
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous organic polymers for CO2 capture, separation and conversion
Kyung Seob
Song†
,
Patrick W.
Fritz†
and
Ali
Coskun
 *
*
Department of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: ali.coskun@unifr.ch
First published on 14th November 2022
Abstract
Porous organic polymers (POPs) have long been considered as prime candidates for carbon dioxide (CO2) capture, separation, and conversion. Especially their permanent porosity, structural tunability, stability and relatively low cost are key factors in such considerations. Whereas heteratom-rich microporous networks as well as their amine impregnation/functionalization have been actively exploited to boost the CO2 affinity of POPs, recently, the focus has shifted to engineering the pore environment, resulting in a new generation of highly microporous POPs rich in heteroatoms and featuring abundant catalytic sites for the capture and conversion of CO2 into value-added products. In this review, we aim to provide key insights into structure–property relationships governing the separation, capture and conversion of CO2 using POPs and highlight recent advances in the field.
 Kyung Seob Song | Kyung Seob Song received his BSc degree in chemical engineering from Chung Nam National University (CNU), Republic of Korea in 2014. He joined the research group of Prof. Ali Coskun in the Graduate School of EEWS at the Korea Advanced Institute of Science and Technology (KAIST) in 2014 and received his MSc degree in 2016. In 2017, He moved to University of Fribourg, Switzerland with Prof. Ali Coskun and received his PhD degree in chemistry in 2022. He works on design and synthesis of porous organic polymers (POPs) for environmental applications such as CO2 capture, separation and conversion, precious metal recovery. Currently, he is a postdoctoral fellow developing POPs for heterogenous catalysis. |
 Patrick W. Fritz | Patrick W. Fritz obtained his BSc in technical chemistry from Technische Universität Wien (TU Wien) in 2018 and received his MSc from the institute of materials chemistry and the institute of applied synthetic chemistry (IMC and IAS) under the supervision of Prof. Miriam M. Unterlass in 2019. In 2020 he joined the group of Prof. Ali Coskun at University of Fribourg, where he works on porous organic polymers and covalent organic frameworks for separation and energy applications. |
 Ali Coskun | Ali Coskun received his PhD degree in chemistry from Middle East Technical University, Ankara, Turkey. He then joined the laboratory of Prof. J. Fraser Stoddart as a postdoctoral research associate at Northwestern University, where he developed dynamic metal–organic frameworks, artificial molecular machines. He started his independent career at Korea Advanced Institute of Science in Technology in 2012. In 2017, he moved to University of Fribourg, Switzerland as a professor. He is currently developing porous organic polymers for CO2 capture, separation and conversion, for environmental remediation as well as high energy density for Li-ion batteries. |
Introduction
Humankinds’ reliance on fossil fuels as a key energy source has resulted in a continuous rise in the atmospheric concentration of carbon dioxide (CO2), reaching record levels of 419 ppm (National Oceanic and Atmospheric Administration, Mauna Loa station, Feb 2022)1 and expected to increase even further in the coming years. Since CO2 is one of the main culprits of global warming, research into remediation technologies is booming. As a result, porous materials have gained considerable interest during the last two decades and various strategies towards inorganic, organic and hybrid porous materials for CO2 capture have been investigated with the common goal to reduce new CO2 emissions and to decrease the overall CO2 concentration in the atmosphere.The main idea behind carbon capture and sequestration/storage (CCS) is to remove CO2 directly at their point sources or to reduce existing emissions from the atmosphere through direct air capture (DAC) and store the captured CO2 underground in deep geological formations. Although the concept of CCS was first introduced in 1977,2 industrial technologies to separate CO2 have been used since the 1920s in the purification of methane from natural gas. The major difference between DAC and CCS lies in the CO2 concentration. Whereas the DAC process needs to operate at low CO2 concentrations of around 400 ppm, CO2 capture from point sources via CCS operate at significantly higher CO2 concentrations. Depending on the source, CO2 concentration in various emission sources can vary significantly from few percent to over 50%. Three main sources namely pre-combustion, post-combustion and oxyfuel combustion are targeted with CCS. Whereas post-combustion CO2 capture from flue gas operates at low CO2 concentrations in the range of 5–15% and requires the separation of CO2/N2 mixtures at 1 bar, pre-combustion (CO2/H2) and oxyfuel combustion (CO2 and water vapor) involve much higher CO2 concentrations.3 Industrially, aqueous amine solutions (often monoethanol amine, MEA) have been employed in such processes. While these solutions are low-cost and offer very high selectivity towards CO2 over other gases, they suffer from low uptake capacities and require a significant amount of energy for their regeneration. Furthermore, MEA poses environmental and human health risks that have to be considered for such large-scale applications.
In this direction, the use of solid sorbents has emerged as a promising alternative as they offer higher thermal stability and potentially higher CO2 capacity, while regeneration can be performed at significantly lower temperatures. For the recovery of the captured CO2 and the regeneration of the sorbents, three main technologies have been established, namely, (i) pressure swing adsorption (PSA), (ii) vacuum swing adsorption (VSA), and (iii) temperature swing adsorption (TSA). PSA is commonly applied for treating flue gas, which is comprised of SO2, Hg, CO, H2O, CO2 and N2, and operates well with CO2 concentrations of 5–15%. Gases are adsorbed within the pores of the sorbent either through physical interactions and/or weak chemical bonds at high pressure and desorbed once the pressure is lowered. Whereas adsorbents such as silica and zeolites have been used in the past, recently, the scope has been extended to porous (metal–)organic materials such as porous organic polymers (POPs) or metal–organic frameworks (MOFs).4,5 Although similar to PSA, VSA operates at near-ambient temperatures and pressures. The main difference is that a pre-adsorption compression step can be avoided, although some studies have shown that gas pressures slightly above atmospheric pressure are favorable towards the uptake capacity. In the VSA process, the desorption of CO2 and sorbent regeneration operates by applying vacuum, which is considered to be the most energy-demanding step of the process.6–8 TSA on the other hand employs a thermal regeneration process, which is attractive for locations with low-grade thermal energy resources.8–10 Each of these three technologies have their own advantages and disadvantages and have also been used in combination to obtain better overall tradeoffs. Another alternative to obtain high-purity CO2 is cryogenic distillation dubbed cryogenic carbon capture (CCC). Cryogenic carbon capture revolves around the physical separation of different gases based on their boiling and re-sublimation temperatures and is commonly used in natural gas purification.11 CCCs biggest advantage lies in the high purity of CO2 that can be obtained and used for several applications including, but not limited to chemical transformations and enhanced oil recovery,12 however, the immense energy cost due to cooling is a major drawback.
Despite the lack of policies and insufficient financial incentives to reduce carbon emissions and to retrofit existing facilities with CCS technologies, the rising public awareness about climate change resulted in new technologies being pioneered by upcoming small businesses. Among them, the combination of CCS/DAC technologies with renewable energy has gained substantial interest. An interesting example of such a technology was brought to the market by “Climeworks” who are using solid sorbents to directly capture CO2 from the air and CO2 is stored till the capacity is reached. Then, CO2 is concentrated by heating the filters using renewable energy sources and subsequently stored underground13 or can be used in green houses to artificially increase the amount of available CO2 for photosynthesis. The cost of this system, however, is still well above 500$ per tonne of CO2, which is rather high. Besides environmental problems, CO2 also poses dangers to the human health, especially in crowded, poorly ventilated rooms, where high concentrations of CO2 can result in headaches, fatigue and poor cognition. Accordingly, the capture of CO2 in such spaces is also expected to be an important research direction.14
Porous materials such as activated carbons, silica, zeolites, covalent organic frameworks (COFs), MOFs and POPs have actively been investigated as adsorbents for CO2. Importantly, key parameters need to be satisfied including high CO2 uptake and working capacity, high CO2 selectivity over other gases (especially CO2/N2 selectivity) under humid conditions, fast adsorption kinetics, low production cost, and low energy consumption during adsorption and regeneration steps.15–17 Besides the molecular properties of the gases or the surface functionalities of the sorbents, textural properties such as the surface area, pore volume, and pore size have to be carefully considered when designing a sorbent for CO2 separation. The adsorption of CO2 can occur via either physisorption or chemisorption process and the latter one involves the formation of a covalent bond between the sorbent and CO2 molecule. The physisorption of CO2 on the surface of porous materials is an exothermic process that occurs through various non-covalent interactions. Critically, fine-tuning the pore size/structure can help to maximize these interactions and to increase the overall CO2 affinity and capacity. Pore sizes <2 nm are referred to as micropores, 2–50 nm as mesopores, and >50 nm as macropores.18 Considering the kinetic diameters of gases, e.g. 3.3 Å of CO2, 3.8 Å of CH4 and 3.64 Å of N2, pore sizes closer to the kinetic diameters of the gases would offer the highest affinity due to the possibility of multiple gas-surface interactions, thus microporous and ultra-microporous (pore size <0.7 nm) structures are preferred for high CO2 affinity.
Considering these aforementioned prerequisites, a key question of “How can CO2-philicity over other gases under humid conditions be governed?” has to be answered.
In organic or metal–organic systems such as POPs, COFs and MOFs, the building blocks determine the pore structure and the functionalities in the pores. CO2 is a highly stable symmetric molecule with a permanent quadrupole moment. Therefore, dipole–quadrupole interactions can be exploited to attract and bind CO2 molecules, thus surface modification with electron-rich or deficient atoms such as N, O, P, S or F are commonly employed in porous materials. As a result, the field has moved to design cheap, heteroatom-rich POPs, COFs and MOFs with high surface areas, high pore volume and high CO2 affinity as key design factors for CO2 capture and separation.
Given suitable functionalization, MOFs, COFs and POPs can deliver high CO2 affinity, however, whereas MOFs and COFs are obtained under thermodynamic conditions, POPs are usually prepared under kinetic conditions and are commonly obtained as amorphous materials. Despite crystallinity being a generally favorable and sought-after property in porous materials, the increased cost associated with their preparation is a major factor hindering their wide-range industrial applications. Nonetheless, since the introduction of MOFs19 and COFs,20 their ordered porosity and wide tunability have attracted considerable interest and the production of some MOFs on an industrial scale has begun. However, in most cases, COFs are still lagging behind as their synthetic conditions require more precise optimization before industrial applications become feasible. At the same time, POPs have made a significant leap forward in terms of the variety of available synthetic strategies and have shown high potential for CO2 capture, separation, and conversion owing to their high tunability, porosity, and exceptional stability under operating conditions.21,22 Although, the amorphous nature of POPs is often referred to as a drawback, the ease of their synthesis has to be considered as a major advantage – especially when compared to their crystalline porous counterparts COFs and MOFs. The fact that POPs are generally obtained as kinetic products allows for easier upscaling as fewer parameters have to be optimized and maintained throughout their preparation. It should however be noted that even in POPs, the synthetic conditions such as concentration, catalyst amount, and solvent can have a profound impact on the porosity of POPs.23 The diversity in structures and suitable synthetic strategies24 that can be envisioned for POPs are rather rich. Suitably functionalized precursors can be polymerized via various strategies and used to tune the desired features of the POPs. Recently even the use of shape-persistent molecules or building blocks such as molecular cages and macrocycles have been investigated and have shown promising results in complex separation tasks.25
Although briefly touched upon already, a wide variety of sorbents can in theory be used and are being used for the remediation of CO2. Among the most used sorbents for CO2 are zeolites, porous carbons, metal oxides, mesoporous silica and their amine functionalized or impregnated analogues. Each of them with specific tradeoffs in terms of capacity, operating conditions, stability, cost of production, and cost of regeneration resulting in a plethora of factors that need to be considered. Taking porous metal oxides as an example one can easily see that their availability, cost, extremely high theoretical capacities, and good reactivity with CO2 under industrial conditions are considerable advantages. However, such systems – similar to amine scrubbers – bind CO2 chemically (e.g. as MgCO3 in the case of MgO) which in turn requires significantly higher reaction temperatures to regenerate the active material. One of the main advantages of POPs over these systems is the structural tunability and precise control over the pore environment for efficient CO2 capture and conversion.
Current CCS and DAC technologies are still costly which hinders their large-scale deployment. To become industrially relevant, a target cost of 50$ per tonne of CO2 should be reached.26 To reduce the cost and to incentivize CCS, a circular economy should be developed by using CO2 as a sustainable C1 carbon source in industrial processes. In organic chemistry, high-purity CO2 is used in various chemical transformations such as the Grignard reaction, methanol and formic acid synthesis, cyclic carbonate formation or carboxylations. One of the main advantages of POPs – and porous materials in general – is that they can serve as hosts or directly as heterogeneous catalysts to simultaneously capture and convert CO2. However, due to the high thermodynamic stability of CO2, its activation is rather difficult, requiring harsh reaction conditions. Therefore, designing POPs for CO2 conversion is significantly more challenging than designing POPs for CO2 capture and separation, and to date, there are significantly fewer POP heterogeneous catalysts for CO2 conversion.
In addition to the research efforts toward the activation of CO2 in purely organic systems, POPs have also been used as porous supports to incorporate atomically dispersed metal ion catalytic sites, single-atom catalysts (SACs), or nanoparticles (Al, Zn, Co, Pd, Ir, Ru and Ni), both of which are herein referred to as metal-containing POPs. Commonly employed ligands such as salens-, bipyridines-, N-heterocyclic carbenes can be used as POP building blocks. Most synthetic approaches toward metal-containing POPs can be classified as follows: (i) polymerization of suitably functionalized molecules featuring complexation sites (e.g. amine-functionalized porphyrins), (ii) in situ formation of metal complexation sites (e.g. salen-based systems), or (iii) wet-impregnation of metal ions and NPs into POPs. In heterogeneous catalysis, POPs acting as porous supports have strong advantages. First of all, elaborate control over the pore structure and heteroatoms contributes to effectively forming stable catalytic sites in POPs and enabling higher catalytic activity. The precise control over the active sites is the most important factor for CO2 conversion. For example, in the cycloaddition of CO2 to epoxides, a bifunctional system is required to activate CO2 while also initiating the ring-opening reaction of the epoxide. In addition, POPs have also been studied in electrochemical27 and photochemical CO2 reduction reactions, which are not the subject of this review article.28
In this review, we provide (Fig. 1) an overview of the progress of promising POPs classes over the past 5 years with a perspective on POPs for CO2 capture, separation, and conversion. First, we discuss heteroatom-rich systems and, in particular, assess different strategies towards their synthesis and the CO2 capture capabilities of these polymers. We also present an in-depth analysis of various parameters such as surface area, microporosity and heteroatom content on the CO2 uptake capacity and provide key insights into structure–property relationships governing CO2 affinity. A perspective on effective control of porosity in POPs by employing specifically designed precursors such as cavitands or cages is also discussed. Subsequently, the use of POPs as heterogeneous catalysts for CO2 conversion is also presented. Later, we discuss how POPs can be used for the simultaneous capture and conversion of CO2 towards value-added products. We highlight metal-free POPs, namely porous organocatalysts, and evaluate their relative performance and how they compare with metal-containing POPs. We also showcase metal-containing POPs in terms of their ability to stabilize single metal atoms while maintaining high porosity and their applications in CO2 conversion beyond cycloaddition reactions.
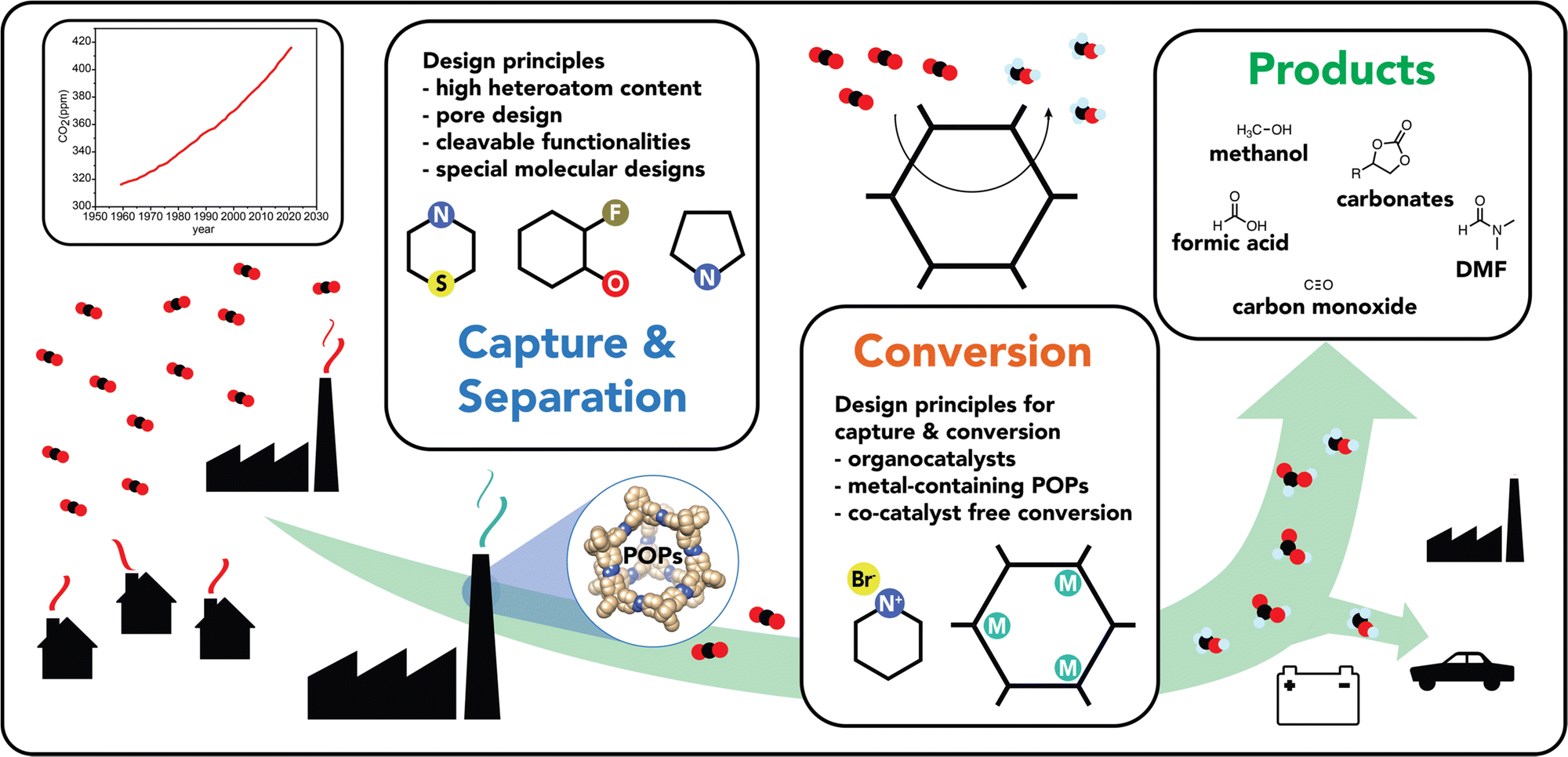 | ||
| Fig. 1 Schematic representation of various CO2 emission sources and a plot of atmospheric CO2 concentration over the years. An overview of as to how POPs can contribute to the CO2 circular economy by the capture/separation and the subsequent conversion of CO2 into value-added products. | ||
POPs for CO2 capture and separation
High surface area, abundant micropores along with the presence of heteroatoms have been considered as the key parameters for POPs with high CO2 selectivity and uptake capacity. Heteroatoms play a central role as high affinity binding sites towards CO2via dipole–quadrupole interactions, in particular, the introduction of nitrogen atoms has been proven to be effective to enhance CO2 affinity over other gases. In this direction, amine grafting and amine impregnation have also been used to boost the CO2 capture performance of POPs.29 In the latter example, corresponding POPs showed very promising CO2 uptake capacities at low partial pressures and warm temperatures (up to 50 °C) under humid conditions, which rendered them suitable for post-combustion CO2 capture and DAC. It should be also emphasized that these systems showed significantly lower regeneration temperatures compared to the conventional aqueous amine solutions.29 However, from an industrial point of view, a high CO2 sorption capacity alone does not render a POP interesting. Rather than that, the heat of adsorption (Qst), working capacity, gas selectivity (especially CO2/N2 selectivity) and operation under humid conditions have to be considered for gas separation tasks. In the following section, heteroatom rich POPs will be discussed and comparatively analysed by considering above mentioned parameters. It should however be noted that there are rather limited number of studies on the CO2 uptake performance of POPs under humid conditions.Heteroatom containing POPs
Heteroatoms such as O, S and F and their various combinations with N have been introduced into POPs upon the reaction of suitably functionalized monomers through cyclization, condensation, or various C–C, C–N bond formation reactions. In this direction, trimerization reactions have been actively investigated, in particular, covalent triazine frameworks (CTFs), which can be synthesized through the trimerization of aromatic nitriles, gained a lot of traction. First reported in 2008 by Thomas et al., CTFs proved to be an intriguing class of materials due to their conjugated nature, high heteroatom content and surface areas.30 Through systematic studies, which involved control over the linkers length, salt amount and reaction temperature, the surface areas and heteroatom contents of CTFs can be controlled.31 It should be, however, noted that CTFs showed significantly lower heteroatom contents compared to the theoretically attainable amounts owing to the high reaction temperatures and irreversible side reactions during their formation. Moreover, partial carbonization is commonly observed for the CTFs synthesized above 350 °C. For example, CTF-0 prepared upon polymerization of 1,3,5-tricyanobenzene under ionothermal conditions at 600 °C showed an exceptionally high surface area of 2011 m2 g−1 however at the expense of crystallinity, formation of defects, increased mesoporosity and depletion of nitrogen content. Nevertheless, the resulting polymer still retained high nitrogen content of 19.3 wt% owing to the high nitrogen amount of the monomer as well as a high micropore content, thus resulting in CO2 uptake capacity of 4.22 mmol g−1 at 273 K, 1 bar.32 In this direction, various aromatic nitriles with high heteroatom contents have been adapted as a general approach to enhance CO2 affinity.33 Besides increasing the nitrogen amount, pairing with other heteroatoms such as O have also been investigated. Dai and coworkers showed that the introduction of methoxy groups onto a rigid hexaazatriphenylene precursor enabled CO2 uptake capacity of 6.3 mmol g−1 at 273 K, 1 bar in the resulting HAT-CTF synthesized at 600 °C with a BET surface area of 1090 m2 g−1. The high CO2 affinity of the HAT-CTF was attributed to the synergistic effect of heteroatoms and their distribution within the polymer network.34 Notably, the HAT-CTF also showed a rather high CO2 uptake capacity of 3 mmol g−1 at 0.15 bar, 273 K, which is more relevant to realistic carbon capture since flue gas contains approximately 15% CO2 at total pressures of around 1 bar. Similarly, Guipeng and coworkers investigated the effect of carboxylic acid and carboxylate moieties on the CO2 affinity of CTFs. Although CTF-CSUs showed significantly reduced surface areas in the range of 326–491 m2 g−1 and relatively low CO2 uptake capacities of 1.56–2.33 mmol g−1 at 273 K, 1 bar when compared to parent CTF-1 (746 m2 g−1 and 2.47 mmol g−1 of CTF-135), they still showed significantly higher CO2Qst of 44.6 kJ mol−1 compared to that of CTF-1, 27.3 kJ mol−1.36 Moreover, Van der Voort et al. also synthesized a series of CTFs bearing aromatic heterocycles such as isoxazoles (isox) and pyrazoles (pyz) and investigated their CO2 affinities. The CTFs showed a marked improvement in the CO2 uptake capacities. Comparative analysis of pyz-CTF-5-500 (CO2 uptake capacity of 3.31 mmol g−1, BET surface area of 1405 m2 g−1, N content of 8.9 wt%, C/N ratio: 7.96) with isox-CTF-5-500 (CO2 uptake capacity of 4.23 mmol g−1, BET surface area of 1537 m2 g−1, N content of 7.9 wt%, C/N ratio: 8.99) revealed the critical role of surface area and higher basicity of isoxazole ring.37 It should be, however, noted that while these functionalization strategies are useful to increase CO2 affinity, they are not expected to perform well under wet conditions owing to the competitive binding of water molecules. In this sense, incorporating F atoms is rather interesting as it would create hydrophobic pockets within the polymer network while maintaining high heteroatom content. The trimerization of (per-)fluorinated precursors results in CTFs bearing C–F moieties in their skeleton. The defluorination of the backbone is commonly observed during the synthesis of CTFs at elevated temperatures. Nevertheless, high heteroatom contents of these polymers located within the ultramicropores (<0.7 nm) enabled high CO2 uptake capacities (Table 1, entry 3–5, 7, 9, 11, 12).35,38–40 Such a CTF was showcased by Han et al. through the polymerization of perfluoroterephthalonitrile. The resulting F-CTF featured a highly microporous structure with a BET surface area of 1535 m2 g−1 and CO2 uptake capacity of 3.41 mmol g−1 at 298 K, 1 bar. Most notably, F-CTF was able to maintain a stable adsorption capacity over multiple adsorption–desorption cycles in the PSA process, which was attributed to the hydrophobicity of the fluorine moieties.35 In a similar fashion, Dai et al. reported a series of tailor-made, extensively fluorinated CTFs with various oxygen containing flexible fluorinated monomers.41 The polymers showed BET surface areas of up to 2085 m2 g−1, well defined ultramicropores (0.5–1.6 nm) and high CO2 adsorption capacities up to 6.58 and 4.33 mmol g−1 at 273 and 298 K, respectively. Interestingly, a deep learning study performed by the authors predicted an optimal F content of (∼4.8 wt%) and an optimal pore size 0.7 nm to result in the highest CO2 uptake capacity. Utilizing a backpropagation algorithm with surface area, micropore volume, mesopore volume, adsorption temperature and pressure as the input parameters, they predicted the CO2 uptake considering each condition. The design of new POPs guided by machine learning is expected to dominate the field in the coming years. The success of this approach, however, depends heavily on the availability of standardized data sets in the POP literature. Recently, Das et al. reported an intriguing approach combining N-rich aromatic moieties with perfluorinated subunits to form Tz-PFCN (Fig. 2). Their idea was not only to use the building blocks to obtain CTFs with high heteroatom contents, but also to deliberately induce thermal defluorination to obtain large amounts of ultra-micropores (Tz-df-CTF). As a result, Tz-df-CTF600 showed a surface area of 2106 m2 g−1 and a remarkable CO2 uptake capacity of 7.65 mmol g−1 at 273 K, 1 bar along with CO2/N2 selectivity of 16.8 under the simulated dry flue gas condition. Moreover, the authors also performed a breakthrough test, in which the Tz-df-CTF600 produced 99% pure N2 (up to 5.210 mmol g−1) and captured 0.987 mmol g−1 of CO2.39 Whereas this material showed the complete loss of its F inventory accompanied by a significant increase in the O content, retaining some of the F moieties could also enable operation under wet conditions.| Sample | S total (m2 g−1) | S micro/Stotal (%) | V total (cm3 g−1) | V micro/Vtotal (%) | Heteroatom contenta (wt%) | CO2 uptake (mmol g−1) | CO2Qst (kJ mol−1) | CO2/N2 selectivityd | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | N | F | O | S | 273 (K) | 298 (K) | — | 273 (K) | |||||||
| a Elemental analysis. b XPS analysis. c EDS analysis. d IAST (ideal adsorbed solution theory) calculation for the flue gas condition CO2/N2: 15/85 (v/v) at 273 K. e At 298 K. | |||||||||||||||
| 1 | CTF-1 | 746 | 0.62 | — | — | 87.1 | 11.4 | — | — | — | 3 | 1.41 | 27.5 | 20 | 35 |
| 2 | CTF-1-600 | 1553 | 0.10 | — | — | 84.6 | 13.6 | — | — | — | 3.82 | 2.25 | 30.0 | 13 | 35 |
| 3 | FCTF-1 | 662 | 0.94 | — | — | 63.3 | 12.0 | 23.8 | — | — | 4.67 | 3.21 | 35.0 | 31 | 35 |
| 4 | FCTF-1-600 | 1535 | 0.49 | — | — | 82.2 | 15.4 | — | — | — | 5.53 | 3.41 | 32.0 | 19 | 35 |
| 5 | F-DCBP-CTF-1 | 1574 | — | 0.51 | 0.34 | 59.7 | 11.3 | 4.2b | — | — | 5.98 | 3.82 | 33.1 | 31e | 38 |
| 6 | CTF-3 | 1454 | — | 0.98 | 0 | — | — | — | — | — | 2.13 | 1.34 | 21.0 | 24.5 | 41 |
| 7 | F12CTF-3 | 1558 | — | 1.32 | 0.44 | — | 5.5b | 3.7b | — | — | 6.58 | 4.33 | 24.5 | 32.4 | 41 |
| 8 | O-CTF-3 | 1450 | — | — | — | — | — | — | — | — | 3.17 | 1.70 | — | — | 41 |
| 9 | O-F12CTF-3 | 1822 | — | — | — | — | — | — | — | — | 5.59 | 3.04 | — | — | 41 |
| 10 | O-CTF-2 | 1130 | — | — | — | 81.4 | 2.7 | — | — | — | 2.65 | 2.06 | — | — | 41 |
| 11 | O-F16CTF-2 | 948 | — | — | — | 53.1 | 5.4 | 16.9 | — | — | 5.10 | 3.30 | — | — | 41 |
| 12 | Tz-df-CTF600 | 2106 | — | 1.43 | 0.64 | 48.4 | 6.2 | — | — | — | 7.65 | 5.08 | 20.0 | 16.8 | 39 |
| 13 | HAT-CTF-450/600 | 1090 | — | — | — | — | 32.8 | — | 9.4b | — | 6.3 | 4.8 | 27.1 | 110 | 34 |
| 14 | Isox-CTF-5-400 | 1683 | — | 0.70 | — | 53.6 | 7.4 | — | 6.0 | — | 4.92 | 2.86 | 29 | 83e | 37 |
| 15 | CTF-CSU38 | 491 | — | 0.44 | 0.24 | 58.8c | 20.6c | — | 15.7b | — | 2.2 | 39.2 | 72.0 | 36 | |
| 16 | CICF-KCl/NaCl-500 | 590 | 0.94 | — | — | 64.1 | 14.1 | — | — | — | 5.9 | 4.04 | 35.9 | — | 47 |
| 17 | CQN-1g | 1870 | 0.68 | 0.93 | — | 63.5 | 24.5 | — | — | — | 7.16 | 4.57 | 40.6 | 42.7e | 42 |
| 18 | BTAP-1 | 750.9 | — | — | 0.86 | 69.8 | 4.9 | — | — | 20.9 | 3.26 | 1.78 | 31.7 | 40 | 24 |
| 19 | BTAP-2 | 445.6 | — | — | 0.77 | 65.6 | 5.4 | — | — | 24.9 | 2.55 | 2.23 | — | 52 | 24 |
| 20 | BTAP-3 | 419.9 | — | — | 0.79 | 65.7 | 5.1 | — | — | 24.3 | 1.58 | 1.41 | 33.8 | 62 | 24 |
| 21 | BTLP-4 | 1011 | — | 0.53 | — | 64.1 | 7.3 | — | — | 16.7 | 4.3 | 2.7 | 28.7 | 41 | 50 |
| 22 | BTLP-5 | 705 | — | 0.41 | — | 62.3 | 6.6 | — | — | 15.0 | 3.2 | 1.98 | 29.1 | 45 | 50 |
| 23 | BOLP-4 | 698 | — | 0.54 | — | 71.7 | 8.2 | — | 16.3 | — | 3.1 | 2.0 | 33.6 | 79 | 50 |
| 24 | BOLP-5 | 759 | — | 0.52 | — | 73.8 | 7.2 | — | 15.0 | — | 2.9 | 1.8 | 32.9 | 95 | 50 |
| 25 | BILP-4 | 1135 | — | 0.65 | — | 67.1 | 14.0 | — | — | — | 5.3 | 3.6 | 28.7 | 79 | 48 |
| 26 | BILP-5 | 599 | — | 0.36 | — | 72.7 | 12.7 | — | — | — | 2.9 | 2.0 | 28.8 | 95 | 48 |
| 27 | Th-1 | 726 | — | 0.45 | 0.49 | 55.2 | 0.0 | — | — | 23.1 | 2.88 | — | 27 | 39 | 57 |
| 28 | Py-1 | 437 | — | 0.38 | 0.37 | 55.9 | 10.9 | — | — | 0.8 | 2.71 | — | 36 | 117 | 57 |
| 29 | Fu-1 | 514 | — | 0.36 | 0.42 | 60.6 | 0.0 | — | — | 0.3 | 2.21 | — | 28 | 50 | 57 |
| 30 | HMC-1 | 855 | — | 0.30 | — | 60.7 | 5.8 | — | — | 27.2 | 5.8 | — | 34.0 | 72 | 58 |
| 31 | HMC-2 | 425 | — | 0.19 | — | 59.9 | 5.1 | — | — | 27.7 | 6.6 | — | 60.0 | 70 | 58 |
| 32 | HMC-3 | 566 | — | 0.16 | — | 60.5 | 5.9 | — | — | 28.0 | 7.1 | — | 52.0 | 23 | 58 |
| 33 | Tp-POP | — | — | — | — | — | — | — | — | — | 1.32 | — | — | — | 59 |
| 34 | Tt-POP-1 | 258 | — | 0.31 | — | 54.1 | 16.2 | — | — | 26.1 | 0.75 | 0.29 | 50.12 | 30 | 59 |
| 35 | Tt-POP-2 | 368 | — | 0.40 | — | 66.3 | 9.8 | — | — | 18.8 | 0.91 | 0.49 | 58.77 | 26 | 59 |
| 36 | Tt-POP-3 | 974 | — | 0.62 | — | 76.2 | 6.4 | — | — | 12.0 | 0.78 | 0.45 | 54.23 | 25 | 59 |
| 37 | TAP1 | 474 | — | 0.74 | — | 78.1b | 11.4b | — | 10.6b | — | 2.2 | 1.4 | 35.6 | 94 | 60 |
| 38 | TAP2 | 772 | — | 1.41 | — | 73.8b | 12.4b | — | 13.9b | — | 3.2 | 2.3 | 37.2 | 112 | 60 |
| 39 | TAP3 | 729 | — | 1.04 | — | 72.2b | 13.7b | — | 14.2b | — | 3.4 | 2.3 | 36.1 | 114 | 60 |
| 40 | PFPOP-1 | 570 | 0.67 | 0.32 | 0.6 | 68.2 | 0.0 | — | — | — | 2.8 | 1.2 | 26.9 | 43.7e | 61 |
| 41 | PFPOP-2 | 630 | 0.65 | 0.35 | 0.54 | 72.53 | 0.0 | — | — | — | 3.4 | 1.5 | 30.2 | 52.1e | 61 |
| 42 | PFPOP-3 | 530 | 0.66 | 0.30 | 0.53 | 68.09 | 1.3 | — | — | — | 3.9 | 1.7 | 32.5 | 56.5e | 61 |
| 43 | PMOP | 1604 | — | 0.885 | 0.65 | — | — | — | — | — | 5.00 | 3.17 | 32.2 | 47.1 | 62 |
| 44 | TBOSBL1 | 649 | — | 0.527 | — | — | — | — | — | — | 4.0 | 2.1 | 35.1 | 68 | 63 |
| 45 | TBOSBL2 | 570 | — | 0.384 | — | — | — | — | — | — | 3.4 | 2.6 | 32.1 | 106 | 63 |
| 46 | TBOSBL3 | 493 | — | 0.467 | — | — | — | — | — | — | 2.8 | 2.2 | 32.7 | 108 | 63 |
| 47 | COP-190H-en | 456 | — | 0.142 | 0.96 | 83.3 | 3.4 | — | 8.7 | — | 3.6 | 2.19 | 100 | 171e | 64 |
| 48 | COP-190H-deta | 72 | — | 0.047 | 0.24 | 83.8 | 3.7 | — | 9.0 | — | 2.7 | 1.79 | 73 | 121e | 64 |
| 49 | COP-190H-CN | 661 | — | 0.21 | 0.95 | 73.6 | 5.0 | — | 13.2 | — | 3.2 | 2.11 | 42 | 91e | 64 |
| 50 | COP-190H-SH | 773 | — | 0.25 | 0.92 | 72.5 | — | — | 10.8 | 7.7 | 3.6 | 2.28 | 37 | 76e | 64 |
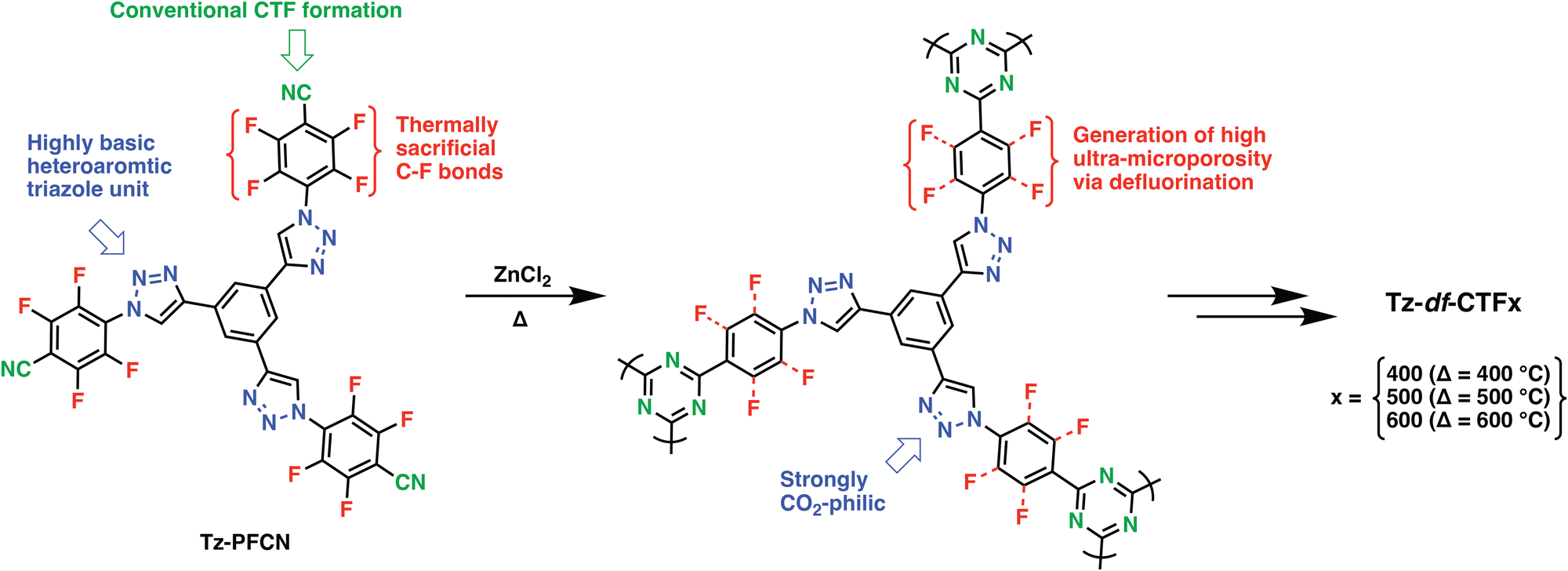 | ||
| Fig. 2 Schematic illustration of the formation of Tz-df-CTFs via a Dual Strategic Approach. Reproduced from ref. 39 with permission of the publisher. | ||
Besides the trimerization of aromatic nitriles, several other trimerization reactions have also been used for the preparation of POPs. A rather intriguing example of such a trimerization reaction in POPs was reported by Buyukcakir et al., who prepared a porous quinazoline network (CQN) through the trimerization of o-aminonitriles (Fig. 3). CQNs showed exceptional stability, partial crystallinity, and surface areas of up to 1870 m2 g−1. This, together with their microporous nature resulted in an outstanding CO2 uptake capacity of 7.16 mmol g−1 at 273 K, 1 bar and a high IAST CO2/N2 selectivity of 74.7 at 298 K under dry flue gas conditions (CO2/N2: 10/90 v/v).42 Interestingly, unlike CTFs, which suffer from the depletion of nitrogen content, CONs showed near ideal C/N ratios in the range of 3.12–3.19 (calculated 3.00) pointing to the efficient stabilization of nitrogen atoms.
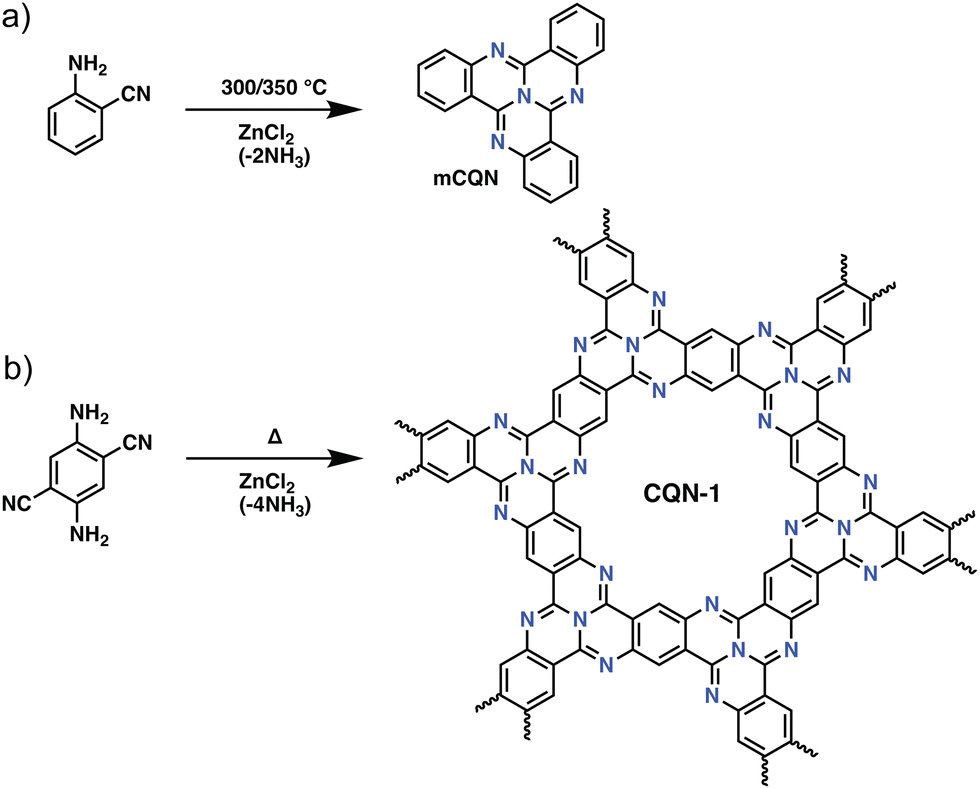 | ||
| Fig. 3 The synthetic route for covalent quinazoline networks (CQNs). (a) Preparation of model compound, tricycloquinazoline (mCQN). (b) The synthesis of CQN-1. Reproduced from ref. 42 with permission of the publisher. | ||
Besides the in situ formation of triazines through a trimerization reaction or condensation, another strategy for triazine-containing polymers is to use cheap and readily available precursors such as melamine or cyanuric acid. Due to the N-rich nature of the derived polymers as well as their easy functionalization and tunability through the choice of the reactants or even post-functionalization, they generally show decent CO2 uptake capacities and allow post-polymerization functionalization through their aminal linkages. For example, Yavuz and coworkers prepared such an amide-linked polymer through the polymerization of melamine with a triacid chloride in different solvents and found that the choice of solvents has a profound effect on the morphology of the polymers and thus their CO2 uptake capacity (DMAc-NMP: 0.134 mmol g−1 at 273 K vs. dioxane: 0.09 mmol g−1 at 273 K).43 Although the uptake capacity in itself is far from top-performing systems, it clearly shows that textural properties have to be carefully monitored and tuned in order to obtain good sorbents. Besides acid chlorides, especially formaldehyde resins have been explored extensively, many of which can reach high CO2 capacities of above 3 mmol g−1 at 273 K.44 Recently, Reimer et al. presented the kilogram-scale preparation two melamine and cyanuric acid–based polymers and their CO2 uptake capacity as pristine materials and after addition of diethylenetriamine. Whereas the non-impregnated system showed rather typical uptake characteristics (0.91 mmol g−1 at 298 K) the amine-treated system showed a much higher capacity of 1.92 mmol g−1 but more importantly, most of the uptake occurred at low pressures below 0.15 bar which is highly relevant for industrial applications. Part of this steep uptake has to be attributed to the chemisorption of CO2 on the supported amine while the authors also partially attribute it to the incorporation of cyanuric acid. The profound effect of the cyanuric acid was also shown through sorbent recycling experiments, where a very stable capacity was found for the cyanuric acid systems whereas a continuous drop in capacity was observed for the systems without the acid.45 One shortcoming of directly using melamine is the exceptionally small pores that are formed, which often results in low surface areas due to pore collapsing. Hence, a variety of research groups explored larger melamine building blocks such as phenyl-spaced diaminotriazines. An example of such an approach was presented by Wang et al. who utilized three different diaminotriazine-derived building blocks to obtain highly nitrogen-rich polymers with surface areas of up to 1228 m2 g−1 and CO2 capacities of 4.61 mmol g−1.46
In an effort to maintain high heteroatom content, Song and coworkers investigated the effect of salt-templation in the formation of covalent isocyanurate frameworks (ClCFs) via the cyclotrimerization of 1,4-phenylene diisocyanate under ionothermal conditions (Fig. 4). The CICFs synthesized using a salt mixture containing KCl/NaCl/ZnCl2 showed near ideal heteroatom content at 500 °C, CICF-KCl/NaCl-500, whereas the ones synthesized using only ZnCl2 exhibited more than 50% depletion of heteroatom content. These results revealed the critical role of the template effect originating from the interactions between alkali metal ions and oxygen atoms of isocyanurate moiety. Interestingly, the authors also observed a significant change in the morphology from micron-sized particles to a sheet-like morphology in the presence of a template. The CICF-KCl/NaCl-500 showed a BET surface area of 590 m2 g−1 along with a CO2 uptake capacity of 5.9 mmol g−1 at 273 K and 1.1 bar owing to its high microporosity and nitrogen content of 14.1 wt%. In stark contrast, CICF-500 with a BET surface area of 1674 m2 g−1 and high mesopore content showed nitrogen content of only 7.8 wt% and a lower CO2 uptake capacity of 3.95 mmol g−1 at 273 K and 1.1 bar.47 These results clearly verify the critical role of the combination of high micropore content and high heteroatom content to realize high CO2 uptake capacity and selectivity. These examples also reveal the potential of in situ formation of heterocyclic units as a powerful strategy in the synthesis of POPs. In this direction, one of the seminal contributions was the synthesis of benzimidazole-linked polymers (BILPs) by El-Kaderi and coworkers. BILPs were readily synthesized by the condensation reaction of aromatic o-diamines with aromatic aldehydes.48 The authors attributed the high CO2 affinity of BILPs to the strong dipole–quadrupole interactions between basic nitrogen sites with the C atom of CO2, which was further reinforced by the hydrogen bonding interactions between O atoms and N–H moieties. This approach is an elegant demonstration of cooperative supramolecular interactions to increase CO2 binding affinity.49 Furthermore, imidazoles provide ample opportunities for post-polymerization modification, thus providing the necessary means to tune the polymer's properties to desired tasks such as the conversion of CO2 to carbonates. In a similar fashion, benzoxazoles and benzothiazole-linked POPs were also investigated.24,50,51 El-Kaderi et al. showcased the synthesis of benzothiazole- and benzoxazole-linked polymers (BTLPs and BOLPs) via condensation reactions, that proceed via Schiff-base reaction and subsequent cyclodehydrogenation. Although the authors set out to showcase the synergistic effects of the different heteroatom-containing linkages on CO2 uptake, no specific effect/correlation on the uptake, the Qst or the selectivity could be elucidated.50 Coskun et al. introduced a solvent and catalyst free approach for the synthesis of an ultra-microporous benzothiazole polymers (BTAPs) using elemental sulfur, which is a major by-product of natural gas purification (Fig. 5).24 The BTAPs were based on different 2D/3D building blocks incorporating p-tolyl and 4-aminophenyl units and were heated to 275 °C in the presence of elemental sulfur. Subsequent heating to 400 °C was employed to remove excess sulfur, while also activating the pores without the need of excessive purification and post-washing steps. The resulting polymers not only contained the desired benzothiazole linkages but also excess sulfur (up to 10 wt%) due to the formation of short sulfur chains and thiols. Despite that, BTAPs showed surface areas of up to 750 m2 g−1 and CO2 uptake capacities up to 3.26 mmol g−1. Although these metrics pale in comparison to some of the best performers (Table 1), BTAPs proved to be exceptional materials after performing breakthrough experiments showing excellent CO2/N2 selectivity, while also being produced in a green and environmentally friendly way. These results also highlight the critical need for the development of sustainable synthetic strategies for the development of POPs. In this direction, hydrothermal synthesis of POPs, pioneered by Unterlass et al., gained significant traction in recent years as it employs only water as a solvent.52 The resulting polyimide POPs showed surface areas of up to 207 m2 g−1 and CO2 uptake capacities of up to 3.57 mmol g−1. Interestingly, post-polymerization processing in the form of sintering did not affect the overall surface area too much resulting in only a slight drop to 203 m2 g−1, indicating that the ultramicropores could be maintained.53
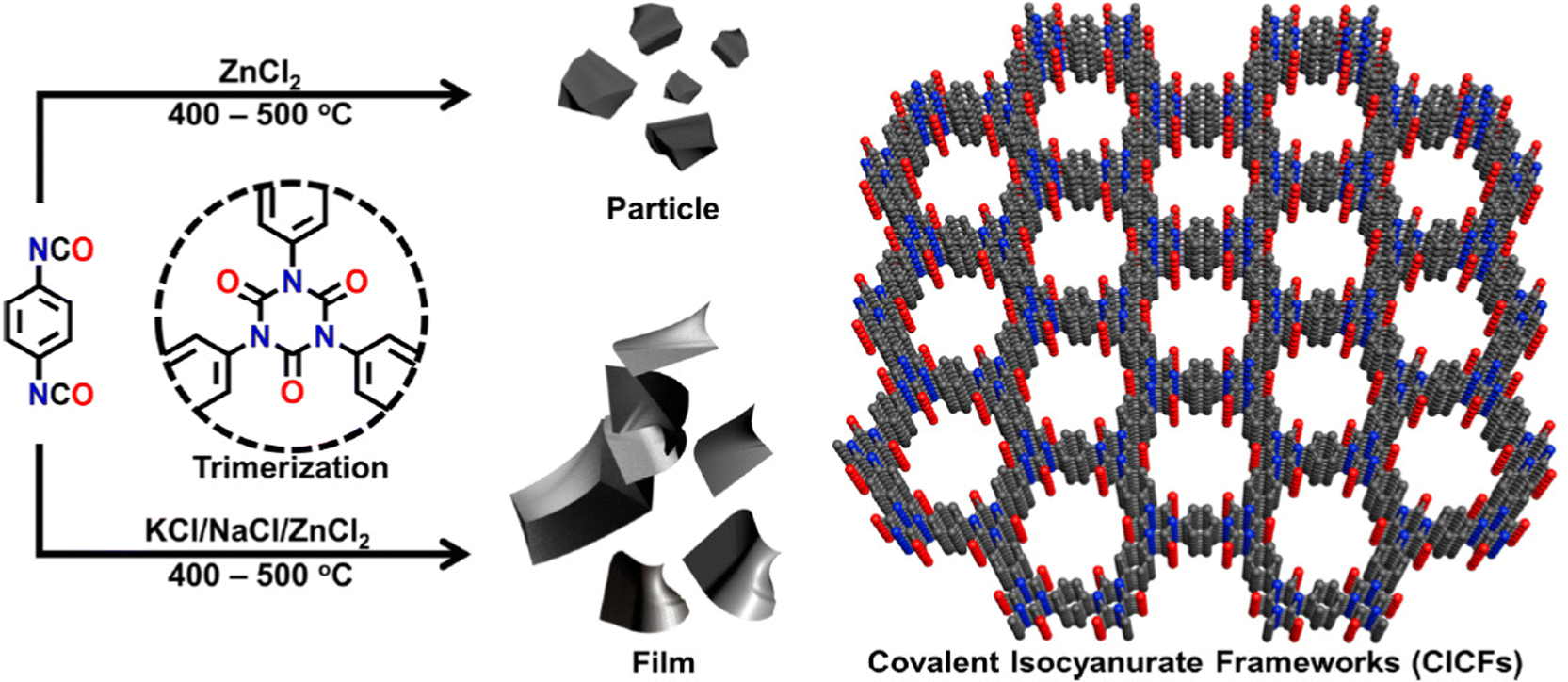 | ||
| Fig. 4 The synthetic route for the preparation of covalent isocyanurate frameworks (CICFs). High heteroatom content improves CO2 uptake capacity and binding affinity. Reproduced from ref. 47 with permission of the publisher. | ||
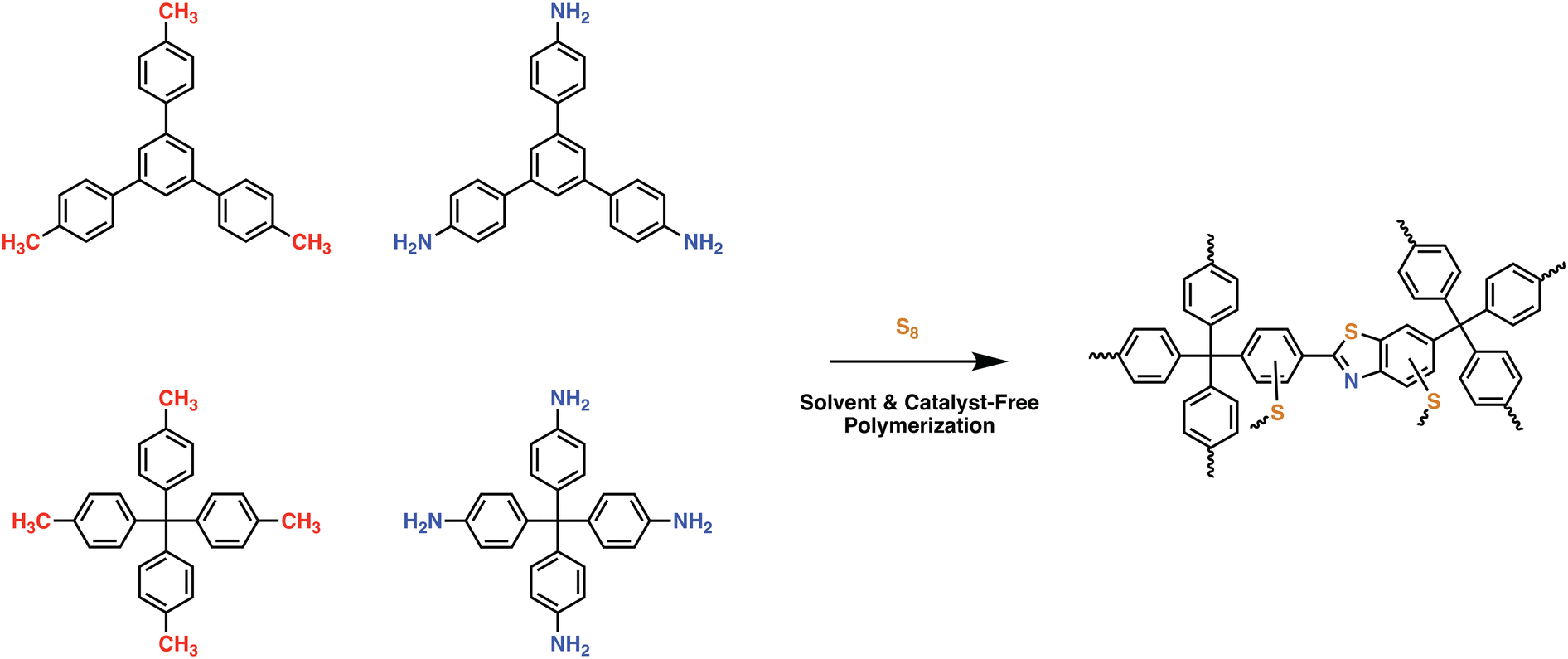 | ||
| Fig. 5 Synthetic scheme for the preparation of ultramicroporous benzothiazole polymers (BTAPs) through environmentally benign conditions without using any solvent or catalyst. BTAPs were synthesized by simply reacting aromatic methyl- (M1 or M2) and amine-substituted monomers (A1 or A2) and elemental sulfur, S8, at 275 °C in quantitative yields, followed by a heating step at 400 °C for pore activation and sulfur impregnation. Reproduced from ref. 24 with permission of the publisher. | ||
Another intriguing method to introduce a large number of heteroatoms into a porous polymer is the utilization of inorganic clusters such as siloxanes. Sun et al. demonstrated such a work in which they utilized octaphenylsilsequioxane in a synthesis with bis(chloromethyl)biphenyl to obtain POPs with surface areas of up to 1170 m2 g−1 and CO2 capacities of up to almost 3 mmol g−1. Although such a system lies on the cusp of what can be defined as an organic polymer it clearly shows the interplay between high heteroatom contents, large surface areas and CO2 uptake.54
Utilizing photo-switchable functional units to govern a polymer's CO2 adsorption capacity is another way to further tune porous polymers for specific applications. Especially azo-groups as part of a porewall-functionalization have been employed by Sun and coworkers to tune POPs and MOFs capabilities for CO2 uptake.55,56 Leveraging the cis/trans isomerization of the azo moiety upon irradiation with UV light, the authors were able to tune the uptake capacitates thus potentially allowing the preparation of switchable membranes.
In an attempt to unveil the parameters to achieve high CO2 affinity, we investigated the impact of surface area, micropore content, heteroatom/C ratios on the CO2 binding. The corresponding performance and structural characteristics of the various POPs rich in heteroatoms are surmised in Table 1. Based on these characteristics, Fig. 6a and b clearly showed the trend that high surface areas and pore volumes as well as a high ratio of micropores results in an overall higher CO2 uptake capacities, highlighting critical role of micropores. A plethora of POPs performing at the highest levels contain combinations of nitrogen and other heteroatoms due to their beneficial interactions with CO2 and thus higher uptake capacities. The correlation between CO2 adsorption enthalpies (Qst) and CO2/N2 selectivities, surface area and heteroatom/C ratios were shown in Fig. 6c and d. For the adsorption enthalpies (Qst) above 40 kJ mol−1, the binding mechanism of CO2 is classified as chemisorption, which is observed for amine functionalized and/or amine impregnated POPs. As the lower surfaces areas generally imply higher amine content, they showed the highest Qst and CO2/N2 selectivity. When we look at the physisorption regime, which is below 40 kJ mol−1, we observed an increase in the Qst and CO2/N2 selectivity with respect to the increasing heteroatom/C ratio. These results further suggest that for an efficient CO2 capture through physisorption, ideal system should feature both high micropore content and high heteroatom/C ratio. Interestingly, we also did not observe a correlation between the CO2Qst and CO2/N2 selectivity within the physisorption regime. The fact that several publications praise fluorine as an attractive heteroatom in POPs for CO2 capture, our analysis revealed no apparent trend between fluorine content and beneficial interactions with CO2. There is an argument to be made, that fluorinated precursors can help with generating nicely microporous materials through thermal defluorination, however, compared to other heteroatoms such as N, O or S the impact of fluorine on a polymer's CO2 uptake and CO2 selectivity is not as significant. On the other hand, the combination of N with O and S consistently showed higher Qst values and CO2/N2 selectivity (Fig. 6e and f), pointing to the positive impact of the synergistic effect of different heteroatoms and in most cases increased basicity of the network.
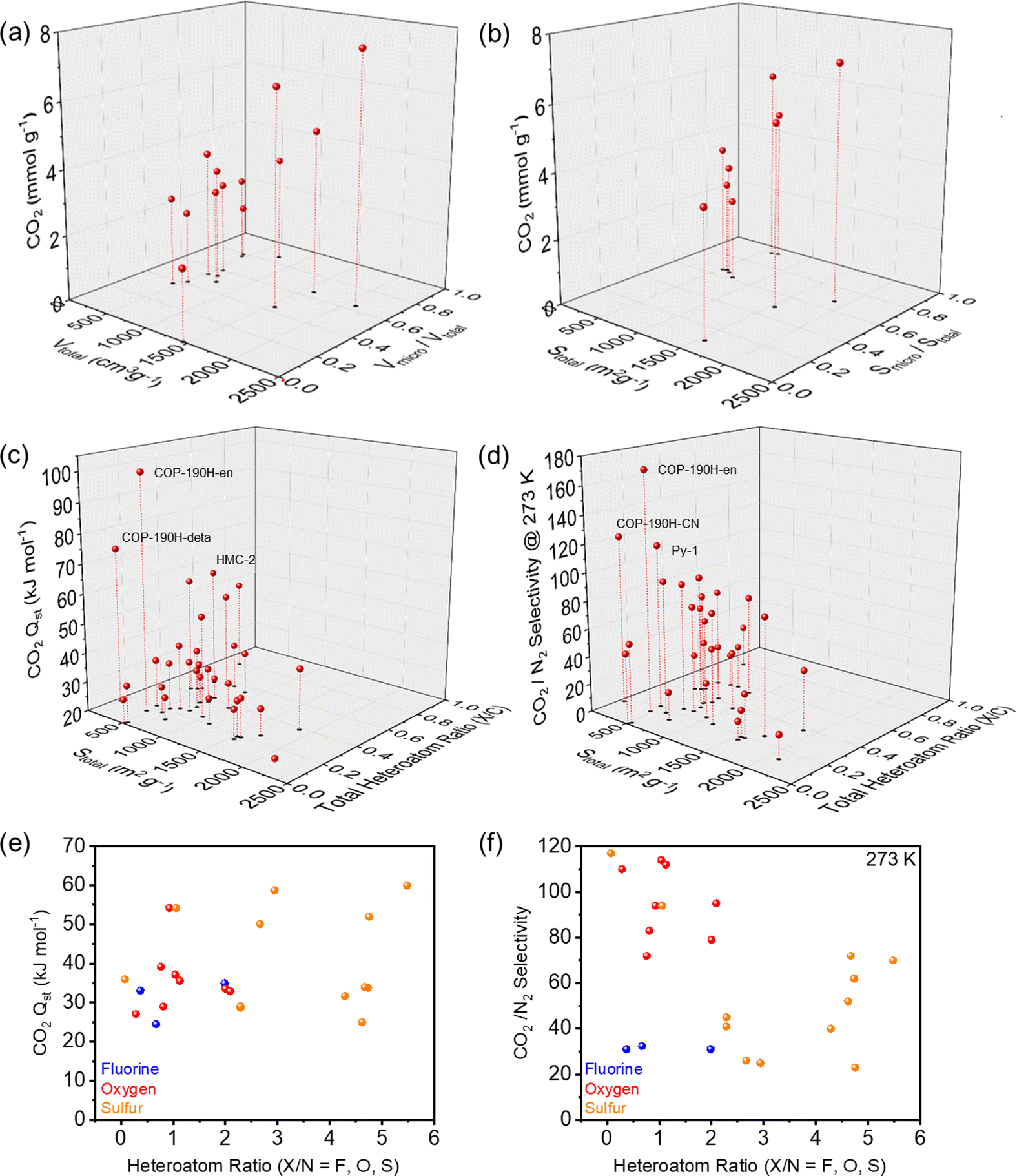 | ||
| Fig. 6 CO2 capture performance of POPs containing heteroatoms with various parameters. (a) CO2 adsorption at 273 K versus total pore volume (Vtotal) and micropore volume ratio (Vmicro/Vtotal). (b) CO2 adsorption at 273 K versus total surface area (Stotal) and micropore surface area ratio (Smicro/Stotal). (c) Heat of adsorption (CO2Qst) and total surface area (Stotal) versus total heteroatom ratio (X/C, X = all heteroatom species in the same POPs) (d) CO2/N2 selectivity at 273 K and total surface area (Stotal) versus total heteroatom ratio (X/C, X = all heteroatom species in the same POPs) (e) heat of adsorption (CO2Qst) versus heteroatom ratio (X/N, X = F, O, S). (f) CO2/N2 selectivity versus heteroatom ratio (X/N, X = F, O, S). All the data is summarized in the Table 1. | ||
POPs based on porous molecular precursors
Until now we have established key functional units and parameters governing high CO2 affinity and selectivity. In this section, we will focus on some intriguing design strategies. Specifically, we will discuss the use of (i) shape persistent monomers such as triptycenes or spirobifluorenes, (ii) pre- and post-polymerization amine functionalized POPs and (iii) cavitands and cages in order to explain why the use of these specific motifs resulted in highly CO2-philic systems and in some cases even in excellent catalysts for the up-conversion of CO2 to value-added products.Although, on a first glance, all the aforementioned systems seem diverse and uncorrelated, many of them combine a specific selection of molecular motifs. For one, they rely on molecular precursors that directly induce a three-dimensional network on a molecular level, thus, in many cases, resulting in large surface area polymers with high accessible pore volumes, given that pore collapsing can be avoided. Secondly, a wide variety of linking functionalities can be employed, however most commonly either amine, ether or C–C based linkages are used, each resulting in specific benefits in terms of stability, CO2-philicity and catalytic activity.
Shape-persistent molecules
In the realm of shape persistent organic precursors especially adamantanes, tetraphenylmethanes (TPMs), spirofluorenes/spirobifluorenes and more recently triptycenes have been employed to obtain higher surface area polymers (Table 2).65 Although many systems based on these precursors suffer from pore collapse, resulting in much lower surface areas than theoretically possible, fine-tuned syntheses and work-up procedures avoid/alleviate these problems. Especially triptycene-based POPs have gathered significant interest in recent years, since the derived POPs often contain a significant amount of (ultra)micropores, which are helpful to capture gases. TPMs are among the most widely used 3D structure inducing precursors in POPs and COFs due to their facile synthesis and ease of functionalization. The high structural tunability allows generating a wide range of linking functionalities/chemistries resulting in highly tunable polymers. To showcase the influence of specifically tuned linking units, Coskun et al. reported a series of TPM-based POPs linked via azo functionalities resulting in excellent CO2-philicity and selectivity over N2. Most notably, the systems showed next to no hysteresis thus showing the polymer's capability for energy-efficient recyclability.23 Besides such conventional linkages, imidazoles, thiazole and thiadiazoles have also proven to be quite useful for CO2 uptake providing both a CO2-philic site that can be further functionalized to facilitate the upconversion of CO2.24,66 On top of the vast amount of linking units, several research groups used side-chain functionalization as a tool to tune the pore structure and environment pre- or post-polymerization.67Whereas TPM-based POPs often suffer from interpenetration or pore collapse, triptycenes have been proven to be a reliable building block to induce molecular dimensionality into a system while maintaining high porosity. Among C–C linked triptycene-based POPs, both the use of stiff, inflexible linkers such as alkyne-linked systems68 or more flexible linkers based on allylhalides68,69 resulted in high surface areas and exceptional CO2 uptake capacities, which can be attributed to the presence of micropores and ultramicropores. However, a major shortcoming of such systems unless provided by a co-monomer70 is the lack of acidic or basic sites within the polymers hindering their potential as porous organic catalysts.
Thus, a variety of triptycene POPs linked via amines71,72 or phenazines73 have been employed to endow the polymers with suitable functional active sites. Both amines and phenazines provide basic sites which help polarizing CO2, thus enabling for example the cycloaddition of epoxides to carbonates. A system nicely showcasing the interplay of dimensionality and CO2 sorption was reported by Baek et al., who employed triptycene hexamine and polymerized it with hexaketocyclohexane to obtain a phenazine-linked POPs. The obtained highly rigid polymer showed a BET surface area of 2247 m2 g−1, while featuring excellent CO2 uptake capacity of 4.98 mmol g−1 at 273 K and 1 bar owing to strong interactions with the heteroatoms located within the ultramicropores of polymer network.73
Pre- and post-functionalized POPs
The wide range of possible pre- and post- functionalization strategies allows chemists to fine tune specific properties of the target porous organic polymers. Among post-polymerization strategies, especially impregnation with amines and the conversion of suitable functional units on the polymer backbone to free amines or amine-containing groups have been heavily investigated. Like previously introduced, amine scrubbing with MEA and others, the amine groups act as chemical sorbents for CO2 resulting in good uptake performance especially at low partial pressures and low CO2 loadings. Many of these post polymerization functionalization strategies revolve around preparing suitably functionalized precursors often based on the shape persistent molecules as building blocks such as adamantanes, TPMs or triptycenes. Such systems often contain halides or allyl halides that are subsequently converted to the desired functional groups. An early example of such functionalization was reported Nguyen et al., who prepared a series of TPM based systems and even used functionalized allyl-TPM precursors for post polymerization modification showcasing the effect of various functional groups on the surface area and gas uptake properties. The free amine functionalized system showed superior performance in terms of CO2 uptake although having one of the lowest surface areas in the series.67 A similar strategy was used by Hopkinson et al., who prepared PIM-1 and further functionalized it via the hydrolysis of the nitrile moieties and subsequent treatment with tris(2-aminoethyl)amine. The resulting amine containing system showed significantly enhanced CO2 uptake capacities of up to 1.62 mmol g−1 at 0.15 bar and 298 K while showing a good cyclability.74 Similarly, Yavuz et al. prepared a triphenylbenzene-based POP (COP-130) and subsequently acylated it with chloroacetyl chloride to obtain a ketone and alkyl halide bearing polymer. The obtained COP-130-Ac allowed derivatization with various amines through Schiff base and nucleophilic amination reactions, thus allowing a wide range of functionalization. Although the functionalization resulted in a significant loss of porosity, the amine functionalized systems showed CO2 uptake capacities up to 4.95 mmol g−1 at 273 K (1.31 mmol g−1 at 273 K and 0.15 bar) while having a low Qst value of 25.6 kJ mol−1 indicating that although free amines are present in the system, next to no chemisorption occurred pointing to good cyclability and low energy regeneration of the sorbents.75 One major advantage of these systems is their ability to operate under wet conditions, which is a serious problem for the POPs owing to the competitive binding of CO2 and H2O molecules. In an attempt to combine TPM or adamantane linkers with non-metalated porphyrins, Wang et al. exploited aldehyde terminated building blocks and reacted them with pyrrole to obtain nicely porous materials with ample Lewis acidic N–H sites in the porphyrin moieties. The obtained polymers showed good CO2 uptake capacities of up to 4.0 mmol g−1 and good CO2/N2 selectivities, thus showcasing how such a simple design approach can lead to highly functional polymers.76Combining N-rich linkages, metal-containing building blocks and amine-functionalized linkers, Echegoyen et al. prepared a phthalocyanine-based POPs via azide Click chemistry utilizing an amine functionalized alkyne linker. The obtained polymer had a relatively low surface area of only 342 m2 g−1 but still showed good CO2 uptake capacity of 3.57 mmol g−1 at 273 K and 1 bar due to the combination of highly CO2-philic building blocks.77 Tetraphenylenes are another class of porosity-inducing precursors that have recently received a lot of attention, however, most of the literature focussed on exploiting the conductive properties of the polymers. Among the few TP-based polymers, Coskun et al. reported the synthesis of a dioxane-linked three-dimensional POPs. Interestingly, pure solvothermal methods did not result in the formation of a nicely porous polymer, while salt templation yielded highly porous polymers with CO2 capacities reaching up to 4.42 mmol g−1.78
Cavitand- and cage-based POPs
Both cavitands and cages have been known for their large intrinsic surface areas for a long time. However, molecular cage/cavitand systems suffered from unpredictable stacking in the solid-state, which in some cases resulted in a loss of surface area due to e.g. phenylene-to-window packing in the case of molecular cages.79 Although directed molecular design allowed to obtain highly porous, selective,80,81 and in some cases catalytically active cages,82 their often complicated synthesis and purification hinders widespread application for CCS and CCC. An intriguing feature of these systems is the possibility to design the pore sizes by choosing the linker length in cages or by adding/removing linking units in case of cavitands. This approach allows fine-tuning of the pores towards specific tasks such as gas sorption by increasing the interactions with a specific gas over another one.79 To avoid a loss of porosity and pore accessibility in the solid-state, researchers developed the “cavitand/cage-to-framework”83 design strategy, wherein suitably functionalized “pre-porous” building blocks are linked via covalent bonds, often utilizing stiff building blocks to endow the polymers with rigidity and structural integrity.25,84 First introduced by Zhang et al. to obtain a porous cage-POPs,83 the strategy was widely adopted and adapted towards cyclodextrins, calixarenes, pillarenes,85,86 resorcin[X]arenes and more specialized systems (e.g. noria-based POPs).87 Whereas cyclodextrins and pillarenes have mostly been used for CO2 uptake and sequestration,85 especially pillarenes and resorcin[X]arenes have been employed both as sorbents and catalysts owing to their abundant phenolic OH functionalities, which can be leveraged to polarize substrates such as epoxides thus facilitating an easier nucleophilic attack by a suitable co-catalyst allowing its conversion to cyclic carbonates. Patra et al. reported a series phenyl resorcin[4]arene-based POPs featuring abundant phenolic OH moieties while being linked through various different functionalities, thus tuning the porosity and gas sorption properties of the systems (Fig. 7). All systems proved to be suitable catalysts for the cycloaddition of CO2 to epoxides, however the azo-linked system showed the highest epoxide conversion even though having the lowest BET surface area in the series exemplifying the role suitable functionalization has on the gas uptake and catalytic performance.88 A recent example of a calixarene-based porous organic polymer was reported by Comotti et al., who prepared a series of differently functionalized calixarenes and reacted them with a bromo-TPM derivative. The obtained polymers exhibited moderate to large surface areas up to 2609 m2 g−1 and CO2 capacities up to 2.91 mmol g−1 at 273 K and 1 bar.89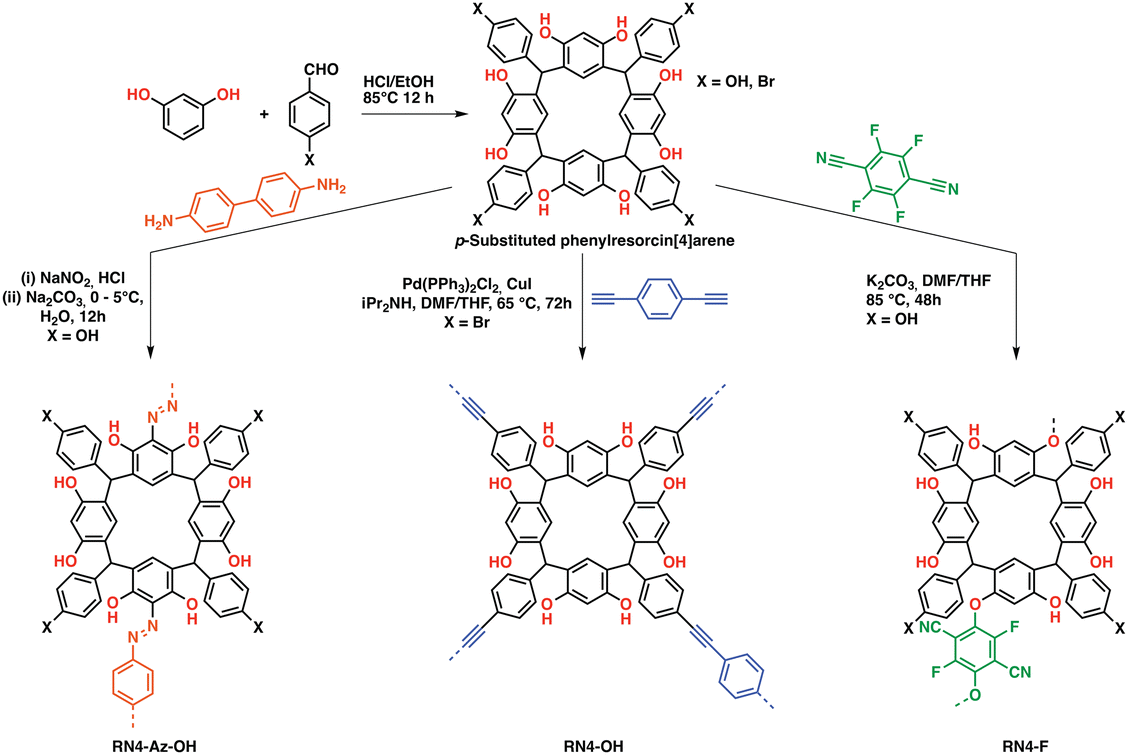 | ||
| Fig. 7 Phenylresorcin-based POPs. (a) Synthetic schemes of C-phenylresorcin[4]arene-based porous organic polymers (POPs). RN4-Az-OH: diazo coupling between p-hydroxyphenylresorcin[4]arene and benzidine, RN4-OH: Sonogashira polycondensation between p-bromophenylresorcin[4]arene and 1,4-diethynylbenzene, and RN4-F: aromatic nucleophilic substitution reaction between p-hydroxyphenylresorcin[4]arene and tetrafluoroterephthalonitrile (12 phenolic OH groups are likely to be equally reactive) digital photographs of the respective POPs in the form of powder Reproduced from ref. 88 with permission of the publisher. | ||
Molecular cages were the first systems for which the cavitand/cage-to-framework strategy was employed on, however, due to the complicated preparation of suitably functionalized cages, significantly fewer systems have been reported to date compared to other systems. In 2015, Coskun and coworkers reported a triazine-based cage that showed exceptional CO2 uptake capacity and high selectivity towards CO2 over other gases thus being highly suitable for CCS.25 Although the system featured basic triazine units and aminal linkages, it was found to be unsuitable for the direct conversion of CO2 without the use of a co-catalyst. Recently, Patra et al. introduced an intriguing new cage precursor dubbed “Noria” that features 24 phenolic hydroxyl groups and seven accessible windows. Improving on their previous design, the use of a stiff linker resulted in a highly porous polymer that showed high affinity of CO2 over N2 and proved to be a suitable catalyst for the conversion of CO2 to a series of carbonates in the presence of tetrabutylammonium bromide (TBAB) as a co-catalyst.87
POPs for CO2 conversion
In organic chemistry, CO2 is a well-known and commonly employed substrate and various chemical, electrochemical, and photochemical strategies have been investigated to utilize CO2 as a C1 building block. In photocatalytic and electrocatalytic applications, semiconducting POPs are of immense interest due to their intrinsic conductivity, broad absorption range, high surface area and structural tunability. Wherein for photocatalysts, the efficacy of CO2 adsorption, the polymers' light absorption range, and charge separation determine the overall efficiency of the POP catalysts, in electrocatalytic applications the efficiency of POPs is mostly determined by the electronic properties, contact to the electrode and the catalysts CO2 affinity. In both cases, the incorporation of precious metal ions and nanoparticles is often employed to increase the overall performance. Due to that, a plethora of high-value products such as CO, methanol, ethanol, methane, and many others are available that can directly be used as substrates or reagents. Although, in recent years the interest in electrochemical and photochemical POPs catalysts has increased, these systems often rely on strict conditions and the use of precious metals. When it comes to the chemical conversion of CO2, especially the cycloaddition of CO2 to epoxides has been explored intensively due to the high atom economy and readily available substrates. Besides that, cyclic carbonates can be directly used as aprotic polar solvents90 in battery electrolytes,91 as precursors for polymers such as polycarbonates92,93 or phenol resins,94 or as precursors in organic synthesis or pharmaceuticals.95,96 Despite research mostly focusing on the conversion of CO2 to cyclic carbonates, various other strategies such as the conversion of CO2 to linear carbonates, cyclic carbamates, to products such as urea or the direct use in carboxylation reactions can be envisioned given a suitable molecular design.Co-catalyst free porous organocatalysts
In the previous section we have presented a variety of design strategies that enabled CO2-philic, highly porous materials, however, only few of these materials were suitable for the direct conversion of CO2 to value-added products without the need of a specific co-catalysts (Table 2). The problem lies in the necessity of providing a range of suitable functionalities near one another to ideally enable the activation of both CO2 and the substrate of choice. Besides incorporating metal ions into the polymers, two strategies allowing fully organic polymers for the conversion of CO2 have been reported: (i) implementing functional units for the activation of both CO2 and the substrate and (ii) utilizing charged moieties such as pyridyls or imidazoles to provide an anion for the nucleophilic attack to the substrate.Yavuz et al. demonstrated the first approach based on a simple polymer featuring pyridyl salicylimines.97 The polymer embodied both an acidic site in the form of phenolic OH and a basic site in the form of pyridine. The intriguing feature of this molecular design lies in the capability of the pyridine moiety to become charged, during the nucleophilic attack on the epoxide removing the necessity of a co-catalyst for the conversion of CO2 to epoxides (Fig. 8). The wide scope of tested substrates and the cyclability of the system nicely demonstrates the interplay between the functional units and showcases how readily available chemicals and thus polymers can be utilized as highly active porous organocatalysts.
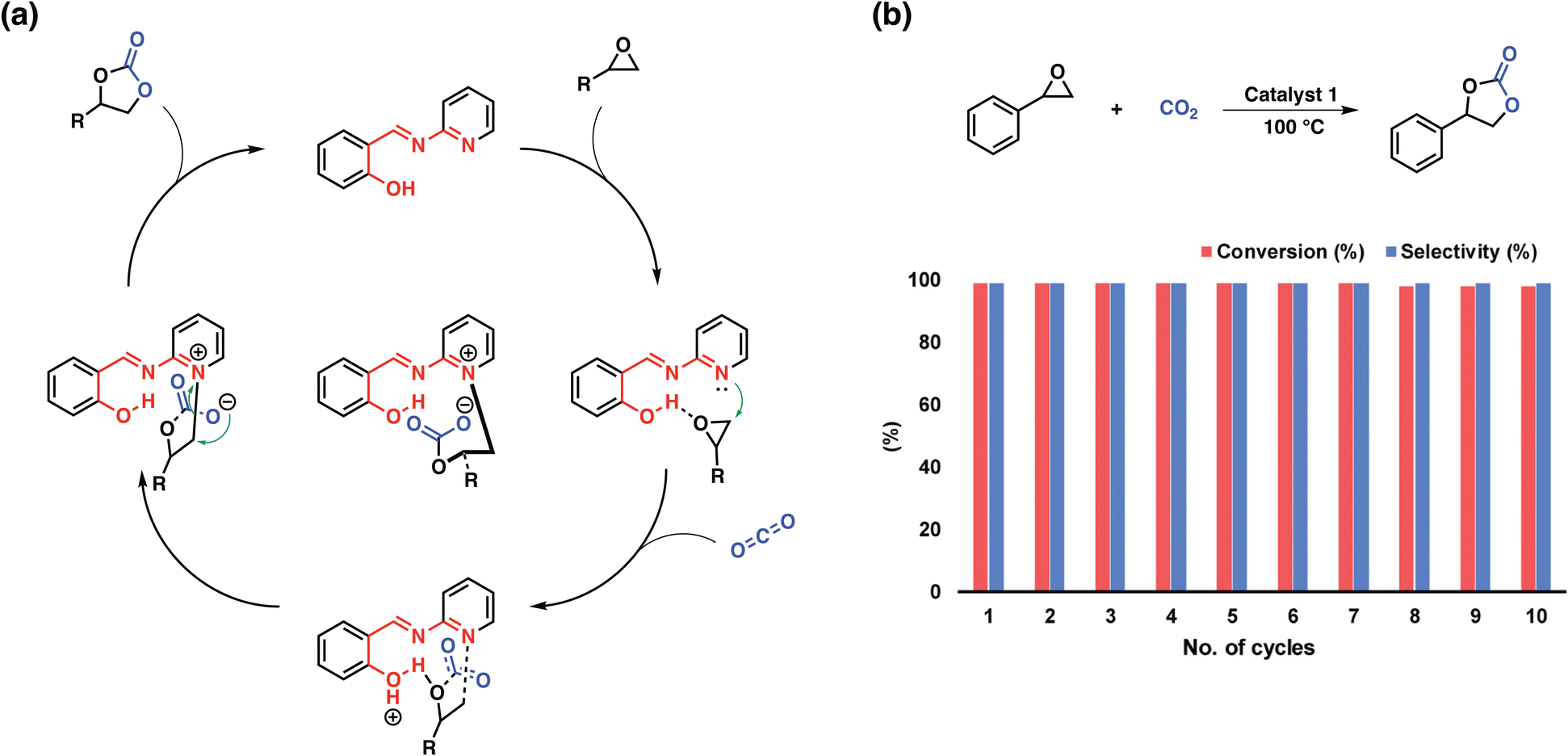 | ||
| Fig. 8 Probable mechanism and recyclability studies. (a) A proposed mechanism for the coupling reaction of an epoxide with CO2 by catalyst 1. (b) Recycling of the catalyst in the coupling reaction of styrene oxide and CO2 under optimized reaction conditions. Reproduced from ref. 97 with permission of the publisher. | ||
Although often resulting in less chemically and thermally stable polymers, the utilization of charged linkers is significantly more common compared to the strategy introduced before. Especially (bi-)pyridyl-linked systems or polymers incorporating or linked by imidazoles have been heavily investigated for that purpose. Besides having a high nitrogen content and thus featuring high CO2-philicity, the charged moiety and the associated counter ion facilitate the nucleophilic attack on the substrates such as epoxides allowing the cycloaddition of CO2. To this end, Coskun and coworkers have reported a CO2-philic CTF-based system containing viologen linkers. The electrostatic interactions of the charged linkers with CO2 molecules facilitated high gas uptake and enabled the cycloaddition of CO2 to a wide range of substrates.98 It should also be noted that when using charged units, the counterion does not only affect the available surface area based on the bulkiness of the ion but also affects the catalytic activity of the system. In their series consisting of Cl−, PF6− and BF4−, the authors demonstrated that the system with chlorine showed the highest CO2 uptake and catalytic activity, which was attributed to the higher nucleophilicity of the anion.99 Along this line, Chen et al. explored the effect and interactions of the anion in their charged cyanovinylene POPs prepared through a Knoevenagel condensation. Despite the mesoporous nature of the polymer, it showed good catalytic activity at low CO2 pressure and moderate reaction temperatures (1 bar and 40–120 °C depending on the substrate). Interestingly, the authors found that the catalytic activity of their polymers was dependent on the in situ formation of HCO3− anions as well as the formation of stable pyridyl radicals.100 Featuring abundant and readily accessible precursors, Yavuz et al. reported an imidazolium-based POPs with exceptional catalytic performance. The charged polymer obtained from the condensation of terephthalaldehyde with ammonium acetate showed high activity for the fixation of CO2 even for challenging substrates, thus showcasing the applicability of their catalyst.101
| Sample | SABETa (m2 g−1) | CO2 at 273 K (mmol g−1) | CO2 at 298 K (mmol g−1) | CO2Qst (kJ mol−1) | CO2/N2 selectivityb | Application | Ref. |
|---|---|---|---|---|---|---|---|
| a Surface area is calculated based on Brunauer–Emmett–Teller (BET) theory. b The CO2/N2 selectivity calculated using IAST (ideal adsorbed solution theory) for the flue gas mixture, CO2/N2:10/90 (v/v) at 298 K. | |||||||
| NPOF-4 | 1249 | 2.50 | 1.40 | 23.2 | 27 | Gas sorption | 65 |
| NPOF-4-NO2 | 337 | 2.42 | 1.56 | 32.5 | 139 | Gas sorption | 65 |
| NPOF-4-NH2 | 554 | 2.90 | 1.88 | 30.1 | 101 | Gas sorption | 65 |
| Azo-COP-1 | 635.8 | 2.44 | 1.48 | 29.3 | 63.7 | Gas sorption | 23 |
| Azo-COP-2 | 729.6 | 2.55 | 1.53 | 24.8 | 109.6 | Gas sorption | 23 |
| Azo-COP-3 | 493.1 | 1.93 | 1.22 | 32.1 | 78.6 | Gas sorption | 23 |
| Azo-COP-4 | 11.1 | 1.75 | 1.12 | 26.8 | 79.3 | Gas sorption | 23 |
| Azo-COP-5 | 127.6 | 2.04 | 1.24 | 27.3 | 73.8 | Gas sorption | 23 |
| Azo-COP-6 | 679.1 | 2.22 | 1.31 | 25.8 | 72.7 | Gas sorption | 23 |
| Azo-COP-7 | 241.6 | 1.91 | 1.16 | 26.1 | 68.5 | Gas sorption | 23 |
| Azo-COP-8 | 472.1 | 2.02 | 1.22 | 25.3 | 70.3 | Gas sorption | 23 |
| Azo-COP-9 | 649.5 | 2.05 | 1.23 | 25.3 | 69.6 | Gas sorption | 23 |
| Azo-COP-10 | 200.2 | 1.91 | 1.15 | 27.9 | 69.6 | Gas sorption | 23 |
| Azo-COP-11 | 336.1 | 2.13 | 1.26 | 27.4 | 77.7 | Gas sorption | 23 |
| PAF-1 | 4100 | 2.46 | — | — | — | Gas sorption | 67 |
| PAF-1-CH3 | 3007 | 2.45 | — | — | — | Gas sorption | 67 |
| PAF-1-CH2OH | 1727 | 2.98 | — | — | — | Gas sorption | 67 |
| PAF-1-phthalimide | 974 | 2.23 | — | — | — | Gas sorption | 67 |
| PAF-1-CH2NH2 | 1263 | 4.37 | — | — | — | Gas sorption | 67 |
| PAF-1-CH2N = CMe2 | 1302 | 3.12 | — | — | — | Gas sorption | 67 |
| 2D-PTNS | 690 | 3.11 | — | 30.7 | 93 | Gas sorption | 68 |
| PNOP-1 | 830 | 4.00 | 2.42 | 30.0 | 52.1 | Gas sorption | 76 |
| PNOP-2 | 729 | 3.04 | 1.96 | 31.3 | 80.1 | Gas sorption | 76 |
| 3D-tPOP | 22 | 2.38 | — | — | — | Gas sorption | 78 |
| 3D-tPOP-NaCl-2.0 | 1058 | 4.42 | 2.62 | 42.1 | — | Gas sorption | 78 |
| CXF1-OMe | 626 | 1.49 | — | 26.0 | — | Gas sorption | 89 |
| CFX1-OH | 540 | 2.20 | 1.5 | 35.0 | 98 | Gas sorption | 89 |
| co-CXF3-OPr | 2609 | 2.40 | — | 23.2 | — | Gas sorption | 89 |
| co-CXF3-OH | 1943 | 2.91 | — | 30.0 | — | Gas sorption | 89 |
| TMP1 | 923 | 3.50 | 2.04 | 22.2 | 64 | Gas sorption | 69 |
| TMP2 | 1094 | 3.70 | 2.18 | 21.1 | 60 | Gas sorption | 69 |
| TMP3 | 1372 | 5.07 | 3.27 | 22.0 | 70 | Gas sorption | 69 |
| TNHCP1 | 848 | 2.89 | 2.20 | 30.8 | 30 | Gas sorption | 70 |
| TNHCP2 | 766 | 3.64 | 2.11 | 32.8 | 42 | Gas sorption | 70 |
| TNHCP3 | 751 | 3.53 | 2.23 | 32.7 | 45 | Gas sorption | 70 |
| NTP | 1067 | 3.45 | 1.82 | 26 | 18 | Gas sorption | 71 |
| 3D-CON | 2247 | 6.07 | 3.90 | 31.87 | — | Gas sorption | 71 |
| PMDI-cage 1a | 522 | 2.91 | — | 27.0 | 26.9 | Gas sorption | 80 |
| Cg-Am | 104 | 0.89 | — | — | — | CO2 conversion | 82 |
| Cage-1 | — | 0.10 | — | — | 138 | Gas sorption | 83 |
| F1 | — | 0.19 | — | — | 64 | Gas sorption | 79 |
| F2 | — | 0,23 | — | — | 42 | Gas sorption | 79 |
| F3 | — | 0,16 | — | — | 213 | Gas sorption | 79 |
| Noria | 221 | 1.77 | — | 30.4 | — | CO2 conversion | 87 |
| NPOP | 773 | 2.09 | 1.36 | 28.9 | 30 | CO2 conversion | 87 |
| HRN4 | 156 | 1.39 | — | — | — | CO2 conversion | 88 |
| RN4-Az-OH | 340 | 2.04 | — | 30.8 | 19 | CO2 conversion | 88 |
| RN4-OH | 720 | 2.25 | — | 29.5 | 20 | CO2 conversion | 88 |
| RN4-F | 1230 | 2.59 | — | 28.5 | 35 | CO2 conversion | 88 |
| cCTF-400 | 744 | 2.86 | 1.89 | 49 | — | CO2 conversion | 98 |
| cCTF-450 | 861 | 2.25 | 1.41 | 46 | — | CO2 conversion | 98 |
| cCTF-500 | 1247 | 3.02 | 1.82 | 43 | — | CO2 conversion | 98 |
| Py-iPOP-1 | 65 | 0.82 | 0.55 | — | — | CO2 conversion | 100 |
| Name | Catalytic site | Metal (wt%) | Coordination ligand site | Preparation method | BET (m2 g−1) | Product | Yield (%) | Conversion condition | TON | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a POPs are employed using high-pressure solvothermal reaction for CO2 conversion. | |||||||||||
| Ir/AP-POPa | Ir cluster | 1.25 | Amide & pyridine moiety | Amide formation/wet impregnation, NaBH4 reduction | 43 | HCOOH | — | 3 MPa CO2 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 135 135 |
— | 115 |
| 3 MPa H2 | |||||||||||
| 80–140 °C | |||||||||||
| Ru@PP-POPa | RuCl3 | 0.81 | Phosphine moiety | Telomerisation of phosphine/wet impregnation | 469 | DMF | 98 | 80 MPa CO2 | 16 × 104 | 29000 |
121 |
| 140 °C | |||||||||||
| POMP-NHC-Ira | Ir+(CO)2BF4− | 2.89–16.12 | N-Heterocyclic carbene moiety | Fridel-craft reaction using bis-NHC-Ir complexes | 19–704 | DMF | 90 | 40 atm 120 °C | 1.58 × 106 | — | 118 |
| MeOH | |||||||||||
| Imine-POP@Pda | Pd NPs | 8.3 | Imine moiety | Hydrothermal imine formation/wet impregnation | 180 | DMF | 97 | 3 MPa CO2 | — | — | 122 |
| 3 MPa H2 | |||||||||||
| 100 °C, K3PO4 | |||||||||||
Metal-containing POPs for CO2 conversion
POPs are ideal hosts for various metal ions to form heterogeneous catalysts for CO2 conversion. First examples of metal-containing POPs were based on metal-complexing building blocks such as metalloporphyrins,102 metallophthalocyanines,103 and salenes.104,105 Whereas POPs incorporating these motifs were most often prepared via Friedel Crafts reactions or Schiff base chemistry, an alternative strategy is to prepare the desired coordination site in situ during the polymerization.106 A third way to introduce large amount of metal ions into POPs is wet impregnation with the desired metal ion, thus introducing the metal ions themselves or inducing the in situ formation of metal nanoparticles. Given suitable functionalization, the high surface areas and the large heteroatom content of POPs make them ideal candidates for the stabilization and distribution of metal ions within the polymer matrix, resulting in a high number of available catalytic active sites. Moreover, POPs can be endowed with single-atom active sites, that can be leveraged for high conversion and selectivity.In this regard, especially phthalocyanines and porphyrins have gained tremendous attention as their easy synthesis allows the introduction of task-specific metal ions. In the following section, we will summarize classical and modern synthetic approaches toward metal-containing POPs and discuss their capabilities as heterogeneous catalysts for the up-conversion of CO2.
Most metal-containing POP-based catalysts for CO2 conversion feature cheap and abundant metal ions/NPs such as Fe, Co, Zn, Mn, Cr, or Al in the oxidation states of either +2 or +3. These metal sites offer a high affinity towards CO2 and help polarize the substrate, a critical step in activating CO2, thus enabling the conversion of an otherwise inert molecule. There have also been various reports on homogeneous catalysts based on rare-earth and early transition metals due to the high Lewis acidity of derived coordination compounds,107 however, given the scarce nature and high cost of these metals, readily available transition metals present a more environmentally conscious and sustainable choice when designing POP-based catalysts.
Metalloporphyrin-based conjugated microporous polymers (M-CMPs)108 and hyper crosslinked polymers (M-HCPs)109 as well as the metal-salene conjugated microporous polymers (M-CMPs, M = Co-, Zn-)110 have been actively investigated for the conversion CO2 into cyclic carbonates. Among these POPs, those containing Co2+, Zn2+, and Fe2+ were reported to be active in the cycloaddition of CO2 to epoxides. A member of this family is the Friedel Crafts derived FePC-POP. The iron-containing phthalocyanine was obtained through a Friedel Crafts reaction with biphenylene.111 The obtained polymers showed moderate surface areas of up to 427 m2 g−1 and high iron content of 5.42 wt%. FePC-POP proved to be suitable for the cycloaddition of CO2 to epoxides showing yields up to 94% at 90 °C and 3 bars. To obtain such high conversions, the authors used DMAP as a coordinating base. Although the catalyst showed good conversion for simple epoxide substrates such as epichlorohydrin, more challenging ones such as styrene oxide or cyclohexane oxide showed significantly lower conversions. This was attributed to electronic reasons in the case of styrene oxide and steric hindrance in the case of cyclohexane oxide. Besides Friedel Crafts reactions, polycondensation reactions are among the most used methods to prepare porous single-atom catalysts. Utilizing an amine-functionalized Co-PC and a benzoquinone linker, Tang and coworkers prepared conjugated microporous polymers (DTTBQ-CMP) with good catalytic activity towards CO2 conversion under mild conditions (1 bar, 25–60 °C) reaching near quantitative yields at low temperature and low CO2 pressure in presence of a TBAB as a co-catalyst. Interestingly, in the case of a simple substrate such as propylene oxide, catalyst-free conversion could be obtained (1 bar, 25 °C), albeit at significantly lower yields. Recently, Jiang and coworkers utilized an amine-based triptycene linker and an aldehyde functionalized phenol to obtain bimetallic salen-based POPs (Fig. 9).112 Metal ions were introduced during the synthesis in the form of different metal salts (M = Al, Co or Ni) and the obtained heterogeneous catalysts showed good activity in the presence of a co-catalyst (TBAB, 25 °C, 1 bar). Triptycenes are often employed to increase the porosity as well as the accessibility of the catalytic sites. Despite showing high metal contents of 8.8, 15.0, and 16.9 wt% for Fe2+, Co2+, and Ni2+ respectively, the polymers showed poor CO2 conversion efficiency, which could be attributed to their relatively low surface areas (142–280 m2 g−1) and the low CO2 uptake capacities (0.74–1.70 mmol g−1). Among these systems, BSPOP-Co showed the highest heat of adsorption for CO2, indicating a good binding affinity of CO2, which together with having a higher surface area and increased accessibility of the catalytic sites compared to the other systems contributed to the overall better catalytic performance. Overall, such bi-metallic systems such as BSPOPs are of immense interest to the field as the two neighboring metal centers are expected to result in a synergistic effect, thus improving the catalytic performance.
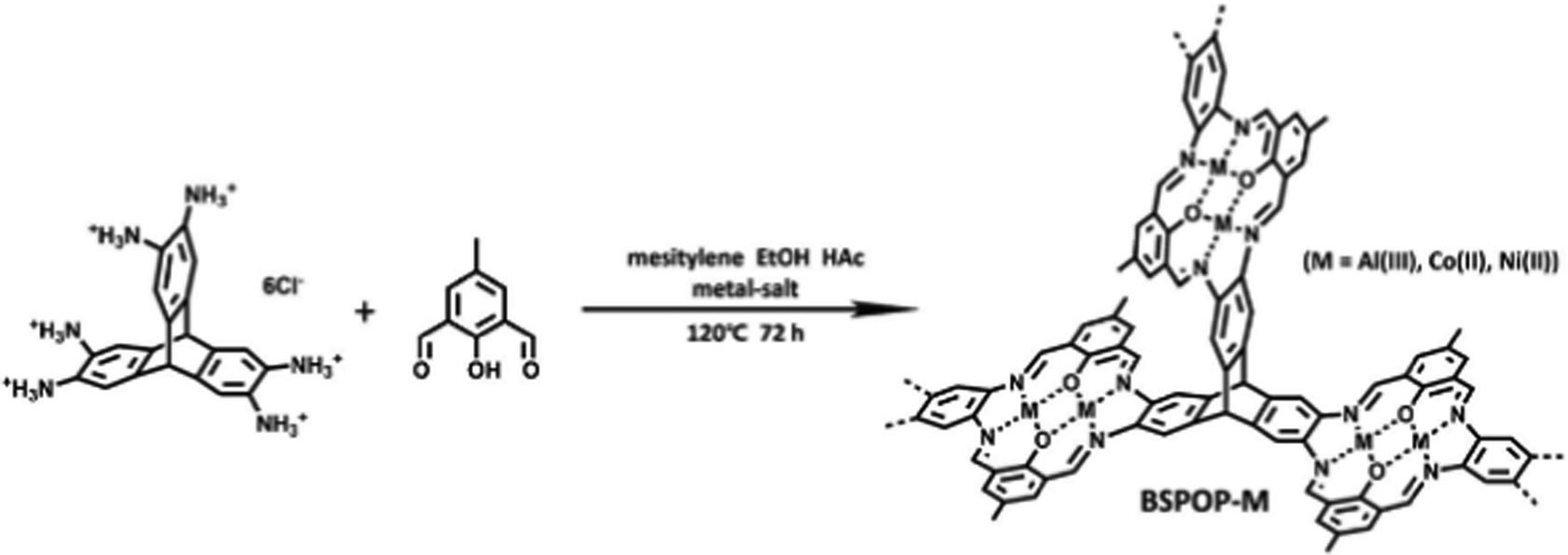 | ||
| Fig. 9 Precise positioning of metal ions in bimetallic POPs. Synthetic scheme of BSPOP-M. Reproduced from ref. 112 with permission of the publisher. | ||
When designing catalysts for the conversion of CO2 to cyclic carbonates not only a Lewis acidic site for the fixation/activation of CO2 is required but also a nucleophilic moiety is needed to facilitate the ring-opening of the epoxide. Accordingly, the design of such bifunctional active sites in POPs is a promising strategy to achieve high catalytic activity. In this direction, both the combination of two different metal ions within a single system or the generation of charged organic moieties (e.g. phosphonium or pyridyl moieties) are promising strategies. As such, a vinyl-functionalized Mg-porphyrin was co-polymerized with a vinyl-functionalized phosphonium salt utilizing free radical polymerization.113 Despite its relatively low surface area (558 m2 g−1), the polymer (Mg-por/pho@POP) showed good catalytic activity in the absence of a co-catalyst. Simple to moderately hindered epoxides ranging from propylene oxide to styrene oxide could be converted at 10–30 bar at 120 °C in moderate to good yields. This result was attributed to the ample amount of functional units endowing the system with Lewis acidic metal sites along with the presence of Br anions, which act as nucleophiles in the ring-opening of the epoxide.109 Ji et al. reported metallosalen-based ionic POPs via a radical polymerization of functionalized salphen-imidazolium precursors.114 Similar to the system reported by Wang et al.,113 a cooperative effect between the different metal ions (Al3+ and Zn2+) in combination with the Br anion of the imidazolium moiety could be observed, resulting in a good catalytic activity for the cycloaddition of CO2 to epoxides and also for the N-formylation of amines under mild conditions. Importantly, the integration of charged units bearing nucleophilic counter anions such as Cl− and Br− naturally eliminates the need for a co-catalyst.
CO2 conversion beyond cycloaddition
Besides the conversion of CO2 into cyclic carbonates, other chemical conversion pathways are also possible using metal-containing POPs (Table 3). Formic acid (HCOOH) as a product of CO2 fixation is considered a key chemical intermediate as it can be further converted into other chemicals (e.g. MeOH). From a molecular design point of view, electron-donating ligands (such as pyridyls or phosphines) are indispensable to provide stability and to modulate the electronic properties of the active site. Iridium and Ruthenium complexes are excellent examples for this type of chemistry and a variety of different supports based on various functional groups have been developed to stabilize such metal species. An example of such a system was recently reported by Zhang et al. who prepared an Ir-based single atom catalyst (Ir-POPs) via a post metalation process. The polymer showed capabilities to produce formic acid in a high-pressure solvothermal procedure.115 AP-POP was easily prepared by reacting diaminopyridine with 1,3,5-benzenetricarbonyl trichloride. The resulting alternating amide and pyridine moieties together with the mesoporous structure proved to be an ideal substrate for wet-impregnation with Ir salts, wherein the Ir3+ ions were coordinated to the carbonyls of the amides and the pyridine N moieties forming Ir/AP-POP (Fig. 10). Interestingly, spectroscopic analysis of Ir/AP-POP supported the formation of single metal ions with a homogeneous distribution of Ir3+ over the entire structure. Comparing their results to activated carbon (AC) or graphitic carbonitride (C3N4) treated in the same way revealed stark differences between the three systems. Whereas Ir/AP-POP showed Ir3+, Ir/AC and IR/C3N4 showed Ir(0) species and the formation of Ir nanoparticles of up to 2 nm in size. As a result, Ir/AP-POP exhibited higher CO2 conversion efficiency to formic acid compared to Ir/AC and Ir/C3N4 while also showing good recyclability. This result showcases that single-atom catalysts based on POPs can show high catalytic activity for CO2 conversion and the design of an appropriate chemical environment with multiple electron-donating groups can efficiently stabilize certain metal species while suppressing the formation of NPs. | ||
Fig. 10 Structure analysis of Ir/AP-POPs. Atomically dispersed and stable Ir metal atoms are analyzed. (A) The solid-state 13C/CP-MAS NMR spectrum of the AP-POP suggests the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–N, and C–Ar groups. (B) The N 1s core level XPS spectrum of the AP-POP distinguishes N in the pyridinic and amide groups in the framework. (C) The UV-vis absorbance spectra of samples with and without Ir indicate the p–p* electron transition of the conjugated polymer and show the supported Ir single atoms result in a red-shift of absorption. (D) An SEM image shows the spherical particles morphology of the AP-POP. (E and F) HAADF-STEM images of fresh (E and F) used 1.25% Ir/AP-POP indicate only the atomically dispersed Ir species on the support, even after the CO2 hydrogenation reaction. Reproduced from ref. 115 with permission of the publisher. O, C–N, and C–Ar groups. (B) The N 1s core level XPS spectrum of the AP-POP distinguishes N in the pyridinic and amide groups in the framework. (C) The UV-vis absorbance spectra of samples with and without Ir indicate the p–p* electron transition of the conjugated polymer and show the supported Ir single atoms result in a red-shift of absorption. (D) An SEM image shows the spherical particles morphology of the AP-POP. (E and F) HAADF-STEM images of fresh (E and F) used 1.25% Ir/AP-POP indicate only the atomically dispersed Ir species on the support, even after the CO2 hydrogenation reaction. Reproduced from ref. 115 with permission of the publisher. | ||
Another industrially important product is N,N-dimethylformamide (DMF), which can be prepared in a base-catalyzed process via an N-formylation of CO with dimethylamine. A more environmentally friendly method of preparing DMF would be to replace CO with CO2. The first catalytic systems capable of this were reported in the 1970s, when Kohnle et al. prepared various metal complexes based on Co, Rh, Ir, Pd, and Ru.116 Following this, various homogeneous catalysts were developed and employed in the synthesis of DMF using CO2 however, in many cases, supercritical media at high pressure (e.g. 130 atm CO2) were required. To circumvent the necessity of such high pressures, heterogeneous catalysts based on POPs were investigated.
Yoon et al. reported a Ru-grafted bisphosphine-based POPs (PP-POPs) via a post metalation procedure utilizing methanolic RuCl3, wherein the Ru-ions are stabilized by the phosphine-backbone. This approach allowed the authors to perform the hydrogenation of CO2 at 80 atm and 140 °C with a TON of up to 160000. Additionally, they showed that the catalyst can be employed industrially in a continuous flow reactor thus showcasing the high potential of Ru-containing POPs.117
N-Heterocyclic carbenes (NHCs) are commonly used as ligands for homogeneous catalysts, where the derived catalysts show some of the highest activities. Bringing NHCs to heterogeneous POPs, Tu et al. prepared an Ir-containing NHC polymer (POMP-NHC-IR) via direct knitting of a robust bis-NHC-Ir complex via a Friedel-Crafts reaction and showcased its utilization towards the N-formylation of amines with CO2 and H2.118 In a previous work, the authors demonstrated that a NHC-Ir catalyst showed excellent catalytic activity. However, the non-porous nature of the linear polymer resulted in a rather low TON of only 730.119 Therefore, they speculated, that a porous matrix could accelerate the N-formylation process due to the well-defined pore-structure and better interactions between the gases. Indeed, their POMP-NHC-Ir catalyst featuring Ir3+ sites resulted in high yield and high selectivity (>99%). Interestingly, the highest yield and selectivity were obtained for the systems with the highest surface areas but with the lowest Ir content and not the systems with the lowest surface area but the highest Ir content, implying that pore and active site accessibility is of tremendous importance when designing highly active catalysts. Moreover, heterogenous POMP-NHC-Ir showed much higher catalytic activity than its corresponding homogenous counterpart (bis-NHC-Ir complex). Remarkably, POMP-NHC-IR showed a record TON (1.58 × 106) in the DMF synthesis, and the solid POMP-NHC-Ir catalysts could be reused for up to 12 runs without obvious losses of its catalytic activity or selectivity even at extremely low catalyst loadings of 20 ppm, highlighting their potential in industrial applications. When it comes to catalytically active single metal site catalysts based on multiple abundant transition metal ions (Cu, Ni or Mn) their interplay is highly important. Zang et al. reported POPs bearing two different catalytic sites by preparing an amine-functionalized Ni–PC that was subsequently reacted with an aldehyde functionalized phenol to obtain the corresponding Ni–PC-salphen hybrid (NiPc-MPOP, M = Ni or Co). The mixed metal system NiPc-CoPOP proved to be an ideal catalyst for the photocatalytic reduction of CO2 to CO (Fig. 11).120 This can be attributed to the well-defined coordination environment of the different metal ions and the availability of multiple active sites. Interestingly the authors were also able to show that the Ni-salphen active site in the NiPc-NiPOP had a higher catalytic activity than the NiPC system for the reduction of CO2 (CO2RR).
 | ||
| Fig. 11 Synthesis example of incorporating two different single metal atom species in porous organic polymers. Stepwise synthesis of Ni-Pc-MPOP by condensing of (NH2)8NiPc and DFP. Reproduced from ref. 120 with permission of the publisher. | ||
Outlook and perspective
Porous organic polymers offer a versatile platform to specifically design and tune polymers towards their desired applications while offering key features such as high stability and porosity. Although a plethora of porous systems offer some sort of capability to store gases such as CO2 due to their heteroatom content or their intrinsic microporosity, proper design allows to directly use POPs as sorbents and organocatalysts for the capture and conversion of the CO2 toward value-added products. Among such products, especially cyclic carbonates have emerged as a common target of chemical conversions due to their relative ease of synthesis, abundant precursors and ample opportunities to use the obtained products as organic solvents, precursors in the synthesis of small molecules and polymers, or as electrolytes in batteries. We believe that it is imperative to focus the design of new POPs to directly capture and convert CO2 in order to have an impact on climate change. Taking inspiration from the best performing systems in literature it is clear that molecular design is key. In fully organic systems a suitably designed catalyst can facilitate the uptake and direct conversion of CO2. Namely, a combination of Lewis acidic or basic sites that polarize CO2 in combination with functional units that enable a nucleophilic attack on e.g. an epoxide substrate should be aimed for. This can be achieved by preparing differently functionalized building blocks and polymerizing them. Although it has been shown that such motifs can work, the spatial separation of the two active sites onto different building blocks can be a problem in rigid, more stable systems. Hence, designing systems to have both functional units in proximity should be prioritized. Additionally, creative molecular design can also allow the generation of charged moieties on the polymer backbone during the catalytic reaction, leading to additional stability since no more labile ionic moieties have to be incorporated directly. Compared to their fully organic counterparts, metal-containing POPs have the advantage, that the metal-complexing moiety directly acts as a Lewis acidic, CO2-philic site and hence only a suitable nucleophile-bearing or stabilizing functional unit is required. Despite that, many metal-containing systems rely on co-catalyst such as TBAB to supply a nucleophile or utilize bases such as DMAP to facilitate substrate activation. In any case, judicious control over porosity, pore size, and textural properties, in general, are of utmost importance to facilitate good mass transfer properties as these will directly influence how CO2 and substrates can interact. This is especially true for systems that require bulky co-catalysts such as TBAB to facilitate the nucleophilic attack on epoxide substrates.Operating conditions reported in literature vary drastically in terms of catalyst amount (wt% vs. mol% or active site vs. total amount), use and amount of co-catalysts, temperature, reaction times, and CO2 pressure. Although this is understandable as different systems offer varying performances under different conditions, as a community we should strive to always test a set of benchmark conditions (e.g. 5 mmol substrate, 5% catalyst, none/5% co-catalyst, 80 °C, 24 h, 1 bar CO2) to enable our peers to more easily compare the performance of POPs and to draw conclusions about the molecular design based on these performance parameters. These issues are often amplified because standard characteristics like TON or TOF are only occasionally discussed in systems where active sites can be easily determined (e.g. active metal sites based on the metal content of a polymer). Besides the operating conditions, recyclability tests are also subject of much ambiguity. The catalysts are often not thoroughly investigated in terms of their recyclability and in many cases, conditions are only described sparingly. This is especially important for metal-containing systems, where different binding motifs result in varying amounts of metal loss. Although some reports in the literature address these points by evaluating the total metal content of the catalysts after each cycle presenting such data might lead to scrutiny during the publication process. Nonetheless, in order to make an impact in the field and on our society, such data is required. Another critical point lies in how such potentially useful polymers are produced. Although the community has shown a continued drive toward more sustainable protocols and even environmentally friendly methods, a plethora of materials are produced with sustainability as an afterthought. Considering that the entire field of carbon capture and conversion is dedicated toward reducing humankinds’ impact on the environment this is a surprising and worrying trend.
To that end, computational materials design can be an exceptional tool in the repertoire of chemists. Not only to reduce the endless amount of potential candidate polymers but also to help optimize reaction conditions through statistical analysis. However, one major culprit of such computational investigations lies in the availability and inhomogeneity of data – the lack of clearly defined benchmark conditions being just one of them – thus making such approaches even more work-intensive, while reducing the potential impact of such investigations.
Especially for industrial purposes the cost of production is a key that is especially hard to assess from an academic point of view. On the one hand a clear differentiation between systems of purely academic interest and systems that are more suitable for industry has to be made. Let's take cage-based polymers as an example. Although such systems can provide good CO2 uptake and, in some cases, even conversion capabilities, their often complex synthetic requirements would make industrial implementation more unfeasible compared to polymers of lower performance but significantly easier synthesis such as functionalized porous melamine formaldehyde resins. That being said, systems such as cage-based polymers or metalated POPs provide invaluable information by studying their interaction with both CO2 and potential substrates due to their defined cavities and distinct metal sites. Taking the recent work by Reimer et al. as an example45 it is clear that POPs can be produced on a kilogram scale – even under lab conditions – while obtaining highly functional polymers with exceptional CO2 uptake at low relative pressures. Especially under these conditions only few systems can outperform their melamine-based POPs; among them are amine scrubbers such as MEA. These amine scrubbers however come at the cost of significantly more energy-intensive purification/regeneration, whereas the shown POPs were easily regenerated and used for several cycles. One should also avoid precious metal catalysts and complex syntheses without sacrificing the performance in order to reach the cost target of 50$ per tonne of CO2.
Conclusions
Within this review article, we have highlighted the key parameters governing CO2 uptake, separation, selectivity and conversion in POPs. The wide range of available building blocks and the vast amount of feasible, industrially relevant syntheses endows POPs with a degree of tunability unlike any other class of materials. The fact that both the pore structure and the chemical environment within the pores can be tuned towards specific applications allows the preparation of highly task-specific polymers. Although most POPs show some capabilities towards CO2 uptake, the importance of specific moieties in POPs for high CO2 uptake and direct conversion must be highlighted. We have presented a plethora of suitable design strategies based on (i) high heteroatom content, (ii) intrinsically porous molecular precursors and (iii) metal-containing systems. Whereas heteroatom-rich systems and systems based on porosity-inducing precursors still often rely on co-catalysts for the conversion of CO2 to value-added products, special design strategies based on charged building blocks or precisely tuned active moieties allow the conversion of CO2 without the need of co-catalysts. POPs incorporating homogenously dispersed metal ions are of special importance as they enable a variety of industrially relevant applications and provide a suitable coordination environment within the system. For these systems, an in-depth understanding of the nature of catalytic sites would be highly beneficial in order to establish a correlation between the structure and the catalytic activity.Overall, POPs are an intriguing platform for CO2 capture, separation, and conversion. However, even though a plethora of POPs have been reported for CO2 uptake, only few have been explored for the conversion of CO2 towards value-added products. With this contribution, we want to stimulate the community towards the design of new POPs aimed at CO2 capture and conversion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was created as part of NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation.References
- E. S. R. L. G. M. Laboratory, Trends in Atmospheric Carbon Dioxide, https://gml.noaa.gov/ccgg/trends/mlo.html, (accessed 28.03.2022, 2022).
- C. Marchetti, Clim. Change, 1977, 1, 59–68 CrossRef CAS.
- A. Basile, A. Gugliuzza, A. Iulianelli and P. Morrone, Advanced Membrane Science and Technology for Sustainable Energy and Environmental Applications, Woodhead Publishing, 2011, pp. 113–159 Search PubMed.
- L. Riboldi and O. Bolland, Energy Procedia, 2017, 114, 2390–2400 CrossRef CAS.
- C. Yang, Y. Xiong, J. Chen, J. Jin and J. Mi, Chem. Eng. J., 2021, 420, 129555 CrossRef CAS.
- A. Andersen, S. Divekar, S. Dasgupta, J. H. Cavka, A. Aarti, A. Nanoti, A. Spjelkavik, A. N. Goswami, M. O. Garg and R. Blom, Energy Procedia, 2013, 37, 33–39 CrossRef CAS.
- H. Yan, Q. Fu, Y. Zhou, D. Li and D. Zhang, Int. J. Greenhouse Gas Control, 2016, 51, 1–10 CrossRef CAS.
- N. Jiang, Y. Shen, B. Liu, D. Zhang, Z. Tang, G. Li and B. Fu, J. CO2 Util., 2020, 35, 153–168 CrossRef CAS.
- J. He, S. Deng, L. Zhao, R. Zhao and S. Li, Energy Procedia, 2017, 142, 3200–3207 CrossRef CAS.
- L. Joss, M. Gazzani and M. Mazzotti, Chem. Eng. Sci., 2017, 158, 381–394 CrossRef CAS.
- C. Font-Palma, D. Cann and C. Udemu, C, 2021, 7, 58 CAS.
- V. Alvarado and E. Manrique, Energies, 2010, 3, 1529–1575 CrossRef.
- Climeworks, https://climeworks.com, (accessed 29.03.2022).
- B. Du, M. C. Tandoc, M. L. Mack and J. A. Siegel, Indoor Air, 2020, 30, 1067–1082 CrossRef CAS.
- A. Samanta, A. Zhao, G. K. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- J. Rouquerol, D. Avnir, C. Fairbridge, D. Everett, J. Haynes, N. Pernicone, J. Ramsay, K. Sing and K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- C. Xu, G. Yu, J. Yuan, M. Strømme and N. Hedin, Mater. Today Adv., 2020, 6, 100052 CrossRef.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith and S. Yuan, Adv. Mater., 2017, 29, 1700229 CrossRef.
- H. A. Patel, S. H. Je, J. Park, Y. Jung, A. Coskun and C. T. Yavuz, Chem. – Eur. J., 2014, 20, 772–780 CrossRef CAS PubMed.
- Sang H. Je, O. Buyukcakir, D. Kim and A. Coskun, Chem, 2016, 1, 482–493 CAS.
- O. Buyukcakir, Y. Seo and A. Coskun, Chem. Mater., 2015, 27, 4149–4155 CrossRef CAS.
- N. Buli, K. Abnett and S. Twidale, 2021, https://www.reuters.com/business/energy/eu-carbon-price-tops-50-euros-first-time-2021-05-04/.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- A. K. Sekizkardes, P. Wang, J. Hoffman, S. Budhathoki and D. Hopkinson, Mater. Adv., 2022, 3, 6668–6686 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef CAS.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- S. Hug, L. Stegbauer, H. Oh, M. Hirscher and B. V. Lotsch, Chem. Mater., 2015, 27, 8001–8010 CrossRef CAS.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS PubMed.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684–3692 RSC.
- Y. Fu, Z. Wang, S. Li, X. He, C. Pan, J. Yan and G. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 36002–36009 CrossRef CAS.
- H. S. Jena, C. Krishnaraj, J. Schmidt, K. Leus, K. Van Hecke and P. Van Der Voort, Chem. – Eur. J., 2020, 26, 1548–1557 CrossRef CAS PubMed.
- G. Wang, K. Leus, H. S. Jena, C. Krishnaraj, S. Zhao, H. Depauw, N. Tahir, Y.-Y. Liu and P. Van Der Voort, J. Mater. Chem. A, 2018, 6, 6370–6375 RSC.
- S. Mukherjee, M. Das, A. Manna, R. Krishna and S. Das, Chem. Mater., 2019, 31, 3929–3940 CrossRef CAS.
- S. Mukherjee, M. Das, A. Manna, R. Krishna and S. Das, J. Mater. Chem. A, 2019, 7, 1055–1068 RSC.
- Z. Yang, S. Wang, Z. Zhang, W. Guo, K. Jie, M. I. Hashim, O. Š. Miljanić, D.-E. Jiang, I. Popovs and S. Dai, J. Mater. Chem. A, 2019, 7, 17277–17282 RSC.
- O. Buyukcakir, R. Yuksel, Y. Jiang, S. H. Lee, W. K. Seong, X. Chen and R. S. Ruoff, Angew. Chem., Int. Ed., 2019, 58, 872–876 CrossRef CAS PubMed.
- S. Zulfiqar, M. I. Sarwar and C. T. Yavuz, RSC Adv., 2014, 4, 52263–52269 RSC.
- R. J. Krupadam and S. S. Rayalu, Emergent Mater., 2021, 4, 545–563 CrossRef CAS.
- H. Mao, J. Tang, G. S. Day, Y. Peng, H. Wang, X. Xiao, Y. Yang, Y. Jiang, S. Chen and D. M. Halat, Sci. Adv., 2022, 8, eabo6849 CrossRef CAS PubMed.
- G. Li, X. Zhou and Z. Wang, J. Phys. Chem. C, 2020, 124, 3087–3094 CrossRef CAS.
- K. S. Song, S. N. Talapaneni, T. Ashirov and A. Coskun, ACS Appl. Mater. Interfaces, 2021, 13, 26102–26108 CrossRef CAS PubMed.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511–1517 CrossRef CAS.
- S. Altarawneh, S. Behera, P. Jena and H. M. El-Kaderi, Chem. Commun., 2014, 50, 3571–3574 RSC.
- M. G. Rabbani, T. Islamoglu and H. M. El-Kaderi, J. Mater. Chem. A, 2017, 5, 258–265 RSC.
- H. A. Patel, D. Ko and C. T. Yavuz, Chem. Mater., 2014, 26, 6729–6733 CrossRef CAS.
- B. Baumgartner, M. Puchberger and M. M. Unterlass, Polym. Chem., 2015, 6, 5773–5781 RSC.
- M. Lahnsteiner, M. Caldera, H. M. Moura, D. A. Cerrón-Infantes, J. Roeser, T. Konegger, A. Thomas, J. Menche and M. M. Unterlass, J. Mater. Chem. A, 2021, 9, 19754–19769 RSC.
- S.-C. Qi, G.-X. Yu, D.-M. Xue, X. Liu, X.-Q. Liu and L.-B. Sun, Chem. Eng. J., 2020, 385, 123978 CrossRef CAS.
- D.-M. Xue, W.-J. Zhang, X.-Q. Liu, S.-C. Qi and L.-B. Sun, Chin. J. Chem. Eng., 2022, 43, 24–30 CrossRef.
- Y. Jiang, P. Tan, S. C. Qi, X. Q. Liu, J. H. Yan, F. Fan and L. B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS PubMed.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- S. K. Kundu and A. Bhaumik, ACS Sustain. Chem. Eng., 2016, 4, 3697–3703 CrossRef CAS.
- N. Das, R. Paul, D. Q. Dao, R. Chatterjee, K. Borah, S. Chandra Shit, A. Bhaumik and J. Mondal, ACS Appl. Nano Mater., 2022, 5, 5302–5315 CrossRef CAS.
- R. Bera, M. Ansari, A. Alam and N. Das, ACS Appl. Polym. Mater., 2019, 1, 959–968 CrossRef CAS.
- S.-H. Jia, X. Ding, H.-T. Yu and B.-H. Han, RSC Adv., 2015, 5, 71095–71101 RSC.
- S. Luo, Q. Zhang, Y. Zhang, K. P. Weaver, W. A. Phillip and R. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 15174–15182 CrossRef CAS PubMed.
- A. Alam, S. Mishra, A. Hassan, R. Bera, S. Dutta, K. Das Saha and N. Das, ACS Omega, 2020, 5, 4250–4260 CrossRef CAS PubMed.
- V. Rozyyev, M. S. Yavuz, D. Thirion, T. S. Nguyen, T. P. N. Nguyen, A.-H. Emwas and C. T. Yavuz, Micropor. Mesopor. Mat., 2021, 328, 111450 CrossRef CAS.
- T. İslamoğlu, M. G. Rabbani and H. M. El-Kaderi, J. Mater. Chem. A, 2013, 1, 10259–10266 RSC.
- M. W. Hussain, S. Bandyopadhyay and A. Patra, Chem. Commun., 2017, 53, 10576–10579 RSC.
- S. J. Garibay, M. H. Weston, J. E. Mondloch, Y. J. Colón, O. K. Farha, J. T. Hupp and S. T. Nguyen, CrystEngComm, 2013, 15, 1515–1519 RSC.
- J.-J. Chen, T.-L. Zhai, Y.-F. Chen, S. Geng, C. Yu, J.-M. Liu, L. Wang, B. Tan and C. Zhang, Polym. Chem., 2017, 8, 5533–5538 RSC.
- R. Bera, S. Mondal and N. Das, Micropor. Mesopor. Mat., 2018, 257, 253–261 CrossRef CAS.
- A. Hassan, S. Goswami, A. Alam, R. Bera and N. Das, Sep. Purif. Technol., 2021, 257, 117923 CrossRef CAS.
- H. Ma, J.-J. Chen, L. Tan, J.-H. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS PubMed.
- A. Alam, R. Bera, M. Ansari, A. Hassan and N. Das, Front. Energy Res., 2019, 7, 141 CrossRef.
- S. Y. Yu, J. Mahmood, H. J. Noh, J. M. Seo, S. M. Jung, S. H. Shin, Y. K. Im, I. Y. Jeon and J. B. Baek, Angew. Chem., Int. Ed., 2018, 57, 8438–8442 CrossRef CAS PubMed.
- A. K. Sekizkardes, S. Hammache, J. S. Hoffman and D. Hopkinson, ACS Appl. Mater. Interfaces, 2019, 11, 30987–30991 CrossRef CAS PubMed.
- P. Jorayev, I. Tashov, V. Rozyyev, T. S. Nguyen, N. A. Dogan and C. T. Yavuz, ChemSusChem, 2020, 13, 6433–6441 CAS.
- J. Yan, B. Zhang, S. Guo and Z. Wang, ACS Appl. Nano Mater., 2021, 4, 10565–10574 CrossRef CAS.
- V. S. P. K. Neti, J. Wang, S. Deng and L. Echegoyen, J. Mater. Chem. A, 2015, 3, 10284–10288 RSC.
- T. Ashirov, K. S. Song and A. Coskun, ACS Appl. Nano Mater., 2022, 5(10), 13711–13719 CrossRef CAS.
- Y. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS PubMed.
- H.-H. Huang, K. S. Song, A. Prescimone, A. Aster, G. Cohen, R. Mannancherry, E. Vauthey, A. Coskun and T. Šolomek, Chem. Sci., 2021, 12, 5275–5285 RSC.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- M. W. Hussain, A. Giri and A. Patra, Sustainable Energy Fuels, 2019, 3, 2567–2571 RSC.
- Y. Jin, B. A. Voss, R. McCaffrey, C. T. Baggett, R. D. Noble and W. Zhang, Chem. Sci., 2012, 3, 874–877 RSC.
- A. Giri, A. Sahoo, T. K. Dutta and A. Patra, ACS Omega, 2020, 5, 28413–28424 CrossRef CAS PubMed.
- S. N. Talapaneni, D. Kim, G. Barin, O. Buyukcakir, S. H. Je and A. Coskun, Chem. Mater., 2016, 28, 4460–4466 CrossRef CAS.
- T. Ogoshi, Y. Shimada, T. Akutsu and T.-A. Yamagishi, Polymer, 2017, 128, 325–329 CrossRef CAS.
- A. Giri, N. N. Patil and A. Patra, Chem. Commun., 2021, 57, 4404–4407 RSC.
- A. Giri, M. W. Hussain, B. Sk and A. Patra, Chem. Mater., 2019, 31, 8440–8450 CrossRef CAS.
- A. Pedrini, J. Perego, S. Bracco, C. X. Bezuidenhout, P. Sozzani and A. Comotti, J. Mater. Chem. A, 2021, 9, 27353–27360 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- F. Ouhib, L. Meabe, A. Mahmoud, B. Grignard, J.-M. Thomassin, F. Boschini, H. Zhu, M. Forsyth, D. Mecerreyes and C. Detrembleur, ACS Appl. Polym. Mater., 2020, 2, 922–931 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed.
- Z. Yi, J. Zhang, S. Zhang, Q. Gao, J. Li and W. Zhang, Polymers, 2016, 8, 159 CrossRef PubMed.
- P. Rollin, L. K. Soares, A. M. Barcellos, D. R. Araujo, E. J. Lenardão, R. G. Jacob and G. Perin, Appl. Sci., 2021, 11, 5024 CrossRef CAS.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- S. Subramanian, J. Park, J. Byun, Y. Jung and C. T. Yavuz, ACS Appl. Mater. Interfaces, 2018, 10, 9478–9484 CrossRef CAS PubMed.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- K. Liu, Z. Xu, H. Huang, Y. Zhang, Y. Liu, Z. Qiu, M. Tong, Z. Long and G. Chen, Green Chem., 2022, 24, 136–141 RSC.
- S. Subramanian, J. Oppenheim, D. Kim, T. S. Nguyen, W. M. H. Silo, B. Kim, W. A. Goddard and C. T. Yavuz, Chem, 2019, 5, 3232–3242 CAS.
- S. Kumar, M. Y. Wani, C. T. Arranja, J. D. A. e Silva, B. Avula and A. J. Sobral, J. Mater. Chem. A, 2015, 3, 19615–19637 RSC.
- W. Ji, T.-X. Wang, X. Ding, S. Lei and B.-H. Han, Coord. Chem. Rev., 2021, 439, 213875 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- S. Bhunia, R. A. Molla, V. Kumari, S. M. Islam and A. Bhaumik, Chem. Commun., 2015, 51, 15732–15735 RSC.
- S. Kramer, N. R. Bennedsen and S. Kegnæs, ACS Catal., 2018, 8, 6961–6982 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D’Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- X. Sheng, H. Guo, Y. Qin, X. Wang and F. Wang, RSC Adv., 2015, 5, 31664–31669 RSC.
- S. Wang, K. Song, C. Zhang, Y. Shu, T. Li and B. Tan, J. Mater. Chem. A, 2017, 5, 1509–1515 RSC.
- Y. Xie, T.-T. Wang, R.-X. Yang, N.-Y. Huang, K. Zou and W.-Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- E. M. Maya, A. Valverde-González and M. Iglesias, Molecules, 2020, 25, 4598 CrossRef CAS PubMed.
- Y. Zheng, X. Wang, C. Liu, B. Yu, W. Li, H. Wang, T. Sun and J. Jiang, Inorg. Chem. Front., 2021, 8, 2880–2888 RSC.
- W. Wang, Y. Wang, C. Li, L. Yan, M. Jiang and Y. Ding, ACS Sustain. Chem. Eng., 2017, 5, 4523–4528 CrossRef CAS.
- R. Luo, Y. Chen, Q. He, X. Lin, Q. Xu, X. He, W. Zhang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 1526–1533 CrossRef CAS PubMed.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
- P. Haynes, L. H. Slaugh and J. F. Kohnle, Tetrahedron Lett., 1970, 11, 365–368 CrossRef.
- G. H. Gunasekar, S. Padmanaban, K. Park, K.-D. Jung and S. Yoon, ChemSusChem, 2020, 13, 1735–1739 CrossRef CAS PubMed.
- Y. Shen, Q. Zheng, Z. N. Chen, D. Wen, J. H. Clark, X. Xu and T. Tu, Angew. Chem., Int. Ed., 2021, 60, 4125–4132 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, H. Zhu and T. Tu, Chem. – Asian J., 2018, 13, 3018–3021 CrossRef CAS PubMed.
- X. Y. Dong, Y. N. Si, Q. Y. Wang, S. Wang and S. Q. Zang, Adv. Mater., 2021, 33, 2101568 CrossRef CAS PubMed.
- G. H. Gunasekar, S. Padmanaban, K. Park, K. D. Jung and S. Yoon, ChemSusChem, 2020, 13, 1735–1739 CrossRef CAS PubMed.
- X. Yu, Z. Yang, S. Guo, Z. Liu, H. Zhang, B. Yu, Y. Zhao and Z. Liu, Chem. Commun., 2018, 54, 7633–7636 RSC.
Footnote |
| † These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2022 |