
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of nanotechnology-mediated precision radiotherapy for anti-metastasis and radioprotection
Yuanbo
Pan
 abcf,
Wei
Tang
e,
Wenpei
Fan
*d,
Jianmin
Zhang
*abc and
Xiaoyuan
Chen
*fghi
abcf,
Wei
Tang
e,
Wenpei
Fan
*d,
Jianmin
Zhang
*abc and
Xiaoyuan
Chen
*fghi
aDepartment of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, 310009, China. E-mail: zjm135@zju.edu.cn
bKey Laboratory of Precise Treatment and Clinical Translational Research of Neurological Diseases, Hangzhou, 310009, Zhejiang, China
cClinical Research Center for Neurological Diseases of Zhejiang Province, Hangzhou, 310009, China
dState Key Laboratory of Natural Medicines and Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University, Nanjing, 210009, China. E-mail: wenpei.fan@cpu.edu.cn
eDepartments of Pharmacy and Diagnostic Radiology, Nanomedicine Translational Research Program, Faculty of Science and Yong Loo Lin School of Medicine, National University of Singapore, Singapore, 117544, Singapore
fDepartments of Diagnostic Radiology, Surgery, Chemical and Biomolecular Engineering, and Biomedical Engineering, Yong Loo Lin School of Medicine and College of Design and Engineering, National University of Singapore, Singapore 119074, Singapore. E-mail: chen.shawn@nus.edu.sg
gClinical Imaging Research Centre, Centre for Translational Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117599, Singapore
hNanomedicine Translational Research Program, NUS Center for Nanomedicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 117597, Singapore
iInstitute of Molecular and Cell Biology, Agency for Science, Technology, and Research (A*STAR), 61 Biopolis Drive, Proteos, Singapore, 138673, Singapore
First published on 10th November 2022
Abstract
Radiotherapy (RT), including external beam RT and internal radiation therapy, uses high-energy ionizing radiation to kill tumor cells. However, ionizing radiation inevitably damages the surrounding normal tissues. Therefore, it is imperative to develop precision RT for improving the treatment outcome and reducing the adverse effects. Recent breakthroughs in nanotechnology have provided a variety of strategies by which RT can precisely and efficiently eradicate local tumors. In this review, we would like to summarize a series of nanotechnology-mediated strategies to achieve precision RT, including tumor-targeted delivery, image-guided precision radiotherapy, and exo/endogenous stimuli-responsive nanomedicines for enhanced tumor accumulation/penetration. In addition, this review will also discuss two representative featured applications of precision RT: RT-induced immunotherapy against cancer metastasis and radioprotection of the surrounding healthy tissues. Since RT is usually thought to be only effective for treating local tumors, this review will interpret the unusual mechanisms of RT-mediated systemic antitumor immunity for eliminating distant cancer metastasis as well as the abscopal effects of RT in combination with other treatments (e.g., photodynamic therapy (PDT), chemodynamic therapy (CDT), etc.). Furthermore, this review will discuss nanotechnology-mediated radioprotection strategies for shielding healthy tissues from radiation damage. Finally, the current challenges and future prospects of precision RT are also elucidated with the intention to accelerate its clinical translation.
 Yuanbo Pan | Yuanbo Pan received his MMed degree from Shanghai Jiao Tong University School of Medicine in 2019. Since then, he has been pursuing his PhD degree at the School of Medicine, Zhejiang University, under the supervision of Prof. Jianmin Zhang. He is currently a joint PhD student at the Yong Loo Lin School of Medicine, National University of Singapore, under the supervision of Prof. Xiaoyuan (Shawn) Chen. His research interests focus on the design and synthesis of multifunctional nanomedicines for brain disease (brain tumors, stroke, traumatic brain injury, etc.) theranostics. |
 Wei Tang | Dr Wei Tang received her BS degree in Chemistry from the University of Science and Technology of China in 2011 and PhD degree in Chemistry from the University of Georgia in 2016. After a postdoctoral training in the Laboratory of Molecular Imaging and Nanomedicine at the National Institutes of Health, she joined National University of Singapore as an Assistant Professor in the Department of Pharmacy and Department of Diagnostic Radiology in 2021. Her research interests focus on the development of safe and effective drug delivery technologies to empower early detection, precise stratification, and personalized therapy for cancer. |
 Wenpei Fan | Prof. Wenpei Fan received his PhD in 2015 from Shanghai Institute of Ceramics, Chinese Academy of Sciences. During 2015–2019, he worked at National Institutes of Health (USA) as a postdoctoral fellow. Since November 2019, he has been a full professor at the Center of Advanced Pharmaceuticals and Biomaterials, China Pharmaceutical University. He has published over 80 papers in reputed journals. His research interest focuses on the design and synthesis of multifunctional theranostic nanomedicines for drug delivery, multimodal imaging, and precision synergistic therapy. |
 Jianmin Zhang | Prof. Jianmin Zhang received his MD degree and PhD degree, in 1983 and 2005, respectively, both from Zhejiang University. He is now a professor and chief physician of the Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University. He has published over 200 papers and numerous books. His current research interests focus on basic and clinical research studies of cerebrovascular diseases and brain tumors as well as the clinical translational application of brain–computer interfaces. |
 Xiaoyuan Chen | Prof. Xiaoyuan (Shawn) Chen received his PhD in Chemistry from the University of Idaho (1999). After being a faculty at the University of Southern California and Stanford University and then Senior Investigator/Lab Chief at the National Institutes of Health, he is now a Nasrat Muzayyin Professor in Medicine and Technology, Yong Loo Lin School of Medicine and College of Design and Engineering, National University of Singapore. His current research interests are mainly theranostics (radiotheranostics, nanotheranostics, immunotheranostics, magnetotheranostics, phototheranostics, etc.) that can be clinically translatable. He has published over 900 papers and numerous books (H index 173 as of September 2022). |
1. Introduction
1.1 Radiotherapy
Cancer remains one of the leading causes of death worldwide, accounting for nearly 10 million deaths every year.1 As an important anticancer treatment, radiotherapy (RT) is delivered to more than 50% of all patients with cancer for both curative and palliative purposes.2,3 There are two main types of RT: external beam RT (EBRT) and internal radiation therapy. ERBT is the most common type of RT that uses high-energy ionizing radiation via a linear accelerator.4 By contrast, internal radiation therapy, including brachytherapy and radioisotope therapy (RIT), makes use of radioactive materials to kill tumor cells.4–6 Generally, RT takes effect by ionizing radiation (e.g., X-ray, γ-ray, etc.) and particle radiation (e.g., carbon ions, electrons, neutrons, α particles, β particles, etc.).7,8 All of these types of radiation are utilized to achieve therapeutic effects to meet various clinical demands. In general, RT damages tumor tissues through direct and indirect actions. Direct action refers to biomolecule damage, especially double-strand breaks in DNA, leading to necrosis or apoptosis. In indirect action, radiolysis of water molecules produces reactive oxygen species (ROS) to destroy biological molecules, such as DNA, proteins, and lipids, to trigger cell apoptosis.8–13 Furthermore, RT is usually used in combination with other treatments, including surgery, chemotherapy, and immunotherapy, to treat tumors. Therefore, RT has always been a mainstay part of cancer treatment.1.2 Precision RT
Precision medicine is an emerging field that considers individual variations in genetics, the environment and lifestyle factors for targeted therapy.14 The precision medicine of cancer patients relies heavily on the development of next generation sequencing and high-throughput data processing technologies.15 However, current RT cannot accurately distinguish tumor tissues from healthy ones, so patients usually suffer from severe radiation damage, such as gastrointestinal syndrome (GIS) and radiation-induced lung injury (RILI), which lowers the long-term life quality of patients and limits the follow-up treatment.16–19 Therefore, precision RT is highly desirable to maximize tumor control and minimize the toxic effects on healthy tissues at the individual patient level.201.3 Emerging nanotechnologies for precision RT
For tumors, only a small proportion of X-ray photon energy can be absorbed. Therefore, high dose radiation is usually required to kill tumor cells, imposing a radiation-related side effect on the surrounding normal tissues.21 Recently, the rapid development of nanotechnology has shown its great value in achieving precision RT. For example, owing to a high photoelectric absorption cross-section, the Au nanoparticles (AuNPs) can enhance local dose deposition in the kilovoltage range (500–300 kVp), which causes the energy of X-ray photons absorbed by AuNPs to be transferred to the surrounding water, resulting in ROS generation.22 In addition, the organic materials, such as protoporphyrin IX (PpIX), were also reported to generate ROS under X-ray irradiation instead of light.23–25 Nowadays, a wide range of multifunctional nanomaterials have been designed for tumor-targeted delivery and radiosensitization through a high-Z element-induced Compton effect or regulation of radioresistant tumor microenvironment (TME).21,26 These nanoradiosensitizers accumulate in tumors via either passive targeting (known as enhanced permeability and retention (EPR) effect) or active targeting mechanisms, including ligand modification-based targeting, biomimetic targeting, magnetic targeting, and even subcellular organelle targeting, allowing for tumor-specific precision RT.27–32 Based on nanoradiosensitizers, low-dose radiation is able to kill tumor cells effectively, lowering side effect on normal tissues. In addition, these nanoradiosensitizers can be endowed with multiple imaging functionalities for image-guided precision RT, including magnetic resonance imaging (MRI), computed tomography (CT) imaging, photoacoustic imaging (PAI), fluorescence imaging (FLI), positron-emission tomography (PET) imaging, single-photon-emission computed tomography (SPECT) imaging, or combination of these imaging modalities. These advanced imaging techniques can provide accurate information of tumor location and track/monitor the pharmacokinetics of nanoradiosensitizers.33–35 Moreover, specific imaging techniques can be used to monitor the TME and tumor responses to therapy, thus beneficial in guiding precision RT.36–38 Furthermore, the exo/endogenous stimuli responsive-nanotechnologies also play an important role in realizing precision RT. For instance, pH-responsive nanoradiosensitizers are able to self-regulate their particle sizes at different pH values for enhanced tumor penetration.39,40Besides destroying the local tumors, precision RT can also suppress distant tumor metastasis. Generally, RT is thought to be only effective in local tumor therapy; however, the nanotechnology-mediated RT is able to elicit systemic antitumor immunity against distant cancer metastasis.41 Moreover, the abscopal effect of RT can be further enhanced by combination with checkpoint blockade therapy or other treatment protocols.42,43 In addition, minimizing the side effects of ionizing radiation on normal healthy tissues is also an important part of precision RT. Several types of ROS-scavenging nanomaterials, including CeO2, fullerenes, graphdiyne, and Bi2Se3, have been synthesized for radioprotection of normal tissues.44–49 Moreover, various nanocarriers have been utilized to deliver molecular radioprotectors to improve their blood circulation and biodistribution. Overall, rapidly advancing nanotechnology enables precision RT to be extended to anti-metastasis and radioprotection.
In this review, the current state-of-the-art strategies for nanotechnology-mediated precision RT are summarized, including tumor-targeted delivery, image-guided positioning, and exo/endogenous stimuli-responsive tumor accumulation/penetration. Besides, this review will also discuss two representative featured applications of precision RT: RT-induced systemic antitumor immunity against distant metastasis and radioprotection of healthy tissues.
2. Tumor-targeted delivery strategies for precision radiotherapy
Tumor-targeted delivery strategies enable nanoradiosensitizers to precisely accumulate in tumor tissues for precision RT. This section will discuss various advanced tumor-targeted nanomedicine delivery strategies for achieving precision RT (Fig. 1), including EPR effect-mediated passive targeting, biological or bioorthogonal ligand-mediated targeting, cell or cell membrane-mediated homing targeting, magnetic targeting, and subcellular organelle targeting (e.g., mitochondrial targeting, nucleus targeting, etc.) strategies.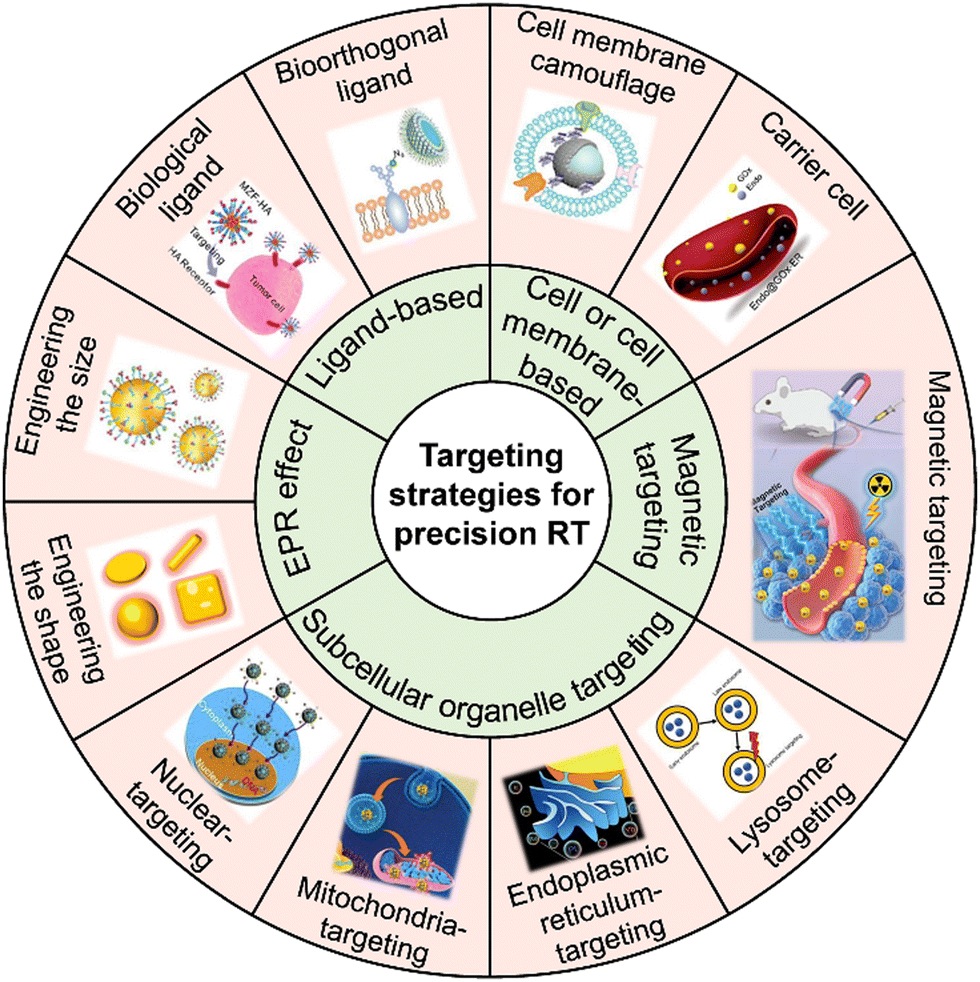 | ||
| Fig. 1 Scheme of the various targeting strategies for precision RT. Adapted with permission from ref. 31, 37 and 50–56 Copyright, American Chemical Society, Wiley, Elsevier, Royal Society of Chemistry, American Scientific Publishers. | ||
2.1 Enhanced permeability and retention (EPR) effect-mediated passive targeting strategy
When administered intravenously, drugs are usually too small thus are easily excreted by the kidneys or too large thus are easily recognized and cleared by the reticuloendothelial system (RES), but nano-sized drugs can achieve long blood circulation for enhanced tumor accumulation.57 Different from healthy vasculatures in normal tissues, tumors have leaky vessels, so nano-sized drugs featured with long blood circulation tend to leak into tumor tissues via the leaky vessels and then stay within the tumors.58,59 This phenomenon, known as the EPR effect, is the primary pathway by which most radiosensitizers accumulate in the tumors.40,60–65For example, Gao et al. used nitrosylated maytansinoid DM1 with the NO releasing group, tert-butyl nitrite (CH3)3CONO, to prepare a prodrug DM1-NO. Considering that DM1-NO could not efficiently accumulate in tumors,66 it was loaded into the poly(lactide-co-glycolic)-block-ploy(ethylene glycol) (PLGA-b-PEG) nanocarrier (DM1-NO-NPs) for efficient delivery to tumors through the EPR effect. The elevated ROS induced by X-ray irradiation led to the cleavage of the S–N bond for release of DM1 and NO. DM1 could arrest the cell cycle at the G2/M phase, sensitizing cancer cells to RT. Moreover, NO could react with radiation-induced ROS to generate more harmful radicals, such as ONOO-, leading to lipid peroxidation and DNA damage. As a result, DM1–NO–NPs plus X-ray irradiation could significantly suppress tumor growth.
Furthermore, the EPR effect is quite heterogeneous within and across tumors and is highly related with cancer type, tumor volume, blood supply, and location.67–72 For instance, Davis and Lewis's group synthesized star polymers (denoted as nanostars). Then, the nanostars were modified with Gd3+ for MR imaging and labeled with 89Zr or 177Lu for in vivo PET imaging or internal RIT.63In vivo PET imaging was performed in two types of tumors with different EPR characteristics by intravenous injection of 89Zr-functionalized nanostars. The result revealed that the nanostars effectively accumulated in CT26 xenografts (high EPR) at 3 days post-injection (14.8 ± 4.0% ID per g). However, the nanostars were observed only at the periphery of BxPC3 xenografts (low EPR) with poor penetration. BxPC3 pancreatic cancer is characterized by a dense extracellular matrix, which might result in a low EPR effect and poor penetration of nanodrugs.73 Finally, the 177Lu-labeled nanostars were utilized for the RT of CT26 tumor-bearing mice. The result showed that the CT26 tumor growth was significantly inhibited when the imposed dose was increased to 3.7 or 7.4 MBq.
For example, Xia and Liu's group doped radioactive 198Au into the crystal lattices of PEGylated Au nanostructures with four different shapes but similar size (∼50 nm), including nanospheres, nanodisks, nanorods, and cubic nanocages (Fig. 2a–d).50 They investigated the biodistribution, tumor accumulation, and intratumoral distribution of the four shapes of 198Au-doped nanostructures in EMT6 breast tumor-bearing mice. The γ radiation of radioactive 198Au was adopted to quantify the biodistribution of these Au nanostructures. However, in vivo tumor uptake was observed by the Cerenkov luminescence derived from the β-emission of 198Au. The autoradiography imaging of tumor slices was utilized to measure the intratumoral distribution of these Au nanostructures. It was observed that Au nanospheres exhibited the longest blood circulation time and highest tumor accumulation of 23.2% ID per g at 24 h post-injection (Fig. 2e). It is worth mentioning that Au nanodisks could accumulate in the lung even at 24 h post-injection (4.9% ID per g) due to their distinct shape, indicating their potential application in lung theranostics. The in vivo luminescence and X-ray imaging further confirmed the highest tumor uptake of Au nanospheres (Fig. 2f). Interestingly, autoradiography imaging of tumor slices at 24 h post-injection revealed that the Au nanocages and nanorods were able to reach the tumor cores while Au nanospheres and nanodisks only stayed at the periphery of tumors. In another similar study, Ma et al. designed three types of PEG-modified Au nanostructures with similar size (about 50 nm) but various shapes, such as Au NPs (GNPs), Au nanorods (GNRs), and Au nanospikes (GNSs).80In vitro cell uptake experiments showed that GNPs presented higher phagocytic efficiency than GNRs and GNSs at 24 h post-incubation. Owing to higher cellular uptake efficiency, GNPs showed better RT enhancement with a sensitization enhancement ratio (SER) of 1.62, much higher than that of GNSs (1.37) and GNRs (1.21). These results suggested that the shape was able to influence the cell uptake efficiency of Au nanostructures and further affect their RT efficacy.
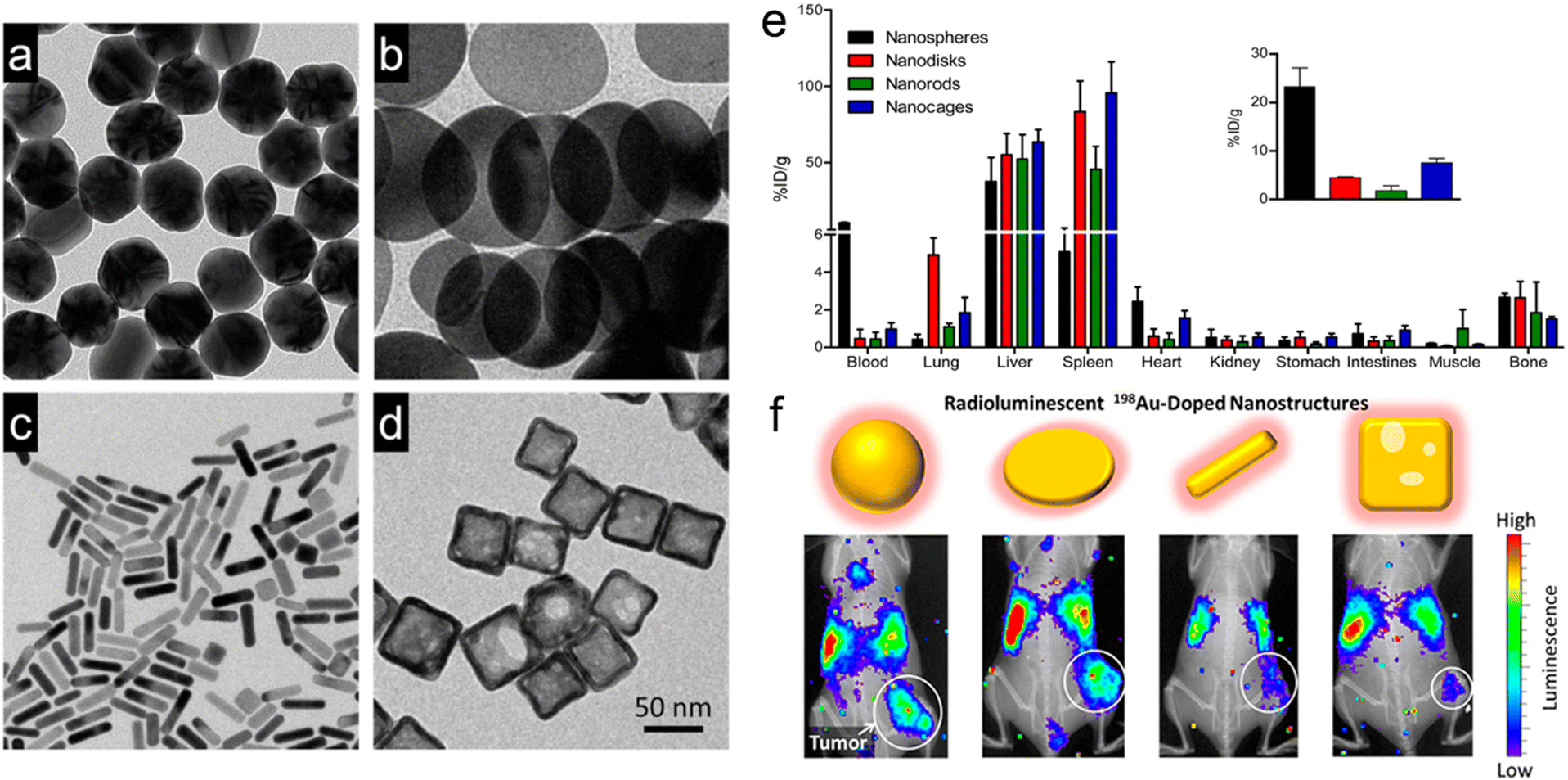 | ||
| Fig. 2 Engineering the shapes of nanoradiosensitizers for better EPR effect. (a–d) TEM images of Au nanospheres (a), Au nanodisks (b), AuNRs (c), and Au cubic nanocages. (e) Biodistribution of various types of 198Au-doped Au nanostructures at 1 day after intravenous administration. Inset: the amount of Au in tumor sites. (f) In vivo radioluminescence images of tumor-bearing mice at 1 day after administration of various types of 198Au-doped Au nanostructures. Reproduced with permission from ref. 50. Copyright 2014, American Chemical Society. | ||
As another example, Wang et al. constructed three types of PEGylated Au nanomaterials (i.e., Au nanohexapods, Au nanorods, and Au nanocages) with similar sizes to investigate their in vitro and in vivo cellular uptake efficiencies.79 The result revealed that Au nanohexapods exhibited higher cellular uptake than Au nanorods and Au nanocages. Moreover, Au nanohexapods and Au nanorods exhibited relatively high tumor accumulation of 7.2 ± 1.2 and 8.4 ± 2.2% ID per g at 24 h post-injection, respectively. However, Au nanocages showed relatively poor accumulation in tumors (2.6 ± 0.8% ID per g at 24 h post-injection).
A previous study reported that the targeting ability of AuNRs could be better than that of Au nanospheres when modified with active targeting ligands. Zhang et al. fabricated two shapes (rod and sphere) of RGD-modified, radioisotope 125I-labeled and cisplatin-loaded Au nanostructures (RGD–125IPt–AuNRs and RGD–125IPt–AuNPs) with similar sizes to evaluate their in vivo biodistribution and chemotherapy/RT efficacy.81 The tumor accumulation of these Au nanostructures was attributed to both the targeting peptides and the EPR effect. The scramble peptide c(RADyC) was used to modify Au nanostructures (RAD–125IPt–AuNRs and RAD–125IPt–AuNPs) as the non-targeting negative control. There was no significant difference in tumor accumulation between RAD–125IPt–AuNRs and RAD–125IPt–AuNPs. Nevertheless, RGD–125IPt–AuNRs exhibited much richer tumor accumulation than RGD–125IPt–AuNPs, suggesting that the shape of Au nanostructures modified with active targeting ligands played a crucial role in their tumor accumulation. The relative tumor volume in the RGD–125IPt–AuNRs plus X-ray group at 21 day post-treatment was only 4.73 compared to 9.8 in the RGD–125IPt–AuNPs plus X-ray group.
The radiosensitizers with the most appropriate size can achieve the richest tumor accumulation for precise RT with very few adverse effects on the adjacent normal tissues.51,83 Zhang et al. synthesized four different sizes (4.8, 12.1, 27.3, and 46.6 nm) of PEG-modified AuNPs to evaluate the size-dependent radiosensitization in vitro and in vivo.84 They investigated the in vitro radiosensitization of AuNPs by measuring the apoptosis and necrosis of HeLa cells after X-ray irradiation (2 Gy) plus AuNPs with different sizes. The apoptosis ratios induced by 4.8, 12.1, 27.3, and 46.6 nm AuNPs plus X-ray irradiation were measured to be 7.65%, 21.86%, 11.19%, and 3.77%, respectively. Meanwhile, necrosis ratios of 6.13%, 15.59%, 6.93%, and 1.09% were induced by 4.8, 12.1, 27.3, and 46.6 nm AuNPs plus X-ray irradiation, respectively. Next, the biodistribution of PEG-modified AuNPs at 24 days after intraperitoneal injection revealed that 12.1 nm and 27.3 nm AuNPs mainly stayed in the liver and spleen, respectively, and that 12.1 nm AuNPs exhibited the richest tumor concentration. However, 4.8 nm or 46.6 nm AuNPs hardly remained in the tumor. The tumor volumes at 24 days post-treatment of 12.1 nm AuNPs + RT and 27.3 nm AuNPs + RT were significantly reduced to 37.1 and 130.3 mm3, respectively, compared to RT alone (394.4 mm3), 4.8 nm AuNPs + RT (349.6 mm3), or 46.6 nm AuNPs + RT (290.4 mm3).
In summary, the tumor accumulation of nanoradiosensitizers through the EPR effect is influenced by various factors, such as particle shape, size, surface modification, tumor type, and location, etc. Therefore, to achieve precise RT, it is necessary to comprehensively consider the above-mentioned factors to design nanoradiosensitizers with optimized EPR effect and maximized tumor accumulation.
2.2 Ligand-mediated active targeting
Folic acid (FA) is one of the most widely used targeting ligands since the corresponding folate receptors are highly expressed in various types of tumors, such as brain, lung, breast, colon, and ovary tumors.85 As such, FA has been commonly employed to engineer radiosensitizers with active tumor targeting for precision RT.86–90 For example, Zhang et al. loaded sorafenib (SOR, a multi-targeted drug for hepatocellular carcinoma (HCC)) into bismuth-based mesoporous nanomaterials (NBOF) with surface modification of PEG–FA (P-FA) to prepare the NBOF@SOR-P-FA NPs for actively targeted chemoradiotherapy of HCC (Fig. 3a).91 Owing to the P-FA coating, NBOF@SOR-P-FA NPs were able to bind with folate receptors overexpressed on cancer cell membranes. Thus, NBOF@SOR-P-FA plus X-ray irradiation could effectively suppress in vitro HCC cell viability to 16.2% (77.2% for NBOF@SOR-P-FA alone group) and increase the cell apoptosis proportion to 51.7% (31.6% for NBOF@SOR-P-FA alone group). Moreover, in vivo treatment with NBOF@SOR-P-FA and X-ray irradiation (6 Gy) could completely inhibit tumor growth, whereas the average tumor volume in the X-ray alone group reached over 500 mm3 at 15 days after treatment.
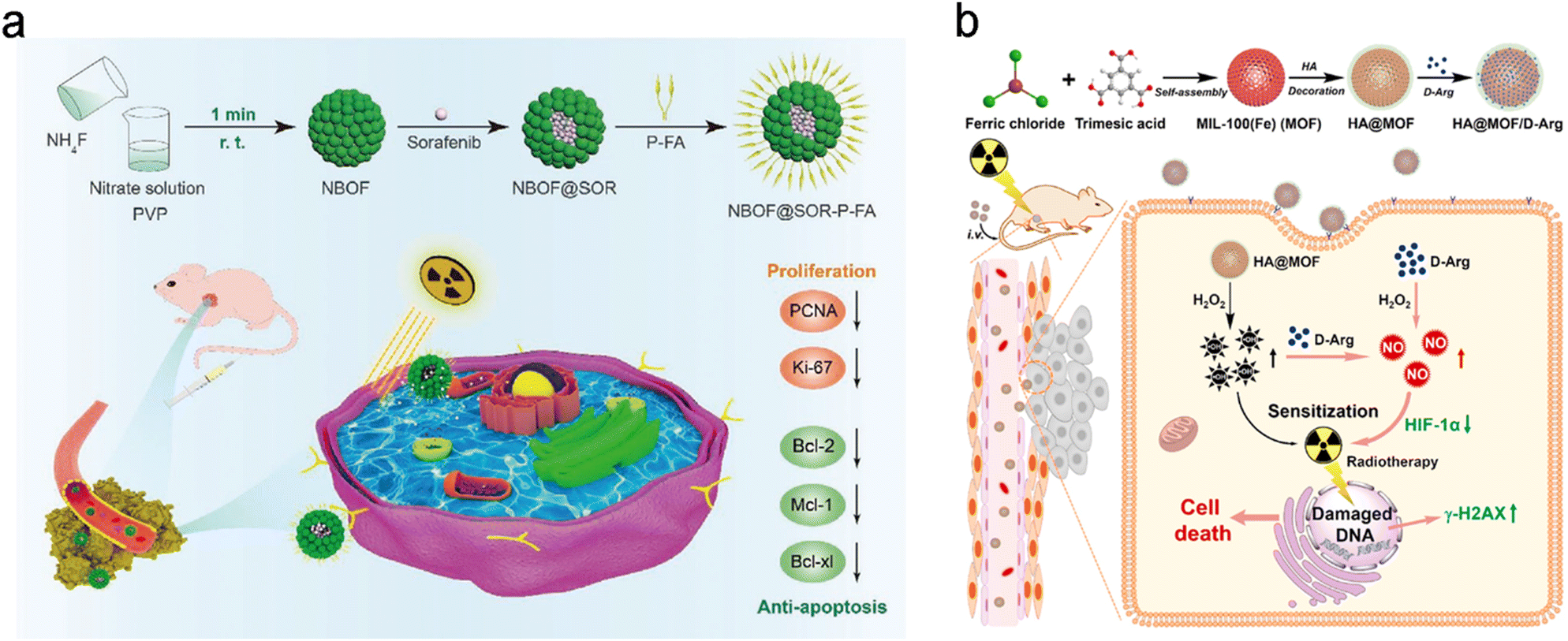 | ||
| Fig. 3 Biological ligand (HA/FA)-based targeting strategies for precision RT. (a) A scheme showing the synthesis process of NBOF@SOR-P-FA and its application for targeted RT/chemotherapy of HCC. Reproduced with permission from ref. 91. Copyright 2020, Wiley. (b) A scheme showing the synthesis process of HA@MOF/D-Arg NPs and their application for targeted RT of osteosarcoma. Reproduced with permission from ref. 97. Copyright 2020, Elsevier. | ||
Hyaluronic acid (HA), a natural high molecular weight polysaccharide in the extracellular matrix and synovial fluid, has been explored as a targeting ligand for nanomedicines.92–94 The surface decoration of HA not only improves the dispersed nature of radiosensitizers but also endows the radiosensitizers with active targeting ability to bind with CD44 (a HA receptor) overexpressed on cancer cells.52,95,96 For example, Du et al. loaded D-arginine (D-Arg) into MIL-100 (Fe) metal–organic frameworks (MOFs) with surface modification of HA to prepare HA@MOF/D-Arg NPs for radiosensitization of osteosarcoma (Fig. 3b).97 After entering cancer cell via receptor-mediated endocytosis, the released D-Arg could generate nitric oxide (NO) and then decrease the expression level of hypoxia-inducible factor-1 alpha (HIF-1α) for attenuating tumor hypoxia. Moreover, the ferric ions were able to convert hydrogen peroxide into hydroxyl radicals (˙OH) which could further react with NO to produce peroxynitrite anions (ONOO−). Both types of ROS could effectively kill cancer cells. The decreased tumor hypoxia and increased ROS yield could sensitize cancer cells to RT. As a result, the HA@MOF/D-Arg NPs plus X-ray irradiation could significantly decrease the survival fraction of K7M2 cells to about 10% under hypoxic conditions compared to over 25% in the D-Arg + X-ray group. In addition, with the modification of HA, HA@MOF/D-Arg plus 8 Gy of X-ray irradiation could efficiently target tumors and completely eradicate the tumor compared to the tumor volume of about 400 mm3 in the D-Arg + X-ray group 20 days after treatment. Furthermore, the co-modification of two targeting ligands can further improve the receptor-mediated endocytosis efficiency. For instance, Askar et al. prepared a HA and FA dual ligand-modified radiosensitizer (2DG@DCA@MgO, DDM) that contained a magnesium oxide (MgO) core and a 2-deoxyglucose (2DG) shell linked to a dichloroacetate (DCA) layer.98 The HA and FA dual-ligand modified DDM could bind with both CD44 and folate receptors overexpressed in breast cancer, allowing for selective tumor targeting/accumulation and effective RT/chemotherapy of breast cancer.
Tripeptide arginine–glycine–aspartic acid (RGD) is a structural recognition motif for αvβ3 and αvβ5 integrins overexpressed on cancer cells.99 The radiosensitizers or radionuclides conjugated with the RGD peptide can specifically recognize the cancer cells that express integrins and then efficiently accumulate in the tumors for precision RT.81,100–103 For example, Chen's group coordinated iridium (Ir) complex on the surface of black phosphorus (BP) nanosheets to construct a powerful nanosystem (Ir@BP) for RT of nasopharyngeal carcinoma.104 In this design, Ir complex was able to improve the photoelectric properties, such as photoinduced carrier dynamic and photocurrent responses, allowing BP to produce more 1O2 upon X-ray excitation. Then, the targeting RGD peptide was successfully decorated onto the Ir complex in the Ir@BP nanosystem, which was confirmed by the 1HNMR spectrum. The western blot result showed that integrins were highly expressed on CNE-2 cancer cells, one type of human nasopharyngeal carcinoma cells. After RGD decoration, the in vitro endocytosis of RGD-Ir@BP in CNE-2 cells was increased from 4.92 μg/106 cells to 12.76 μg/106 cells. Due to high cell uptake, RGD-Ir@BP could significantly inhibit tumor growth and result in the tumor volume of 50 mm3 30 days after treatment compared to 450 mm3 in the untargeted group (BP + X-ray). In addition to the above-mentioned commonly used biological ligands, other ligands, such as prostate specific membrane antigen,105 transferrin,27 chimeric L6 monoclonal antibody,106 epidermal growth factor,107 and low-density lipoprotein receptor-related protein-1-targeting peptide,108 have been also used to modify radiosensitizers and radionuclides for tumor-targeted precision RT.
The bioorthogonal click reaction has also been applied to engineer radiosensitizers for precision RT. For example, Liu et al. designed a coordination polymer NP (Hf–AIE–PEG) composed of hafnium tetrachloride (HfCl4) and 2,2′-(((2-(4′-(2,2-dicyano-1-phenylvinyl)-[1,1′-biphenyl]-4-yl)-2-phenylethene-1,1-diyl)bis(4,1-phenylene))bis(oxy)) diacetic acid (TPEDC-DAC, a photosensitizer with aggregation-induced emission (AIE)) for radio- and radiodynamic therapy (RT-RDT) (Fig. 4a).53 Hf4+ could not only deposit X-ray energy as a high-Z element to increase ˙OH generation but also convert X-ray radiation into light for activating TPEDC-DAC to produce 1O2 for RDT. Moreover, bioorthogonal click reaction was applied for improving tumor targeting and accumulation of NPs. Briefly, N-azidoacetylmannosamine-tetraacylated (Ac4ManNAz), an azido-containing metabolic glycoprotein labeling reagent, was first used to label azide groups onto the cell membranes. Next, NPs (Hf–AIE–PEG–DBCO) decorated with dibenzocyclooctyne (DBCO, a bioorthogonal ligand) were added for bioorthogonal click reaction with the azide groups on membranes (Fig. 4b). In vitro confocal fluorescence images revealed that the fluorescence of Hf–AIE–PEG–DBCO was obviously observed at 1 h after incubation with 4T1 cells prelabeled with Ac4ManNAz. Moreover, the fluorescence of Hf–AIE–PEG–DBCO was co-localized with the red fluorescence of the membrane tracker, indicating that Hf–AIE–PEG–DBCO was labeled on the cancer cell membrane via the bioorthogonal click reaction with azide groups (Fig. 4c). However, 1 h of incubation was too short for Hf–AIE–PEG to be uptaken by cells, leading to weaker fluorescence in cells. Then, the in vivo targeting efficiency of Hf–AIE–PEG–DBCO was evaluated using a bilateral tumor-bearing mouse model (Fig. 4d). The bilateral tumors were intratumorally pretreated with Ac4ManNAz and PBS for 3 days, which were denoted T2 and T1, respectively. Biodistribution analyses through measuring the concentration of Hf ions revealed that the accumulation of Hf–AIE–PEG–DBCO in T2 tumor was 1.83- and 3.17-fold higher than that in T1 tumor at 1 and 3 days post-injection, respectively (Fig. 4e). These results indicated that the tumor accumulation and retention were remarkably enhanced through bioorthogonal click reaction. Finally, with the help of bioorthogonal ligand-mediated targeting, the tumor volume in the Hf–AIE–PEG–DBCO + Ac4ManNAz + X-ray (8 Gy) group was effectively inhibited and smaller than the initial volume (Fig. 4f). However, the Hf–AIE–PEG + X-ray (8 Gy) group without the bioorthogonal click reaction showed more than 2-fold increase in the relative tumor volume after treatment. Furthermore, the bioorthogonal click reaction has also been applied for radioimmunotherapy, a selective internal RT based on radiolabeled antibodies.123–126 Overall, the bioorthogonal click reaction could be a promising targeting strategy for precision RT owing to its highly specific recognition.
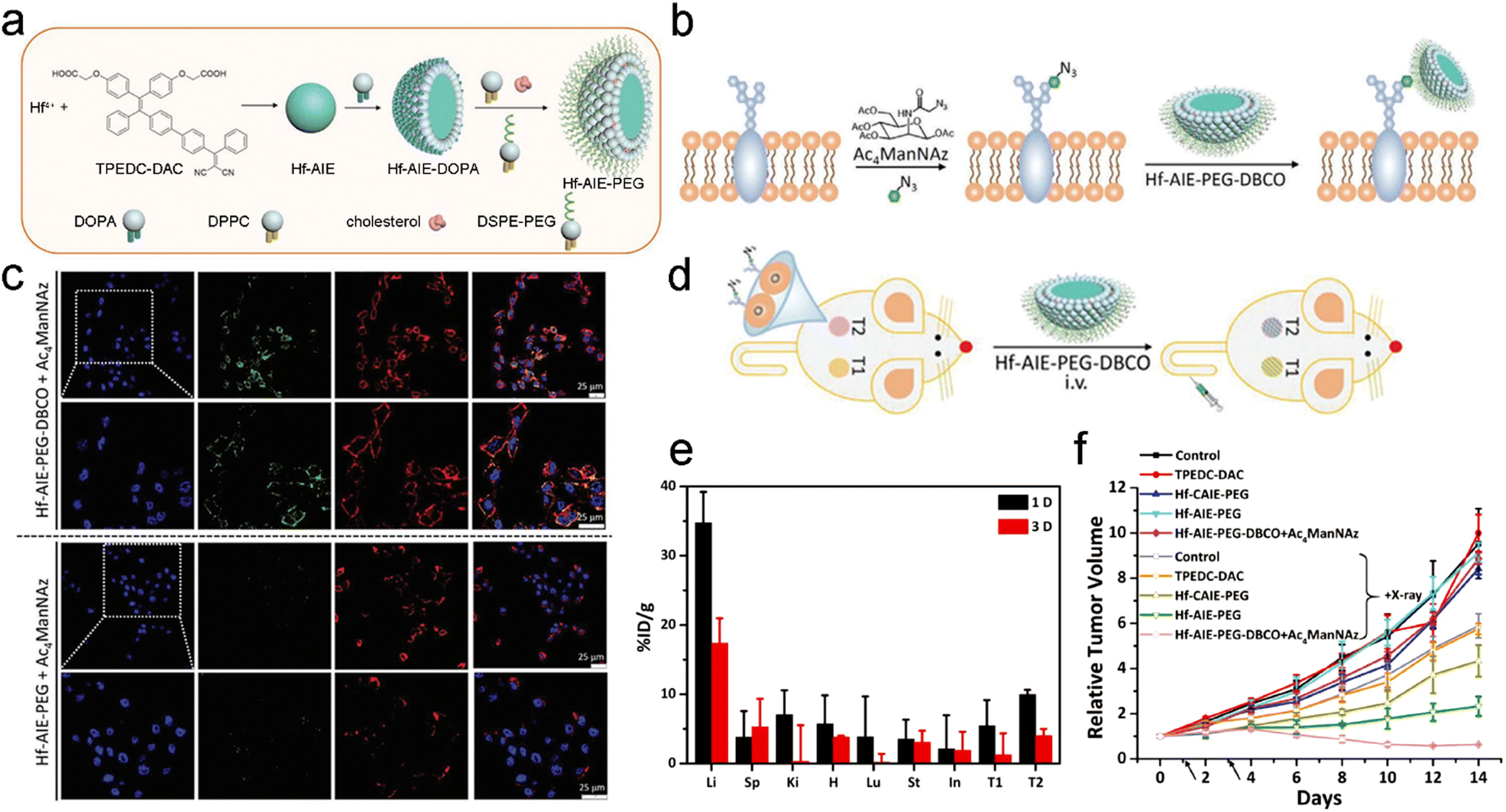 | ||
| Fig. 4 Bioorthogonal ligand-based targeting strategies for precision RT. (a) A scheme of the synthesis process of Hf–AIE–PEG NPs. (b) Schematic illustration of azide expression on tumor cell membrane glycan, and bioorthogonal labeling of Hf–AIE–PEG–DBCO. (c) Fluorescence images of Ac4ManNAz pre-cultured 4T1 cells after Hf–AIE–PEG and Hf–AIE–PEG–DBCO bioorthogonal labeling for 1 h and CellMask Deep Red treatment. (d) Schematic illustration of the experimental process for bioorthogonal design on mouse. T1: tumors without Ac4ManNAz pretreatment. T2: tumors with Ac4ManNAz pretreatment. (e) Biodistribution of Hf–AIE–PEG–DBCO in tumor bearing mice at 1 and 3 days after intravenous administration. (f) Tumor growth curves of mice in different treatment groups. Black arrows indicate radiation time points. Reproduced with permission from ref. 53. Copyright 2021, Wiley. | ||
2.3 Cell or cell membrane-mediated targeting
The synthetic nanomaterials, whether organic or inorganic, have been found to be more or less immunogenic or toxic after intravenous administration. Recent studies have explored the possibility of utilizing natural cells or cell membrane-based vesicles for the delivery of radiosensitizers, including cell membrane camouflaged NPs, exosomes, and whole cells. These vesicles with the natural membrane structure can be regarded as the “self” and exhibit excellent biocompatibility, longer blood circulation, and rich tumor accumulation. Moreover, several types of cells or cell membranes possess inherent homotypic binding ability, allowing particles or drugs to homologously target tumors. In this section, the natural cell or cell membrane-mediated targeting strategies for radiosensitizer delivery will be introduced (Table 1).| Targeting strategy | Nanoparticles | Cell/membrane type | Coating or loading method | Functions | Ref. |
|---|---|---|---|---|---|
| Cell membrane camouflage-based tumor targeting | TiO2@MnO2-GOx@C | B16-F10 cancer cell membrane | Co-stirring at a low temperatures | Homologous targeting | 54 |
| GNR@Mem | Oral squamous KB cancer cell membrane | Co-extruding | Homologous targeting, improving blood circulation | 131 | |
| CQM | 4T1 cancer cell membrane | NA | Homologous targeting | 133 | |
| CMC | 4T1 cancer cell membrane | Co-extruding | Homologous targeting, improving blood circulation | 130 | |
| Au@MC38 | MC38 cancer cell membrane | Biosynthesis | Homologous targeting | 30 | |
| TDSP-Exos | CT26 cancer cell exosome | Exocytosis | Homologous targeting, immune escaping | 151 | |
| PFC@PLGA-RBCM | Red blood cell membrane | Co-stirring at 4 °C | Improving blood circulation | 29 | |
| F-RBC bismuth NPs | Red blood cell membrane | Co-extruding | Improving blood circulation | 135 | |
| CM-EM-GNCs@DOX | Membrane from MCF-7 cancer cell and red blood cell | Co-extruding | Homologous targeting, immune escaping, improving blood circulation | 134 | |
| PLT/CANS | Platelet membrane | Co-extruding | Target anomalous vessels in tumors, immune escaping | 137 | |
| BMSNR@PM | Platelet membrane | Sonicating | Tumor targeting, immune escaping | 136 | |
| Cyp-PMAA-Fe@MSCs | Mesenchymal stem cell membrane | Sonicating | Immune escaping, improving blood circulation | 138 | |
| Carrier cell-based tumor targeting | Au-Hb@PLT | Platelet carrier | Sonicating | Tumor targeting | 150 |
| Endo@GOx-ER | Erythrocyte carrier | Hypotonic dialysis | Improving blood circulation | 37 |
Cancer cell membrane camouflage endows biomimetic nanomedicines with homologous adhesion, immune escaping, and deep tumor penetration abilities.142 For example, Pan et al. constructed glucose oxidase (GOx)-loaded TiO2@MnO2 core–shell nanoreactors that were further coated with B16-F10 cancer cell membrane (TiO2@MnO2-GOx@C) via a co-extrusion approach for homologous targeting and enhanced RT (Fig. 5a).54 The GOx in these nanoreactors could catalyze the oxidization of glucose in cancer cells to produce gluconic acid and H2O2. Then, the MnO2 shell could catalyze the decomposition of H2O2 into O2. Moreover, the high-Z element of TiO2 and O2 generation could contribute to radiosensitization. The cancer cell membrane camouflage was able to improve homologous targeting abilities of TiO2@MnO2-GOx@C nanoreactors. In vivo biodistribution showed that the Mn element in tumors at 24 h post-injection of TiO2@MnO2-GOx@C was 4.5-fold higher than that in tumor treated with TiO2@MnO2-GOx, suggesting the great targeting capability of the biomimetic nanoreactors (Fig. 5b). Next, X-ray irradiation at a dose of 4 Gy was imposed at 24 h after injection. The lung metastases of melanoma (B16-F10) were completely eliminated 14 days after treatment with TiO2@MnO2-GOx@C plus X-ray; however, the metastatic tumors in other groups were still observed. In addition, no mice in the TiO2@MnO2-GOx@C plus X-ray group died even after 40 days, whereas the mice in the other groups all died within 28 days (Fig. 5c).
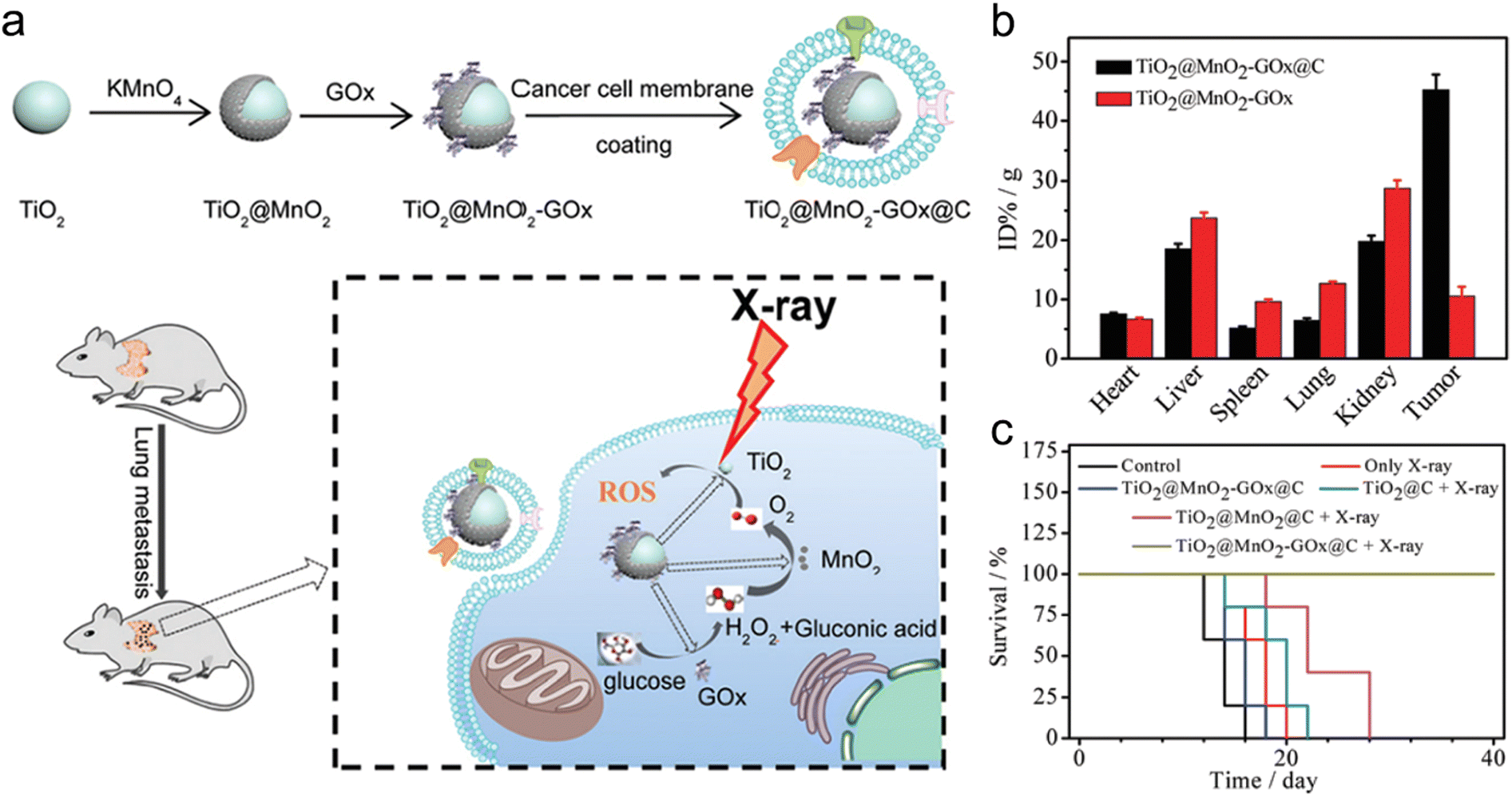 | ||
| Fig. 5 Cell membrane camouflage-based tumor targeting. (a) A scheme of synthesis process of TiO2@MnO2-GOx@C and its application in the RT of cancer metastasis. (b) Biodistribution of Mn in major organs and tumors of mice after injection of TiO2@MnO2 or TiO2@MnO2-GOx@C NPs. (c) Survival curves of melanoma-bearing mice in different treatment groups. Reproduced with permission from ref. 54. Copyright 2020, Royal Society of Chemistry. | ||
Membranes of erythrocytes have also been used to camouflage radiosensitizers for prolonged blood circulation.143 For example, Liu's group coated red blood cell (RBC) membrane on perfluorocarbon (PFC)-loaded PLGA (PFC@PLGA-RBCM) to prepare an artificial RBC nanosystem for hypoxia relief-enhanced RT. Interestingly, PFC@PLGA-RBCM exhibited a much longer blood circulation half-life of 13.93 h compared to naked PFC@PLGA. However, the RBC membrane camouflage alone can only prolong circulation half-life while the tumor accumulation of NPs still relies on passive targeting. It is worth noting that fusion of multiple cell membrane types can integrate their respective strengths. For instance, Sun et al. utilized fusion membrane from both MCF-7 cancer cells and erythrocytes to coat DOX-loaded gold nanocages (CM-EM-GNCs@DOX) for PTT/RT/chemotherapy of breast cancer.134 Due to the hybrid membrane coating, the nanocages possessed both homologous targeting and improved immune-escape abilities. Thus, the in vitro cell uptake of CM-GNCs and CM-EM-GNCs in MCF-7 cells was about 3.9- and 4.1-fold higher than those in MCF 10A cells, respectively, owing to the cancer cell membrane-mediated targeting ability. Besides, due to the erythrocyte membrane-mediated immune-escape ability, the in vitro macrophage uptake of both EM-GNCs and CM-EM-GNCs was significantly lower than that in the CM-GNC group. Finally, effective RT-based combination treatment significantly suppressed the breast cancer growth in vivo. Similar to RBCs, bone marrow mesenchymal stem cells (MSCs) with low immunogenicity have also been utilized to camouflage NPs for prolonging the blood circulation time and evading immune surveillance. For example, Yin et al. decorated Fe(III) ions and cypate co-loaded polymethacrylic acid (PMAA) NPs with the MSC membrane to prepare Cyp-PMAA-Fe@MSCs for RT/PTT of lung cancer.138
Furthermore, platelet (PLT) membrane camouflage is another strategy for preparing biomimetic radiosensitizers. For example, Lyu et al. fabricated core–shell Au@AuPd nanospheres and then coated the nanospheres with the PLT membrane (PLT/CANS) for brachytherapy (BT) of colon cancer.137 The PLT membrane camouflage enables these NPs to enhance immune evasion and actively target anomalous vessels in tumors, leading to rich tumor accumulation of PLT/CANS. Palladium (Pd)-based NPs could catalyze the decomposition of H2O2 into O2 to alleviate hypoxic TME for radiosensitization. Moreover, high-Z element-based X-ray deposition could also sensitize BT. The CANS NPs coated with the RBC membrane (RBC/CANS) were regarded as the control group. The in vivo circulation profiles of these NPs revealed that both PLT/CANS and RBC/CANS NPs could prolong the blood circulation time compared to naked CANS. Moreover, with the PLT membrane decoration, PLT/CANS could actively target tumor and show a 1.9-fold richer tumor accumulation than that in RBC/CANS group at 24 h post-injection, which was confirmed by ICP-AES and in vivo FL imaging. Finally, the survival of colon tumor-bearing mice in the PLT/CANS + BT group was 100% at 30 days after treatment compared to 20% and 40% of BT and RBC/CANS + BT groups, respectively. In summary, the cell membrane camouflage as a promising tumor-targeting strategy can endow radiosensitizers with diverse excellent biological capabilities for precision RT of cancer.
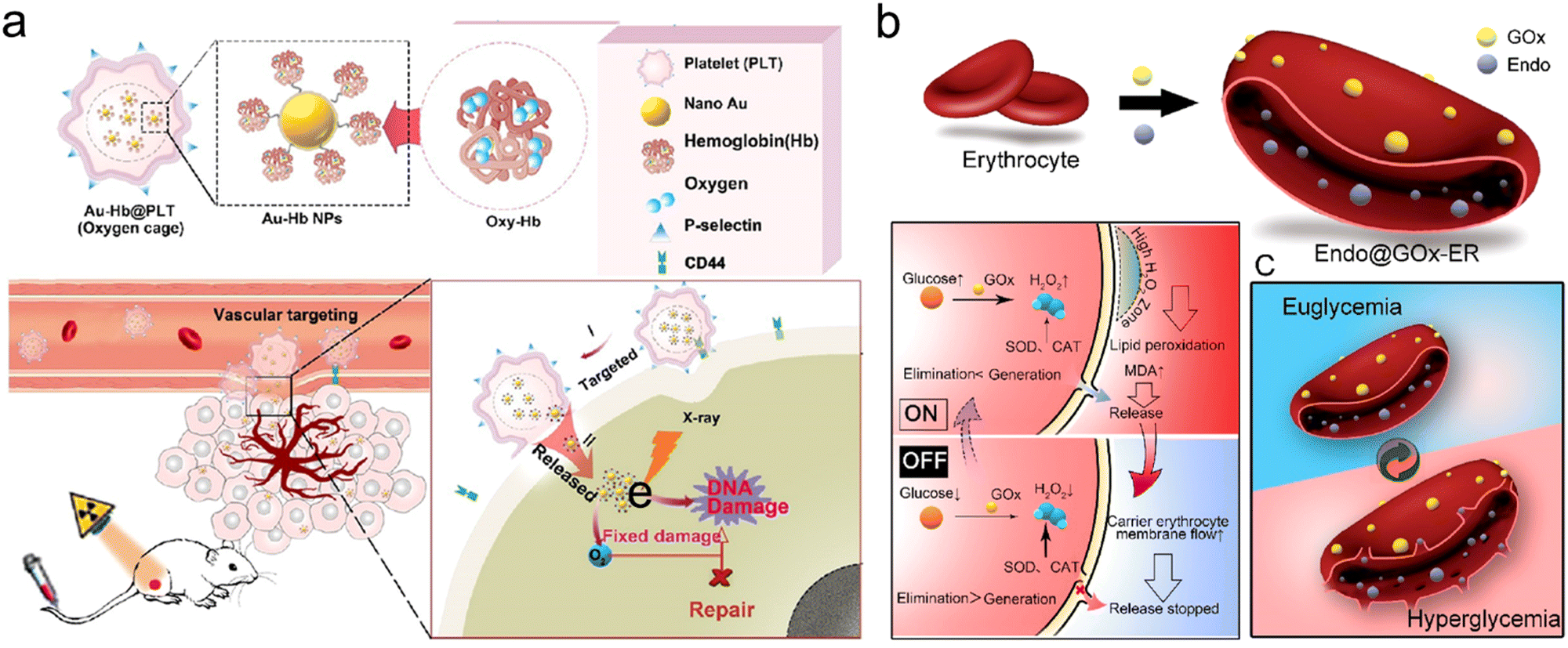 | ||
| Fig. 6 Carrier cell-based tumor targeting. (a) Schematic illustration of synthetic procedures of Au-Hb@PLT and its applications in targeting tumor vessel and enhancing RT. Reproduced with permission from ref. 150. Copyright 2020, American Chemical Society. (b) Schematic illustration showing synthetic procedures of Endo@GOx-ER and closed loop glucose-regulated release of Endo from Endo@GOx-ER. Endo is stored in the ER under euglycemic conditions, whereas it is released during hyperglycemia. Reproduced with permission from ref. 37. Copyright 2020, American Chemical Society. | ||
Furthermore, Huang et al. developed endostar (Endo)-loaded GOx-modified erythrocytes (Endo@GOx-ER) for glucose-regulated drug release.37 The erythrocyte (ER) was chosen as a carrier for the delivery of Endo, an inhibitor of angiogenesis which could normalize tumor vasculature for overcoming tumor hypoxia. The TEM and confocal images showed that Endo@GOx-ER maintained normal size and shape. In this nanosystem, the release rate could be controlled by the blood glucose level (BGL). The Endo@GOx-ER could function as a physiological ER (off-state) under normoglycemia where the Endo was kept inside, reducing immune clearance. However, during hyperglycemia, GOx could effectively catalyze the decomposition of glucose to generate H2O2 for promoting ER membrane perforation, allowing Endo release (on-state) (Fig. 6b). In addition, naked Endo could be cleared from the body at 10 h post intravenous injection, but the Endo@GOx-ER significantly prolonged the serum circulation time of Endo to over 24 h. Overall, the Endo@GOx-ER could result in the normalization of tumor vessel and long-term alleviation of TME hypoxia, which could potentiate repeated RT. The PA imaging of 4T1 tumor-bearing mice displayed that the oxygenation levels were significantly increased even at 6 and 12 days after intravenous injection of Endo@GOx-ER, much higher than those in mice treated with free Endo. With the strategy of overcoming long-term tumor hypoxia, the survival of tumor-bearing mice in the Endo@GOx-ER + RT group was 90% at 45 days after treatment, whereas all the mice in the PBS + RT group died by day 40.
2.4 Magnetic targeting
Magnetic targeting is a promising targeting strategy based on drug-loaded magnetic materials under an external magnetic field (MF).152,153 The magnetic field could guide the magnetic materials to the target tumors or organs where the materials could release drug, nuclear acid, or bioactive molecules for cancer therapy.154 This strategy is able to largely avoid unwanted distribution of drugs in normal organs and thus remarkably lower side effects. For example, Lyu et al. prepared core–shell nanozymes, Fe3O4@MnO2, for magnetic targeting and RT.31 The Fe3O4@MnO2 NPs were used in combination with GOx for enhanced RT. In this design, GOx could catalyze the oxidation of intratumoral glucose to generate H2O2 which then reacted with the MnO2 shell to produce O2 for alleviating tumor hypoxia. Meanwhile, the MnO2 shell could also deplete the overexpressed GSH to generate Mn2+ for T1-weighted MRI. Besides, the GSH depletion could greatly enhance RT efficacy by reducing ROS consumption. Importantly, the Fe3O4 core could be utilized for magnetic targeting and T2-weighted MRI. As a result, the brighter or darker signals in the tumor regions of T1- or T2-weighted MR images were observed at 6 h after intravenous administration of Fe3O4@MnO2 NPs under the assistance of MF, indicating successful tumor accumulation of Fe3O4@MnO2 NPs. However, in the absence of MF, only a slight contrast change in the tumor site was observed after intravenous administration of Fe3O4@MnO2 NPs. With the MF-mediated targeting, the average tumor volume in the Fe3O4@MnO2 NPs + GOx + RT + MF group was remarkably suppressed and smaller than the initial volume at 18 days after treatment, whereas the average tumor volume of mice more than tripled in the Fe3O4@MnO2 NPs + RT group. Besides, the magnetic targeting strategy has also been applied in radioisotope therapy.155–1592.5 Subcellular organelle targeting
Subcellular organelles (e.g., mitochondria, nucleus, endoplasmic reticulum, lysosome, etc.) play an important role in maintaining normal cellular physiological processes.160 In recent years, a lot of radiosensitizers that specifically target various subcellular organelles have been explored. Selective delivery of radiosensitizers to subcellular organelles is able to induce cell death through different signaling pathways, reduce radioresistance of tumors and significantly enhance the RT efficacy.The type 1 human immunodeficiency virus (HIV-1) transactivator of transcription protein (TAT) has been evidenced to function as a nucleus-targeting molecule.161 Shi and Bu proposed a novel “intranuclear biophotonics” strategy by the smart design of silicon phthalocyanine dihydroxide (SPCD) and PpIX co-loaded upconversion NPs modified with PEG and a nuclear targeting peptide TAT (UCSPs-PEG/TAT) (Fig. 7a).55 Based on the fluorescence resonance energy transfer (FRET), the UCNP core was able to convert NIR light into visible light which further activated both two photosensitizers (SPCD and PpIX) to produce singlet oxygen (1O2). Moreover, PpIX could act as a radiosensitizer to convert water into superoxide radicals (O2−) and ˙OH upon X-ray radiation. High-resolution and 2D/3D CLSM images revealed that UCSPs-PEG/TAT (green and red luminescence emitted by the UCNP core) could co-localize with the nucleus stained with DAPI (blue) while UCSP-PEG without the conjugation of TAT could not enter the nucleus (Fig. 7b). Due to nucleus-targeting ability, the generated ROS from PDT and RT was able to remarkably induce DNA breakage and further trigger substantial cell death. Therefore, the nuclear-targeting synergistic RT/PDT strategy (UCSPs-PEG/TAT + NIR + X-ray) decreased the relative tumor volume to around 0.5 compared to over 3.5 in the RT alone group.
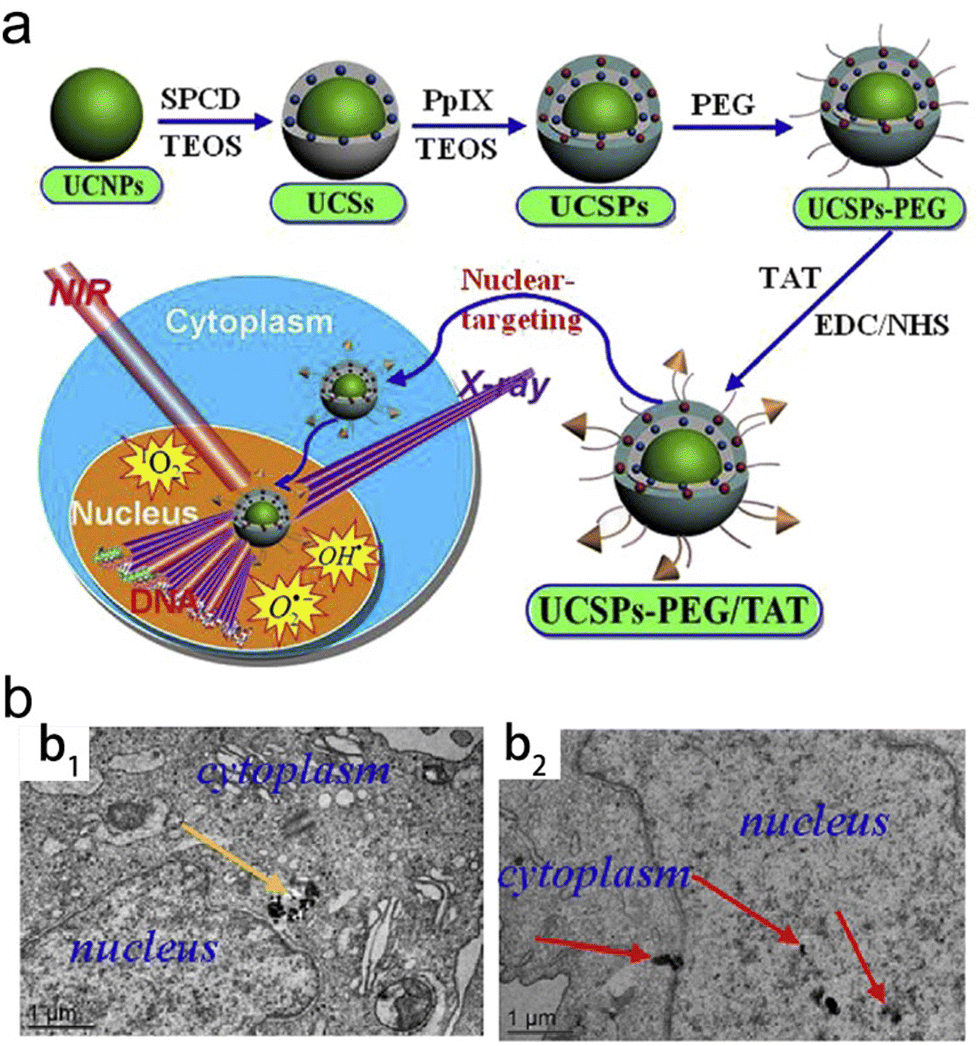 | ||
| Fig. 7 Nuclear-targeting. (a) A scheme of synthetic process of UCSPs-PEG/TAT and its application in nuclear-targeted RT/PDT of tumors. (b) Bio-TEM images of HT-1080 cells at 24 h after treatment with UCSPs-PEG (b1) and UCSPs-PEG/TAT (b2). Reproduced with permission from ref. 55. Copyright 2015, Elsevier. | ||
Furthermore, several studies proposed a dual-targeting strategy in which cancer cell-targeting moieties and TAT were co-used to modify radiosensitizers for enhanced tumor cell nucleus-targeting RT.162–164 For example, Pan et al. loaded 7-ethyl-10-hydroxy-camptothecin (SN-38) into mesoporous TiO2 NPs (MTiO2(SN-38) NPs). The NPs were then anchored with TAT and RGD peptide (MTiO2(SN-38)-TAT-RGD NPs) for nucleus-targeted RT.164 The RGD peptide allowed the MTiO2(SN-38)-TAT-RGD NPs to bind with the αvβ3 integrin overexpressed in several types of cancer cells. Next, the MTiO2(SN-38)-TAT-RGD NPs could further target the nucleus of cancer cells thanks to the decoration of TAT. SN-38 could selectively target topoisomerase I in the nucleus and control the cell cycle of 4T1-Luc cells in the radiosensitive G2/M phase. The in vivo anticancer experiments revealed that the average tumor volume in the MTiO2(SN-38)-TAT-RGD + X-ray group 21 days after treatment was about half of that in the MTiO2(SN-38)-TAT + X-ray or MTiO2(SN-38)-RGD + X-ray group. Overall, this strategy of cancer cell nucleus-targeted delivery of radiosensitizers is able to remarkably enhance the RT efficacy and reduce the side effect by lowering X-ray irradiation doses and reducing the irradiation time.
Triphenylphosphonium (TPP), a moiety able to selectively target the inner mitochondrial membrane owing to its excellent cationic and lipophilic properties, has been widely used to engineer NPs for mitochondria-targeted delivery.165–169 For example, Tang and colleagues modified Gd-doped titanium dioxide NPs (TiO2(Gd) NPs) with TPP to construct a mitochondria-targeting radiosensitizer.170 The confocal images revealed that IR806-labeled TiO2(Gd)-TPP NPs could effectively target the mitochondria and co-localize with Mito-Tracker Green stained mitochondria of MCF-7 cells compared to TiO2(Gd) NPs. Moreover, TiO2(Gd)-TPP NPs were able to generate a great deal of ROS upon X-ray irradiation, resulting in mitochondrial collapse and irreversible cell apoptosis. Moreover, antitumor evaluation revealed that TiO2(Gd)-TPP NPs plus X-ray irradiation (6 Gy) completely eliminated MCF-7 xenograft tumors at 14 days post-treatment; however, the tumor volume was still over 30 mm3 after treatment with TiO2(Gd) plus X-ray. Besides, other mitochondria-targeting moieties have also been used to modify radiosensitizers. For instance, Fang et al. fabricated peptide-templated Au nanoclusters (AuNCs) for mitochondria-targeting radiosensitization.56 Briefly, they synthesized a new peptide (CCYKFR) containing two domains Cys–Cys–Tyr (CCY) and Dmt–D-Arg–Phe–Lys–NH2 (KFR) for peptide-templated AuNC synthesis. In the new peptide, the CCY peptide segment could reduce the Au ions into nanoclusters by the phenolic group of Tyr (Y) as well as stabilize these nanoclusters through the sulfhydryl groups of Cys (C). Moreover, the KFR peptide segment could function as a mitochondria-targeting moiety, which has been reported in other studies.171
Recently, cationic ruthenium (Ru)-based complexes have shown mitochondria-targeting capability without coupling exogenous targeting molecules like TPP.172,173 On the basis of Ru, Lin's group designed a mitochondria-targeted nanoscale metal–organic framework (nMOF) for radiotherapy-radiodynamic therapy (RT-RDT).174 They conformed Ru(bpy)32+ into the nMOF to synthesize Hf-DBB-Ru [DBB-Ru = bis(2,2′-bipyridine)(5,5′-di(4-benzoato)-2,2′-bipyridine)ruthenium(II) chloride] with excellent mitochondria-targeting ability. Hf6SBUs could effectively deposit X-ray energy to produce H2O2 for enhanced RT and transfer energy to Ru(bpy)32+-based bridging ligands to produce 1O2 for RDT. Owing to the presence of cationic [DBB-Ru]2+, Hf-DBB-Ru displayed a strong positive potential (38.9 ± 3.1 mV). The mitochondria uptake of positively charged Hf-DBB-Ru and neutral nMOF (Hf-DBA) was evaluated. The mitochondria of MC38 cells incubated with Hf-DBB-Ru or Hf-DBA were extracted for quantitative analyses via ICP-MS. The result showed that Hf-DBB-Ru rapidly accumulated in the mitochondria and reached the maximum (more than 90%) at 4 h post-incubation. However, only 18% of Hf-DBA was internalized into the mitochondria at 4 h post-incubation. The in vivo antitumor evaluation demonstrated that the treatment/control (T/C) ratios in MC38 tumor-bearing mice treated with Hf-DBB-Ru plus X-ray (6 Gy) irradiation was 3.0% at day 22 after treatment, much lower than 42.1% of the Hf-DBA + X-ray groups.
Several studies have reported the use of increased ER stress to sensitize cancer cells to RT. For example, tunicamycin (TM) was utilized to trigger ER stress in human esophageal cancer cell line EC109.179 The combination treatment of TM and RT effectively arrested the G2/M phase and induced cancer cell apoptosis. The in vitro and in vivo experiment results showed that TM could significantly sensitize cancer cells to RT via apoptosis and autophagy. Besides, ER-targeting radiosensitizers have also been designed for enhanced RT. Klein et al. proposed that ultrasmall aminosilanized oxidized silicon NPs (NH2-SiNPs) with positive potential could accumulate in the membranes of ER and mitochondria of 3T3 cells.180 Moreover, NH2-SiNPs and SiNPs could serve as radiosensitizers. Due to ER targeting, the ROS concentrations of 3T3 and MCF-7 cells incubated with NH2-SiNPs could increase to 120% and 180% after X-ray irradiation, whereas X-ray irradiation showed almost no impact on the ROS level of3T3 and MCF-7 cells incubated with SiNPs.
In addition, ER targeting is also a promising strategy to amplify the effects of ICD. Chen et al. loaded an ER-targeting photosensitizer TCPP-TER (4,4′,4′′,4′′′-(porphyrin-5,10,15,20-tetrayl)tetrakis(N-(2-(methylphenyl)sulfonamido)-ethyl)benzamide) into GSH-responsive Ds-sP (PEG-s-s-1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino-(polyethylene glycol)-2000]) NPs for ER-targeting PDT which was able to effectively induce ER stress and amplify ICD, resulting in enhanced cancer immunotherapy efficacy.181
For example, Simonet et al. fabricated a class of gadolinium-based NPs (GBNs), AGulX® (Activation and Guidance of Irradiation by X-ray), for the radiosensitization of head and neck squamous cell carcinoma (HNSCC).185 Interestingly, the AGulX® preferred to accumulate into lysosomes rather than mitochondria or nucleus after the uptake by SQ20B J.L. cells. The cancer cells pre-treated with AGulX® could enhance RT efficacy, leading to severe damage of DNA and autophagic cell death. Likewise, other high-Z metallic NPs have also shown lysosome-targeting ability. Hullo et al. evaluated the radio-enhancement effect of platinum NPs (PtNPs) in breast cancer cell lines.186 They found that the PtNPs were able to accumulate in lysosomes and multivesicular bodies after internalization. The lysosome-localized PtNPs could deposit X-ray energy to effectively damage DNA and kill cancer cells. In another study, Lacombe and colleagues fabricated label-free GBNs for radiosensitization of U87 glioblastoma cells.187 The bio-TEM and CLSM images found that GBNs were internalized into the cells and then accumulated in the lysosomes rather than nucleus or mitochondria. In addition, clonogenic assay measurements revealed that the incubation of GBNs (0.5 mM) could significantly improve irradiation-induced cell killing effects with 23% of enhancing factor. The RT enhancement based on lysosome-targeting radiosensitizers was attributed to radiation-induced lysosomal perturbations (e.g., lysosomal overload, phospholipidosis, etc.) that further induced autophagy.
3. Exo/endogenous stimuli-responsive strategies for precision radiotherapy
Endogenous stimuli (e.g., pH, GSH, H2O2, hypoxia, enzyme, etc.) and/or exogenous stimuli (e.g., X-ray, NIR irradiation, ultrasound, etc.) can cause size or shape change, chemical degradation, surface property change, and heating up of responsive NPs, resulting in enhanced tumor accumulation/penetration and controllable drug release. This section will discuss exo/endogenous stimuli-responsive strategies for precision RT, including exogenous stimuli, endogenous stimuli in the tumor microenvironment, and a combination of these stimuli (Table 2).| Stimuli | Nanoparticles | Responsiveness | Outcomes | Treatment | Sensitive part | Ref. |
|---|---|---|---|---|---|---|
| pH | Au-NNP(RTX) | Size transformation, drug release | Disassemble into ultrasmall AnNPs to enhance tumor penetration, enhance RTX delivery efficiency | Enhance RT and chemotherapy | N,N-Dibutylethylenediamine, 1-(2-aminoethyl) pyrrolidine | 39 |
| pH | Se@SiO2@Bi NCs | Se NP release | Enhance tumor accumulation of Se NPs | Enhance RT and PTT, reduce side effect of radiation | Bi NPs | 300 |
| pH | Pt@HSA/CA NPs | Drug release | Enhance CA delivery efficiency | Enhance RT and chemotherapy | pH-responsive imine bond | 193 |
| pH | mTa2O5-PEG/DOX | Drug release | Enhance DOX delivery efficiency | Enhance RT and chemotherapy | Surface polymer | 301 |
| GSH | MNPs | Drug release | Enhance TPE-Pt and PPy delivery efficiency | Enhance RT and chemotherapy | RGD-POEGMA-b-PAZMB | 201 |
| GSH | Bi2Se3 HNC-s-s-HA/GA | Drug release | Enhance GA delivery efficiency | Enhance RT and PTT | S–S bond | 202 |
| GSH | GdW10@CSsiRNA nanospheres | Gene delivery | GSH depletion, enhance gene delivery efficiency | Enhance RT and genetherapy | Polyoxometalates | 302 |
| ROS | ACF@MnO2 NPs | O2 generation, drug release | Hypoxia relief, enhance ACF delivery efficiency | Enhance RT and immunotherapy | MnO2 | 216 |
| ROS | Au@SA-QBA | Drug release | Enhance 8HQ delivery efficiency | Enhance RT | QBA | 217 |
| Enzyme | Au@Tat-R-EK | Tat peptide exposure | Enhance nuclear targeting of AuNPs | Enhance RT | Cathepsin B responsive peptide (CFLG) | 227 |
| Enzyme | Bac@BNP | Bi2S3 NPs release | Enhance tumor accumulation of Bi2S3 NPs | Enhance RT | MMP-2 responsive peptide (PLGVR) | 228 |
| Hypoxia | ALP-(MIs)n/DOX | Drug release | Enhance DOX delivery efficiency | Enhance RT and chemotherapy | P-(MIs)n | 255 |
| pH/GSH | PLGA-SS-D@BPQDs | Size expansion and surface-charge-switching (pH), BPQDs release (GSH) | Enhance tumor cell uptake, enhance tumor accumulation of BPQDs | Enhance RT | Amide linkages of DMMA (pH), S–S bond (GSH) | 103 |
| GSH/enzyme | Ce6-Leu@Mn2+ | Size transformation | Enhance tumor accumulation and penetration | Enhance RT and PDT | S–S bond (GSH), leucine motif (LAP) | 257 |
| GSH/hypoxia | HA-Fe-NIs-DOX | Drug and radiosensitizer release | Enhance DOX delivery efficiency, enhance radiosensitization | Enhance RT and chemotherapy | Ferrocenium ion (GSH), NIs (hypoxia) | 256 |
| X-ray | 131I-HSA | Upregulation of Caveolin-1 | Improve the cancer cell uptake of 131I-HSA NPs | Enhance RT | Cancer cells | 264 |
| X-ray | mPEG-b-P(LG-co-CELG) | Drug release | Enhance DOX delivery efficiency | Enhance RT and chemotherapy | Se–Se bond | 277 |
| Light | Ir@liposome | NIR light controllable catalases | Alleviate tumor hypoxia | Enhance RT | Ir nanocrystals | 284 |
| Light | CuS/131I-PEGDA/AIPH | In situ gelation | Enhance tumor retention, reduce leakage to the surrounding blood or tissues | Enhance RT | AIPH | 285 |
| Light | dAuNP-FA | In situ crosslinking | Enhance tumor accumulation and retention | Enhance RT | DA group | 303 |
| Ultrasound | Nano-PFC | Oxygen release | Alleviate tumor hypoxia | Enhance RT | PFC | 291 |
| pH/light | Cs–Au–ICG | Size-transformation | Assemble into larger-sized aggregates (pH) for enhancing tumor accumulation, disassemble into ultrasmall AuNCs (light) for deep tumor penetration | Enhance RT and PTT | Surface charge (pH), AuNCs (light) | 292 |
| pH/ROS/light | M/H-D | Degrade to release ultrasmall HfO2 NPs | Enhance HfO2 delivery efficiency, enhance tumor accumulation and penetration | Enhance RT and PTT | MoS2 nanosheets | 293 |
3.1 Endogenous stimuli-responsive strategies
Endogenous stimuli, such as pH, GSH, H2O2, hypoxia, enzyme, etc., have gradually become tumor-specific biotargets to trigger drug release, size/shape transformation, chemical degradation, or surface chemical/physical property change, which may contribute to enhanced tumor accumulation/penetration of radiosensitizers for precision RT.Yuan's group constructed an intelligent targeting system on the basis of pH-responsive self-assembly and disassembly of AuNPs. In this system, the targeting ligands could be protected inside the assembled AuNPs at the physiological pH level (pH = 7.4), but exposed when the self-assembled AuNPs disassembled at the tumor extracellular pH level (pH = 6.8), which resulted in prolonged blood circulation, reduced RES clearance and enhanced tumor accumulation.191,192 In another study by Yuan's group, they decorated small-sized AuNPs with PEG linked raltitrexed (RTX, chemotherapeutics and targeting ligand) and two small tertiary amine molecules (N,N-dibutylethylenediamine and 1-(2-aminoethyl) pyrrolidine denoted as NR1 and NR2) via lipoic acid (LA) to prepare self-assembled AuNPs (Au-NNP(RTX), Fig. 8a). The Au-NNP(RTX) was relatively stable with a large size of 160 nm at pH 7.4–7.0 but rapidly disassembled into ultrasmall AuNPs with a size of 6 nm under pH 6.8 conditions (Fig. 8b). After pH-responsive disassembly, the emerging ultrasmall AuNPs were able to penetrate into deeper tumor tissues. Simultaneously, the exposed RTX could target the FA receptor on the tumor cell surface and serve as a chemotherapeutic drug (Fig. 8c). Due to the excellent penetration and targeting abilities of small-sized AuNPs after pH-responsive disassembly, in vitro penetration evaluation on CT26 tumor spheroids showed that Au-NNP(RTX) could penetrate deep into the spheroids at pH 6.8 compared to Au-NP(RTX) that could not disassemble. The in vivo tumor accumulation/penetration investigation via CLSM of tumor slices revealed that the fluorescence signals of Au-NP(RTX) stayed around blood vasculatures at 12 h after intravenous injection while the fluorescence signals of Au-P(RTX) were partly around and away from vasculatures. However, the fluorescence signals of Au-NNP(RTX) were found both around and far away from the blood vasculatures, indicating that the pH-responsive targeting strategy allowed for better penetration into tumors. As a result, the tumor-bearing mice receiving X-ray irradiation (4 Gy) at 2 h and 12 h after intravenous injection of Au-NNP(RTX) demonstrated a much higher inhibition rate of 95.4% at 14 days after treatment, which is 24% higher than that of the Au-NNP(RTX) group. Overall, the pH-responsive disassembly strategy enhanced tumor retention/penetration of radiosensitizers and targeted delivery of surviving gene, which was beneficial to achieve precision RT/gene therapy of tumors.
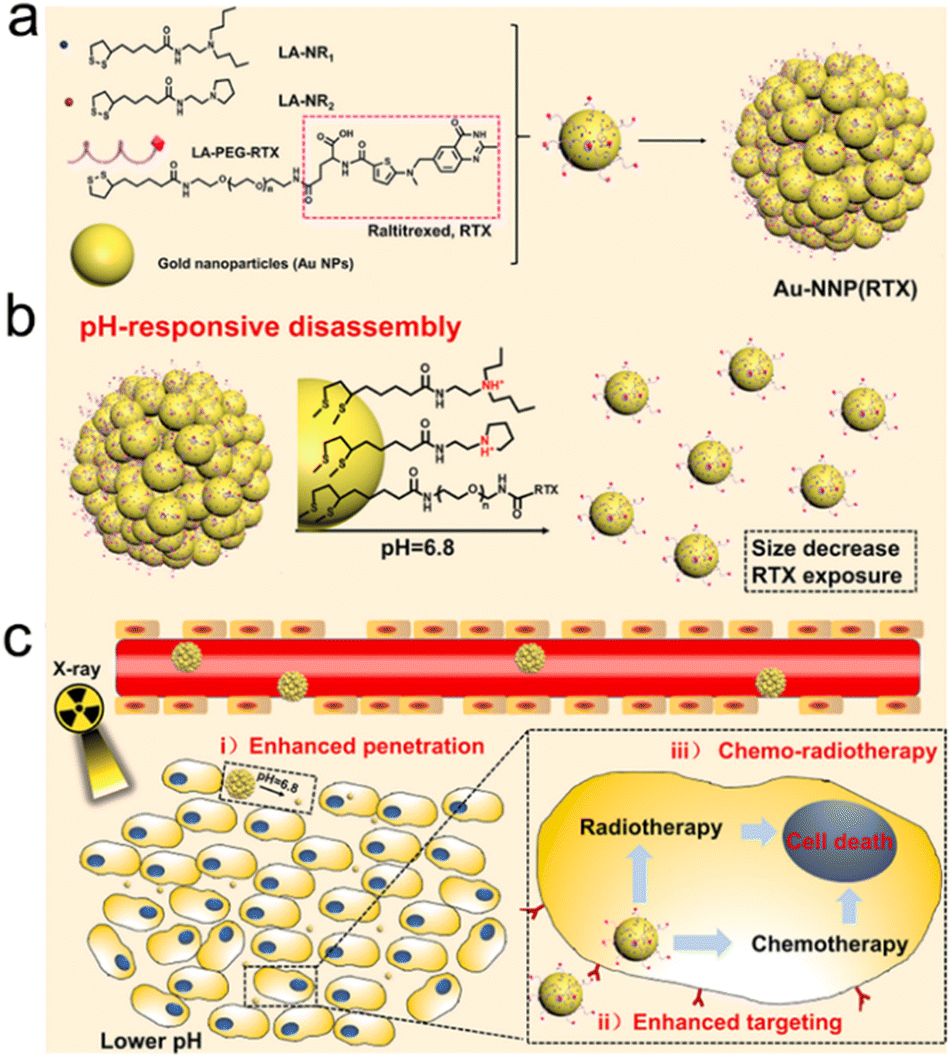 | ||
| Fig. 8 pH responsiveness. (a) A scheme of Au-NNP(RTX) nanoassembly. (b) A scheme showing pH-responsive disassembly of Au-NNP(RTX). (c) A scheme showing RT/chemotherapy of tumor based on Au-NNP(RTX) nanoassembly. Reproduced with permission from ref. 39. Copyright 2021, Elsevier. | ||
The low pH-responsive drug release strategy has also been used for precision RT. For example, Yu et al. designed a pH-responsive platinum (Pt)-based radiosensitizer by loading cinnamic aldehyde (CA) into Pt@human serum albumin NPs (Pt@HSA/CA NPs) for enhanced RT of tumors.193 After the Pt@HSA/CA NPs entered the tumor cells, CA was released due to the break of pH-responsive imine bond at pH 5.5. The released CA could break the intracellular redox homeostasis, reduce antioxidant contents, and increase the concentration of H2O2. Pt could further catalyze the decomposition of H2O2 into O2 for sensitizing the tumor cell to X-ray irradiation. In addition, the in vivo tumor inhibition rate of mice in the Pt@HSA/CA + X-ray group was 91.2%, which was much higher than 74.3% in the Pt@HSA + X-ray group. Thus, this pH-responsive precision RT strategy was able to enhance the RT efficacy against tumors with negligible side effects on healthy tissues.
In addition, Bu's group utilized 2-nitroimidazole, 1H-imidazole-4-carbonitrile and Zn2+ to construct pH-sensitive ZIF-82 nanocrystals for hypoxic prostate cancer therapy.194 On entering the cancer cells, the ZIF-82 nanocrystals can be degraded into electrophilic ligands and Zn2+. The low-energy electrons generated by X-ray irradiation can be captured by these electrophilic ligands to produce nitrite (NO2−). The NO2− is able to augment intratumoral nitrosative stress and inhibit autophagy to enhance X-ray therapeutic efficacy. In addition, the released Zn2+ can suppress migration and invasion of prostate cancer cells via ion interference. Overall, the promising strategy of X-ray-induced nitrosative stress showed a significant inhibitory effect on hypoxic prostate tumor.
Recently, Ding et al. synthesized an NIR discrete metallacycle (M) using tetraphenylethylene-based di-Pt(II) organometallic precursor (TPE-Pt) and perylene bisimide fluorophore (PPy) (Fig. 9a) for the chemoradiotherapy of tumor.201 Next, they used a GSH-responsive copolymer (RGD-POEGMA-b-PAZMB) to encapsulate M for the construction of M-loaded NPs (MNPs) (Fig. 9b). Due to GSH-triggered elimination of AZMB groups, the MNPs could rapidly release 51.5% and 86.4% of M at 48 h post-incubation with 1 mM and 10 mM GSH, respectively (Fig. 9c). TPE-Pt in MNPs could serve as both a chemotherapeutic drug and a radiosensitizer. Based on GSH-responsive radiosensitization, MNPs plus X-ray irradiation (6 Gy) showed the highest tumor inhibition rate (83.2%) compared to 42.3% of the X-ray alone group (Fig. 9d).
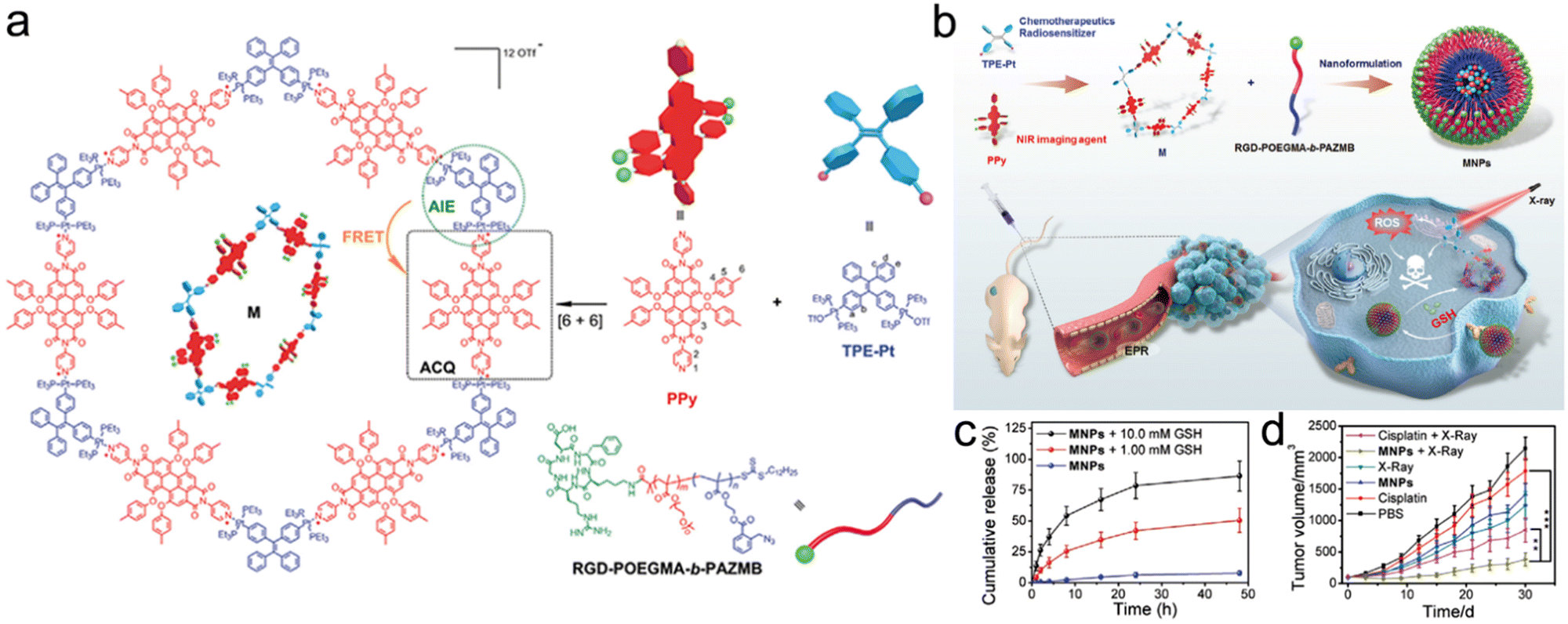 | ||
| Fig. 9 GSH responsiveness. (a) Chemical structures of PPy, TPE-Pt, M, and RGD-POEGMA-b-PAZMB. (b) Schematic illustration of MNPs self-assembled from M and RGD-POEGMA-b-PAZMB, as well as their application in RT/chemotherapy. (c) Release profiles of MNPs with or without GSH at various concentrations. (d) Tumor growth curves of mice in different treatment groups. Reproduced with permission from ref. 201. Copyright 2020, Wiley. | ||
Disulfide (S–S) bond has been used to prepare GSH-responsive nanomedicines. For example, Song et al. decorated HA onto Bi2Se3 hollow nanocubes (HNCs) via the S–S bond, followed by loading of gambogic acid (GA, a heat shock protein inhibitor) for redox-responsive and tumor-targeted RT/PTT.202 The Bi2Se3 HNC-s-s-HA/GA could actively target CD44-overexpressed cancer cells due to HA modification. Moreover, due to GSH-responsive cleavage of the S–S bond, the Bi2Se3 HNC-s-s-HA/GA could collapse in the presence of GSH and then release GA to downregulate the expression level of heat shock protein for eliminating cancer cells’ resistance to PTT. Considering that the enhanced RT could kill deep tumor cells, the synergistic low-temperature PTT and RT based on the GSH-responsive Bi2Se3 HNC-s-s-HA/GA decreased the relative tumor volume to 0.2 at 14 days after treatment, much lower than 1.1 in the non-responsive group (Bi2Se3 HNC/GA + NIR +RT).
The ROS-responsive strategy has been applied in the design of radiosensitizers for precision RT.215–217 For example, Meng et al. designed a ROS-responsive nanoplatform by loading acriflavine (ACF, a cationic and hydrophilic HIF-1 inhibitor) into MnO2 NPs (ACF@MnO2 NPs) for enhanced RT against primary and metastatic tumors (Fig. 10a and b).216 The ACF was loaded onto the basal template MnO2 NPs (MnO2-ACF intermediate) via electrostatic adsorption. Next, after additional MnO2 redeposition, an external shell was constructed onto the MnO2-ACF intermediate to obtain final ACF@MnO2 NPs. After arriving in the lysosomes of tumor cells, ACF@MnO2 NPs could react with acidic H2O2 to generate O2 and Mn2+ for alleviating tumor hypoxia and increasing T1-weighted MR imaging contrast, which could be used for radiosensitization and MR-guided RT, respectively. The UV absorption of ACF@MnO2 NP solution ([MnO2 = 400 μM]) at 400 nm was gradually reduced when the H2O2 concentration was increased from 0 to 400 μM, indicating the rapid ROS-responsiveness of ACF@MnO2 (Fig. 10c). The H2O2-responsive degradation of MnO2 enabled the rapid release of ACF (Fig. 10d), which could suppress the transcription of HIF-1 to inhibit downstream signaling molecules. They found that alleviating tumor hypoxia and inhibiting transcription of HIF-1 could downregulate the PD-L1 expression level and then relieve T-cell exhaustion. Excitingly, the infiltration of CD8+ T cells in tumors treated with ACF@MnO2 plus RT was similar to that in tumors treated with anti-PD-L1 plus RT. The bilateral CT26 tumor-bearing mice treated with ACF@MnO2 plus RT showed excellent tumor inhibition rates of both primary tumors (88.72%) and distant tumors (78.9%), much better than those in the anti-PD-L1 plus RT group (65.66% for primary tumors and 67.59% for distant tumors) (Fig. 10e and f). Furthermore, the ACF@MnO2 plus RT could also inhibit the metastases of the lung and liver of 4T1 breast tumor-bearing mice. All these results suggest that ROS-responsive ACF@MnO2 in combination with X-ray irradiation could effectively suppress the growth of primary tumors and activate immune responses against abscopal tumors through alleviation of tumor hypoxia and HIF-1 functional suppression.
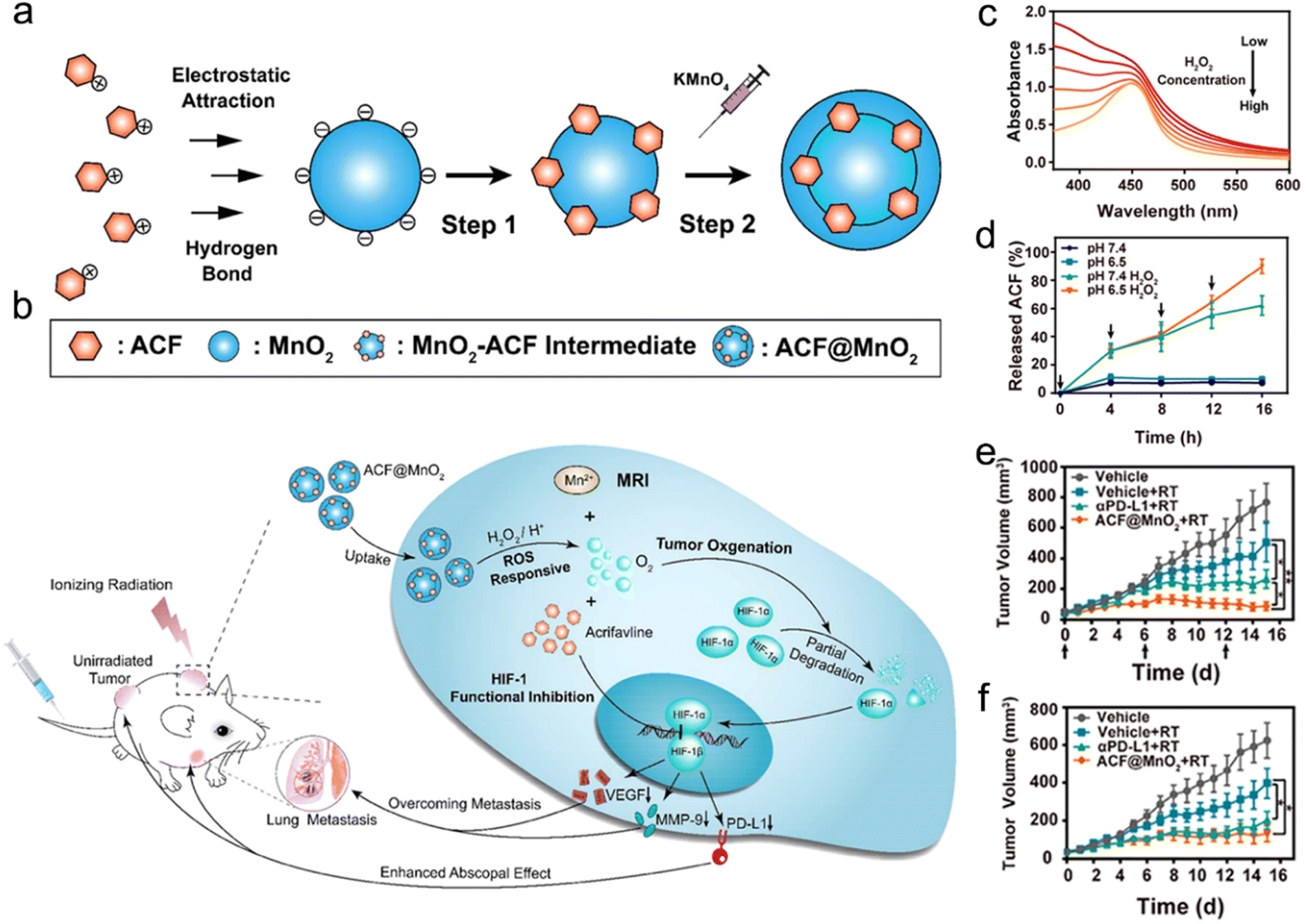 | ||
| Fig. 10 ROS responsiveness. (a) A scheme of synthetic procedures of ACF@MnO2. (b) Schematic illustration showing the mechanism of ACF@MnO2 for RT and abscopal effect. (c) UV/vis absorption spectra of ACF@MnO2 treated with H2O2 at different concentrations. (d) ACF release profiles of ACF@MnO2 with or without H2O2 under pH 7.4 or pH 6.5. (e and f) Primary (e) and distant (f) tumor growth curves of mice that received different treatments. Reproduced with permission from ref. 216. Copyright 2018, American Chemical Society. | ||
The radiosensitizers in response to diverse types of enzymes have been reported to specifically enhance RT and reduce adverse effects on healthy tissues for precision RT.226–228 For example, Ding et al. designed multifunctional responsive peptide-modified AuNPs (Au@Tat-R-EK) for tumor-specific targeted RT (Fig. 11a).227 The peptide was composed of three building blocks. A Tat peptide section (GRKKRRQRRRPQ) as the first unit acquired from HIV-1 transactivator of transcription, was directly decorated onto the AuNPs through the Au–S bonds for cell penetration and nucleus targeting.229,230 The second unit was a cathepsin B-cleavable peptide (GFLG).231–233 The outer peptide, a zwitterionic peptide (EKEKEKEKEK), endowed the NPs with outstanding biocompatibility and passive tumor targeting capability.234,235 Cathepsin B, one of the lysosomal proteases, is significantly upregulated in the TME of various cancers, and the enzyme is secreted and bound to the cell surface.236,237 The peptide (CFLG) of Au@Tat-R-EK could be selectively cleaved in response to cathepsin B in the TME. After specific split, the Tat peptide section was exposed, which adjusted the surface charge of AuNPs (Au@Tat) to be positive (Fig. 11b), suggesting successful enzyme responsiveness. Moreover, the Au@Tat NPs were able to enter cancer cells and target the nucleus, realizing nuclear accumulation of AuNPs. To evaluate the enzyme-responsive cell uptake, the cathepsin B-irresponsive peptide was used to prepare Au@Tat-I-EK as a counterpart of Au@Tat-R-EK. The in vitro cellular uptake results showed that the intracellular content of Au@Tat-I-EK was quite low even with additional cathepsin B. Addition of cathepsin B to culture medium significantly increased LM3 cell endocytosis of Au@Tat-R-EK. However, after adding GM6001, an inhibitor of cathepsin B, into the medium, the cellular uptake of Au@Tat-R-EK was remarkably inhibited. As a result, Au@Tat-R-EK exhibited great cytotoxicity in vitro, which could be enhanced or declined after adding cathepsin B or GM6001, respectively. In addition, the biodistribution of AuNPs by ICP-MS revealed that the tumor-bearing mice treated with Au@Tat-R-EK showed a higher tumor accumulation of 9.6% ID per g at 24 h post-injection than those in the Au@Tat-I-EK group (2.7% ID per g) (Fig. 11c). Finally, the tumor inhibition rate caused by Au@Tat-I-EK-mediated radiosensitization was 5.3 times that of the RT alone group.
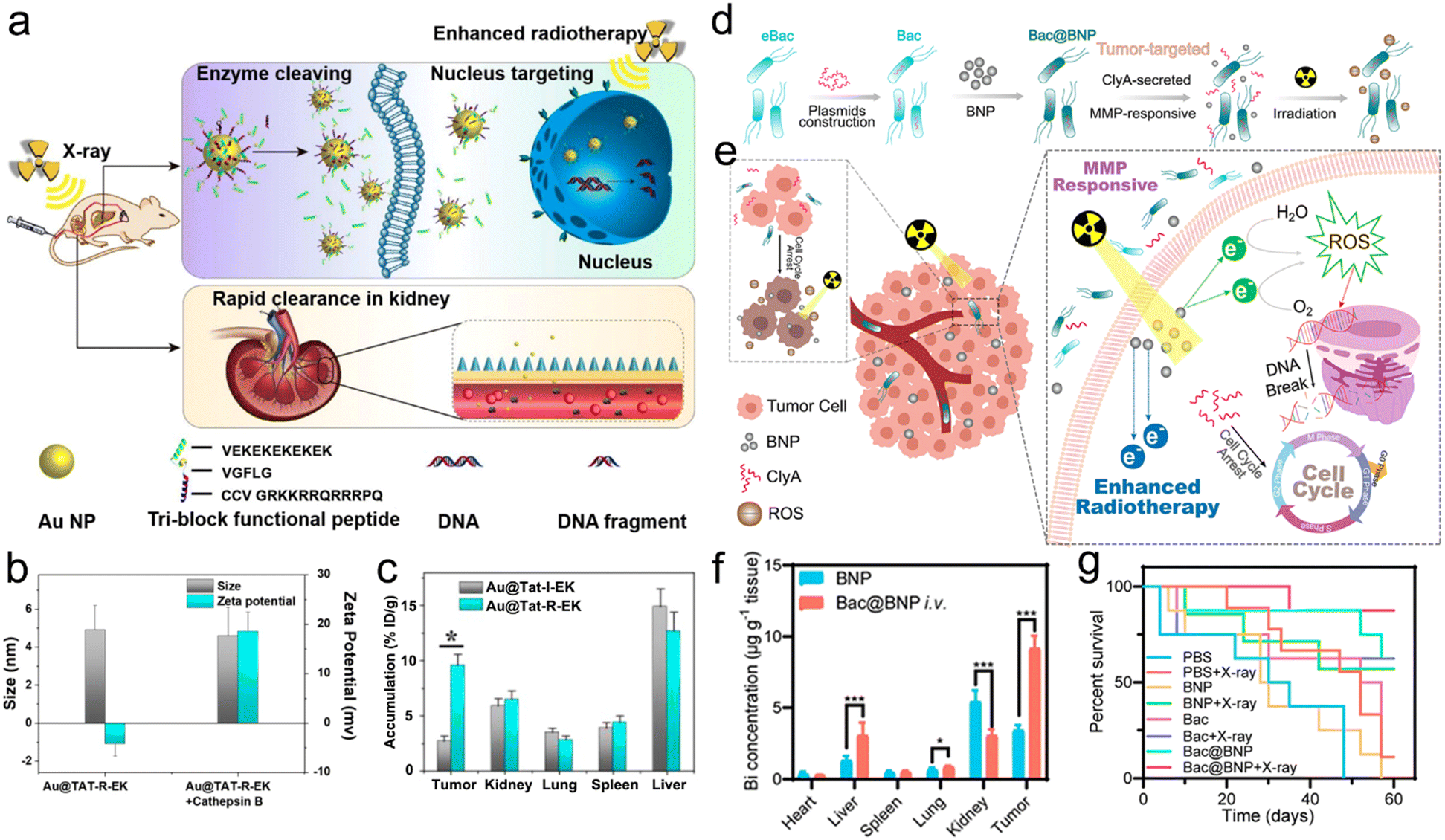 | ||
| Fig. 11 Enzyme responsiveness. (a) A scheme showing the enzyme cleaving, nucleus targeting, and rapid renal clearance of Au@Tat-R-EK NPs. (b) Hydrodynamic sizes and zeta potentials of Au@Tat-R-EK NPs treated with or without cathepsin B. (c) Biodistribution of Au NPs in the major organs and tumors of mice at 24 h after intravenous administration of Au@Tat-I-EK or Au@Tat-R-EK NPs. Reproduced with permission from ref. 227. Copyright 2020, Ivyspring International Publisher. (d) A scheme of synthetic procedures and enzyme-responsive process of the Bac@BNP nanosystem. (e) Schematic illustration showing the MMP-responsive Bac@BNP nanosystem for enhanced RT. (f) Biodistribution of the Bi element in major organs and tumors of mice at 48 h after intravenous administration of BNP or Bac@BNP. (g) Survival curves of tumor-bearing mice in different treatment groups. Reproduced with permission from ref. 228. Copyright 2020, American Chemical Society. | ||
Matrix metalloproteinases (MMPs), enzymes secreted by cancer cells, were reported to be extracellularly overexpressed in various malignant tumors, such as colon, breast, and brain cancers.238–241 Accordingly, the elevated expression of MMPs has been widely used to design enzyme-responsive nanosystems for cancer theranostics.242–244 For example, the Zhang's group developed a smart biomimetic nanosystem composed of engineered bacteria (eBac) and Bi2S3 NPs (BNPs) for enhanced RT of breast carcinoma (Fig. 11d).228 Concretely, the Escherichia coli MG1655 was first transferred with pBAD18-ClyA plasmid for upregulation of the ClyA protein. Next, the matrix metalloproteinases-2 (MMP-2)-responsive peptide (PLGVR)-modified BNPs were chemically conjugated onto the eBac surface to construct Bac@BNP. The Bac@BNP was able to target the tumors owing to the tropism of bacteria. When Bac@BNP accumulated at the tumor sites, BNPs with a high-Z element were released to deposit X-ray energy, and the upregulated ClyA in Bac was secreted to arrest the cell cycle for radiosensitization (Fig. 11e). The in vitro cell cycle analyses via flow cytometry showed that the incubation of 4T1 cells with Bac and L-arabinose (an activator for ClyA expression) could significantly increase the G2/M phase from 21.06% to 45.23% and reduce the S phase from 36.89% to 3.04%. However, the cell cycles of 4T1 cells incubated with plasmid-transferred Bac (pBac) or Bac without L-arabinose showed no significant change. The in vitro drug release result revealed that Bac@BNP could rapidly release the BNPs in the presence of MMP-2, whereas the addition of MMP-2 inhibitor could significantly inhibit the BNP release. Moreover, the in vivo biodistribution evaluation via ICP-MS analyses of the Bi element showed that the tumor accumulation of BNPs at 48 h after intravenous injection of Bac@BNP was almost 3 times that treated with BNPs alone (Fig. 11f). Therefore, the 4T1-luc tumor-bearing mice in the Bac@BNP + X-ray group showed 87.5% of survival proportion even at 60 days after treatment, whereas the mice in the X-ray alone group all died within 30 days (Fig. 11g). Overall, the endogenous enzyme-responsive radiosensitizers can improve the RT effectiveness. However, the presence of different enzyme subtypes with a similar cleavage site should be considered during the design of enzyme-responsive radiosensitizers to prevent off-target effect or serious side effect.
For instance, Hua et al. developed a hypoxia-responsive angiopep-2-lipid-poly(metronidazoles)n (ALP-(MIs)n) to specifically sensitize hypoxic tumor cells to RT.255 The ALP-(MIs)n NPs were composed of three ingredients: (1) the inner side was the hydrophobic poly(metronidazoles)n (P-(MIs)n) core which could load with small molecule drugs; (2) the nitro groups in P-(MIs)n could be transformed into hydrophilic amino groups in response to hypoxia, leading to drug release; (3) the outer side was a lipid layer modified with angiopep-2 (Fig. 12a–c). They also prepared AL-PLGA and AL-PLGA/DOX as negative controls. Then, ALP-(MIs)n (n = 25, 48) NPs were loaded with DOX for hypoxia-responsive RT/chemotherapy of glioma. ALP-(MIs)25 with uniform spherical morphology under normoxic conditions was decomposed in response to hypoxic conditions, whereas the AL-PLGA NPs showed no change under hypoxic conditions (Fig. 12d). Moreover, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were stable under normoxic conditions but precipitated under hypoxic conditions since the nitro groups transformed into amino groups in response to hypoxia. With the hypoxia responsiveness, ALP-(MIs)25/DOX and ALP-(MIs)48/DOX could rapidly release most DOX within 4 h of incubation with hypoxic PBS. Besides, the angiopep-2, a ligand for low density lipoprotein receptor-related protein 1 (LRP-1) expressed in brain microvascular endothelial cells, enabled ALP-(MIs)n to cross the blood–brain barrier (BBB) and then accumulate in the glioma. In vivo DOX accumulation investigation revealed that the DOX concentrations in brain tissues of mice treated with ALP-(MIs)25/DOX and ALP-(MIs)48/DOX were significantly higher than those treated with LP-(MIs)n/DOX (without angiopep-2 modification) or AL-PLGA/DOX (without hypoxia-responsiveness) (Fig. 12e). Due to higher glioma accumulation and hypoxia-responsive DOX release, the orthotopic C6-GFP-Luci glioma-bearing mice receiving 2 Gy of RT after injection of ALP-(MIs)25/DOX (57 days) or ALP-(MIs)48/DOX (59 days) exhibited a longer median survival time than those in LP-(MIs)25/DOX + RT (49 days) and LP-(MIs)48/DOX + RT groups (49.5 days). Overall, the design of endogenous stimuli-responsive radiosensitizers based on hypoxia, a distinctive feature of tumor tissues, is a promising strategy for precision RT. In further research, the stronger tumor penetration ability needs to be considered in radiosensitizer design since the degree of hypoxia in deep tumor tissues is much higher.
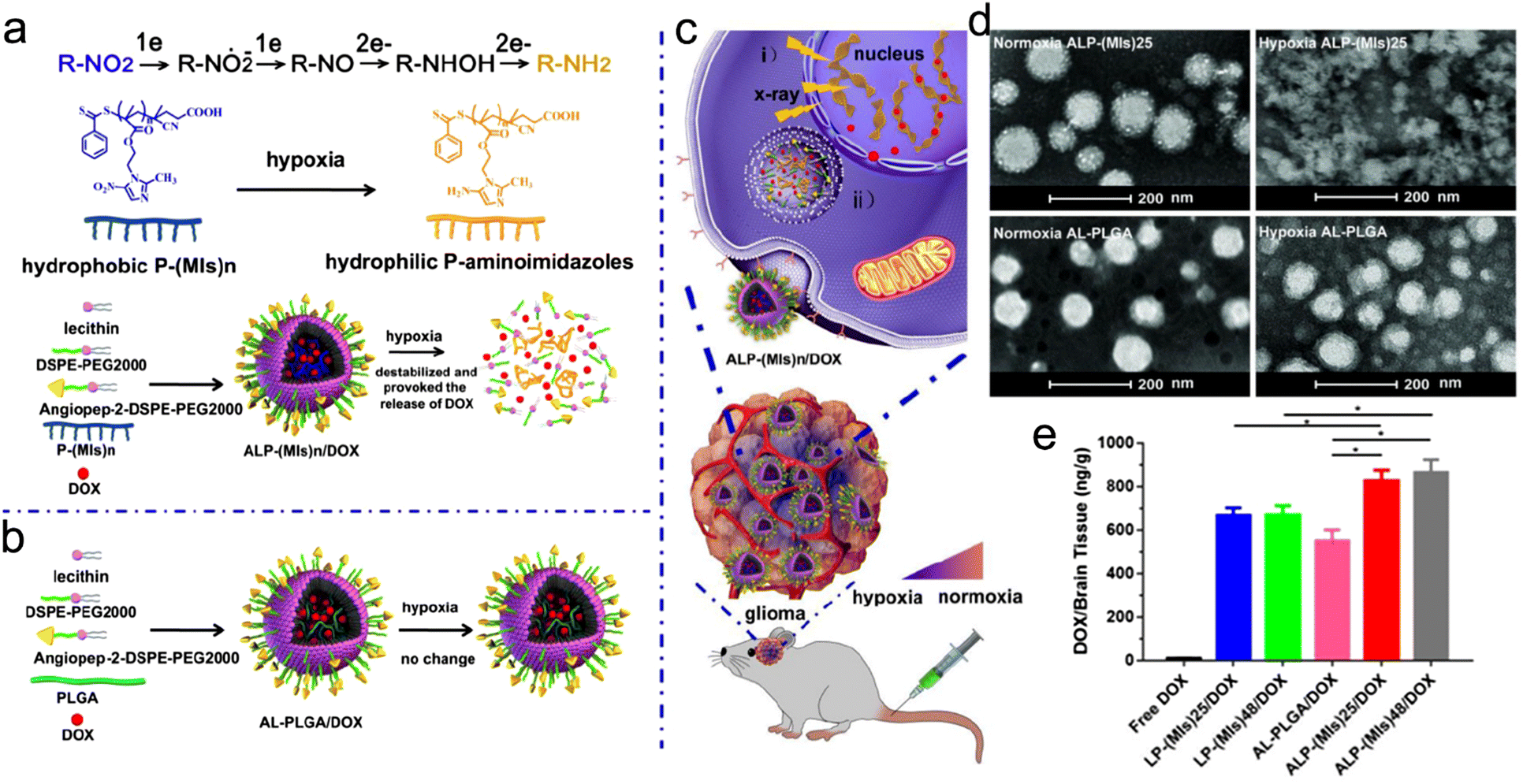 | ||
| Fig. 12 Hypoxia responsiveness. (a) A scheme of ALP-(MIs)n for hypoxia-responsive DOX release and radiosensitization. (b) Construction of AL-PLGA/DOX as the negative control. (c) A scheme showing the applications of ALP-(MIs)n: (i) radiosensitization in hypoxic cells; (ii) hypoxia-responsive DOX release. (d) TEM images of ALP-(MIs)25 and AL-PLGA under normoxic or hypoxic conditions. (e) DOX content in mouse brain of different treatment groups. Reproduced with permission from ref. 255. Copyright 2018, Ivyspring International Publisher. | ||
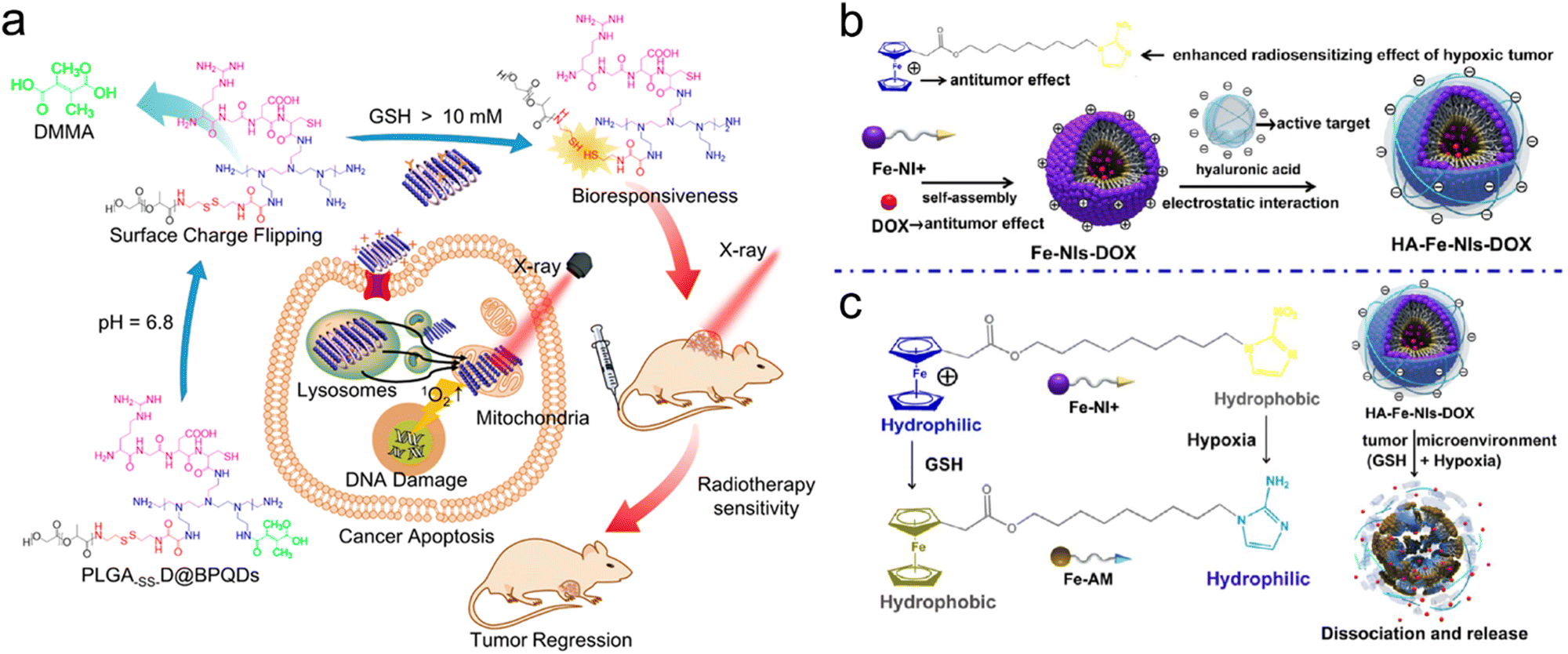
For example, Chan et al. designed a sequentially pH/GSH-responsive delivery nanosystem (PLGA-SS-D@BPQDs) based on black phosphorus quantum dots (BPQDs) and poly(lactic-co-glycolic acid) (PLGA) for precise RT of tumors. The NPs were coated with several shells of polymers, including polyethylenimine (PEI), 2,3-dimethylmaleic anhydride (DMMA), and RGD polypeptide, for sequential pH/GSH-responsive delivery (Fig. 13a).103 First, the conjugation of RGD polypeptide endowed the PLGA-SS-D@BPQD NPs with active tumor-targeting capability. Next, after reaching the TME, the NPs could gradually expand in response to acidic conditions (pH = 6.8) due to the hydrolyzation of amide linkages of DMMA, which changed the surface potential of NPs from −7.8 mV to +35.5 mV due to the exposure of the PEI layer. The positive charges potentiated tumor cell uptake of the NPs. Moreover, the average size of the NPs expanded from 157 nm to over 2500 nm in both pH 6.8 solution and tumor homogenate. Then, the disulfide bonds between the PEI layers and the PLGA were broken by intracellular GSH, which decreased the size of expanded NPs to 139 nm and released the ultrasmall BPQDs that sensitized tumor cells to X-ray irradiation. For antitumor efficacy investigation, the tumor inhibition rate of the PLGA-SS-D@BPQDs + X-ray group was 2.8 times that of the BPQDs + X-ray group.
 | ||
| Fig. 13 Dual endogenous stimuli responsiveness. (a) Schematic illustration of synthetic procedures of PLGA-SS-D@BPQDs and their application in RT of tumors. Reproduced with permission from ref. 103. Copyright 2018, American Chemical Society. (b) A scheme of synthetic procedures of HA-Fe-NIs-DOX micelles. (c) Schematic illustration showing the mechanism of HA-Fe-NIs-DOX micelles under hypoxia and GSH. Reproduced with permission from ref. 256. Copyright 2018, American Chemical Society. | ||
In addition to the sequential dual-responsive strategy, synergistic dual-responsive strategy has been also reported to enhance precision RT. For example, Mao et al. fabricated amphiphilic ferrocenium-hexane-nitroimidazoles (Fe-NIs) as a GSH/hypoxia dual-responsive nanocarrier for chemoradiotherapy (Fig. 13b).256 The Fe-NI micelles were loaded with DOX to construct HA-Fe-NIs-DOX micelles for chemotherapy and decorated with HA via electrostatic interactions for active tumor targeting. It is worth mentioning that the HA-Fe-NIs-DOX micelles were able to dissociate and release DOX in response to dual endogenous GSH/hypoxia stimuli (Fig. 13c). On the one hand, the hydrophilic ferrocenium ion was converted into hydrophobic ferrocene with effective antitumor effect under GSH conditions.261 On the other hand, NIs could function as a radiosensitizer, and the hydrophobic NIs in the micelles could be selectively reduced to hydrophilic aminoimidazoles in the presence of hypoxia.262,263 HI-Fe-DOX micelles without radiosensitization were prepared as the negative control. The TEM images showed a well-dispersed micelle structure of HA-Fe-NIs-DOX. Then, the in vitro GSH/hypoxia dual-responsiveness of HA-Fe-NIs micelles was investigated by TEM imaging. The HAase was first used to degrade the HA shell to expose the Fe-NI micelles. After incubation under GSH (10 mM), HAase and hypoxic conditions, the HA-Fe-NI micelles were rapidly disassembled and the yellow precipitates were observed. The in vitro drug release experiments revealed that more than 80% DOX was released from the HA-Fe-NIs-DOX micelles within 6 h of incubation with hypoxia, GSH, or hypoxia plus GSH while only 37% DOX was released even after 12 h in pH 7.4 PBS buffer. Finally, due to excellent chemotherapeutic efficacy and radiosensitive effect, HA-Fe-NIs-DOX plus X-ray irradiation effectively inhibited the tumors with 0.89 of relative tumor volume at 20 days after treatment, much lower than that in the HA-Fe-DOX + X-ray group (4.03) without a hypoxic radiosensitizer.
Overall, dual endogenous stimuli-responsive strategy may indeed realize the precise delivery of radiosensitizers and lessen the side effect on healthy tissues, but it may also result in poor RT efficacy due to the complicated response process. In addition, the strength of endogenous stimuli varies spatiotemporally since it is affected by various factors in the body, including tumor type, progression, location, and individual differences between patients, resulting in unpredictable and uncontrollable antitumor effects. Furthermore, the cost of making endogenous stimuli-responsive radiosensitizers, including synthesis of specific cleavable peptide and other responsive small-molecules, also need to be considered to promote the translational use of these radiosensitizers.
3.2 Exogenous stimuli-responsive strategies
Exogenous stimuli, such as X-ray, light, ultrasound, etc., have attracted extensive interest as a remote switch to spatially and temporally trigger morphology transformation of nanostructures or drug release. With exogenous stimuli, the location, timing, radiosensitizer dosage and tumor accumulation/retention can be controlled for precision RT. Moreover, these exogenous stimuli can also endow radiosensitizers with multiple functions beyond RT or radiosensitizer delivery, such as PA imaging, ultrasound imaging, CT imaging and photothermal therapy.X-Ray irradiation on the tumor site can significantly enhance the tumor accumulation/retention of many different types of radiosensitizers and meanwhile reduce their efflux through various mechanisms, thus leading to precision RT.264–266 For example, Yang's group proposed an X-ray-optimized delivery strategy by enhancing tumor accumulation/retention of radionuclide-labeled human serum albumin (HSA) NPs upon X-ray irradiation (Fig. 14a).264 First, the expression levels of Caveolin-1 in various cancer cells were upregulated at 24 h after X-ray irradiation. Previous studies reported that the upregulated expression levels of Caveolin-1 in cancer cells could increase the uptake of HSA-based NPs.267,268 The flow cytometry of Cy5.5-labeled HSA revealed that the cell uptake of HSA NPs was remarkably increased after 6 Gy of X-ray irradiation. Next, the HSA NPs were labeled with 131I (131I-HSA) to evaluate their in vivo tumor accumulation using γ imaging. The bilateral CT26 tumor-bearing mice were intravenously injected with 131I-HSA NPs, followed by γ imaging. The γ images showed that the radioactivity in the tumor pre-exposed to X-rays was still detectable at 72 h post-injection while the radioactivity in the control tumor could not be detected after 48 h, indicating enhanced tumor retention of 131I-HSA under X-ray irradiation (Fig. 14b). In addition, the prolonged tumor retention time after X-ray exposure could potentiate the radionuclide therapy of 131I-HSA. The in vivo antitumor efficacy evaluation revealed that the tumor inhibition rate of CT26 tumor-bearing mice injected with 131I-HSA at 24 h after X-ray irradiation was 1.5 times that of mice receiving X-ray irradiation at 24 h post-injection of 131I-HSA, further suggesting that X-ray-optimized tumor retention could improve the radiosensitizing effect.
 | ||
| Fig. 14 X-ray stimuli responsiveness. (a) Schematic illustration showing the mechanism of HSA delivery under X-ray irradiation. (b) In vivo γ imaging of mice bearing bilateral CT26 tumors at various time points after intravenous administration of 131I-HSA. The tumor 1 (left) was preirradiated. Reproduced with permission from ref. 264. Copyright 2020, Elsevier. (c) Schematic illustration of synthetic procedures of RBS-T-SCNPs and its application in X-ray-controlled ONOO− generation for tumor RT. Reproduced with permission from ref. 272. Copyright 2018, Wiley. | ||
Furthermore, X-ray-responsive drug release is another strategy to sensitize cancer cells to RT.269–271 For example, Du et al. developed an X-ray-triggered peroxynitrite (ONOO−) production nanoplatform for improved radiosensitizing effect (Fig. 14c).272 First, the Ce-doped LiLuF4 was chosen as scintillating NPs (SCNPs) to be conjugated with tocopheryl polyethylene glycol 1000 succinate (TPGS) through electrostatic interaction. Next, the as-prepared T-SCNPs were loaded with Roussin's black salt (RBS, a photo-responsive NO donor) via electrostatic attraction to produce RBS-T-SCNPs. The LiLuF4:Ce3+ in the RBS-T-SCNPs could function as a radiosensitizer to generate ROS, such as O2˙−, upon X-ray irradiation. In addition, the SCNPs were able to convert X-ray into UV light to trigger the decomposition of RBS to generate NO. Thus, the X-ray-triggered generation of O2˙− and NO resulted in the production of ONOO−. The ONOO− could contribute to radiosensitization by effectively aggravating DNA damage and downregulating DNA-repair enzyme. The in vitro NO release under X-ray irradiation presented an “on/off” profile, suggesting X-ray-responsive NO release. Flow cytometry revealed that the fluorescence intensity of 3-amino-4-aminomethyl-2′,7′-difluorescein, diacetate (DAF-FM, a NO fluorescence indicator) in the A549 cells treated with RBAS-T-SCNPs plus X-ray irradiation was much higher than those treated with RBAS-T-SCNPs or X-ray alone. Therefore, RBAS-T-SCNPs exhibited almost no cytotoxicity whereas effectively decreased in vitro cell viability to only 5.9% compared to 52.1% in the X-ray alone group. Besides, the in vivo tumor inhibition rate of the RBAS-T-SCNPs + X-ray group after 20 days of treatment was more than 4 times higher than that of X-ray alone group. These results indicate that the X-ray-triggered sufficient release of ONOO− from RBS-T-SCNPs could significantly enhance RT efficacy.
Recently, various X-ray-responsive nanoplatforms based on the intrinsic weakness of certain chemical bonds have been designed for precision RT. For instance, S–S (240 kJ mol−1) and diselenide (Se–Se, 172 kJ mol−1) bonds have been harnessed to construct X-ray-responsive nanosystems for controllable drug release due to X-ray-induced cleavage of the S–S or Se–Se bonds.273–276 Wang et al. synthesized an X-ray responsive DOX-loaded polypeptide nanogel (PNG/DOX) for on-demand controllable DOX release and synergistic RT/chemotherapy of human non-small-cell lung carcinoma (NSCLC).277 The PNG was prepared by crosslinking poly(ethylene glycol)-block-poly(L-glutamic acid-co-γ-2-chloroethyl-L-glutamate) (mPEG-b-P(LG-co-CELG)) with an X-ray responsive Se–Se bond, which was further loaded with DOX as a model drug. The in vitro drug release result showed that DOX was rapidly released due to the Se–Se bond break upon X-ray irradiation and the released DOX amount was increased with the elevated X-ray dose. The X-ray triggered rapid release of DOX could improve the radiosensitizing effect and result in 2.4 of relative tumor volume after 24 days of treatment compared to 3.6 in the DOX + X-ray group (negative group).
The light stimulus has been shown to improve the radiosensitizing effect. For example, Feng et al. fabricated iridium nanocrystal (IrNC)-encapsulated liposomes (Ir@liposome) to regulate tumor oxygenation for radiosensitization.284 Concretely, the Ir@liposome NPs could effectively catalyze oxygen generation from H2O2 to alleviate tumor hypoxia under NIR light-triggered mild hyperthermia (Fig. 15a). Next, the Ir as a high-Z element was able to deposit X-ray energy, which could combine with tumor hypoxia relief to improve the radiosensitizing effect. The as-prepared Ir@liposome NPs showed stronger catalytic activity with elevated temperature in vitro, whereas the activity of biological catalase showed almost no change after heating (Fig. 15b and c). Moreover, the NIR light at 1 W cm−2 could significantly improve the oxygen generation from Ir@liposome due to the photothermal heating effect, indicating the temperature-dependent catalytic activity of Ir@liposome NPs (Fig. 15d). Since GSH could effectively inhibit the catalytic activity of IrNCs, the Ir-GSH@liposome was set as the negative control. The ex vivo immunofluorescence staining of tumor tissues collected from mice treated with Ir@liposome plus NIR light irradiation revealed that the hypoxia area in tumor slices was significantly reduced, indicating the great catalase activity of Ir@liposome (Fig. 15e). However, the hypoxia area in tumors treated with Ir-GSH@liposome did not change significantly even with laser irradiation. Based on NIR light controllable hypoxia relief and the high-Z element of Ir@liposome, tumors receiving NIR light and X-ray irradiation post-injection of Ir@liposome were remarkably eliminated, whereas the relative tumor volume was almost 4 in the X-ray alone group after 2 weeks of treatment.
 | ||
| Fig. 15 Light stimuli responsiveness. (a) A scheme of NIR light controllable theranostic nanozyme (Ir@liposome) and its application in PA imaging, NIR-enhanced tumor oxygenation, and radiosensitization. (b) A scheme showing heating and NIR light controllable oxygenation through Ir@liposome mediated H2O2 decomposition. (c) Relative catalytic activities of catalase and Ir@liposome toward H2O2 decomposition at various temperatures for 1 min. (d) Oxygen generation profiles of H2O2 solutions with or without Ir@liposome under 785 nm laser irradiation (1 W cm−2) or not. (e) Quantification analysis of tumor hypoxia regions in tumor sections with different treatments. Reproduced with permission from ref. 284. Copyright 2018, Elsevier. (f) Schematic illustration of localized synthesis of the CuS/131I-PEGDA hydrogel at the tumor site. (g) Schematic illustration showing the dispersion of CuS/131I-PEGDA/AIPH in the tumor region after local injection. (h) In vivo γ imaging of mice at different time points after local injection of various NPs with 915 nm laser preirradiation for gelation or not. Reproduced with permission from ref. 285. Copyright 2018, American Chemical Society. | ||
Besides, external light can trigger morphology/size transformation of nanomaterials to enrich their tumor accumulation/retention for enhanced RT. For example, Liu's group proposed a light-triggered localized gelation strategy by making use of a hybrid hydrogel system for repeated photothermal brachytherapy (Fig. 15f).285 The hydrogel system (CuS/131I-PEGDA/AIPH) was fabricated by using 131I-labeled copper sulfide (CuS/131I) NPs as both a radionuclide therapeutic drug and a photothermal agent, PEG double acrylates (PEGDAs) as a polymeric matrix, and 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (AIPH) as a thermal activator. With great photothermal conversion performance, the gelation of CuS-PEGDA NPs could be formed in vitro when the temperature reached 43 °C upon 915 nm laser (2.0 W cm−2) irradiation in the presence of AIPH (a thermal initiator). After local injection into the tumor sites, CuS/131I-PEGDA/AIPH could gradually diffuse into the whole tumor area in 10 min, validated by in vivo PA imaging (Fig. 15g). Next, the subcutaneous 4T1 tumors were irradiated with 915 nm laser (1.0 W cm−2, 12 min) for 10 min post-injection of CuS/131I-PEGDA and AIPH. Upon laser irradiation, the tumor temperature increased to 43 °C which further triggered gelation. The gelation could fix the CuS/131I-PEGDA NPs in the tumors with less leakage to the surrounding blood or tissues. The in vivo γ imaging of tumor-bearing mice revealed that the tumors receiving laser irradiation after local injection of CuS/131I-PEGDA/AIPH exhibited significantly higher retention of radioactivity even at 192 h post-injection, while the radioactivity in tumors of other groups was almost undetectable at 96 h post-injection (Fig. 15h). The light-triggered localized gelation enabled long-term tumor retention of the CuS/131I-PEGDA/AIPH hydrogel system. Moreover, mild hyperthermia has been reported to effectively improve the blood supply of tumor for relieving tumor hypoxia.286,287 The immunofluorescence staining of tumor slices indicated that the repeated mild PTT at 10 min, 48 h, and 96 h after a single injection of CuS/131I-PEGDA/AIPH remarkably reduced the tumor hypoxic area compared to single hypothermia heating at 10 min post-injection. Furthermore, the long-term tumor hypoxia relief would significantly augment the radiosensitizing effect. The 4T1 tumors receiving multiple rounds of mild PTT after in situ gelation of CuS/131I-PEGDA/AIPH were rapidly regressed in 10 days, leading to mouse survival even at 60 days after treatment. However, all the mice in the CuS/131I-PEGDA/AIPH + single round of mild PTT were dead within 26 days.
 | ||
| Fig. 16 Ultrasound stimuli responsiveness. (a) Schematic illustration showing the mechanism of US-triggered tumor oxygenation and enhanced RT/PDT using nano-PFC as the oxygen shuttle. (b) In vitro oxygen concentration in water or PFC@O2 solution with or without US treatment. (c) In vivo PA imaging of Hb/HbO2 (ex = 750/850 nm) tumors before and 30 min after various treatments. (d) Quantification analysis of oxygen concentration in the tumor before and after various treatments. (e and f) Tumor growth curves (e) and tumor weight (f) of mice in different treatment groups. Reproduced with permission from ref. 291. Copyright 2016, American Chemical Society. | ||
3.3 Exo/endogenous dual stimuli-responsive strategies
Beside single exogenous or endogenous stimulus-responsive strategy, exo/endogenous dual stimuli-responsive strategies are also trending. The exo/endogenous dual stimuli-responsive strategies are able to integrate the merits of auto-response to TME and remote “on-off” switchable control, thus improving the precision of responsiveness. In addition, the complementarity of endogenous and exogenous stimuli can significantly enhance the treatment efficacy and reduce the adverse effect on healthy tissues to enable precision RT.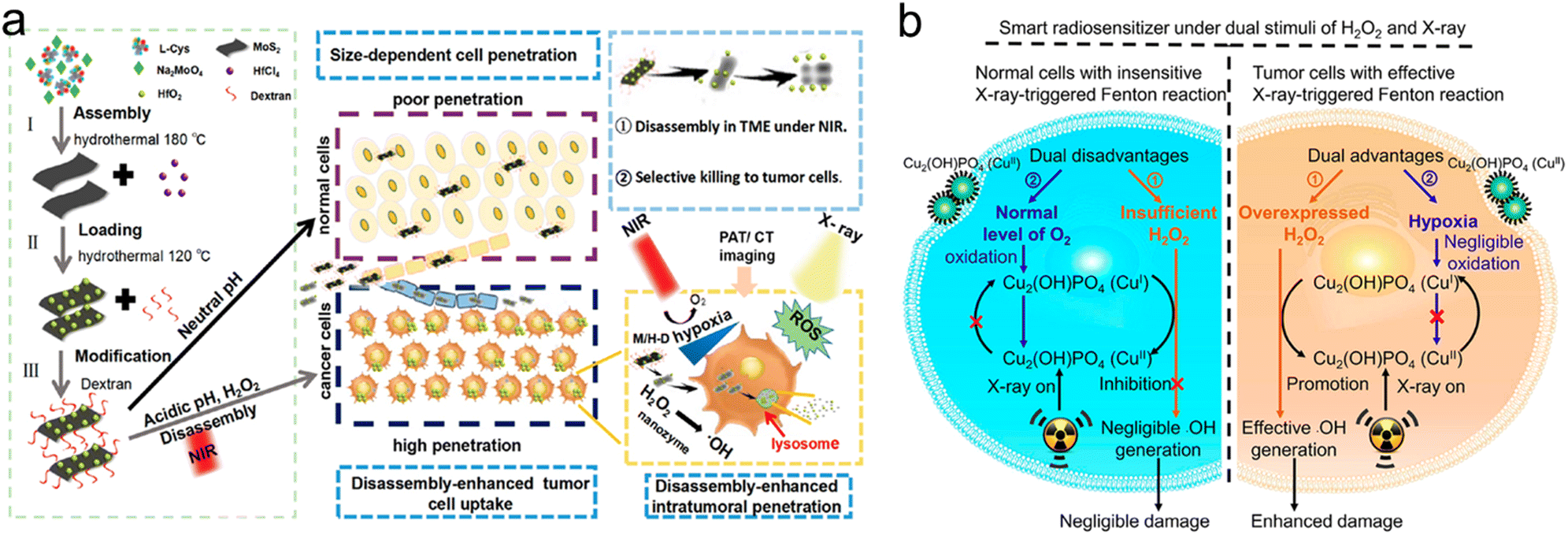 | ||
| Fig. 17 Exo/endogenous dual stimuli-responsive strategies. (a) Schematic illustration of the synthetic procedures of M/H-D and its disassembly in TME under NIR laser irradiation. The disassembly of M/H-D can enhance tumor penetration. M/H-D can function as a peroxidase-like nanocatalyst to catalyze H2O2 to produce ˙OH for tumor cell killing. Reproduced with permission from ref. 293. Copyright 2020, American Chemical Society. (b) A scheme showing the mechanism of Cu2(OH)PO4@PAAS NCs for enhanced RT via X-ray-triggered Fenton reaction. Reproduced with permission from ref. 294. Copyright 2019, American Chemical Society. | ||
Overall, the exo/endogenous dual stimuli-responsive radiosensitizers were able to integrate the merits of remote controllability and flexibility of auto-response to TME, thereby improving precision of responsiveness. However, the multi stimuli-responsive strategy for precision RT is currently too complicated for clinical translation.299
4. Image-guided techniques for precision RT
Multifunctional imaging techniques, including MRI, SPECT, CT, PET, FLI, and PAI, can provide important information on tumor location, boundaries and responses to RT. Diverse image-guided precision RT techniques, such as FL image-guided RT, MR image-guided RT, PA image-guided RT, US image-guided RT, CT image-guided RT, and nuclear image-guided RT, will be discussed in this section (Table 3). In addition, bimodal and multimodal imaging can compensate for the drawbacks of each single imaging modality and take advantage of each imaging technique.| Imaging modality | Nanoparticles | Imaging component | Source | Tumor models | Guiding strategy | Ref. |
|---|---|---|---|---|---|---|
| FL imaging | CAT-SAHA@PLGA-IR775 | IR775 | IVIS imaging system (Ex = 745 nm, Em = 820 nm) | CT26 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) for RT | 34 |
| DiI/Au-DOX@PO-ANG NPs | DiI | IVIS imaging system | U87MG orthotopic glioblastoma-bearing mice | Determine peak concentration in tumor (24 h) for RT | 312 | |
| Mn-doped Ag2Se–RGD–PEG | Ag2Se QDs | NIR-II small animal imaging system (Ex = 808 nm, 980 nm long-pass filter) | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (2 h) for RT | 35 | |
| CPPDA-Hf@Poloxamer | CPPDA | NIR-II small animal imaging system (Ex = 808 nm) | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) for RT | 317 | |
| R-AIE-Au | Au cluster | IVIS imaging system (Ex = 570 nm, Em = 585 nm) | U87MG subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) for RT | 38 | |
| MR imaging | CDs | Gd | MRI scanner | Heps subcutaneous tumor-bearing mice | Locate tumor (T1 MR imaging) | 322 |
| NSC@SiO2-SNO | NA | 3.0 T MRI scanner | 4T1 subcutaneous tumor-bearing mice | Monitor oxygen level and radiosensitivity of tumor (BOLD/DWI fMRI) | 36 | |
| Hb-Lipo | NA | 3.0 T MRI scanner | CT26 subcutaneous tumor-bearing mice | Monitor the tumor oxygenation (BOLD fMRI) | 396 | |
| MR-CA | Mn2+ | 7.0 T MRI scanner | Ultrasmall subcutaneous BxPC3 pancreatic tumor-bearing mice, spontaneous BxPC3 pancreatic tumor-bearing mice | Stratify the degree of tumor hypoxia for precision RT (T1 MR imaging) | 335 | |
| US imaging | NDr(Au + PFOB + O2) | PFOB | US machine with a sector transducer | EMT-6 subcutaneous tumor-bearing mice | Determine peak oxygen concentration in tumor (12 h) for RT | 338 |
| PA imaging | Ir-R/T NCs | Ir NCs | PA imaging system | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (4 h) for RT | 230 |
| Au-TiO2@ZnS | Au NRs | PA imaging system | MC38 orthotopic liver cancer-bearing mice | Determine peak concentration in tumor (24 h) for RT | 342 | |
| PFC@PLGA-RBCM | HbO2 | PA imaging system | 4T1 subcutaneous tumor-bearing mice | Determine peak oxygen concentration in tumor (24 h) for RT | 29 | |
| Cu2−xSe@PtSe | HbO2 | PA imaging system | 4T1 subcutaneous tumor-bearing mice | Determine peak oxygen concentration in tumor (5 h) for RT | 343 | |
| CT imaging | BNTs | Bi | CT scanner | Huh-7 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (9 h) for RT | 40 |
| dAuNP-FA | AuNP | CT scanner | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (8 h) for RT | 352 | |
| Nuclear medicine imaging | 64Cu-Eu/VBBO lipo | 64Cu (PET) | PET scanner | FaDu subcutaneous tumor-bearing mice | Monitor biodistribution of radioisotope | 370 |
| 131I-AuNFs | 131I (SPECT) | Infinia GE SPECT | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) | 364 | |
| BSO/GA-(99mTc)Fe(II)@liposome | 99mTc4+ (SPECT) | Animal SPECT imaging system | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) | 370 | |
| 89Zr-TiO2-Tf NPs | 89Zr (PET) | Inveon small-animal PET/CT scanner | MM1.S bearing SCID mice | Monitor biodistribution of radioisotope | 380 | |
| MR/CT imaging | NSC@mSiO2-SNO/ICG NPs | Eu3+-doped NaGdF4 scintillating nanocrystal | 3.0 T MRI scanner, CT scanner | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) | 64 |
| MR/FL imaging | Cyp-PMAA-Fe@MSCs | Fe (T1/2 MRI), Cypate (FL) | Optical and X-ray small imaging system (Ex = 780 nm, Em = 845 nm), 7.0 T MRI scanner | LLC1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (28 h) | 138 |
| MR/PA imaging | Biomimetic CMC nanoplatform | Mn2+(T1 MRI), HbO2 (PA) | 4.7 T MRI scanner, PA imaging system | 4T1 subcutaneous tumor-bearing mice | Monitor tumor concentration of NPs and oxygen | 130 |
| PA/CT imaging | WS2 QDs | WS2 (PA/CT) | PA imaging system, CT scanner | BEL-7402 tumor-bearing mice | Determine peak concentration in tumor (2 h) | 382 |
| Ti2C3@Au | Au (PA/CT) | PA imaging system, CT scanner | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (24 h) | 397 | |
| PA/FL imaging | Lf-Liposome-DiR | DiR (FL), HbO2 (PA) | PA imaging system, IVIS imaging system (Ex = 748 nm, Em = 780 nm) | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration of NPs and oxygen in tumor (24 h) | 383 |
| CV@CaP | ChlorophyII (FL/PA), HbO2 (PA) | PA imaging system, IVIS imaging system | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration of NPs and oxygen in tumor (4 h) | 384 | |
| CT/PA/SPECT imaging | 99mTc-Bi2Se3 nanodots | Bi2Se3 (CT/PA), 99mTc4+ (SPECT) | PA imaging system, SPECT imaging system, CT scanner | 4T1 subcutaneous tumor-bearing mice | Determine peak concentration in tumor (12 h) | 384 |
4.1 Fluorescence (FL) image-guided precision RT
Featuring the advantages of high sensitivity, non-invasiveness, real-time detection, short imaging acquisition time, and minimal toxicity to normal tissues, FL imaging has become one of the most commonly used imaging modalities. Nevertheless, the in vivo application of FL imaging is restricted by strong light–tissue interaction (e.g., absorption, autofluorescence, scattering, etc.) within the UV-vis range.304–306 Thus, to overcome these limitations, fluorescence probes within the near-infrared (NIR) window have been developed to enhance tissue penetration and reduce background noise. Besides, FL imaging has been used to provide precise information of biodistribution and accumulation of NPs to guide precision radiotherapy.34,61,136,307–312For example, Wang et al. doped Mn(II) ions into Ag2Se QDs with NIR-II fluorescence to fabricate a novel nanoprobe (Mn-doped Ag2Se QDs) for NIR-II FL imaging-guided RT (Fig. 18a).35 The nanoprobe was then conjugated with the RGD peptide for tumor-targeted delivery. The well-prepared Mn-doped Ag2Se–RGD–PEG nanoprobes could catalyze the decomposition of H2O2 into O2 to alleviate hypoxic TME for radiosensitization. After intravenous injection of the Mn-doped Ag2Se–RGD–PEG nanoprobes, the high-resolution NIR-II FL images revealed that the nanoprobes could selectively accumulate inside tumor boundaries (Fig. 18b). The tumor-to-normal tissue (T/NT) signal ratios gradually increased to the maximum (1.5–1.6) at 2 h post-injection (Fig. 18c). Assisted by the FL imaging guidance, 12 Gy of X-ray irradiation at 2 h post-injection of Mn-doped Ag2Se–RGD–PEG nanoprobes completely eradicated tumors in 2 weeks. However, the tumor volume in the X-ray alone group was still above 350 mm3 2 weeks after treatment.
 | ||
| Fig. 18 FL imaging-guided precision RT. (a) A scheme showing the NIR-II FL imaging-guided RT of tumors and elicitation of antitumor immunity based on Mn-doped nanoprobes. (b) In vivo FL imaging of tumor-bearing mice at different time points after intravenous administration of Mn-doped nanoprobes. Scale bars = 10 mm. (c) The corresponding T/NT signal ratio curve after intravenous administration of Mn-doped nanoprobes. Reproduced with permission from ref. 35. Copyright 2021, Wiley. (d) Schematic illustration of synthetic procedures of R-AIE-Au. (e) TEM images of GCs and AIE-GCs. (f) Luminescence spectra of GCs and AIE-GCs. (g) In vivo FL imaging of tumor-bearing mice at different time points after intravenous administration of R-AIE-Au. Reproduced with permission from ref. 38. Copyright 2020, Wiley. | ||
Featuring unique modifiability and photoelectric properties, semiconducting polymer NPs (SPNs) have been developed as biosensors or bioimaging agents.313,314 NIR-absorbing SPNs can even emit NIR-II fluorescence for NIR-II FL imaging.315,316 Dai and colleagues designed a metal–polyphenolic nanosystem (CPPDA-Hf@Poloxamer) for NIR-II FL image-guided synergistic RT and photothermal therapy (PTT). Specifically, the dopamine-modified semiconducting polymer (CPPDA) was chelated with hafnium (Hf) ions and then coated with an amphiphilic polymer (Poloxamer).317 CPPDA with broad NIR absorption could be used for FL imaging and PTT. The Hf as a high-Z element was able to promote X-ray deposition for radiosensitization. The CPPDA-Hf@Poloxamer presented strong NIR-II emission under both 808 nm and 1064 nm laser excitation. The in vivo NIR-II FL imaging under 808 nm irradiation revealed that CPPDA-Hf@Poloxamer gradually accumulated in the tumor over time and peaked at 24 h post-injection. Finally, the combined irradiation of X-ray and a 1064 nm laser at 24 h post-injection of CPPDA-Hf@Poloxamer effectively eliminated subcutaneous tumors, whereas the tumor volume in the X-ray alone group was about 400 mm3 at 15 days post-treatment.
Unfortunately, many organic dyes suffer from the aggregation-caused quenching (ACQ) effect, which seriously attenuates the FL imaging performance. To overcome this issue, Sun et al. developed aggregation-induced emission (AIE) gold clustoluminogens (AIE-Au) for FL image-guided RT and PDT.38 First, glutathione-protected gold clusters were self-assembled into AIE-GCs through a cationic polymer-mediated approach (Fig. 18e). Moreover, the X-ray-excited optical luminescence (XEOL) peak of AIE-GCs was 5.2-fold stronger than that of GCs (Fig. 18f). Next, the rose bengal (RB, a clinically used photosensitizer) conjugated GCs were self-assembled into AIE-Au and then modified with a tumor-targeting RGD peptide (R-AIE-Au) (Fig. 18d). The in vivo FL imaging showed that the fluorescence signals in tumors increased over time and reached the maximum at 24 h post-injection (Fig. 18g). Guided by FL imaging, low-dose X-rays (0.5 Gy) were used to irradiate the tumor-bearing mice at 24 h post-injection and resulted in a much higher tumor inhibition rate of 97.1% compared to ∼30% in the X-ray alone group.
Despite the superiority of FL imaging in guiding RT, the penetration depth and spatial resolution are still much poorer compared to other imaging techniques, such as MRI, CT, or PET.
4.2 MR image-guided precision RT
MR imaging has been extensively utilized in clinic for diagnosis due to the advantages of unlimited penetration, non-invasiveness, lack of ionizing radiation, and high spatial resolution. In vivo MR imaging can be used to monitor the concentration of contrast agents in various organs or tumor.318 Furthermore, MR imaging can also show the accurate tumor location and adjacent tissues.Wang et al. modified self-assembled Mn–Zn ferrite magnetic NPs with hyaluronic acid (HA) to construct a block copolymer micelle (MZF-HA) for MR image-guided RT/hyperthermia therapy (HT).52 MZF-HA could target CD44-overexpressing tumor cells due to the selective recognition of CD44 receptors by HA. Besides, MZF-HA could heat up tumor under an alternating magnetic field (AMF) for HT. HT could also improve tumor oxygen levels and contribute to radiosensitization (Fig. 19a). Moreover, MZF-HA could be used for MRI as a T2-weighted contrast agent with an r2 value of 331 mM−1 s−1. After intravenous injection, MR imaging showed gradually darkening signals in A459 xenograft tumor at 24 h and 48 h after i.v. injection, suggesting the tumor accumulation of MZF-HA (Fig. 19b). Guided by MR imaging, RT/HT remarkably reduced the relative tumor ratio to 49.6% by day 13, much lower than ∼70% in the RT alone group.
 | ||
| Fig. 19 MR imaging-guided precision RT. (a) Schematic illustration of the synthetic process of MZF-HA and its application in MR imaging-guided RT and hyperthermia. (b) In vivo T1-weighted MR imaging of tumor-bearing mice before and after administration of MZF-HA. Reproduced with permission from ref. 52. Copyright 2020, American Chemical Society. (c) Schematic illustration of the synthetic process of Gd-doped CDs. (d) In vivo T1-weighted MR imaging of subcutaneous tumor-bearing mice before and after intravenous administration of Gd-doped CDs (10 mg kg−1). (e) The SNRpost/SNRpre ratio of tumor region before and after intravenous administration. (f) Tumor growth curves of tumor-bearing mice in different treatment groups. Reproduced with permission from ref. 322. Copyright 2020, Wiley. | ||
Nowadays, the clinical translation of T2-weighted contrast agents has been hampered by several limitations. For instance, the dark images based on T2-weighted contrast agents are not preferred for observation.75 With the bright tumor images, T1-weighted contrast agents have attracted wide attention for guiding precision RT.33,319–323 Gd-chelator complexes are the most commonly used T1 contrast agents in the clinic even though they have several shortcomings, such as nephrogenic toxicity and low longitudinal relaxivity. Additionally, even small amount of Gd has been reported to remarkably improve the RT efficacy.324 For example, Du et al. designed Gd-doped carbon dots (CDs) with high longitudinal relaxivity and a long circulation time for MR image-guided RT (Fig. 19c).322 The r1 value of Gd-doped CDs (6.45 mM−1 s−1) was much larger than that of clinically used Magnevist (4.05 mM−1 s−1). In vivo MR imaging revealed that Gd-doped CDs could significantly brighten the tumor (Fig. 19d). The values of SNRpost/SNRpre in the tumor region reached 1.74 and 1.93 with transverse and coronal scanning, respectively (Fig. 19e). Finally, guided by T1-weighted MR imaging, the relative tumor volume in the Gd-doped CDs + X-ray group after 7 days of treatment was about half of that in the X-ray alone group (Fig. 19f).
As opposed to structural imaging, functional imaging is a medical imaging technique that detects physiological activities within a specific tissue or organ, such as blood supply, metabolism, chemical composition, etc.325 The functional imaging techniques include CT perfusion imaging, functional MR imaging (fMRI), PET, magnetoencephalography (MEG), magnetic source imaging (MSI), near infrared spectroscopy (NIRS), etc.326–328 fMRI can provide biological molecular information based on the anatomical structure and has attracted wide attention from basic and clinical researchers.329 A lot of fMRI techniques have been used for in vivo evaluation of various biological processes in tissues, thus allowing for the presentation of spatiotemporal variations in tumor biological response to RT.330,331 Diffusion-weighted imaging (DWI) is an fMRI technique that does not require a contrast agent. DWI can detect the random motion water protons (water diffusion). The water diffusion is closely related to tissue cellularity and cell membranes’ integrity.331 The apparent diffusion coefficient (ADC) can be used to characterize the diffusion. Compared to normal tissues with low cellularity, most tumors are hypercellular and contain lots of intact cell membranes, which restrict water diffusion and show a high signal. Since effective treatments can decrease cell density in tumor, the therapeutic effects can be monitored and evaluated in real time by DWI.332 Blood-oxygen-level-dependent (BOLD) fMRI is a noninvasive technique for the assessment of tissue hypoxia based on endogenous paramagnetic deoxyhemoglobin levels.333,334 An increased level of deoxyhemoglobin in hypoxic tumors leads to decreased 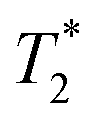 signal. Therefore, BOLD fMRI can be used to monitor and assess the dynamic changes of tumor oxygen levels in real time.
signal. Therefore, BOLD fMRI can be used to monitor and assess the dynamic changes of tumor oxygen levels in real time.
For example, Dou et al. proposed a dual fMRI (BOLD/DWI imaging) strategy for noninvasive monitoring of tumor hypoxia and radiosensitization efficacy (Fig. 20a).36 The NaGdF4:Eu3+ NPs were coated with a mesoporous silica layer and then loaded with S-nitrosothiol (SNO, a nitric oxide (NO) donor) (NSC@SiO2-SNO). NSC@SiO2-SNO could release a great deal of NO upon X-ray irradiation. The released NO was able to effectively alleviate tumor hypoxia. Next, in vivo BOLD/DWI imaging was used to dynamically monitor tumor oxygen levels and radiosensitivity during treatment by measuring the tumor 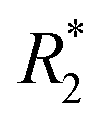 and ADC values. T2-weighted MRI showed that RT plus the NSC@SiO2-SNO could effectively inhibit tumor growth compared to the control or RT group (Fig. 20b). The tumor
and ADC values. T2-weighted MRI showed that RT plus the NSC@SiO2-SNO could effectively inhibit tumor growth compared to the control or RT group (Fig. 20b). The tumor 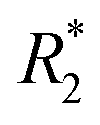 value of both the control and RT alone groups increased slightly overtime, indicating that rapid tumor growth might lead to insufficiency of tumor blood supply and decrease of oxygen levels. However, the NSC@SiO2-SNO-mediated RT could remarkably decrease the tumor
value of both the control and RT alone groups increased slightly overtime, indicating that rapid tumor growth might lead to insufficiency of tumor blood supply and decrease of oxygen levels. However, the NSC@SiO2-SNO-mediated RT could remarkably decrease the tumor 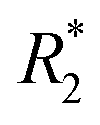 value from 40.25 s−1 at the beginning to 33.39 and 21.06 s−1 at day 7 and day 14, respectively, suggesting significant hypoxia relief. In addition, the tumor ADC value in the NSC@SiO2-SNO + RT group significantly increased from the initial 0.973 × 10−3 mm2 s−1 to 2.325 × 10−3 mm2 s−1 at day 14, indicating that NSC@SiO2-SNO-mediated radiosensitization could effectively kill tumor cells and decrease cell density. However, the tumor ADC value in the RT alone group only increased from 0.898 × 10−3 mm2 s−1 to 1.095 × 10−3 mm2 s−1.
value from 40.25 s−1 at the beginning to 33.39 and 21.06 s−1 at day 7 and day 14, respectively, suggesting significant hypoxia relief. In addition, the tumor ADC value in the NSC@SiO2-SNO + RT group significantly increased from the initial 0.973 × 10−3 mm2 s−1 to 2.325 × 10−3 mm2 s−1 at day 14, indicating that NSC@SiO2-SNO-mediated radiosensitization could effectively kill tumor cells and decrease cell density. However, the tumor ADC value in the RT alone group only increased from 0.898 × 10−3 mm2 s−1 to 1.095 × 10−3 mm2 s−1.
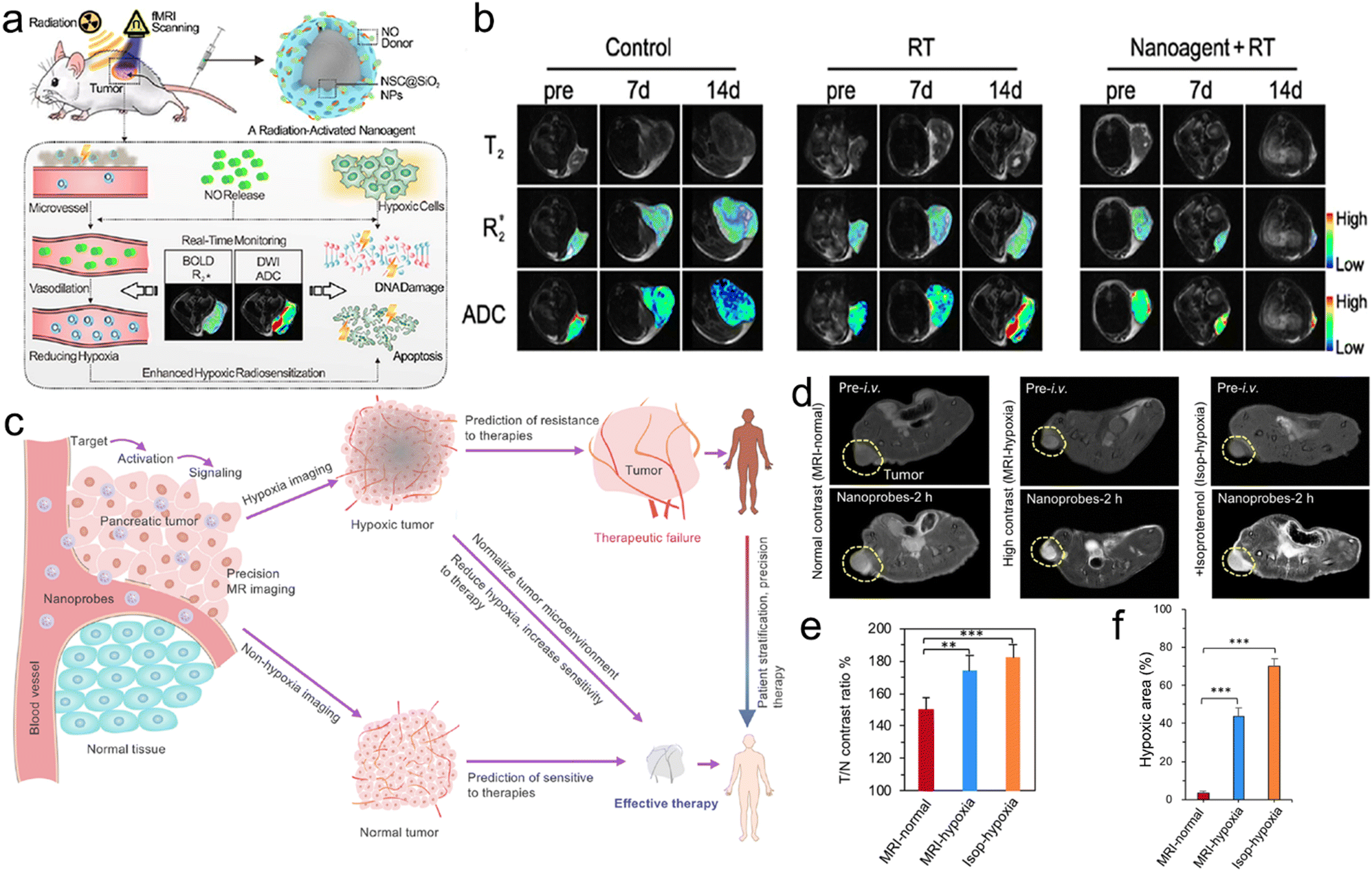 | ||
Fig. 20 Functional MR imaging-guided precision RT. (a) Schematic illustration of NO-induced tumor oxygenation and radiosensitization via a radiation-activated NP under real-time monitoring of BOLD/DWI imaging. (b) In vivo T2-weighted,  -mapping, and ADC-mapping images before and after different treatments. Reproduced with permission from ref. 36. Copyright 2021, American Chemical Society. (c) A scheme of MR-CA nanoprobes for MR imaging of pancreatic tumors and tumor hypoxia to realize the prediction of tumor responses to RT and immunotherapy. (d) In vivo T1-weighted MR imaging of subcutaneous BxPC3 pancreatic tumor-bearing mice after intravenous administration of MR-CA nanoprobes to sort tumor according to signal intensities. (e) Corresponding T/N ratios of the MR images. (f) Quantitative measurement of the hypoxia area from the immunofluorescence staining images of tumors. Reproduced with permission from ref. 335. Copyright 2021, American Chemical Society. -mapping, and ADC-mapping images before and after different treatments. Reproduced with permission from ref. 36. Copyright 2021, American Chemical Society. (c) A scheme of MR-CA nanoprobes for MR imaging of pancreatic tumors and tumor hypoxia to realize the prediction of tumor responses to RT and immunotherapy. (d) In vivo T1-weighted MR imaging of subcutaneous BxPC3 pancreatic tumor-bearing mice after intravenous administration of MR-CA nanoprobes to sort tumor according to signal intensities. (e) Corresponding T/N ratios of the MR images. (f) Quantitative measurement of the hypoxia area from the immunofluorescence staining images of tumors. Reproduced with permission from ref. 335. Copyright 2021, American Chemical Society. | ||
Besides fMRI, nanotechnology-based MR imaging can also be utilized to monitor tumor response. Mi and co-workers designed MR contrast amplification (MR-CA) nanoprobes for the early diagnosis of pancreatic tumor and prediction of tumor sensitivity to RT (Fig. 20c).335 The MR-CA nanoprobes were fabricated based on PEGylated polyanions and self-assembly of Mn2+-doped CaP NPs. The nanoprobes could degrade in response to the low pH value of TME and rapidly release Mn2+ to boost higher molecular relaxivity (r1) via binding with surrounding proteins.336 Owing to acid-responsive Mn2+ release, the MR-CA nanoprobe was able to sensitively and selectively image ultrasmall, orthotopic, and spontaneous pancreatic tumors compared to the clinically applied Gd-DOTA (Fig. 20d). Besides, the tumor-to-normal tissue (T/N) contrast ratio in the MR-CA group was much higher than that in the Gd-DOTA group (Fig. 20e). The precise MR imaging of ultrasmall pancreatic tumors allowed early detection and precision therapy of the tumors. In addition, MR-CA nanoprobe-based MR imaging could stratify the degree of tumor hypoxia so as to predict the tumor response to RT and tumor prognosis. Isoproterenol hydrochloride was injected intraperitoneally into BxPC3 tumor-bearing mice in order to establish hypoxic tumors as the positive control (denoted as isop-hypoxia). Based on T/N contrast ratios, the other BxPC3 tumors were divided into two groups: MRI-normal (normoxic tumors with the ratios lower than 160%) and MRI-hypoxia (hypoxic tumors with the ratios higher than 160%). The immunofluorescence staining of these tumors showed that MRI-hypoxic tumors exhibited around 43% hypoxic area in tumor regions compared to 3.5% and 70% in MRI-normal tumors and the positive group (isop-hypoxia), respectively (Fig. 20f). For in vivo anti-tumor evaluation, the MRI-hypoxic tumor showed lower sensitivity to RT and over 600 mm3 of tumor volume after treatment, whereas the RT showed effective inhibition of tumor growth in MRI-normal tumors with nearly 200 mm3 of tumor volume. In short, MR imaging can be used to guide RT by providing anatomical structure information, tumor accumulation of radiosensitizers, and molecular biological information of TME.337 However, MR imaging is limited by high concentration of contrast agents and relatively long acquisition time required for imaging. Therefore, future research needs to focus on substantially improving the MR imaging sensitivity based on the contrast agents.
4.3 US image-guided precision RT
US imaging has been commonly used in clinical practice due to the advantages of low cost, real-time imaging, and noninvasiveness. US can be applied to guide precision RT by dynamically monitoring the tumor accumulation of NPs. For example, Jiang et al. encapsulated ultrasmall AuNPs and a liquid perfluorooctyl bromide (PFOB) core to construct a nanodroplet (NDr) for US image-guided precision RT. The NDr was then oxygenated to generate hierarchical multiplexing NDr(Au + PFOB + O2) (Fig. 21a).338 Next, the NDr(Au + PFOB + O2) was doped with N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl)ammonium bromide (BHEM-Chol) for a slightly positive charge, thus allowing the NDrs to target the tumor vessel and accumulate in the tumor. Moreover, the AuNPs inside the NDrs could deposit X-ray energy for radiosensitization. Furthermore, under US stimulation, the NDrs could break and rapidly release a lot of O2 for enhanced RT (Fig. 21b and c). In addition, the US imaging could real-time track the NDrs. The in vivo US imaging showed that echo signals in tumor regions gradually increased over time and peaked at 12 h after intravenous injection (Fig. 21d–f), indicating that the 12 h post-injection could be the optimal time point for further RT. Finally, the NDr(Au + PFOB + O2) + US + RT group displayed the highest tumor inhibition rate of 93.04% after 20 days of treatment compared to 49.62% of the RT alone group (Fig. 21g). In this study, the US served as both an imaging modality for real-time tracking and a trigger for O2 release, realizing precision oxygen-elevated RT.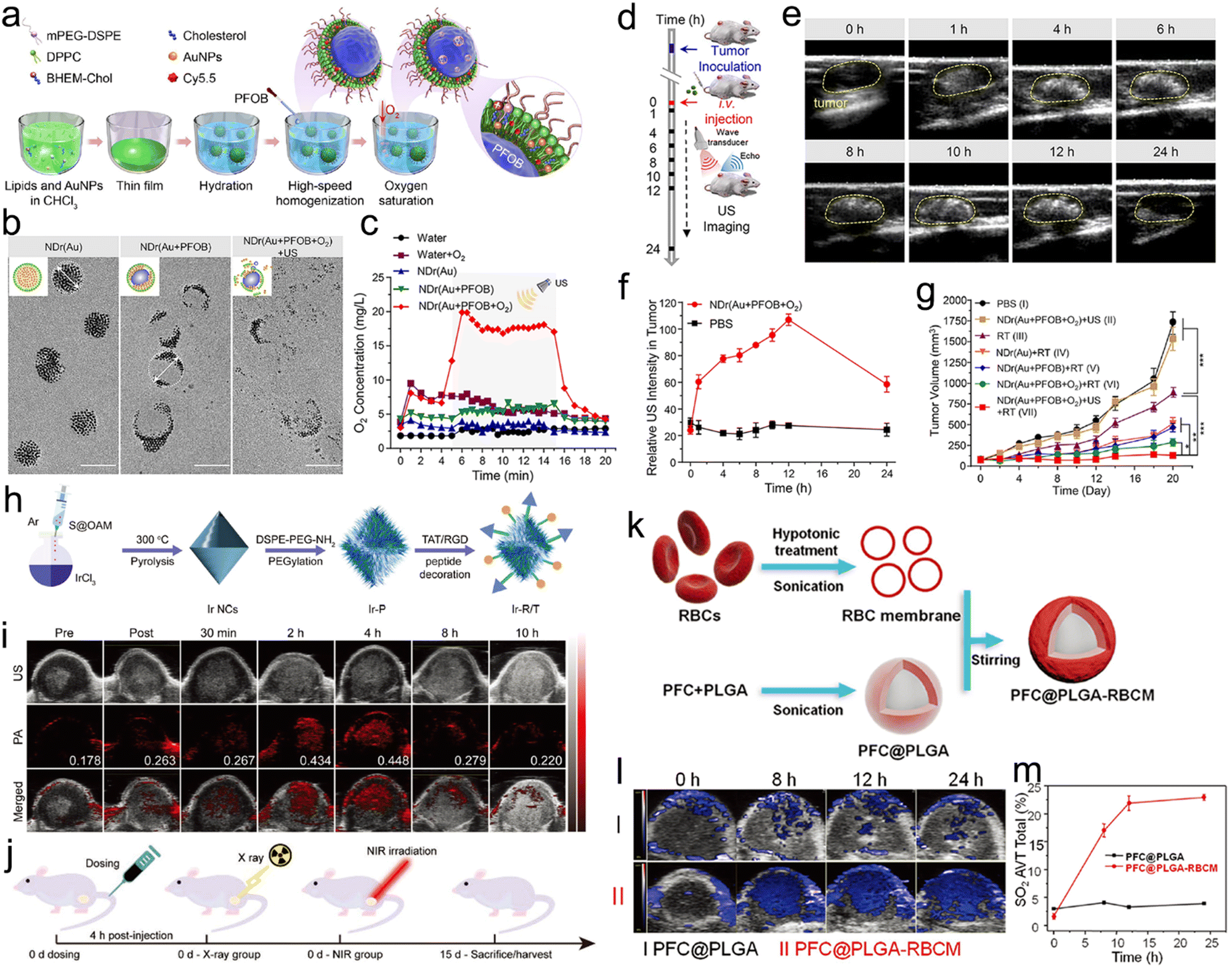 | ||
| Fig. 21 US or PA imaging-guided precision RT. (a) A scheme of synthetic procedures of hierarchical multiplexing NDrs. (b) TEM images of NDr(Au), NDr(Au + PFOB), and NDr(Au + PFOB + O2) under US (130 W, 20 kHz, 5 min). (c) In vitro O2 release profiles of NDr(Au), NDr(Au + PFOB), and NDr(Au + PFOB + O2) treated with US. (d) US imaging schedule of EMT-6 tumor-bearing mice. (e) US images of tumor-bearing mice at different time points after intravenous administration of NDr(Au + PFOB + O2). Yellow dash circles: tumor regions. (f) Relative US intensities in the tumor region at different time points after intravenous administration of NDr(Au + PFOB + O2). (g) Tumor growth curves of mice in different treatment groups. Reproduced with permission from ref. 338. Copyright 2018, American Chemical Society. (h) A scheme of synthetic procedures of Ir-R/T NCs. (i) In vivo PA imaging of tumors at different time points after administration of Ir-R/T NCs (8 mg kg−1). (j) Treatment schedule of in vivo RT/PTT of tumors. Reproduced with permission from ref. 230. Copyright 2019, Wiley. (k) A scheme of synthetic procedures of PFC@PLGA-RBCM NPs. (l) In vivo PA imaging of tumors at different time points after administration of PFC@PLGA and PFC@PLGA-RBCM NPs. (m) Quantification of oxyhemoglobin saturation concentrations based on PA images of tumors. Reproduced with permission from ref. 29. Copyright 2017, Wiley. | ||
4.4 PA image-guided precision RT
PAI as a hybrid imaging technique integrates high spatial resolution of US imaging and high contrast and sensitivity of optical imaging.339 PA imaging contrast agents, including Au nanostars, Au nanorods, AuNPs, ICG, methylene blue, and transition-metal chalcogenides, etc., have been extensively applied for tumor visualization and image-guided therapy.340 Besides, several studies have utilized PA imaging to real-time track the tumor accumulation of radiosensitizers and monitor tumor oxygen levels for guiding precision RT.29,37,230,341–344Wang et al. fabricated dual targeting peptide (RGD and TAT)-modified ultrasmall iridium (Ir) nanocrystals (Ir-R/T NCs) for PA imaging-guided RT (Fig. 21h).230 Ir in the nanocrystals as a high-Z element could deposit X-ray energy for radiosensitization. Moreover, based on the dual targeting peptides, the Ir-R/T NCs could target 4T1 tumor cells and subsequently target the nucleus. The intrinsic NIR absorption of Ir-R/T NCs enabled PA imaging and photothermal capability. The in vivo PA imaging of 4T1 tumor-bearing mice displayed that the PA signals of Ir-R/T NCs in the tumor region reach the maximum at 4 h after intravenous administration (Fig. 21i). Guided by PA imaging, the X-ray and NIR irradiations were imposed on subcutaneous tumors at 4 h post-injection for synergistic RT/PTT, resulting in complete tumor eradication (Fig. 21j). However, the tumor volume in the X-ray alone group was still 520 mm3 after 15 days of treatment.
Besides tracking tumor accumulation of radiosensitizers, the PA imaging has also been utilized to determine the tumor oxygenation status by taking advantages of the difference in the optical absorption spectrum of deoxygenated haemoglobin (λ = 750 nm) and oxygenated hemoglobin (λ = 850 nm).345 Importantly, hypoxic TME in many types of solid tumors is closely related to radioresistance and severely hampers RT efficacy. Liu and co-workers encapsulated PFC into poly(D,L-lactide-co-glycolide) (PFC@PLGA) and then coated the PFC@PLGA with a red blood cell membrane (PFC@PLGA-RBCM NPs) (Fig. 21k).29 The PFC@PLGA-RBCM NPs with excellent blood circulation could accumulate in the interior region of solid tumors. Moreover, PFC@PLGA-RBCM NPs with high oxygen carrying capability could effectively alleviate tumor hypoxia. In vivo PA imaging revealed that the total oxygenation levels within the whole tumor region (sO2 Tot) increased from 1.6% of pre-injection to 24% at 24 h post-injection (Fig. 21l and m), which were further confirmed by immunofluorescence staining of tumor slices with hypoxyprobe and anti-HIF-1α. Finally, guided by PA imaging, the X-ray radiation (8 Gy) was given to the 4T1 tumor-bearing mice at 24 h post-injection of PFC@PLGA-RBCM for precision RT, resulting in tumor weight about half that of the X-ray alone group after 2 weeks of treatment.
4.5 CT image-guided precision RT
As a noninvasive medical imaging test, CT imaging has advantages of short acquisition time, deep penetration and low cost. Nevertheless, the clinically used iodinated compound-based CT contrast agents have shown limited success due to their extremely short blood circulation half-life.346 Furthermore, patients need to be injected with a large amount of iodine-based contrast agents owing to the low X-ray attenuation effect of iodine.347 Inorganic nanomaterials with high X-ray attenuation coefficients have been regarded as potential alternative CT contrast agents, such as bismuth (Bi), gold (Au), tantalum (Ta), and platinum (Pt).348–351 In addition, these high-Z metallic nanomaterials can not only serve as CT contrast agents but also deposit X-ray energy for radiosensitization.91,352–355The U.S. Food and Drug Administration (FDA) claims that renal clearance of injected metal nanomaterials is essential to avoid side effects associated with their long-term retention.356 However, the renal-clearable nanomaterials with ultrasmall sizes cannot effectively accumulate in tumor site due to their rapid clearance in vivo. To address this issue, Hu et al. proposed a controlled assembly strategy by assembling ultrasmall (BiO)2CO3 nanoclusters (BNCs, 1.5 nm) into hollow (BiO)2CO3 nanotubes (BNTs) (Fig. 22a).40 Due to the higher X-ray attenuation coefficient of Bi, the in vitro CT values (Hounsfield unit, HU) of BNTs were significantly higher than those of iohexol at equal molar concentrations. The in vivo reconstructed 3D CT imaging of tumor-bearing mice showed that the CT value in the tumor region increased over time and peaked at 9 h post-injection of BNT (Fig. 22b and c). However, the BNC group displayed a relatively low enhancement signal at the tumor site (Fig. 22b and c). Importantly, the BNTs could be disassembled into nanoclusters under acidic TME and then cleared via kidneys. In addition, the BNTs were loaded with DOX (loading efficiency of ∼53%) for CT image-guided synergistic RT/chemotherapy. Guided by CT imaging, BNTs/DOX plus RT achieved a tumor suppression rate of ∼83.5%, much higher than that of the BNTs plus RT group (42.4%) and BNTs/DOX alone group (32.2%) (Fig. 22d).
 | ||
| Fig. 22 CT image-guided precision RT. (a) A scheme of synthetic procedures of BNTs. (b) In vivo reconstructed 3D CT imaging of tumor-bearing mice at various time points after intravenous administration of BNTs and BNCs. (c) In vivo CT value of tumor sites at various time points after intravenous administration of BNTs and BNCs. (d) Tumor growth curves of mice received different treatments. Reproduced with permission from ref. 40. Copyright 2018, American Chemical Society. | ||
4.6 Radionuclide image-guided precision RT
SPECT and PET, two major molecular imaging modalities in nuclear medicine, can detect radioactive signals from radioisotopes and offer functional imaging, thus allowing clinical diagnosis, delivery of targeted therapeutics, and assessment of response to treatment. For PET imaging, the positrons emitted by radionuclides interact with the nearby electrons to produce annihilation and then release energy in the form of two gamma ray photons in the opposite directions. The gamma ray photons can be detected by PET. SPECT can detect the gamma ray photons emitted by radioisotopes during radioactive decay.357 SPECT/PET imaging can play multiple roles in cancer management, such as determining tumor volume, providing metabolic information of TME, and offering biodistribution information of radiolabeling drugs.358–360A lot of studies have utilized SPECT imaging based on various radioisotopes (e.g., 131I, 99mTc, and 177Lu) to observe pharmacokinetics and biodistribution of radioactive drugs.361–363 For instance, Cheng et al. designed radioisotope 131I-labeled Au nanoframeworks (131I-AuNFs) for synergistic RIT/PTT of breast cancer.364 Specifically, the AuNFs were coated with polydopamine (PDA) and further chelated with 131I to construct 131I-AuNFs (Fig. 23a). 131I with the advantages of long half-period and suitable radiation energy was used for SPECT imaging and RIT. The as-prepared 131I-AuNFs exhibited steady radioactivity and great photothermal conversion efficiency. The in vivo SPECT imaging was used to observe the biodistribution of 131I in 4T1 tumor-bearing mice, showing that the radio-signals in the tumor regions of 131I-AuNF groups gradually increased and peaked at 24 h post-injection (Fig. 23b). Guided by in vivo SPECT imaging, the NIR-II laser (1 W cm−2, 10 min) was irradiated on subcutaneous 4T1 tumor at 24 h post-injection for PTT. The in vivo SPECT imaging-guided RIT/PTT strategy showed a tumor inhibition rate of 100%, much higher than 57.8% of the AuNFs plus the NIR-II laser group.
 | ||
| Fig. 23 PET/SPECT imaging-based RT. (a) A scheme of synthetic procedures of 133I-AuNFs. (b) In vivo SPECT imaging of tumor-bearing mice at different time points after intravenous administration of free 133I and 133I-AuNFs. Reproduced with permission from ref. 364. Copyright 2021, Royal Society of Chemistry. (c) In vivo PET imaging of FaDu tumor-bearing mice at various time points after intravenous administration of 64Cu-Eu/VBBO lipo (upper panel: maximal intensity projection (MIP); middle panel: coronal view; lower panel: surface plot for the tumor region from the coronal image). Reproduced with permission from ref. 370. Copyright 2020, American Chemical Society. | ||
PDT as an effective antitumor treatment is hampered by limited tissue penetration depth of light and poor tumor-targeting ability of photosensitizer (PS).365 To overcome these limitations, the scintillating materials (e.g., terbium (Tb), europium (Eu), etc.) can convert ionizing radiation into visible light to trigger PS to generate ROS (X-ray-induced PDT).275,366,367 Likewise, instead of X-ray irradiation, the Cerenkov luminescence emitted by RIT can also be used to induce PDT.368,369 Lee et al. designed a 64Cu-labeled Eu-diethylenetriaminepentaacetic acid (Eu-DTPA)/Victoria blue-BO (VBBO, a PS) co-loaded liposome (64Cu-Eu/VBBO lipo) for in vivo PET imaging and radioisotope-mediated PDT.370 The efficiency of luminescence resonance energy transfer between Eu-DTPA and VBBO was 6-fold higher than that in Cerenkov luminescence energy transfer (CLET). Moreover, in vivo PET imaging was used to track 64Cu-Eu/VBBO lipo in mice and revealed that the 64Cu-Eu/VBBO lipo with 20.15 h of circulation half-life showed a high tumor uptake of up to 19.29% ID per g at 48 h post-injection (Fig. 23c). Furthermore, guided by PET imaging, 64Cu-Eu/VBBO lipo could effectively eliminate the FaDu tumors. The tumor volume ratio in the 64Cu-Eu/VBBO lipo group was about half of that in the 64Cu-VBBO lipo group. In addition, other radioisotopes, such as 89Zr, 86Y, and 64Cu, have also been used for PET image-guided RT.371–374
Although PET and SPECT can provide functional information with high sensitivity and specificity at the molecular level, PET or SPECT alone is not the most proper imaging technique for cancer diagnosis due to the lack of anatomical information. Therefore, the PET or SPECT is usually combined with a high-resolution anatomic imaging modality (CT or MRI) to provide both functional and anatomical information.375,376 Liu and co-workers encapsulated ultrasmall gallic acid-ferrous (GA-Fe(II)) nanocomplexes and L-buthionine sulfoximine (BSO, an inhibitor of GSH) into a liposome (BSO/GA-Fe(II)@liposome).377 Next, the BSO/GA-Fe(II)@liposome was chelated with 99mTc4+ radioisotope with a high radiolabeling efficiency and steady radioactivity for SPECT/CT image-guided RT/chemotherapy. The in vivo SPECT/CT imaging revealed that the BSO/GA-Fe(II)@liposome could effectively accumulate at the tumor site and reach the maximum at 24 h post-injection. Therefore, the 24 h post-injection was considered as the optimal time-point for RT of tumors. Besides, the PET/CT imaging based on various radionuclides, such as 86Zr and 64Cu, was also utilized to guide precision RT.378–381 For example, Achilefu and colleagues coated titanium dioxide NPs (TiO2 NPs) with transferrin (Tf) and further labeled the NPs with radionuclide 89Zr (89Zr-TiO2-Tf NPs) for bone targeting and PET/CT imaging-guided Cerenkov radiation-induced therapy (CRIT) of multiple myeloma (MM, a type of bone-related tumor).380
In summary, after radiolabeling, the nuclear medicine imaging can monitor the distribution of drugs in tumors or organs and further guide precision RT. However, the PET imaging is costly while the SPECT imaging is much cheaper but the latter produces images with lower resolution. Besides, both the imaging techniques present radiation hazard, which may hamper their wide applications.
4.7 Dual-modal imaging-guided RT
Each imaging method has its own advantages and disadvantages. Nowadays, more and more nanomedicines have been designed to incorporate two or more imaging modalities, which allows for integration of merits of various imaging techniques.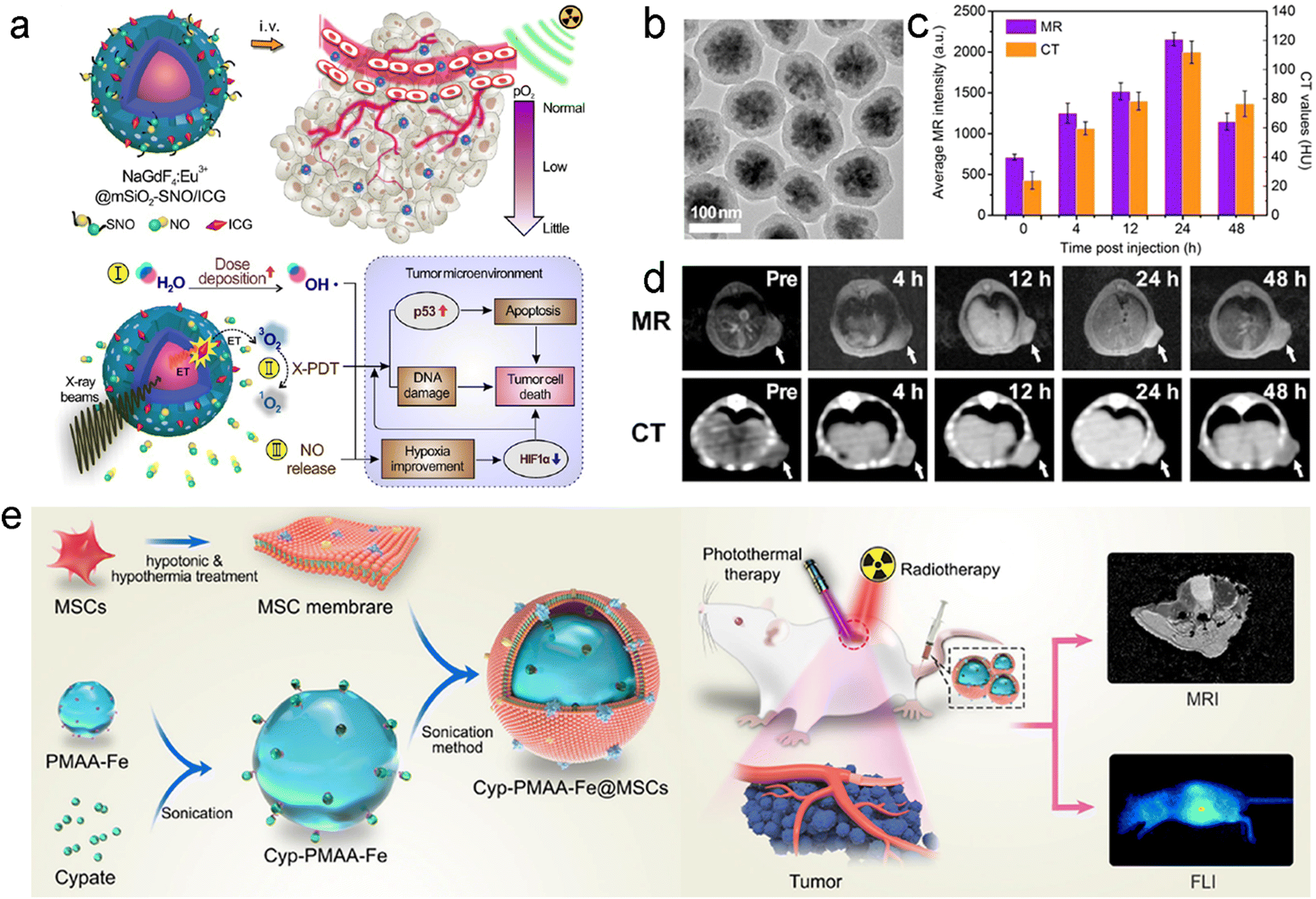 | ||
| Fig. 24 MR-based bimodal imaging-guided RT. (a) Schematic illustration showing several responses induced by X-ray radiation on NSC@mSiO2-SNO/ICG NPs. (b) TEM image of NSC@mSiO2-SNO/ICG NPs. (c and d) In vivo MR/CT imaging (d) and quantification analysis (c) of tumor-bearing mice at different time points after intravenous administration of NSC@mSiO2-SNO/ICG NPs. Reproduced with permission from ref. 64. Copyright 2018, Ivyspring International Publisher. (e) A scheme of synthetic procedures of Cyp-PMAA-Fe@MSCs NP and its application in MR/FL imaging-guided RT/PTT of NSCLC. Reproduced with permission from ref. 138. Copyright 2021, Springer Nature. | ||
Furthermore, the NPs with bimodal MR and FL imaging capabilities have also been designed to guide precision RT. Yin et al. coordinated cypate (Cyp, a derivative of ICG with bis-carboxyl groups) with Fe(III)-loaded polymethacrylic acid (PMAA) NPs (Cyp-PMAA-Fe NPs). Then, the Cyp-PMAA-Fe NPs were coated with mesenchymal stem cell membranes (Cyp-PMAA-Fe@MSCs NPs) for MR/FL image-guided RT/PTT of non-small cell lung cancer (NSCLC) (Fig. 24e).138 To assess the advantages of MSC cell membrane in inducing immune escape and prolonging circulation time, the Cyp-PMAA-Fe NPs were coated with red blood cell membranes (RBCs) to construct Cyp-PMAA-Fe@RBCs as negative control. The in vivo FL imaging of LLC1 tumor-bearing mice revealed that the FL signals of tumors at 28 h post-injection of Cyp-PMAA-Fe@MSCs were 21% higher than those in the Cyp-PMAA-Fe@RBC group, suggesting more effective tumor targeting and accumulation of Cyp-PMAA-Fe@MSCs owing to the MSC membranes. Likewise, the T1-weighted MR imaging showed a 30.01% decrease of the T1 signal at the tumor sites at 28 h after intravenous injection of Cyp-PMAA-Fe@MSCs. Guided by the MR/FL imaging, the mice were irradiated with NIR laser and X-rays at 28 h post-injection of Cyp-PMAA-Fe@MSCs for synergistic RT/PTT. The relative tumor volume in the Cyp-PMAA-Fe@MSCs + NIR + RT group was 0.63, much lower than 3.32 in the RT alone group.
4.8 Tri- and tetra-modal imaging-guided RT
Tri-modal imaging-based techniques, such as CT/MRI/PAI,386,387 CT/FLI/PAI,388 MRI/PAI/PET,389 MRI/PAI/SPECT,390 PAI/US/PET,391 CT/PA/SPECT,392,393 and CT/MRI/FLI,394 have also been reported to guide RT. For example, Li and colleagues constructed biocompatible Bi2Se3 nanodots for CT/PA/SPECT tri-modal imaging-guided synergistic RT/PTT of tumor.393 These Bi2Se3 nanodots with relatively high photothermal conversion efficiency (η = 50.7%) could function as theranostic agents for PTT and PA imaging. Besides, these nanodots could also deposit X-ray energy and serve as a radiosensitizer. The in vivo PA imaging of 4T1 tumor-bearing mice revealed that the PA signals at the tumor sites were enhanced over time and reached the peak at 12 h post-injection of Bi2Se3 nanodots. Then the tumoral CT value increased gradually from the initial 50 HU to the peak of 94 HU at 12 h after intravenous injection of the nanodots, which was consistent with the PA imaging results. In addition, these nanodots were chelated with radioactive 99mTc for evaluation of their biodistribution via SPECT. The SPECT/CT images showed remarkable tumor accumulation of Bi2Se3 nanodots within 24 h of intravenous injection. Guided by the tri-modal imaging, the tumor-bearing mice received X-ray (6 Gy) and 808 nm laser (1 W cm−2, 10 min) irradiations at 12 h post-injection of Bi2Se3 nanodots, resulting in a survival rate of 100% even at 36 days after treatment. However, all the mice in the RT alone group died within 32 days.Besides, Kang et al. even integrated four imaging modalities (MRI/CT/PAI/FLI) into one nanoplatform for tetra-modal imaging-guided RT/chemotherapy.395 They encapsulated rare-earth down-conversion (DC) NPs, copper bismuth sulfide (CBS) NPs, and DOX into zeolitic imidazolate framework-8 (ZIF8) to construct multifunctional nanocomposites (CBS@DC-ZIF8@DOX). After intratumoral injection of CBS@DC-ZIF8@DOX nanocomposites, the in vivo CT and MR images showed significant contrast enhancement. Moreover, the PA imaging at both wavelengths of 700 nm and 808 nm revealed remarkably increased PA signals in tumors after intratumoral injection. Likewise, the FL imaging of tumors after intratumoral injection of the CBS@DC-ZIF8 nanocomposites displayed a strong FL signal under 808 nm laser irradiation. For the anticancer evaluation, the 4T1 tumor-bearing mice were intratumorally administered with CBS@DC-ZIF8@DOX nanocomposites and then received X-ray radiation (6 Gy), resulting in a ∼1.4 of relative tumor volume at day 20. However, the relative tumor volume in the X-ray alone group was 7.5. These multi-modal imaging techniques can integrate the advantages of each imaging modality and provide a comprehensive information for guiding precision RT.
5. Feature application of precision RT for anti-metastasis
Precision treatment of tumors includes not only primary (local) tumors, but also distant tumors and metastasis. RT has been mainly utilized to treat local tumors rather than distant metastasis. Previous studies have revealed that RT is able to produce a certain level of abscopal effects; however, they are far from meeting clinical demands and only rare clinical cases have been reported over the years.398–402 Even with the help of immune checkpoint blockades, therapeutic efficacies of RT for distant and metastatic tumors are still unsatisfactory.403–407 The reasons why conventional RT cannot induce systemic antitumor immunity against metastasis include inefficient induction of immunogenic cell death (ICD), immunosuppressive TME, poor presentation efficiency of tumor-specific antigens, etc.408 Recently, the rapidly evolving nanotechnologies enable RT to induce potent systemic immunity through various mechanisms, thereby extending the application of RT in the treatment of metastases.409 Besides, multifunctional NPs enable RT to be combined with other therapies (e.g., ICB therapy, immunoadjuvant, PTT, PDT, CDT, etc.) to activate systemic antitumor immune responses against distant metastasis. The representative nanotechnologies for inducing abscopal effects of RT are summarized in Table 4.| Nanoparticles | Local treatment | Mechanisms of RT-mediated abscopal effects | ICB/immunoadjuvant | Cancer type | Tumor models | Ref. |
|---|---|---|---|---|---|---|
| DBP-Hf nMOF | RT-RDT | ICD | IDOi | Primary, distant, and re-challenged tumors | SQ20B/U87MG/PC-3/CT26/TUBO tumor-bearing mice | 507 |
| Hb@Hf-Ce6 | RT-RDT | ICD, reverse the immunosuppressive TME | Anti-PD-1 antibody | Primary and distant tumors, lung metastasis | 4T1 tumor-bearing mice | 505 |
| 4PI-Zn@CaCO3 NPs | RT | Reverse the immunosuppressive TME | IDO-1 inhibitor | Primary, distant, and re-challenged tumors | CT26 tumor-bearing mice | 533 |
| PLGA/STING@EPBM | RT | Promote DC maturation and antigen presentation | STING agonist | Primary tumor, lung metastasis | B16-OVA/TC1/4T1 tumor-bearing mice | 132 |
| PSeR NPs | RT/chemotherapy | Enhance the NK cell functions | NA | Primary tumor, lung metastasis | MDA-MB-231 tumor-bearing mice | 270 |
| PLGA-R837@Cat NPs | RT | ICD, reverse the immunosuppressive TME | R837, anti-CTLA4 antibody | Primary, distant, metastatic and re-challenged tumors | CT26 tumor-bearing mice, 4T1 orthotopic breast tumor metastasis | 432 |
| ALG-ATP-Aptamer/CpG-cAptamer | RT or chemotherapy | ICD | CpG, anti-PD-1 antibody | Primary, distant, and re-challenged tumors, lung metastasis | CT26 tumor-bearing mice, orthotopic 4T1 breast tumor-bearing mice | 517 |
| 131I-Cat/ALG | RIT | ICD | CpG, anti-CTLA4 antibody | Primary, distant, metastatic and re-challenged tumors | 4T1/CT26 tumor-bearing mice, metastatic orthotopic 4T1 breast tumor-bearing mice, VX2 tumor-bearing rabbits, and patients’ prostate tumor bearing mice | 42 |
| WO2.9-WSe2-PEG NPs | RT/PTT | ICD | Anti-PD-L1 antibody | Primary, distant, and re-challenged tumors, lung metastasis | 4T1 tumor-bearing mice | 528 |
| Au@MC38 | RT | ICD | Anti-PD-1 antibody | Primary and distant tumors, lung metastasis | MC38 tumor-bearing mice | 30 |
| CpG@Au NPs | RT | ICD, repolarize TAMs | CpG, anti-PD-1 antibody | Primary and distant tumors | GL261 tumor-bearing mice | 458 |
| ZGd-NRs | RT | ICD, deplete TAMs | Anti-PD-L1 antibody | Primary and distant tumors, lung metastasis | 4T1/CT26 tumor-bearing mice | 459 |
| H@Gd-NCPs | RT | ICD | Anti-PD-L1 antibody | Primary and distant tumors, lung metastasis | 4T1/CT26 tumor-bearing mice | 41 |
| Cu-NCPs | RT/CDT | ICD | Anti-PD-L1 antibody | Primary and distant tumors, lung metastasis | 4T1/CT26 tumor-bearing mice | 532 |
| PLGA AC-NP | RT | Deliver antigen to DCs | Anti-PD-1 antibody | Primary and distant tumors | B16-F10 tumor-bearing mice | 486 |
5.1 Mechanisms of RT-mediated abscopal effect by advanced nanotechnologies
In this section, the mechanisms of RT-mediated abscopal effect by advanced nanotechnologies will be introduced, including ICD induction, repolarizing or depleting tumor-associated macrophages (TAMs), enhancing NK cell function, enhancing antigen presentation of dendritic cells, reprograming peripheral neutrophils as antigen-presenting cells, etc.A higher ROS yield by tumor accumulation of high-Z elements can induce a stronger ICD to elicit potent immune responses. For example, Qin et al. designed a tumor cell-reactor to produce biogenetic gold NPs (Au@MC38) for radiosensitization and abscopal effect.30 Coated with the MC38 cell membrane, the Au@MC38 NPs could effectively target and stay in tumors for more than 72 h after injection. Moreover, Au@MC38 containing a high-Z element could significantly enhance radiosensitizing effect on tumors and thereby induce a high tumor inhibition rate of 87.7% under X-ray irradiation compared to 66.5% in the RT alone group. In addition, the Au@MC38-mediated radiosensitization was able to induce potent ICD of tumor cells and further elicit systematic immune responses, resulting in a higher lung metastasis inhibition rate (53.5%) compared to 34.9% in the RT alone group.
GSH depletion could also augment RT-induced ROS generation and induce more potent ICD for antitumor immunity activation. Huang et al. used 5′-guanosine monophosphate (5′-GMP) and gadolinium to form nanoscale coordination polymers (NCPs) and further loaded Hemin into the NCPs (H@Gd-NCPs) for boosting RT-induced ICD.41 The H@Gd-NCPs could effectively deposit X-ray energy to elevate the intracellular ROS level due to the high-Z element. Moreover, Hemin could function as peroxidase to deplete GSH in TME and thereby augment RT-induced oxidative stress. The immunofluorescence signal of CRT in CT26 cells treated with H@Gd-NCPs plus RT (8 Gy) was significantly stronger than that in Gd-NCPs + RT and RT alone groups. Western blot assay of CT26 tumor tissues indicated that in vivo treatment of H@Gd-NCPs + RT (6 Gy) could remarkably upregulate HMGB1 expression. Furthermore, flow cytometry analysis revealed that the tumor-draining lymph nodes (TDLNs) of mice in the H@Gd-NCPs + RT group showed a higher percentage (57.85%) of mature DCs than those in the RT alone (37.42%) and Gd-NCPs + RT groups (43.45%), suggesting that H@Gd-NCP-mediated radiosensitization was able to induce potent ICD and DC maturation. Due to high immunogenicity, the H@Gd-NCP-mediated RT would potentiate checkpoint blockade immunotherapy. The treatment of H@Gd-NCPs + RT could remarkably eliminate the primary tumors with or without αPD-L1. Moreover, the H@Gd-NCPs + RT + αPD-L1 could also eradicate the distant tumors, whereas the average tumor volume in RT + αPD-L1 was over 800 mm3 21 days after treatment. Further mechanism study revealed that RT + αPD-L1 could enhance the ratios of CD4+/CD8+ T cells to 2.65%/0.91% and 2.64%/1.04% in irradiated and unirradiated tumors, respectively; however, the ratios in the H@Gd-NCPs + RT + αPD-L1 group were 5.95%/1.94% and 5.98%/2.31%. In addition, IFN-γ and infiltrating memory T cells were increased in bilateral tumors of mice treated with H@Gd-NCPs plus RT.
Furthermore, Wang et al. loaded FDA-approved physcion (Phy, an inhibitor of the pentose phosphate pathway (PPP)) onto PEG-decorated layered gadolinium hydroxide (PLGdH) nanosheets to prepare Phy@PLGdH nanosheets for boosting RT-mediated ICD (Fig. 25a).435 The Phy was reported to inhibit 6-phosphogluconate dehydrogenase (6PGD) and further reduce two main products of PPP: nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate (ribose 5-P), leading to reduced intracellular antioxidants and imbalance of nucleotide homeostasis (Fig. 25b).436–439 As such, both Phy and high-Z elements could enhance the radiosensitizing effect of Phy@PLGdH nanosheets on tumors and thereby induce stronger ICD. The CT26 cancer cells treated with Phy@PLGdH + RT rapidly exposed CRT and released HMGB1/ATP compared to the RT, PLGdH + RT, or Phy@PLGdH group. To evaluate the activation of antitumor immunity, the CT26 cancer cells treated with Phy@PLGdH with or without RT were inoculated subcutaneously in the left flank of mice. Then, these mice were rechallenged with CT26 cancer cells on the right flank for observation of tumor growth. Interestingly, vaccination with CT26 cancer cells (Phy@PLGdH + RT) could significantly inhibit the growth of rechallenged tumors and led to a 70% cancer-free state over 50 days compared to 20% in the RT alone group. Nevertheless, the vaccination of CT26 cancer cells showed no significant inhibition effect on other cancer types, such as breast cancer and renal cancer, indicating that the antitumor immunity induced by Phy@PLGdH-mediated RT was tumor-specific. Besides, Phy@PLGdH + RT + αPD-L1 could significantly eliminate lung metastasis, whereas the counts of lung foci were over 40 in the RT alone group.
 | ||
| Fig. 25 Inducing ICD. (a) A scheme of synthetic procedures of Phy@PLGdH nanosheets. (b) Schematic illustration showing in situ tumor vaccination of Phy@PLGdH nanosheets. Reproduced with permission from ref. 435. Copyright 2022, Wiley. | ||
So far, lots of nanomedicines have been designed for TAM repolarization. For example, Ma et al. revealed that graphene oxide (GO) promoted the polarization of M1 TAMs by interaction with toll-like receptors (TLRs) of macrophages and activation of NF-κB pathways.452 Zanganeh et al. reported that FDA-approved iron oxide NPs (Ferumoxytol) were able to improve the proportion of M1 phenotype TAMs in tumor tissues after intravenous administration.453 Kim's group developed M1 macrophage-derived exosome-mimetic nanovesicles to re-educate M2 phenotype TAM to M1 macrophages in vitro and in vivo.454 Shi et al. utilized NP-based ROS photogeneration to repolarize TAMs to M1 phenotype.455
Recently, several nanoradiosensitizers have been designed to regulate TAMs and further elicit antitumor immunity for suppression of distant, metastatic, and recurrent tumors.456,457 For example, Cao et al. modified AuNPs with CpG (an agonist of TLR9) to form CpG@Au NPs as a TAM repolarizing nanosystem for enhanced radioimmunotherapy (Fig. 26a and b).458 Bone marrow-derived macrophages (BMDMs) treated with CpG@Au NPs showed significant upregulation of M1 type markers (iNOS and CD86) and downregulation of M2 marker CD206. The reeducating TAMs could reduce radioresistance, and AuNPs as a high-Z element could enhance RT efficacy. The GL261 tumor-bearing mice were intratumorally injected with CpG@Au NPs, followed by X-ray irradiation (2 Gy). Immunohistochemistry (IHC) and flow cytometry analyses showed that CpG@Au + RT treatment could significantly downregulate CD206 and upregulate CD86 expression in tumors (Fig. 26c). As a result, the relative tumor volume in the CpG@Au + RT group was over 2 compared to 6 in the RT alone group (Fig. 26d). In addition, the immune checkpoint blockade (ICB) was introduced into the TAM reeducation-mediated RT to investigate whether the elicited systemic immunity was able to induce the abscopal effect of RT. The bilateral GL261 tumor model was used to evaluate the abscopal effect of RT (Fig. 26e). The results displayed that the CpG@Au + RT + anti-PD-1 treatment was able to significantly suppress the growth of both primary and distant tumors (Fig. 26f and g). However, anti-PD-1+ RT treatment showed almost no inhibition effect on distant tumors. Overall, the TAM repolarization could trigger potent systemic antitumor immunity and improve the abscopal effect of RT in cooperation with ICB.
 | ||
| Fig. 26 Repolarizing TAMs. (a) A scheme of synthetic process of CpG@Au NPs. (b) A scheme showing mechanism of TAM reeducation and enhanced RT/ICB based on CpG@Au NPs. (c) Flow cytometry analysis showing CD206 and CD86 expression of GL261 tumors in different treatment groups. (d) Tumor growth curves of mice receiving various treatments. (e) Schematic illustration of treatment schedule. Tumors on the left and right sides were regarded as distant and primary tumors, respectively. (f and g) Primary (f) and distant (g) tumor growth curves of mice in different treatment groups. Reproduced with permission from ref. 458. Copyright 2021, American Chemical Society. | ||
Besides, Huang et al. combined TAM depletion with ICD induction to boost RT-induced antitumor immunity.459 They constructed bifunctional coordination polymer nanorods (ZGd-NRs) through self-assembly of zoledronic acid (Zol) and gadolinium. With the high-Z element, ZGd-NRs could improve the radiosensitizing effect and thereby induce strong ICD in CT26 tumor cells. Meanwhile, the percentage of mature DCs in TDLN of mice treated with ZGd-NR plus RT was increased to 33.6%, much higher than 14.1% of the RT alone group. In addition, Zol was able to inhibit the mevalonate pathway and thereby induce apoptosis of macrophages.460 The treatment of ZGd-NRs + RT could reprogram immunosuppressive TME by depleting intratumoral F4/80+CD11b+ TAMs and decreasing secretion of IL-10, TGF-β1, and VEGFA. Therefore, the ZGd-NR-mediated RT could remarkably suppress distant tumor growth while RT alone had a negligible inhibitory effect on distant tumors. Besides, the distant tumors could be completely eradicated in the ZGd-NR + RT + αPD-L1 group. Further mechanism experiments showed that ZGd-NR-mediated RT could increase the levels of infiltrated CD4+/CD8+ T cells to 3.97%/1.37% and 4.14%/1.41% in primary and distant tumors, respectively, much higher than those in the RT alone group (1.46%/0.28% and 1.75%/0.29% for primary and distant tumors). These results indicated that ZGd-NR-mediated RT could activate systemic antitumor immune responses by TAM depletion and ICD induction.
A previous study found that inorganic selenium compound selenite (SeO32−) was able to block the HLA-E expression of tumors at the posttranscriptional level and thereby improve sensitization of tumor cells to CD94/NKG2A-positive NK cells, suggesting that selenite could enhance NK cell-based immunotherapy.471 Recently, organic seleninic acid was also reported to activate NK cells for immunotherapy.270,472 In addition, the diselenide bond can be oxidized into seleninic acid upon γ-ray irradiation.473–475 Li et al. assembled cytosine-containing diselenides (Cyt–SeSe–Cyt) with pemetrexed (a clinically used chemotherapeutics) through hydrogen bonds to construct diselenide-pemetrexed (Pem/Se) assemblies for RT/chemotherapy/immunotherapy.472 Upon γ-ray irradiation, the diselenide bonds and hydrogen bonds were cleaved, resulting in the disassembly of Pem/Se assemblies and pemetrexed release. The MDA-MB-231 cells treated with Pem/Se assemblies and γ-ray irradiation (5 Gy) showed significant downregulation of HLA-E due to the generation of seleninic acid. The tumor inhibition rate of MDA-MB-231 tumor-bearing mice in the Pem/Se + γ-ray radiation group was about 1.5 times that of the Pem + γ-ray radiation group. The immunofluorescence staining of tumor slices revealed that the Pem/Se + γ-ray radiation treatment significantly reduced HLA-E expression and upregulated the expression levels of IFN-γ and CD49b, suggesting elevated immunoactivity of NK cells in tumor tissues. In addition, the NK cells in metastases were also activated after the Pem/Se + γ-ray radiation treatment, and the induced systemic immune response could inhibit lung metastases.
In another similar study, Gao et al. proposed a NK cell-based immunotherapy strategy using radiation-sensitive NPs (PSeR NPs).270 The PSeR NPs were loaded with DOX and then decorated with RGD-modified PEG (Fig. 27a). The TEM images further showed that the PSeR NPs with spherical morphology gradually collapsed into irregular structures upon elevated doses of γ-ray irradiation owing to the oxidation of diselenide bonds (Fig. 27b). Due to structural collapse, DOX could be rapidly released from PSeR NPs. Moreover, they found that seleninic acid (RSeO(OH), oxidation products of diselenides) could react with intracellular GSH to affect the redox balance in cancer cells. The GSH/GSSG ratio and expression levels of glutathione peroxidase (GPx) and GSH-transferase (GST) were remarkably decreased in MDA-MB-231 cells treated with PSeR NPs + γ-ray radiation (5 Gy). Whereas, CAT was upregulated after treatment of PSeR NPs + γ-ray radiation. These intracellular alterations could potentiate efficacy of RT and chemotherapy. Besides, the PSeR NPs + γ-ray radiation (5 Gy) treatment could induce the NK cell-mediated cytotoxicity through blocking HLA-E expression and releasing Granzyme B (GZB)/IFN-γ. Next, the MDA-MB-231 tumor-bearing nude mice with normal NK cell but defective CD8+ T cells were used to evaluate the efficacy of NK cell-based immunotherapy induced by PSeR/DOX and RT. The results showed that the tumor inhibition rate in the PSeR/Dox + RT group was almost 2-fold higher than that in the DOX + RT group (Fig. 27c). The confocal images of tumor tissues displayed upregulated HLA-E expression after combined treatment (Fig. 27d). Simultaneously, GZB and IFN-γ were significantly increased in serum and tumors (Fig. 27e and f). These results suggested that the excellent antitumor efficacy was attributed to synergistic RT/chemotherapy and NK cell-mediated immunotherapy. Furthermore, to evaluate the in vivo anti-metastasis efficacy of NK cell-mediated immunotherapy, the MDA-MB-231 cells treated with or without PSeR + radiation were intravenously injected into nude mice to establish the human breast cancer lung metastasis model. The mice receiving MDA-MB-231 cells treated with PSeR + radiation showed almost no metastatic foci in lung tissues compared to the control groups.
 | ||
| Fig. 27 Enhancing NK cell function. (a) A scheme of the synthetic process of PSeR NPs and its application in synergistic RT/chemotherapy and inducing NK cell-mediated immunotherapy. (b) TEM images of PSeR NPs under various doses of γ-ray irradiation. (c) Tumor growth curves of mice in different treatment groups. (d) Quantification analysis of HLA-E fluorescence intensities in tumors of different treatment groups. (e and f) The serum IFN-γ (e) and GZB (f) concentrations of mice in different treatment groups. Reproduced with permission from ref. 270. Copyright 2020, Wiley. | ||
Min et al. designed antigen-capturing NPs (AC-NPs) to enhance antigen presentation of DCs for augmenting the abscopal effect of RT.486 They utilized PLGA to form a polymer and then decorated the polymer with various molecules to bind the TDPAs, including methoxypolyethylene glycol (mPEG AC-NPs, negative control), 1,2-dioleoyl-3-trimethylammoniopropane (DOTAP AC-NPs), amine polyethylene glycol (NH2 AC-NPs), and maleimide polyethylene glycol (Mal AC-NPs). After incubating with irradiated B16F10 melanoma cell lysates, the unmodified PLGA AC-NPs and DOTAP AC-NPs captured most proteins compared to other types of AC-NPs. Further experiments indicated that all the AC-NPs except for mPEG AC-NPs were able to capture neoantigens and damage-associated molecular pattern proteins (DAMPs, a series of pro-inflammatory proteins). The AC-NPs could deliver these antigens toward DCs to enhance antigen presentation and thereby induce potent immune responses. Next, to evaluate the enhanced immunotherapy of AC-NPs, bilateral B16F10 melanoma-bearing mice treated with αPD-1 were utilized. Interestingly, the PLGA AC-NPs and Mal AC-NPs produced significant abscopal effects on the secondary tumors, whereas the RT alone showed negligible inhibition effect on the secondary tumors. Further mechanism study revealed that the PLGA AC-NPs and Mal AC-NPs were transported to the adjacent lymph nodes and mainly accumulated in CD11c+ DCs, F4/80+ macrophages, and B220+ B-cells at 16 h after intratumoral injection into the irradiated tumors. Moreover, the increased CD8+ T cells and decreased immunosuppressive CD4+FOXP3+ Treg cells were shown in secondary tumors treated with PLGA AC-NPs or Mal AC-NPs, indicating potent antitumor immune responses in these secondary tumors. More importantly, unlike traditional immunotherapy strategies by using several “chosen” antigens to elicit immune responses, these AC-NPs could realize personalized treatment by delivering patient-specific tumor antigens to DCs, which could facilitate the abscopal effect of RT and extend RT from local tumors to metastasis treatment.
For example, Guo et al. proposed an nMOF-mediated RT-RDT strategy to reprogram peripheral neutrophils as non-canonical APCs for enhancing antitumor immune responses.498 They metalated the porphyrin center with Pt(II) to construct Hf-DBP-Pt (Hf-Pt-5,15-di(p-benzoato)porphyrin) as a nanoradiosensitizer for effective RT-RDT. The in vitro clonogenic assays showed that Hf-DBP-Pt exhibited stronger radiotherapeutic effects on MC38, HepG2, CT26, AsPC-1, and Panc02 cells compared to Hf-DBP, which might be attributed to stronger energy deposition and radiodynamic effect through facilitating intersystem crossing. To investigate the in vivo antitumor efficacy of RT-RDT, MC38 or CT26 tumor-bearing mice were intratumorally administered with Hf-DBP or Hf-DBP-Pt, followed by X-ray radiation. The results showed that both Hf-DBP and Hf-DBP-Pt could significantly inhibit tumor growth under X-ray irradiation. Further flow cytometry analysis of treated MC38 tumors indicated that Hf-DBP-Pt plus RT could recruit CD11b+Ly6G+ neutrophils into the tumors. The phenotype analysis revealed that these recruited neutrophils were reprogrammed to hybrid neu-DC characterized by upregulation of CD11c. Hf-DBP-Pt + RT elevated the proportion of neu-DC in the whole CD11b+Ly6G+ neutrophils to 36.4%, which was 10-fold higher than that in the RT alone group. In addition, the hybrid neu-DC exhibited elevated expression levels of co-stimulatory CD86, CD80, and MHC II markers, indicating that neu-DC could function as APC to present tumor antigen. In addition, the hybrid neu-DC induced by RT-RDT could remarkably augment cross-presentation of tumor antigens and enhance both innate and adaptive antitumor immune responses.
5.2 Strategies for synergistic induction of abscopal effect by RT and other therapies
This section will introduce strategies for synergistic induction of abscopal effect by RT and other therapies, including ICB therapy, immunoadjuvant, and other therapies.For example, Dai and colleagues combined RT-RDT with ICB therapy to trigger systemic antitumor immunity against primary tumors and distant metastasis (Fig. 28a).505 First, chlorin e6 (Ce6) and catechol were conjugated onto PEG to form Ce6-PEG-polyphenols. The Ce6-PEG-polyphenols could self-assemble with Hf and were further loaded with Hb (Hb@Hf-Ce6 NPs). The Hf, a high-Z metal, could not only act as a radiosensitizer to enhance RT but also play a scintillator role in efficient radioluminescence. Moreover, Hb in the nanoplatform could deliver oxygen to alleviate tumor hypoxia and reverse the immunosuppressive TME (Fig. 28a). The bilateral 4T1 tumor-bearing mice were utilized to investigate the treatment efficacy of RT-RDT and immunotherapy (Fig. 28b). Hb@Hf-Ce6 NP-mediated RT-RDT could significantly inhibit the growth of both primary and distant tumors (Fig. 28c and d). Notably, the primary and distant tumor volumes in the Hb@Hf-Ce6 NP-mediated RT-RDT + αPD-1 group were about 1/6 and 1/2 of those in the RT + αPD-1 group, respectively. Next, the immune mechanism study revealed that combination of Hb@Hf-Ce6 NP-mediated RT-RDT and αPD-1 therapy could remarkably reduce M2 phenotype TAMs, regulatory T cells, and immunosuppressive IL-10 secretion as well as increase effector T cells, memory T cells, and cytokines (e.g., IL-6, IL-12, IFN-γ, TNF-α, etc.) (Fig. 28e). In addition, Hb@Hf-Ce6 NP-mediated RT-RDT plus αPD-1 could also inhibit lung metastases with a mouse survival rate of 100%, 20 days after treatment compared to 50% in the RT + αPD-1 group (Fig. 28f).
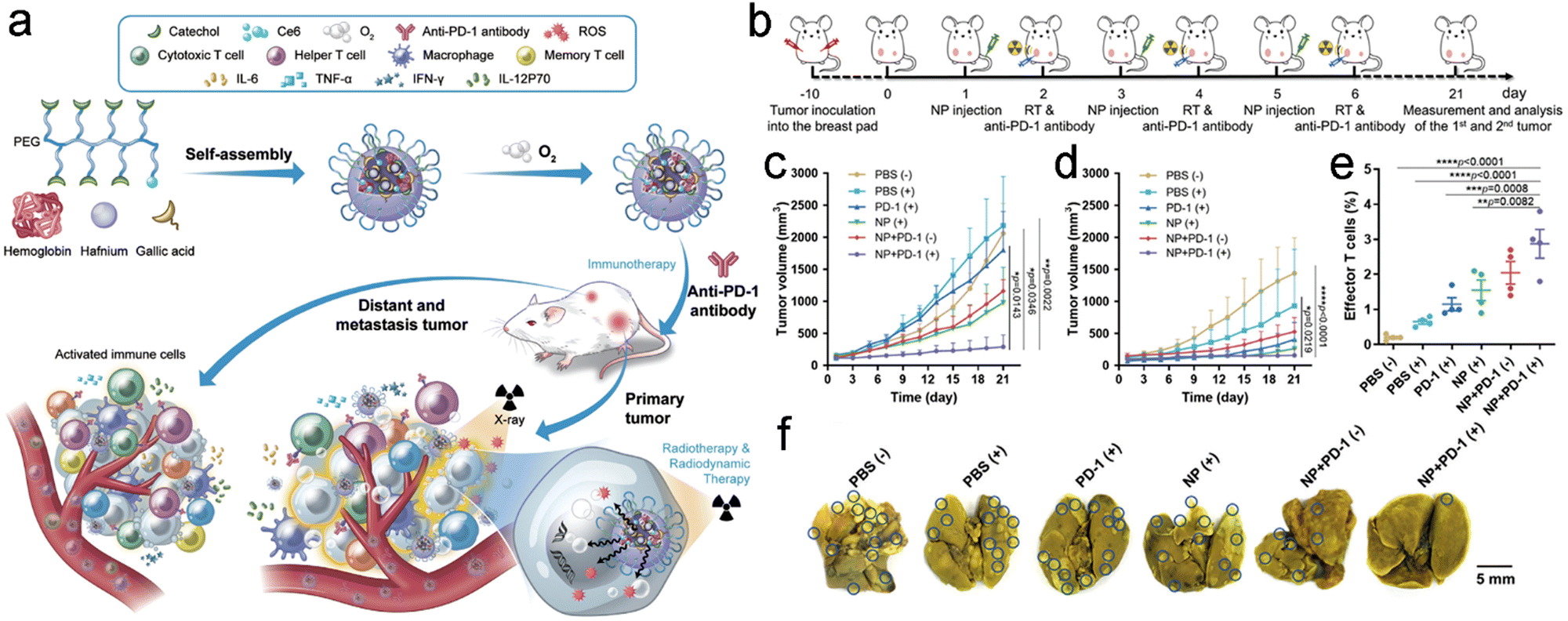 | ||
| Fig. 28 Combination of RT and ICB therapy. (a) A scheme showing the synthetic process of Hb@Hf-Ce6 NP and its application in synergistic RT-RDT/αPD-1 immunotherapy against primary tumors and distant metastasis. (b) A scheme of treatment schedule for bilateral breast cancer mice. (c and d) Primary (c) and distant (d) tumor growth curves of mice in different treatment groups. (e) The percentages of effector T cells in distant tumors of mice received different treatments. (f) Representative photos of lung metastases in 4T1 tumor metastasis mice received different treatments. Reproduced with permission from ref. 505. Copyright 2020, Wiley. | ||
In addition, checkpoint blockade antibody could also be encapsulated into NPs. Choi et al. developed snowflake-like Au nanocarriers (S-AuNC) and loaded αPD-L1 into the S-AuNC for RT/ICB therapy of tumors.271 The S-AuNC could be decomposed under radiation to release αPD-L1. Moreover, the S-AuNC with high-Z elements could enhance the radiosenstizing effect and thereby induce strong ICD after intratumoral injection. This radiation-responsive in situ αPD-L1 release strategy enabled a controllable spatiotemporal combination of RT and ICB immunotherapy, leading to activation of potent antitumor immunity against tumor growth.
For example, Patel et al. designed bacterial membrane-coated NPs (BNPs) to promote RT to elicit systemic antitumor immunity against local tumors and metastatic relapse.518 The BNPs were composed of four components. The inner polyplex core contained two immunological adjuvants: CpG (a TLR-9 agonist) and acid-responsive PC7A polymer for endosomal escape of antigens (Fig. 29a). The core was then coated with bacterial membranes that could activate innate immunity and DCs. Furthermore, the Mal groups were modified onto the bacterial membranes for tumor antigen capturing. The neoantigen released by irradiated tumors could be captured by BNPs (Fig. 29b and c). The neoantigen-carrying BNPs could further enhance the DC uptake owing to the bacterial membranes. After internalization by DCs, the BNPs could escape from the endosomes owing to the pH-responsive PC7A polymer. The PC7A could activate the stimulator of the interferon gene (STING) pathway to increase mature DCs, NK cells and T cells.519,520 Furthermore, the released adjuvant CpG could activate TLR-9 on the inner membrane of endosomes and further promote DC maturation. Moreover, the intracellular neoantigens were degraded into antigen peptides for antigen presentation. Then, mature DCs could present antigens to T cells and thereby activate antitumor immunity (Fig. 29b and c). The combined treatment of RT and BNPs could completely eliminate tumors in both “cold” B78 melanoma and “hot” NXS2 neuroblastoma mouse models. In addition, the mice with complete regression of tumors were inoculated with the same cancer cells to evaluate immune memory. All the mice previously treated with RT + BNPs in the B78 tumor group eradiated the re-challenged tumors. Moreover, 75% of the NXS2 mice in the RT + BNP group eradiated the re-challenged tumor, compared to 50% in mice receiving BNPs alone and 15% in the control group. Further immune mechanism experiments indicated that the treatment of RT + BNPs could increase tumor infiltrating levels of CD4+/CD8+ T cells and secretion of IFN-γ. Therefore, the treatment of BNPs and RT could induce strong tumor-specific immune responses and long-term immune memory against primary and re-challenged tumors.
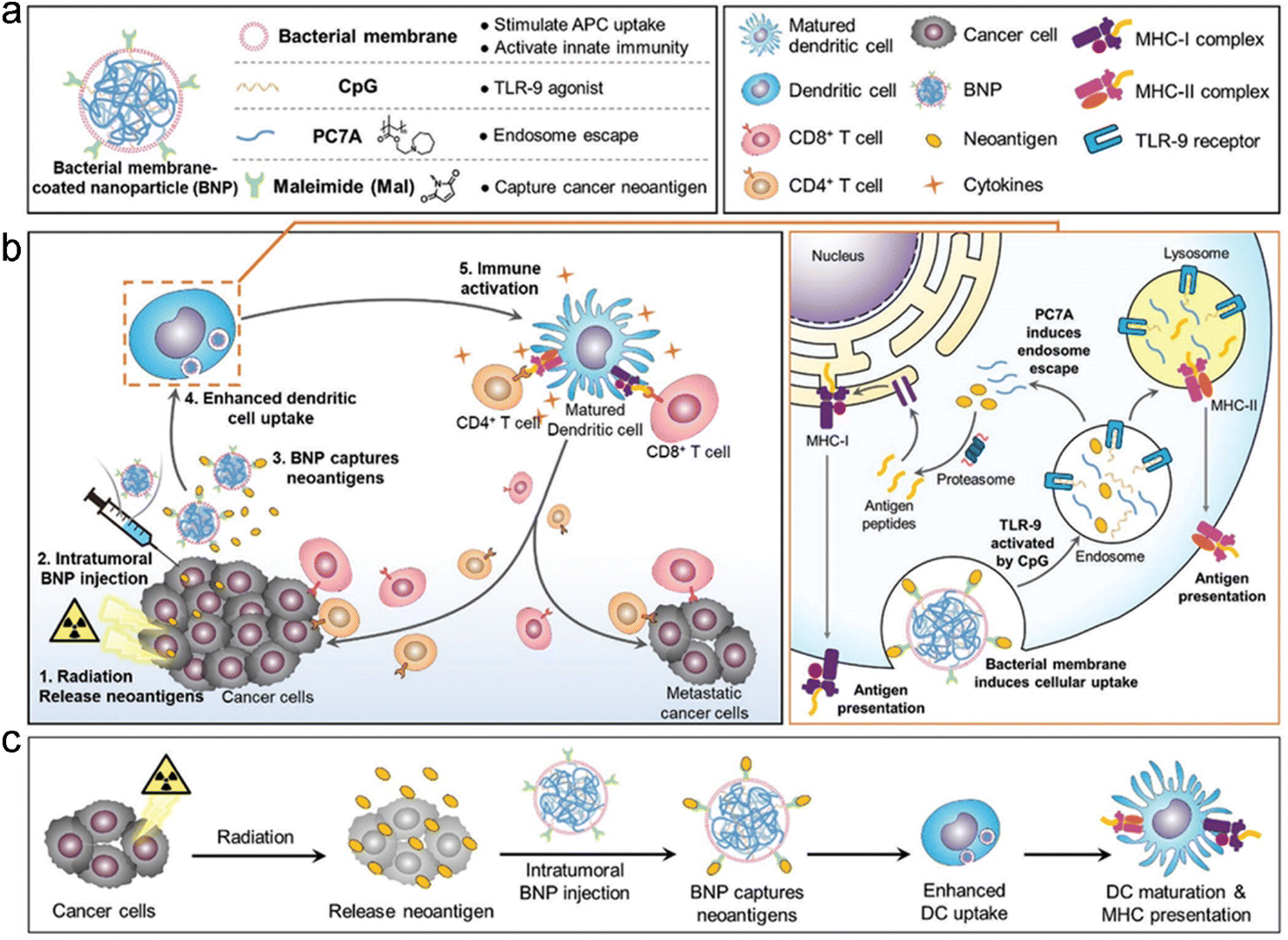 | ||
| Fig. 29 Combination of RT and immunoadjuvant. (a) A scheme showing components of BNPs and the corresponding functions. (b) A scheme of antitumor immune responses induced by BNP + RT against primary and metastatic tumors. (c) Schematic illustration of the process by which BNPs interact with neoantigens to promote DC uptake and maturation. Reproduced with permission from ref. 518. Copyright 2019, Wiley. | ||
Indoleamine 2,3-dioxygenase (IDO, an immunoregulatory enzyme) is able to catalyze the conversion of Trp into Kyn and thereby cause T cell apoptosis and tumor immune escape, thus resulting in immunosuppression.521–524 IDO inhibitors have been widely recognized as immunometabolic adjuvants to augment antitumor immunity.525–527 Lu et al. designed 5,15-di(p-benzoato)porphyrin-Hf (DBP-Hf) based nMOFs for RT-RDT of tumor.507 Hf clusters could absorb X-ray photons to generate ˙OH radicals for RT and to excite porphyrin-based photosensitizer ligands for RDT. The nMOF-mediated RT-RDT could effectively inhibit tumor growth and induce ICD upon low-dose X-ray irradiation. Then, the nMOFs were loaded with IDOi (9.4 wt%) to prepare IDOi@DBP-Hf for yielding abscopal effects. The bilateral CT26 tumor-bearing mice were utilized to evaluate antitumor efficacy. The local administration of IDOi@DBP-Hf and X-ray radiation could significantly eradicate both irradiated and unirradiated tumors. However, both IDOi@DBP-Hf alone and IDOi + RT showed negligible inhibition effect on the untreated tumors. In addition, to investigate the immune memory, the tumors on the right flanks of mice were first treated with IDOi@DBP-Hf and X-ray irradiation. Then, new TUBO tumor cells were inoculated on the left flanks of these treated mice to establish re-challenged tumors. The mice in the IDOi@DBP-Hf + X-ray irradiation group exhibited no growth of the re-challenged tumors for 2 months, suggesting a potent antitumor immune memory. In contrast, the mice in the PBS group showed no suppressive effect on the re-challenged tumors.
CDT is an emerging therapy modality that converts intracellular H2O2 into more harmful ˙OH to kill cancer cells.129 For example, Wang et al. combined RT with CDT to activate systemic antitumor immunity against primary and metastatic tumors.532 They self-assembled 5’-GMP and Cu2+ to prepare Cu-based NCPs (Cu-NCPs). The Cu-NCPs contained mixed valence (Cu+/Cu2+) since part of Cu2+ was reduced to Cu+. Cu2+ in the Cu-NCPs could deplete GSH to reduce ROS scavenging, and the Cu+-mediated Fenton-like reaction could convert H2O2 to ˙OH for CDT of tumors. With these capabilities, the Cu-NCPs could potentiate RT-induced oxidative stress. Cu-NCP-mediated synergistic RT/CDT could induce strong ICD. The flow cytometry analysis revealed that the synergistic RT/CDT triggered significant DC maturation (35.1%) in TDLNs, much higher than that in the RT alone (19.7%) or Cu-NCPs alone (23.1%) group. Unsurprisingly, RT alone showed almost no inhibition effect on distant tumors of bilateral CT26 tumor-bearing mice. However, Cu-NCP-mediated synergistic RT/CDT could suppress both primary and distant tumor growth, resulting in 25% complete regression of distant tumors. After combination with PD-L1 checkpoint blockade, the rate of complete regression of distant tumors was elevated to 62.5%. The infiltrating CD4+/CD8+ T cell and IFN-γ secretion were significantly increased in both primary and distant tumors of the Cu-NCP-mediated RT/CDT group. In addition, the Cu-NCP-mediated RT/CDT plus PD-L1 checkpoint blockade showed a significant inhibition effect on lung metastases of 4T1 tumors and resulted in a 90% survival rate after 100 days. However, all the mice in the RT + PD-L1 group died within 75 days. Consequently, these results indicated that synergistic RT/CDT could activate potent systemic antitumor immunity and potentiate ICB against primary tumors and distant metastases.
Phototherapy has been also used to enhance the abscopal effect of RT. Dong et al. constructed the semiconductor heterojunction WO2.9-WSe2-PEG NPs (WSP NPs) for local RT/PTT of tumor.528 The WSP NPs could effectively deposit X-ray energy to enhance the radiosensitizing effect of tumors owing to the high-Z element. Moreover, the heterojunction structure could promote the separation of electrons and holes, which further increased ROS production. Furthermore, the WSP NPs could also be used for PTT under NIR laser irradiation, which would directly induce ICD and increase tumor blood supply to sensitize RT. Consequently, the WSP NP-mediated RT/PTT could trigger potent ICD and potentiate checkpoint blockade immunotherapy. WSP NPs + NIR laser + X-ray + αPD-L1 could significantly inhibit both primary and distant tumor growth, leading to 80% complete regression of distant tumors. However, RT alone showed almost no suppressive effect on distant tumors. Further flow cytometry analysis indicated that synergistic RT/PTT/αPD-L1 remarkably elevated infiltrating levels of CD4+/CD8+ T cells in distant tumors, indicating that the combined treatment was able to elicit systemic antitumor immune responses. Besides, the WSP NP-mediated synergistic RT/PTT/αPD-L1 could also induce long-term immune memory to eradicate re-challenged tumors.
6. Feature application of precision radiotherapy for radioprotection
RT based on high energy ionizing radiation can not only kill cancer cells but also simultaneously damage surrounding normal tissues, including direct damage caused by interaction between radiation and DNA as well as ROS-mediated indirect damage through interaction of radiation and water molecules.13,534,535 Maximizing the radiosensitizing effect of tumors while minimizing side effects of ionizing radiation on healthy tissues is one aim of precision RT. Hence, a lot of radioprotectors have been designed to reduce the ionizing radiation-induced damages to healthy tissues.536–538 Until now, most radioprotectors are organic molecular agents, such as naturally occurring compounds and chemical compounds.536,539–542 For example, amifostine (Ethyol®), the only small-molecular chemical radioprotector approved by U.S. Food and Drug Administration (FDA) so far, shows limited clinical efficacy due to its high toxicity, short blood elimination half-time (<10 min), rapid renal clearance from the body, and narrow administration window.543,544 With the rapid development of nanotechnology, the nanomaterial-based radioprotectors are expected to overcome these drawbacks of conventional radioprotection. This section will discuss nanotechnology-mediated radioprotection of healthy tissues, including nanoradioprotectors (e.g., cerium-based nanomaterials, transition-metal dichalcogenide, etc.) and delivery of molecular radioprotectors.6.1 Nanomaterials for radioprotection
Recent studies have reported that several types of nanomaterials, also known as nanoradioprotectors, possess the intrinsic radioprotective activities to scavenge excessive ROS generated from ionizing radiation. The nanoradioprotectors exhibit longer blood circulation time and poorer RES clearance compared to small-molecular radioprotectors. The nanoradioprotectors can be divided into the following six categories: (1) carbon-based nanoradioprotectors; (2) noble metal-based nanoradioprotectors; (3) metal oxide-based nanoradioprotectors; (4) transition-metal dichalcogenide (TMDC)-based nanoradioprotectors; (5) MXene-based nanoradioprotectors; and (6) organic nanoradioprotectors (Fig. 30 and Table 5).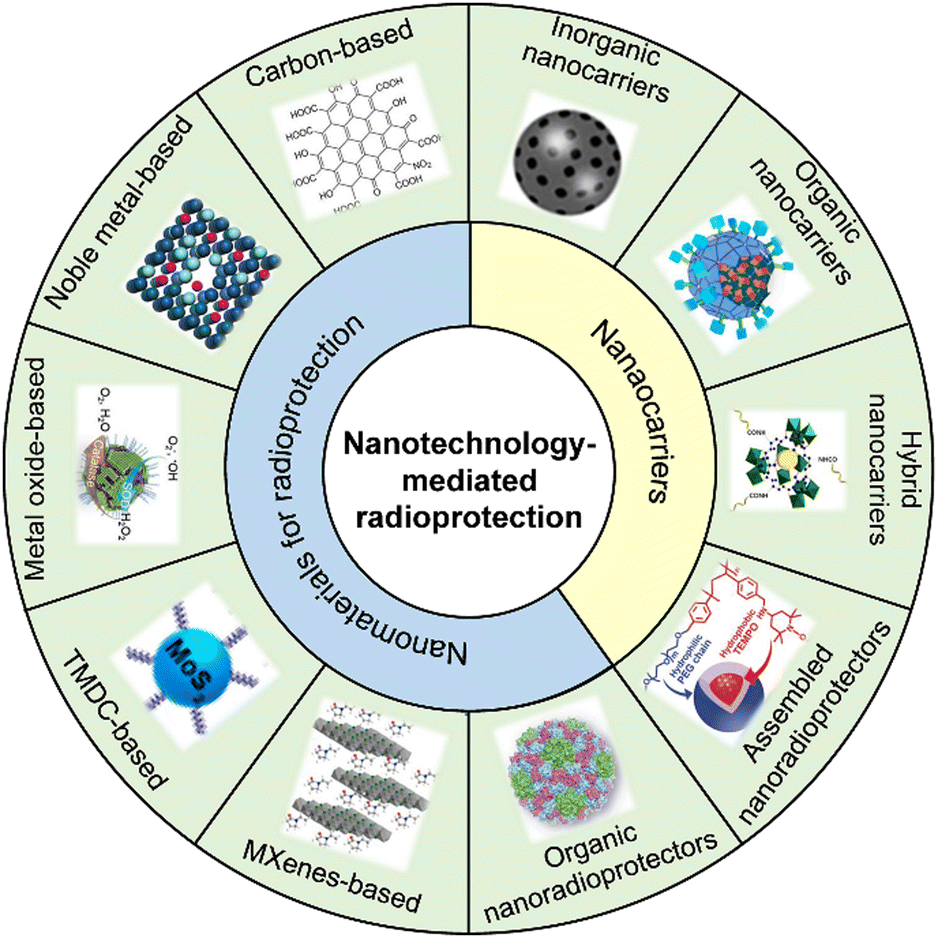 | ||
| Fig. 30 Scheme of general nanotechnology-mediated radioprotection. Adapted with permission.545–554 Copyright, Wiley, American Chemical Society, and Elsevier. | ||
| Type | Nanoradioprotectors | Route of administration | Timing of administration | Experimental subject | Radiation type/dose | Ref. |
|---|---|---|---|---|---|---|
| Carbon-based | Fullerenol C60(OH)24 | Intraperitoneal administration | 30 min before irradiation | Adult Wister male rats | X-ray (7 and 8 Gy) | 569 |
| Carbon-based | F-NaHA | External use on the skin | 1 h before irradiation | Adult BALB/c male mice | NA | 571 |
| Carbon-based | DF-1 | Co-incubation | 5, 15, 30 min after irradiation | Zebrafish embryos | γ-ray (0–40 Gy) | 684 |
| Carbon-based | M@Cs | Intravenous administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.5 Gy) | 585 |
| Carbon-based | Graphdiyne-BSA | Intravenous administration | Before irradiation | Adult BALB/c male mice | X-ray (6.5 Gy) | 594 |
| Carbon-based | CNSI | Oral administration | Before irradiation | Adult BALB/c male mice | X-ray (4.5 Gy) | 545 |
| Noble metal-based | PtPd nanocubes | Intraperitoneal administration | Before irradiation | Adult C57BL/6 male mice | γ-ray (7.2 Gy) | 609 |
| Noble metal-based | PtPdMo nanocubes | Intraperitoneal administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.2 Gy) | 610 |
| Noble metal-based | PtPdRh nanocubes | Intraperitoneal administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.3 Gy) | 546 |
| Noble metal-based | Ag14 clusterzymes | Intravenous administration | 30 min before irradiation | Adult BALB/c female mice | γ-ray (7 Gy) | 685 |
| Metal oxide-based | Ceria NPs | Intravenous administration | 1 d before irradiation, weekly dose after irradiation for one month | Adult C57BL/6 male mice | X-ray (2.5, 5, and 10 Gy) | 623 |
| Metal oxide-based | CeO2/Mn3O4 | Intraperitoneal administration | 1 h before irradiation | Adult ICR mice | γ-ray (13 Gy) | 547 |
| Metal oxide-based | Mn12 | Intraperitoneal administration | 30 min before irradiation | Adult BALB/c mice | γ-ray (6.5 Gy) | 686 |
| TMDC-based | PVP-Bi2Se3@Sec | Intratumoral injection | Before irradiation | BEL-7402 tumor-bearing BALB/c nude mice | X-ray (6 Gy) | 45 |
| TMDC-based | Bi2Se3 | Intraperitoneal administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.5 Gy) | 648 |
| TMDC-based | Cys-MoS2 dots | Intraperitoneal administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.5 Gy) | 548 |
| TMDC-based | Cys-WSe2 dots | Intravenous administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (7.5 Gy) | 687 |
| TMDC-based | Au-MoS2 clusters | Intraperitoneal administration | 30 min before irradiation | Adult C57BL/6 male mice | γ-ray (5 Gy) | 688 |
| MXene-based | Nb2C-PVP | Intravenous administration | 1 d before irradiation | Adult BALB/c male mice | γ-ray (6.5 Gy) | 549 |
| Organic nanoradioprotectors | PHA-L | Intraperitoneal administration | Once per day for 3 days before and 7 days after irradiation | Adult C57BL/6 male mice | γ-ray (7.2 Gy) | 683 |
6.1.1.1 Fullerene-based nanoradioprotectors. Fullerenes are a family of cage-like molecules that contain various numbers of carbon atoms, including C20, C40, C60, C70, and C84.555–557 The fullerenes are able to react with free radicals since they have a large number of π-conjugated bonds and an unoccupied molecular orbital with the lowest energy.558 Thus, fullerenes can act as radioprotectors to scavenge ROS, including H2O2, ˙OH, hydroperoxy radicals (HO2) and O2−.559–564
Fullerenol (C60(OH)n; n = 12–26, one type of C60 fullerene derivatives) was prepared by introducing hydroxyl groups into the structure of fullerene.565,566 Fullerenol has attracted wide attention in the radioprotection field owing to its excellent free radical scavenging capability.567 For example, Zhao et al. investigated the radioprotective effect of fullerenols on γ-ray-irradiated Stylonychia mytilus (S. mytilus) cells.568 They found that fullerenols exhibited great anti-oxidative and ROS scavenging activities, thus increasing the survival fraction of γ-ray-irradiated S. mytilus. In addition, the radioprotective efficacy was associated with both the irradiation dose and fullerenol concentration. Trajković et al. revealed that intraperitoneal administration of fullerenol C60(OH)24 (100 mg kg−1) into rats exposed to X-ray irradiation (8 Gy) could provide a 60% survival rate 30 days after treatment.569 However, the irradiated rats in the control group died within 30 days.
Radiodermatitis, one of the most common side effects of cancer RT, not only reduces the life quality of patients, but also increases the risk of infection.570 Zhao et al. systematically evaluated the radioprotective effect of fullerenols on radiodermatitis.571 They proposed an environmentally friendly and gram-scale (>20 g) synthesis method for mass production of fullerenols (Fig. 31a). Then the evaluation of free radical scavenging capabilities showed that fullerenols presented much higher 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonate) (ABTS) and 1,1-diphenyl-2-picrylhydrazyl (DPPH) scavenging performances than those of superoxide dismutase (SOD) at the same concentrations. Besides the two model radicals, fullerenols also exhibited great scavenging effects on ˙OH and O2−. Furthermore, fullerenols (25 μg per Ml) could provide a survival fraction (56.34%) of human keratinocyte (HaCaT) cells under 8 Gy of X-ray irradiation, much higher than 33.95% in the X-ray alone group. Moreover, the further mechanism exploration indicated that fullerenols were able to decrease lipid peroxidation and reduce mitochondrial dysfunction and DNA damage (Fig. 31b). For in vivo protective effect evaluation, fullerenols were utilized to construct fullerenol-sodium hyaluronate (F-NaHA) hydrogels that could form a thin film on the skin surface for radiation protection. The commercial SOD salves were used as a positive control. The back skin of healthy BALB/c mice was irradiated with X-rays to induce radiodermatitis. Compared to X-ray alone and X-ray + SOD salve groups, the irradiated skin pre-smeared with F-NaHA hydrogels presented an obviously less inflammatory infiltration area at 4, 9, 16, and 25 d after irradiation, suggesting that F-NaHA hydrogels possess excellent radioprotective capacity for radiodermatitis in vivo.
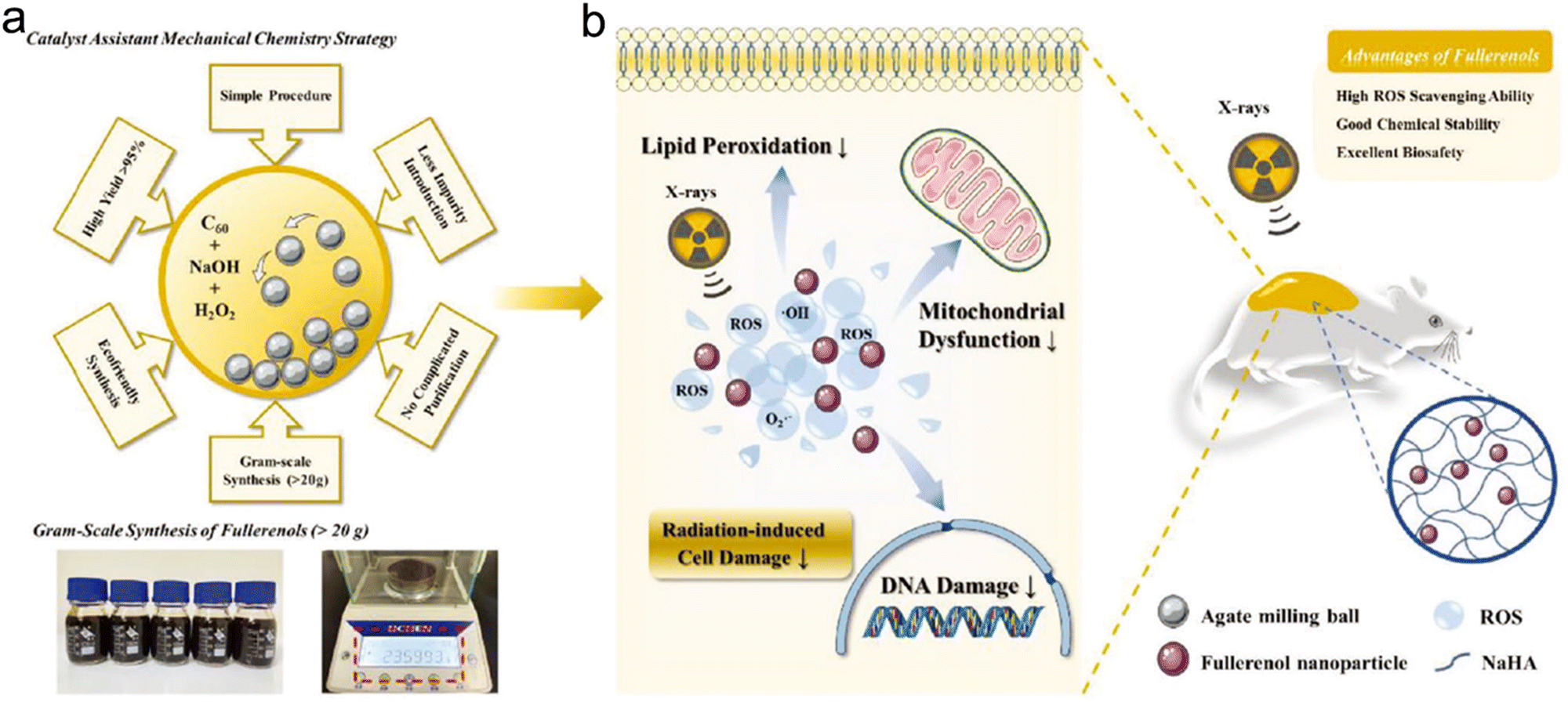 | ||
| Fig. 31 Fullerene/graphene-based nanoradioprotectors. (a) Gram-scale synthesis of fullerenes using an ecofriendly method. (b) Fullerenes with excellent free radical scavenging properties for skin radioprotection. Reproduced with permission from ref. 571. Copyright 2021, Wiley. | ||
6.1.1.2 Graphene-based nanoradioprotectors. Graphene and its derivatives have been previously reported to scavenge free ROS owing to their unique chemical structures, such as pristine sp2 carbon domains.572–577 For example, Nilewski et al. previously reported that graphene quantum dots (GQDs) possessed the capability of SOD enzyme and could scavenge O2− and ˙OH to protect bEnd.3 murine endothelioma cells against H2O2.578 Song et al. reported that carboxyl-decorated GO (GO-COOH) possessed peroxidase-like nature and could catalyze the oxidization of peroxidase substrate 3,3,5,5-tetramethylbenzidine (TMB) in the presence of H2O2.579
Graphene-encapsulated metal nanohybrids have been reported to possess excellent catalytic activities of oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) processes, indicating that these nanohybrids are able to scavenge oxygen free radicals.580–584 Wang et al. evaluated the in vivo radioprotective efficiency of a series of single-layer graphene-encapsulated metal nanoshields (M@Cs), including graphene-encapsulated Fe and CoNi (denoted as Fe@C and CoNi@C).585 The TEM images showed that the metal cores were coated with a single layer of graphene (about 3.4 Å). Next, density functional theory (DFT) calculations were utilized to interpret the ROS scavenging processes. Specifically, graphene could adsorb ˙OH and then react with a proton to generate H2O during process 1. The O2− could transfer an electron to the surface of CoNi@C and be converted into O2 during process 2. The O2− on the surface of Fe@C could be converted into H2O during process 3. Moreover, the ˙O, ˙OOH, as well as the formed *O and *OOH adsorbed on the surface could be converted into H2O2. The pretreatment of Fe@C or CoNi@C nanoshields at 30 min before irradiation could remarkably rescue the ROS level and DNA damage, leading to a higher cell survival proportion (over 70%). However, the survival fraction of CHO cells in the RT alone group was about 50%. Additionally, to evaluate the in vivo radioprotective effect, healthy male C57BL/6 mice were intravenously administered with amifostine, Fe@C or CoNi@C 30 min before receiving whole-body γ-ray (7.5 Gy). Pretreatment with CoNi@C or Fe@C could significantly reduce the ROS level in irradiated organs and further increase 3,4-methylenedioxyamphetamine (MDA)/SOD contents in the lungs and liver. Therefore, the irradiated mice in the control group showed a 10% survival rate for 30 days; however, the mice pretreated with CoNi@C and Fe@C exhibited great radioprotective efficacy with 80% and 90% survival rates, respectively, which was even better than that in irradiated mice treated with amifostine (80% survival rate).
6.1.1.3 Graphdiyne-based nanoradioprotectors. Graphdiyne, a type of carbon network material, is composed of acetylenic and benzene moieties. Due to its unique chemical and electronic properties, graphdiyne has been widely applied in various fields, including photocatalysis,586,587 solar cells,588–591 energy storage,592,593etc.
Owing to its π-conjugated structure and diacetylenic linkages, graphdiyne has also been reported to possess ROS scavenging capability. Xie et al. fabricated bovine serum albumin (BSA)-decorated graphdiyne NPs (graphdiyne-BSA NPs) to investigate their radiation protection effect.594 The graphdiyne-BSA NPs could effectively scavenge two model free radicals (DPPH and ABTS) in a dose-dependent manner. To investigate the in vivo radioprotective effect, healthy BALB/c male mice irradiated with X-rays (6.5 Gy) after intravenous injection of graphdiyne-BSA NPs (200 μg each mouse) were collected for SOD, MDA, and bone marrow DNA measurements. The DNA content in bone marrow is an important indicator of ionizing radiation damage. The bone marrow DNA detection assays indicated that graphdiyne-BSA NPs were able to reduce the severe DNA damage induced by X-ray irradiation at day 1, 3, and 7. Furthermore, MDA, lipoxidation products induced by ROS, could be elevated in irradiated liver and lungs. Pretreatment with graphdiyne-BSA NPs could significantly decrease the MDA content to around normal levels. SOD, an antioxidant enzyme, was able to convert O2− into H2O2 or O2. X-ray irradiation on mice could remarkably reduce the SOD contents in liver and lungs, while graphdiyne-BSA NPs were able to obviously rescue the SOD levels. These results suggested that graphdiyne-BSA NPs could act as a promising nanoradioprotector with excellent biocompatibility.
6.1.1.4 CNSI-based nanoradioprotectors. CNSI is the first China Food and Drug Administration (CFDA)-approved carbon NP for clinical application in lymph node mapping.595 Recently, CNSI is found to possess free radical scavenging activity due to its delocalized π-conjugated structure. Moreover, CNSI shows great chemical tolerance toward intestinal conditions, making it possible for oral administration.
Wang et al. investigated the intestinal radioprotection performance of CNSI via oral administration.545 CNSI is a graphene analog with 12 benzene rings conjugated and carbonylated. The CNSI was decorated with polyvinylpyrrolidone (PVP) to improve its biocompatibility and water solubility. The CNSI showed broad-spectrum free radical scavenging performance and great chemical stability. Specifically, the CNSI presented better ˙OH and O2− scavenging abilities than amifostine at the same concentration. The UV absorption of CNSI showed no change under simulated gastric juice (pH = 1 acid conditions), which further confirmed its potential for oral administration.596 The in vitro and in vivo radioprotection investigation indicated that CNSI could obviously remove intracellular ROS induced by X-ray irradiation and thereby suppress the apoptosis of crypt stem cells and small intestinal epithelial cells. In addition, CNSI could keep the balance of intestinal flora. CNSI could decrease the damage of ROS to the intestinal flora. Besides, CNSI was able to protect the intestinal mechanical barrier from ROS damage to suppress proliferation of pathogenic bacteria. Thus, CNSI as a CFDA-approved carbon material could be a potential intestinal nanoradioprotector to improve the quality of life of cancer patients.
For example, Xu et al. utilized ultrasmall Pt clusters to scavenge free radicals induced by γ-ray exposure. The Pt clusters could rescue the SOD and bone marrow DNA levels as well as effectively improve the survival time of mice receiving γ-ray irradiation.608 Long et al. found that hollow PtPd bimetal nanocubes could reduce the free radical levels and recover the SOD/MDA levels in γ-ray-irradiated mice.609 Besides, Long et al. further introduced Mo into the PtPd nanocubes to improve their ROS scavenging ability.610 The prepared ternary alloy PtPdMo nanocubes could effectively protect healthy cells from γ-ray irradiation and improve the survival rate of irradiated mice by 50%, which was higher than those pre-treated with Pt (30%) and PtPd (40%) counterparts. In addition, Wang et al. incorporated rhodium (Rh) into PtPd nanocubes to prepare ternary PtPdRh nanocubes for investigation of their free radical scavenging capability (Fig. 32a).546 The energy dispersive spectroscopy (EDS) mapping confirmed the distribution of Pd, Pt, and Rh elements in the nanocubes. The atomic ratio of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt:Rh in PtPdRh nanocubes was 4:2:1. In this work, the ternary PtPdRh nanocubes could not only clear excessive ˙OH, but also serve as catalase and peroxidase to scavenge H2O2 and O2−, respectively. Moreover, the free radical scavenging ability of PtPdRh nanotubes was better than that of Pt or PtPd nanotubes at the same concentration. The CHO cells pre-treated with PtPdRh nanotubes (1.2 μg mL−1) followed by γ-ray irradiation (4 Gy) showed significantly improved cell viability (94%), much higher than that in the radiation alone group (Fig. 32b). To investigate the in vivo radioprotection, healthy C57BL/6 mice were intraperitoneally injected with Pt, PtPd, or PtPdRh nanocubes (1 mg each mouse) 30 min before γ-ray irradiation (7.3 Gy). The results showed that pretreatment of PtPdRh nanocubes remarkably improved the survival rate of irradiated mice to 50% on the 30th day, compared to 30% and 40% in the Pt and PtPd groups, respectively (Fig. 32c).
:Pt:Rh in PtPdRh nanocubes was 4:2:1. In this work, the ternary PtPdRh nanocubes could not only clear excessive ˙OH, but also serve as catalase and peroxidase to scavenge H2O2 and O2−, respectively. Moreover, the free radical scavenging ability of PtPdRh nanotubes was better than that of Pt or PtPd nanotubes at the same concentration. The CHO cells pre-treated with PtPdRh nanotubes (1.2 μg mL−1) followed by γ-ray irradiation (4 Gy) showed significantly improved cell viability (94%), much higher than that in the radiation alone group (Fig. 32b). To investigate the in vivo radioprotection, healthy C57BL/6 mice were intraperitoneally injected with Pt, PtPd, or PtPdRh nanocubes (1 mg each mouse) 30 min before γ-ray irradiation (7.3 Gy). The results showed that pretreatment of PtPdRh nanocubes remarkably improved the survival rate of irradiated mice to 50% on the 30th day, compared to 30% and 40% in the Pt and PtPd groups, respectively (Fig. 32c).
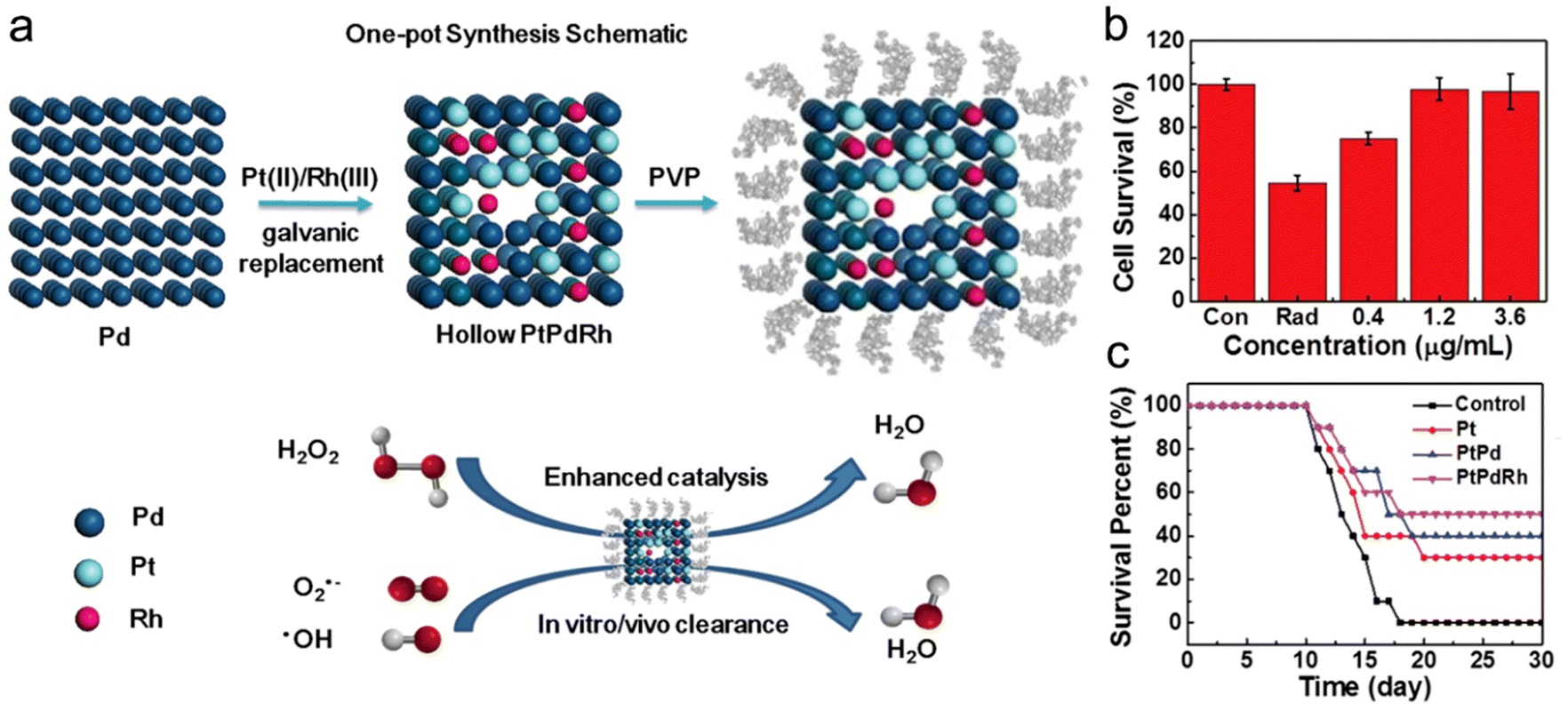 | ||
| Fig. 32 Noble metal-based nanoradioprotectors. (a) A scheme showing the synthesis and catalysis of hollow PtPdRh nancubes. (b) Survival proportion of CHO-K1 cells under γ-ray radiation with or without the treatment of hollow PtPdRh nancubes at various concentrations. (c) Survival rates of mice receiving different treatments before γ-ray irradiation (7.3 Gy). Pt, PtPd, and PtPdRh nancubes: 1 mg each mouse. Reproduced with permission from ref. 546, 2018, Wiley. | ||
Furthermore, Ag-based nanomaterials have also been shown as one type of nanoradioprotectors. Chandrasekharan et al. complexed silver NPs (SN) with glycyrrhizic acid (GLY) to form the SN-GLY complex.611 Oral administration of SN-GLY at 60 min before γ-ray irradiation (8 Gy) could improve the survival rate of irradiated mice to 40% on the 30th day. However, all the irradiated mice in the control group died within 9 days.
CeO2 is the most commonly used antioxidant metal oxide since its ability to reversibly shift between the two oxidation states (Ce3+ and Ce4+) endows it with CAT and SOD mimetic activities. CeO2 can catalytically scavenge diverse free radicals, including H2O2, O2−, and ˙OH.434,621 Therefore, several studies have utilized CeO2 NPs to protect radiation injury.622–626
To further strengthen the ROS scavenging capability, Han et al. grew manganese ions onto CeO2 nanocrystals to form heterostructured CeO2/Mn3O4 nanocrystals for enhanced radioprotection (Fig. 33a).547 The UV Raman spectroscopy results revealed that the CeO2/Mn3O4 nanocrystals had a higher oxygen vacancy level and stronger free radical scavenging ability than CeO2 nanocrystals. To further improve the biocompatibility and dispersibility, the CeO2/Mn3O4 nanocrystals were modified with PEG. Next, a mouse intestinal organoid (mIO) model constructed from leucine-rich repeat-containing G-protein coupled receptor 5-green fluorescence protein (LGR5-GFP) transgenic mice was used to evaluate the radioprotective effects of CeO2/Mn3O4 nanocrystals. The irradiated mIOs (8 Gy) pre-treated with CeO2/Mn3O4 nanocrystals showed more crypt buds and Ki67 positive cells than those pretreated with CeO2 or Mn3O4 alone (Fig. 33b). LGR5-positive intestinal stem cells could recover the irradiation-induced intestinal structure damage and thus were regarded as an indicator of radioprotection.627 The fluorescence-activated cell sorting (FACS) results showed around 14% LGR5 + mIOs under normal conditions while the proportion dropped sharply to 0.17% post-irradiation. Pretreatment with CeO2/Mn3O4 nanocrystals could recover the proportion of LGR5 + mIOs by 9.78%, much higher than that in the CeO2 (4.82%) or Mn3O4 group (2.54%). To investigate the in vivo radioprotective effect, healthy mice were intraperitoneally administered with various radioprotectors at 1 h before receiving a lethal dose of total body irradiation (TBI, 13 Gy). All the mice in the radiation alone group died within 13 days. The mice pre-treated with amifostine at a dose of 250 mg kg−1 showed an increased survival rate by 20% after 30 days post-TBI. However, 60% mice treated with a higher dose of amifostine (400 mg kg−1) died instantly due to relatively high systemic toxicity. In addition, pretreatment with CeO2/Mn3O4 nanocrystals (0.55 mg kg−1) could significantly improve the survival rate by 67% even at 150 days post-TBI compared to the survival rate in the CeO2 (20%) or Mn3O4 (30%) group (Fig. 33c).
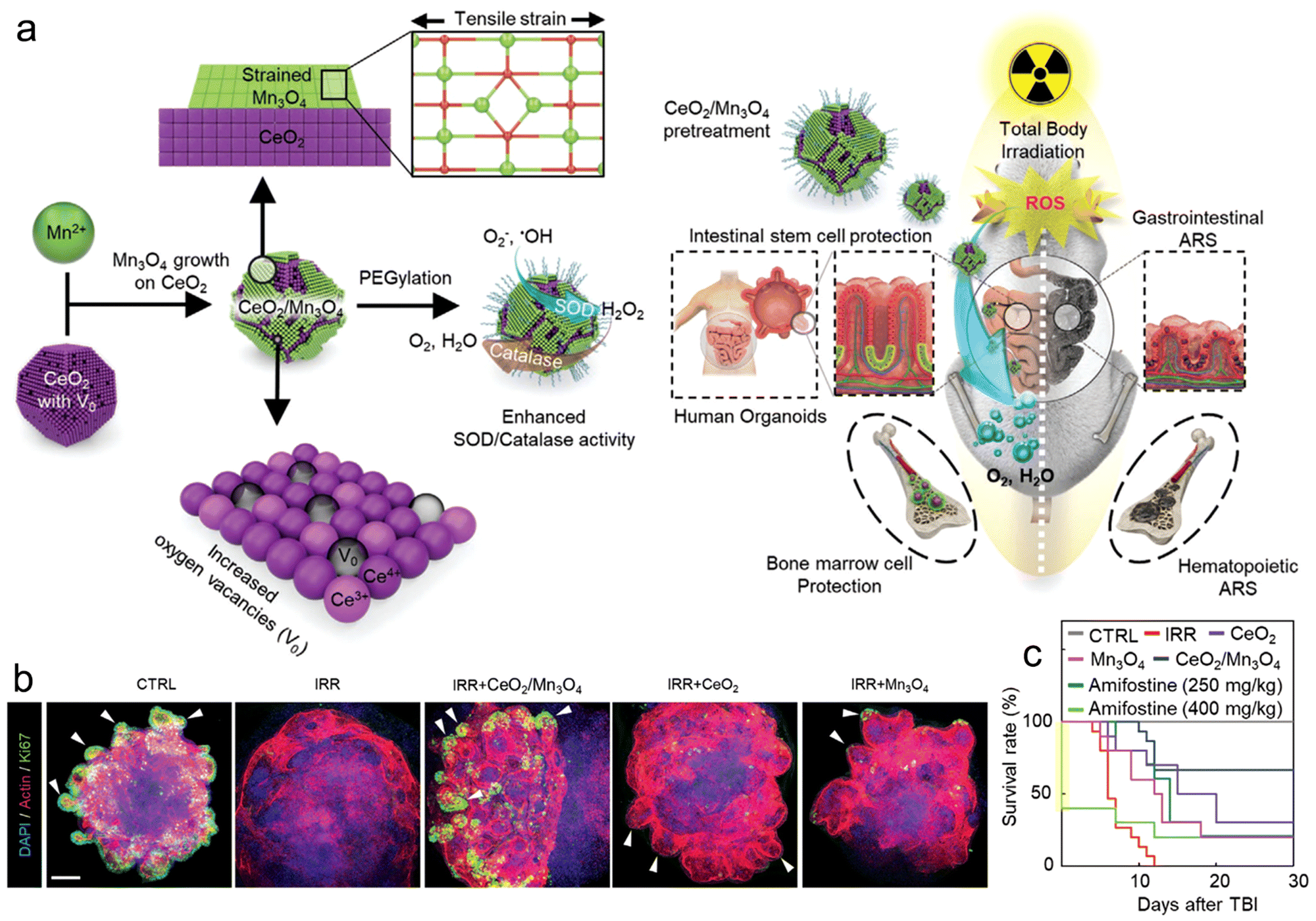 | ||
| Fig. 33 Metal oxide-based nanoradioprotectors (a) A scheme of CeO2/Mn3O4 nanocrystals for radioprotection. (b) Immunofluorescence staining images (DAPI, Actin, and Ki67) of cells after different treatments. The white arrow heads indicate representative crypt buds. Scale bar = 200 μm. (c) Survival rates of irradiated mice (13 Gy) treated with CeO2, Mn3O4 and CeO2/Mn3O4 nanocrystals (0.55 mg kg−1), and amifostine (250 and 400 mg kg−1). Reproduced with permission from ref. 547, 2020, Wiley. | ||
Recently, several TMDCs were reported to exhibit high catalytic activities, endowing them with potential radioprotective function. For instance, Zhang et al. found that Bi2Se3 NPs could be oxidized easily in air, PBS solution, and even blood.647 Inspired by this, they employed PVP-modified Bi2Se3 NPs to clear the ROS induced by γ-ray radiation.648 Healthy C57BL/6 mice were intraperitoneally administered with Bi2Se3 NPs at 30 min after γ-ray (a lethal dose of 7.5 Gy) irradiation. All the mice in the radiation alone group died within two weeks. Treatment with Bi2Se3 NPs (0.2 and 1 mg each mouse) could obviously improve the 30 day survival rate by 46% and 71%, respectively. Moreover, administration of Bi2Se3 NPs could also recover the radiation-induced changes in the blood cell levels, such as red blood cells, platelets, white blood cells, etc.
Ultrasmall nanoradioprotectors show rapid clearance through urine excretion and lead to low systemic toxicity. For example, Zhang's group developed ultrasmall MoS2 dots and then modified the dots with a cysteine protection layer to improve their water-solubility and biocompatibility.548 The prepared cysteine-protected MoS2 dots at the concentration of 140 μg mL−1 showed no obvious cytotoxicity to 3T3/A31 cells. The in vitro experiments revealed that the cysteine-protected MoS2 dots could effectively scavenge intracellular H2O2 and O2−. Moreover, the in vivo radioprotection was evaluated on high-dose (7.5 Gy) γ-ray irradiated C57BL/6 mice. The cysteine-protected MoS2 dots could remarkably rescue radiation-induced DNA damage as well as the SOD and MDA contents in liver and lungs. All the mice receiving radiation alone died within 14 days. However, pretreatment with various concentrations (10, 20, and 50 mg kg−1) of cysteine-protected MoS2 dots via intraperitoneal injection at 30 min before radiation could significantly improve the 30 day survival rates to 7.1%, 42.9%, and 78.6%, respectively. In addition, the in vivo pharmacokinetics results indicated that the cysteine-protected MoS2 dots with a blood circulation half-time of 2.1 h could be removed around 80% via renal clearance within 1 day owing to the ultrasmall hydrodynamic size of 3.1 nm. The biodistribution analysis showed almost no accumulation of MoS2 dots in various organs at 30 d post-injection. The rapid renal clearance and great radioprotective effect make the ultrasmall MoS2 dots potential for clinical translation.
Ren et al. designed ultrathin two-dimensional (2D) niobium carbide (Nb2C) MXenes for radioprotection.549 The prepared Nb2C MXenes were decorated with PVP (denoted as Nb2C-PVP) to improve their dispersibility and biocompatibility (Fig. 34a). The DFT calculations suggested that the ROS scavenging capability of Nb2C-PVP nanosheets (NSs) was due to the continuous ˙OH attack. The ˙OH attack resulted in the dehydration of hydroxyl groups on the surface and formation of an oxygenic layer above Nb nanosheets (NbOx species) (Fig. 34b and c). The Nb2C-PVP NSs exhibited great antioxidant activities to scavenge O2−, H2O2, and ˙OH in a concentration-dependent manner. The in vitro experiments revealed that Nb2C-PVP NSs showed no cytotoxicity to the 3T3/A31 cells even at a concentration of 200 μg mL−1, and that pretreatment with Nb2C-PVP NSs at different concentrations (50 and 100 μg mL−1) could remarkably reduce 3T3/A31 cell death induced by X-ray irradiation at various doses (Fig. 34d). To evaluate the in vivo radioprotective effect, healthy BALB/C mice were intravenously administered with Nb2C-PVP NSs at 1 d before receiving γ-ray TBI at a lethal dose of 6.5 Gy. Pretreatment with 5, 10, and 20 mg kg−1 of Nb2C-PVP NSs significantly improved the survival rate by 30%, 50%, and 81% at day 30. However, the mice in the TBI alone group all died within 20 days. The bone marrow mononuclear cell (BM-MNC) count was utilized to investigate the radioprotective effect on the hematopoietic system. The results showed that the BM-MNC count in irradiated mice pre-treated with Nb2C-PVP NSs was obviously higher than that in the TBI only group (Fig. 34e). Moreover, the BM-MNC count in the Nb2C-PVP NS-treated mice was totally rescued to the normal level at 30 d after irradiation, suggesting the great radioprotective effect of Nb2C-PVP NSs on the hematopoietic system. In addition, the biodistribution analysis showed that Nb2C-PVP NSs could be effectively metabolized through kidneys and liver with almost no accumulation in the organs at 14 d after intravenous injection (Fig. 34f).
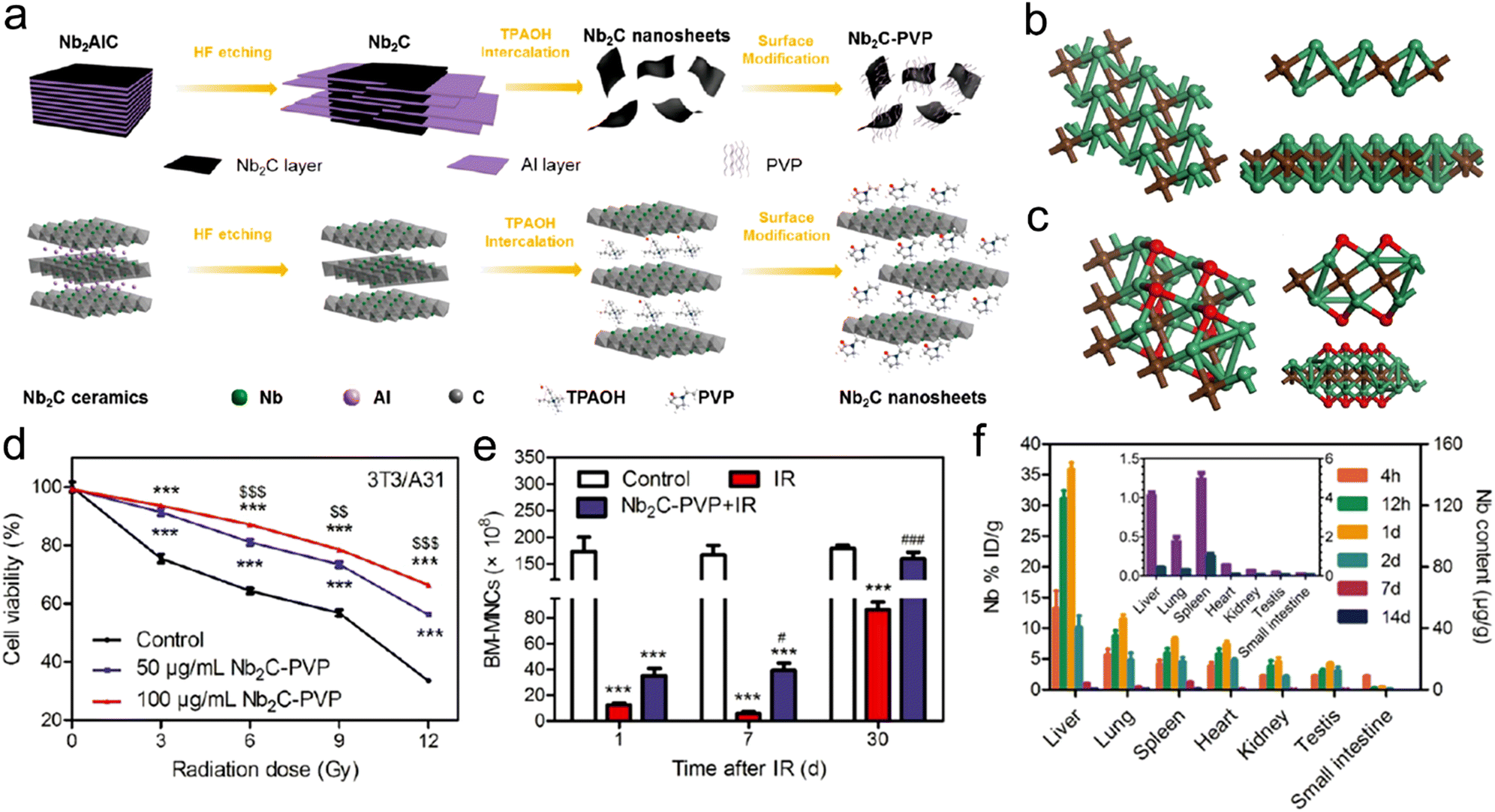 | ||
| Fig. 34 MXene-based nanoradioprotectors. (a) A scheme of synthesis process and exfoliation process of ultrathin Nb2C-PVP NSs. (b) Geometrically optimized single-layer Nb2C nanostructure viewed from the orthogonal, front, and left sides. (c) Geometrically optimized full nanostructure of oxidized Nb2C viewed from the orthogonal, front, and left sides. (d) Viability of 3T1/A31 cells treated with Nb2C-PVP NSs (50 and 100 μg mL−1) under various radiation doses. (e) BM-MNC counts of irradiated mice (5 Gy) at 1, 7, and 30 days after treatment with Nb2C-PVP NSs (20 mg kg−1). (f) Biodistribution of Nb in various organs at 4 h, 12 h, 1, 2, 7, and 14 days after intravenous injection of Nb2C-PVP NSs (20 mg kg−1). Reproduced with permission from ref. 549, 2019, American Chemical Society. | ||
Besides, protein NPs based on natural biomolecules have gained considerable interest owing to their intrinsic biocompatibility, biodegradability, and low toxicity.672–674 The protein NPs can not only serve as drug carriers but also target the receptors on the cell membrane to activate intracellular signaling pathways.675–679
Previous studies have revealed that several certain proteins were able to function as protectors against radiation damage.680–682 Long et al. designed phytohemagglutinin (PHA-L) protein NPs to activate toll-like receptor 5 (TLR5) for radioprotection.683 Due to spontaneous biocompatibility, the PHA-L protein NPs showed no significant cytotoxicity to CHO-K1 cells at a concentration of 100 μg mL−1. The PHA-L protein NPs could target the TLR5 receptor on the cell surface and then activate the TLR5/NF-kB pathway to inhibit radiation-induced CHO-K1 cell death. Moreover, the PHA-L protein NPs could also induce the expression of TLR5 receptor on innate immune cells to modulate immune responses. Then, to investigate the in vivo radioprotective efficacy, the healthy C57BL/6 mice were intraperitoneally administered with PHA-L protein NPs before γ-ray TBI (7.2 Gy). The treatment of 5 and 10 mg PHA-L protein NPs significantly improved the 30 day survival rate by 38% and 69%, respectively. However, all the mice in the TBI alone group died within 20 days. In addition, PHA-L protein NPs could obviously rescue various radiation-induced injury parameters, including bone marrow DNA content, hematopoietic system, gastrointestinal tract injury, etc. To further confirm the protective mechanisms, the TLR5−/− mice were used to evaluate the radioprotection of the PHA-L protein NPs. The results showed that the 30 day survival rate of irradiated TLR5−/− mice pre-treated with PHA-L protein NPs was 0%, suggesting that the TLR5 receptor was necessary for the radioprotection of PHA-L protein NPs. Furthermore, about 70% of PHA-L protein NPs could be rapidly excreted from the organs via kidneys within 48 h of injection. Overall, compared to those inorganic nanoradioprotectors, the organic nanoradioprotectors with superior biocompatibility and rapid renal-clearance exhibit more potential for clinical translation.
6.2 Nanocarriers to deliver small molecular radioprotectors
So far, numerous small molecular radioprotectors of both natural and synthetic origin have been developed to protect normal tissues from radiation, such as amifostine, curcumin, glycyrrhizic acid, melanin, caffeic acid, baicalein, dihydropyridines, MnTnBuOE-2-PyP5+, etc.689–695 However, these small molecular radioprotectors exhibit poor bioavailability and short blood circulation half-life, resulting in limited protective efficacy.696 Advanced nanotechnology promises to overcome these challenges. Besides acting as nanoradioprotectors to scavenge free ROS, the nanomaterials have also been utilized as carriers to improve the protective efficacy of small molecular radioprotectors. These nanocarriers are divided into the following four categories: (1) inorganic nanocarriers; (2) organic nanocarriers; (3) organic–inorganic hybrid nanocarriers; and (4) assembled nanoradioprotectors.For example, Dadachova's group utilized silica NPs as carriers to deliver melanin into the bone marrow for radioprotection of the hematopoietic system.702 The intravenous injection of MNs (50 mg kg−1) at 3 h before TBI with 137Cs radiation (125 cGy) could effectively rescue the hematopoietic damage caused by TBI. Chandrasekharan et al. complexed silver NPs (SN, <50 nm) with glycyrrhizic acid (SN-GLY, a radioprotective molecule) to reduce radiation-induced injuries. The in vivo experiments revealed that oral administration of SN-GLY before γ-ray TBI was able to rescue bone marrow DNA damage and decrease peripheral blood cells.703–705
Curcumin, a hydrophobic polyphenol drug derived from turmeric, has gained considerable interest in biomedicine, including tumor therapy, antibacterial, antiviral, antioxidant, etc.706–713 Besides, curcumin is able to serve as both a radiosensitizer for various tumors and a radioprotector for healthy tissues.714 However, the poor water solubility and biocompatibility of curcumin result in short blood circulation half-time and low tissue accumulation, thus limiting its biomedical application.715,716 Various nanocarriers have been utilized to load curcumin for longer blood circulation half-life and better tissue distribution.717–721 For example, Xie et al. modified bamboo charcoal NPs (BCNPs) with D-α-tocopherol polyethylene glycol succinate (TPGS) and then loaded curcumin into the NPs (TPGS-BCNPs@curcumin) for both radiosensitization and radioprotection.550 The TPGS-BCNPs exhibited good biocompatibility and high tumor accumulation. With a large surface area of 259.31 m2 g−1, curcumin could be efficiently loaded into TPGS-BCNPs with a loading ratio of 36.15%. Moreover, the TPGS-BCNPs@curcumin possessed high photothermal conversion efficiency, and the curcumin could be released on demand upon NIR laser stimulation. In addition, the TGPS, a P-glycoprotein (P-gp) inhibitor, could suppress the efflux of curcumin and further enhance chemotherapy efficacy. Besides, the TPGS-BCNPs@curcumin could deliver considerable curcumin into normal cells. The curcumin exhibited radioprotective effect by effectively clearing intracellular ROS content and reducing radiation-generated DNA damage.
Gastrointestinal tract, one of the organs most vulnerable to radiation, can progress to severe gastrointestinal syndrome under radiation at a dose higher than 8–10 Gy.740,741 Thus, radioprotection of the gastrointestinal tract is urgent and important. However, systemic administration of radioprotectors causes poor distribution in the gastrointestinal tract. Besides, conventional molecular radioprotectors administered orally may fail in the specific environment of gastrointestinal tract, especially the gastric acid.742 In addition, the radioprotective agents would be rapidly removed by the mucus layer even if they reach to the gastrointestinal tract.743 To overcome these challenges, Zhang et al. loaded molecular radioprotectors into organic nanocarriers for small intestine radioprotection.737 Since a previous study revealed that deficiency in Absent-In-Melanoma 2 (AIM2) protein could reduce the radiation-induced gastrointestinal injury,744 in this work, the arginine-chitosan (Arg-CS) polymer was loaded with thalidomide (THA, an AIM2 inhibitor) and then coated with the polydopamine layer (PDA@Arg-CS(THA) NPs) for water-resistant adhesion in small intestine (Fig. 35a).745Ex vivo intestinal crypt 3D model was used to evaluate biocompatibility and radioprotection efficacy. The results indicated that PDA@Arg-CS(THA) showed no significant toxicity to the intestinal crypts and PDA@Arg-CS(THA) could effectively protect the intestinal crypts from radiation. Next, the SEM, FT-IR, and DLS analyses revealed that the morphology, chemical structure, and hydrodynamic size of PDA@Arg-CS(THA) were stable in simulated gastric acid and intestinal medium due to the acid-resistance of the PDA layer (Fig. 35b). Moreover, the zeta potential of PDA@Arg-CS(THA) was neutral in a simulated intestinal medium, which could reduce the electrostatic interactions with mucus and further enhance the diffusion of NPs (Fig. 35c). Interestingly, PDA@Arg-CS(THA) exhibited a pH-switchable release profile. The THA release was inhibited in the simulated gastric acid (pH 1.2) for the first 2 h, whereas the THA was rapidly released in the next 48 h an a simulated intestinal medium (pH 6.0) owing to the neutral surface charge of PDA@Arg-CS(THA) in an intestinal environment (Fig. 35d). Furthermore, the PDA@Arg-CS(THA) was labeled with Cy5.5 to investigate the in vivo biodistribution, which showed that Cy5.5-PDA@Arg-CS(THA) was mainly distributed in the small intestine after oral administration (Fig. 35e). In addition, the Cy5.5-PDA@Arg-CS(THA) showed a longer retention time than Cy5.5-Arg-CS(THA) due to PDA coating (Fig. 35f). To evaluate the in vivo radioprotective effect, the mice were orally administered with vehicle, PDA@Arg-CS, free THA, and PDA@Arg-CS(THA) 12 h before receiving whole abdominal irradiation at a lethal dose. The results showed that PDA@Arg-CS(THA) could effectively improve the survival rate by 45% compared to 15% in the free THA group (Fig. 35g).
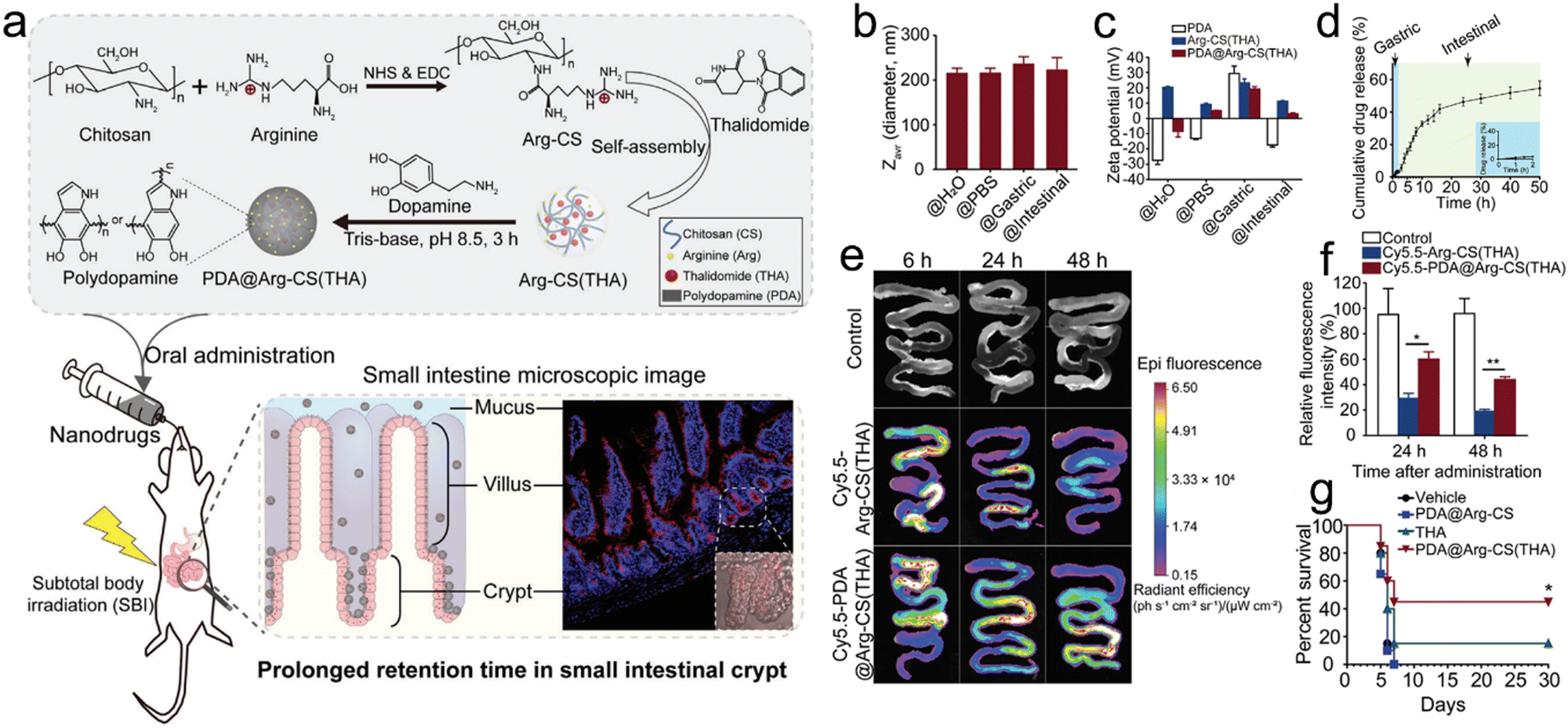 | ||
| Fig. 35 Organic nanocarriers to deliver small molecular radioprotectors. (a) A scheme showing the synthetic process of PDA@Arg-CS(THA) and its radioprotective effect on small intestine. (b) Hydrodynamic sizes of PDA@Arg-CS(THA)in H2O, PBS, and simulated gastric and intestinal media. (c) Zeta potentials of self-polymerized PDA, Arg-CS(THA), and PDA@Arg-CS(THA) in different media. (d) Drug release profile of PDA@Arg-CS(THA) in simulated gastric and intestinal media. Inset: THA release profile of PDA@Arg-CS(THA) in a simulated gastric medium in the first 2 h. (e) Ex vivo FL images of small intestine at 6, 24, and 48 h after oral administration of PDA@Arg-CS(THA), Cy5.5-Arg-CS(THA), or Cy5.5-PDA@Arg-CS(THA). (f) Relative FL intensity of the small intestine at 24 and 48 h after oral administration. (g) Survival rate of irradiated mice (14 Gy) treated with free THA, Arg-CS(THA), or PDA@Arg-CS(THA). Reproduced with permission from ref. 737, 2020, Wiley. | ||
MOFs, one type of hybrid materials composed of metal centers and organic linkers, have gained considerable interest in biomedical application due to their large surface areas, tunable pore sizes, and great biodegradability.750–752 For example, Cao et al. used PEGylated MIL-101(Cr) MOFs to load WR-1065 (the major active metabolite of amifostine) (WR@PEG-MIL-101(Cr)) for hematopoietic radioprotection.552 With a large surface area of over 1300 m2 g−1, the MOF NPs were loaded with WR-1065 (47.2 wt%) to improve blood circulation and tissue distribution. To investigate the in vivo radioprotective efficacy, healthy C57/BL male mice were orally administered with radioprotective agents at 1 h before γ-ray TBI at a lethal dose of 8.0 Gy. Administration of 150 and 265 mg kg−1 of WR@PEG-MIL-101(Cr) significantly improved the 30 day survival rate by 50% and 80%, respectively, compared to 30% in the free WR-1065 group (125 mg kg−1) and 0% in the control group.
4-Amino-2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), one type of low-molecular-weight (LMW) nitroxide compound, is able to serve as an antioxidant to scavenge oxidants. Previous studies have reported that 4-amino-TEMPO could function as a radioprotector in vitro. However, free TEMPO could lead to severe off-target effects and disrupt the redox balance in normal cells.753–755 In addition, free TEMPO could be rapidly removed from the body due to its extremely poor bioavailability.756 Nagasaki's group conjugated 4-amino-TEMPO to PEG-b-poly(chloromethylstyrene) (PEG-b-PCMS, a diblock copolymer) to form PEG-b-PMNT. Then, the PEG-b-PMNT could self-assemble into redox NPs (RNPs) (Fig. 36a).553 The self-assembly of RNPs could not only avoid leakage of TEMPO molecules but also increase blood circulation half-life and tissue retention time. The RNPs were prepared to alleviate excessive ROS in healthy tissues induced by indirect damage of radiation (Fig. 36b and c). To evaluate the in vivo radioprotective effect of RNPs, the tumor-bearing mice were subcutaneously injected with RNPs (TEMPO = 200 mg kg−1) at 1 d before X-ray irradiation at various doses (10, 20, 25, and 30 Gy). The tumor-bearing mice subcutaneously injected with amifostine (500 mg kg−1) at 2 h before radiation were used as a positive control. The results showed that pre-administration of RNPs could significantly prolong the median survival time of mice receiving 10, 20, and 25 Gy of X-ray irradiation compared to the amifostine group. Moreover, the treatment of RNPs could also remarkably rescue the decline of body weight in X-ray-irradiated tumor mice compared to the PBS or amifostine group. In addition, both RNPs and amifostine could rescue the abnormal changes of various hematological and biochemical indicators, including RBC, white blood cells, PLT, creatinine (CRE), blood urea nitrogen (BUN), alanine aminotransferase (ALT), aspartate transaminase (AST), and total bilirubin (TBIL), indicating that the RNPs were able to reduce off-target damage of radiation to the hematopoietic system, kidneys, and liver. Overall, both nanocarriers and self-assembled nanoradioprotectors can effectively improve blood circulation and tissue retention of small molecular radioprotectors, thus enhancing radioprotective efficacy.
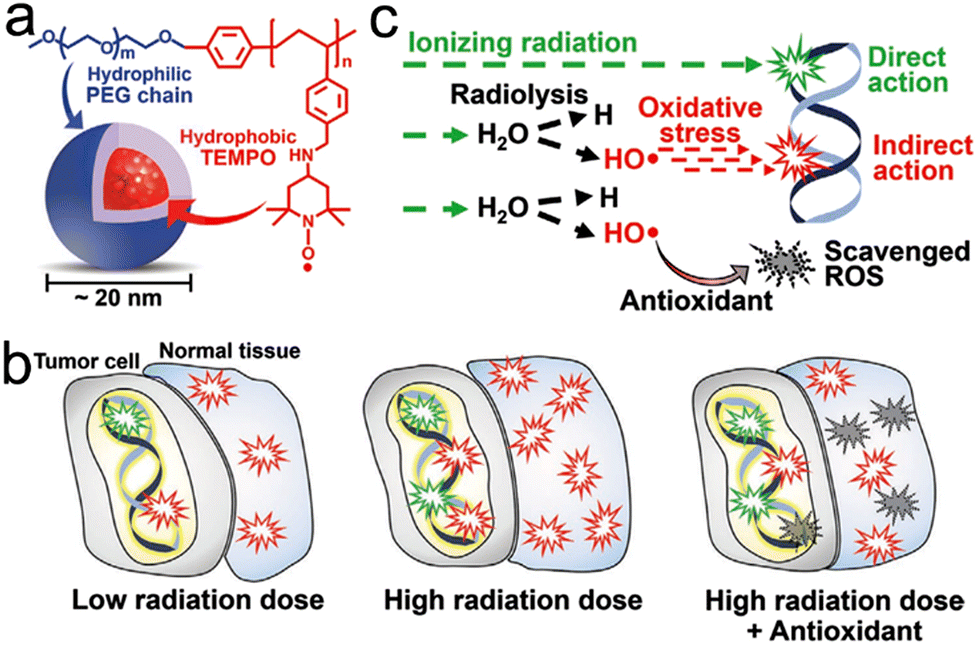 | ||
| Fig. 36 Assembled nanoradioprotectors by small molecular radioprotectors. (a) Chemical structure of the RNP. (b) Antioxidant clears most of radiation-induced ROS in normal tissues. (c) Antioxidant removes ROS produced by the indirect action of ionizing radiation. Reproduced with permission from ref. 553, 2020, Royal Society of Chemistry. | ||
7. Prospects and challenges
In this review, we systematically summarized the strategies for maximizing RT efficacy of tumors, including tumor-targeted delivery, exo/endogenous stimuli-responsive strategies, and imaging-guided precision RT. Besides, we also reviewed two representative featured applications of precision RT: the nanomaterial-mediated RT-induced abscopal effect against distant metastasis, and nanomaterial-mediated radioprotection of the healthy tissues. However, there are a lot of challenges that still require to be overcome.Various tumor-targeted strategies, including EPR effect-based passive targeting, ligand-based targeting, biomimetic targeting, magnetic targeting, and subcellular organelle targeting, were utilized to enhance the tumor accumulation of nanoradiosensitizers. For the EPR effect, several studies have revealed that the particle sizes and shapes are quite important for tumor accumulation and retention. However, the specific mechanism for this phenomenon has not been deeply discussed. Biological ligand-based modification is one of the most common active targeting strategies for nanoradiosensitizers. The density of targeting ligands on the surfaces of NPs needs to be adjusted to balance targeting efficiency and potential instability of NPs, especially in a complex biological environment. Due to the highly specific recognition, bioorthogonal ligand-based targeting has been a promising strategy for targeted theranostics. However, there are still some issues that need to be considered in the future research. For example, the antibodies cannot be completely eliminated from the body even with administration of the clearing agents. Biomimetic targeting as an emerging strategy utilizes cells or cell membrane to load or coat nanoradiosensitizers for targeted delivery. However, these biomimetic nanoradiosensitizers cannot be stored for a long time, which may affect their potential for clinical translation. Moreover, the efficiency of magnetic targeting is mainly limited by the distance between the magnets and the target sits. Future research needs to take the geometry of the magnetic field into account. In addition, most current nanoradiosensitizers act on traditional physical enhancement, such as X-ray energy deposition by high-Z elements. However, the toxicity of these heavy metals is unavoidable. Bu's group systematically reviewed the development of catalytic radiosensitization on the basis of radio–nano interactions and catalysis–biological interactions.757 Catalytic radiosensitization refers to the development of nanocatalysts to enhance X-ray radiation-induced chemical reactions to produce active species or molecules for treatment, thus improving RT efficacy. The catalytic radiosensitization might provide more opportunities for biological applications of different nanocatalysts.
Exogenous and/or endogenous stimuli-responsive strategies are able to induce size/shape change, structural degradation, and drug release, for precision RT. However, there are still several challenges. First, the exogeneous stimuli, such as X-rays, light, and US, can achieve spatiotemporally precise control of responsive nanoradiosensitizers. However, the efficiency of responsiveness may be limited. The endogenous stimuli-based responsiveness may be more efficient but less controllable. Therefore, combining exogenous and endogenous stimuli is able to realize spatiotemporally controllable and efficient responsiveness for precision RT. Second, endogenous stimuli-responsive nanoradiosensitizers can respond to tumor tissues based on the differences in biomarker levels (e.g., pH, GSH, ROS, hypoxia, and enzymes, etc.), between tumors and normal tissues. However, these nanoradiosensitizers will still exhibit a certain degree of off-target side effects owing to the prevalence of these biomarkers in normal tissues. Third, there are also several shortcomings of exogeneous stimuli in clinical applications. For example, light stimuli do not work in deep tumors due to poor tissue penetration. The US stimuli are more suitable to superficial tumors rather than deep tissues. Thus, considering the advantages and disadvantages of different exogenous and/or endogenous stimuli, responsive strategies will be proposed according to various clinical applications to achieve precision RT in future studies. In addition, other exogenous (e.g., magnetic field, hyperthermia, etc.) and endogenous stimuli (e.g., glucose, nucleic acids, ATP, etc.) can be considered to develop responsive nanoradiosensitizers in future research.
In clinical practice of RT, the imaging techniques, such as MRI and CT, were commonly used for precise organ/tumor delineation and thereby minimize the radiation dose to normal tissues. For example, magnetic resonance-guided radiotherapy (MRgRT) has become clinically available, which offers excellent soft-tissue contrast for precise observation of interfractional changes in tumor anatomy.758,759 The MRgRT allows for on-table treatment adaptation that enables dose escalation and reducing toxicity. So far, many types of imaging techniques, including FLI, PAI, MRI, CT, US, PET, and SPECT, have been applied to guide precision RT. However, these studies mainly utilized imaging methods to monitor the biodistribution and tumor accumulation of nanoradiosensitizers, which could help select the optimal time point for RT. In addition, the imaging methods could provide ample information of tumors, such as TME, and tumor response to therapy. The functional imaging techniques, including functional CT, fMRI, and PET, allow visualizing various pathophysiological characteristics of tumors, including perfusion, hypoxia, proliferation, and metabolism.760 Thus, functional imaging-based guidance can provide more opportunities for individualized RT treatment. For example, dynamic contrast enhanced CT (DCE-CT) as a functional CT technique can be utilized to monitor tissue perfusion.761 The BOLD/DWI fMRI was used to monitor tumor radiosensitivity and oxygen concentration.36 Liu et al. developed a quantitative MRI-based diagnostic system to predict tumors’ responses to RT. The system could stratify pancreatic patients into resistant and sensitive groups.335 Moreover, the PA and US imaging could also be applied for monitoring tumor oxygenation. Therefore, more future efforts are required to extend various imaging techniques to provide information on tumor location, boundary, response to therapy, and TME (e.g., hypoxia, GSH, etc.).
The antitumor immune responses induced by RT alone are inefficient to inhibit distant and metastatic tumors. However, nanomaterial-based RT is able to induce potent ICD and thereby elicit systemic antitumor immunity against metastasis. The anti-metastasis efficacy could be further improved in combination with ICB therapy, immunoadjuvant, or other treatment modalities, such as PTT, PDT, CDT, gas therapy, chemotherapy, etc. Besides, nanoimmunotherapeutics could be designed to exert immunotherapeutic action only in response to external stimuli, such as X-ray irradiation, resulting in more precise anti-tumor immune activation and less incidence of immune-related adverse events.762 Animal models are crucial for investigation of RT-mediated antitumor immune responses. First, the tumors established with cancer cell lines are generally homogenous, which makes it difficult to stimulate heterogenous tumors of clinical patients. Second, most subcutaneous tumors used in these studies, although easy to monitor and operate, do not represent appropriate sites for human tumors. Thus, the subcutaneous tumor model is less predictive when used to test response to RT and immunotherapy. Third, the human cancer cell lines or patient-derived xenograft can only be inoculated into immunodeficiency animals, which makes it difficult to measure systemic immune responses. Moreover, immunocompetent tumor-bearing mice based on mouse cancer cell lines cannot stimulate human tumors well, since the immune system in mice is quite different from that in human. Therefore, more suitable animal models used for immunotherapy evaluation need to developed in the future. In addition, lung metastasis is the most common metastatic model in most previous studies. However, the metastasis in clinical practice can appear in various organs, such as liver, bones, lungs, brain, and peritoneal cavity. The response of different organ metastases to immunotherapy varies greatly. Therefore, various metastatic models need to be considered in future studies.
Amifostine as a phosphorothioate is able to diffuse into cells after being dephosphorylated by alkaline phosphatase and then function as a free radical scavenger.763 Previous studies have revealed that amifostine could rapidly accumulate in normal tissues rather than tumors, which is believed to result from intratumoral blood flow, acidic tumor microenvironment, and the low level of alkaline phosphatase in tumors.538,764 Likewise, the nanoradioprotectors can be designed to exert radioprotective effect only in response to specific intratumoral stimuli, such as acidosis, hypoxia, high GSH, H2O2 levels, etc., so that the nanoradioprotectors can more concentrate in and protect normal tissues rather than tumor tissues. In addition, targeting strategies, such as ligand-based targeting and magnetic targeting, can also be used to enhance accumulation of normal tissues. Moreover, the nanoradioprotectors are mainly administered intraperitoneally or intravenously to protect healthy tissues from whole body radiation, which might, however, induce systemic toxicity. Typically, the RT only irradiates surrounding tissues or organs when treating local tumors. Thus, more future efforts need to be focused on other routes of nanoradioprotector administration that are easier to be translated to the clinic. For example, the oral or rectal route can be used for radioprotection of gastrointestinal tracts. Inhalation (intratracheal) route is more suitable to protect lung from RILI. External route may be suitable for radioprotection of skin. Moreover, the intrathecal or topical route can be applied for radioprotection of specific tissues, such as brain, spinal, eyes, etc. In addition, most studies have been focused on the evaluation of in vitro and in vivo radioprotective performance of nanoradioprotectors. However, the specific radioprotective mechanisms have not been explored in depth, which requires further investigation in future studies. Furthermore, the choice of animal models to investigate the radioprotective performance is important for clinical translation. Most studies utilized mouse models to evaluate the radioprotective effect of nanomaterials. Large animals can be considered as models for in vivo investigation of protective efficacy of nanoradioprotectors.
Besides, a lot of challenges still remain before the clinical translation of these nanoradiosensitizers. Nanoradiosensitizers that can be produced in a large-scale have more potential for clinical translation. Moreover, most studies have performed in vivo biosafety evaluations of nanoradiosensitizers. However, most evaluations were short-term rather than long-term. Moreover, these biosafety evaluations were focused on major organs without considering that these nanoradiosensitizers could also accumulate in other tissues, such as eyes, brain, and muscle. Thus, more comprehensive and long-term biosafety evaluations of the nanoradiosensitizers are required before clinical translation. In addition, the choice of nanomaterials directly affects the time required for clinical translation. The nanoradiosensitizers composed of FDA-approved drugs or materials could be applied in clinical practice faster.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (82071287, 81870916), the MOE Frontier Science Center for Brain Science & Brain-Machine Integration, Zhejiang University, the Funding of Double First-Rate Discipline Innovation Team of China Pharmaceutical University (CPUQNJC22_04), the National University of Singapore Start-up Grant (NUHSRO/2020/133/Startup/08), the NUS School of Medicine Nanomedicine Translational Research Programme (NUHSRO/2021/034/TRP/09/Nanomedicine), National University of Singapore (NUS Startup Fund: A-0008499-00-00 and A-0008505-00-00 to W. T.), the Singapore Ministry of Education (MOE Tier 1 grant: A-0008503-00-00 to W. T.), and the Singapore National Medical Research Council (NMRC OF-YIRG grant: A-8000675-00-00 to W. T.). Y. P. thanks China Scholarship Council (CSC) for a scholarship allowing him to study in Singapore.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- G. Petroni, L. C. Cantley, L. Santambrogio, S. C. Formenti and L. Galluzzi, Nat. Rev. Clin. Oncol., 2022, 19, 114–131 CrossRef CAS PubMed.
- R. A. Chandra, F. K. Keane, F. E. M. Voncken and C. R. Thomas Jr., Lancet, 2021, 398, 171–184 CrossRef.
- C. F. Dunne-Daly, Cancer Nurs., 1994, 17, 355–366 CAS.
- H. Westerveld, N. Nesvacil, L. Fokdal, C. Chargari, M. P. Schmid, M. Milosevic, U. M. Mahantshetty and R. A. Nout, Lancet Oncol., 2020, 21, e157–e167 CrossRef CAS PubMed.
- P. Pei, T. Liu, W. Shen, Z. Liu and K. Yang, Mater. Horiz., 2021, 8, 1348–1366 RSC.
- A. M. Chinnaiyan, U. Prasad, S. Shankar, D. A. Hamstra, M. Shanaiah, T. L. Chenevert, B. D. Ross and A. Rehemtulla, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1754–1759 CrossRef CAS PubMed.
- R. Imai, T. Kamada, N. Araki, S. Abe, Y. Iwamoto, T. Ozaki, C. Kanehira, M. Kaya, K. Takahashi, H. Chuman, H. Tsujii, M. Tsuneyoshi, Y. Nishida, H. Hiraga, T. Hiruma, R. Machinami, A. Matsumine, S. Matsumoto, H. Morioka, T. Yamaguchi and T. Yonemoto, Int. J. Radiat. Oncol. Biol. Phys., 2016, 95, 322–327 CrossRef PubMed.
- S. Hall, S. Rudrawar, M. Zunk, N. Bernaitis, D. Arora, C. M. McDermott and S. Anoopkumar-Dukie, Antioxidants, 2016, 5, 22 CrossRef PubMed.
- T. Kusumoto, R. Ogawara, K. Igawa, K. Baba, T. Konishi, Y. Furusawa and S. Kodaira, J. Radiat. Res., 2020, 62, 86–93 CrossRef PubMed.
- H. Liew, S. Mein, J. Debus, I. Dokic and A. Mairani, Int. J. Mol. Sci., 2020, 21, 3471 CrossRef CAS PubMed.
- C. Borek, J. Nutr., 2004, 134, 3207S–3209S CrossRef CAS PubMed.
- H. Wang, X. Mu, H. He and X.-D. Zhang, Trends Pharmacol. Sci., 2018, 39, 24–48 CrossRef CAS PubMed.
- R. Hodson, Nature, 2016, 537, S49–S49 CrossRef CAS PubMed.
- Y.-B. Pan, S. Wang, B. Yang, Z. Jiang, C. Lenahan, J. Wang, J. Zhang and A. Shao, J. Cell. Mol. Med., 2020, 24, 3901–3916 CrossRef CAS PubMed.
- S. H. Abid, V. Malhotra and M. C. Perry, Curr. Opin. Oncol., 2001, 13, 242–248 CrossRef CAS PubMed.
- R. A. Rosiello and W. W. Merrill, Clin. Chest Med., 1990, 11, 65–71 CrossRef CAS PubMed.
- D. G. Kirsch, P. M. Santiago, E. d Tomaso, J. M. Sullivan, W.-S. Hou, T. Dayton, L. B. Jeffords, P. Sodha, K. L. Mercer, R. Cohen, O. Takeuchi, S. J. Korsmeyer, R. T. Bronson, C. F. Kim, K. M. Haigis, R. K. Jain and T. Jacks, Science, 2010, 327, 593–596 CrossRef CAS PubMed.
- P. B. Romesser, A. S. Kim, J. Jeong, A. Mayle, L. E. Dow and S. W. Lowe, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20672–20678 CrossRef CAS PubMed.
- R. G. Bristow, B. Alexander, M. Baumann, S. V. Bratman, J. M. Brown, K. Camphausen, P. Choyke, D. Citrin, J. N. Contessa, A. Dicker, D. G. Kirsch, M. Krause, Q. T. Le, M. Milosevic, Z. S. Morris, J. N. Sarkaria, P. M. Sondel, P. T. Tran, G. D. Wilson, H. Willers, R. K. S. Wong and P. M. Harari, Lancet Oncol., 2018, 19, e240–e251 CrossRef PubMed.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed.
- B. J. Choi, K. O. Jung, E. E. Graves and G. Pratx, Nanotechnology, 2018, 29, 504001 CrossRef PubMed.
- T. Hasegawa, J. Takahashi, S. Nagasawa, M. Doi, A. Moriyama and H. Iwahashi, Int. J. Mol. Sci., 2020, 21, 2302 CrossRef CAS PubMed.
- A. Moriyama, T. Hasegawa, L. Jiang, H. Iwahashi, T. Mori and J. Takahashi, Sci. Rep., 2019, 9, 18163 CrossRef CAS PubMed.
- J. Takahashi, M. Murakami, T. Mori and H. Iwahashi, Sci. Rep., 2018, 8, 2728 CrossRef PubMed.
- J. Xie, L. Gong, S. Zhu, Y. Yong, Z. Gu and Y. Zhao, Adv. Mater., 2019, 31, e1802244 CrossRef PubMed.
- X. Song, C. Liang, L. Feng, K. Yang and Z. Liu, Biomater. Sci., 2017, 5, 1828–1835 RSC.
- J. Zang, Q. Liu, H. Sui, R. Wang, O. Jacobson, X. Fan, Z. Zhu and X. Chen, J. Nucl. Med., 2020, 61, 1772–1778 CrossRef CAS PubMed.
- M. Gao, C. Liang, X. Song, Q. Chen, Q. Jin, C. Wang and Z. Liu, Adv. Mater., 2017, 29, 1701429 CrossRef PubMed.
- X. Qin, C. Yang, H. Xu, R. Zhang, D. Zhang, J. Tu, Y. Guo, B. Niu, L. Kong and Z. Zhang, Small, 2021, 17, e2103984 CrossRef PubMed.
- M. Lyu, D. Zhu, X. Kong, Y. Yang, S. Ding, Y. Zhou, H. Quan, Y. Duo and Z. Bao, Adv. Healthcare Mater., 2020, 9, e1901819 CrossRef PubMed.
- C. Y. Y. Yu, H. Xu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, X. Li, S. Krishnan, D. Ding and B. Z. Tang, Adv. Mater., 2017, 29, 1606167 CrossRef PubMed.
- G. Le Duc, I. Miladi, C. Alric, P. Mowat, E. Bräuer-Krisch, A. Bouchet, E. Khalil, C. Billotey, M. Janier, F. Lux, T. Epicier, P. Perriat, S. Roux and O. Tillement, ACS Nano, 2011, 5, 9566–9574 CrossRef CAS PubMed.
- Z. Zhang, L. Wang, Y. Ding, J. Wu, Y. Hu and A. Yuan, Biomater. Sci., 2020, 8, 4739–4749 RSC.
- M. Wang, H. Li, B. Huang, S. Chen, R. Cui, Z. J. Sun, M. Zhang and T. Sun, Adv. Healthcare Mater., 2021, 10, e2100090 CrossRef PubMed.
- Y. Dou, F. Zhao, X. Li and Y. Guo, ACS Biomater. Sci. Eng., 2021, 7, 5242–5254 CrossRef CAS PubMed.
- H. Huang, C. Zhang, X. Wang, J. Shao, C. Chen, H. Li, C. Ju, J. He, H. Gu and D. Xia, Nano Lett., 2020, 20, 4211–4219 CrossRef CAS PubMed.
- W. Sun, L. Luo, Y. Feng, Y. Cai, Y. Zhuang, R. J. Xie, X. Chen and H. Chen, Angew. Chem., Int. Ed., 2020, 59, 9914–9921 CrossRef CAS PubMed.
- X. Li, L. Yu, C. Zhang, X. Niu, M. Sun, Z. Yan, W. Wang and Z. Yuan, Bioact. Mater., 2022, 7, 377–388 CrossRef CAS PubMed.
- X. Hu, J. Sun, F. Li, R. Li, J. Wu, J. He, N. Wang, J. Liu, S. Wang, F. Zhou, X. Sun, D. Kim, T. Hyeon and D. Ling, Nano Lett., 2018, 18, 1196–1204 CrossRef CAS PubMed.
- Z. Huang, Y. Wang, D. Yao, J. Wu, Y. Hu and A. Yuan, Nat. Commun., 2021, 12, 145 CrossRef CAS PubMed.
- Y. Chao, L. Xu, C. Liang, L. Feng, J. Xu, Z. Dong, L. Tian, X. Yi, K. Yang and Z. Liu, Nat. Biomed. Eng., 2018, 2, 611–621 CrossRef CAS PubMed.
- T. I. Liu, T. Y. Lu, Y. C. Yang, S. H. Chang, H. H. Chen, I. L. Lu, A. Sabu and H. C. Chiu, Biomaterials, 2020, 257, 120229 CrossRef CAS PubMed.
- J. Xie, M. Zhao, C. Wang, S. Zhu, W. Niu, Y. Yong, L. Zhao and Z. Gu, Chem. Eng. J., 2022, 430, 132866 CrossRef CAS.
- J. Du, Z. Gu, L. Yan, Y. Yong, X. Yi, X. Zhang, J. Liu, R. Wu, C. Ge, C. Chen and Y. Zhao, Adv. Mater., 2017, 29, 1701268 CrossRef PubMed.
- A. P. Brown, E. J. Chung, M. E. Urick, W. P. Shield, A. L. Sowers, A. Thetford, U. T. Shankavaram, J. B. Mitchell and D. E. Citrin, Radiat. Oncol., 2010, 5, 34 CrossRef PubMed.
- J. Colon, S. Seal, S. Konduri, A. Limaye, M. Abdelrahim and C. Baker, Cancer Res., 2007, 67, 754 Search PubMed.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Nano Lett., 2005, 5, 2573–2577 CrossRef CAS PubMed.
- C. A. Theriot, R. C. Casey, V. C. Moore, L. Mitchell, J. O. Reynolds, M. Burgoyne, R. Partha, J. L. Huff, J. L. Conyers, A. Jeevarajan and H. Wu, Radiat. Environ. Biophys., 2010, 49, 437–445 CrossRef CAS PubMed.
- K. C. Black, Y. Wang, H. P. Luehmann, X. Cai, W. Xing, B. Pang, Y. Zhao, C. S. Cutler, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2014, 8, 4385–4394 CrossRef CAS PubMed.
- Y. Yang, L. Zhang, J. Cai, X. Li, D. Cheng, H. Su, J. Zhang, S. Liu, H. Shi, Y. Zhang and C. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 1718–1732 CrossRef CAS PubMed.
- Y. Wang, L. Zou, Z. Qiang, J. Jiang, Z. Zhu and J. Ren, ACS Biomater. Sci. Eng., 2020, 6, 3550–3562 CrossRef CAS PubMed.
- J. Liu, F. Hu, M. Wu, L. Tian, F. Gong, X. Zhong, M. Chen, Z. Liu and B. Liu, Adv. Mater., 2021, 33, e2007888 CrossRef PubMed.
- W. Pan, B. Cui, P. Gao, Y. Ge, N. Li and B. Tang, Chem. Commun., 2020, 56, 547–550 RSC.
- W. Fan, B. Shen, W. Bu, X. Zheng, Q. He, Z. Cui, D. Ni, K. Zhao, S. Zhang and J. Shi, Biomaterials, 2015, 69, 89–98 CrossRef CAS PubMed.
- X. Fang, Y. Wang, X. Ma, Y. Li, Z. Zhang, Z. Xiao, L. Liu, X. Gao and J. Liu, J. Mater. Chem. B, 2017, 5, 4190–4197 RSC.
- H. Kobayashi and M. W. Brechbiel, Adv. Drug Delivery Rev., 2005, 57, 2271–2286 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- H. Kobayashi, R. Watanabe and P. L. Choyke, Theranostics, 2013, 4, 81–89 CrossRef PubMed.
- Z. He, H. Yan, W. Zeng, K. Yang and P. Rong, J. Mater. Chem. B, 2021, 9, 1625–1637 RSC.
- K. Kim, K. S. Oh, D. Y. Park, J. Y. Lee, B. S. Lee, I. S. Kim, K. Kim, I. C. Kwon, Y. K. Sang and S. H. Yuk, J. Controlled Release, 2016, 228, 141–149 CrossRef CAS PubMed.
- C. H. Chang, M. G. Stabin, Y. J. Chang, L. C. Chen, M. H. Chen, T. J. Chang, T. W. Lee and G. Ting, Cancer Biother. Radiopharm., 2008, 23, 749–758 CrossRef CAS PubMed.
- J. Goos, A. Cho, L. M. Carter, T. R. Dilling, M. Davydova, K. Mandleywala, S. Puttick, A. Gupta, W. S. Price, J. F. Quinn, M. R. Whittaker, J. S. Lewis and T. P. Davis, Theranostics, 2020, 10, 567–584 CrossRef CAS PubMed.
- Y. Dou, Y. Liu, F. Zhao, Y. Guo, X. Li, M. Wu, J. Chang and C. Yu, Theranostics, 2018, 8, 5870–5889 CrossRef CAS PubMed.
- Q. Zhang, J. Chen, J. Shen, S. Chen, K. Liang, H. Wang and H. Chen, Theranostics, 2019, 9, 2779–2790 CrossRef CAS PubMed.
- S. Gao, W. Zhang, R. Wang, S. P. Hopkins, J. C. Spagnoli, M. Racin, L. Bai, L. Li, W. Jiang, X. Yang, C. Lee, K. Nagata, E. W. Howerth, H. Handa, J. Xie, Q. Ma and A. Kumar, ACS Nano, 2020, 14, 1468–1481 CrossRef CAS PubMed.
- R. Duncan, Y.-N. Sat-Klopsch, A. M. Burger, M. C. Bibby, H. H. Fiebig and E. A. Sausville, Cancer Chemother. Pharmacol., 2013, 72, 417–427 CrossRef CAS PubMed.
- D. Huang, L. Sun, L. Huang and Y. Chen, J. Pers. Med., 2021, 11, 124 CrossRef PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- A. Nel, E. Ruoslahti and H. Meng, ACS Nano, 2017, 11, 9567–9569 CrossRef CAS PubMed.
- U. Prabhakar, H. Maeda, R. K. Jain, E. M. Sevick-Muraca, W. Zamboni, O. C. Farokhzad, S. T. Barry, A. Gabizon, P. Grodzinski and D. C. Blakey, Cancer Res., 2013, 73, 2412–2417 CrossRef CAS PubMed.
- G. Song, H. Wu, K. Yoshino and W. C. Zamboni, J. Liposome Res., 2012, 22, 177–192 CrossRef CAS PubMed.
- A. Dimou, K. N. Syrigos and M. W. Saif, Ther. Adv. Med. Oncol., 2012, 4, 271–279 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- C. Kinnear, T. L. Moore, L. Rodriguez-Lorenzo, B. Rothen-Rutishauser and A. Petri-Fink, Chem. Rev., 2017, 117, 11476–11521 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, A. Roy and S.-D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- Y. Wang, K. C. L. Black, H. Luehmann, W. Li, Y. Zhang, X. Cai, D. Wan, S.-Y. Liu, M. Li, P. Kim, Z.-Y. Li, L. V. Wang, Y. Liu and Y. Xia, ACS Nano, 2013, 7, 2068–2077 CrossRef CAS PubMed.
- N. Ma, F. G. Wu, X. Zhang, Y. W. Jiang, H. R. Jia, H. Y. Wang, Y. H. Li, P. Liu, N. Gu and Z. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 13037–13048 CrossRef CAS PubMed.
- L. Zhang, H. Su, H. Wang, Q. Li, X. Li, C. Zhou, J. Xu, Y. Chai, X. Liang, L. Xiong and C. Zhang, Theranostics, 2019, 9, 1893–1908 CrossRef CAS PubMed.
- A. Wilhelm and Q. D. Tavares, Nat. Rev. Mater., 2016, 1, 16014 CrossRef.
- D. B. Chithrani, S. Jelveh, F. Jalali, M. van Prooijen, C. Allen, R. G. Bristow, R. P. Hill and D. A. Jaffray, Radiat. Res., 2010, 173, 719–728 CAS.
- X.-D. Zhang, D. Wu, X. Shen, J. Chen, Y.-M. Sun, P.-X. Liu and X.-J. Liang, Biomaterials, 2012, 33, 6408–6419 CrossRef CAS PubMed.
- P. S. Low and A. C. Antony, Adv. Drug Delivery Rev., 2004, 56, 1055–1058 CrossRef CAS PubMed.
- H. Nosrati, Y. Baghdadchi, R. Abbasi, M. Barsbay, M. Ghaffarlou, F. Abhari, A. Mohammadi, T. Kavetskyy, S. Bochani, H. Rezaeejam, S. Davaran and H. Danafar, J. Mater. Chem. B, 2021, 9, 4510–4522 RSC.
- Y. Huang, Y. Luo, W. Zheng and T. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19217–19228 CrossRef CAS PubMed.
- M. E. Werner, S. Karve, R. Sukumar, N. D. Cummings, J. A. Copp, R. C. Chen, T. Zhang and A. Z. Wang, Biomaterials, 2011, 32, 8548–8554 CrossRef CAS PubMed.
- M. E. Werner, J. A. Copp, S. Karve, N. D. Cummings, R. Sukumar, C. Li, M. E. Napier, R. C. Chen, A. D. Cox and A. Z. Wang, ACS Nano, 2011, 5, 8990–8998 CrossRef CAS PubMed.
- X. Liu, Z. Yuan, Z. Tang, Q. Chen, J. Huang, L. He and T. Chen, Biomater. Sci., 2021, 9, 4691–4700 RSC.
- G. C. Zhang, J. Liu, X. N. Yu, Y. Deng, Y. Sun, T. T. Liu, L. Dong, C. F. Zhu, X. Z. Shen, J. M. Zhu, S. Q. Weng and Y. Li, Adv. Healthcare Mater., 2020, 9, e2000650 CrossRef PubMed.
- L. Lapcık Jr, L. Lapcık, S. De Smedt, J. Demeester and P. Chabrecek, Chem. Rev., 1998, 98, 2663–2684 CrossRef PubMed.
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS PubMed.
- K. Y. Choi, K. H. Min, H. Y. Yoon, K. Kim, J. H. Park, I. C. Kwon, K. Choi and S. Y. Jeong, Biomaterials, 2011, 32, 1880–1889 CrossRef CAS PubMed.
- Y. Chong, J. Huang, X. Xu, C. Yu, X. Ning, S. Fan and Z. Zhang, Bioconjugate Chem., 2020, 31, 1756–1765 CrossRef CAS PubMed.
- C. S. Chiang, I. J. Shih, P. W. Shueng, M. Kao, L. W. Zhang, S. F. Chen, M. H. Chen and T. Y. Liu, Acta Biomater., 2021, 125, 300–311 CrossRef CAS PubMed.
- C. Du, M. Zhou, F. Jia, L. Ruan, H. Lu, J. Zhang, B. Zhu, X. Liu, J. Chen, Z. Chai and Y. Hu, Biomaterials, 2021, 269, 120642 CrossRef CAS PubMed.
- M. A. Askar, N. M. Thabet, G. S. El-Sayyad, A. I. El-Batal, M. Abd Elkodous, O. E. El Shawi, H. Helal and M. K. Abdel-Rafei, Cancers, 2021, 13, 5571 CrossRef CAS PubMed.
- Y. Sun, C. Kang, F. Liu, Y. Zhou, L. Luo and H. Qiao, Drug Delivery Res., 2017, 78, 283–291 CAS.
- Y. Ding, X. Xiao, L. Zeng, Q. Shang, W. Jiang, S. Xiong, X. Duan, J. Shen, R. Wang, J. Guo and Y. Pan, Bioact. Mater., 2021, 6, 4707–4716 CrossRef CAS PubMed.
- P. Li, Y. W. Shi, B. X. Li, W. C. Xu, Z. L. Shi, C. Zhou and S. Fu, J. Nanobiotechnol., 2015, 13, 52 CrossRef PubMed.
- L. Zhao, H. Chen, Z. Guo, K. Fu, L. Yao, L. Fu, W. Guo, X. Wen, O. Jacobson, X. Zhang, L. Sun, H. Wu, Q. Lin and X. Chen, Mol. Cancer Ther., 2020, 19, 2034–2043 CrossRef CAS PubMed.
- L. Chan, P. Gao, W. Zhou, C. Mei, Y. Huang, X. F. Yu, P. K. Chu and T. Chen, ACS Nano, 2018, 12, 12401–12415 CrossRef CAS PubMed.
- L. Chan, X. Chen, P. Gao, J. Xie, Z. Zhang, J. Zhao and T. Chen, ACS Nano, 2021, 15, 3047–3060 CrossRef CAS PubMed.
- D. Luo, X. Wang, S. Zeng, G. Ramamurthy, C. Burda and J. P. Basilion, Small, 2019, 15, e1900968 CrossRef PubMed.
- S. J. DeNardo, G. L. DeNardo, L. A. Miers, A. Natarajan, A. R. Foreman, C. Gruettner, G. N. Adamson and R. Ivkov, Clin. Cancer Res., 2005, 11, 7087s–7092s CrossRef CAS PubMed.
- E. Thomas, J. U. Menon, J. Owen, I. Skaripa-Koukelli, S. Wallington, M. Gray, C. Mannaris, V. Kersemans, D. Allen, P. Kinchesh, S. Smart, R. Carlisle and K. A. Vallis, Theranostics, 2019, 9, 5595–5609 CrossRef CAS PubMed.
- K. J. Lee, E. J. Ko, Y. Y. Park, S. S. Park, E. J. Ju, J. Park, S. H. Shin, Y. A. Suh, S. M. Hong, I. J. Park, K. P. Kim, J. J. Hwang, S. J. Jang, J. S. Lee, S. Y. Song, S. Y. Jeong and E. K. Choi, Biomaterials, 2020, 255, 120151 CrossRef CAS PubMed.
- W. Sun, Q. Hu, W. Ji, G. Wright and Z. Gu, Physiol. Rev., 2017, 97, 189–225 CrossRef.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2012, 64, 206–212 CrossRef.
- S. N. Ekdawi, D. A. Jaffray and C. Allen, Nano Today, 2016, 11, 402–414 CrossRef CAS.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- L. Du, H. Qin, T. Ma, T. Zhang and D. Xing, ACS Nano, 2017, 11, 8930–8943 CrossRef CAS PubMed.
- Y. Han, H. Pan, W. Li, Z. Chen, A. Ma, T. Yin, R. Liang, F. Chen, Y. Ma, Y. Jin, M. Zheng, B. Li and L. Cai, Adv. Sci., 2019, 6, 1900251 CrossRef PubMed.
- S. Hou, J.-S. Choi, M. A. Garcia, Y. Xing, K.-J. Chen, Y.-M. Chen, Z. K. Jiang, T. Ro, L. Wu, D. B. Stout, J. S. Tomlinson, H. Wang, K. Chen, H.-R. Tseng and W.-Y. Lin, ACS Nano, 2016, 10, 1417–1424 CrossRef CAS PubMed.
- L.-L. Huang, W. Nie, J. Zhang and H.-Y. Xie, Acc. Chem. Res., 2020, 53, 276–287 CrossRef CAS PubMed.
- B. Li, P. Liu, H. Wu, X. Xie, Z. Chen, F. Zeng and S. Wu, Biomaterials, 2017, 138, 57–68 CrossRef CAS PubMed.
- Q. Wang, Y. Wang, J. Ding, C. Wang, X. Zhou, W. Gao, H. Huang, F. Shao and Z. Liu, Nature, 2020, 579, 421–426 CrossRef CAS PubMed.
- H. Y. Yoon, M. L. Shin, M. K. Shim, S. Lee, J. H. Na, H. Koo, H. Lee, J.-H. Kim, K. Y. Lee, K. Kim and I. C. Kwon, Mol. Pharm., 2017, 14, 1558–1570 CrossRef CAS PubMed.
- F. Hu, D. Mao, Kenry, X. Cai, W. Wu, D. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 57, 10182–10186 CrossRef CAS PubMed.
- J.-S. Ni, P. Zhang, T. Jiang, Y. Chen, H. Su, D. Wang, Z.-Q. Yu, R. T. K. Kwok, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2018, 30, 1805220 CrossRef PubMed.
- Y. Tu, Y. Dong, K. Wang, S. Shen, Y. Yuan and J. Wang, Biomaterials, 2020, 259, 120298 CrossRef CAS PubMed.
- R. Membreno, B. E. Cook, K. Fung, J. S. Lewis and B. M. Zeglis, Mol. Pharm., 2018, 15, 1729–1734 CrossRef CAS PubMed.
- J. L. Houghton, R. Membreno, D. Abdel-Atti, K. M. Cunanan, S. Carlin, W. W. Scholz, P. B. Zanzonico, J. S. Lewis and B. M. Zeglis, Mol. Cancer Ther., 2017, 16, 124–133 CrossRef CAS PubMed.
- S. Poty, L. M. Carter, K. Mandleywala, R. Membreno, D. Abdel-Atti, A. Ragupathi, W. W. Scholz, B. M. Zeglis and J. S. Lewis, Clin. Cancer Res., 2019, 25, 868–880 CrossRef CAS PubMed.
- A. Rondon, S. Schmitt, A. Briat, N. Ty, L. Maigne, M. Quintana, R. Membreno, B. M. Zeglis, I. Navarro-Teulon, J.-P. Pouget, J.-M. Chezal, E. Miot-Noirault, E. Moreau and F. Degoul, Theranostics, 2019, 9, 6706–6718 CrossRef CAS PubMed.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana, D. Dehaini, P. Nguyen, P. Angsantikul, C. H. Wen, A. V. Kroll, C. Carpenter, M. Ramesh, V. Qu, S. H. Patel, J. Zhu, W. Shi, F. M. Hofman, T. C. Chen, W. Gao, K. Zhang, S. Chien and L. Zhang, Nature, 2015, 526, 118–121 CrossRef CAS PubMed.
- Y. Pan, C. Xu, H. Deng, Q. You, C. Zhao, Y. Li, Q. Gao, O. U. Akakuru, J. Li, J. Zhang, A. Wu and X. Chen, Nano Today, 2022, 43, 101435 CrossRef CAS.
- D. Zhu, M. Lyu, W. Jiang, M. Suo, Q. Huang and K. Li, J. Mater. Chem. B, 2020, 8, 5312–5319 RSC.
- Q. Sun, J. Wu, L. Jin, L. Hong, F. Wang, Z. Mao and M. Wu, J. Mater. Chem. B, 2020, 8, 7253–7263 RSC.
- S. Gou, W. Liu, S. Wang, G. Chen, Z. Chen, L. Qiu, X. Zhou, Y. Wu, Y. Qi and Y. Gao, Nano Lett., 2021, 21, 9939–9950 CrossRef CAS PubMed.
- C. Huang, T. Chen, D. Zhu and Q. Huang, Front. Chem., 2020, 8, 225 CrossRef CAS PubMed.
- M. Sun, Y. Duan, Y. Ma and Q. Zhang, Int. J. Nanomed., 2020, 15, 6749–6760 CrossRef CAS PubMed.
- J. Deng, S. Xu, W. Hu, X. Xun, L. Zheng and M. Su, Biomaterials, 2018, 154, 24–33 CrossRef CAS PubMed.
- Y. Chen, G. Zhao, S. Wang, Y. He, S. Han, C. Du, S. Li, Z. Fan, C. Wang and J. Wang, Biomater. Sci., 2019, 7, 3450–3459 RSC.
- M. Lyu, M. Chen, L. Liu, D. Zhu, X. Wu, Y. Li, L. Rao and Z. Bao, Theranostics, 2021, 11, 7589–7599 CrossRef PubMed.
- Y. Yin, Y. Li, S. Wang, Z. Dong, C. Liang, J. Sun, C. Wang, R. Chai, W. Fei, J. Zhang, M. Qi, L. Feng and Q. Zhang, J. Nanobiotechnol., 2021, 19, 80 CrossRef CAS PubMed.
- W. Zai, L. Kang, T. Dong, H. Wang, L. Yin, S. Gan, W. Lai, Y. Ding, Y. Hu and J. Wu, ACS Nano, 2021, 15, 15381–15394 CrossRef CAS PubMed.
- M. H. Chen, T. Y. Liu, Y. C. Chen and M. H. Chen, Nanomaterials, 2021, 11, 1661 CrossRef CAS PubMed.
- C. Huang, F. B. Wang, L. Liu, W. Jiang, W. Liu, W. Ma and H. Zhao, Adv. Healthcare Mater., 2021, 10, e2002207 CrossRef PubMed.
- R. J. C. Bose, R. Paulmurugan, J. Moon, S.-H. Lee and H. Park, Drug Discovery Today, 2018, 23, 891–899 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, e1706759 CrossRef PubMed.
- E. Zocchi, M. Tonetti, C. Polvani, L. Guida, U. Benatti and A. D. Flora, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2040–2044 CrossRef CAS PubMed.
- N. Talwar and N. K. Jain, Drug Dev. Ind. Pharm., 1992, 18, 1799–1812 CrossRef CAS.
- G. V. Shavi, R. C. Doijad, P. B. Deshpande, F. Manvi, S. R. Meka, N. Udupa, R. Omprakash and K. Dhirendra, Pak. J. Pharm. Sci., 2010, 23, 194–200 CAS.
- M. J. Mitchell and M. R. King, Expert Opin. Drug Delivery, 2015, 12, 375–392 CrossRef CAS PubMed.
- C. G. Millán, M. a L. S. Marinero, A. Z. Castañeda and J. M. Lanao, J. Controlled Release, 2004, 95, 27–49 CrossRef PubMed.
- Y. Lu, Q. Hu, C. Jiang and Z. Gu, Curr. Opin. Biotechnol., 2019, 58, 81–91 CrossRef CAS PubMed.
- D. Xia, D. Hang, Y. Li, W. Jiang, J. Zhu, Y. Ding, H. Gu and Y. Hu, ACS Nano, 2020, 14, 15654–15668 CrossRef PubMed.
- D. Zhu, M. Lyu, Q. Huang, M. Suo, Y. Liu, W. Jiang, Y. Duo and K. Fan, ACS Appl. Mater. Interfaces, 2020, 12, 36928–36937 CrossRef CAS PubMed.
- A. Kumar, P. K. Jena, S. Behera, R. F. Lockey, S. Mohapatra and S. Mohapatra, Nanomedicine, 2010, 6, 64–69 CrossRef CAS PubMed.
- A. Amirfazli, Nat. Nanotechnol., 2007, 2, 467–468 CrossRef CAS PubMed.
- J. Dobson, Drug Delivery Res., 2006, 67, 55–60 CAS.
- U. Häfeli, G. Pauer, S. Failing and G. Tapolsky, J. Magn. Magn. Mater., 2001, 225, 73–78 CrossRef.
- U. O. Häfeli, S. M. Sweeney, B. A. Beresford, J. L. Humm and R. M. Macklis, Nucl. Med. Biol., 1995, 22, 147–155 CrossRef.
- S. Liang, Y. Wang, J. Yu, C. Zhang, J. Xia and D. Yin, J. Mater. Sci.: Mater. Med., 2007, 18, 2297–2302 CrossRef CAS PubMed.
- J. Cao, Y. Wang, J. Yu, J. Xia, C. Zhang, D. Yin and U. O. Häfeli, J. Magn. Magn. Mater., 2004, 277, 165–174 CrossRef CAS.
- Z. Chunfu, C. Jinquan, Y. Duanzhi, W. Yongxian, F. Yanlin and T. Jiajü, Appl. Radiat. Isot., 2004, 61, 1255–1259 CrossRef PubMed.
- J. G. Huang, T. Leshuk and F. X. Gu, Nano Today, 2011, 6, 478–492 CrossRef CAS.
- G. Wan, Y. Cheng, J. Song, Q. Chen, B. Chen, Y. Liu, S. Ji, H. Chen and Y. Wang, Chem. Eng. J., 2020, 380, 122458 CrossRef CAS.
- B. Hoang, R. M. Reilly and C. Allen, Biomacromolecules, 2012, 13, 455–465 CrossRef CAS PubMed.
- L. Wang, T. Zhang, M. Huo, J. Guo, Y. Chen and H. Xu, Small, 2019, 15, 1903254 CrossRef CAS PubMed.
- W. Pan, S. Gong, J. Wang, L. Yu, Y. Chen, N. Li and B. Tang, Chem. Commun., 2019, 55, 8182–8185 RSC.
- X.-H. Wang, H.-S. Peng, L. Yang, F.-T. You, F. Teng, A.-W. Tang, F.-J. Zhang and X.-H. Li, J. Mater. Chem. B, 2013, 1, 5143–5152 RSC.
- Y. Wang, G. Wei, X. Zhang, X. Huang, J. Zhao, X. Guo and S. Zhou, Small, 2018, 14, 1702994 CrossRef PubMed.
- H. Yu, J.-M. Li, K. Deng, W. Zhou, C.-X. Wang, Q. Wang, K.-H. Li, H.-Y. Zhao and S.-W. Huang, Theranostics, 2019, 9, 7033–7050 CrossRef CAS PubMed.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., Int. Ed., 2019, 58, 7657–7661 CrossRef CAS PubMed.
- W. Deng, K. J. McKelvey, A. Guller, A. Fayzullin, J. M. Campbell, S. Clement, A. Habibalahi, Z. Wargocka, L. Liang, C. Shen, V. M. Howell, A. F. Engel and E. M. Goldys, ACS Cent. Sci., 2020, 6, 715–726 CrossRef CAS PubMed.
- Y. Chen, N. Li, J. Wang, X. Zhang, W. Pan, L. Yu and B. Tang, Theranostics, 2019, 9, 167–178 CrossRef CAS PubMed.
- Z. Zhang, L. Zhou, Y. Zhou, J. Liu, X. Xing, J. Zhong, G. Xu, Z. Kang and J. Liu, Biomaterials, 2015, 65, 56–65 CrossRef CAS PubMed.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS PubMed.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS PubMed.
- K. Ni, G. Lan, S. S. Veroneau, X. Duan, Y. Song and W. Lin, Nat. Commun., 2018, 9, 4321 CrossRef PubMed.
- M. Michalak, J. M. Robert Parker and M. Opas, Cell Calcium, 2002, 32, 269–278 CrossRef CAS PubMed.
- K. F. Ferri and G. Kroemer, Nat. Cell Biol., 2001, 3, E255–E263 CrossRef CAS PubMed.
- S. C. Chiu, S. P. Chen, S. Y. Huang, M. J. Wang, S. Z. Lin, H. J. Harn and C. Y. Pang, PLoS One, 2012, 7, e33742 CrossRef CAS PubMed.
- S. Chen, Y. Zhang and D. Zhang, Sci. Rep., 2015, 5, 14723 CrossRef CAS PubMed.
- X.-L. Pang, G. He, Y.-B. Liu, Y. Wang and B. Zhang, World J. Gastroenterol., 2013, 19, 1736–1748 CrossRef CAS PubMed.
- S. Klein, M. L. Dell’Arciprete, M. Wegmann, L. V. Distel, W. Neuhuber, M. C. Gonzalez and C. Kryschi, Biochem. Biophys. Res. Commun., 2013, 434, 217–222 CrossRef CAS PubMed.
- H. Deng, Z. Zhou, W. Yang, L.-S. Lin, S. Wang, G. Niu, J. Song and X. Chen, Nano Lett., 2020, 20, 1928–1933 CrossRef CAS PubMed.
- Z. Zhang, P. Yue, T. Lu, Y. Wang, Y. Wei and X. Wei, J. Hematol. Oncol., 2021, 14, 79 CrossRef PubMed.
- H. Appelqvist, P. Wäster, K. Kågedal and K. Öllinger, J. Mol. Cell Biol., 2013, 5, 214–226 CrossRef CAS PubMed.
- X. Zhang, J. Wang, X. Li and D. Wang, Cancer Lett., 2018, 439, 39–46 CrossRef CAS PubMed.
- S. Simonet, C. Rodriguez-Lafrasse, D. Beal, S. Gerbaud, C. Malesys, O. Tillement, F. Lux, H. Fayyad-Kazan, W. Rachidi and D. Ardail, J. Biomed. Nanotechnol., 2020, 16, 111–124 CrossRef CAS PubMed.
- M. Hullo, R. Grall, Y. Perrot, C. Mathé, V. Ménard, X. Yang, S. Lacombe, E. Porcel, C. Villagrasa, S. Chevillard and E. Bourneuf, Int. J. Mol. Sci., 2021, 22, 4436 CrossRef CAS PubMed.
- L. Štefančíková, E. Porcel, P. Eustache, S. Li, D. Salado, S. Marco, J.-L. Guerquin-Kern, M. Réfrégiers, O. Tillement, F. Lux and S. Lacombe, Cancer Nanotechnol., 2014, 5, 6 CrossRef PubMed.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- D. Rotin, P. Wan, S. Grinstein and I. Tannock, Cancer Res., 1987, 47, 1497–1504 CAS.
- H. Lee, W. Akers, K. Bhushan, S. Bloch, G. Sudlow, R. Tang and S. Achilefu, Bioconjugate Chem., 2011, 22, 777–784 CrossRef CAS PubMed.
- Z. Tian, C. Yang, W. Wang and Z. Yuan, ACS Appl. Mater. Interfaces, 2014, 6, 17865–17876 CrossRef CAS PubMed.
- Z. Hu, J. Ma, F. Fu, C. Cui, X. Li, X. Wang, W. Wang, Y. Wan and Z. Yuan, J. Controlled Release, 2017, 268, 1–9 CrossRef CAS PubMed.
- L. Yu, X. Zhang, X. Li, Z. Zhang, X. Niu, X. Wang, W. Wang and Z. Yuan, Nanoscale, 2021, 13, 13735–13745 RSC.
- Y. Li, T. Gong, H. Gao, Y. Chen, H. Li, P. Zhao, Y. Jiang, K. Wang, Y. Wu, X. Zheng and W. Bu, Angew. Chem., Int. Ed., 2021, 60, 15472–15481 CrossRef CAS PubMed.
- B. A. Arrick and C. F. Nathan, Cancer Res., 1984, 44, 4224–4232 CAS.
- X. Guo, Y. Cheng, X. Zhao, Y. Luo, J. Chen and W.-E. Yuan, J. Nanobiotechnol., 2018, 16, 74 CrossRef PubMed.
- R. Li and Y. Xie, J. Controlled Release, 2017, 251, 49–67 CrossRef CAS PubMed.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman and M. F. Clarke, Nature, 2009, 458, 780–783 CrossRef CAS PubMed.
- D. Siemann and K. Beyers, Br. J. Radiol., 1993, 68, 1071–1079 CAS.
- Y. Ding, Z. Tong, L. Jin, B. Ye, J. Zhou, Z. Sun, H. Yang, L. Hong, F. Huang, W. Wang and Z. Mao, Adv. Mater., 2021, 34, e2106388 CrossRef PubMed.
- Y. Song, Y. Wang, Y. Zhu, Y. Cheng, Y. Wang, S. Wang, F. Tan, F. Lian and N. Li, Adv. Healthcare Mater., 2019, 8, e1900250 CrossRef PubMed.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- C. Zhang, K. Zhao, W. Bu, D. Ni, Y. Liu, J. Feng and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 1770–1774 CrossRef CAS PubMed.
- H. Pelicano, W. Lu, Y. Zhou, W. Zhang, Z. Chen, Y. Hu and P. Huang, Cancer Res., 2009, 69, 2375–2383 CrossRef CAS PubMed.
- X. Chen, M. Song, B. Zhang and Y. Zhang, Oxid. Med. Cell. Longev., 2016, 2016, 1580967 Search PubMed.
- C. Xu, H. Hong, Y. Lee, K. S. Park, M. Sun, T. Wang, M. E. Aikins, Y. Xu and J. J. Moon, ACS Nano, 2020, 14, 13268–13278 CrossRef CAS PubMed.
- Y. He, S. Guo, L. Wu, P. Chen, L. Wang, Y. Liu and H. Ju, Biomaterials, 2019, 225, 119501 CrossRef CAS PubMed.
- C. Yue, Y. Yang, C. Zhang, G. Alfranca, S. Cheng, L. Ma, Y. Liu, X. Zhi, J. Ni, W. Jiang, J. Song, J. M. de la Fuente and D. Cui, Theranostics, 2016, 6, 2352–2366 CrossRef CAS PubMed.
- J. Wang, Y. Zhang, E. Archibong, F. S. Ligler and Z. Gu, Adv. Biosyst., 2017, 1, 1700084 CrossRef PubMed.
- S. J. Lee, T. B. Neugut, K. L. Rosenblum, R. M. Tolman, W. J. Travis and M. H. Walker, Child. Youth Serv. Rev., 2013, 35, 908–915 CrossRef.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- Y. Yu, Z. Feng, J. Liu, X. Hou, X. Zhou, J. Gao, W. Wang, Y. Zhang, G. Li and J. Liu, ACS Omega, 2021, 6, 19445–19457 CrossRef CAS PubMed.
- L. Meng, Y. Cheng, X. Tong, S. Gan, Y. Ding, Y. Zhang, C. Wang, L. Xu, Y. Zhu, J. Wu, Y. Hu and A. Yuan, ACS Nano, 2018, 12, 8308–8322 CrossRef CAS PubMed.
- X. Wang, X. Niu, W. Sha, X. Feng, L. Yu, Z. Zhang, W. Wang and Z. Yuan, Biomater. Sci., 2021, 9, 6308–6324 RSC.
- R. Chandrawati, Exp. Biol. Med., 2016, 241, 972–979 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- J. R. Graff, B. W. Konicek, J. A. Deddens, M. Chedid, B. M. Hurst, B. Colligan, B. L. Neubauer, H. W. Carter and J. H. Carter, Clin. Cancer Res., 2001, 7, 3857–3861 CAS.
- J. Ge, D. Lu, C. Yang and Z. Liu, Macromol. Rapid Commun., 2011, 32, 546–550 CrossRef CAS PubMed.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Soft Matter, 2011, 7, 6222–6230 RSC.
- Z.-H. Peng and J. Kopeček, J. Am. Chem. Soc., 2015, 137, 6726–6729 CrossRef CAS PubMed.
- R. Zou, Q. Gong, Z. Shi, J. Zheng, J. Xing, C. Liu, Z. Jiang and A. Wu, Nanoscale, 2020, 12, 14870–14881 RSC.
- J. Mu, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2018, 47, 5554–5573 RSC.
- Z. Zhang, Q. Zhang, J. Xie, Z. Zhong and C. Deng, Biomater. Sci., 2021, 9, 6915–6926 RSC.
- Y. Ding, Z. Sun, Z. Tong, S. Zhang, J. Min, Q. Xu, L. Zhou, Z. Mao, H. Xia and W. Wang, Theranostics, 2020, 10, 5195–5208 CrossRef CAS PubMed.
- P. Pan, X. Dong, Y. Chen, X. Zeng and X. Z. Zhang, ACS Nano, 2022, 16, 801–812 CrossRef CAS PubMed.
- L. Wu, B. Lin, H. Yang, J. Chen, Z. Mao, W. Wang and C. Gao, Acta Biomater., 2019, 86, 363–372 CrossRef CAS PubMed.
- L. Wang, T. Zhang, M. Huo, J. Guo, Y. Chen and H. Xu, Small, 2019, 15, e1903254 CrossRef PubMed.
- E. S. Olson, T. Jiang, T. A. Aguilera, Q. T. Nguyen, L. G. Ellies, M. Scadeng and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4311–4316 CrossRef CAS PubMed.
- S. Yu, Y. Duan, X. Zuo, X. Chen, Z. Mao and C. Gao, Biomaterials, 2018, 180, 193–205 CrossRef CAS PubMed.
- Y. Yuan, C. J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- T. Ren, S. Yu, Z. Mao and C. Gao, Biomaterials, 2015, 56, 58–67 CrossRef CAS PubMed.
- S. Chen, Z. Cao and S. Jiang, Biomaterials, 2009, 30, 5892–5896 CrossRef CAS PubMed.
- C. S. Gondi and J. S. Rao, Expert Opin. Ther. Targets, 2013, 17, 281–291 CrossRef CAS PubMed.
- S. Roshy, B. F. Sloane and K. Moin, Cancer Metastasis Rev., 2003, 22, 271–286 CrossRef CAS PubMed.
- L. Yin, H. Sun, H. Zhang, L. He, L. Qiu, J. Lin, H. Xia, Y. Zhang, S. Ji, H. Shi and M. Gao, J. Am. Chem. Soc., 2019, 141, 3265–3273 CrossRef CAS PubMed.
- V. K. Garripelli, J. K. Kim, S. Son, W. J. Kim, M. A. Repka and S. Jo, Acta Biomater., 2011, 7, 1984–1992 CrossRef CAS PubMed.
- L. Blavier, A. Lazaryev, F. Dorey, G. M. Shackleford and Y. A. DeClerck, Cancer Res., 2006, 66, 2691–2699 CrossRef CAS PubMed.
- L. E. Samuelson, R. L. Scherer, L. M. Matrisian, J. O. McIntyre and D. J. Bornhop, Mol. Pharm., 2013, 10, 3164–3174 CrossRef CAS PubMed.
- M. Shahriari, M. Zahiri, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2019, 308, 172–189 CrossRef CAS PubMed.
- W.-X. Qiu, L.-H. Liu, S.-Y. Li, Q. Lei, G.-F. Luo and X.-Z. Zhang, Small, 2017, 13, 1603956 CrossRef PubMed.
- G. Wei, Y. Wang, X. Huang, H. Hou and S. Zhou, Small Methods, 2018, 2, 1700358 CrossRef.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- C. Chu, H. Lin, H. Liu, X. Wang, J. Wang, P. Zhang, H. Gao, C. Huang, Y. Zeng, Y. Tan, G. Liu and X. Chen, Adv. Mater., 2017, 29, 1605928 CrossRef PubMed.
- X. Zhang, S. Wang, G. Cheng, P. Yu, J. Chang and X. Chen, Matter, 2021, 4, 26–53 CrossRef CAS PubMed.
- K. Kiyose, K. Hanaoka, D. Oushiki, T. Nakamura, M. Kajimura, M. Suematsu, H. Nishimatsu, T. Yamane, T. Terai, Y. Hirata and T. Nagano, J. Am. Chem. Soc., 2010, 132, 15846–15848 CrossRef CAS PubMed.
- L. Cui, Y. Zhong, W. Zhu, Y. Xu, Q. Du, X. Wang, X. Qian and Y. Xiao, Org. Lett., 2011, 13, 928–931 CrossRef CAS PubMed.
- S. Im, J. Lee, D. Park, A. Park, Y. M. Kim and W. J. Kim, ACS Nano, 2019, 13, 476–488 CrossRef CAS PubMed.
- Z. Yuan, C. Lin, Y. He, B. Tao, M. Chen, J. Zhang, P. Liu and K. Cai, ACS Nano, 2020, 14, 3546–3562 CrossRef CAS PubMed.
- G. Yang, S. Z. F. Phua, W. Q. Lim, R. Zhang, L. Feng, G. Liu, H. Wu, A. K. Bindra, D. Jana, Z. Liu and Y. Zhao, Adv. Mater., 2019, 31, e1901513 CrossRef PubMed.
- K. Zhang, X. Meng, Z. Yang, H. Dong and X. Zhang, Biomaterials, 2020, 258, 120278 CrossRef CAS PubMed.
- L. Hua, Z. Wang, L. Zhao, H. Mao, G. Wang, K. Zhang, X. Liu, D. Wu, Y. Zheng, J. Lu, R. Yu and H. Liu, Theranostics, 2018, 8, 5088–5105 CrossRef CAS PubMed.
- H. Mao, Y. Xie, H. Ju, H. Mao, L. Zhao, Z. Wang, L. Hua, C. Zhao, Y. Li, R. Yu and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 33923–33935 CrossRef CAS PubMed.
- A. Wang, J. Fang, S. Ye, Q. Mao, Y. Zhao, C. Cui, Y. Zhang, Y. Feng, J. Li, L. He, L. Qiu and H. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 59787–59802 CrossRef CAS PubMed.
- Z. Zhang, X. Niu, X. Feng, X. Wang, L. Yu, W. Wang and Z. Yuan, ACS Biomater. Sci. Eng., 2021, 7, 3434–3445 CrossRef CAS PubMed.
- J. Weng, Z. Huang, X. Pu, X. Chen, G. Yin, Y. Tian and Y. Song, Colloids Surf., B, 2020, 191, 110943 CrossRef CAS PubMed.
- J. Liu, J. Zhang, K. Song, J. Du, X. Wang, J. Liu, B. Li, R. Ouyang, Y. Miao, Y. Sun and Y. Li, Small, 2021, 17, e2101015 CrossRef PubMed.
- Y. Chang, K. Yang, P. Wei, S. Huang, Y. Pei, W. Zhao and Z. Pei, Angew. Chem., Int. Ed., 2014, 53, 13126–13130 CrossRef CAS PubMed.
- C. Qian, J. Yu, Y. Chen, Q. Hu, X. Xiao, W. Sun, C. Wang, P. Feng, Q.-D. Shen and Z. Gu, Adv. Mater., 2016, 28, 3313–3320 CrossRef CAS PubMed.
- J. Yu, Y. Zhang, Y. Ye, R. Di Santo, W. Sun, D. Ranson, F. S. Ligler, J. B. Buse and Z. Gu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8260–8265 CrossRef CAS PubMed.
- X. Yi, H. Zhou, Z. Zhang, S. Xiong and K. Yang, Biomaterials, 2020, 233, 119764 CrossRef CAS PubMed.
- C. Ju, Y. Wen, L. Zhang, Q. Wang, L. Xue, J. Shen and C. Zhang, Small, 2019, 15, e1804191 CrossRef PubMed.
- X. Yi, L. Chen, J. Chen, D. Maiti, Z. Chai, Z. Liu and K. Yang, Adv. Funct. Mater., 2018, 28, 1705161 CrossRef.
- M. Pucci, V. Bravatà, G. I. Forte, F. P. Cammarata, C. Messa, M. C. Gilardi and L. Minafra, Cancer Genomics Proteomics, 2015, 12, 143–152 CAS.
- M. Chatterjee, E. Ben-Josef, R. Robb, M. Vedaie, S. Seum, K. Thirumoorthy, K. Palanichamy, M. Harbrecht, A. Chakravarti and T. M. Williams, Cancer Res., 2017, 77, 5925–5937 CrossRef CAS PubMed.
- M. Bouché, Y. C. Dong, S. Sheikh, K. Taing, D. Saxena, J. C. Hsu, M. H. Chen, R. D. Salinas, H. Song, J. A. Burdick, J. Dorsey and D. P. Cormode, ACS Biomater. Sci. Eng., 2021, 7, 3209–3220 CrossRef PubMed.
- S. Gao, T. Li, Y. Guo, C. Sun, B. Xianyu and H. Xu, Adv. Mater., 2020, 32, e1907568 CrossRef PubMed.
- B. Choi, H. Choi, B. Yu and D. H. Kim, ACS Nano, 2020, 14, 13115–13126 CrossRef PubMed.
- Z. Du, X. Zhang, Z. Guo, J. Xie, X. Dong, S. Zhu, J. Du, Z. Gu and Y. Zhao, Adv. Mater., 2018, 30, e1804046 CrossRef PubMed.
- L. Zhang, S. Zhang, J. Xu, Y. Li, J. He, Y. Yang, T. Huynh, P. Ni, G. Duan, Z. Yang and R. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 43398–43407 CrossRef CAS PubMed.
- D. Shao, F. Zhang, F. Chen, X. Zheng, H. Hu, C. Yang, Z. Tu, Z. Wang, Z. Chang, J. Lu, T. Li, Y. Zhang, L. Chen, K. W. Leong and W. F. Dong, Adv. Mater., 2020, 32, e2004385 CrossRef PubMed.
- X. Chen, J. Song, X. Chen and H. Yang, Chem. Soc. Rev., 2019, 48, 3073–3101 RSC.
- K. Tanabe, T. Asada, T. Ito and S.-i Nishimoto, Bioconjugate Chem., 2012, 23, 1909–1914 CrossRef CAS PubMed.
- J. Wang, W. Xu, N. Zhang, C. Yang, H. Xu, Z. Wang, B. Li, J. Ding and X. Chen, J. Controlled Release, 2021, 332, 1–9 CrossRef CAS PubMed.
- B. Tao, C. Lin, Z. Yuan, Y. He, M. Chen, K. Li, J. Hu, Y. Yang, Z. Xia and K. Cai, Chem. Eng. J., 2021, 403, 126182 CrossRef CAS.
- H. P. Lee, G. Lokhande, K. A. Singh, M. K. Jaiswal, S. Rajput and A. K. Gaharwar, Adv. Mater., 2021, 33, 2101238 CrossRef CAS PubMed.
- Y. Wang, L. Shi, W. Wu, G. Qi, X. Zhu and B. Liu, Adv. Funct. Mater., 2021, 31, 2010241 CrossRef CAS.
- T. He, Y. Yuan, C. Jiang, N. T. Blum, J. He, P. Huang and J. Lin, Angew. Chem., Int. Ed., 2021, 133, 6112–6119 CrossRef.
- X. Zhou, Y. Li, Y. Xing, J. Li and X. Jiang, Dalton Trans., 2019, 48, 15068–15073 RSC.
- S. Li, B. A. Moosa, J. G. Croissant and N. M. Khashab, Angew. Chem., Int. Ed., 2015, 127, 6908–6912 CrossRef.
- L. Feng, Z. Dong, C. Liang, M. Chen, D. Tao, L. Cheng, K. Yang and Z. Liu, Biomaterials, 2018, 181, 81–91 CrossRef CAS PubMed.
- Z. Meng, Y. Chao, X. Zhou, C. Liang, J. Liu, R. Zhang, L. Cheng, K. Yang, W. Pan, M. Zhu and Z. Liu, ACS Nano, 2018, 12, 9412–9422 CrossRef CAS PubMed.
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed.
- G. Song, C. Liang, X. Yi, Q. Zhao, L. Cheng, K. Yang and Z. Liu, Adv. Mater., 2016, 28, 2716–2723 CrossRef CAS PubMed.
- H.-J. Kim, H. Matsuda, H. Zhou and I. Honma, Adv. Mater., 2006, 18, 3083–3088 CrossRef CAS.
- N. Y. Rapoport, A. M. Kennedy, J. E. Shea, C. L. Scaife and K. H. Nam, J. Controlled Release, 2009, 138, 268–276 CrossRef CAS PubMed.
- T. Porter, D. Kricsfeld, S. Cheatham and S. Li, J. Am. Soc. Echocardiogr., 1998, 11, 421–425 CrossRef CAS PubMed.
- X. Song, L. Feng, C. Liang, K. Yang and Z. Liu, Nano Lett., 2016, 16, 6145–6153 CrossRef CAS PubMed.
- S. Hua, J. He, F. Zhang, J. Yu, W. Zhang, L. Gao, Y. Li and M. Zhou, Biomaterials, 2021, 268, 120590 CrossRef CAS PubMed.
- W. Fu, X. Zhang, L. Mei, R. Zhou, W. Yin, Q. Wang, Z. Gu and Y. Zhao, ACS Nano, 2020, 14, 10001–10017 CrossRef CAS PubMed.
- C. Zhang, L. Yan, X. Wang, X. Dong, R. Zhou, Z. Gu and Y. Zhao, Nano Lett., 2019, 19, 1749–1757 CrossRef CAS PubMed.
- X. Zhou, Y. Zhang, C. Wang, X. Wu, Y. Yang, B. Zheng, H. Wu, S. Guo and J. Zhang, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS PubMed.
- P. Hu, T. Wu, W. Fan, L. Chen, Y. Liu, D. Ni, W. Bu and J. Shi, Biomaterials, 2017, 141, 86–95 CrossRef CAS PubMed.
- Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC.
- L. Ju, Z. Chen, L. Fang, W. Dong, F. Zheng and M. Shen, J. Am. Ceram. Soc., 2011, 94, 3418–3424 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- N. Zheng, Q. Wang, C. Li, X. Wang, X. Liu, X. Wang, G. Deng, J. Wang, L. Zhao and J. Lu, Adv. Healthcare Mater., 2021, 10, e2002024 CrossRef PubMed.
- Y. Chen, G. Song, Z. Dong, X. Yi, Y. Chao, C. Liang, K. Yang, L. Cheng and Z. Liu, Small, 2017, 13, 1602869 CrossRef PubMed.
- Y. Yong, C. Zhang, Z. Gu, J. Du, Z. Guo, X. Dong, J. Xie, G. Zhang, X. Liu and Y. Zhao, ACS Nano, 2017, 11, 7164–7176 CrossRef CAS PubMed.
- X. Cheng, R. Sun, H. Xia, J. Ding, L. Yin, Z. Chai, H. Shi and M. Gao, Nanomedicine, 2019, 14, 2941–2955 CrossRef CAS PubMed.
- F. Ding, J. Feng, X. Zhang, J. Sun, C. Fan and Z. Ge, Adv. Drug Delivery Rev., 2021, 173, 141–163 CrossRef CAS PubMed.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef CAS.
- V. Ntziachristos, J. Ripoll, L. V. Wang and R. Weissleder, Nat. Biotechnol., 2005, 23, 313–320 CrossRef CAS PubMed.
- Z. Duan, Q. Luo, L. Gu, X. Li, H. Zhu, Z. Gu, Q. Gong, H. Zhang and K. Luo, Nanoscale, 2021, 13, 13681–13692 RSC.
- C. Yao, J. Li, X. Cao, J. R. Gunn, M. Wu, S. Jiang and B. W. Pogue, ACS Appl. Mater. Interfaces, 2020, 12, 44383–44392 CrossRef CAS PubMed.
- S. Chen, S. Yu, Z. Du, X. Huang, M. He, S. Long, J. Liu, Y. Lan, D. Yang, H. Wang, S. Li, A. Chen, Y. Hao, Y. Su, C. Wang and S. Luo, J. Med. Chem., 2021, 64, 3381–3391 CrossRef CAS PubMed.
- Y. Li, M. H. Cho, S. S. Lee, D. E. Lee, H. Cheong and Y. Choi, J. Controlled Release, 2020, 325, 100–110 CrossRef CAS PubMed.
- A. Mizrachi, Y. Shamay, J. Shah, S. Brook, J. Soong, V. K. Rajasekhar, J. L. Humm, J. H. Healey, S. N. Powell, J. Baselga, D. A. Heller, A. Haimovitz-Friedman and M. Scaltriti, Nat. Commun., 2017, 8, 14292 CrossRef CAS PubMed.
- C. He, Z. Zhang, Y. Ding, K. Xue, X. Wang, R. Yang, Y. An, D. Liu, C. Hu and Q. Tang, J. Nanobiotechnol., 2021, 19, 29 CrossRef CAS PubMed.
- Z. Yang, Y. Dai, C. Yin, Q. Fan, W. Zhang, J. Song, G. Yu, W. Tang, W. Fan, B. C. Yung, J. Li, X. Li, X. Li, Y. Tang, W. Huang, J. Song and X. Chen, Adv. Mater., 2018, 30, 1707509 CrossRef PubMed.
- F. Feng, L. Liu, Q. Yang and S. Wang, Macromol. Rapid Commun., 2010, 31, 1405–1421 CrossRef CAS PubMed.
- J. Li, R. Jiang, Q. Wang, X. Li, X. Hu, Y. Yuan, X. Lu, W. Wang, W. Huang and Q. Fan, Biomaterials, 2019, 217, 119304 CrossRef CAS PubMed.
- K. Shou, Y. Tang, H. Chen, S. Chen, L. Zhang, A. Zhang, Q. Fan, A. Yu and Z. Cheng, Chem. Sci., 2018, 9, 3105–3110 RSC.
- J. Li, W. Li, L. Xie, W. Sang, G. Wang, Z. Zhang, B. Li, H. Tian, J. Yan, Y. Tian, Z. Li, Q. Fan, L. Yu and Y. Dai, Chem. Commun., 2021, 57, 11473–11476 RSC.
- Y. B. Pan, S. Wang, X. He, W. Tang, J. Wang, A. Shao and J. Zhang, J. Mater. Chem. B, 2019, 7, 7683–7689 RSC.
- H. Wang, D. Jia, D. Yuan, X. Yin, F. Yuan, F. Wang, W. Shi, H. Li, L. M. Zhu and Q. Fan, J. Nanobiotechnol., 2021, 19, 138 CrossRef CAS PubMed.
- W. Liu, J. Deacon, H. Yan, B. Sun, Y. Liu, D. Hegan, Q. Li, D. Coman, M. Parent, F. Hyder, K. Roberts, R. Nath, O. Tillement, D. Engelman and P. Glazer, Transl. Oncol., 2020, 13, 100839 CrossRef PubMed.
- Y. Fan, J. Zhang, M. Shi, D. Li, C. Lu, X. Cao, C. Peng, S. Mignani, J. P. Majoral and X. Shi, Nano Lett., 2019, 19, 1216–1226 CrossRef CAS PubMed.
- F. Du, L. Zhang, L. Zhang, M. Zhang, A. Gong, Y. Tan, J. Miao, Y. Gong, M. Sun, H. Ju, C. Wu and S. Zou, Biomaterials, 2017, 121, 109–120 CrossRef CAS PubMed.
- C. Wu, R. Cai, T. Zhao, L. Wu, L. Zhang, J. Jin, L. Xu, P. Li, T. Li, M. Zhang and F. Du, Nanoscale Res. Lett., 2020, 15, 94 CrossRef CAS PubMed.
- S. Dufort, G. Le Duc, M. Salomé, V. Bentivegna, L. Sancey, E. Bräuer-Krisch, H. Requardt, F. Lux, J.-L. Coll, P. Perriat, S. Roux and O. Tillement, Sci. Rep., 2016, 6, 29678 CrossRef CAS PubMed.
- R. M. Martens, D. P. Noij, M. Ali, T. Koopman, J. T. Marcus, M. R. Vergeer, H. de Vet, M. C. de Jong, C. R. Leemans, O. S. Hoekstra, R. de Bree, P. de Graaf, R. Boellaard and J. A. Castelijns, Oral Oncol., 2019, 88, 75–83 CrossRef PubMed.
- A. Hillebrand, K. D. Singh, I. E. Holliday, P. L. Furlong and G. R. Barnes, Hum. Brain Mapp., 2005, 25, 199–211 CrossRef PubMed.
- P. Rolfe, Annu. Rev. Biomed. Eng., 2000, 2, 715–754 CrossRef CAS PubMed.
- J. W. Wheless, E. Castillo, V. Maggio, H. L. Kim, J. I. Breier, P. G. Simos and A. C. Papanicolaou, Neurologist, 2004, 10, 138–153 CrossRef PubMed.
- A. Shen, X. Meng, X. Gao, X. Xu, C. Shao, Z. Tang, Y. Liu, W. Bu and P. Wang, Adv. Funct. Mater., 2019, 29, 1803832 CrossRef.
- J. M. Lupo and S. J. Nelson, Semin. Radiat. Oncol., 2014, 24, 248–258 CrossRef PubMed.
- D. M. Lambregts, M. Maas, M. P. Stokkel and R. G. Beets-Tan, Semin. Radiat. Oncol., 2016, 26, 193–198 CrossRef PubMed.
- J. Ye, G. Fu, X. Yan, J. Liu, X. Wang, L. Cheng, F. Zhang, P. Z. Sun and G. Liu, Nanoscale, 2018, 10, 5864–5868 RSC.
- M. Liu, X. Guo, S. Wang, M. Jin, Y. Wang, J. Li and J. Liu, Eur. Radiol., 2013, 23, 3221–3227 CrossRef PubMed.
- R. Fusco, V. Granata, P. Pariante, V. Cerciello, C. Siani, M. Di Bonito, M. Valentino, M. Sansone, G. Botti and A. Petrillo, Magn. Reson. Imaging, 2021, 75, 51–59 CrossRef CAS PubMed.
- J. Liu, H. Cabral, B. Song, I. Aoki, Z. Chen, N. Nishiyama, Y. Huang, K. Kataoka and P. Mi, ACS Nano, 2021, 15, 13526–13538 CrossRef CAS PubMed.
- P. Mi, D. Kokuryo, H. Cabral, H. Wu, Y. Terada, T. Saga, I. Aoki, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2016, 11, 724–730 CrossRef CAS PubMed.
- J. Hörner-Rieber, S. Klüter, J. Debus, G. Adema, M. Ansems and M. Verheij, Front. Oncol., 2020, 10, 615697 CrossRef PubMed.
- W. Jiang, Q. Li, L. Xiao, J. Dou, Y. Liu, W. Yu, Y. Ma, X. Li, Y. Z. You, Z. Tong, H. Liu, H. Liang, L. Lu, X. Xu, Y. Yao, G. Zhang, Y. Wang and J. Wang, ACS Nano, 2018, 12, 5684–5698 CrossRef CAS PubMed.
- M. Xu and L. V. Wang, Rev. Sci. Instrum., 2006, 77, 041101 CrossRef.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- D. Jiao, K. Wu, K. Xu, Y. Liu, D. Zhao, X. Han and R. Fan, Front. Bioeng. Biotechnol., 2021, 9, 764531 CrossRef PubMed.
- Y. Wu, L. Su, M. Yuan, T. Chen, J. Ye, Y. Jiang, J. Song and H. Yang, Angew. Chem., Int. Ed., 2021, 60, 12868–12875 CrossRef CAS PubMed.
- X. Zhou, H. Liu, Y. Zheng, Y. Han, T. Wang, H. Zhang, Q. Sun and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 4231–4240 CrossRef CAS PubMed.
- R. Zhou, X. Liu, Y. Wu, H. Xiang, J. Cao, Y. Li, W. Yin, Y. Zu, J. Li, R. Liu, F. Zhao, Z. Liu, C. Chen, Z. Gu, L. Yan and Y. Zhao, ACS Nano, 2020, 14, 13016–13029 CrossRef CAS PubMed.
- H. F. Zhang, K. Maslov, G. Stoica and L. V. Wang, Nat. Biotechnol., 2006, 24, 848–851 CrossRef CAS PubMed.
- F. Hyafil, J.-C. Cornily, J. E. Feig, R. Gordon, E. Vucic, V. Amirbekian, E. A. Fisher, V. Fuster, L. J. Feldman and Z. A. Fayad, Nat. Med., 2007, 13, 636–641 CrossRef CAS PubMed.
- C. Haller and I. Hizoh, Investig. Radiol., 2004, 39, 149–154 CrossRef CAS PubMed.
- O. Rabin, J. Manuel Perez, J. Grimm, G. Wojtkiewicz and R. Weissleder, Nat. Mater., 2006, 5, 118–122 CrossRef CAS PubMed.
- L. Zhang, X.-Q. Yang, J.-S. Wei, X. Li, H. Wang and Y.-D. Zhao, Theranostics, 2019, 9, 5424–5442 CrossRef CAS PubMed.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. a Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed.
- M. H. Oh, N. Lee, H. Kim, S. P. Park, Y. Piao, J. Lee, S. W. Jun, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 5508–5515 CrossRef CAS PubMed.
- J. Ding, Q. Mao, M. Zhao, Y. Gao, A. Wang, S. Ye, X. Wang, W. Xie and H. Shi, Nanoscale, 2020, 12, 22963–22969 RSC.
- F. Zhang, S. Liu, N. Zhang, Y. Kuang, W. Li, S. Gai, F. He, A. Gulzar and P. Yang, Nanoscale, 2020, 12, 19293–19307 RSC.
- J. Liu, J. Chen, H. Liu, K. Zhang, Q. Zeng, S. Yang, Z. Jiang, X. Zhang, T. Chen, D. Li and H. Shan, ACS Appl. Mater. Interfaces, 2021, 13, 42473–42485 CrossRef CAS PubMed.
- D. Huo, S. Liu, C. Zhang, J. He, Z. Zhou, H. Zhang and Y. Hu, ACS Nano, 2017, 11, 10159–10174 CrossRef CAS PubMed.
- E. B. Ehlerding, F. Chen and W. Cai, Adv. Sci., 2016, 3, 1500223 CrossRef PubMed.
- J. Pellico, P. J. Gawne and R. T. M. de Rosales, Chem. Soc. Rev., 2021, 50, 3355–3423 RSC.
- J. Ge, Q. Zhang, J. Zeng, Z. Gu and M. Gao, Biomaterials, 2020, 228, 119553 CrossRef CAS PubMed.
- M. Plotkin, U. Gneveckow, K. Meier-Hauff, H. Amthauer, A. Feußner, T. Denecke, M. Gutberlet, A. Jordan, R. Felix and P. Wust, Int. J. Hyperthermia, 2006, 22, 319–325 CrossRef CAS PubMed.
- P. Garrigue, J. Tang, L. Ding, A. Bouhlel, A. Tintaru, E. Laurini, Y. Huang, Z. Lyu, M. Zhang, S. Fernandez, L. Balasse, W. Lan, E. Mas, D. Marson, Y. Weng, X. Liu, S. Giorgio, J. Iovanna, S. Pricl, B. Guillet and L. Peng, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11454–11459 CrossRef CAS PubMed.
- N. Song, L. Zhao, X. Xu, M. Zhu, C. Liu, N. Sun, J. Yang, X. Shi and J. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 12395–12406 CrossRef CAS PubMed.
- X. Yi, M. Xu, H. Zhou, S. Xiong, R. Qian, Z. Chai, L. Zhao and K. Yang, ACS Nano, 2018, 12, 9142–9151 CrossRef CAS PubMed.
- X. Shi, Q. Li, C. Zhang, H. Pei, G. Wang, H. Zhou, L. Fan, K. Yang, B. Jiang, F. Wang and R. Zhu, J. Nanobiotechnol., 2021, 19, 337 CrossRef CAS PubMed.
- D. Cheng, J. Gong, P. Wang, J. Zhu, N. Yu, J. Zhao, Q. Zhang and J. Li, J. Mater. Chem. B, 2021, 9, 9316–9323 RSC.
- L. Luo, W. Sun, Y. Feng, R. Qin, J. Zhang, D. Ding, T. Shi, X. Liu, X. Chen and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 12591–12599 CrossRef CAS PubMed.
- S. Wang, R. Tian, X. Zhang, G. Cheng, P. Yu, J. Chang and X. Chen, Adv. Mater., 2021, 33, e2007488 CrossRef PubMed.
- F. Ahmad, X. Wang, Z. Jiang, X. Yu, X. Liu, R. Mao, X. Chen and W. Li, ACS Nano, 2019, 13, 10419–10433 CrossRef CAS PubMed.
- B. Cline, I. Delahunty and J. Xie, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1541 Search PubMed.
- D. Ni, C. A. Ferreira, T. E. Barnhart, V. Quach, B. Yu, D. Jiang, W. Wei, H. Liu, J. W. Engle, P. Hu and W. Cai, J. Am. Chem. Soc., 2018, 140, 14971–14979 CrossRef PubMed.
- W. Lee, M. Jeon, J. Choi, C. Oh, G. Kim, S. Jung, C. Kim, S. J. Ye and H. J. Im, ACS Nano, 2020, 14, 13004–13015 CrossRef CAS PubMed.
- Z. Wang, O. Jacobson, R. Tian, R. C. Mease, D. O. Kiesewetter, G. Niu, M. G. Pomper and X. Chen, Bioconjugate Chem., 2018, 29, 2309–2315 CrossRef CAS PubMed.
- C. A. Ferreira, S. Goel, E. B. Ehlerding, Z. T. Rosenkrans, D. Jiang, T. Sun, E. Aluicio-Sarduy, J. W. Engle, D. Ni and W. Cai, Nano Lett., 2021, 21, 4692–4699 CrossRef CAS PubMed.
- D. Chen, D. Yang, C. A. Dougherty, W. Lu, H. Wu, X. He, T. Cai, M. E. Van Dort, B. D. Ross and H. Hong, ACS Nano, 2017, 11, 4315–4327 CrossRef CAS PubMed.
- Z. He, X. Huang, C. Wang, X. Li, Y. Liu, Z. Zhou, S. Wang, F. Zhang, Z. Wang, O. Jacobson, J. J. Zhu, G. Yu, Y. Dai and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 8752–8756 CrossRef CAS PubMed.
- D. W. Townsend, J. P. J. Carney, J. T. Yap and N. C. Hall, J. Nucl. Med., 2004, 45, 4S–14S Search PubMed.
- G. Delso, S. Fürst, B. Jakoby, R. Ladebeck, C. Ganter, S. G. Nekolla, M. Schwaiger and S. I. Ziegler, J. Nucl. Med., 2011, 52, 1914–1922 CrossRef PubMed.
- Z. Dong, L. Feng, Y. Chao, Y. Hao, M. Chen, F. Gong, X. Han, R. Zhang, L. Cheng and Z. Liu, Nano Lett., 2019, 19, 805–815 CrossRef CAS PubMed.
- E. Bindini, M. L. A. Ramirez, X. Rios, U. Cossío, C. Simó, V. Gomez-Vallejo, G. Soler-Illia, J. Llop and S. E. Moya, Small, 2021, 17, e2101519 CrossRef PubMed.
- Y. Shi, Q. Fu, J. Li, H. Liu, Z. Zhang, T. Liu and Z. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 55564–55573 CrossRef CAS PubMed.
- R. Tang, A. Zheleznyak, M. Mixdorf, A. Ghai, J. Prior, K. C. L. Black, M. Shokeen, N. Reed, P. Biswas and S. Achilefu, ACS Nano, 2020, 14, 4255–4264 CrossRef CAS PubMed.
- A. R. Muslimov, D. O. Antuganov, Y. V. Tarakanchikova, M. V. Zhukov, M. A. Nadporojskii, M. V. Zyuzin and A. S. Timin, ACS Appl. Mater. Interfaces, 2021, 13, 25599–25610 CrossRef CAS PubMed.
- Y. Yong, X. Cheng, T. Bao, M. Zu, L. Yan, W. Yin, C. Ge, D. Wang, Z. Gu and Y. Zhao, ACS Nano, 2015, 9, 12451–12463 CrossRef CAS PubMed.
- Z. Zhang, J. Yang, Q. Min, C. Ling, D. Maiti, J. Xu, L. Qin and K. Yang, Small, 2019, 15, e1803703 CrossRef PubMed.
- D. Zhong, W. Li, S. Hua, Y. Qi, T. Xie, Y. Qiao and M. Zhou, Theranostics, 2021, 11, 3580–3594 CrossRef CAS PubMed.
- W. Li, D. Zhong, S. Hua, Z. Du and M. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 44541–44553 CrossRef CAS PubMed.
- M. Lyu, D. Zhu, Y. Duo, Y. Li and H. Quan, Biomaterials, 2020, 233, 119656 CrossRef PubMed.
- Z. Wang, G. Wang, T. Kang, S. Liu, L. Wang, H. Zou, Y. Chong and Y. Liu, J. Nanobiotechnol., 2021, 19, 90 CrossRef CAS PubMed.
- Q. Dan, D. Hu, Y. Ge, S. Zhang, S. Li, D. Gao, W. Luo, T. Ma, X. Liu, H. Zheng, Y. Li and Z. Sheng, Biomater. Sci., 2020, 8, 973–987 RSC.
- L. Xia, X. Meng, L. Wen, N. Zhou, T. Liu, X. Xu, F. Wang, Z. Cheng, Z. Yang and H. Zhu, Small, 2021, 17, e2100378 CrossRef PubMed.
- Z. Guo, M. Chen, C. Peng, S. Mo, C. Shi, G. Fu, X. Wen, R. Zhuang, X. Su, T. Liu, N. Zheng and X. Zhang, Biomaterials, 2018, 179, 134–143 CrossRef CAS PubMed.
- N. Lu, W. Fan, X. Yi, S. Wang, Z. Wang, R. Tian, O. Jacobson, Y. Liu, B. C. Yung, G. Zhang, Z. Teng, K. Yang, M. Zhang, G. Niu, G. Lu and X. Chen, ACS Nano, 2018, 12, 1580–1591 CrossRef CAS PubMed.
- Q. Huang, S. Zhang, H. Zhang, Y. Han, H. Liu, F. Ren, Q. Sun, Z. Li and M. Gao, ACS Nano, 2019, 13, 1342–1353 CAS.
- F. Mao, L. Wen, C. Sun, S. Zhang, G. Wang, J. Zeng, Y. Wang, J. Ma, M. Gao and Z. Li, ACS Nano, 2016, 10, 11145–11155 CrossRef CAS PubMed.
- X. Li, J. Li, C. Li, Q. Guo, M. Wu, L. Su, Y. Dou, X. Wu, Z. Xiao and X. Zhang, J. Mater. Chem. B, 2021, 9, 7530–7543 RSC.
- Y. Kang, X. Yu, X. Fan, Aodenggerile, S. Zhao, C. Tu, Z. Yan, R. Wang, W. Li and H. Qiu, ACS Nano, 2020, 14, 4336–4351 CrossRef CAS PubMed.
- C. Zhu, X. Guo, L. Luo, Z. Wu, Z. Luo, M. Jiang, J. Zhang, B. Qin, Y. Shi, Y. Lou, Y. Qiu and J. You, ACS Appl. Mater. Interfaces, 2019, 11, 46536–46547 CrossRef CAS PubMed.
- W. Tang, Z. Dong, R. Zhang, X. Yi, K. Yang, M. Jin, C. Yuan, Z. Xiao, Z. Liu and L. Cheng, ACS Nano, 2019, 13, 284–294 CrossRef CAS PubMed.
- S. C. Formenti and S. Demaria, Lancet Oncol., 2009, 10, 718–726 CrossRef PubMed.
- J. Antoniades, L. W. Brady and D. A. Lightfoot, Int. J. Radiat. Oncol. Biol. Phys., 1977, 2, 141–147 CrossRef CAS.
- G. Ehlers and M. Fridman, Br. J. Radiol., 1973, 46, 220–222 CrossRef CAS PubMed.
- K. Ohba, K. Omagari, T. Nakamura, N. Ikuno, S. Saeki, I. Matsuo, H. Kinoshita, J. Masuda, H. Hazama, I. Sakamoto and S. Kohno, Gut, 1998, 43, 575–577 CrossRef CAS PubMed.
- G. J. Rees and C. M. Ross, Br. J. Radiol., 1983, 56, 63–66 CrossRef CAS PubMed.
- S. Demaria, B. Ng, M. L. Devitt, J. S. Babb, N. Kawashima, L. Liebes and S. C. Formenti, Int. J. Radiat. Oncol. Biol. Phys., 2004, 58, 862–870 CrossRef PubMed.
- S. Demaria, N. Kawashima, A. M. Yang, M. L. Devitt, J. S. Babb, J. P. Allison and S. C. Formenti, Clin. Cancer Res., 2005, 11, 728–734 CrossRef CAS PubMed.
- M. Z. Dewan, A. E. Galloway, N. Kawashima, J. K. Dewyngaert, J. S. Babb, S. C. Formenti and S. Demaria, Clin. Cancer Res., 2009, 15, 5379–5388 CrossRef CAS PubMed.
- W. Ngwa, O. C. Irabor, J. D. Schoenfeld, J. Hesser, S. Demaria and S. C. Formenti, Nat. Rev. Cancer, 2018, 18, 313–322 CrossRef CAS PubMed.
- R. R. Weichselbaum, H. Liang, L. Deng and Y.-X. Fu, Nat. Rev. Clin. Oncol., 2017, 14, 365–379 CrossRef CAS PubMed.
- E. Deutsch, C. Chargari, L. Galluzzi and G. Kroemer, Lancet Oncol., 2019, 20, e452–e463 CrossRef CAS PubMed.
- J. Li, Y. Luo and K. Pu, Angew. Chem., Int. Ed., 2021, 60, 12682–12705 CrossRef CAS PubMed.
- E. E. Sweeney, J. Cano-Mejia and R. Fernandes, Small, 2018, 14, 1800678 CrossRef PubMed.
- W. Yang, F. Zhang, H. Deng, L. Lin, S. Wang, F. Kang, G. Yu, J. Lau, R. Tian, M. Zhang, Z. Wang, L. He, Y. Ma, G. Niu, S. Hu and X. Chen, ACS Nano, 2020, 14, 620–631 CrossRef CAS PubMed.
- Y. Yin, X. Jiang, L. Sun, H. Li, C. Su, Y. Zhang, G. Xu, X. Li, C. Zhao, Y. Chen, H. Xu and K. Zhang, Nano Today, 2021, 36, 101009 CrossRef CAS.
- G. Zhan, Q. Xu, Z. Zhang, Z. Wei, T. Yong, N. Bie, X. Zhang, X. Li, J. Li, L. Gan and X. Yang, Nano Today, 2021, 38, 101195 CrossRef CAS.
- E. Golden, I. Pellicciotta, S. Demaria, M. H. Barcellos-Hoff and S. Formenti, Front. Oncol., 2012, 2, 404–408 Search PubMed.
- E. B. Golden and L. Apetoh, Semin. Radiat. Oncol., 2015, 25, 11–17 CrossRef PubMed.
- S. Chen, D. Li, X. Du, X. He, M. Huang, Y. Wang, X. Yang and J. Wang, Nano Today, 2020, 35, 100924 CrossRef CAS.
- W. Wu, Y. Pu, H. Yao, H. Lin and J. Shi, Nano Today, 2022, 42, 101377 CrossRef CAS.
- X. Xiong, J. Zhao, R. Su, C. Liu, X. Guo and S. Zhou, Nano Today, 2021, 39, 101225 CrossRef CAS.
- F. Zhou, J. Gao, Z. Xu, T. Li, A. Gao, F. Sun, F. Wang, W. Wang, Y. Geng, F. Zhang, Z. P. Xu and H. Yu, Nano Today, 2021, 36, 101025 CrossRef CAS.
- W. Chen, S. Wang, Y. Wu, X. Shen, Z. Guo, Q. Li and D. Xing, Microb. Pathog., 2020, 141, 103983 CrossRef CAS PubMed.
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 17, 97–111 CrossRef CAS PubMed.
- A. Takasu, A. Masui, M. Hamada, T. Imai, S. Iwai and Y. Yura, Cancer Gene Ther., 2016, 23, 107–113 CrossRef CAS PubMed.
- P.-J. Tian, B.-L. Li, Y.-J. Shan, J.-N. Zhang, J.-Y. Chen, M. Yu and L.-W. Zhang, Int. J. Mol. Sci., 2015, 16, 20033–20049 CrossRef CAS PubMed.
- M. Obeid, A. Tesniere, F. Ghiringhelli, G. M. Fimia, L. Apetoh, J. L. Perfettini, M. Castedo, G. Mignot, T. Panaretakis, N. Casares, D. Métivier, N. Larochette, P. van Endert, F. Ciccosanti, M. Piacentini, L. Zitvogel and G. Kroemer, Nat. Med., 2007, 13, 54–61 CrossRef CAS PubMed.
- P. Scaffidi, T. Misteli and M. E. Bianchi, Nature, 2002, 418, 191–195 CrossRef CAS PubMed.
- R. Spisek, A. Charalambous, A. Mazumder, D. H. Vesole, S. Jagannath and M. V. Dhodapkar, Blood, 2007, 109, 4839–4845 CrossRef CAS PubMed.
- J. Fucikova, P. Kralikova, A. Fialova, T. Brtnicky, L. Rob, J. Bartunkova and R. Spísek, Cancer Res., 2011, 71, 4821–4833 CrossRef CAS PubMed.
- A. D. Garg, D. V. Krysko, P. Vandenabeele and P. Agostinis, Cancer Immunol. Immunother., 2012, 61, 215–221 CrossRef CAS PubMed.
- A. D. Garg, D. V. Krysko, T. Verfaillie, A. Kaczmarek, G. B. Ferreira, T. Marysael, N. Rubio, M. Firczuk, C. Mathieu, A. J. Roebroek, W. Annaert, J. Golab, P. de Witte, P. Vandenabeele and P. Agostinis, EMBO J., 2012, 31, 1062–1079 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- Q. Chen, J. Chen, Z. Yang, J. Xu, L. Xu, C. Liang, X. Han and Z. Liu, Adv. Mater., 2019, 31, e1802228 CrossRef PubMed.
- A. Bansal and M. C. Simon, J. Cell Biol., 2018, 217, 2291–2298 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- Y. Wang, J. Chen, R. Duan, R. Gu, W. Wang, J. Wu, H. Lian, Y. Hu and A. Yuan, Adv. Mater., 2022, 34, 2109726 CrossRef CAS PubMed.
- C. Cosentino, D. Grieco and V. Costanzo, EMBO J., 2011, 30, 546–555 CrossRef CAS PubMed.
- Q. Li, T. Qin, Z. Bi, H. Hong, L. Ding, J. Chen, W. Wu, X. Lin, W. Fu, F. Zheng, Y. Yao, M.-L. Luo, P. E. Saw, G. M. Wulf, X. Xu, E. Song, H. Yao and H. Hu, Nat. Commun., 2020, 11, 1456 CrossRef CAS PubMed.
- S. J. Moon, W. Dong, G. N. Stephanopoulos and H. D. Sikes, Bioeng. Transl. Med., 2020, 5, e10184 CAS.
- K. C. Patra and N. Hay, Trends Biochem. Sci., 2014, 39, 347–354 CrossRef CAS PubMed.
- C. Mills, Crit. Rev. Immunol., 2012, 32, 463–488 CrossRef CAS PubMed.
- A. Sica, P. Larghi, A. Mancino, L. Rubino, C. Porta, M. G. Totaro, M. Rimoldi, S. K. Biswas, P. Allavena and A. Mantovani, Semin. Cancer Biol., 2008, 18(5), 349–355 CrossRef CAS PubMed.
- A. Gulzar, J. Xu, C. Wang, F. He, D. Yang, S. Gai, P. Yang, J. Lin, D. Jin and B. Xing, Nano Today, 2019, 26, 16–56 CrossRef CAS.
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed.
- A. Zhang, Y. Xu, H. Xu, J. Ren, T. Meng, Y. Ni, Q. Zhu, W.-B. Zhang, Y.-B. Pan, J. Jin, Y. Bi, Z. B. Wu, S. Lin and M. Lou, Theranostics, 2021, 11, 3839–3852 CrossRef CAS PubMed.
- B. Ruffell and Lisa M. Coussens, Cancer Cell, 2015, 27, 462–472 CrossRef CAS PubMed.
- A. Oweida, M. K. Hararah, A. Phan, D. Binder, S. Bhatia, S. Lennon, S. Bukkapatnam, B. Van Court, N. Uyanga, L. Darragh, H. M. Kim, D. Raben, A. C. Tan, L. Heasley, E. Clambey, R. Nemenoff and S. D. Karam, Clin. Cancer Res., 2018, 24, 5368–5380 CrossRef CAS PubMed.
- E. Persa, A. Balogh, G. Sáfrány and K. Lumniczky, Cancer Lett., 2015, 368, 252–261 CrossRef CAS PubMed.
- G. Genard, S. Lucas and C. Michiels, Front. Immunol., 2017, 8, 828 CrossRef PubMed.
- L. Seifert, G. Werba, S. Tiwari, N. N. Giao Ly, S. Nguy, S. Alothman, D. Alqunaibit, A. Avanzi, D. Daley, R. Barilla, D. Tippens, A. Torres-Hernandez, M. Hundeyin, V. R. Mani, C. Hajdu, I. Pellicciotta, P. Oh, K. Du and G. Miller, Gastroenterology, 2016, 150, 1659–1672.e5 CrossRef PubMed.
- Y. Meng, M. A. Beckett, H. Liang, H. J. Mauceri, N. van Rooijen, K. S. Cohen and R. R. Weichselbaum, Cancer Res., 2010, 70, 1534–1543 CrossRef CAS PubMed.
- T. Inoue, S. Fujishima, E. Ikeda, O. Yoshie, N. Tsukamoto, S. Aiso, N. Aikawa, A. Kubo, K. Matsushima and K. Yamaguchi, Eur. Respir. J., 2004, 24, 49–56 CrossRef CAS PubMed.
- J. Ma, R. Liu, X. Wang, Q. Liu, Y. Chen, R. P. Valle, Y. Y. Zuo, T. Xia and S. Liu, ACS Nano, 2015, 9, 10498–10515 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- Y. W. Choo, M. Kang, H. Y. Kim, J. Han, S. Kang, J.-R. Lee, G.-J. Jeong, S. P. Kwon, S. Y. Song, S. Go, M. Jung, J. Hong and B.-S. Kim, ACS Nano, 2018, 12, 8977–8993 CrossRef CAS PubMed.
- C. Shi, T. Liu, Z. Guo, R. Zhuang, X. Zhang and X. Chen, Nano Lett., 2018, 18, 7330–7342 CrossRef CAS PubMed.
- L. Tian, X. Yi, Z. Dong, J. Xu, C. Liang, Y. Chao, Y. Wang, K. Yang and Z. Liu, ACS Nano, 2018, 12, 11541–11551 CrossRef CAS PubMed.
- X. Song, J. Xu, C. Liang, Y. Chao, Q. Jin, C. Wang, M. Chen and Z. Liu, Nano Lett., 2018, 18, 6360–6368 CrossRef CAS PubMed.
- Y. Cao, S. Ding, L. Zeng, J. Miao, K. Wang, G. Chen, C. Li, J. Zhou, X. W. Bian and G. Tian, ACS Appl. Mater. Interfaces, 2021, 13, 53504–53518 CrossRef CAS PubMed.
- Z. Huang, D. Yao, Q. Ye, H. Jiang, R. Gu, C. Ji, J. Wu, Y. Hu and A. Yuan, ACS Nano, 2021, 15, 8450–8465 CrossRef CAS PubMed.
- S. P. Luckman, D. E. Hughes, F. P. Coxon, R. G. G. Russell and M. J. Rogers, J. Bone Miner. Res., 1998, 13, 581–589 CrossRef CAS PubMed.
- M. A. Caligiuri, Blood, 2008, 112, 461–469 CrossRef CAS PubMed.
- L. E. Lowry and W. A. Zehring, Front. Immunol., 2017, 8, 1061 CrossRef PubMed.
- C. J. Chan, M. J. Smyth and L. Martinet, Cell Death Differ., 2014, 21, 5–14 CrossRef CAS PubMed.
- S. Paul and G. Lal, Front. Immunol., 2017, 8, 1124 CrossRef PubMed.
- E. Vivier, E. Tomasello, M. Baratin, T. Walzer and S. Ugolini, Nat. Immunol., 2008, 9, 503–510 CrossRef CAS PubMed.
- J. Bi and Z. Tian, Front. Immunol., 2017, 8, 760 CrossRef PubMed.
- E. L. Monaco, E. Tremante, C. Cerboni, E. Melucci, L. Sibilio, A. Zingoni, M. R. Nicotra, P. G. Natali and P. Giacomini, Neoplasia, 2011, 13, 822-IN814 CrossRef PubMed.
- C. Sun, J. Xu, Q. Huang, M. Huang, H. Wen, C. Zhang, J. Wang, J. Song, M. Zheng, H. Sun, H. Wei, W. Xiao, R. Sun and Z. Tian, Oncoimmunology, 2017, 6, e1264562 CrossRef PubMed.
- J.-Y. Kim, Y.-O. Son, S.-W. Park, J.-H. Bae, J. S. Chung, H. H. Kim, B.-S. Chung, S.-H. Kim and C.-D. Kang, Exp. Mol. Med., 2006, 38, 474–484 CrossRef CAS PubMed.
- K. M. Au, S. I. Park and A. Z. Wang, Sci. Adv., 2020, 6, eaba8564 CrossRef CAS PubMed.
- M. Enqvist, G. Nilsonne, O. Hammarfjord, R. P. A. Wallin, N. K. Björkström, M. Björnstedt, A. Hjerpe, H.-G. Ljunggren, K. Dobra, K.-J. Malmberg and M. Carlsten, J. Immunol., 2011, 187, 3546–3554 CrossRef CAS PubMed.
- T. Li, S. Pan, S. Gao, W. Xiang, C. Sun, W. Cao and H. Xu, Angew. Chem., Int. Ed., 2020, 59, 2700–2704 CrossRef CAS PubMed.
- W. Cao, X. Zhang, X. Miao, Z. Yang and H. Xu, Angew. Chem., Int. Ed., 2013, 52, 6233–6237 CrossRef CAS PubMed.
- N. Ma, H. Xu, L. An, J. Li, Z. Sun and X. Zhang, Langmuir, 2011, 27, 5874–5878 CrossRef CAS PubMed.
- J. Xia, T. Li, C. Lu and H. Xu, Macromolecules, 2018, 51, 7435–7455 CrossRef CAS.
- J. K. Chan, J. Roth, J. J. Oppenheim, K. J. Tracey, T. Vogl, M. Feldmann, N. Horwood and J. Nanchahal, J. Clin. Invest., 2012, 122, 2711–2719 CrossRef CAS PubMed.
- J. F. Curtin, N. Liu, M. Candolfi, W. Xiong, H. Assi, K. Yagiz, M. R. Edwards, K. S. Michelsen, K. M. Kroeger, C. Liu, A. K. M. G. Muhammad, M. C. Clark, M. Arditi, B. Comin-Anduix, A. Ribas, P. R. Lowenstein and M. G. Castro, PLoS Med., 2009, 6, e1000010 CrossRef PubMed.
- A. B. Sharabi, M. Lim, T. L. DeWeese and C. G. Drake, Lancet Oncol., 2015, 16, e498–e509 CrossRef PubMed.
- L. Apetoh, F. Ghiringhelli, A. Tesniere, M. Obeid, C. Ortiz, A. Criollo, G. Mignot, M. C. Maiuri, E. Ullrich, P. Saulnier, H. Yang, S. Amigorena, B. Ryffel, F. J. Barrat, P. Saftig, F. Levi, R. Lidereau, C. Nogues, J.-P. Mira, A. Chompret, V. Joulin, F. Clavel-Chapelon, J. Bourhis, F. André, S. Delaloge, T. Tursz, G. Kroemer and L. Zitvogel, Nat. Med., 2007, 13, 1050–1059 CrossRef CAS PubMed.
- R. H. Fang, A. V. Kroll and L. Zhang, Small, 2015, 11, 5483–5496 CrossRef CAS PubMed.
- M. S. Goldberg, Cell, 2015, 161, 201–204 CrossRef CAS PubMed.
- D. J. Irvine, M. C. Hanson, K. Rakhra and T. Tokatlian, Chem. Rev., 2015, 115, 11109–11146 CrossRef CAS PubMed.
- J. Kim, W. A. Li, Y. Choi, S. A. Lewin, C. S. Verbeke, G. Dranoff and D. J. Mooney, Nat. Biotechnol., 2015, 33, 64–72 CrossRef CAS PubMed.
- K. Shao, S. Singha, X. Clemente-Casares, S. Tsai, Y. Yang and P. Santamaria, ACS Nano, 2015, 9, 16–30 CrossRef CAS PubMed.
- D. M. Smith, J. K. Simon and J. R. Baker Jr, Nat. Rev. Immunol., 2013, 13, 592–605 CrossRef CAS PubMed.
- Y. Min, K. C. Roche, S. Tian, M. J. Eblan, K. P. McKinnon, J. M. Caster, S. Chai, L. E. Herring, L. Zhang, T. Zhang, J. M. DeSimone, J. E. Tepper, B. G. Vincent, J. S. Serody and A. Z. Wang, Nat. Nanotechnol., 2017, 12, 877–882 CrossRef CAS PubMed.
- M. L. Broz, M. Binnewies, B. Boldajipour, A. E. Nelson, J. L. Pollack, D. J. Erle, A. Barczak, M. D. Rosenblum, A. Daud, D. L. Barber, S. Amigorena, L. J. van’t Veer, A. I. Sperling, D. M. Wolf and M. F. Krummel, Cancer Cell, 2014, 26, 638–652 CrossRef CAS PubMed.
- S. LeibundGut-Landmann, J.-M. Waldburger, C. R. e Sousa, H. Acha-Orbea and W. Reith, Nat. Immunol., 2004, 5, 899–908 CrossRef CAS PubMed.
- R. A. Franklin, W. Liao, A. Sarkar, M. V. Kim, M. R. Bivona, K. Liu, E. G. Pamer and M. O. Li, Science, 2014, 344, 921–925 CrossRef CAS PubMed.
- K. D. Moynihan and D. J. Irvine, Cancer Res., 2017, 77, 5215–5221 CrossRef CAS PubMed.
- F. Veglia, M. Perego and D. Gabrilovich, Nat. Immunol., 2018, 19, 108–119 CrossRef CAS PubMed.
- V. Cortez-Retamozo, M. Etzrodt, A. Newton, P. J. Rauch, A. Chudnovskiy, C. Berger, R. J. H. Ryan, Y. Iwamoto, B. Marinelli, R. Gorbatov, R. Forghani, T. I. Novobrantseva, V. Koteliansky, J.-L. Figueiredo, J. W. Chen, D. G. Anderson, M. Nahrendorf, F. K. Swirski, R. Weissleder and M. J. Pittet, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2491–2496 CrossRef CAS PubMed.
- T. N. Mayadas, X. Cullere and C. A. Lowell, Annu. Rev. Pathol.: Mech. Dis., 2014, 9, 181–218 CrossRef CAS PubMed.
- C. Nathan, Nat. Rev. Immunol., 2006, 6, 173–182 CrossRef CAS PubMed.
- T. Engeman, A. V. Gorbachev, D. D. Kish and R. L. Fairchild, J. Leukoc. Biol., 2004, 76, 941–949 CrossRef CAS PubMed.
- A. Mantovani, M. A. Cassatella, C. Costantini and S. Jaillon, Nat. Rev. Immunol., 2011, 11, 519–531 CrossRef CAS PubMed.
- M. Vono, A. Lin, A. Norrby-Teglund, R. A. Koup, F. Liang and K. Loré, Blood, 2017, 129, 1991–2001 CrossRef PubMed.
- N. Guo, K. Ni, T. Luo, G. Lan, A. Arina, Z. Xu, J. Mao, R. R. Weichselbaum, M. Spiotto and W. Lin, ACS Nano, 2021, 15, 17515–17527 CrossRef CAS PubMed.
- L. Deng, H. Liang, B. Burnette, M. Beckett, T. Darga, R. R. Weichselbaum and Y. X. Fu, J. Clin. Invest., 2014, 124, 687–695 CrossRef CAS PubMed.
- E. Lauret Marie Joseph, A. Kirilovsky, B. Lecoester, C. El Sissy, L. Boullerot, L. Rangan, A. Marguier, F. Tochet, M. Dosset, J. Boustani, P. Ravel, R. Boidot, L. Spehner, N. Haicheur-Adjouri, F. Marliot, J. R. Pallandre, F. Bonnefoy, V. Scripcariu, M. Van den Eynde, E. Cornillot, C. Mirjolet, F. Pages and O. Adotevi, J. Immunother. Cancer, 2021, 9, 687–695 CrossRef.
- S. J. Dovedi, A. L. Adlard, G. Lipowska-Bhalla, C. McKenna, S. Jones, E. J. Cheadle, I. J. Stratford, E. Poon, M. Morrow, R. Stewart, H. Jones, R. W. Wilkinson, J. Honeychurch and T. M. Illidge, Cancer Res., 2014, 74, 5458–5468 CrossRef CAS PubMed.
- C. Twyman-Saint Victor, A. J. Rech, A. Maity, R. Rengan, K. E. Pauken, E. Stelekati, J. L. Benci, B. Xu, H. Dada, P. M. Odorizzi, R. S. Herati, K. D. Mansfield, D. Patsch, R. K. Amaravadi, L. M. Schuchter, H. Ishwaran, R. Mick, D. A. Pryma, X. Xu, M. D. Feldman, T. C. Gangadhar, S. M. Hahn, E. J. Wherry, R. H. Vonderheide and A. J. Minn, Nature, 2015, 520, 373–377 CrossRef CAS PubMed.
- D. Chen, H. B. Barsoumian, G. Fischer, L. Yang, V. Verma, A. I. Younes, Y. Hu, F. Masropour, K. Klein, C. Vellano, J. Marszalek, M. Davies, M. A. Cortez and J. Welsh, J. Immunother. Cancer, 2020, 8, e000289 CrossRef PubMed.
- J. Zeng, A. P. See, J. Phallen, C. M. Jackson, Z. Belcaid, J. Ruzevick, N. Durham, C. Meyer, T. J. Harris, E. Albesiano, G. Pradilla, E. Ford, J. Wong, H. J. Hammers, D. Mathios, B. Tyler, H. Brem, P. T. Tran, D. Pardoll, C. G. Drake and M. Lim, Int. J. Radiat. Oncol., Biol., Phys., 2013, 86, 343–349 CrossRef CAS PubMed.
- W. Sang, L. Xie, G. Wang, J. Li, Z. Zhang, B. Li, S. Guo, C. X. Deng and Y. Dai, Adv. Sci., 2021, 8, 2003338 CrossRef CAS PubMed.
- Y. Wang, N. Shen, Y. Wang, M. Li, W. Zhang, L. Fan, L. Liu, Z. Tang and X. Chen, Biomater. Sci., 2021, 9, 3019–3027 RSC.
- K. Lu, C. He, N. Guo, C. Chan, K. Ni, G. Lan, H. Tang, C. Pelizzari, Y. X. Fu, M. T. Spiotto, R. R. Weichselbaum and W. Lin, Nat. Biomed. Eng., 2018, 2, 600–610 CrossRef CAS PubMed.
- T. Seremet, F. Brasseur and P. G. Coulie, Cancer J., 2011, 17, 325–330 CrossRef CAS PubMed.
- F. R. Vogel, Ann. N. Y. Acad. Sci., 1995, 754, 153–160 CrossRef CAS PubMed.
- P. Aiyer-Harini, H. Ashok-Kumar, G. P. Kumar and N. Shivakumar, J. Vaccines Vaccination, 2013, 4, 1000167 Search PubMed.
- R. L. Coffman, A. Sher and R. A. Seder, Immunity, 2010, 33, 492–503 CrossRef CAS PubMed.
- Y. Yoshizaki, E. Yuba, N. Sakaguchi, K. Koiwai, A. Harada and K. Kono, Biomaterials, 2017, 141, 272–283 CrossRef CAS PubMed.
- A. Banstola, J.-H. Jeong and S. Yook, Acta Biomater., 2020, 114, 16–30 CrossRef CAS PubMed.
- M. Li, Z. Wang, X. Liu, N. Song, Y. Song, X. Shi, J. Liu, J. Liu and Z. Yu, J. Controlled Release, 2021, 340, 35–47 CrossRef CAS PubMed.
- F. Castro, M. L. Pinto, C. L. Pereira, K. Serre, M. A. Barbosa, K. Vermaelen, F. Gärtner, R. M. Gonçalves, O. De Wever and M. J. Oliveira, Biomaterials, 2020, 257, 120218 CrossRef CAS PubMed.
- J. Wei, D. Wu, S. Zhao, Y. Shao, Y. Xia, D. Ni, X. Qiu, J. Zhang, J. Chen, F. Meng and Z. Zhong, Adv. Sci., 2022, 9, 2103689 CrossRef CAS PubMed.
- L. Sun, F. Shen, L. Tian, H. Tao, Z. Xiong, J. Xu and Z. Liu, Adv. Mater., 2021, 33, e2007910 CrossRef PubMed.
- R. B. Patel, M. Ye, P. M. Carlson, A. Jaquish, L. Zangl, B. Ma, Y. Wang, I. Arthur, R. Xie, R. J. Brown, X. Wang, R. Sriramaneni, K. Kim, S. Gong and Z. S. Morris, Adv. Mater., 2019, 31, 1902626 CrossRef CAS PubMed.
- M. Luo, Z. Liu, X. Zhang, C. Han, L. Z. Samandi, C. Dong, B. D. Sumer, J. Lea, Y.-X. Fu and J. Gao, J. Controlled Release, 2019, 300, 154–160 CrossRef CAS PubMed.
- M. Luo, H. Wang, Z. Wang, H. Cai, Z. Lu, Y. Li, M. Du, G. Huang, C. Wang, X. Chen, M. R. Porembka, J. Lea, A. E. Frankel, Y.-X. Fu, Z. J. Chen and J. Gao, Nat. Nanotechnol., 2017, 12, 648–654 CrossRef CAS PubMed.
- E. Albini, A. Coletti, F. Greco, M. T. Pallotta, G. Mondanelli, M. Gargaro, M. L. Belladonna, C. Volpi, R. Bianchi, U. Grohmann, A. Macchiarulo and C. Orabona, Biochem. Pharmacol., 2018, 158, 286–297 CrossRef CAS PubMed.
- J. B. Katz, A. J. Muller and G. C. Prendergast, Immunol. Rev., 2008, 222, 206–221 CrossRef CAS PubMed.
- A. L. Mellor and D. H. Munn, Nat. Rev. Immunol., 2004, 4, 762–774 CrossRef CAS PubMed.
- D. H. Munn, M. Zhou, J. T. Attwood, I. Bondarev, S. J. Conway, B. Marshall, C. Brown and A. L. Mellor, Science, 1998, 281, 1191–1193 CrossRef CAS PubMed.
- G. C. Prendergast, A. Mondal, S. Dey, L. D. Laury-Kleintop and A. J. Muller, Trends Cancer, 2018, 4, 38–58 CrossRef CAS PubMed.
- T. Miyazaki, K. Moritake, K. Yamada, N. Hara, H. Osago, T. Shibata, Y. Akiyama and M. Tsuchiya, J. Neurosurg., 2009, 111, 230–237 CAS.
- T. Singh and R. K. Goel, Eur. J. Pharmacol., 2016, 784, 111–120 CrossRef CAS PubMed.
- X. Dong, R. Cheng, S. Zhu, H. Liu, R. Zhou, C. Zhang, K. Chen, L. Mei, C. Wang, C. Su, X. Liu, Z. Gu and Y. Zhao, ACS Nano, 2020, 14, 5400–5416 CrossRef CAS PubMed.
- W. Huang, L. He, Z. Zhang, S. Shi and T. Chen, ACS Nano, 2021, 15, 20225–20241 CrossRef CAS PubMed.
- X. Zhou, Z. Meng, J. She, Y. Zhang, X. Yi, H. Zhou, J. Zhong, Z. Dong, X. Han, M. Chen, Q. Fan, K. Yang and C. Wang, Nano-Micro Lett., 2020, 12, 100 CrossRef CAS PubMed.
- Q. Zhang, X. Guo, Y. Cheng, L. Chudal, N. K. Pandey, J. Zhang, L. Ma, Q. Xi, G. Yang, Y. Chen, X. Ran, C. Wang, J. Zhao, Y. Li, L. Liu, Z. Yao, W. Chen, Y. Ran and R. Zhang, Signal Transduction Targeted Ther., 2020, 5, 58 CrossRef CAS PubMed.
- Y. Wang, Y. Ding, D. Yao, H. Dong, C. Ji, J. Wu, Y. Hu and A. Yuan, Small, 2021, 17, e2006231 CrossRef PubMed.
- C. Wang, Z. Dong, Y. Hao, Y. Zhu, J. Ni, Q. Li, B. Liu, Y. Han, Z. Yang, J. Wan, K. Yang, Z. Liu and L. Feng, Adv. Mater., 2021, 34, 2106520 CrossRef PubMed.
- A. Shirazi, G. Ghobadi and M. Ghazi-Khansari, J. Radiat. Res., 2007, 48, 263–272 CrossRef CAS PubMed.
- E. J. Moding, M. B. Kastan and D. G. Kirsch, Nat. Rev. Drug Discovery, 2013, 12, 526–542 CrossRef CAS PubMed.
- G.-I. Mun, S. Kim, E. Choi, C. S. Kim and Y.-S. Lee, Arch. Pharm. Res., 2018, 41, 1033–1050 CrossRef CAS PubMed.
- C. K. K. Nair, D. K. Parida and T. Nomura, J. Radiat. Res., 2001, 42, 21–37 CrossRef CAS PubMed.
- D. Citrin, A. P. Cotrim, F. Hyodo, B. J. Baum, M. C. Krishna and J. B. Mitchell, Oncologist, 2010, 15, 360–371 CrossRef PubMed.
- E. González, M. P. Cruces, E. Pimentel and P. Sánchez, Environ. Toxicol. Pharmacol., 2018, 62, 210–214 CrossRef PubMed.
- F. Tabeie, S. M. Tabatabaei, A. Mahmoud-Pashazadeh and M. Assadi, J. Clin. Diagn. Res., 2017, 11, TC08 CAS.
- T. K. Mandal, L. A. Bostanian, R. A. Graves, S. R. Chapman and I. Womack, Drug Dev. Ind. Pharm., 2002, 28, 339–344 CrossRef CAS PubMed.
- I. Ahmad, M. Anwar, S. Akhter, P. Thakur, R. Chawla, R. K. Sharma, A. Ali and F. J. Ahmad, J. Pharm. Innov., 2016, 11, 308–322 CrossRef.
- S. J. Hosseinimehr, Drug Discovery Today, 2007, 12, 794–805 CrossRef CAS PubMed.
- M. Cheki, E. Mihandoost, A. Shirazi and A. Mahmoudzadeh, J. Cancer Res. Ther., 2016, 12, 1234–1242 CrossRef CAS PubMed.
- C. Wang, J. Xie, X. Dong, L. Mei, M. Zhao, Z. Leng, H. Hu, L. Li, Z. Gu and Y. Zhao, Small, 2020, 16, e1906915 CrossRef PubMed.
- J.-Y. Wang, X. Mu, Y. Li, F. Xu, W. Long, J. Yang, P. Bian, J. Chen, L. Ouyang, H. Liu, Y. Jing, J. Wang, L. Liu, H. Dai, Y. Sun, C. Liu and X.-D. Zhang, Small, 2018, 14, 1703736 CrossRef PubMed.
- S. I. Han, S. W. Lee, M. G. Cho, J. M. Yoo, M. H. Oh, B. Jeong, D. Kim, O. K. Park, J. Kim, E. Namkoong, J. Jo, N. Lee, C. Lim, M. Soh, Y. E. Sung, J. Yoo, K. Park and T. Hyeon, Adv. Mater., 2020, 32, e2001566 CrossRef PubMed.
- X. D. Zhang, J. Zhang, J. Wang, J. Yang, J. Chen, X. Shen, J. Deng, D. Deng, W. Long, Y. M. Sun, C. Liu and M. Li, ACS Nano, 2016, 10, 4511–4519 CrossRef CAS PubMed.
- X. Ren, M. Huo, M. Wang, H. Lin, X. Zhang, J. Yin, Y. Chen and H. Chen, ACS Nano, 2019, 13, 6438–6454 CrossRef CAS PubMed.
- J. Xie, Y. Yong, X. Dong, J. Du, Z. Guo, L. Gong, S. Zhu, G. Tian, S. Yu, Z. Gu and Y. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 14281–14291 CrossRef CAS PubMed.
- T. Liu, Q. Yang, H. Zheng, H. Jia, Y. He, X. Zhang, J. Zheng, Y. Xi, H. Zhang, R. Sun, X. Chen and W. Shan, Biomaterials, 2021, 277, 121103 CrossRef CAS PubMed.
- J. Cao, X. Peng, H. Li, L. Ren, T. Xu, K. Sun, Y. Zhang and D. Li, Mater. Sci. Eng., C, 2021, 129, 112369 CrossRef CAS PubMed.
- A. Kim, C. Yonemoto, C. P. Feliciano, B. Shashni and Y. Nagasaki, Small, 2021, 17, e2008210 CrossRef PubMed.
- N. M. Molino and S.-W. Wang, Curr. Opin. Biotechnol., 2014, 28, 75–82 CrossRef CAS PubMed.
- C. F. Richardson, D. I. Schuster and S. R. Wilson, Org. Lett., 2000, 2, 1011–1014 CrossRef CAS PubMed.
- G. Seifert, K. Vietze and R. Schmidt, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, 5183–5192 CrossRef CAS.
- Y. Ueno and S. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 085403 CrossRef.
- J. Grebowski, A. Krokosz, A. Konarska, M. Wolszczak and M. Puchala, Radiat. Phys. Chem., 2014, 103, 146–152 CrossRef CAS.
- S. S. Ali, J. I. Hardt, K. L. Quick, J. Sook Kim-Han, B. F. Erlanger, T.-t Huang, C. J. Epstein and L. L. Dugan, Free Radical Biol. Med., 2004, 37, 1191–1202 CrossRef CAS PubMed.
- P. J. Krusic, E. Wasserman, P. N. Keizer, J. R. Morton and K. F. Preston, Science, 1991, 254, 1183–1185 CrossRef CAS PubMed.
- X. Cheng, X. Ni, R. Wu, Y. Chong, X. Gao, C. Ge and J. J. Yin, Nanomedicine, 2018, 13, 733–747 CrossRef CAS PubMed.
- H. Ma, J. Zhao, H. Meng, D. Hu, Y. Zhou, X. Zhang, C. Wang, J. Li, J. Yuan and Y. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 16104–16113 CrossRef CAS PubMed.
- J. Tong, M. C. Zimmerman, S. Li, X. Yi, R. Luxenhofer, R. Jordan and A. V. Kabanov, Biomaterials, 2011, 32, 3654–3665 CrossRef CAS PubMed.
- J.-J. Yin, F. Lao, P. P. Fu, W. G. Wamer, Y. Zhao, P. C. Wang, Y. Qiu, B. Sun, G. Xing, J. Dong, X.-J. Liang and C. Chen, Biomaterials, 2009, 30, 611–621 CrossRef CAS PubMed.
- A. Djordjevic, B. Srdjenovic, M. Seke, D. Petrovic, R. Injac and J. Mrdjanovic, J. Nanomater., 2015, 2015, 567073 Search PubMed.
- S.-E. Zhu, F. Li and G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7535–7570 RSC.
- J. Vesna, J. Danica, K. Kamil, D.-S. Viktorija, D. Silva, T. Sanja, B. Ivana, S. Zoran, M. Zoran, B. Dubravko and D. Aleksandar, J. Appl. Biomed., 2016, 14, 285–297 CrossRef.
- Q. Zhao, Y. Li, J. Xu, R. Liu and W. Li, Int. J. Radiat. Biol., 2005, 81, 169–175 CrossRef CAS PubMed.
- S. Trajković, S. Dobrić, V. Jaćević, V. Dragojević-Simić, Z. Milovanović and A. Dordević, Colloids Surf., B, 2007, 58, 39–43 CrossRef PubMed.
- M. Singh, A. Alavi, R. Wong and S. Akita, Am. J. Clin. Dermatol., 2016, 17, 277–292 CrossRef PubMed.
- M. Zhao, C. Wang, J. Xie, C. Ji and Z. Gu, Small, 2021, 17, e2102035 CrossRef PubMed.
- L. Zhao, Y. Wang and Y. Li, Nanomaterials, 2019, 9, 1708 CrossRef CAS PubMed.
- W. Xia, H. Xue, J. Wang, T. Wang, L. Song, H. Guo, X. Fan, H. Gong and J. He, Carbon, 2016, 101, 315–323 CrossRef CAS.
- E. Kolanthai, S. Bose, K. S. Bhagyashree, S. V. Bhat, K. Asokan, D. Kanjilal and K. Chatterjee, Phys. Chem. Chem. Phys., 2015, 17, 22900–22910 RSC.
- M. Mahmudzadeh, H. Yari, B. Ramezanzadeh and M. Mahdavian, J. Hazard. Mater., 2019, 371, 609–624 CrossRef CAS PubMed.
- H. Cho, M. R. Jones, S. C. Nguyen, M. R. Hauwiller, A. Zettl and A. P. Alivisatos, Nano Lett., 2017, 17, 414–420 CrossRef CAS PubMed.
- C. Zhang, S. Chen, P. J. J. Alvarez and W. Chen, Carbon, 2015, 94, 531–538 CrossRef CAS.
- L. Nilewski, K. Mendoza, A. S. Jalilov, V. Berka, G. Wu, W. K. A. Sikkema, A. Metzger, R. Ye, R. Zhang, D. X. Luong, T. Wang, E. McHugh, P. J. Derry, E. L. Samuel, T. A. Kent, A.-L. Tsai and J. M. Tour, ACS Appl. Mater. Interfaces, 2019, 11, 16815–16821 CrossRef CAS PubMed.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS PubMed.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- M. Gong, D.-Y. Wang, C.-C. Chen, B.-J. Hwang and H. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- J. Wang, X. Cui, H. Li, J. Xiao, J. Yang, X. Mu, H. Liu, Y.-M. Sun, X. Xue, C. Liu, X.-D. Zhang, D. Deng and X. Bao, Nano Res., 2018, 11, 2821–2835 CrossRef CAS.
- S. Wang, L. Yi, J. E. Halpert, X. Lai, Y. Liu, H. Cao, R. Yu, D. Wang and Y. Li, Small, 2012, 8, 265–271 CrossRef CAS PubMed.
- J. Li, J. Xu, Z. Xie, X. Gao, J. Zhou, Y. Xiong, C. Chen, J. Zhang and Z. Liu, Adv. Mater., 2018, 30, 1800548 CrossRef PubMed.
- Z. Jin, M. Yuan, H. Li, H. Yang, Q. Zhou, H. Liu, X. Lan, M. Liu, J. Wang, E. H. Sargent and Y. Li, Adv. Funct. Mater., 2016, 26, 5284–5289 CrossRef CAS.
- C. Kuang, G. Tang, T. Jiu, H. Yang, H. Liu, B. Li, W. Luo, X. Li, W. Zhang, F. Lu, J. Fang and Y. Li, Nano Lett., 2015, 15, 2756–2762 CrossRef CAS PubMed.
- H. Ren, H. Shao, L. Zhang, D. Guo, Q. Jin, R. Yu, L. Wang, Y. Li, Y. Wang, H. Zhao and D. Wang, Adv. Energy Mater., 2015, 5, 1500296 CrossRef.
- J. Xiao, J. Shi, H. Liu, Y. Xu, S. Lv, Y. Luo, D. Li, Q. Meng and Y. Li, Adv. Energy Mater., 2015, 5, 1401943 CrossRef.
- H. Du, H. Yang, C. Huang, J. He, H. Liu and Y. Li, Nano Energy, 2016, 22, 615–622 CrossRef CAS.
- C. Huang, S. Zhang, H. Liu, Y. Li, G. Cui and Y. Li, Nano Energy, 2015, 11, 481–489 CrossRef CAS.
- J. Xie, N. Wang, X. Dong, C. Wang, Z. Du, L. Mei, Y. Yong, C. Huang, Y. Li, Z. Gu and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 2579–2590 CrossRef CAS PubMed.
- P. Xie, S.-T. Yang, T. He, S. Yang and X.-H. Tang, Int. J. Mol. Sci., 2017, 18, 2562 CrossRef PubMed.
- E. M. Pridgen, F. Alexis and O. C. Farokhzad, Expert Opin. Drug Delivery, 2015, 12, 1459–1473 CrossRef PubMed.
- Y. Yang, L. Guo, Z. Wang, P. Liu, X. Liu, J. Ding and W. Zhou, Biomaterials, 2021, 264, 120390 CrossRef CAS PubMed.
- A. Watanabe, M. Kajita, J. Kim, A. Kanayama, K. Takahashi, T. Mashino and Y. Miyamoto, Nanotechnology, 2009, 20, 455105 CrossRef PubMed.
- T. Hamasaki, T. Kashiwagi, T. Imada, N. Nakamichi, S. Aramaki, K. Toh, S. Morisawa, H. Shimakoshi, Y. Hisaeda and S. Shirahata, Langmuir, 2008, 24, 7354–7364 CrossRef CAS PubMed.
- M. Kajita, K. Hikosaka, M. Iitsuka, A. Kanayama, N. Toshima and Y. Miyamoto, Free Radical Res., 2007, 41, 615–626 CrossRef CAS PubMed.
- W. Luo, C. Zhu, S. Su, D. Li, Y. He, Q. Huang and C. Fan, ACS Nano, 2010, 4, 7451–7458 CrossRef CAS PubMed.
- Y. Hu, H. Cheng, X. Zhao, J. Wu, F. Muhammad, S. Lin, J. He, L. Zhou, C. Zhang, Y. Deng, P. Wang, Z. Zhou, S. Nie and H. Wei, ACS Nano, 2017, 11, 5558–5566 CrossRef CAS PubMed.
- M. Cui, Y. Zhao, C. Wang and Q. Song, Mikrochim. Acta, 2017, 184, 3113–3119 CrossRef CAS.
- M. Cui, J. Zhou, Y. Zhao and Q. Song, Sens. Actuators, B, 2017, 243, 203–210 CrossRef CAS.
- D.-Y. Zhang, M. R. Younis, H. Liu, S. Lei, Y. Wan, J. Qu, J. Lin and P. Huang, Biomaterials, 2021, 271, 120706 CrossRef CAS PubMed.
- T. Wen, W. He, Y. Chong, Y. Liu, J.-J. Yin and X. Wu, Phys. Chem. Chem. Phys., 2015, 17, 24937–24943 RSC.
- Z. Jia, X. Yuan, J.-a Wei, X. Guo, Y. Gong, J. Li, H. Zhou, L. Zhang and J. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 49602–49613 CrossRef CAS PubMed.
- F. Xu, X. Mu, J. Wang, P. Bian, L. Liu, H. Liu, Y. Jing, W. Long, C. Liu and X. D. Zhang, J. Biomed. Nanotechnol., 2017, 13, 1512–1521 CrossRef CAS PubMed.
- W. Long, J. Wang, F. Xu, H. Wu, X. Mu, J. Wang, Y. Sun and X.-D. Zhang, Chin. Chem. Lett., 2020, 31, 269–274 CrossRef CAS.
- W. Long, X. Mu, J.-Y. Wang, F. Xu, J. Yang, J. Wang, S. Sun, J. Chen, Y.-M. Sun, H. Wang and X.-D. Zhang, Front. Chem., 2019, 7, 784 CrossRef CAS PubMed.
- D. K. Chandrasekharan and C. K. Nair, Cancer Biother. Radiopharm., 2012, 27, 642–651 CrossRef CAS PubMed.
- L. He, G. Huang, H. Liu, C. Sang, X. Liu and T. Chen, Sci. Adv., 2020, 6, eaay9751 CrossRef CAS PubMed.
- Q. Bao, P. Hu, Y. Xu, T. Cheng, C. Wei, L. Pan and J. Shi, ACS Nano, 2018, 12, 6794–6805 CrossRef CAS PubMed.
- K. E. Peloi, B. A. Ratti, C. V. Nakamura, C. J. Neal, T. S. Sakthivel, S. Singh, S. Seal and S. de Oliveira Silva Lautenschlager, J. Biomed. Mater. Res., 2021, 109, 2570–2579 CrossRef CAS PubMed.
- S. Wang, H. Zheng, L. Zhou, F. Cheng, Z. Liu, H. Zhang and Q. Zhang, Biomaterials, 2020, 260, 120314 CrossRef CAS PubMed.
- W. Sun, Y. Xu, Y. Yao, J. Yue, Z. Wu, H. Li, G. Shen, Y. Liao, H. Wang and W. Zhou, J. Nanobiotechnol., 2022, 20, 88 CrossRef CAS PubMed.
- C. Li, Z. Zhao, Y. Luo, T. Ning, P. Liu, Q. Chen, Y. Chu, Q. Guo, Y. Zhang, W. Zhou, H. Chen, Z. Zhou, Y. Wang, B. Su, H. You, T. Zhang, X. Li, H. Song, C. Li, T. Sun and C. Jiang, Adv. Sci., 2021, 8, e2101526 CrossRef PubMed.
- W. Hou, Y. Jiang, G. Xie, L. Zhao, F. Zhao, X. Zhang, S. K. Sun, C. Yu and J. Pan, Nanoscale, 2021, 13, 8531–8542 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- Z. Chen, J.-J. Yin, Y.-T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001–4012 CrossRef CAS PubMed.
- M. Soh, D. W. Kang, H. G. Jeong, D. Kim, D. Y. Kim, W. Yang, C. Song, S. Baik, I. Y. Choi, S. K. Ki, H. J. Kwon, T. Kim, C. K. Kim, S. H. Lee and T. Hyeon, Angew. Chem., Int. Ed., 2017, 56, 11399–11403 CrossRef CAS PubMed.
- A. Briggs, S. Corde, S. Oktaria, R. Brown, A. Rosenfeld, M. Lerch, K. Konstantinov and M. Tehei, Nanomedicine, 2013, 9, 1098–1105 CrossRef CAS PubMed.
- S. Das, C. J. Neal, J. Ortiz and S. Seal, Nanoscale, 2018, 10, 21069–21075 RSC.
- N. Abdi Goushbolagh, R. Abedi Firouzjah, K. Ebrahimnejad Gorji, M. Khosravanipour, S. Moradi, A. Banaei, A. Astani, M. Najafi, M. H. Zare and B. Farhood, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S1215–S1225 CrossRef CAS PubMed.
- A. Salvetti, G. Gambino, L. Rossi, D. De Pasquale, C. Pucci, S. Linsalata, A. Degl'Innocenti, S. Nitti, M. Prato, C. Ippolito and G. Ciofani, Mater. Sci. Eng., C, 2020, 115, 111113 CrossRef CAS PubMed.
- Z. Zal, A. Ghasemi, S. Azizi, H. Asgarian-Omran, A. Montazeri and S. J. Hosseinimehr, Curr. Radiopharm., 2018, 11, 109–115 CrossRef CAS PubMed.
- C. Metcalfe, Noelyn M. Kljavin, R. Ybarra and Frederic J. de Sauvage, Cell Stem Cell, 2014, 14, 149–159 CrossRef CAS PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- R. Lv, H. Terrones, A. L. Elías, N. Perea-López, H. R. Gutiérrez, E. Cruz-Silva, L. P. Rajukumar, M. S. Dresselhaus and M. Terrones, Nano Today, 2015, 10, 559–592 CrossRef CAS.
- H. Zhang, M. Chhowalla and Z. Liu, Chem. Soc. Rev., 2018, 47, 3015–3017 RSC.
- R. G. Dickinson and L. Pauling, J. Am. Chem. Soc., 1923, 45, 1466–1471 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- K. F. Mak and J. Shan, Nat. Photonics, 2016, 10, 216–226 CrossRef CAS.
- J. Xu, J. Zhang, W. Zhang and C.-S. Lee, Adv. Energy Mater., 2017, 7, 1700571 CrossRef.
- Q. Yun, L. Li, Z. Hu, Q. Lu, B. Chen and H. Zhang, Adv. Mater., 2020, 32, 1903826 CrossRef CAS PubMed.
- D. Er, H. Ye, N. C. Frey, H. Kumar, J. Lou and V. B. Shenoy, Nano Lett., 2018, 18, 3943–3949 CrossRef CAS PubMed.
- L. Lin, P. Sherrell, Y. Liu, W. Lei, S. Zhang, H. Zhang, G. G. Wallace and J. Chen, Adv. Energy Mater., 2020, 10, 1903870 CrossRef CAS.
- R. J. Toh, Z. Sofer and M. Pumera, J. Mater. Chem. A, 2016, 4, 18322–18334 RSC.
- R. Kumar, N. Goel, M. Hojamberdiev and M. Kumar, Sens. Actuators, A, 2020, 303, 111875 CrossRef CAS.
- E. Lee, Y. S. Yoon and D.-J. Kim, ACS Sens., 2018, 3, 2045–2060 CrossRef CAS PubMed.
- D. Sarkar, X. Xie, J. Kang, H. Zhang, W. Liu, J. Navarrete, M. Moskovits and K. Banerjee, Nano Lett., 2015, 15, 2852–2862 CrossRef CAS PubMed.
- A. Daus, S. Vaziri, V. Chen, Ç. Köroğlu, R. W. Grady, C. S. Bailey, H. R. Lee, K. Schauble, K. Brenner and E. Pop, Nat. Electron., 2021, 4, 495–501 CrossRef CAS.
- C. Gong, H. Zhang, W. Wang, L. Colombo, R. M. Wallace and K. Cho, Appl. Phys. Lett., 2013, 103, 053513 CrossRef.
- Y. Chen, Y. He, H. Xu, C. Du, X. Wu and G. Yang, Nano Today, 2022, 43, 101421 CrossRef CAS.
- S. Zhu, L. Gong, J. Xie, Z. Gu and Y. Zhao, Small Methods, 2017, 1, 1700220 CrossRef.
- X.-D. Zhang, J. Chen, Y. Min, G. B. Park, X. Shen, S.-S. Song, Y.-M. Sun, H. Wang, W. Long, J. Xie, K. Gao, L. Zhang, S. Fan, F. Fan and U. Jeong, Adv. Funct. Mater., 2014, 24, 1718–1729 CrossRef CAS.
- X. D. Zhang, Y. Jing, S. Song, J. Yang, J. Y. Wang, X. Xue, Y. Min, G. Park, X. Shen, Y. M. Sun and U. Jeong, Nanomedicine, 2017, 13, 1597–1605 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- W. Meng, X. Liu, H. Song, Y. Xie, X. Shi, M. Dargusch, Z.-G. Chen, Z. Tang and S. Lu, Nano Today, 2021, 40, 101273 CrossRef CAS.
- J. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. Liu and M. H. Rummeli, Chem. Soc. Rev., 2019, 48, 72–133 RSC.
- Y. A. J. Al-Hamadani, B.-M. Jun, M. Yoon, N. Taheri-Qazvini, S. A. Snyder, M. Jang, J. Heo and Y. Yoon, Chemosphere, 2020, 254, 126821 CrossRef CAS PubMed.
- X. Xie, C. Chen, N. Zhang, Z.-R. Tang, J. Jiang and Y.-J. Xu, Nat. Sustain, 2019, 2, 856–862 CrossRef.
- P. Salles, D. Pinto, K. Hantanasirisakul, K. Maleski, C. E. Shuck and Y. Gogotsi, Adv. Funct. Mater., 2019, 29, 1809223 CrossRef.
- S. J. Kim, H.-J. Koh, C. E. Ren, O. Kwon, K. Maleski, S.-Y. Cho, B. Anasori, C.-K. Kim, Y.-K. Choi, J. Kim, Y. Gogotsi and H.-T. Jung, ACS Nano, 2018, 12, 986–993 CrossRef CAS PubMed.
- S. Sun, M. Wang, X. Chang, Y. Jiang, D. Zhang, D. Wang, Y. Zhang and Y. Lei, Sens. Actuators, B, 2020, 304, 127274 CrossRef CAS.
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491–8494 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- B. Zhou, Y. Pu, H. Lin, W. Yue, H. Yin, Y. Yin, W. Ren, C. Zhao, Y. Chen and H. Xu, J. Mater. Chem. B, 2020, 8, 5257–5266 RSC.
- B. Zhou, H. Yin, C. Dong, L. Sun, W. Feng, Y. Pu, X. Han, X. Li, D. Du, H. Xu and Y. Chen, Adv. Sci., 2021, 8, 2101043 CrossRef CAS PubMed.
- K. Rasool, M. Helal, A. Ali, C. E. Ren, Y. Gogotsi and K. A. Mahmoud, ACS Nano, 2016, 10, 3674–3684 CrossRef CAS PubMed.
- M. A. Unal, F. Bayrakdar, L. Fusco, O. Besbinar, C. E. Shuck, S. Yalcin, M. T. Erken, A. Ozkul, C. Gurcan, O. Panatli, G. Y. Summak, C. Gokce, M. Orecchioni, A. Gazzi, F. Vitale, J. Somers, E. Demir, S. S. Yildiz, H. Nazir, J.-C. Grivel, D. Bedognetti, A. Crisanti, K. C. Akcali, Y. Gogotsi, L. G. Delogu and A. Yilmazer, Nano Today, 2021, 38, 101136 CrossRef CAS PubMed.
- F. Bu, M. M. Zagho, Y. Ibrahim, B. Ma, A. Elzatahry and D. Zhao, Nano Today, 2020, 30, 100803 CrossRef CAS.
- R. B. Rakhi, P. Nayak, C. Xia and H. N. Alshareef, Sci. Rep., 2016, 6, 36422 CrossRef CAS PubMed.
- S. Iravani and R. S. Varma, Mater. Adv., 2021, 2, 2906–2917 RSC.
- M. Caldas, A. C. Santos, F. Veiga, R. Rebelo, R. L. Reis and V. M. Correlo, Acta Biomater., 2020, 105, 26–43 CrossRef CAS PubMed.
- Y. Liu, K. Ai, X. Ji, D. Askhatova, R. Du, L. Lu and J. Shi, J. Am. Chem. Soc., 2017, 139, 856–862 CrossRef CAS PubMed.
- G. Zhong, X. Yang, X. Jiang, A. Kumar, H. Long, J. Xie, L. Zheng and J. Zhao, Nanoscale, 2019, 11, 11605–11616 RSC.
- M. M. Rageh, R. H. El-Gebaly, H. Abou-Shady and D. G. Amin, Mol. Cell. Biochem., 2015, 399, 59–69 CrossRef CAS PubMed.
- N. T. Le Na, S. Duc Loc, N. L. Minh Tri, N. T. Bich Loan, H. Anh Son, N. Linh Toan, H. Phuong Thu, H. T. My Nhung, N. Lai Thanh, N. T. Van Anh and N. Dinh Thang, Materials, 2019, 12, 1725 CrossRef CAS PubMed.
- M. M. Rageh and R. H. El-Gebaly, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2018, 828, 15–22 CrossRef CAS PubMed.
- W. Lohcharoenkal, L. Wang, Y. C. Chen and Y. Rojanasakul, Biomed Res. Int., 2014, 2014, 180549 Search PubMed.
- L. P. Herrera Estrada and J. A. Champion, Biomater. Sci., 2015, 3, 787–799 RSC.
- M. J. Hawkins, P. Soon-Shiong and N. Desai, Adv. Drug Delivery Rev., 2008, 60, 876–885 CrossRef CAS PubMed.
- M. Bellini, B. Riva, V. Tinelli, M. A. Rizzuto, L. Salvioni, M. Colombo, F. Mingozzi, A. Visioli, L. Marongiu, G. Frascotti, M. S. Christodoulou, D. Passarella, D. Prosperi and L. Fiandra, Small, 2020, 16, e2001450 CrossRef PubMed.
- K. Fan, X. Jia, M. Zhou, K. Wang, J. Conde, J. He, J. Tian and X. Yan, ACS Nano, 2018, 12, 4105–4115 CrossRef CAS PubMed.
- L. He, X. Lin, Y. Wang, C. Abraham, C. Sou, T. Ngo, Y. Zhang, I. A. Wilson and J. Zhu, Sci. Adv., 2021, 7, eabf1591 CrossRef PubMed.
- W. Wang, B. Huang, Y. Zhu, W. Tan and M. Zhu, Cell. Mol. Immunol., 2021, 18, 749–751 CrossRef CAS PubMed.
- J. Zhang, D. Cheng, J. He, J. Hong, C. Yuan and M. Liang, Nat. Protoc., 2021, 16, 4878–4896 CrossRef CAS PubMed.
- A. Haimovitz-Friedman, N. Balaban, M. McLoughlin, D. Ehleiter, J. Michaeli, I. Vlodavsky and Z. Fuks, Cancer Res., 1994, 54, 2591–2597 CAS.
- M. Li, A. Du, J. Xu, Y. Ma, H. Cao, C. Yang, X.-D. Yang, C.-G. Xing, M. Chen, W. Zhu, S. Zhang and J. Cao, Sci. Rep., 2016, 6, 30180 CrossRef CAS PubMed.
- Y. Shi, W. Wu, H. Qiao, L. Yue, L. Ren, S. Zhang, W. Yang and Z. Yang, Sci. Rep., 2016, 6, 26664 CrossRef CAS PubMed.
- W. Long, J. Wang, J. Yang, H. Wu, J. Wang, X. Mu, H. He, Q. Liu, Y. M. Sun, H. Wang and X. D. Zhang, J. Biomed. Nanotechnol., 2019, 15, 62–76 CrossRef CAS PubMed.
- B. Daroczi, G. Kari, M. F. McAleer, J. C. Wolf, U. Rodeck and A. P. Dicker, Clin. Cancer Res., 2006, 12, 7086–7091 CrossRef CAS PubMed.
- J. Guo, H. Yang, Y. Liu, W. Liu, R. Zhao, H. Li, W. Long, W. Xu, M. Guo and X. Zhang, J. Nanobiotechnol., 2021, 19, 377 CrossRef CAS PubMed.
- S. Lv, W. Long, J. Chen, Q. Ren, J. Wang, X. Mu, H. Liu, X. D. Zhang and R. Zhang, Nanoscale, 2020, 12, 548–557 RSC.
- H. Liu, J. Wang, Y. Jing, J. Yang, X. Bai, X. Mu, F. Xu, X. Xue, L. Liu, Y.-M. Sun, Q. Liu, H. Dai, C. Liu and X.-D. Zhang, Part. Part. Syst. Charact., 2017, 34, 1700035 CrossRef.
- P. Bian, J. Zhang, J. Wang, J. Yang, J. Wang, H. Liu, Y. Sun, M. Li and X.-D. Zhang, Sci. Bull., 2018, 63, 925–934 CrossRef CAS.
- R. S. Patwardhan, D. Sharma, R. Checker and S. K. Sandur, Free Radicals Biol. Med., 2014, 68, 52–64 CrossRef CAS PubMed.
- S. L. Schlichte, S. Romanova, K. Katsurada, E. A. Kosmacek, T. K. Bronich, K. P. Patel, R. E. Oberley-Deegan and M. C. Zimmerman, Redox. Biol., 2020, 36, 101610 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, Y. Li, F. Wang, F. Yang and W. Xu, Int. J. Mol. Sci., 2017, 18, 2233 CrossRef PubMed.
- U. Anand, P. Biswas, V. Kumar, D. Ray, P. Ray, V. I. P. Loake, R. Kandimalla, A. Chaudhary, B. Singh, N. K. Routhu, Z.-S. Chen, J. Proćków and A. Dey, Biomed. Pharmacother., 2022, 146, 112555 CrossRef CAS PubMed.
- C. N. Andreassen, C. Grau and J. C. Lindegaard, Semin. Radiat. Oncol., 2003, 13, 62–72 CrossRef PubMed.
- N. M. Gandhi, D. K. Maurya, V. Salvi, S. Kapoor, T. Mukherjee and C. K. K. Nair, J. Radiat. Res., 2004, 45, 461–468 CrossRef CAS PubMed.
- H.-J. Sim, G. Bhattarai, J. Lee, J.-C. Lee and S.-H. Kook, Aging Dis., 2019, 10, 1320–1327 CrossRef PubMed.
- J. Yi, J. Zhu, C. Zhao, Q. Kang, X. Zhang, K. Suo, N. Cao, L. Hao and J. Lu, Food Funct., 2021, 12, 5204–5218 RSC.
- S. Klein, L. V. R. Distel, W. Neuhuber and C. Kryschi, Nanomaterials, 2021, 11, 1167 CrossRef CAS PubMed.
- C. C. Perry, S. M. Urata, M. Lee, J. A. Aguilera and J. R. Milligan, Radiat. Environ. Biophys., 2012, 51, 457–468 CrossRef CAS PubMed.
- S. Klein, M. Smuda, C. Harreiß, C. Menter, L. V. R. Distel and C. Kryschi, ACS Appl. Mater. Interfaces, 2019, 11, 39613–39623 CrossRef CAS PubMed.
- J. Lin, M. Wang, H. Hu, X. Yang, B. Wen, Z. Wang, O. Jacobson, J. Song, G. Zhang, G. Niu, P. Huang and X. Chen, Adv. Mater., 2016, 28, 3273–3279 CrossRef CAS PubMed.
- G. Lin, P. Mi, C. Chu, J. Zhang and G. Liu, Adv. Sci., 2016, 3, 1600134 CrossRef PubMed.
- A. D. Schweitzer, E. Revskaya, P. Chu, V. Pazo, M. Friedman, J. D. Nosanchuk, S. Cahill, S. Frases, A. Casadevall and E. Dadachova, Int. J. Radiat. Oncol. Biol. Phys., 2010, 78, 1494–1502 CrossRef CAS PubMed.
- D. K. Chandrasekharan, P. K. Khanna and C. K. K. Nair, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2011, 723, 51–57 CrossRef CAS PubMed.
- D. K. Chandrasekharan and C. K. K. Nair, Int. J. Low Radiat., 2010, 7, 453–466 CrossRef CAS.
- D. K. Chandrasekharan and C. K. K. Nair, Cancer Biother. Radiopharm., 2012, 27, 642–651 CrossRef CAS PubMed.
- T. Ak and İ. Gülçin, Chem.-Biol. Interact., 2008, 174, 27–37 CrossRef CAS PubMed.
- Y. Cui, M. Zhang, F. Zeng, H. Jin, Q. Xu and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 32159–32169 CrossRef CAS PubMed.
- W.-H. Lee, C.-Y. Loo, P. M. Young, D. Traini, R. S. Mason and R. Rohanizadeh, Expert Opin. Drug Delivery, 2014, 11, 1183–1201 CrossRef CAS PubMed.
- K. Mansouri, S. Rasoulpoor, A. Daneshkhah, S. Abolfathi, N. Salari, M. Mohammadi, S. Rasoulpoor and S. Shabani, BMC Cancer, 2020, 20, 1–11 CrossRef PubMed.
- E. SchraufstÄTter and H. Bernt, Nature, 1949, 164, 456–457 CrossRef PubMed.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Drug Discovery Today, 2012, 17, 71–80 CrossRef CAS PubMed.
- S. Zorofchian Moghadamtousi, H. Abdul Kadir, P. Hassandarvish, H. Tajik, S. Abubakar and K. Zandi, Biomed Res. Int., 2014, 2014, 186864 Search PubMed.
- J. Trujillo, Y. I. Chirino, E. Molina-Jijón, A. C. Andérica-Romero, E. Tapia and J. Pedraza-Chaverrí, Redox. Biol., 2013, 1, 448–456 CrossRef CAS PubMed.
- V. Verma, World J. Clin. Oncol., 2016, 7, 275–283 CrossRef PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharm., 2007, 4, 807–818 CrossRef CAS PubMed.
- E. Babaei, M. Sadeghizadeh, Z. M. Hassan, M. A. H. Feizi, F. Najafi and S. M. Hashemi, Int. Immunopharmacol., 2012, 12, 226–234 CrossRef CAS PubMed.
- F. Akhtar, M. M. Rizvi and S. K. Kar, Biotechnol. Adv., 2012, 30, 310–320 CrossRef CAS PubMed.
- Y. Chen, C. Chen, X. Zhang, C. He, P. Zhao, M. Li, T. Fan, R. Yan, Y. Lu, R. J. Lee, M. W. Khan, M. Sarfraz, X. Ma, T. Yang and G. Xiang, Acta Pharm. Sin. B, 2020, 10, 1106–1121 CrossRef CAS PubMed.
- F. D. S. Feltrin, T. Agner, C. Sayer and L. M. F. Lona, Adv. Colloid Interface Sci., 2022, 300, 102582 CrossRef CAS PubMed.
- G. Jia, Y. Han, Y. An, Y. Ding, C. He, X. Wang and Q. Tang, Biomaterials, 2018, 178, 302–316 CrossRef CAS PubMed.
- M. M. Yallapu, S. Khan, D. M. Maher, M. C. Ebeling, V. Sundram, N. Chauhan, A. Ganju, S. Balakrishna, B. K. Gupta, N. Zafar, M. Jaggi and S. C. Chauhan, Biomaterials, 2014, 35, 8635–8648 CrossRef CAS PubMed.
- H. Kang, J. Gravier, K. Bao, H. Wada, J. H. Lee, Y. Baek, G. El Fakhri, S. Gioux, B. P. Rubin, J.-L. Coll and H. S. Choi, Adv. Mater., 2016, 28, 8162–8168 CrossRef CAS PubMed.
- Y. Peng, J. Bariwal, V. Kumar, C. Tan and R. I. Mahato, Adv. Ther., 2020, 3, 1900136 CrossRef.
- V. López-Dávila, A. M. Seifalian and M. Loizidou, Curr. Opin. Pharmacol., 2012, 12, 414–419 CrossRef PubMed.
- H. A. Joshi, R. S. Patwardhan, D. Sharma, S. K. Sandur and P. V. Devarajan, Int. J. Pharm., 2021, 595, 120181 CrossRef CAS PubMed.
- S. H. Choi, D. Y. Lee, S. Kang, M. K. Lee, J. H. Lee, S. H. Lee, H. L. Lee, H. Y. Lee and Y. I. Jeong, Int. J. Mol. Sci., 2021, 22, 6347 CrossRef CAS PubMed.
- X. Lin, Y. Shi, S. Yu, S. Li, W. Li, M. Li, S. Chen, Y. Wang and M. Cong, Front. Chem., 2020, 8, 212 CrossRef CAS PubMed.
- M. H. Nguyen, N. D. Pham, B. Dong, T. H. Nguyen, C. B. Bui and K. Hadinoto, Int. J. Radiat. Biol., 2017, 93, 1267–1273 CrossRef CAS PubMed.
- H. Nosrati, H. Danafar, H. Rezaeejam, N. Gholipour and M. Rahimi-Nasrabadi, Bioorg. Chem., 2020, 100, 103891 CrossRef CAS PubMed.
- Y. Liu, L. Miao, Y. Guo, R. Yuan, X. Li, X. Wang, X. Lin and H. Tian, ACS Biomater. Sci. Eng., 2021, 7, 2496–2507 CrossRef CAS PubMed.
- Y. Zhou, S. Hua, J. Yu, P. Dong, F. Liu and D. Hua, J. Mater. Chem. B, 2015, 3, 2931–2934 RSC.
- J. Kinoda, M. Ishihara, S. Nakamura, M. Fujita, K. Fukuda, Y. Sato and H. Yokoe, J. Radiat. Res., 2018, 59, 27–34 CrossRef CAS PubMed.
- S. Y. Wu, V. Parasuraman, T. Hsieh Chih, V. Arunagiri, S. Gunaseelan, H. Y. Chou, R. Anbazhagan, J. Y. Lai and N. R. Prasad, Int. J. Pharm., 2020, 579, 119161 CrossRef CAS PubMed.
- R. H. El-Gebaly, M. M. Rageh and I. K. Maamoun, J. X-Ray Sci. Technol., 2019, 27, 83–96 CAS.
- S. Arany, D. S. Benoit, S. Dewhurst and C. E. Ovitt, Mol. Ther., 2013, 21, 1182–1194 CrossRef CAS PubMed.
- S. Kumar, R. Meena and P. Rajamani, J. Agric. Food Chem., 2016, 64, 6024–6034 CrossRef CAS PubMed.
- Y. Zhang, L. Wang, M. Xu, T. Zhao, L. Kuang and D. Hua, Adv. Healthcare Mater., 2020, 9, e1901778 CrossRef PubMed.
- S. Pamujula, V. Kishore, B. Rider, K. C. Agrawal and T. K. Mandal, Int. J. Radiat. Biol., 2008, 84, 900–908 CrossRef CAS PubMed.
- S. G. Lee, T. M. Kalidindi, H. Lou, K. Gangangari, B. Punzalan, A. Bitton, C. J. Lee, H. A. Vargas, S. Park, L. Bodei, M. G. Kharas, V. K. Singh, N. V. Kishore Pillarsetty and S. M. Larson, J. Nucl. Med., 2021, 62, 584–590 CrossRef CAS PubMed.
- A. Dubois and R. I. Walker, Gastroenterology, 1988, 95, 500–507 CrossRef CAS.
- S. Saha, P. Bhanja, L. Liu, A. A. Alfieri, D. Yu, E. R. Kandimalla, S. Agrawal and C. Guha, PLoS One, 2012, 7, e29357 CrossRef CAS PubMed.
- Y. Lee, T. E. Deelman, K. Chen, D. S. Y. Lin, A. Tavakkoli and J. M. Karp, Nat. Mater., 2018, 17, 834–842 CrossRef CAS PubMed.
- F. Laffleur and A. Bernkop-Schnürch, Nanomedicine, 2013, 8, 2061–2075 CrossRef CAS PubMed.
- B. Hu, C. Jin, H.-B. Li, J. Tong, X. Ouyang, N. M. Cetinbas, S. Zhu, T. Strowig, F. C. Lam, C. Zhao, J. Henao-Mejia, O. Yilmaz, K. A. Fitzgerald, S. C. Eisenbarth, E. Elinav and R. A. Flavell, Science, 2016, 354, 765–768 CrossRef CAS PubMed.
- L. Zitvogel, O. Kepp, L. Galluzzi and G. Kroemer, Nat. Immunol., 2012, 13, 343–351 CrossRef CAS PubMed.
- W. Park, H. Shin, B. Choi, W.-K. Rhim, K. Na and D. Keun Han, Prog. Mater. Sci., 2020, 114, 100686 CrossRef CAS.
- L. Wang and Y. Yan, Macromol. Biosci., 2021, 21, 2100183 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-C. Liu, H. Sun and D.-S. Guo, Coord. Chem. Rev., 2019, 395, 46–62 CrossRef CAS.
- G. Chen, Y. Qian, H. Zhang, A. Ullah, X. He, Z. Zhou, H. Fenniri and J. Shen, Appl. Mater. Today, 2021, 23, 101003 CrossRef.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- X. Liu, T. Liang, R. Zhang, Q. Ding, S. Wu, C. Li, Y. Lin, Y. Ye, Z. Zhong and M. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 9643–9655 CrossRef CAS PubMed.
- R. F. Mendes, F. Figueira, J. P. Leite, L. Gales and F. A. Almeida Paz, Chem. Soc. Rev., 2020, 49, 9121–9153 RSC.
- L. B. Vong, T. Yoshitomi, H. Matsui and Y. Nagasaki, Biomaterials, 2015, 55, 54–63 CrossRef CAS PubMed.
- L. B. Vong, M. Kobayashi and Y. Nagasaki, Mol. Pharm., 2016, 13, 3091–3097 CrossRef CAS PubMed.
- C. P. Feliciano and Y. Nagasaki, ACS Biomater. Sci. Eng., 2019, 5, 5631–5636 CrossRef CAS PubMed.
- C. P. Feliciano, K. Tsuboi, K. Suzuki, H. Kimura and Y. Nagasaki, Biomaterials, 2017, 129, 68–82 CrossRef CAS PubMed.
- Y. Wang, H. Zhang, Y. Liu, M. H. Younis, W. Cai and W. Bu, Mater. Today, 2022, 57, 262–278 CrossRef CAS.
- S. Chin, C. L. Eccles, A. McWilliam, R. Chuter, E. Walker, P. Whitehurst, J. Berresford, M. Van Herk, P. J. Hoskin and A. Choudhury, J. Med. Imaging Radiat. Oncol., 2020, 64, 163–177 CrossRef PubMed.
- S. Corradini, F. Alongi, N. Andratschke, C. Belka, L. Boldrini, F. Cellini, J. Debus, M. Guckenberger, J. Hörner-Rieber, F. J. Lagerwaard, R. Mazzola, M. A. Palacios, M. E. P. Philippens, C. P. J. Raaijmakers, C. H. J. Terhaard, V. Valentini and M. Niyazi, Radiat. Oncol., 2019, 14, 92 CrossRef CAS PubMed.
- S. M. Bentzen and V. Gregoire, Semin. Radiat. Oncol., 2011, 21, 101–110 CrossRef PubMed.
- D. Thorwarth, Br. J. Radiol., 2015, 88, 20150056 CrossRef CAS PubMed.
- C. Zhang and K. Pu, Chem. Soc. Rev., 2020, 49, 4234–4253 RSC.
- P. M. Calabro-Jones, R. C. Fahey, G. D. Smoluk and J. F. Ward, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1985, 47, 23–27 CrossRef CAS PubMed.
- J. M. Yuhas, Cancer Res., 1980, 40, 1519–1524 CAS.
| This journal is © The Royal Society of Chemistry 2022 |