
Separation of intact proteins by capillary electrophoresis†
Sarah
Meyer
 a,
David
Clases
b,
Raquel
Gonzalez de Vega
b,
Matthew P.
Padula
c and
Philip A.
Doble
*a
a,
David
Clases
b,
Raquel
Gonzalez de Vega
b,
Matthew P.
Padula
c and
Philip A.
Doble
*a
aThe Atomic Medicine Initiative, University of Technology Sydney, Sydney, NSW, Australia. E-mail: philip.doble@uts.edu.au
bInstitute of Chemistry, University of Graz, Graz, Austria
cSchool of Life Sciences and Proteomics, Lipidomics and Metabolomics Core Facility, Faculty of Science, University of Technology Sydney, Sydney, NSW, Australia
First published on 25th May 2022
Abstract
This work introduces novel and universal workflows for the analysis of intact proteins by capillary electrophoresis and presents guidelines for the targeted selection of appropriate background electrolytes (BGEs) by consideration of the target proteins’ isoelectric point (pI). The suitability of neutral dimethyl polysiloxane (PDMS) capillaries with dynamic coatings of cationic cetyltrimethylammonium bromide (CTAB) or anionic sodium dodecyl sulfate (SDS), and bare fused silica (BFS) capillaries were systematically evaluated for the analysis of histidine and seven model proteins in six BGEs with pH values between 3.0 and 9.6. Multiple capillary and BGE combinations were suitable for the analysis of all proteins with molecular weights ranging from 13.7–150 kDa, and pIs between 4.7 and 9.6. The CTAB-PDMS capillary was best suited for low pH BGEs, while the SDS-PDMS and BFS capillary were superior for high pH BGEs. These combinations consistently resulted in sharp peak shapes and rapid migration times. pH values of BGEs closer to the proteins’ pI produced poorer peak shapes and decreased effective mobilities due to suppressed ionisation. Plots of mobility vs. pH crossed at approximately the pI of the protein in most cases. The workflow was applied to the analysis of caseins and whey proteins in milk for the separation of the seven most abundant proteins, including the isoforms of A1 and A2 β-casein and β-lactoglobulin A and B.
Introduction
Proteomics may be defined as the study of the entirety of proteomes. The proteome is a collection of proteins produced in a species or organ and typically contains many thousands of proteins of potential interest that require identification and quantification to gain insights into fundamental biological processes and biomarker discovery.1 Capillary Electrophoresis (CE) is an ideal modality for the separation and analysis of intact proteins due to its high separation efficiency, compatibility with wide mass ranges, suitability for small sample volumes, and facile sample preparation prior to analysis. However, separations by CE are often hampered by undesirable electrostatic and hydrophobic interactions between proteins and the fused silica capillary surface,2–5 leading to potential sample loss, band broadening, peak tailing, and poor reproducibility.6,7Regardless, most CE protein separations are performed using bare fused silica (BFS) capillaries. The net charge of any given protein may be inferred by consideration of the isoelectric point (pI) and the pH of the CE background electrolyte (BGE). When the pH of the BGE is below that of the pI, the protein will have a net positive charge, and at pH values above, a net negative charge. Furthermore, at BGE pH values above 3, the silanol groups at the surface of the capillary are deprotonated and negatively charged, typically limiting the selection of suitable BGEs to a narrow pH range. At high pH, electrostatic interactions are minimised by the repulsive negative charges on both the capillary surface and the protein. This limited pH selection renders some protein analyses unfeasible due to solubility or stability issues, or insufficient electrophoretic mobility when the pH of the BGE is close to the isoelectric point of the protein of interest. Attempts to mitigate adsorption include increasing the ionic strength of the BGE to compete with proteins for available binding sites on the capillary surface; the addition of ion pairing reagents and surfactants to the BGE to lower the net protein charge;7 or by inclusion of organic solvents into the BGE.4
Modifications of the BFS capillary surface have also shown some success at preventing protein interactions to improve the separation efficiency and reproducibility, and the practicable pH range for separation. These approaches typically involve the addition of dynamic coating reagents to the BGE; or the manufacture of static coatings by chemically modifying the capillary surface, or adsorption of a coating agent.7 These coatings must be compatible with the sample matrix, be stable over time, and allow the use of a variety of BGEs with a wide range of concentrations. Ideally, they should be easy to prepare and inexpensive.7
Various neutral, cationic, and anionic BGE additives have been employed to dynamically coat the surface of BFS capillaries. The charge on the surface of the capillary may be neutralised by addition of non-ionic polymeric surfactants including polyoxyethylene ether (Brij-35),8 or hydroxypropylmethylcellulose,9 which mask the silica surface and deactivate the silanol groups, and slow down or eliminate the electroosmotic flow (EOF). Suitable cationic additives include polyamines,10 quaternary ammonium surfactants, and polysaccharides,11 which electrostatically bind to the capillary surface and suppress or reverse the EOF. Anionic additives, such as dextran sulfate12 or poly(vinyl sulfonic acid)13 generate a negative charge at the surface of the capillary over a wide pH range. These dynamic coatings offer facile optimisation of separation selectivity as the modifier is added to the BGE, and improved repeatability by continuous regeneration of the coating during each analysis. However, separation efficiencies may be negatively impacted by undesirable interactions between the additive and the analyte or matrix.7
Static-covalent and static-adsorbed coatings offer greater stability than their dynamic counterparts. Static-covalent coatings are typically manufactured in situ by polymerisation or silanisation reactions. Static-adsorbed coatings are prepared by flushing the capillary with polymers containing cationic amine functional groups, or polymers with a polyacrylamide backbone. The high charge density of these reagents produce sufficiently stable coatings by strong electrostatic adsorption to the silanol surface of the capillary.14 Other examples of cationic coating reagents include polyethylenimine,15 polybrene16 and polyarginine,17 or the application of successive multiple ionic polymer layers.18 Anionic coatings may also be fabricated with bilayers of polybrene and dextran sulfate19 or polybrene and polyvinyl sulfonic acid.20 Similarly, neutral capillary surfaces may be prepared with polyvinyl alcohol,21 and linear polyacrylamide.6,22 Further details and applications of capillary coatings are available in comprehensive reviews by Horvath and Dolník2 and Huhn et al.7
An area of investigation that has had little attention is the transfer of the column technology of Gas Chromatography (GC). GC capillary columns are manufactured from the same fused silica and outer polyimide coating typically employed with CE separations, are easily obtained and of low cost. GC capillary columns are internally coated with a bonded liquid film of varying polarities such as neutral dimethyl polysiloxane (PDMS) and (phenyl)-methylpolysiloxane, or highly polar polyethylene glycol. Reports of the application of GC columns for CE include the analysis of anions,23–25 low-molecular-mass RNA,26 amino acids,27 nucleotides and coenzyme A compounds,28 cysteine and thiols,29 proteins in white wine,30 glycoproteins,31 and oligosaccharides.32 While some groups reported reliable and reproducible outcomes, others experienced difficulties with BGE pH selection, concentration, or incompatible additives.
The analysis of intact proteins remains challenging due to the variety of molecular weights (MW), hydrophobicities, and pIs, rendering the selection of suitable CE conditions tedious and/or ad hoc. This work introduces a novel and universal experimental workflow for the analysis of intact proteins with any MW or pI, as well as guidelines for targeted selection of appropriate BGEs. Here, we evaluated the suitability of neutral GC PDMS (DB-1) capillaries with dynamic coatings of cationic cetyltrimethylammonium bromide (CTAB) or anionic sodium dodecyl sulfate (SDS), and BFS capillaries, for the separation of seven model proteins with a range of MWs and pIs using six different BGEs with pH values between 3.0–9.5. Finally, we applied the workflow and guidelines to select the most suitable BGE for the analysis of caseins and whey proteins in cow's milk.
Experimental section
Chemicals and consumables
Ultrapure water was obtained from an Arium Pro system (Sartorius Stedim Plastics GmbH, Germany). The proteins, bovine serum albumin (fraction V), α-casein (from bovine milk), β-casein (from bovine milk), creatine kinase (from rabbit muscle), human serum albumin, α-lactalbumin (from bovine milk), β-lactoglobulin (from bovine milk), β-lactoglobulin A (from bovine milk), β-lactoglobulin B (from bovine milk), myoglobin (from equine heart), ribonuclease A (from bovine pancreas), and transferrin (human, 98%, low endotoxin) were purchased from Sigma Aldrich (Castle Hill, New South Wales, Australia). An immunoglobulin G antibody (raised in sheep) was acquired from Abcam (Melbourne, Victoria, Australia). Citric acid (CA), tris(hydroxymethyl)aminomethane (TRIS), di-sodium hydrogen orthophosphate dodecahydrate (Na2HPO4·12H2O), sodium dihydrogen orthophosphate (NaH2PO4·2H2O), sodium tetraborate decahydrate, cetyltrimethylammonium bromide (CTAB), sodium dodecyl sulfate (SDS) and acetone were obtained from Sigma Aldrich (Castle Hill, New South Wales, Australia). Reduced fat (lite) milk was bought from a local supermarket (Coles, Sydney, Australia). The bare fused silica (BFS) capillary was purchased from CM Scientific (Silsden, United Kingdom) and the DB-1 Capillary (part number 126-1012, internal diameter 0.05 mm, film thickness 0.2 μm) was acquired from Agilent Technologies (Mulgrave, Victoria, Australia).Capillary dimensions and coatings
The experiments for the systematic study were performed via consideration of three capillary configurations; a CTAB coated PDMS capillary (effective length 51 cm, total length 59 cm, internal diameter 50 μm), a SDS coated PDMS capillary (effective length 56 cm, total length 64 cm, internal diameter 50 μm) and a bare fused silica capillary (effective length 56 cm, total length 64 cm, internal diameter 50 μm). The analyses of the milk samples were executed using a CTAB coated PDMS capillary with an increased capillary length (effective length 110 cm, total length 103 cm, internal diameter 50 μm).Prior to using PDMS capillaries for the first time, they were conditioned with the respective surfactant to establish a consistent coating of the PDMS capillary walls. The capillaries were flushed with 1 mM CTAB (or 1 mM SDS) for four hours and subsequently equilibrated with 0.1 mM CTAB (or 0.01 mM SDS) for two hours.
BGE, standards and sample preparation
| BGE composition | pH (BGE with 0.1 mM CTAB) | pH (BGE with 0.01 mM SDS) | pH (BGE without surfactant) | |
|---|---|---|---|---|
| “—” Not used for systematic evaluation. | ||||
| BGE I | 10 mM CA + 5 mM TRIS (+surfactant) | 3.2 | 3.0 | 3.1 |
| BGE II | 10 mM CA + 15 mM TRIS (+surfactant) | 4.5 | 4.5 | 4.5 |
| BGE III | 10 mM CA + 25 mM TRIS (+surfactant) | 5.9 | 6.1 | 6.2 |
| BGE IV | 10 mM phosphate (+surfactant) | 7.0 | 7.0 | 7.0 |
| BGE V | 10 mM CA + 60 mM TRIS (+surfactant) | 8.2 | 8.4 | 8.3 |
| BGE VI | 10 mM tetraborate (+surfactant) | 9.3 | 9.6 | 9.3 |
| BGE M-I | 20 mM CA + 10 mM TRIS (+surfactant) | 3.2 | — | — |
| BGE M-II | 20 mM CA + 20 mM TRIS (+surfactant) | 3.8 | — | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and stored at 5 °C. Prior to analysis the supernatant was diluted 50:50 with ultrapure water and 3.3% Acetone was added as an EOF marker. Protein identification in the milk sample was performed by individual spiking each protein standard and visual evaluation of the resultant electropherograms (Fig. SI 4†).
000 rpm) and stored at 5 °C. Prior to analysis the supernatant was diluted 50:50 with ultrapure water and 3.3% Acetone was added as an EOF marker. Protein identification in the milk sample was performed by individual spiking each protein standard and visual evaluation of the resultant electropherograms (Fig. SI 4†).
CE experimental parameters
The CE analyses were performed using an Agilent CE 7100 system (Agilent, Santa Clara, CA, USA). The cartridge temperature was 30 °C and the sample storage carousel was maintained at 20 °C. Absorption was monitored at 200 nm for proteins and 240 nm for acetone with an acquisition rate of 10 Hz. Prior to each analysis, the capillary was rinsed with the BGE for 2 min for the systematic study, and 5 min for the milk analysis. The sample was injected by applying a pressure of 0.725 psi for 10 s and separated at −30 kV (CTAB Capillary) or +30 kV (SDS Capillary, Fused Silica Capillary) for 8–80 min. Analyses were performed in triplicate to determine standard deviations.Data analysis
The electropherograms were integrated using the Agilent ChemStation software. The migration time for the EOF marker acetone, and the proteins were obtained using the peak maximum at 240 nm and 200 nm, respectively. The electroosmotic mobility μEOF and the apparent mobility of the analyte μa were calculated using eqn (1) and (2). The effective mobility μe was calculated using eqn (3);33 | (1) |
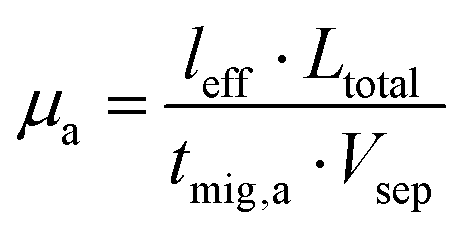 | (2) |
| μe = μa − μEOF | (3) |
Results and discussion
Suitability of PDMS capillaries
Typical CE applications use BFS capillaries with internal diameters of 10–75 μm and capillary lengths of 20–100 cm.34 Fortunately, GC capillary columns are available with CE compatible internal diameters of 50–100 μm and lengths between 5–100 m.35 Accordingly, GC columns may be purchased off-the-shelf and cut to size as required. A window for UV detection may be simply fabricated by burning away the capillary polyimide outer coating at the appropriate position for custom fits to any CE instrument. GC analyses separate volatile molecules using an inert gas as the mobile phase. The injection of water is usually avoided on polar GC columns to maintain repeatability and to prevent adsorption of water molecules on the inner capillary thin film. Neutral GC capillaries are unaffected by exposure to water. The successful transfer of GC capillary columns for CE applications requires the internal coating to be resilient to the BGE at varying pH values, and stability of the coating during electrophoresis at high voltages to provide a constant current. Therefore, we evaluated the stability of a neutral PDMS capillary using various aqueous buffers that encompassed standard CE pH working ranges from approximately 3 to 9. The PDMS capillary was cut to a length of 60 cm and flushed with each of the buffers for 10 min prior to applying high voltage. The current remained stable in all buffers in a single PDMS capillary for over 100 h of operation. Table 2 lists the recorded average currents and relative standard deviation (RSD) for the six BGEs when applying a voltage of +30 kV to the inlet electrode for 10 min. The recorded currents varied due to different ionic strengths between 5 and 24 μA, the RSDs, however, were below 0.5% for each BGE. This result indicated that the PDMS film was stable in aqueous buffers and was a candidate for evaluation of the separation of intact proteins.| Average current [μA] | RSD [%] | |
|---|---|---|
| BGE I | 5.94 | 0.48 |
| BGE II | 10.54 | 0.12 |
| BGE III | 19.58 | 0.18 |
| BGE IV | 19.48 | 0.39 |
| BGE V | 23.80 | 0.23 |
| BGE VI | 19.88 | 0.30 |
Dynamic coating of PDMS capillaries
The suitability of the PDMS capillary was initially evaluated with a standard solution consisting of an EOF marker, acetone, the amino acid, histidine (His), and the small protein, transferrin (Tf), using BGE I (10 mM CA + 5 mM TRIS, pH = 3.2) at +30 kV applied to the inlet electrode. Via consideration of the isoelectric points of 7.636 (His) and 5.437 (Tf), it was expected that both analytes were positively charged at pH 3.2, and would migrate towards the detector placed at the cathode.As anticipated, an EOF was not observed due to the neutral capillary surface, and His migrated as a sharp peak at 6.5 min. However, despite having a net positive charge, a peak for Tf was not observed during the acquisition because of complete hydrophobic adsorption on the neutral capillary coating (Fig. 1A, top). The capillary was then flushed for ten min with the same BGE, this time with the addition of CTAB (10 mM CA, 5 mM TRIS, 0.1 mM CTAB, pH = 3.2). The polarity of the voltage was reversed to −30 kV at the inlet electrode. 10 min of initial equilibration with the new BGE was sufficient to generate a stable and rapid anodic EOF as measured by the acetone peak at 2.8 min, the Tf migrated as a distinct peak at 4.8 min, and His migrated as a narrow and symmetrical peak at 5.2 min (Fig. 1A, bottom). CTAB is a quaternary ammonium surfactant with a long alkyl chain of 16 carbon atoms. This hydrophobic chain adsorbed to the neutral capillary surface, whilst the positive ammonium functional group orientated towards the aqueous bulk electrolyte. This surfactant layer effectively eliminated the hydrophobic adsorption of the protein by repulsive positive charges on both the PDMS film and the protein. The CTAB in the BGE was below the critical micellar concentration (CMC) to minimize potential interactions between the surfactant and the analytes, yet at sufficient concentration for effective maintenance of the coating of the capillary. The separation of His and Tf occurred counter to the direction of the EOF. The migration time of all three analytes stabilised after the fourth injection indicating that the surfactant layer reached equilibrium in less than 50 min of continuous acquisition (Fig. SI 1A†).
 | ||
| Fig. 1 Analysis of a standard comprising of acetone, His and Tf on uncoated and coated PDMS capillaries at pH 3.2 (A) and 8.6 (B). At pH 3.2, all analytes were positively charged, however only His was detected when +30 kV was applied to the inlet electrode of the uncoated PDMS capillary. The EOF was not observed due to the neutral capillary surface, and a peak for Tf was absent due to hydrophobic absorption (A, top). This was resolved by adding 0.1 mM CTAB to the BGE and flushing the capillary for 10 min. At −30 kV applied to the inlet electrode, all three analytes rapidly migrated with narrow and symmetrical peak shapes (A, bottom). The surfactant's carbon chain adsorbed to the PDMS capillary walls and produced a positively charged internal coating. In the equivalent experiments at pH 8.4, neither of the three analytes were detected on the uncoated capillary at −30 kV (B, top). The EOF and Tf were again absent due to the neutral capillary surface and hydrophobic absorption, while His was not observed. Adding 0.01 mM SDS to the BGE and flushing the capillary for 10 min and application of +30 kV produced an EOF and co-migrating His peak, whilst Tf was not observed (B, bottom). | ||
The longevity of the surfactant layer was evaluated by removal of CTAB from the BGE and successive injections of the three analytes. As shown in Fig. SI 2A,† acetone, Tf and His were only observed as narrow peaks when the BGE contained CTAB (red line). Subsequent analyses without CTAB and a three min BGE flush before each injection resulted in adsorption of the protein to the capillary surface and increasing migration times and larger peak widths of the EOF marker and His (Fig. SI 1B†). This indicated that the surfactant layer was rapidly washed from the PDMS film and that incorporation of CTAB in the BGE was essential.
An equivalent reciprocal investigation was conducted above the isoelectric points of the target analytes at pH 8.4. Application of −30 kV with BGE V (10 mM CA + 60 mM TRIS, pH = 8.4) resulted in no observable peaks due to an absence of EOF, hydrophobic absorption of Tf, and insufficient mobility of His for migration within a 60 min acquisition window (Fig. 1B, top). Addition of the anionic surfactant, sodium dodecyl sulfate (SDS) at 0.01 mM, flushing for 10 min, and application of +30 kV produced a stable cathodic current with the EOF and His migrating at 3.1 min, whilst Tf was not observed (Fig. 1B, bottom). In contrast to the CTAB coated capillary, repeated coating and standard injection produced ever increasing migration times for both acetone and His (Fig. SI 1B†). This instability indicated that the SDS coating was compromised by adsorption of Tf, which may be resolved by coating the capillary for an extended time prior to injection. The stability of the coating was similar to that of CTAB, with rapid wash off when removed from the BGE.
These observations implied that PDMS capillaries had excellent potential for tuneable analyses of proteins.
Systematic evaluation of BGEs and capillaries for protein analyses
Both surfactants remain charged across the entire practicable CE BGE pH range, allowing manipulation of pH and EOF direction for predictable control of selectivity and analyte mobility. Accordingly, we conducted a systematic evaluation of three possible scenarios; CTAB coated PDMS capillaries with anodic EOF; SDS coated PDMS capillaries with cathodic EOF; and the more traditional BFS capillaries with cathodic EOF. These three scenarios offered the possibility to tune and optimise the separation of any protein with known pI without hydrophobic or electrostatic adsorption. We chose seven model proteins (and the amino acid His) with a variety of MWs and pIs as illustrated in Fig. 2. The MWs and pIs of these proteins are shown in Table SI 1A.† β-lactoglobulin (β-Lg), myoglobin (Mb) and ribonuclease A (RNase) represented small proteins (<30 kDa) with pIs of 5.1, 7.1 (±0.3) and 8.6, respectively. Medium sized proteins (60–100 kDa) consisted of human serum albumin (HSA), transferrin (Tf) and creatine kinase (CK) with pIs of 4.7, 5.4, and 7.2, respectively. An immunoglobulin G antibody (IgG) was selected as a representative of large proteins (>100 kDa) with a pI of 6.0 (±1.0). Acetone was added to each protein standard for determination of the EOF. Six different BGEs were prepared and had pH values ranging from 3.0 to 9.6 as shown in Table 1. For clarification, the surfactants were only added to the BGEs for analyses using the PDMS capillary columns, but not for the BFS capillary. | ||
| Fig. 2 Molecular masses and isoelectric points of selected model proteins. The amino acid His was added as a marker due to its charge over a wide pH range. The analytes were selected to cover a wide range of MWs and pIs. | ||
The versatility of the proposed workflow may be best illustrated by evaluation of the migration behaviour of the simplest analyte, His. His has a pI of 7.6 and was charged over a wide pH range and had negligible interaction with the surface of the capillaries under all separation conditions.
Consider the electropherograms obtained from the CTAB coated PDMS capillary as shown in Fig. 3A1. As expected, there was stable anodic EOF (negative polarity) under all conditions due to the positive charge on the ammonium functional group of CTAB irrespective of the BGE pH. However, the mobility of the EOF varied between the BGE systems due to necessary changes of ionic strengths of the BGEs for pH selection. At pH 3.2, His was positively charged and migrated as a sharp symmetrical peak after the EOF, i.e., counter to the direction of the EOF. As the pH was increased to 4.5, the ionisation of His was suppressed and migrated as an asymmetrical peak at a considerably later migration time. At pH 5.9, the ionisation of His was further suppressed with lengthy migration and similar peak asymmetry. At pH 7.0, His had negligible charge and migrated with the EOF. At pH 8.2, His was negatively charged and migrated before the EOF peak, i.e., in the same direction as the EOF. At pH 9.3, His was fully ionised and negatively charged and migrated as a symmetrical narrow peak before the EOF. The electropherograms obtained from the reciprocal scenarios with the SDS coated PDMS capillary, or the free zone BFS capillary are shown in Fig. 3A1. As expected, stable cathodic EOFs (positive polarity) were generated for the SDS-PDMS combination across all BGEs, whereas the EOF was negligible for the BFS capillary at low pH due to suppression of ionisation of the silanol groups at the capillary surface. As before, the mobility of the EOF varied between the BGEs due to varying ionic strengths. The migration of His was consistent across the two systems, and opposite to that of the CTAB-PDMS combination, i.e., His migrated in the same direction as the EOF at low pH, and a counter direction at high pH. At pH 7.0, His migrated with the EOF in both systems. This predictable and tuneable manipulation of the selectively of His across all scenarios is visualised in Fig. 3A2, where the mobility of His was plotted against the pH of the BGE. There was excellent consistency of the mobility of His across all three capillaries. A line of best fit crossed the x axis at approximately the pI of His, indicating that the electrophoretic mobility and therefore selectivity may be estimated via knowledge of the pI.
 | ||
| Fig. 3 Systematic evaluation of the CTAB-PDMS, SDS-PDMS and BFS capillary employing BGEs with pH values ranging from 3.0 to 9.6. The electropherograms for His (A1) and Tf (B1) were obtained at −30 kV for the CTAB-PDMS capillary and at +30 kV for the SDS-PDMS and BFS capillary. The arrows indicate the EOF (E) and system peaks are marked with a S. Generally, the CTAB-PDMS capillary was more suited for low pH BGEs, while the SDS-PDMS and BFS capillaries were superior for high pH BGEs. The tuneable manipulation of the selectivity may be monitored when plotting the analytes effective mobility against the pH value of the BGE (A2 and B2). A high consistency was observed across all three capillaries. A line of best fit crosses the x axis at approximately the pI of the protein, signalling that the electrophoretic mobility and therefore selectivity may be estimated by knowledge of the analytes’ pI. | ||
The same approach was used to evaluate Tf (pI 5.4) as shown in Fig. 3B1. Like His, the CTAB-PDMS system produced sharp symmetrical Tf peaks at pH 3.2 and 4.5 after the EOF, and at pH 5.9, 7.0 and 8.2 before the EOF. At pH 9.3, Tf was not detected, likely due to a global negative charge across the protein and electrostatic adsorption to the positively charged CTAB coating, or ion pair formation between the protein and free CTAB molecules from the dynamic coating formulation of the BGE. Tf was observed at BGE pHs above the pI of the protein after the EOF (counter EOF), with increasing peak symmetry with increasing pH for both the SDS-PDMS system and the BFS capillary. Interestingly, the peak symmetry in the SDS-PDMS system was generally superior to that of the BFS separation at the same pH, likely due to improved mitigation of hydrophobic adsorption. Again, the mobility of Tf was plotted against the pH of the BGEs and is shown in Fig. 3B2. As before, there was consistency of mobility within all three systems.
The workflow was applied to the selected panel of representative proteins that encompassed a range of MWs and pIs to demonstrate the universality of the BGEs and capillary combinations for the separation of any intact protein. Fig. SI 3A–F† details the electropherograms and mobility vs. pH plots. Multiple suitable combinations of the three systems were apparent for all target proteins. As expected, the CTAB capillary was more suited to low pH BGEs, whilst the SDS and BFS capillaries were superior at high pH as shown in Fig. 4. For example, the peaks for HSA, Tf and CK were symmetrical and adsorption free with the SDS-PDMS capillary. The BFS capillary was the better choice for all other analytes with sufficient efficiency for separation of the two isoforms of β-Lg A and β-Lg B. These differences were likely due to various interactions between the buffer components and the proteins. Regardless, the simple mechanism of separation proposed here is an effective practical approach for unprecedented control of EOF at all pH values, minimisation of unwanted adsorption of proteins, and facile method development and manipulation of selectivity.
 | ||
| Fig. 4 CE analysis of eight model analytes with various MWs and pIs at low (A, CTAB-PDMS capillary) and high pH (B, SDS-PDMS capillary; C, BFS capillary). A fast EOF and symmetrical peaks for all analytes were produced with the CTAB-PDMS capillary (A). The peaks HSA, Tf and CK were symmetrical and adsorption free with the SDS capillary (B). The BFS capillary was superior for all other analytes with a sufficient separation efficiency for the isoforms β-Lg A and β-Lg A (C). | ||
Proof-of-principle: analysis of proteins in milk
To showcase the possibilities to predict the behaviour and separation of proteins, the workflow was applied to the analysis of proteins in cow's milk. The total protein content of cow's milk typically varies between 3.0–3.5%, of which 80% are genetic variants of casein (CN) and 15% are whey proteins.38 The major caseins are isoforms of α-casein (α-CN), β-casein (β-CN) and κ-casein (κ-CN).39 Prominent whey proteins are isoforms of α-lactalbumin A (α-Lac A) and β-Lg.40 The pIs of these proteins range between 4.4 and 5.1, and are listed together with their respective MWs in Table SI 1B.† Consideration of the proteins pIs and the outcomes of the systematic evaluation, indicated that the most suitable CE separation was a combination of an acidic BGE and the CTAB capillary.Fig. 5A shows the analysis of the milk sample with BGE M-I (20 mM CA, 10 mM TRIS, 0.1 mM CTAB, pH = 3.2) utilizing a CTAB capillary with a total length of 110 cm and an effective length of 103 cm. The capillary length was extended to improve protein separation efficiency, and the BGE concentration was adjusted to maintain sufficient ionic strength for constant current (two-fold concentration compared to BGE I). The proteins in the milk sample were identified via spiking individual protein standards to the sample (Fig. SI 4†). The initial BGE M-I provided baseline separation of casein and whey proteins consisting of isoforms of A1 β-CN and A2 β-CN, BSA, α-CN and α-Lac. However, the two isoforms of β-Lg A and β-Lg B were only partially separated. The isoelectric points of β-Lg A and β-Lg B are 5.1 and 5.2, slightly higher than the other proteins of interest. Accordingly, the pH of the BGE was increased to 3.8 (BGE M-II, 20 mM CA, 20 mM TRIS, 0.1 mM CTAB) to impart a charge difference between the two isoforms. Baseline separation of β-Lg A and β-Lg B was observed with slightly less separation of the other components (Fig. 5B).
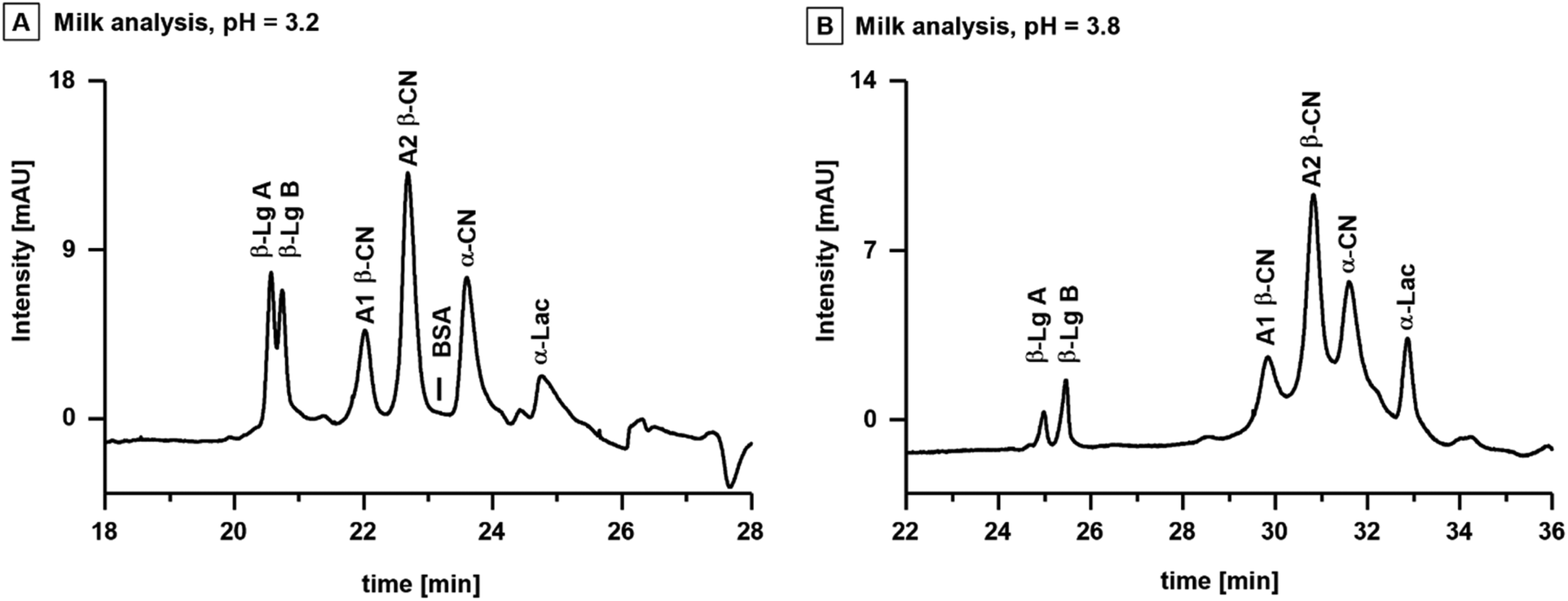 | ||
| Fig. 5 Simultaneous analysis of caseins and whey proteins in fresh cow's milk employing a CTAB-PDMS capillary (total length = 110 cm, effective length = 103 cm) and two BGEs. BGE M-I (pH = 3.2) is suitable for the separation of total two isoforms A1 β-CN and A2 β-CN, BSA, α-CN and α-Lac (A). The analysis of the two isoforms β-Lg A and β-Lg B requires BGE M-II (pH 3.8) with a slightly higher pH value due the higher pI of the two analytes (B). | ||
Conclusions
This work presented a novel and universal workflow for the analysis of intact proteins with any MW or pI via CE and introduced guidelines for the selection of appropriate BGEs based on the knowledge of the proteins’ pI. The suitability of GC PDMS (DB-1) capillaries with dynamic coatings of CTAB and SDS, and BFS capillaries were evaluated for the analysis of seven model proteins in six BGEs with pH values between 3.0–9.6. The CTAB-PDMS capillary was most suited for low pH BGEs with a low concentration of 0.1 mM CTAB added to the BGE. The SDS-PDMS capillary and BFS capillary were superior at high pH BGEs, with 0.01 mM SDS added to the BGE for the SDS-PDMS capillary. Multiple capillary and BGE combinations were applicable for separation of each of the model proteins with MWs between 13.7–150 kDa and pIs ranging from 4.7–9.6. The proteins’ effective mobility was plotted against the BGEs pH value, and a line of best fit crossed the x axis at approximately the pI of the protein, while fast mobilities were observed at pH values distant from the proteins pI. This was consistent across all capillaries, indicating that the electrophoretic mobility and therefore selectivity may be estimated via knowledge of the pI. Finally, the guidelines were used to select the most suitable capillary – BGE combination for the analysis of caseins and whey proteins in cow's milk.Author contributions
S. Meyer and P. Doble conceptualized and designed the study. S. Meyer conducted the experiments, performed the analyses, and drafted the manuscript. P. Doble supervised the project and contributed to the writing, review, and editing of the manuscript. D. Clases, R. Gonzalez de Vega and M. Padula took part in the discussion of the results and assisted with the manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. A. D. is supported by the Australian Research Council Discovery Project (DP190102361). The Atomic Medicine Initiative gratefully acknowledges philanthropic support from the Miklos Family.References
- Y. Zhang, B. R. Fonslow, B. Shan, M.-C. Baek and J. R. Yates, Chem. Rev., 2013, 113, 2343–2394 Search PubMed.
- J. Horvath and V. Dolník, Electrophoresis, 2001, 22, 644–655 Search PubMed.
- I. Rodriguez and S. F. Y. Li, Anal. Chim. Acta, 1999, 383, 1–26 Search PubMed.
- A. Staub, S. Comte, S. Rudaz, J. L. Veuthey and J. Schappler, Electrophoresis, 2010, 31, 3326–3333 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- Z. Zhang, E. H. Peuchen and N. J. Dovichi, Anal. Chem., 2017, 89, 6774–6780 CrossRef CAS PubMed.
- C. Huhn, R. Ramautar, M. Wuhrer and G. W. Somsen, Anal. Bioanal. Chem., 2010, 396, 297–314 CrossRef CAS PubMed.
- B. P. Salmanowicz, Chromatographia, 1995, 41, 99–106 CrossRef CAS.
- H. Lindner, W. Helliger, B. Sarg and C. Meraner, Electrophoresis, 1995, 16, 604–610 CrossRef CAS PubMed.
- F. Bedia Erim, J. Chromatogr. A, 1997, 768, 161–167 CrossRef.
- M. Nakatani, A. Shibukawa and T. Nakagawa, J. Chromatogr. A, 1994, 672, 213–218 CrossRef CAS PubMed.
- H. Katayama, Y. Ishihama and N. Asakawa, Anal. Chem., 1998, 70, 5272–5277 CrossRef CAS PubMed.
- C. Stathakis, E. A. Arriaga, D. F. Lewis and N. J. Dovichi, J. Chromatogr. A, 1998, 817, 227–232 CrossRef CAS PubMed.
- C. A. Lucy, A. M. MacDonald and M. D. Gulcev, J. Chromatogr. A, 2008, 1184, 81–105 CrossRef CAS PubMed.
- X. Han, Y. Wang, A. Aslanian, B. Fonslow, B. Graczyk, T. N. Davis and J. R. Yates, J. Proteome Res., 2014, 13, 6078–6086 CrossRef CAS PubMed.
- C. Berardet, J. Kaffy, S. Ongeri and M. Taverna, J. Chromatogr. A, 2018, 1578, 83–90 CrossRef CAS PubMed.
- Y. Li, P. D. Compton, J. C. Tran, I. Ntai and N. L. Kelleher, Proteomics, 2014, 14, 1158–1164 CrossRef PubMed.
- A. Stolz, Y. Hedeland, L. Salzer, J. Römer, R. Heiene, L. Leclercq, H. Cottet, J. Bergquist and C. Neusüβ, Anal. Chem., 2020, 92, 10531–10539 CrossRef CAS PubMed.
- E. Dominguez-Vega, T. De Vijlder, E. P. Romijn and G. W. Somsen, Anal. Chim. Acta, 2017, 982, 122–130 CrossRef CAS PubMed.
- J. R. Catai, J. Sastre Toraño, P. M. J. M. Jongen, G. J. de Jong and G. W. Somsen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 852, 160–166 CrossRef CAS PubMed.
- A. Marie, E. Dominguez-vega, F. Saller, J. Plantier, D. Borgel, N. T. Tran and G. W. Somsen, Anal. Chim. Acta, 2016, 947, 58–65 CrossRef CAS PubMed.
- S. Hjertén, J. Chromatogr., 1985, 347, 191–198 CrossRef.
- T. Soga, Y. Inoue and G. A. Ross, J. Chromatogr. A, 1995, 718, 421–428 CrossRef CAS.
- G. W. Tindall and R. L. Perry, J. Chromatogr. A, 1995, 696, 349–352 CrossRef CAS.
- T. Soga, Y. Ueno, H. Naraoka, K. Matsuda, M. Tomita and T. Nishioka, Anal. Chem., 2002, 74, 6224–6229 CrossRef CAS PubMed.
- E. Katsivela and M. G. Höfle, J. Chromatogr. A, 1995, 717, 91–103 CrossRef CAS.
- J. H. Lee, O. K. Choi, H. S. Jung, K. R. Kim and D. S. Chung, Electrophoresis, 2000, 21, 930–934 CrossRef CAS PubMed.
- T. Soga, Y. Ohashi, Y. Ueno, H. Naraoka, M. Tomita and T. Nishioka, J. Proteome Res., 2003, 2, 488–494 CrossRef CAS PubMed.
- S. H. Kang, J. W. Kim and D. S. Chung, J. Pharm. Biomed. Anal., 1997, 15, 1435–1441 CrossRef CAS PubMed.
- M. Dizy and L. F. Bisson, Am. J. Enol. Vitic., 1999, 50, 120–127 CAS.
- M. Kinoshita, E. Murakami, Y. Oda, T. Funakubo, D. Kawakami, K. Kakehi, N. Kawasaki, K. Morimoto and T. Hayakawa, J. Chromatogr. A, 2000, 866, 261–271 CrossRef CAS PubMed.
- S. Kamoda, C. Nomura, M. Kinoshita, S. Nishiura, R. Ishikawa, K. Kakehi, N. Kawasaki and T. Hayakawa, J. Chromatogr. A, 2004, 1050, 211–216 CrossRef CAS PubMed.
- D. Heiger, High performance capillary electrophoresis, Agilent Technologies, 2000 Search PubMed.
- X. Shen, Z. Yang, E. N. McCool, R. A. Lubeckyj, D. Chen and L. Sun, TrAC, Trends Anal. Chem., 2019, 120, 115644 CrossRef CAS PubMed.
- H. M. Mcnair and J. M. Miller, Basic Gas Chromatography, John Wiley & Sons, Inc., New York, 1967 Search PubMed.
- A. S. Pérez, F. L. Conde and J. H. Méndez, J. Electroanal. Chem. Interfacial Electrochem., 1976, 74, 339–346 CrossRef.
- T. Marquardt and J. Denecke, Eur. J. Pediatr., 2003, 162, 359–379 CrossRef CAS PubMed.
- R. J. P. Walstra, Dairy chemistry and physics, John Wiley and Sons, New York, 1984 Search PubMed.
- M. Strickland, M. E. Johnson and J. R. Broadbent, Electrophoresis, 2001, 22, 1510–1517 CrossRef CAS PubMed.
- J. Zhao and Z. Xu, Anal. Methods, 2021, 13, 801–808 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular masses and isoelectric points of all analytes. Coating time for CTAB-PDMS and SDS-PDMS capillary. Longevity of the surfactant layer. Electropherograms of all analytes using the three different capillaries and six different background electrolytes. Effective mobility depending on the pH value for all analytes. Electropherograms of spiking experiments for protein identification in milk analysis. See DOI: https://doi.org/10.1039/d2an00474g |
| This journal is © The Royal Society of Chemistry 2022 |