
A labeling strategy with effective preservation of fluorophores for expansion single-molecule localization microscopy (Ex-SMLM)
Weibing
Kuang
 a,
Bo
Xin
b,
Zhen-Li
Huang
c and
Bing
Shi
*c
a,
Bo
Xin
b,
Zhen-Li
Huang
c and
Bing
Shi
*c
aBritton Chance Center and MoE Key Laboratory for Biomedical Photonics, School of Engineering Sciences, Wuhan National Laboratory for Optoelectronics-Huazhong University of Science and Technology, Wuhan 430074, China
bWuhan Institute for Food and Cosmetic Control, Wuhan 430040, China
cKey Laboratory of Biomedical Engineering of Hainan Province, School of Biomedical Engineering, Hainan University, Haikou 570228, China. E-mail: shibing@hainanu.edu.cn
First published on 22nd November 2021
Abstract
Expansion microscopy (ExM) significantly improves the resolution of conventional diffraction-limited optical microscopy by using physically expanding biological samples. Combining ExM with single-molecule localization microscopy (SMLM) could further enhance the resolving power of SMLM, which is typically in the order of 20–30 nm. However, to make this combination successful, we need to solve three key issues related to sample preparation, including mainly hydrogel shrinking in an ionic photoswitching buffer, fluorescence photobleaching due to a free-radical reaction and reduced labelling efficiency from protease digestion. Re-embedding polyacrylamide gel or using an improved photoswitching buffer with a low ionic strength is able to minimize or even solve the hydrogel shrinking problem, while the development of post-expansion labelling approaches avoids fluorescence bleaching. However, the preservation of protein epitopes (which determines the labelling efficiency) remains to be challenging. In this paper, we propose to tackle this challenge by introducing the highly selective and stable biotin–streptavidin interaction into the post-expansion labelling strategy. After upgrading the popular immunolabelling linkage scheme from Epitope–Primary antibody–Secondary antibody–Fluorophores to Epitope–Primary antibody–Secondary antibody–Biotin–Streptavidin–Fluorophores, we were able to label protein epitopes with biotin, which was stable during the expansion process, and thus avoid the troublesome problem in preserving protein epitopes or antibodies. We demonstrate that combining Ex-SMLM with the new post-expansion linkage scheme enables new possibilities in resolving the detailed arrangement of Nup133 proteins in the nuclear pore complex, which helps researchers to observe a clearer structure. This study provides new opportunities for studying the ultrastructural details of subcellular organelles or even biomacromolecules, using the conventional SMLM system.
Introduction
Expansion microscopy (ExM) achieves super-resolution imaging from physical expansion of biological samples rather than a complex control of optical systems.1 To make this technique successful, researchers need to find a suitable strategy to covalently link fluorophores or proteins to a cross-linked network of a swellable hydrogel. The expansion of the hydrogel magnifies the distance of the fluorophores or proteins to a level that can be resolved by conventional optical microscopy. ExM was originally designed to use DNA-labelled antibodies for fluorescence retention and it achieved a resolution of 70 nm through physically expanding samples ∼4 times.1 After several years of improvement in sample preparation, now it is possible to realize ExM with conventional antibodies and fluorescent proteins.2,3 Researchers also tried to improve the resolution of ExM by increasing the expansion factor to 10 times4 or even 16 times,5 thus achieving a final resolution of 25 nm or better.ExM is simple and low cost because it realizes super-resolution imaging mainly by sample treatment. In typical ExM, most of the proteins inside biological samples are digested to minimize the effect of tissue scattering on spatial resolution. Therefore, ExM is only suitable for fixed samples. Furthermore, the power of ExM could be further strengthened after combining ExM with other advanced optical microscopy techniques. For example, the combination of ExM with light sheet microscopy is able to realize super-resolution imaging in large tissues,6 while combining ExM with Stimulated Emission Depletion Microscopy (STED) or Structured Illumination Microscopy (SIM) further improves the resolution.7,8
The combination of ExM with single-molecule localization microscopy (SMLM) enables a powerful technique called Ex-SMLM (or Ex-STORM),9–11 which is especially effective in resolving the distribution of closely-packed fluorophores that cannot be resolved by conventional SMLM alone. In this way, Ex-SMLM further pushes the resolution of SMLM from tens of nanometers to below 20 nm, with an ultimate goal to approach the molecular resolution of electron microscopy.9 However, three key issues should be solved to make Ex-SMLM successful. The first issue is hydrogel shrinking due to ionic interactions between the photoswitching buffer and hydrogel, and in the worst case, the hydrogel shrinks to its initial size.9 Sauer et al. solved this issue by re-embedding the charged hydrogel with an uncharged hydrogel, while Cang et al. proved that the use of a photoswitching buffer with a low ionic strength can keep the expansion ratio to about four times.10 The second issue is fluorescence photobleaching because free-radical polymerization for sample expansion could irreversibly destroy fluorophores.2,9 For example, the best fluorophore for SMLM, Alexa Fluor 647, is easily destroyed during expansion (only 10% survived).3,9 Fortunately, the development of post-labelling strategies pushes irreversible fluorophore destruction to an acceptable level. But this kind of strategy should be combined with good preservation of protein epitopes during expansion.9 The third issue related to sample preparation is the reduced labelling efficiency from protease digestion.2,3,12 Recently, Sauer et al.9 and Huang et al.12 tried to solve this issue using upgraded protocols, but these methods still suffer from partial loss of protein epitopes.
Here we propose a simple but efficient approach, which is based on the highly selective biotin–streptavidin interaction, to improve the labelling efficiency. In this approach, we firstly used a biotin-labelled antibody to link biotin covalently to protein epitopes. Then we performed sample expansion using well-established protocols. Later, we used fluorophore-labelled streptavidin to interact with the biotin in the hydrogel. Finally, we used a photoswitching buffer with a low ionic strength, as reported by Cang et al.,10 to soak the expanded hydrogels so that sample shrinking can be reduced, and thus obtained fluorophore-labelled expanded cell samples for further Ex-SMLM imaging. Using microtubules and nuclear pore complexes as two representative examples, we demonstrated the performance of our post-labelling strategy in Ex-SMLM. We achieved a resolution of ∼13 nm with Ex-SMLM and realized good visualization on the distribution of Nup133 proteins (representing low-density protein epitopes) inside the nuclear pore complex. This proposed method effectively improves the situation of insufficient resolution of conventional SMLM and optimizes the existing scheme of low efficiency of Ex-SMLM labeling.
Results and discussion
As illustrated in Fig. 1, the procedures for preparing expanded samples include gelation, digestion and expansion. Immunolabelling can be performed before gelation (thus called the pre-labelling strategy, Fig. 1b) or after expansion (thus called the post-labelling strategy, Fig. 1a). Compared to the pre-labelling strategy, the post-labelling strategy could avoid the fluorescence photobleaching problem during gelation. However, a fraction of protein epitopes or pre-labelled antibodies may be lost in the protease digestion step,9 resulting in reduced labelling efficiency. Here we aim to improve the labelling efficiency by introducing the well-established biotin–streptavidin interaction into the labelling scheme, based on the assumption that biotin is a small and stable chemical compound that has a better chance to survive in the gelation and digestion steps, and that labelling sites could be increased using secondary antibodies. Combining the biotin–streptavidin interaction with the popular immunolabelling strategy, we propose an upgraded post-labelling linkage scheme: Epitope–Primary antibody–Secondary antibody–Biotin–Streptavidin–Fluorophores.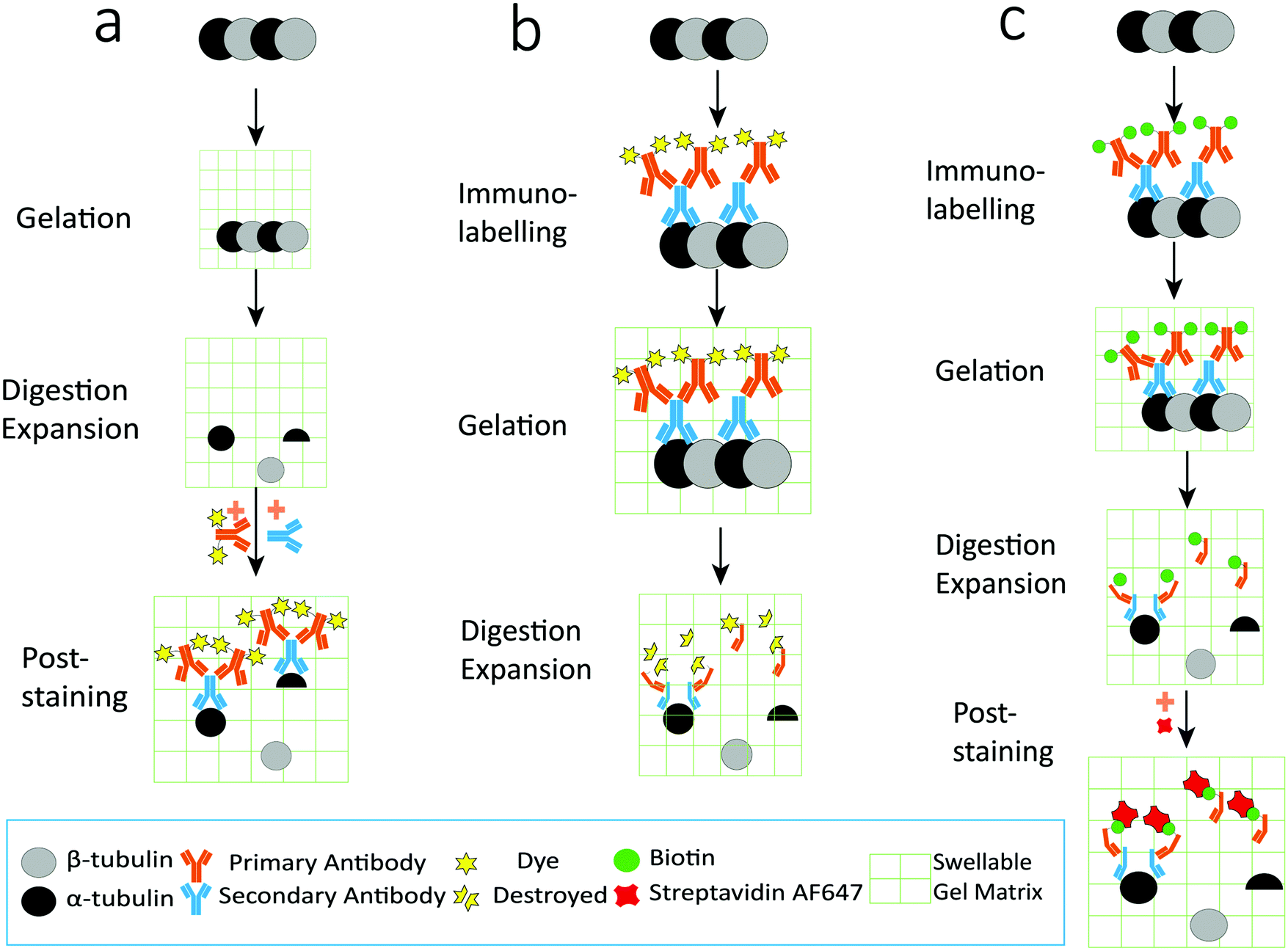 | ||
| Fig. 1 Schematic illustration of the labelling strategies in ExM. (a) Post-labelling strategy: protein epitopes were immunolabelled with fluorophores after all expansion steps, including gelation, digestion and expansion. (b) Pre-labelling strategy: protein epitopes were immunolabelled with fluorophores before the gelation step. (c) Epitopes were labelled with the primary antibody and biotin-conjugated secondary antibody, and then fluorophores were introduced by dye-conjugated streptavidin after gelation, digestion, and expansion. | ||
On the other hand, it is important to quantify the expansion ratio before performing Ex-SMLM experiments. Note that the resolution improvement in ExM is closely related to the expansion ratio, while the latter is determined by the type of hydrogel and the composite of monomers in expansion.1 A greater expansion ratio means a better resolution, but it also results in a hydrogel that can easily change its shape, which is unfavourable in the following SMLM experiments. Note that the most popular formula for sample expansion is from Chen et al.1 In this paper, we calculated the expansion ratio using microtubules immunolabelled with Alexa Fluor 546, because this fluorophore was reported to have good fluorescence preservation during the expansion process. Note that the expansion ratio should be quantified with the same medium used in Ex-SMLM and that any changes in the components of the photoswitching buffer would result in differences in the expansion ratio.
Fig. 2 shows wide-field fluorescence images of unexpanded (Fig. 2a) and expanded (Fig. 2b) U2OS cells. Note that Fig. 2b shows expanded cells that were fully incubated with the photoswitching buffer. We calculated the expansion ratio using structural features from several samples and obtained a mean expansion ratio of 2.37 ± 0.040 (mean ± SD). Not surprisingly, we obtained clearer cellular structures from expanded samples, proving the effectiveness of ExM in improving the spatial resolution of SMLM. Furthermore, we found that the cell shapes were almost the same before and after expansion, without abnormal structures such as cracks. These findings indicate that the formula and procedures used in sample expansion are efficient enough for further use in Ex-SMLM.
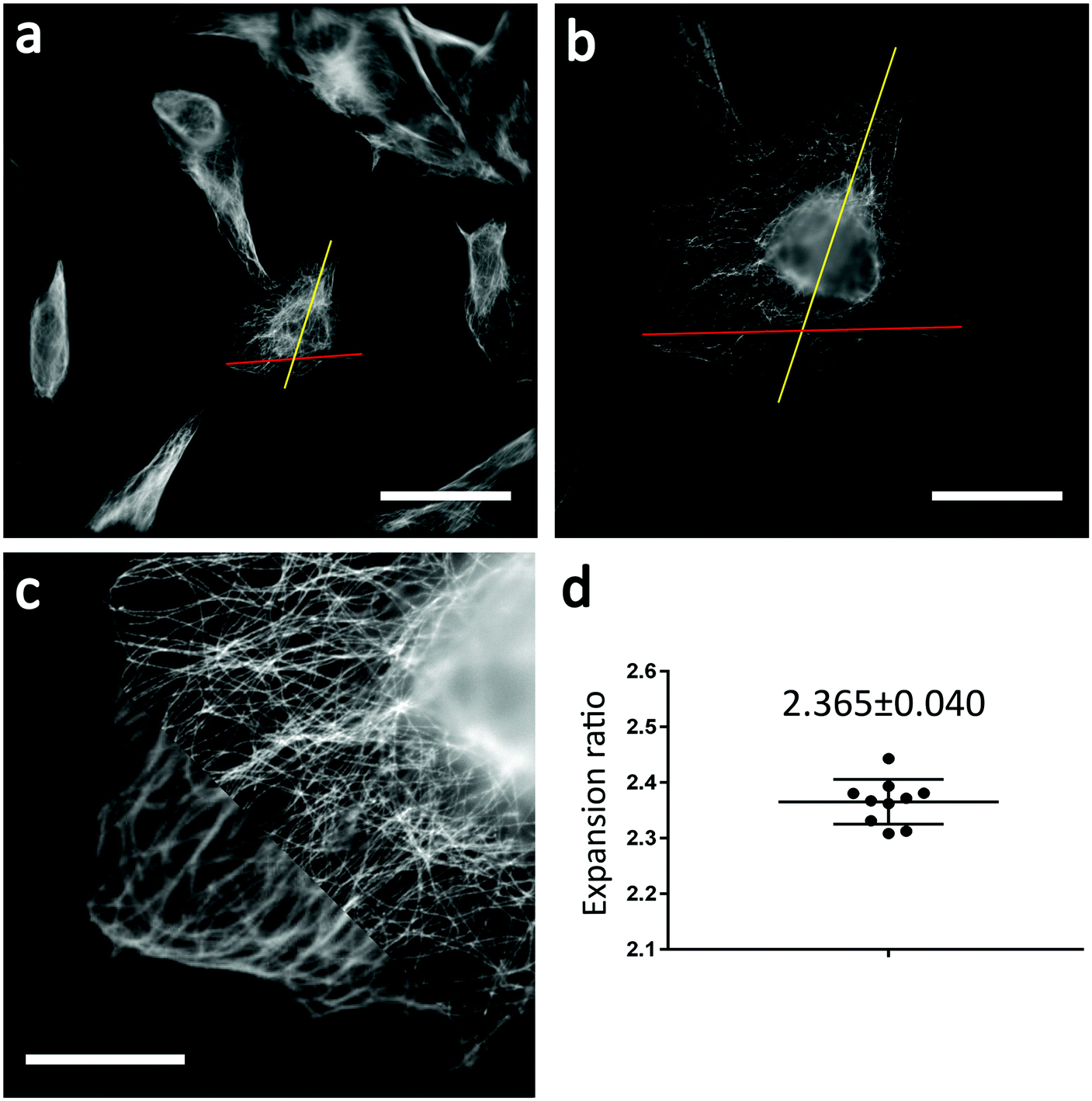 | ||
| Fig. 2 Determining the expansion ratio. (a) Wide-field image of unexpanded U2OS cells where microtubules were immunolabelled with Alexa Fluor 546. (b) Wide-field image of the same cells in (a) after expansion. The red line and the yellow line in (a) and (b) were used to calculate the expansion ratio. (c) Overlay of the wide-field images of the same cells before (lower left corner) and after (upper right corner) expansion. (d) The calculated expansion ratio with a mean value of 2.365. Note that the expansion ratio was calculated from five different cells. Each cell was measured twice in two different directions to minimize the measurement error. Scale bars: 50 μm (a and b) and 10 μm (c). | ||
We verified the effectiveness of our upgraded post-labelling strategy with a well-studied biological model in SMLM, i.e., microtubules in U2OS cells. We labelled the microtubules with a popular SMLM fluorophore called Alexa Fluor 647, using an upgraded post-labelling linkage scheme – Epitope–Primary antibody–Secondary antibody–Biotin–Streptavidin–Alexa Fluor 647 (see the Experimental section for details). Briefly, we labelled microtubules in U2OS cells with a primary antibody against α-tubulin and then used a secondary antibody to link biotin to the microtubules. After performing all sample expansion steps, we labelled the microtubules with Alexa Fluor 647 via biotin–streptavidin interaction. Using a reported photoswitching buffer,10 we imaged the expanded cells and were able to observe that most of the signals on the microtubules were well preserved in the wide-field image (Fig. 3a). We obtained similar signal brightness between expanded and unexpanded cell samples. We did not observe a notable sample drift after coating the coverslip with poly-L-lysine (0.1%) and sealing hydrogels with agarose. If necessary, we could use a redundant cross-correlation algorithm13 to correct the small sample drift in super-resolution images. We checked the SMLM images and found clear and continuous microtubular structures (Fig. 3b and c), proving that our upgraded post-labelling strategy is working properly in Ex-SMLM. We further investigated the zoom-in images of the microtubules and observed hollow structures (Fig. 3d–f) that can be hardly seen from unexpanded cell samples. The total size of an expanded protein was determined by the original size and linkage error. According to the previous works,11,14 the size of streptavidin was ∼5 nm. Streptavidin was added after expansion, and the linkage error was 2.1 nm when divided by the expansion factor, which had a little impact on the estimation of the protein size. But the use of biotin–streptavidin interaction was irreplaceable, which effectively preserved the fluorophores and location information of the protein. Besides, biotin could directly label the target proteins in the previous work,11 which could significantly reduce the linkage error. However, this method needed specialized sample preparation technologies which complicated the protocol and were more time-consuming.
 | ||
| Fig. 3 Ex-SMLM imaging of expanded U2OS cells where microtubules were immunolabelled with Alexa Fluor 647. (a) Wide-field image. (b) SMLM image. (c) Zoom-in image of the yellow boxed region in (b). (d and e) Zoom-in images of the blue and red boxed regions in (c). (f) Cross-sectional profile of the green line shown in (d). Scale bars: 10 μm (a and b) and 2 μm (c). | ||
We further evaluated the performance of our post-labelling strategy in Ex-SMLM imaging of the nuclear pore complex (NPC). The NPC is a large protein complex consisting of approximately thirty nucleoporins (Nups).15 Here we chose to image the distribution of Nup133, which forms an eight-fold rotationally symmetric structure in nuclear membranes.16 The Nup133 protein is a representative low-density protein epitope. In previous studies, although some researchers saw Nup133 through SMLM, it was limited by the resolution and could not be seen very clearly, as shown in Fig. 6. Therefore, if we compare the super-resolution images of Nup133 proteins between unexpanded and expanded cells, we would be able to determine whether ExM brings resolution enhancement to SMLM.
We firstly used conventional SMLM to image Nup133 proteins under optimal imaging conditions, but without sample expansion and the results are shown in Fig. 4. NPCs imaged by wide-field fluorescence microscopy (Fig. 4a and b) are blurred and overlapped, the central channel in NPCs cannot be visualized, and the number of NPCs cannot be counted precisely. These situations were improved significantly in the SMLM images of NPCs (Fig. 4c and d), where the ring-shaped structure of NPCs can be seen, but the rotationally symmetric structure of Nup133 proteins is still not observed.
 | ||
| Fig. 4 SMLM imaging of unexpanded U2OS cells where the Nup133 proteins in NPCs were immunolabelled with Alexa Fluor 647. (a) Wide-field image. (b) Zoom-in image of the yellow boxed region in (a). (c) SMLM image of the same cell in (a). (d) Zoom-in image of the yellow boxed region in (c). Scale bars: 5 μm (a and c) and 1 μm (b and d). | ||
Then, we applied Ex-SMLM imaging to Nup133 in NPCs using expanded cell samples. The imaging was performed under optimal experimental conditions, and the results are shown in Fig. 5. The point-like distribution of NPCs can be seen in the wide-field images of the expanded samples (Fig. 5a and b), but the central channel in NPCs is still not visible. The wide-field fluorescence images of NPCs look bright, indicating a good labelling efficiency from our post-labelling strategy. Furthermore, the Ex-SMLM images of NPCs (Fig. 5c and d) clearly show a ring-shaped structure with a central channel, and the rotationally symmetric structure of Nup133 proteins is also visualized, although some of the Nup133 proteins may be missing. Note that the expansion ratio of the sample has been taken into account for the scale bars as shown in Fig. 5.
 | ||
| Fig. 5 Ex-SMLM imaging of expanded U2OS cells where the Nup133 proteins in NPCs were immunolabelled with Alexa Fluor 647. (a) Wide-field image. (b) Zoom-in image of the yellow boxed region in (a). (c) Ex-SMLM image of the same cell in (a). (d) Zoom-in image of the yellow boxed region in (c). Scale bars: 10 μm (a and c) and 2 μm (b and d). | ||
Finally, we compared the super-resolution images of Nup133 proteins between unexpanded and expanded NPCs, and quantified the resolution enhancement from SMLM to Ex-SMLM. The super-resolution images of Nup133 proteins were selected based on the image quality and structure completeness. From the SMLM images of unexpanded NPCs, Nup133 proteins are still overlapped (Fig. 6a and b) and the rotationally symmetric distribution of Nup133 proteins is hard to observe. The diameter of the NPC in Fig. 6 was found to be 140 nm (Fig. 6a), and the FWHM resolution was calculated to be ∼34.3 nm using the SMLM image of an isolated Nup133 protein (Fig. 6b). From the Ex-SMLM images of expanded NPCs, the diameter of the NPC was calculated to be 330 nm before considering the expansion ratio, and 140 nm after considering the expansion ratio of 2.37. The FWHM resolution of Ex-SMLM was calculated to be 13.2 nm since the FWHM resolution was 31.4 nm and the expansion ratio was 2.37. Nup133 proteins separate clearly in the Ex-SMLM images of expanded NPCs, and the number of Nup133 proteins can even be counted (Fig. 6c). Therefore, not surprisingly, the resolution of Ex-SMLM shows 2.6-foldimprovement (34.3 nm/13.2 nm) over the resolution of SMLM. Furthermore, it is interesting to further compare the measured diameter of the NPC with the reported values. After considering the FWHM resolution of 13.2 nm and the linkage error of 19.0 nm, the diameter of the NPC from Ex-SMLM was approximately 108 nm, which is close to the reported value of 110 nm.15 Besides, a few signals on microtubules and Nup133 proteins were lost. According to the previous work,11 we considered that it was caused by some unanchored proteins in the gel.
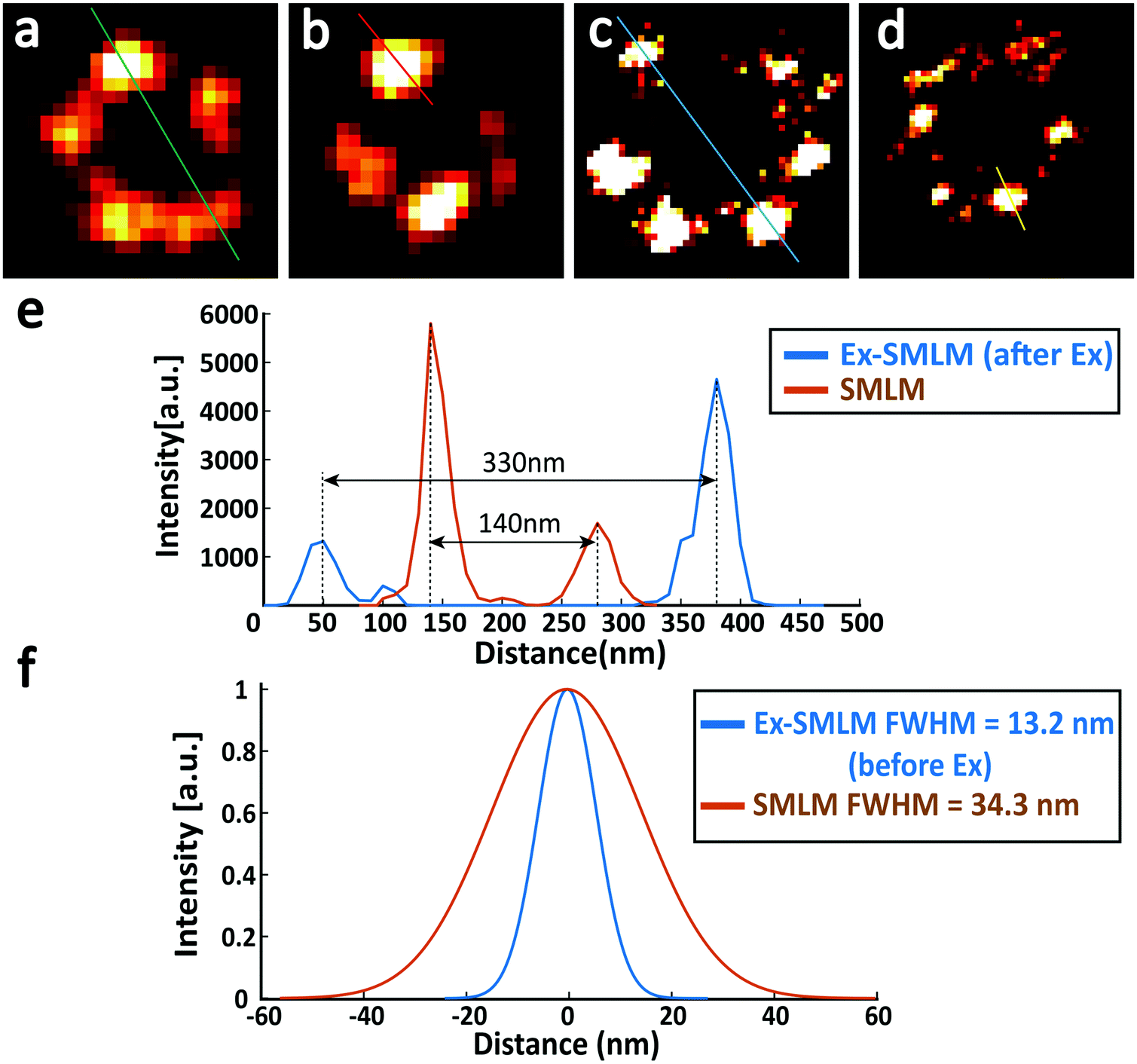 | ||
| Fig. 6 The resolution improvement in Ex-SMLM imaging of U2OS cells where Nup133 proteins were labelled with Alexa Fluor 647. (a and b) SMLM image of unexpanded U2OS cells. (c and d) Ex-SMLM image of expanded U2OS cells. (e) The orange line and the blue line represent the cross-sectional profile of the green line shown in (a) and the blue line shown in (c), respectively. (f) The orange line and the blue line represent the cross-sectional profile of the red line shown in (b) and the yellow line shown in (d), respectively. Gaussian fits were used in (f), and the FWHM resolution was 34.3 nm and 13.2 nm for unexpanded and expanded (divided by the expansion factor 2.37×) Nup133 proteins. | ||
Conclusions
Expansion microscopy achieves super-resolution imaging through physical expansion of samples, and it is proved to be compatible with existing super-resolution microscopy techniques, including but not limited to SIM, STED and SMLM. However, if we want to combine ExM with SMLM to further enhance the resolving power of SMLM, we need to solve three key issues related to sample preparation, including mainly hydrogel shrinking, fluorescence photobleaching and reduced labelling efficiency. After years of efforts from different groups, the first two issues have almost been solved. This study aims to solve the last issue by introducing the well-known biotin–streptavidin interaction into the established post-expansion labelling strategy. Using SMLM and Ex-SMLM imaging of microtubules and nuclear pore complexes, we demonstrate that combining Ex-SMLM with the new post-labelling strategy provides a better resolving power than conventional SMLM. We performed a detailed analysis on the super-resolution images of Nup133 proteins in nuclear pore complexes, a representative example of low-density protein epitopes, and found out that the resolution of Ex-SMLM was improved to be 13.2 nm, which is 2.6 times the resolution of SMLM. In this study, the labelling efficiency of the current post-labelling strategies is improved, and the study provides new opportunities for studying the distribution of low-density protein epitopes using the accessible SMLM system.Experimental section
Reagents
Methacrylic acid N-hydroxysuccinimide ester (MA-NHS) (730300), acrylamide (A9099), N,N′-methylenebis (146072), sodium acrylate (408220), tris base (T1503), glucose (G8270), glucase oxidase (G2133), catalase (C1345), cysteamine (MEA) (30070) and Triton X-100(X100-100ML) were purchased from Sigma-Aldrich. Ammonium persulfate (17874), tetramethylethylenediamine (17919), and proteinase K (EO0492) were purchased from Thermo Scientific. TAE buffer (10×) (15558042) and guanidine hydrochloride (15502016) were purchased from Invitrogen. McCoy's 5A medium (16600082), Dulbecco's Modified Eagle's Medium (DMEM) (11965092), fetal bovine serum (11965092), trypsin-EDTA (25200072) and Phosphate-Buffered Saline (PBS) (10010023) were purchased from Gibco. 8% Paraformaldehyde (PFA) aqueous solution (157-8) and 8% glutaraldehyde (GA) EM Grade (16020) were purchased from Electron Microscopy Sciences. Bovine Serum Albumin (BSA) (AB_2336946) was purchased from Jackson ImmunoResearch. Sodium chloride (10019318) and sodium bicarbonate (10018960) were purchased from Sinopharm Chemical Reagent. All of the reagents were used as received.Antibodies and labelling reagents
The mouse anti-α-tubulin monoclonal antibody (primary antibody) (T5168) was purchased from Sigma-Aldrich. The rabbit anti-Nup133 polyclonal antibody (primary antibody) (PA5-63774), Alexa Fluor 647 labelled goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (A-21235), Alexa Fluor 546 labelled goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (A-11003), Alexa Fluor 647 labelled F(ab’)2-goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (A-21246), Biotin-XX labelled goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (B-2763), Biotin-XX labelled goat anti-rabbit IgG (H + L) cross-adsorbed secondary antibody (B-2763), and Alexa Fluor 647 conjugated streptavidin (S32357) were purchased from Invitrogen.Cell culture
U2OS and HeLa cells (purchased from Boster Biological Technology, Wuhan) were cultured at 37 °C under 5% CO2 in McCoy's 5A medium (U2OS) or DMEM medium (HeLa), with fetal bovine serum (10%), penicillin (100 U ml−1) and streptomycin (0.1 mg ml−1). Cells were seeded on a glass bottom dish (MatTek P35G-1.5-14-C) and grown for 24 hours before use.Immunostaining of unexpanded cells
The culture medium in the glass bottom dish was discarded and U-2OS or HeLa cells were rinsed once with PBS. Cells were then fixed with a pre-warmed fixation solution (3% PFA, 0.02% GA, and 0.2% Triton X-100) for 15 min. The fixation solution was discarded and the cells were washed with PBS three times for 5 min each. Next, the cells were incubated with a permeabilization solution (0.2% Triton X-100 in PBS) for 10 min. Then, the permeabilization solution was discarded, and the cells were treated with a blocking solution (3% BSA and 0.05% Triton X-100 in PBS) for 1 h. After discarding the blocking solution, the cells were incubated with the primary antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :400 dilution for the anti-α-tubulin monoclonal antibody, or 1:100 dilution for the Nip133 polyclonal antibody) at room temperature for 1 hour or kept at 4 °C for 12 hours. The cells were washed with PBS three times for 10 min each with gentle shaking and then incubated with the secondary antibody (1:400 dilution) at room temperature for 1 hour. After washing three times with PBS for 10 min each, the cells were ready for fluorescence imaging.
:400 dilution for the anti-α-tubulin monoclonal antibody, or 1:100 dilution for the Nip133 polyclonal antibody) at room temperature for 1 hour or kept at 4 °C for 12 hours. The cells were washed with PBS three times for 10 min each with gentle shaking and then incubated with the secondary antibody (1:400 dilution) at room temperature for 1 hour. After washing three times with PBS for 10 min each, the cells were ready for fluorescence imaging.
Cell expansion
Cells were incubated with MA-NHS (25 mM in PBS) at room temperature for 1 hour and washed with PBS three times. Gelation was performed on a glass chamber made from a standard microscope slide and two rectangular coverslips. The coverslips were placed on both sides of the microscope slide to make a groove that was slightly smaller than the diameter of the glass bottom of the cell culture dish. ExM monomer solution (8.625% (w/w) sodium acrylate, 2.5% (w/w) acrylamide, 0.15% (w/w) N,N′-methylenebis, and 2 M NaCl in PBS) supplemented with 0.2% APS and 0.2% TEMED, was added to the groove. The glass bottom containing cultured cells was pulled off carefully from the cell culture dish, and placed on top of the glass chamber with cells facing down. Cells were incubated for 1 hour at room temperature for chemical crosslinking. After gelation, samples were treated with proteinase K in a digestion buffer (0.5% Triton X-100, 0.8 M guanidine hydrochloride, and 10% 10× TAE) for 12 hours, and then expanded using a large amount of PBS. PBS was exchanged three times (30 min each). Here PBS was used to ensure a consistent expansion ratio in the following SMLM imaging. The expanded hydrogels were treated twice with a photoswitching buffer (dSTORM buffer: 10% (w/v) glucose, 100 mM cysteamine, 0.5 mg mL−1 glucose oxidase, and 40 μg mL−1 catalase in Tris-HCl (pH 8.0)). Hydrogels were transferred to a poly-L-lysine (0.1%) coated coverslip with cells facing down, and sealed with agarose to minimize the drift between hydrogels and the coverslip.Immunostaining of expanded cells
Expanded hydrogels labelled with the biotin-labelled antibody were incubated with Alexa Fluor 647 conjugated streptavidin for 1.5 hours and washed with a large amount of PBS three times (30 min each) to remove excess streptavidin.Cell imaging and image processing
Cells were imaged on a customized SMLM system built from an Olympus IX73 microscope. A 532 nm excitation laser was used to excite Alexa Fluor 546, and a 640 nm excitation laser (∼5 kW cm−2) was used to excite Alexa Fluor 647. Both lasers were purchased from LaserWave, China, and controlled to have an intensity of ∼5 kW cm−2 at the sample plane. Fluorescence emission was collected using an Olympus 60×/NA1.42 oil-immersion objective and captured using a Hamamatsu Flash 4.0 V3 camera with an exposure time of 10 ms. A 405 nm activation laser (LaserWave, China) was used to control the density of activated emitters. Raw images were processed using a home-built software called QC-STORM,17 and the axial drift was corrected by the cross-correlation method. The cross-sectional profile analysis was used to determine the diameter of microtubules or nuclear pore complexes.Expansion factor determination
The expansion factor was determined by measuring the dimension of each pair of cellular structures before and after expansion, using fluorescence images from U2OS cells where microtubules were labelled with Alexa Fluor 546. Note that the fluorescence signal from Alexa Fluor 546 can be well preserved during the expansion process. To reduce the effort in finding paired structures, samples were placed in the same direction and fluorescence images taken before expansion were used as a guide to search for the paired images after expansion. Furthermore, the expansion factor was calculated using images from different directions.Author contributions
B.X. conducted the experiments. W.K and B.X. analysed the data. W.K., Z.H. and B.S. prepared the manuscript with help from B.X. Z.H. and B.S. conceived and supervised the project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (81827901) and Start-up Fund from Hainan University (KYQD(ZR)-20077). We also thank the Optical Bioimaging Core Facility of WNLO-HUST for technical support.References
- F. Chen, P. W. Tillberg and E. S. Boyden, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- T. J. Chozinski, A. R. Halpern, H. Okawa, H.-J. Kim, G. J. Tremel, R. O. L. Wong and J. C. Vaughan, Nat. Methods, 2016, 13, 485–488 CrossRef CAS PubMed.
- P. W. Tillberg, F. Chen, K. D. Piatkevich, Y. Zhao, C.-C. Yu, B. P. English, L. Gao, A. Martorell, H.-J. Suk, F. Yoshida, E. M. DeGennaro, D. H. Roossien, G. Gong, U. Seneviratne, S. R. Tannenbaum, R. Desimone, D. Cai and E. S. Boyden, Nat. Biotechnol., 2016, 34, 987–992 CrossRef CAS PubMed.
- S. Truckenbrodt, M. Maidorn, D. Crzan, H. Wildhagen, S. Kabatas and S. O. Rizzoli, EMBO Rep., 2018, 19, e45836 CrossRef PubMed.
- J.-B. Chang, F. Chen, Y.-G. Yoon, E. E. Jung, H. Babcock, J. S. Kang, S. Asano, H.-J. Suk, N. Pak, P. W. Tillberg, A. T. Wassie, D. Cai and E. S. Boyden, Nat. Methods, 2017, 14, 593–599 CrossRef CAS PubMed.
- J. Buergers, I. Pavlova, J. E. Rodriguez-Gatica, C. Henneberger, M. Oeller, J. A. Ruland, J. P. Siebrasse, U. Kubitscheck and M. K. Schwarz, Neurophotonics, 2019, 6, 015005 CAS.
- M. Gao, R. Maraspini, O. Beutel, A. Zehtabian, B. Eickholt, A. Honigmann and H. Ewers, ACS Nano, 2018, 12, 4178–4185 CrossRef CAS PubMed.
- Y. Wang, Z. Yu, C. K. Cahoon, T. Parmely, N. Thomas, J. R. Unruh, B. D. Slaughter and R. S. Hawley, Nat. Protoc., 2018, 13, 1869–1895 CrossRef CAS PubMed.
- F. U. Zwettler, S. Reinhard, D. Gambarotto, T. D. M. Bell, V. Hamel, P. Guichard and M. Sauer, Nat. Commun., 2020, 11, 3388 CrossRef CAS PubMed.
- H. Xu, Z. Tong, Q. Ye, T. Sun, Z. Hong, L. Zhang, A. Bortnick, S. Cho, P. Beuzer, J. Axelrod, Q. Hu, M. Wang, S. M. Evans, C. Murre, L.-F. Lu, S. Sun, K. D. Corbett and H. Cang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18423–18428 CrossRef CAS PubMed.
- M. Kang, J. Lee, S. Ko and S.-H. Shim, ChemBioChem, 2021, 22, 1396–1399 CrossRef CAS PubMed.
- X. Shi, Q. Li, Z. Dai, A. A. Tran, S. Feng, A. D. Ramirez, Z. Lin, X. Wang, T. T. Chow, I. B. Seiple and B. Huang, bioRxiv, 2019, 687954 Search PubMed.
- Y. Wang, J. Schnitzbauer, Z. Hu, X. Li, Y. Cheng, Z.-L. Huang and B. Huang, Opt. Express, 2014, 22, 15982–15991 CrossRef PubMed.
- L. Zhang, K. Dammann, S. C. Bae and S. Granick, Soft Matter, 2007, 3, 551–553 RSC.
- A. von Appen, J. Kosinski, L. Sparks, A. Ori, A. L. DiGuilio, B. Vollmer, M.-T. Mackmull, N. Banterle, L. Parca, P. Kastritis, K. Buczak, S. Mosalaganti, W. Hagen, A. Andres-Pons, E. A. Lemke, P. Bork, W. Antonin, J. S. Glavy, K. H. Bui and M. Beck, Nature, 2015, 526, 140–143 CrossRef CAS PubMed.
- A. Szymborska, A. de Marco, N. Daigle, V. C. Cordes, J. A. G. Briggs and J. Ellenberg, Science, 2013, 341, 655–658 CrossRef CAS PubMed.
- L. Li, B. Xin, W. Kuang, Z. Zhou and Z.-L. Huang, Opt. Express, 2019, 27, 21029–21049 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |