit the energy transfer, leading to the gradual enhancement of fluorescence intensity during spin-state conversions from the LS to the HS state. Consequently, the synergetic modulation between fluorescence and SCO in 1 could be realized. Photo-monitored structural analyses together with UV-vis spectroscopic studies demonstrated that the LIESST and RE-LIESST processes changed energy transfer efficiency from the TPA fluorophore to the metal-centered charge transfer bands by switching the spin states of FeII, which ultimately contributed to the modulation of fluorescence intensities.
 | ||
| Fig. 6 Schematic diagram of energy transfer between the energy donor fluorophore and the energy acceptor FeII SCO center. | ||
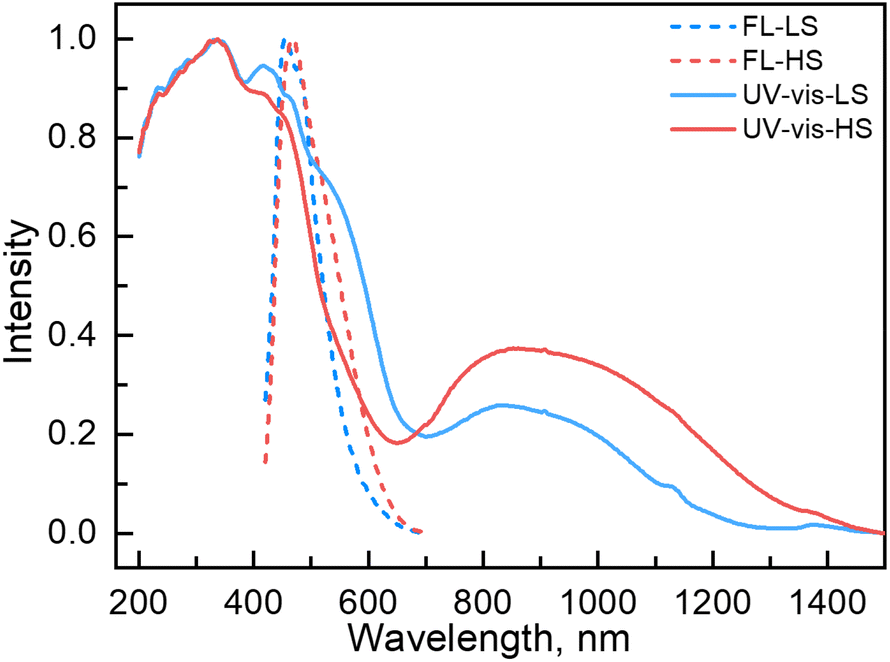 | ||
| Fig. 7 UV-vis (experimental and normalized) and fluorescence emission (experimental and normalized) spectra of 1 in the LS and HS states. | ||
Conclusions
To summarize, a new 3D Hofmann type SCO-MOF, {Fe(TPA-diPy)[Ag(CN)2]2}·2EtOH (1), was synthesized based on the strategy to control the fluorescence intensity by spin-state conversions of the FeII ion. It exhibited gradual and incomplete spin crossover centered at T1/2 = 161 K with synergetic fluorescence–SCO properties. Meanwhile, its fluorescence and magnetism at 10 K can be effectively and reversibly modulated by alternating 532 and 808 nm lasers. Photo-monitored X-ray crystal structural analyses combined with the spectroscopic studies confirmed that the photoinduced spin-state conversions changed energy transfer efficiency from the TPA fluorophore to the metal-centered charge transfer bands via FRET, leading to the switching of fluorescence intensities. To our knowledge, it is the first example to exhibit reversible photo-switches of fluorescence by manipulating the LIESST and RE-LIESST effects. This work proposes a valuable strategy for designing function-coupled SCO materials and opens up new avenues for the applications of MOFs in fluorescence thermometry and innovative photo-switchable materials.Data availability
The data supporting the findings of this work are available within the main article and the ESI.†Author contributions
T. Liu conceived the project and designed the experiment. F.-F. Yan synthesized the complex and performed all characterizations. W.-J. Jiang and L. Zhao help to perform the photomagnetic studies. N.-T. Yao and P.-D. Mao helped to perform the ultra-low temperature X-ray diffraction experiment. H.-Y. Sun helped to draw figures and structures. F.-F. Yan and Y.-S. Meng analyzed the data. F.-F. Yan, Y.-S. Meng and T. Liu wrote the draft. All authors analyzed and discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the National Natural Science Foundation of China (Grant No. 22025101, 22222103, 91961114, 21871039, 22173015, 21801037 and 07120002) and the Fundamental Research Funds for the Central Universities of China (DUT22LAB606). We wish to acknowledge Dr Rui Cai at the Instrumental Analysis center of Dalian University of Technology for assistance with luminescence measurements and helpful discussions.References
- Y. Lu, J. Zhao, R. Zhang, Y. Liu, D. Liu, E. M. Goldys, X. Yang, P. Xi, A. Sunna, J. Lu, Y. Shi, R. C. Leif, Y. Huo, J. Shen, J. A. Piper, J. P. Robinson and D. Jin, Nat. Photonics, 2013, 8, 32–36 CrossRef.
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- H. Zhang, Y. Fan, P. Pei, C. Sun, L. Lu and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10153–10157 CrossRef CAS PubMed.
- R. Yang, Y. Jiao, B. Wang, B. Xu and W. Tian, J. Phys. Chem. Lett., 2021, 12, 1290–1294 CrossRef CAS PubMed.
- Y. Jiao, R. Yang, Y. Luo, L. Liu, B. Xu and W. Tian, CCS Chem., 2022, 4, 132–140 CrossRef CAS.
- M. Zhao, B. Li, H. Zhang and F. Zhang, Chem. Sci., 2021, 12, 3448–3459 RSC.
- Y. Matsuo, K. Kanaizuka, K. Matsuo, Y.-W. Zhong, T. Nakae and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5016–5017 CrossRef CAS PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- S. Hernot, L. van Manen, P. Debie, J. S. D. Mieog and A. L. Vahrmeijer, Lancet Oncol., 2019, 20, e354–e367 CrossRef CAS PubMed.
- H.-B. Cheng, Y. Li, B. Z. Tang and J. Yoon, Chem. Soc. Rev., 2020, 49, 21–31 RSC.
- X. Chai, H.-H. Han, A. C. Sedgwick, N. Li, Y. Zang, T. D. James, J. Zhang, X.-L. Hu, Y. Yu, Y. Li, Y. Wang, J. Li, X.-P. He and H. Tian, J. Am. Chem. Soc., 2020, 142, 18005–18013 CrossRef CAS PubMed.
- J. Huang and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 11717–11731 CrossRef CAS PubMed.
- M. Engeser, L. Fabbrizzi, M. Licchelli and D. Sacchi, Chem. Commun., 1999, 13, 1191–1192 RSC.
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed.
- M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552–2557 RSC.
- M. S. Kwon, J. Gierschner, S.-J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487–5492 CrossRef CAS PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- J. Kim, D. T. McQuade, S. K. McHugh and T. M. Swager, Angew. Chem., Int. Ed., 2000, 39, 3868–3872 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2008, 47, 5175–5178 CrossRef CAS PubMed.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. S. Kwon, J. H. Jordahl, A. W. Phillips, K. Chung, S. Lee, J. Gierschner, J. Lahann and J. Kim, Chem. Sci., 2016, 7, 2359–2363 RSC.
- X. Wang, G. Pan, H. Ren, J. Li, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2022, 61, e202114264 CAS.
- R. Yang, X. Ren, L. Mei, G. Pan, X.-Z. Li, Z. Wu, S. Zhang, W. Ma, W. Yu, H.-H. Fang, C. Li, M.-Q. Zhu, Z. Hu, T. Sun, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2022, 61, e202117158 CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed.
- K. S. Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- F. J. Valverde-Muñoz, M. Seredyuk, M. Meneses-Sánchez, M. C. Muñoz, C. Bartual-Murgui and J. A. Real, Chem. Sci., 2019, 10, 3807–3816 RSC.
- W.-W. Wu, S.-G. Wu, Y.-C. Chen, G.-Z. Huang, B.-H. Lyu, Z.-P. Ni and M.-L. Tong, Chem. Commun., 2020, 56, 4551–4554 RSC.
- W. Lan, F. J. Valverde-Muñoz, X. Hao, Y. Dou, M. C. Muñoz, Z. Zhou, H. Liu, Q. Liu, J. A. Real and D. Zhang, Chem. Commun., 2019, 55, 4607–4610 RSC.
- J.-Y. Ge, Z. Chen, L. Zhang, X. Liang, J. Su, M. Kurmoo and J.-L. Zuo, Angew. Chem., Int. Ed., 2019, 58, 8789–8793 CrossRef CAS PubMed.
- S. Ghosh, S. Kamilya, T. Pramanik, M. Rouzières, R. Herchel, S. Mehta and A. Mondal, Inorg. Chem., 2020, 59, 13009–13013 CrossRef CAS PubMed.
- M. K. Javed, A. Sulaiman, M. Yamashita and Z.-Y. Li, Coord. Chem. Rev., 2022, 467, 214625 CrossRef CAS.
- I. Sánchez-Molina, D. Nieto-Castro, A. Moneo-Corcuera, E. Martínez-Ferrero and J. R. Gálan-Mascarós, J. Phys. Chem. Lett., 2021, 12, 10479–10485 CrossRef PubMed.
- K. Sun, J.-P. Xue, Z.-S. Yao and J. Tao, Dalton Trans., 2022, 51, 16044–16054 RSC.
- J. Wang, M. Kong, X.-J. Song, Y. Jing, Y. Zhao and Y. Song, Inorg. Chem., 2022, 61, 20923–20930 CrossRef CAS PubMed.
- X.-R. Wu, Z.-K. Liu, M. Zeng, M.-X. Chen, J. Tao, S.-Q. Wu and H.-Z. Kou, Sci. China: Chem., 2022, 65, 1569–1576 CrossRef CAS.
- C. Yi, Y.-S. Meng, L. Zhao, N.-T. Yao, Q. Liu, W. Wen, R.-X. Li, Y.-Y. Zhu, H. Oshio and T. Liu, CCS Chem., 2023, 5, 915–924 CrossRef CAS.
- W. Zhang, K. S. Kjær, R. Alonso-Mori, U. Bergmann, M. Chollet, L. A. Fredin, R. G. Hadt, R. W. Hartsock, T. Harlang, T. Kroll, K. Kubiček, H. T. Lemke, H. W. Liang, Y. Liu, M. M. Nielsen, P. Persson, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, T.-C. Weng, D. Zhu, K. Wärnmark, V. Sundström and K. J. Gaffney, Chem. Sci., 2017, 8, 515–523 RSC.
- S. Zerdane, L. Wilbraham, M. Cammarata, O. Iasco, E. Rivière, M.-L. Boillot, I. Ciofini and E. Collet, Chem. Sci., 2017, 8, 4978–4986 RSC.
- A. Marino, P. Chakraborty, M. Servol, M. Lorenc, E. Collet and A. Hauser, Angew. Chem., Int. Ed., 2014, 53, 3863–3867 CrossRef CAS PubMed.
- T. Delgado, M. Meneses-Sánchez, L. Piñeiro-López, C. Bartual-Murgui, M. C. Muñoz and J. A. Real, Chem. Sci., 2018, 9, 8446–8452 RSC.
- J.-L. Wang, Q. Liu, Y.-S. Meng, X. Liu, H. Zheng, Q. Shi, C.-Y. Duan and T. Liu, Chem. Sci., 2018, 9, 2892–2897 RSC.
- H. J. Shepherd, C. M. Quintero, G. Molnár, L. Salmon and A. Bousseksou, Luminescent Spin-Crossover Materials, in Spin-Crossover Materials: Properties and Application, ed. M. A. Halcrow, John Wiley & Sons Ltd, Chichester, 2003, pp. 347–373 Search PubMed.
- J.-P. Xue, Y. Hu, B. Zhao, Z.-K. Liu, J. Xie, Z.-S. Yao and J. Tao, Nat. Commun., 2022, 13, 3510 CrossRef CAS PubMed.
- N.-T. Yao, L. Zhao, H.-Y. Sun, C. Yi, Y.-H. Guan, Y.-M. Li, H. Oshio, Y.-S. Meng and T. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208208 CAS.
- K.-P. Xie, Z.-Y. Ruan, B.-H. Lyu, X.-X. Chen, X.-W. Zhang, G.-Z. Huang, Y.-C. Chen, Z.-P. Ni and M.-L. Tong, Angew. Chem., Int. Ed., 2021, 60, 27144–27150 CrossRef CAS PubMed.
- Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- M. J. Murphy, K. A. Zenere, F. Ragon, P. D. Southon, C. J. Kepert and S. M. Neville, J. Am. Chem. Soc., 2017, 139, 1330–1335 CrossRef CAS PubMed.
- M. Meneses-Sánchez, L. Piñeiro-López, T. Delgado, C. Bartual-Murgui, M. C. Muñoz, P. Chakraborty and J. A. Real, J. Mater. Chem. C, 2020, 8, 1623–1633 RSC.
- N. F. Sciortino, K. A. Zenere, M. E. Corrigan, G. J. Halder, G. Chastanet, J.-F. Létard, C. J. Kepert and S. M. Neville, Chem. Sci., 2017, 8, 701–707 RSC.
- K. A. Zenere, S. G. Duyker, E. Trzop, E. Collet, B. Chan, P. W. Doheny, C. J. Kepert and S. M. Neville, Chem. Sci., 2018, 9, 5623–5629 RSC.
- R. G. Miller and S. Brooker, Chem. Sci., 2016, 7, 2501–2505 RSC.
- C. Bartual-Murgui, A. Akou, H. J. Shepherd, G. Molnár, J. A. Real, L. Salmon and A. Bousseksou, Chem. – Eur. J., 2013, 19, 15036–15043 CrossRef CAS PubMed.
- R. Kulmaczewski, E. Trzop, L. J. Kershaw Cook, E. Collet, G. Chastanet and M. A. Halcrow, Chem. Commun., 2017, 53, 13268–13271 RSC.
- A. L. Thompson, A. E. Goeta, J. A. Real, A. Galet and M. Carmen Muñoz, Chem. Commun., 2004, 12, 1390–1391 RSC.
- J.-F. Létard, C. Carbonera, E. Courcot and J. S. Costa, Bull. Mater. Sci., 2006, 29, 567–571 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescence spectra of ligand TPA-diPy, details of complex 1 including TGA traces, X-ray single crystal data and structures, PXRD patterns, fluorescence spectra and supplementary magnetic data. CCDC 2218477–2218479 and 2218494–2218496. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01217d. |
| This journal is © The Royal Society of Chemistry 2023 |