
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of an amino acid sequence-dependent analytical method for peptides using near-infrared spectroscopy†
Mika
Ishigaki
 *ab,
Atsushi
Ito
c,
Risa
Hara
c,
Shun-ichi
Miyazaki
*c,
Kodai
Murayama
c,
Sana
Tusji
a,
Miho
Inomata
a,
Keisuke
Yoshikiyo
a,
Tatsuyuki
Yamamoto
ab and
Yukihiro
Ozaki
d
*ab,
Atsushi
Ito
c,
Risa
Hara
c,
Shun-ichi
Miyazaki
*c,
Kodai
Murayama
c,
Sana
Tusji
a,
Miho
Inomata
a,
Keisuke
Yoshikiyo
a,
Tatsuyuki
Yamamoto
ab and
Yukihiro
Ozaki
d
aInstitute of Agricultural and Life Sciences, Academic Assembly, Shimane University, 1060 Nishikawatsu, Matsue, Shimane 690-8504, Japan. E-mail: ishigaki@life.shimane-u.ac.jp
bRaman Project Center for Medical and Biological Applications, Shimane University, 1060 Nishikawatsu, Matsue, Shimane 690-8504, Japan
cResearch and Development Department, Yokogawa Electric Corporation, 2-9-32 Nakacho, Musashino, Tokyo 180-8750, Japan. E-mail: shun-ichi.miyazaki@yokogawa.com
dSchool of Biological and Environmental Sciences, Kwansei Gakuin University, 2-1 Gakuen-Uegahara, Sanda, Hyogo 669-1330, Japan
First published on 15th June 2022
Abstract
We aimed to develop an amino acid sequence-dependent analytical method using near-infrared (NIR) spectroscopy. The detailed analysis of the NIR spectra of eight different amino acid aqueous solutions (glycine, alanine, serine, glutamine, lysine, phenylalanine, tyrosine, and proline) revealed different spectral patterns characteristic of different amino acid residues in the 6200–5700 and 5000–4200 cm−1 regions, and the amino acids were identified based on the patterns. The spectra in the region of 5000–4500 cm−1 for tripeptide organic solutions that were composed of the aforementioned eight amino acids clearly showed the spectral differences depending on the amino acid species and amino acid sequences. Namely, tripeptide species were clearly differentiated from each other based on the spectral pattern of NIR bands due to the combinations of N–H stretching and amide II/III modes and those derived from the first overtones of amide II and amide I. The quantitative evaluation of changes in the concentrations of dipeptides and tripeptides composed of two different amino acids, glycine and proline was performed using partial least squares regression (PLSR) analysis and a combination of bands for amide modes. The calibration and validation results with high determination coefficients (R2 ≥ 0.99) were successfully obtained based on the amino acid sequences. The results not only revealed the usefulness of NIR spectroscopy as a process analytical technology (PAT) tool for synthesizing peptides in a micro flow reactor but also proposed a general method for quantitatively analyzing NIR spectra obtained in the course of chemical synthesis.
Introduction
Special peptide drugs are considered in the pharmaceutical field as next-generation breakthrough pharmaceuticals,1 and a possible method for their synthesis has been sought using a micro flow reactor.2–7 The micro flow reactor has the following advantages: raw materials can be flowed to the columns step by step, and the targeted compound is obtained without isolating and purifying the intermediates.4,7–9 Moreover, the method has a high reaction efficiency because raw materials are reacted in fine tubes. Thus, the reaction conditions are precisely controlled by fine tuning, and a stable system of chemical synthesis is feasible using the method. The United States Food and Drug Administration has recommended the continuous flow method to be adopted as a process analytical technology (PAT) in the last two decades.10,11 Monitoring technology for chemical reactions in real time must be established to apply a micro flow reactor for special peptide drug synthesis and utilize all of its advantages.Near-infrared (NIR) spectroscopy has been partially used as one of the powerful tools for PAT.11–19 NIR spectroscopy is based on the absorption and reflection of light in the 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–4000 cm−1 region.20–23 NIR light has excellent transparency because the light energy corresponds to the forbidden transition of molecular vibrations, such as overtones and combinations of fundamentals. Thus, NIR spectra of samples on the order of millimeter thickness can be measured in a nondestructive and noninvasive manner without preprocessing. Furthermore, since NIR light has a high transmission efficiency through an optical fiber, NIR spectroscopy is ideal for application in in-line or on-line analysis during production in manufacturing. Many studies have attempted to quantitatively evaluate active pharmaceutical ingredients (APIs) using NIR spectroscopy.13–17 For example, Blanco et al. reported quantitative evaluations of the API content within tablets using partial least squares regression (PLSR) and discriminant partial least squares (DPLS) classification models.13 The NIR method showed high prediction accuracies for API concentrations within 5% error from HPLC measurements. Wahl et al. assessed the content uniformity of tablets using an NIR probe.14 They successfully developed a PLSR model for API and two excipients with high accuracy and a coefficient of determination (R2) of 0.97. In our previous study, a further possible application of NIR spectroscopy to monitor the peptide synthesis reaction in a micro flow reactor was investigated.19 The increase in the number of amide bonds with the elongation of the chain length of peptides assumed in peptide synthesis, such as glycine, diglycine, triglycine, and tetraglycine, was quantitatively evaluated using PLSR based on key NIR bands due to the combinations of amide A and amide II/III modes with very high accuracy, R2 ≈ 0.99. Based on these advantages of the NIR method, the real-time monitoring of a micro flow reactor is expected to be feasible at multiple points using NIR spectroscopy.
500–4000 cm−1 region.20–23 NIR light has excellent transparency because the light energy corresponds to the forbidden transition of molecular vibrations, such as overtones and combinations of fundamentals. Thus, NIR spectra of samples on the order of millimeter thickness can be measured in a nondestructive and noninvasive manner without preprocessing. Furthermore, since NIR light has a high transmission efficiency through an optical fiber, NIR spectroscopy is ideal for application in in-line or on-line analysis during production in manufacturing. Many studies have attempted to quantitatively evaluate active pharmaceutical ingredients (APIs) using NIR spectroscopy.13–17 For example, Blanco et al. reported quantitative evaluations of the API content within tablets using partial least squares regression (PLSR) and discriminant partial least squares (DPLS) classification models.13 The NIR method showed high prediction accuracies for API concentrations within 5% error from HPLC measurements. Wahl et al. assessed the content uniformity of tablets using an NIR probe.14 They successfully developed a PLSR model for API and two excipients with high accuracy and a coefficient of determination (R2) of 0.97. In our previous study, a further possible application of NIR spectroscopy to monitor the peptide synthesis reaction in a micro flow reactor was investigated.19 The increase in the number of amide bonds with the elongation of the chain length of peptides assumed in peptide synthesis, such as glycine, diglycine, triglycine, and tetraglycine, was quantitatively evaluated using PLSR based on key NIR bands due to the combinations of amide A and amide II/III modes with very high accuracy, R2 ≈ 0.99. Based on these advantages of the NIR method, the real-time monitoring of a micro flow reactor is expected to be feasible at multiple points using NIR spectroscopy.
In the present study, the possibility of using NIR spectroscopy to monitor the synthesis of peptides with different amino acid sequences was verified. In a micro flow reactor, raw materials are mixed in multiple steps to obtain an objective peptide.2,3 Thus, the NIR spectral features of amino acids and peptides as raw materials and products must be investigated, and marker bands derived from each substance should be identified. Therefore, eight different amino acids with characteristic amino acid residues were selected, and their NIR absorption profiles were examined in detail. Their NIR spectra showed specific spectral patterns arising from each of the residues. Furthermore, NIR spectra of tripeptides containing those amino acids also showed the characteristic strength and shape of the bands due to the combinations of N–H stretching and amide II/III modes and those derived from the first overtones of amide II and amide I in the 5000–4500 cm−1 region, in addition to the characteristic bands derived from the amino acid residues. Second, the spectral features of dipeptides and tripeptides composed of the same amino acids with different sequences were studied. Glycine (Gly) and proline (Pro) were selected as the constituents of the peptides, and the spectral differences in dipeptides (GlyPro and ProGly) and tripeptides (GlyGlyPro, GlyProGly, ProGlyGly, GlyProPro, ProGlyPro, and ProProGly) depending on the sequence of amino acids were investigated. An astonishing finding was that the NIR spectra of the peptides showed systematically different patterns, depending on the sequences.
Finally, the potential for monitoring peptide synthesis was discussed based on the results that the spectral features of dipeptides and tripeptides with different amino acid sequences were significantly different, although they were composed of the same amino acids. As the peptide synthesis reaction progresses, the concentrations of amino acids and peptides as raw materials and products decrease and increase within those mixed solutions, respectively. Mixed dimethyl sulfoxide (DMSO) solutions containing raw materials and products were prepared by changing the concentrations of the components, and the quantitative accuracies of their concentrations within mixed DMSO solutions were estimated using PLSR analysis. Since the results showed very high accuracy (R2 ≥ 0.99), they proved the possibility of monitoring peptide synthesis in a manner dependent on the amino acid sequences. Furthermore, a quantitative evaluation model for monitoring peptide synthesis may be built if the profiles of NIR spectra of raw materials and products were obtained in advance.
An NIR spectral analysis of both aqueous and organic solutions of amino acids and peptides was performed in an unprecedentedly thorough manner in this study. Based on the results obtained here, we successfully showed that the NIR bands attributed to the combination bands of amide modes showed different spectral patterns, depending on the amino acid sequences, and peptide synthesis can be monitored during the reaction time, based on these results. Furthermore, a new analytical method for NIR spectra using the second derivative spectra was also denoted. The present study not only showed the usefulness of NIR spectroscopy as a PAT tool for monitoring the synthesis of peptides with different amino acid sequences but also suggested the expansion of applications for general synthetic chemistry as a spectral analytical method.
Materials and methods
Sample preparation
A stock solution of each amino acid was prepared at a concentration of 200 mM by dissolving it in ultrapure water (214-01301, FUJIFILM Wako Pure Chemical Co., Osaka, Japan), 0.5 mol L−1 NaOH (199-02185, FUJIFILM Wako Pure Chemical Co.), or DMSO (043-07216, FUJIFILM Wako Pure Chemical Co.). Eight different amino acids (glycine, alanine, serine, glutamine, lysine, phenylalanine, tyrosine, and proline) were selected from those with the most basic structure to characteristic amino acid residues. Generally, reactive functional groups are converted to inactive groups to protect them during organic synthesis. Furthermore, a butyl group is often combined with the side chain of an amino acid to increase its solubility in an organic solvent. Thus, amino acids with an N-tert-butoxycarbonyl (Boc) group and a tertiary butyl (tBu) group at the N-terminus and a side chain, respectively, were also investigated. As examples, the chemical structures of (I) glycine and (II) Boc-protected triglycine are shown in Fig. S1 in ESI 1.† The stock solutions of various amino acids and peptides were diluted with each solvent to final concentrations of 50, 100, 150 and 200 mM. Detailed information on the amino acids and peptides used in this study is presented in ESI 1.†Fig. 1 shows a schematic of a micro flow reactor. The products are obtained by flowing raw materials, and the multiple NIR sensors installed on a reaction column detect the decrease and increase in the concentrations of raw materials and products within mixed solutions, respectively. Mixed DMSO solutions were prepared to reproduce the concentration gradients of the constituents that occurred in the micro flow reactor in the course of peptide synthesis. For example, a tripeptide (Boc-GlyGlyPro-OH) was synthesized by dehydration condensation of Boc-protected diglycine (Boc-GlyGly-OH) and a naked proline (H-Pro-OH). Stock solutions used as raw materials were prepared at a concentration of 800 mM, and those used as products were prepared at 400 mM. The detailed recipe for mixing DMSO solutions is shown in Table S1 in ESI,† and the changes in each solution over time are designated 1–9. Amino acids without protection groups were not dissolved in DMSO. Therefore, glycine tBu ester hydrochloride (H-Gly-OtBu·HCl) and proline tBu ester hydrochloride (H-Pro-OtBu·HCl) were used as model substances after desalting. The detailed procedure for desalting is presented in ESI 2,† and the comparison of the second derivative spectra of H-Gly-OtBu·HCl, H-Gly-OtBu, and (Boc)-protected glycine is presented in Fig. S2.†
 | ||
| Fig. 1 Schematic of a microflow reactor. | ||
In the present study, peptide synthesis was monitored using the combination bands due to the amide modes in the 5000–4500 cm−1 region. Since the tBu group does not produce any absorption bands in the wavenumber region of the combination bands, the addition of the group to amino acids does not affect the quantitative model for peptide synthesis. For simplicity, raw materials and products with a tBu group were displayed as its omitted manner.
NIR measurements and spectral analysis
A Spectrum One FT-NIR system (PerkinElmer, Waltham, MA, USA) was used for NIR measurements. The optical path lengths of the quartz cells used were 0.5 mm and 1 mm for aqueous and DMSO solutions, respectively. The wavenumber region measured was 10000–4000 cm−1, the spectral resolution was 4 cm−1, and 128 spectra were accumulated. Every measurement was repeated three times, and the mean spectrum was calculated using them.
The second derivative spectra were calculated using the Savitzky–Golay (SG) method with second order polynomial set of the window size as 15 (totally 31) after smoothing with the same SG conditions. The second derivative spectra of organic solutions exhibited strong peaks due to DMSO. The difference spectra of the second derivatives were calculated to remove the contributions of DMSO, and the processed spectra were used as “background-subtracted second derivative spectra” in the present study. A detailed description of the process used to calculate the difference spectra is provided in ESI 3,† and the comparison of the second derivative spectra with and without background subtraction is shown in Fig. S3.† The temperature of the quartz cells was controlled by a bath circulator (NESLAB RTE7, Thermo Scientific, Waltham, MA, USA) operated at 25 and 30 °C for the aqueous and DMSO solutions, respectively. Since amino acid and peptide samples in DMSO were sometimes precipitated and not completely dissolved at 25 °C, the temperature for measuring DMSO solutions was set as 30 °C. The stability of the temperature controller was ±0.1 °C. For the spectral analysis, the chemometrics software Unscrambler X 10.3 (Camo Analytics, Oslo, Norway) and OriginPro 6.1 (OriginLab Corporation, Massachusetts, USA) were used.
Results and discussion
NIR spectra of aqueous solutions of eight amino acids
Fig. S4A† depicts an NIR absorbance spectrum in the 10000–4000 cm−1 region for a 200 mM aqueous glycine solution. Two broad peaks at approximately 6900 and 5230 cm−1 are assigned to the combination of the antisymmetric and symmetric O–H stretching modes and that of the antisymmetric O–H stretching and O–H bending modes of water, respectively.21,24 Second derivative spectra were calculated to specify the NIR bands attributed to amino acids. Fig. 2A and B show the second derivative spectra in the 5000–4200 and 6200–5700 cm−1 regions for eight aqueous solutions of amino acids, respectively. For glycine and alanine, three peaks in the 4450–4300 cm−1 region were assigned to the combination of the C–H stretching and bending modes,24–26 although the peak positions were slightly different between the two spectra. The difference in the chemical structure between the two amino acids is the presence of a methyl group. Three peak patterns due to the combination of the C–H stretching and bending modes were characterized by a decrease in the peak intensity at 4448 cm−1 and increases at 4370 and 4296 cm−1 in the case of the alanine spectrum compared to the glycine spectrum. The changes in intensity may be because C–H components were included in the methyl or methylene group. Furthermore, in the higher wavenumber region, a new peak appeared at 5934 cm−1 in the alanine spectrum in addition to a stronger intensity of the peak at 5984 cm−1. The peak at 5934 cm−1 is due to the first overtone of the C–H stretching mode of the methyl group.24 Namely, the difference in the chemical structural difference, whether an amino acid contains a methyl group, is viewed as the difference in the spectrum in the vicinity of 5940 cm−1. Serine contains a hydroxymethyl group on a side chain, and the second derivative spectral pattern due to the C–H component was significantly different from those of glycine and alanine (Fig. 2A and B).
 | ||
| Fig. 2 The second derivative spectra in the (A) 5000–4200 and (B) 6200–5750 cm−1 regions for 200 mM aqueous solutions of eight amino acids. (C) Chemical structures of glutamine (in 0.5 mol L−1 NaOH), lysine (in pure water) and tyrosine (in 0.5 mol L−1 NaOH). | ||
Glutamine and lysine are acidic and basic amino acids with two carboxyl and amino groups, respectively. Since glutamine was dissolved in a NaOH aqueous solution, it exists as a divalent anion with two COO− groups and an amino group, as shown in Fig. 2C. Lysine, on the other hand, was diluted in pure water, and thus it exists as a dipolar ion with one COO− group and one NH3+ group in addition to an amino group (Fig. 2C). In the 4450–4300 cm−1 region, glutamine and lysine showed peaks derived from C–H components because they contain two and four methylene groups, respectively (Fig. 2A). Lysine, like glycine and alanine in pure water, exhibited three prominent peaks at 4455, 4370, and 4282 cm−1 in the 4500–4200 cm−1 region, but glutamine in a basic solution showed one peak at 4370 cm−1 with a weaker peak at 4272 cm−1, which was a different spectral pattern compared to that of lysine. The spectral variations due to C–H components depending on pH are consistent with those reported in our previous study examining the pH dependency of a glycine solution.16 Moreover, glutamine and lysine showed common peaks at approximately 4940 and 4730 cm−1. The corresponding peaks were also observed in the second derivative spectrum of tyrosine in a basic solution. The common chemical structures among glutamine, lysine, and tyrosine were an amino group, as shown in Fig. 2C, and the peaks were expected to be derived from this group. In our previous study, these peaks were consistently detected in a basic glycine solution.19 The primary amines display unique absorption peaks in the wavenumber region of the combinations at approximately 5000 cm−1.24,27 For example, n-butyl amine shows strong doublet absorption bands at 5025 and 4950 cm−1, which are not observed in the NIR spectra of di-isobutyl amine as the second amine.27 The peak at 4940 cm−1 in the second derivative spectra (Fig. 2A), which appeared without overlapping with the band arising from water, likely corresponded to the band observed in the NIR spectra of the primary amine. Therefore, a reasonable approach is to assign these two peaks (4940 and 4730 cm−1) characteristic of glutamine, lysine, and tyrosine to the amino group.
Amino acids with an aromatic ring, such as phenylalanine and tyrosine, showed a few bands in the 4700–4500 cm−1 region, including peaks at 4680, 4623, and 4582 cm−1 for phenylalanine and more complicated patterns for tyrosine with a hydroxyl group on the ring. These peaks are assigned to the combination of C–C and C–H stretching modes in a benzene ring.24,28 A pyrrolidine within proline did not produce peaks in the 4700–4500 cm−1 region but displayed several peaks in the 4500–4300 cm−1 region. In the 6150–5900 cm−1 region, phenylalanine and tyrosine produced doublet peaks at approximately 6000 and 5900 cm−1 due to the combination of C–H stretching vibrations in a benzene ring, which are expressed as ν12 + ν1 and ν15 + ν5, respectively.24,28 Proline also showed strong peaks due to the first overtone of C–H stretching at 6022, 5956, and 5826 cm−1.24
Furthermore, NIR spectra of four aqueous solutions of amino acids with a tBu group attached to a side chain, Ser(OtBu), Glu(OtBu), Lys(tBu), and Tyr(tBu), were also investigated (Fig. S4B and S4C in ESI 4†). The peaks derived from C–H components in the methyl group were clearly observed in the 6000–5900 and 4450–4200 cm−1 regions compared with amino acids without the tBu group (Fig. 2A and B).
Thus, amino acids were identified in aqueous solutions based the peak patterns for different amino acids in the 6200–5700 and 5000–4200 cm−1 regions.
DMSO solutions of Boc-protected tripeptides
The second derivative spectra for four Boc-protected tripeptides with and without an additional tBu group were investigated to understand the spectral features of the amino acids in DMSO. The main difference in the tripeptide samples in DMSO from amino acids in aqueous solutions was whether a protecting group was attached to the N-terminus, where an additional amide bond was constructed on the site (Fig. SI1†). Since DMSO contains two butyl groups, its strong absorbance due to the C–H components in the 4450–4200 cm−1 region disturbed the spectral analysis of amino acids. Therefore, the spectra of DMSO solutions were examined by excluding the wavenumber region lower than 4500 cm−1.Fig. 3 and S4D† show the background-subtracted second derivative spectra in the 5000–4500 and 6200–5700 cm−1 regions for eight Boc-protected tripeptides in DMSO, respectively. The spectra of aqueous amino acid solutions contained no peaks in the 5000–4500 cm−1 region, except for those attributed to an aromatic ring and an amino group (Fig. 2A and S4B†). DMSO solutions, on the other hand, showed some common peaks at approximately 4830, 4700, and 4600 cm−1, which were assigned to the combination of N–H stretching (or amide A) and amide II, the combination of the first overtone of amide II and amide I, and the combination of N–H stretching and amide III, respectively.20,24,26 In particular, the bands at 4840 and 4600 cm−1 were proven to be good candidates for the quantitative estimation of the number of amide bonds that varied with the elongation of the peptide chain.19 Notably, the peak shapes, peak positions, and peak intensities of these amide bands differ slightly, depending on the species of amino acids constituting tripeptides (Fig. 3). The peak intensity of the tripeptide with a proline at approximately 4840 cm−1 was particularly lower than those of the other tripeptides, and the peak position at approximately 4600 cm−1 shifted to a lower wavenumber. Amides II and III consist of coupling modes of C–N stretching and N–H in-plane bending.24,29 Since the N–H within a pyrrolidine contributes to the amide bond in the tripeptide containing proline, the differences in the molecular structure in three dimensions may have caused the changes in the vibrational modes of amide II and III.
 | ||
| Fig. 3 The background-subtracted second derivative spectra in the 5000–4500 cm−1 region for 200 mM solution of eight amino acids in DMSO. | ||
The strong peaks were detected in the background-subtracted second derivative spectra of phenylalanine and tyrosine in the same wavenumber region (4700–4500 cm−1) due to an aromatic ring, similar to their corresponding aqueous solutions (Fig. 2A and 3B). Since the intense peaks from C–H components present in a Boc group were observed in the 6200–5700 cm−1 region (Fig. S4D†) in addition to those derived from DMSO, the identification of the amino acid species was difficult in this wavenumber region. Thus, spectral differences of DMSO solutions based on the amino acid species were analyzed by focusing the wavenumber region (5000–4500 cm−1) where the combination bands of amide modes appeared.
Spectral differences in dipeptides and tripeptides based on the amino acid sequence
The amino acids were identified using the combination bands due to amide modes in the DMSO solutions. Thus, we examined whether the dipeptides and tripeptides containing the same amino acids with different sequences were distinguishable. Two Boc-protected dipeptides, Boc-GlyPro-OH (GlyPro) and Boc-ProGly-OH (ProGly), were selected as candidate dipeptides. We selected these peptides because glycine is the simplest amino acid and proline, on the other hand, has one of the most characteristic molecular structures and differs from glycine. The two dipeptides contain two amide bonds, and the molecular weights are exactly the same, but the amino acid sequence within the peptides is reversed. Six Boc-protected tripeptides consisting of at least one glycine and proline were comprehensively examined.Fig. 4 depicts the background-subtracted second derivative spectra in the 5000–4500 cm−1 region for (A) Boc-protected glycine, Boc-protected proline, two dipeptides (GlyPro and ProGly) and (B) six tripeptides (GlyGlyPro, GlyProGly, ProGlyGly, GlyProPro, ProGlyPro, and ProProGly). In Fig. 4A, bands due to the combination bands of amide modes were observed at approximately 4850, 4710, and 4600 cm−1, as mentioned above. Near the peak at approximately 4850 cm−1, doublet peaks were detected at 4870 and 4838 cm−1, and the relative peak intensities varied based on the amino acids. Furthermore, the peak positions of amide bands at approximately 4700 and 4600 cm−1 differed, depending on the amino acid sequence. Fig. 4B shows more complicated spectral patterns for amino acid sequences of tripeptides than those of dipeptides.
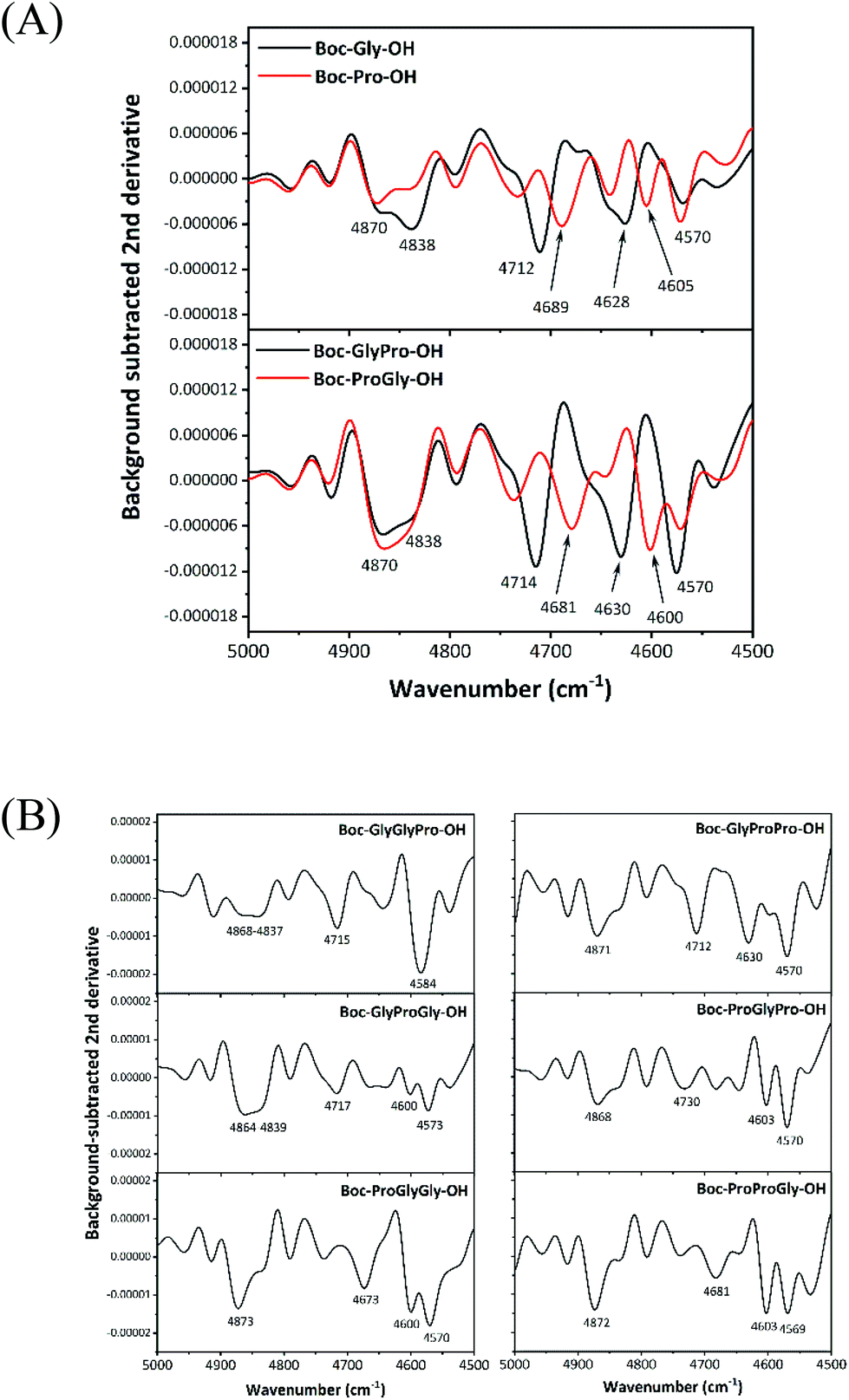 | ||
| Fig. 4 Background-subtracted second derivative spectra in the 5000–4500 cm−1 region for (A) Boc-protected glycine, Boc-protected proline, dipeptides (GlyPro and ProGly) and (B) six Boc-protected tripeptides (GlyGlyPro, GlyProGly, ProGlyGly, GlyProPro, ProGlyPro, and ProProGly) in DMSO with a 200 mM concentration. | ||
Principal component analysis (PCA) of the dataset for the second derivative spectra of Boc-protected amino acids (glycine and proline) and two dipeptides (GlyPro and ProGly) at 50, 100, 150, and 200 mM concentrations was performed to systematically understand the spectral features. Fig. 5A and B show score plots of principal component (PC) 1 vs. PC 2 and PC 3 vs. PC 4, respectively. The data were classified into two groups, (Gly, GlyPro) and (Pro, ProGly), by PC1 depending on the amino acid (glycine or proline) adjacent to the protecting group. In the loading plot of PC1, the peaks at 4716 and 4627 cm−1 and those at 4685 and 4604 cm−1 were observed to be negative and positive, respectively. These peak positions corresponded to the characteristic peaks attributed to the combination bands of amide modes shown in Fig. 4A that depended on the amino acid adjacent to the protecting group. As expected, the second derivative spectra were confirmed to mainly reflect the amino acid adjacent to the protecting group. Since the values of the PC2 score increased in proportion to the concentration of each amino acid, PC2 seemed to contain information about the average spectrum, regardless of the amino acid. In fact, the PC2 loading plot exhibited peaks at 4870–4840 and 4574 cm−1 that were out of the abovementioned region (4700–4600 cm−1), where the characteristic peaks attributed to GlyPro or ProGly were located. Furthermore, PC3 separates the dataset into [Gly, ProGly] and [Pro, GlyPro]. Notably, in the PC3 loading plot, the combination bands of amide modes at approximately 4838 cm−1 and 4574 cm−1 were negative and positive, respectively. The result reflected that the second derivative intensities of two samples (glycine and ProGly) were higher and lower at 4838 and 4574 cm−1 than those of the other samples, respectively. PC4 scores varied according to the dipeptide concentrations. The dataset of the tripeptide spectra was also systematically classified into groups primarily reflecting the amino acid adjacent to the protection group followed by the amino acid bound to the first amino acid and the next amino acid.
 | ||
| Fig. 5 Score plots of (A) PC1 vs. PC2 and (B) PC3 vs. PC4. (C) Loading plots of PC1, PC2, PC3, and PC4. | ||
In the present study, peptides consisted of combinations of glycine and proline, which have the most basic molecular structure and the most characteristically different structure, respectively. Thus, differences in the spectra based on the amino acid sequences were likely to be clearly observed. Tripeptides composed of glycine and alanine, which have similar molecular structures except for the presence or absence of a methyl group, respectively, also showed different spectral patterns in the wavenumber region of the combination bands of amide modes (Fig. S5 in ESI 4†). Based on these results, dipeptides and tripeptides composed of the same amino acids but with different sequences were identified by the spectral patterns of the combination bands of amide modes in the 5000–4500 cm−1 region.
Quantitative evaluation of the amino acid concentrations in a model of peptide synthesis
The possibility of monitoring peptide synthesis using NIR spectroscopy was verified. In mixed DMSO solutions consisting of amino acids and peptides assumed to be raw materials and products in the course of peptide synthesis, the concentration of each substance was quantitatively estimated using PLSR analysis. Two types of raw materials and one product were assumed to be present in chemical reactions designed to synthesize dipeptides with one glycine and one proline (a, b) and tripeptides with two glycine and one proline (c–e) or one glycine and two prolines (f–h), as listed in Table S2 in ESI.†Fig. 6A depicts the background-subtracted second derivative spectra of peptide synthesis (a) and (b) changes over time, as defined in Table S2,† which were prepared using the recipe shown in Table S1.† As the concentrations of the constituents changed, the intensities of the combination bands of amide modes at approximately 4860, 4740, and 4600 cm−1 were confirmed to vary. PLSR analysis was conducted on the dataset of the second derivative spectra of (a) defined in Table S2† in the 5000–4500 cm−1 region, and the concentrations of Boc-protected glycine, naked proline, and GlyPro in the mixed DMSO solutions were quantitatively evaluated using a leave-one-out cross validation method. In this method, one spectral data point was left out, and the calibration model was built using the remaining data. The first removed data point was applied to the calibration model, and the prediction residual was calculated for validation. This process was repeated until all the spectral data points were excluded once. The accuracy of the models was assessed by calculating the coefficient of determination (R2) and root mean square error (RMSE) for both calibration (in black) and validation (in red). Fig. 6B shows the results of the calibration and validation curves for the concentration of Boc-protected glycine in the mixed DMSO solutions. The results in Table S3 in ESI† show that R2 values were always greater than 0.99 and that the contribution of Factor 1 to explaining the variance was dominant and greater than 99%. Fig. 6C(a) shows the loading plots of Factor 1 that were extracted to evaluate the concentration of Boc-protected glycine in a mixed DMSO solution. The ratio of decreasing concentrations of both Boc-protected glycine and naked proline as raw materials was the same, and the ratio of an increasing concentration of GlyPro as the product was also the same as those of raw materials, but with an opposite sign. Thus, Factor 1 in the PLSR analysis for evaluating one constituent was revealed to include changes in the concentrations of the other two constituents (a naked proline and GlyPro) in addition to those of Boc-protected glycine. Namely, only one loading plot was sufficient to quantitatively evaluate the concentrations of all three constituents. In fact, Factor 1 in the PLSR model of a naked proline showed the same spectral pattern as Boc-protected glycine shown in Fig. 6C(a), and the model for estimating GlyPro showed the inverse spectral pattern in Fig. 6C(a).
 | ||
| Fig. 6 (A) The background-subtracted second derivative spectra in the 5000–4500 cm−1 region for mixed DMSO solutions (a) and (b) defined in Table S2.† (B) Plots of the calibration (in black) and validation (in red) results for the concentration of Boc-protected glycine obtained using the second derivative spectral data from (a) in Table S2† in the 5000–4500 cm−1 region. Validation plots of the concentration of GlyPro (in blue) estimated by applying the data from (b) into the calibration curve built using the data from (a) in Table S2.† (C) (Left panel) The loading plots of Factor 1 in the PLSR model for evaluating the concentration of Boc-protected (a) glycine and (b) proline in Table S2.† (Right panel) the spectra of CCF for (a) and (b). | ||
The PLSR results calculated for the dipeptide dataset with the different amino acid sequences (b) defined in Table S2† also provided very high quantitative accuracies (Table S3†). The loading plot of Factor 1 extracted to calculate the concentration of Boc-protected proline is shown in Fig. 6C(b), and it showed a different spectral pattern than that presented in Fig. 6C(a). The loading plot reflects the characteristic peaks of the constituents in the mixed DMSO solutions; the raw materials and the products were negative and positive, respectively. For example, the peaks appearing on the background-subtracted spectra of GlyPro (4630 and 4570 cm−1) and ProGly (4680 and 4600 cm−1) shown in Fig. 4A were extracted as positive peaks in the loading plots in Fig. 6C(a) and C(b), respectively. Namely, Factor 1 reflects the characteristic spectral pattern of raw materials and products, and the chemical synthetic reaction of peptides with different amino acid sequences should be monitored in a sequence-dependent manner. The dataset in (b) was applied as a test set (in blue) to the calibration model (in black) built using the dataset shown in (a), and the concentrations of GlyPro within the mixed DMSO solution were predicted to quantitatively confirm the results (Fig. 6B). The value of R2 was 0.228, and the concentration of GlyPro was unable to be evaluated using the calibration model built based on the dataset of the dipeptide with different amino acid sequences (ProGly) compared to its own dataset.
The PLSR results obtained for the model samples in which six tripeptides were synthesized (c–h), as defined in Table S2,† were also revealed to have very high accuracies, with R2 > 0.99, as shown in Table S4.† The loading plots of Factor 1 extracted to calculate the concentration of raw material 1 in Table S2† are shown in Fig. S6,† and they showed different spectral patterns reflecting the characteristic spectral features of raw materials and products, as in the case of dipeptides.
Here, the detailed spectral analysis of the loading plots for Factor 1 in the PLSR model was performed by examining whether Factor 1 in the PLSR model that contributed more than 99% to the explained variance was extracted by the spectral calculation as a calculated contributing factor (CCF). In a DMSO solution prepared to reproduce peptide synthesis, the equal concentrations of two raw materials decreased, and the concentration of one product increased by an equal amount, as mentioned above. Therefore, the CCF was expected to be calculated by summing the second derivative spectra of two raw materials and by subtracting those of one product and a solvent. Double counted DMSO components in summation of the raw material spectra were removed by subtracting the DMSO spectra in addition to those of the product. The right side of Fig. 6C shows the CCF spectra extracted using the second derivative spectra of raw materials and products with 200 mM concentrations of (a) and (b) defined in Table S2† and those of DMSO. They showed a spectral pattern similar to that of the loading plot of Factor 1 in the PLSR model, as shown on the left side of Fig. 6C. Namely, the CCF to monitor peptide synthesis can be extracted only if the spectral patterns of raw materials, products, and a solution are obtained in advance.
Conclusions
The analytical method for monitoring peptides with different amino acid sequences was investigated using NIR spectroscopy. The NIR second derivative spectra of amino acids displayed different spectral patterns, depending on the amino acid residues, and the peptides with different amino acid sequences were discriminated based on the differences in spectral patterns due to the combination bands of amide modes. Using the results, peptide synthesis was quantitatively monitored with very high accuracy. The main factor contributing to the PLSR model was calculated using the second derivative spectra of raw materials, products, and an organic solvent. Thus, not all calibration curves needed to be built, based on the experimental data obtained for each peptide synthesis reaction. Since the verification of the quantitative accuracy for peptide synthesis based on the CCF is beyond the scope of this study, further discussions of this parameter are not mentioned. However, the possibility of universally constructing a quantitative model for monitoring peptide synthesis by understanding the NIR spectra of raw materials and products for synthesizing peptides is a great advance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST-Mirai Program Grant Number JPMJMI18G7, Japan and “Shimane University Grants for Joint Research Project led by Female Researchers” under the MEXT “Initiative for Realizing Diversity in the Research Environment (Collaboration Type)”.References
- M. Castanho and S. Nuno, Peptide drug discovery and development: translational research in academia and industry, John Wiley & Sons, New Yolk, 2011 Search PubMed.
- S. Fuse, Y. Mifune and T. Takahashi, Angew. Chem., Int. Ed., 2014, 53, 851–855 CrossRef CAS PubMed.
- S. Fuse, Y. Otake and H. Nakamura, Chem. – Asian J., 2018, 13, 3818–3832 CrossRef CAS PubMed.
- S. Ramesh, P. Cherkupally, G. Beatriz, T. Govender, H. G. Kruger and F. Albericio, Amino Acids, 2014, 46, 2091–2104 CrossRef CAS PubMed.
- A. S. Myerson, M. Krumme, M. Nasr, H. Thomas and R. D. Braatz, J. Pharm. Sci., 2015, 104, 832–839 CrossRef CAS PubMed.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- G. Allison, Y. T. Cain, C. Cooney, T. Garcia, T. G. Bizjak, O. Holte, N. Jagota, B. Komas, E. Korakianiti, D. Kourti, R. Madurawe, E. Morefield, F. Montgomery, M. Nasr, W. Randolph, J. Robert, D. Rudd and D. Zezza, J. Pharm. Sci., 2015, 104, 803–812 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- D. J. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- Food and Drug Administration, PAT—Framework for Innovative-Pharmaceutical Development, Manufacturing, and Quality Assurance. 2004, 2004 Search PubMed.
- K. A. Bakeev, Process analytical technology: spectroscopic tools and implementation strategies for the chemical and pharmaceutical industries, John Wiley & Sons, New Yolk, 2010 Search PubMed.
- E. W. Ciurczak and B. Igne, Pharmaceutical and medical applications of near-infrared spectroscopy, CRC Press, Boca Raton, 2014 Search PubMed.
- M. Blanco, M. Alcalá, J. M. González and E. Torras, J. Pharm. Sci., 2006, 95, 2137–2144 CrossRef CAS PubMed.
- P. R. Wahl, G. Fruhmann, S. Sacher, G. Straka, S. Sowinski and J. G. Khinast, Eur. J. Pharm. Biopharm., 2014, 87, 271–278 CrossRef CAS PubMed.
- F. F. Gouveia, J. P. Rahbek, A. R. Mortensen, M. T. Pedersen, P. M. Felizardo, R. Bro and M. J. Mealy, Anal. Bioanal. Chem., 2017, 409, 821–832 CrossRef CAS PubMed.
- C. Schaefer, C. Lecomte, D. Clicq, A. Merschaert, E. Norrant and F. Fotiadu, J. Pharm. Biomed. Anal., 2013, 83, 194–201 CrossRef CAS PubMed.
- R. B. Shah, M. A. Tawakkul and M. A. Khan, J. Pharm. Sci., 2007, 96, 1356–1365 CrossRef CAS PubMed.
- K. Murayama, D. Ishikawa, T. Genkawa and Y. Ozaki, Appl. Spectrosc., 2018, 72, 551–561 CrossRef CAS PubMed.
- M. Ishigaki, A. Ito, R. Hara, S. Miyazaki, K. Murayama, K. Yoshikiyo, T. Yamamoto and Y. Ozaki, Anal. Chem., 2020, 93, 2758–2766 CrossRef PubMed.
- M. Ishigaki and Y. Ozaki, in Vibrational Spectroscopy in Protein Research, ed. Y. Ozaki, M. Baranska, I. K. Lednev and B. R. Wood, Academic Press, Massachusetts, 2020 Search PubMed.
- Y. Ozaki, C. Huck, S. Tsuchikawa and S. B. Engelsen, Near-Infrared Spectroscopy: Theory, Spectral Analysis, Instrumentation, and Applications, Springer, Berlin/Heidelberg, 2021 Search PubMed.
- Y. Ozaki, C. W. Huck and K. B. Beć, in Molecular and Laser Spectroscopy, ed. V. P. Gupta, Elsevier, Amsterdam, 2017 Search PubMed.
- Y. Ozaki, Anal. Sci., 2012, 28, 545–563 CrossRef CAS PubMed.
- J. Workman Jr. and L. Weyer, Practical guide and spectral atlas for interpretive near-infrared spectroscopy, CRC Press, Boca Raton, 2012 Search PubMed.
- W. Hug, J. M. Chalmers and P. R. Griffith, Handbook of vibrational spectroscopy, John Wiley & Sons, New Yolk, 2002 Search PubMed.
- P. Wu and H. W. Siesler, J. Near Infrared Spectrosc., 1999, 7, 65–76 CrossRef CAS.
- J. E. Sinsheimer and A. M. Keuhnelian, Anal. Chem., 1974, 46, 89–93 CrossRef CAS.
- W. Kaye, Spectrochim. Acta, 1954, 6, 257–287 CrossRef CAS.
- Y. Sugawara, A. Y. Hirakawa and M. Tsuboi, J. Mol. Spectrosc., 1984, 108, 206–214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Information about amino acids and peptides, the procedure used in the desalting operation, the procedure used to calculate the background-subtracted second derivative spectra, supporting tables, and figures for NIR spectra. See DOI: https://doi.org/10.1039/d2an00895e |
| This journal is © The Royal Society of Chemistry 2022 |