
Enabling rapid pseudocapacitive multi-electron reactions by heterostructure engineering of vanadium oxide for high-energy and high-power lithium storage†
Feng
Su
ab,
Feifei
Xing
ab,
Xiao
Wang
a,
Fangyan
Liu
a,
Liangzhu
Zhang
a and
Zhong-Shuai
Wu
 *ac
*ac
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: wuzs@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, 19 A Yuquan Road, Shijingshan District, Beijing 100049, China
cDalian National Laboratory for Clean Energy, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China
First published on 12th November 2022
Abstract
Charge storage reactions with multi-electron transfer represent an effective approach to obtaining higher energy density. V2O5 is a potential multi-electron reaction material, but suffers from irreversible phase transformation and sluggish kinetics upon deep discharge. Herein, we report a rational strategy of constructing a two-dimensional heterostructure of V2O5 and graphene for realizing reversible and fast multi-electron reactions. The ultrathin hybrid structure with abundant heterointerfaces leads to the reversible structure transition of V2O5 and facilitates ion/electron transport and interfacial charge transfer, thus enabling high-rate multi-electron transfer lithium storage with a significant pseudocapacitive contribution. The heterostructure delivers a high capacity of 361 mA h g−1 at 1C and retains 175 mA h g−1 at an ultrahigh rate of 100C, outperforming most intercalation metal oxides. Furthermore, through decoupling such a multi-electron reaction with high capacity and a wide potential window, a symmetric full cell with prelithiated V2O5/graphene serving as both the anode and the cathode is constructed, showing superb energy/power performance and cyclability up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. This work suggests that creating a heterostructure is a reliable strategy for achieving high-rate multi-electron reactions in redox-active electrode materials.
000 cycles. This work suggests that creating a heterostructure is a reliable strategy for achieving high-rate multi-electron reactions in redox-active electrode materials.
Broader contextThe pursuit of high-performance electrochemical energy storage devices has highlighted the need for innovative electrode materials with both high capacity and rate capability. The theoretical capacity of an electrode material closely correlates to the number of electrons transferred in each redox center. To this end, the concept of multi-electron reactions has been developed to break through the bottleneck of one-electron transfer in conventional battery chemistry, but remains challenging due to the increased complexity in both thermodynamics and kinetics for multi-electron transfer processes. In this work, we report a rational strategy of constructing a two-dimensional heterostructure of V2O5 and graphene to overcome the limitations of reversibility and kinetics for multi-electron reactions by virtue of its ultrathin nanosheet morphology and abundant heterogeneous interfaces, enabling pseudocapacitive multi-electron transfer lithium storage with both high capacity and rate capability, which are among the best of the reported results for intercalation metal oxides. Moreover, to explore the application of multi-electron reactions, we present a new paradigm of decoupling the full reaction into two regions with equivalent capacities to construct symmetric cells, which exhibit superb energy/power performance and ultralong cycle life. This work underscores the potential importance of creating a heterostructure as a strategy for achieving high-rate multi-electron transfer charge storage in redox-active materials. |
Introduction
Increasing energy supply challenges have intensified the pursuit of innovative electrochemical energy storage technologies with higher energy density, higher power density and other new performance benchmarks.1–4 The concept of multi-electron reactions that refers to the transfer of more than one electron (per redox center) during the charge storage process has been developed to break through the bottleneck of low specific capacity of electrode materials in conventional battery chemistry, which is critical for realizing high energy density but faces the challenges of reaction reversibility and kinetics.5–7 In principle, multi-electron redox electrode materials can be classified into intercalation, conversion and alloying types, among which intercalation metal oxides have been underscored for the potential advantages in terms of structural stability and reversibility.8,9Vanadium oxide (V2O5) is a feasible candidate to achieve multi-electron transfer processes due to the rich redox chemistry of the vanadium element.10,11 However, irreversible phase transition into a rock-salt structure (ω-phase) occurs when more than two lithium ions are inserted into bulk orthorhombic V2O5, which would cause structural collapse and fast capacity fading.12 In contrast, nanostructured xerogels or aerogels derived from bilayered V2O5 can reversibly accommodate/release multiple lithium ions, even 3 Li per unit formula corresponding to a theoretical capacity of 442 mA h g−1.13–15 However, the rate performance of V2O5 xerogels/aerogels is still unsatisfactory due to their low intrinsic electrical conductivity and slow lithium-ion diffusion. To overcome the above limitations, a well-designed morphology and structural engineering are required to improve the reaction reversibility and kinetics. Recently, studies have shown that two-dimensional (2D) nanosheets with a smaller thickness could exhibit enhanced charge storage ability (that is, increased reversibility and capacity) relating to multi-electron transfer processes.16–18 Moreover, coupling two different nanosheets to form heterostructures could generate heterogeneous interfaces with a built-in electric field, thus accelerating interfacial charge transfer.19–21 More importantly, with substantially improved electron and ion transport by rational nanostructuring, the electrochemical signature of a battery material can be transformed into a pseudocapacitive one, which is free from solid-state diffusion limitation and exhibits rapid surface-controlled kinetics.22,23 In particular, a pseudocapacitive reaction with greater than one-electron redox is extremely attractive due to its ability to achieve high capacity and rate capability simultaneously, which, however, has been rarely reported and remains highly challenging owing to the increased complexity in both thermodynamics and kinetics.23
Symmetric batteries with two identical electrodes serving as the cathode and the anode show unique advantages including a simplified fabrication process, lower manufacturing cost and buffered electrode volume change.24,25 Also, the same electrode material can facilitate electrode kinetics balance for enhancing the electrochemical performance of the full cell.26,27 However, the development of symmetric cells has been limited due to the lack of suitable electrode materials. In general, symmetric cells are based on certain bipolar materials with two distinct potential plateaus.28,29 However, these battery materials usually exhibit sluggish kinetics, limiting the power performance of symmetric cells. Alternatively, pseudocapacitive multi-electron reaction materials that display linear galvanostatic charge/discharge (GCD) profiles with high capacity and a wide potential window could offer a different paradigm (Fig. S1, ESI†). By decoupling the full charge storage reaction into a positive region and a negative region with equivalent capacities and appropriate potential ranges, a symmetric cell that benefits from the rapid pseudocapacitive kinetics could be constructed using the pseudocapacitive multi-electron reaction material as both the cathode and the anode.30,31
In this work, we rationally designed and synthesized a 2D heterostructure of bilayered V2O5 and graphene (V2O5/graphene) to realize reversible and fast multi-electron reactions for high-energy and high-power lithium-ion storage. The ultrathin nanosheet morphology can shorten ion diffusion paths and enable facile strain relaxation upon lithium-ion insertion/extraction, thus improving the reaction kinetics and facilitating fast reversible structural transformation. Meanwhile, hybridization with graphene not only enhances electronic conductivity, but also generates abundant heterogeneous interfaces with boosted charge transfer, as revealed by the electrochemical measurements and density functional theory (DFT) calculations. The above structural merits contribute to the pseudocapacitive multi-electron transfer lithium-ion storage with high capacity and rate capability of the 2D V2O5/graphene heterostructure. Furthermore, a symmetric full cell based on prelithiated V2O5/graphene was constructed by decoupling the multi-electron reaction, exhibiting high energy/power density and excellent cycle performance.
Results and discussion
A 2D V2O5/graphene heterostructure was synthesized using a well-controlled two-step process involving freeze–drying and subsequent thermal annealing (Fig. S2, ESI†). Through rapid freeze–drying, graphene oxide (GO) nanosheets as a 2D template can uniformly load the vanadium source. During the subsequent thermal annealing process, the deoxygenation of GO and the decomposition of NH4VO3 occurred, and the heterostructure (Fig. 1a) was finally obtained. It is noted that NH3 derived from the decomposition of NH4VO3 may also function as a reducing agent for GO. From the scanning electron microscopy (SEM) images (Fig. 1b and Fig. S3a, ESI†), a fluffy material composed of smooth nanosheets with no segregation of big V2O5 particles is observed. Elemental mapping (Fig. S3b, ESI†) clearly demonstrates the homogeneous distribution of V and O in the nanosheets. The transmission electron microscopy (TEM) image (Fig. 1c) shows the heterostructure featuring a 2D sheet-like morphology, in which the weak contrast indicates an ultrathin thickness. The morphology was further examined by atomic force microscopy (AFM), revealing a small thickness of ∼2.8 nm of the heterostructure (Fig. 1d and Fig. S4, ESI†). Moreover, the high-resolution TEM (HRTEM) images (Fig. 1e and Fig. S3c, ESI†) validate the existence of a sheet-on-sheet heterostructure. | ||
| Fig. 1 Characterization of the 2D V2O5/graphene heterostructure: (a) schematic illustration, (b) SEM image, (c) TEM image, (d) AFM image; the inset shows the height profile, (e) HRTEM image, (f) SAED pattern showing diffraction rings from bilayered V2O5 (brown) and graphene (blue), and (g) XRD pattern. (h) Raman spectra of the 2D V2O5/graphene heterostructure, orthorhombic V2O5 and V2O5 cryogels. (i) N2 adsorption–desorption isotherm of the 2D V2O5/graphene heterostructure. | ||
The crystalline structure of the 2D V2O5/graphene heterostructure was investigated by selected area electron diffraction (SAED) and powder X-ray diffraction (XRD). As shown in Fig. 1f and g, the two patterns can be indexed to the monoclinic bilayered V2O5 structure, which is consistent with those of the V2O5 xerogel or aerogel.15 Besides, the SAED pattern also displays typical diffraction rings from graphene (Fig. 1f), further identifying the heterostructure of bilayered V2O5 and graphene. Fig. 1h shows a comparison of the Raman spectra of the 2D V2O5/graphene heterostructure, orthorhombic V2O5 (Fig. S5, ESI†) and V2O5 cryogel (Fig. S6, a typical type of aerogel prepared by freeze-drying of the V2O5 hydrogel; see details in the ESI†). It is noteworthy that the as-prepared heterostructure exhibits the characteristic peaks of both V2O5 and graphene, confirming the composition of the heterostructure. The mass ratio of V2O5 in the heterostructure is ∼73% according to thermogravimetric analysis (Fig. S7, ESI†). In addition, the N2 adsorption/desorption isotherm reveals a high Brunauer–Emmett–Teller specific surface area of 108 m2 g−1 (Fig. 1i), which can contribute to improving surface-controlled extrinsic pseudocapacitance. Overall, the above characterization methods disclose an ultrathin 2D heterostructure of bilayered V2O5 and graphene, which may be conducive to promoting strain relaxation and ion/electron transport as well as boosting charge transfer through the rich heterointerfaces.
The lithium-ion storage performance of the 2D V2O5/graphene heterostructure was first evaluated in a half-cell configuration. Fig. 2a and Fig. S8 (ESI†) show the initial galvanostatic charge/discharge (GCD) profiles and cyclic voltammetry (CV) curves of the 2D V2O5/graphene heterostructure, respectively. Notably, different from orthorhombic V2O5 with four irreversible discharge plateaus corresponding to the phase transformations of α, ε, δ, γ and ω during the first cycle (Fig. S9a, ESI†),12 the 2D V2O5/graphene heterostructure displays nearly overlapped GCD profiles and CV curves during the initial cycles, indicative of a highly reversible charge storage process. The enhanced reversibility of the 2D V2O5/graphene heterostructure is also demonstrated by a high initial coulombic efficiency of ∼99%, which is superior to those of the V2O5 cryogel and orthorhombic V2O5 (Fig. S9, ESI†). The corresponding differential capacity analysis of the 2D V2O5/graphene heterostructure reveals three peaks at 2.84, 2.51, and 1.61 V (Fig. 2b), suggestive of at least three distinct types of lithium-ion intercalation sites.15
 | ||
| Fig. 2 Lithium-ion storage performance of the 2D V2O5/graphene heterostructure. (a) GCD profiles at 1C of the initial two cycles. (b) dQ/dV plots (Q, capacity; V, voltage). (c) Rate performance of the 2D V2O5/graphene heterostructure, orthorhombic V2O5 and V2O5 cryogels. (d) GCD profiles of the 2D V2O5/graphene heterostructure at different current rates. (e) Energy density based on the mass of the active material and (f) rate capability of the 2D V2O5/graphene heterostructure in comparison with the reported results. | ||
The rate performance of the 2D V2O5/graphene heterostructure, orthorhombic V2O5 and V2O5 cryogel was measured at charge/discharge rates ranging from 1 to 150C (Fig. 2c). Remarkably, the 2D V2O5/graphene heterostructure delivers a high capacity of 361 mA h g−1 at 1C (350 mA g−1) within a potential window of 1.4–4 V vs. Li+/Li (Fig. 2d). It is noted that this capacity is beyond the theoretical value for two lithium-ion storage in V2O5 (∼294 mA h g−1), suggestive of a multi-electron transfer charge storage process. The calculated energy density based on the electrode material is as high as 840 W h kg−1, which is superior to those of conventional cathode materials in lithium-ion batteries including LiMn2O4 (<600 W h kg−1), LiFePO4 (<600 W h kg−1) and LiNi0.8Co0.15Al0.05O2 (700–820 W h kg−1), and exceeds the values reported for other typical multi-electron reaction materials (Fig. 2e), such as Cu2.33V4O11 (729 W h kg−1),32 FeS (714 W h kg−1),33 Li1.3V0.4Nb0.3O2 (∼715 W h kg−1)34 and Li3V2(PO4)3 (614 W h kg−1).35 With stepwise increased charge/discharge rates, the 2D V2O5/graphene heterostructure still affords high capacities of 319, 262, 216 and 175 mA h g−1 at 5, 20, 50 and 100C, respectively, which significantly outperforms the V2O5 cryogel and orthorhombic V2O5 (Fig. S10, ESI†). The ultrahigh rate capability of the 2D V2O5/graphene heterostructure is among the best of the reported results for V2O5 derived materials13,14,36 and also surpasses those of other representative vanadium-based metal oxides (Fig. 2f),37–39 implying that heterostructure engineering substantially improves the reaction kinetics. Besides, the 2D V2O5/graphene heterostructure also exhibits stable cycling performance with a capacity retention of 89% after 1000 cycles (Fig. S11, ESI†), suggestive of excellent structural stability.
To understand the effect of heterostructure engineering on the reaction kinetics, kinetic analyses were conducted through electrochemical impedance spectroscopy (EIS), galvanostatic intermittent titration technique (GITT) and CV. Fig. 3a shows the Nyquist plots featuring a depressed semicircle at a high frequency (related to charge transfer resistance) and an oblique line at a low frequency (related to ion diffusion), and the inset shows the equivalent circuit adopted to fit the plots. Importantly, the 2D V2O5/graphene heterostructure presents much lower charge transfer resistance (23.4 Ω) and Warburg impedance (23.1 Ω) compared with the V2O5 cryogel (154.8 and 221.3 Ω, respectively), demonstrative of greatly improved charge transfer and ion transport kinetics (Fig. 3b). Furthermore, the lithium-ion diffusion coefficient calculated from the EIS results is 2.5 × 10−10 cm2 s−1 for the 2D V2O5/graphene heterostructure (Fig. S12, ESI†), which is much higher than that of the V2O5 cryogel (5.6 × 10−11 cm2 s−1). The whole reaction resistance consisting of ohmic resistance and polarization impedance involving charge transfer and ion diffusion was evaluated using GITT (Fig. 3c and Fig. S13, ESI†).40 The reaction resistance calculated by dividing the overpotential by the pulse current density is shown in Fig. 3d. As expected, the 2D V2O5/graphene heterostructure exhibits lower reaction resistance than the V2O5 cryogel. Interestingly, the reaction resistance of the V2O5 cryogel increases dramatically to 6.8 Ω g in the latter half of the discharge process, while the 2D V2O5/graphene heterostructure maintains a low resistance below 1.6 Ω g. The results reveal severely deteriorated kinetics upon deep discharge for the V2O5 cryogel and meanwhile demonstrate the significance of constructing a heterostructure for breaking through the limitation of the reaction kinetics to realize multi-electron transfer charge storage reactions.
 | ||
| Fig. 3 Kinetics analysis for lithium-ion storage in the 2D V2O5/graphene heterostructure and the V2O5 cryogel. (a) Nyquist plots obtained at the open circuit potential; the inset shows the corresponding equivalent circuit. (b) Calculated charge transfer resistance and Warburg impedance. (c) GITT profiles and (d) corresponding reaction resistances. (e) CV curves and (f) capacitive contribution ratios of the 2D V2O5/graphene heterostructure at different scan rates. | ||
CV curves at varying scan rates were obtained to further clarify the lithium-ion storage kinetics of the 2D V2O5/graphene heterostructure. The power-law relationship between the peak current (i) and the scan rate (ν) was used to qualitatively evaluate the charge storage kinetics. According to the equation of i = aνb, the b value is determined from the slope of the fitted logi versus logν plot (Fig. S14, ESI†). As shown in Fig. 3e, the calculated b values of the 2D V2O5/graphene heterostructure are 0.65, 0.74 and 0.80 for the cathodic peaks and 0.69, 0.83 and 0.77 for the anodic peaks, which suggests a combination of surface- and diffusion-controlled charge storage processes since b values of 1.0 and 0.5 are considered as indicators for the surface-controlled capacitive process and diffusion-controlled process, respectively.41 To quantitatively figure out the contribution from each process, the total current at a fixed potential is separated into capacitive (k1ν) and diffusion-controlled (k2ν1/2) components (Fig. S15, ESI†).42 The calculated capacitive contribution varies from 76% to 85% at scan rates ranging from 1.0 to 3.0 mV s−1 (Fig. 3f). Considering the relatively low specific surface area of the 2D V2O5/graphene heterostructure for electric double-layer capacitance, it can be concluded that the charge storage in the heterostructure is mainly contributed by pseudocapacitance, which correlates closely with its ability to achieve high capacity and high rate capability simultaneously.
To elucidate the lithium-ion storage mechanism of the 2D V2O5/graphene heterostructure, ex situ X-ray photoelectron spectroscopy (XPS), TEM and XRD were carried out. The high-resolution XPS spectra of V 2p were obtained from electrodes at different states to access changes in the valence state of vanadium element in the 2D V2O5/graphene heterostructure upon lithiation/delithiation (Fig. 4a). The V 2p3/2 spectrum of the pristine electrode can be deconvoluted into two peaks at 517.7 and 516.3 eV, corresponding to V5+ and V4+, respectively.43 Upon discharging to 1.4 V, two new peaks appear at lower binding energies of 515.2 and 513.9 eV, which are assigned to V3+ and V2+, respectively.31 When further charging back to 4 V, the electrode almost returns to the pristine V 2p3/2 with no residual V3+/V2+ states and a slightly increased V4+ content. The increased proportion of V4+ after the first recharging may be attributed to some residual Li+, which can function as the stabilizing agent to preserve the bilayered structure of vanadium oxide.44 The results validate that the vanadium element in the 2D V2O5/graphene heterostructure undergoes reversible multi-electron transfer during charge/discharge. Ex situ TEM was conducted to clarify the structural evolution of the 2D V2O5/graphene heterostructure. A highly disordered form is observed from the HRTEM image of the fully discharged sample (Fig. 4b), which is also identified by the SAED pattern with only two diffraction rings from graphene (Fig. 4c). Upon charging back to 4 V, the well-defined monoclinic bilayered structure fully recovers (Fig. 4d and e), demonstrating that the lithiation/delithiation of the 2D V2O5/graphene heterostructure is highly reversible. After 20 charge/discharge cycles, the 2D V2O5/graphene heterostructure still retains its structure with no agglomeration of V2O5 (Fig. S16, ESI†), indicative of high structural stability. The ex situ XRD results (Fig. 4f) also confirm the reversible phase transformation and excellent structural stability of the 2D V2O5/graphene heterostructure, which is likely attributed to the facile strain relaxation benefiting from the ultrathin thickness.45,46
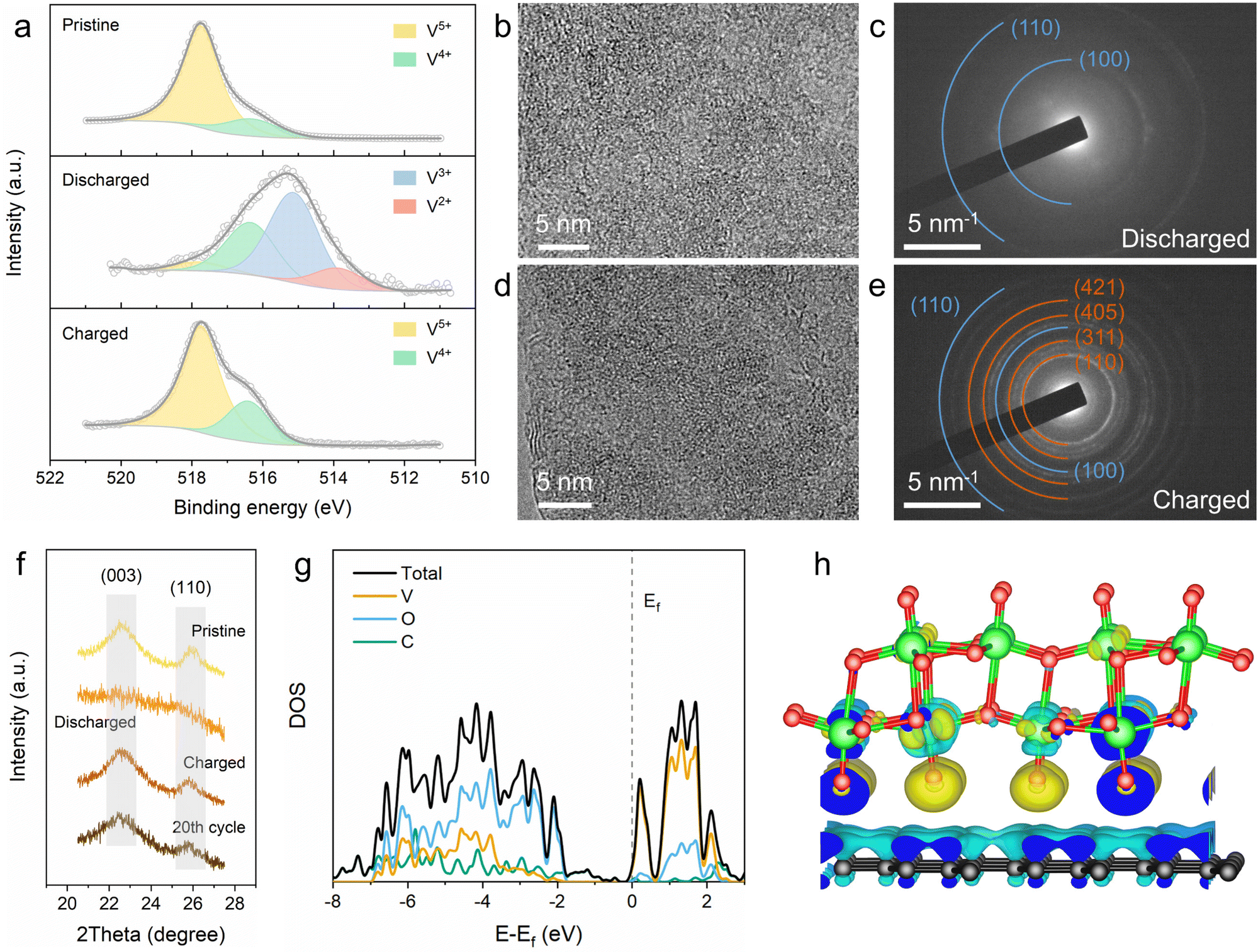 | ||
| Fig. 4 Charge storage mechanism study of the 2D V2O5/graphene heterostructure. (a) V 2p3/2 spectra in the pristine, fully discharged and charged states. (b) HRTEM image and (c) SAED pattern of the 2D V2O5/graphene heterostructure discharged to 1.4 V. (d) HRTEM image and (e) SAED pattern of the 2D V2O5/graphene heterostructure charged back to 4 V. The brown and blue diffraction rings in the SAED patterns correspond to bilayered V2O5 and graphene, respectively. (f) Ex situ XRD patterns of the V2O5/graphene electrodes in different states. (g) Density of states of the 2D V2O5/graphene heterostructure. (h) Charge density difference at the interface of the 2D V2O5/graphene heterostructure. Yellow and cyan represent electron accumulation and depletion, respectively. | ||
DFT calculations were conducted to investigate the electronic structure of the 2D V2O5/graphene heterostructure. To this end, a simplified structural model for the heterostructure was employed (Fig. S17, ESI†), which displays an optimized distance of 0.29 nm between the bilayered V2O5 and graphene. The calculated densities of states (DOSs) of the heterostructure and pure bilayered V2O5 are shown in Fig. 4g and Fig. S18 (ESI†), respectively. Both bulk and a single layer of pure bilayered V2O5 are identified as semiconductors with a notable band gap of ∼2.1 eV, suggestive of insufficient electronic conductivity for high-rate charge storage. In contrast, the 2D V2O5/graphene heterostructure exhibits significantly enhanced DOS near the Fermi level, which indicates improved electronic conductivity due to energy gap contraction.20,47 Besides, the shift of the Fermi level toward the conduction band in the heterostructure suggests lowered work function and augmented concentration of electrons in the conduction band, which further demonstrate the enhanced electronic conductivity of n-type vanadium oxide.48,49 The charge density difference depicted in Fig. 4h reveals that electron accumulation and depletion occur at the interface of the 2D V2O5/graphene heterostructure. This charge redistribution with electrons transferred from graphene to V2O5 will induce the formation of a built-in electric field across the atomic heterointerface, which can greatly reduce the interfacial resistance and boost the charge transfer process upon charge/discharge.50,51
The quasi-linear GCD profiles with a wide potential window and high capacity obtained from the reversible pseudocapacitive multi-electron reaction of the 2D V2O5/graphene heterostructure suggest a new approach for designing symmetric energy storage devices. On the basis of decoupling the whole potential window into two appropriate potential ranges with equivalent capacities, a symmetric full cell can be constructed using the prelithiated V2O5/graphene heterostructure as both the anode and the cathode (Fig. 5a). Accordingly, the working potential window of the 2D V2O5/graphene heterostructure can be separated into a positive region of 2.45–4 V and a negative region of 1.4–2.45 V (Fig. 5b), and hence the electrodes were prelithiated to 2.45 V vs. Li+/Li before being assembled into the full cell. The rate performance of the symmetric full cell at different current densities ranging from 0.1 to 10 A g−1 is shown in Fig. 2c and Fig. S19 (ESI†). The full cell delivers a reversible capacity of 82 mA h g−1 at 0.1 A g−1 within a voltage range of 0–2.5 V. At increased current densities of 0.5, 2, 5 and 10 A g−1, the full cell still offers high capacities of 74, 64, 54 and 44 mA h g−1, respectively. The outstanding rate capability of the full cell is ascribed to the pseudocapacitance-dominant lithium-ion storage behavior of the 2D V2O5/graphene heterostructure. CV and EIS measurements were conducted to study the kinetics of the full cell. The b values at different voltages calculated according to the power-law relationship (i = aνb) are shown in Fig. S20 (ESI†). Notably, the full cell exhibits b values almost higher than 0.8 throughout the whole charge/discharge process. In particular, the b values of the two peaks are calculated to be 0.90 and 0.88. The high b values imply highly rapid electrochemical kinetics. The EIS plots obtained at different charge/discharge states reveal a stable low charge transfer resistance of 4–5 Ω (Fig. 5e and Table S1, ESI†), further indicating the superb kinetics of the full cell.
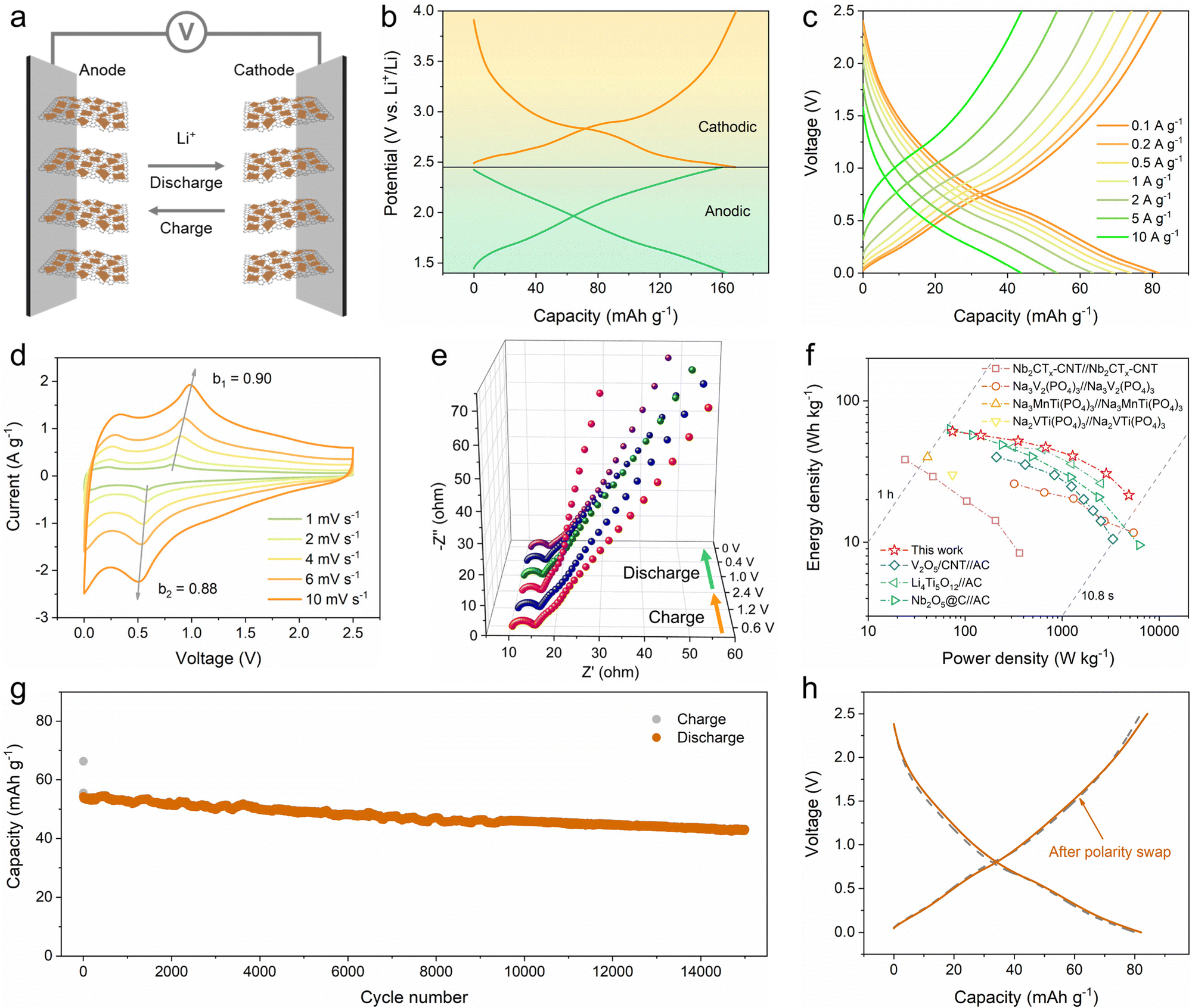 | ||
| Fig. 5 Electrochemical performance of the symmetric full cell based on the 2D V2O5/graphene heterostructure. (a) Schematic illustration of the full cell. (b) GCD profiles of the 2D V2O5/graphene heterostructure within the potential ranges of 2.45–4 V and 1.4–2.45 V. (c) GCD profiles of the full cell at various current densities. (d) CV curves at various scan rates. (e) Nyquist plots obtained at different charge/discharge states. (f) Ragone plot in comparison with the reported results. (g) Long-term cycle performance at 5 A g−1. (h) Comparison of GCD profiles before and after polarity swap. | ||
To demonstrate the practical applicability of the symmetric full cell, the energy and power densities based on the combined mass of active materials in both electrodes were calculated from the GCD curves, and the Ragone plot in comparison with the reported results is shown in Fig. 5f. Importantly, the full cell achieves energy densities of 61, 57, 52, 47, 41, 31 and 22 W h kg−1 at power densities of 74, 145, 351, 677, 1283, 2849 and 4875 W kg−1, respectively. This performance is superior to those of the previously reported symmetric systems based on Nb2CTx,30 Na3V2(PO4)3,28 Na3MnTi(PO4)324 and Na2VTi(PO4)329 and surpasses the results reported for several typical lithium-ion capacitors, such as V2O5//activated carbon (AC),52 Li4Ti5O12//AC53 and Nb2O5//AC.54 The long-term cycling performance of the full cell was measured at a current density of 5 A g−1, as shown in Fig. 5g. Impressively, 79% of the initial capacity is maintained after 15000 cycles. The GCD profiles during the 5th and 15000th cycles are presented in Fig. S21 (ESI†) for comparison, which show nearly identical shapes. Furthermore, when the polarity of the pristine symmetric full cell is reversed, the resulting full cell exhibits a stable GCD curve almost the same as the pristine one (Fig. 5h), demonstrating the unique superiority of symmetric energy storage devices.
Conclusions
In summary, we developed a novel 2D heterostructure of bilayered V2O5 and graphene with the ability to realize rapid pseudocapacitive multi-electron transfer lithium storage. The structural merits of ultrathin nanosheet morphology and abundant heterogeneous interfaces contribute to breaking through the limitations of reversibility and kinetics for the multi-electron reaction by affording fast reversible phase transformation and promoting electron/ion transport and interfacial charge transfer, thus endowing the 2D V2O5/graphene heterostructure with high capacity and rate capability simultaneously. Moreover, a symmetric full cell based on prelithiated V2O5/graphene was constructed by decoupling the multi-electron reaction, exhibiting superb energy/power performance and ultralong cycle life. This work underscores the potential importance of creating a heterostructure as a strategy for overcoming the limitations of reaction reversibility and kinetics to achieve high-rate multi-electron transfer charge storage in redox-active materials and provides a new paradigm to construct symmetric full cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grants 22125903, 51872283, 22005298, 22209176, 22209173), the “Transformational Technologies for Clean Energy and Demonstration” Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA21000000), the Dalian Innovation Support Plan for High Level Talents (2019RT09), the Dalian National Laboratory For Clean Energy (DNL), CAS, DNL Cooperation Fund, CAS (DNL201912, DNL201915, DNL202016, DNL202019), DICP (DICP I2020032), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (YLU-DNL Fund 2021002, YLU-DNL 2021009), and the China Postdoctoral Science Foundation (2021M703145).References
- X. Y. Shi, P. Das and Z.-S. Wu, ACS Energy Lett., 2022, 7, 267–281 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- F. F. Xing, Z. H. Bi, F. Su, F. Y. Liu and Z.-S. Wu, Adv. Energy Mater., 2022, 12, 2200594 CrossRef CAS.
- X. P. Gao and H. X. Yang, Energy Environ. Sci., 2010, 3, 174–189 RSC.
- Y. X. Huang, F. Wu and R. J. Chen, Natl. Sci. Rev., 2020, 7, 1367–1386 CrossRef CAS PubMed.
- X. R. Wang, G. Q. Tan, Y. Bai, F. Wu and C. Wu, Electrochem. Energy Rev., 2021, 4, 35–66 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- M. Z. Chen, Q. N. Liu, Z. Hu, Y. Y. Zhang, G. C. Xing, Y. X. Tang and S. L. Chou, Adv. Energy Mater., 2020, 10, 2002244 CrossRef CAS.
- X. Xu, F. Xiong, J. Meng, Q. An and L. Mai, Mater. Today Nano, 2020, 10, 100073 CrossRef.
- J. H. Yao, Y. W. Li, R. C. Masse, E. Uchaker and G. Z. Cao, Energy Storage Mater., 2018, 11, 205–259 CrossRef.
- Q. Liu, Z. F. Li, Y. Liu, H. Zhang, Y. Ren, C. J. Sun, W. Lu, Y. Zhou, L. Stanciu, E. A. Stach and J. Xie, Nat. Commun., 2015, 6, 6127 CrossRef.
- A. Moretti, F. Maroni, I. Osada, F. Nobili and S. Passerini, ChemElectroChem, 2015, 2, 529–537 CrossRef CAS.
- A. Moretti and S. Passerini, Adv. Energy Mater., 2016, 6, 1600868 CrossRef.
- S. T. Geng, T. Zhou, M. Y. Jia, X. Y. Shen, P. B. Gao, S. Tian, P. F. Zhou, B. Liu, J. Zhou, S. P. Zhuo and F. Li, Energy Environ. Sci., 2021, 14, 3184–3193 RSC.
- J. Kang, Y. Xue, J. Yang, Q. Hu, Q. Zhang, L. Gu, A. Selloni, L. M. Liu and L. Guo, J. Am. Chem. Soc., 2022, 144, 8969–8976 CrossRef CAS PubMed.
- C. Xia, Z. Lin, Y. Zhou, C. Zhao, H. Liang, P. Rozier, Z. Wang and H. N. Alshareef, Adv. Mater., 2018, 30, 1803594 CrossRef PubMed.
- W. J. Liu, X. Zhang, Y. N. Xu, L. Wang, Z. Li, C. Li, K. Wang, X. Z. Sun, Y. B. An, Z. S. Wu and Y. W. Ma, Adv. Funct. Mater., 2022, 32, 2202342 CrossRef CAS.
- X. Lu, Y. Shi, D. Tang, X. Lu, Z. Wang, N. Sakai, Y. Ebina, T. Taniguchi, R. Ma, T. Sasaki and C. Yan, ACS Nano, 2022, 16, 4775–4785 CrossRef CAS PubMed.
- M. Ma, S. Zhang, L. Wang, Y. Yao, R. Shao, L. Shen, L. Yu, J. Dai, Y. Jiang, X. Cheng, Y. Wu, X. Wu, X. Yao, Q. Zhang and Y. Yu, Adv. Mater., 2021, 33, 2106232 CrossRef CAS PubMed.
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. E. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS PubMed.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2019, 5, 5–19 CrossRef.
- H. Gao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 12768–12772 CrossRef CAS PubMed.
- L. Zhang, S. X. Dou, H. K. Liu, Y. Huang and X. Hu, Adv. Sci., 2016, 3, 1600115 CrossRef PubMed.
- J. X. Liang and D. W. Wang, Adv. Energy Mater., 2022, 12, 2200920 CrossRef CAS.
- F. Su, J. Q. Qin, P. Das, F. Zhou and Z.-S. Wu, Energy Environ. Sci., 2021, 14, 2269–2277 RSC.
- Z. L. Jian, V. Raju, Z. F. Li, Z. Y. Xing, Y. S. Hu and X. L. Ji, Adv. Funct. Mater., 2015, 25, 5778–5785 CrossRef CAS.
- H. Wang, T. Zhang, C. Chen, M. Ling, Z. Lin, S. Zhang, F. Pan and C. Liang, Nano Res., 2017, 11, 490–498 CrossRef.
- A. Byeon, A. M. Glushenkov, B. Anasori, P. Urbankowski, J. W. Li, B. W. Byles, B. Blake, K. L. Van Aken, S. Kota, E. Pomerantseva, J. W. Lee, Y. Chen and Y. Gogotsi, J. Power Sources, 2016, 326, 686–694 CrossRef CAS.
- D. Chao, R. DeBlock, C. H. Lai, Q. Wei, B. Dunn and H. J. Fan, Adv. Mater., 2021, 33, 2103736 CrossRef CAS PubMed.
- M. Morcrette, P. Rozier, L. Dupont, E. Mugnier, L. Sannier, J. Galy and J. M. Tarascon, Nat. Mater., 2003, 2, 755–761 CrossRef CAS PubMed.
- J. Zou, K. Yuan, J. Zhao, B. Wang, S. Chen, J. Huang, H. Li, X. Niu and L. Wang, Energy Storage Mater., 2021, 43, 579–584 CrossRef.
- X. Zhao, Y. Tian, Z. Lun, Z. Cai, T. Chen, B. Ouyang and G. Ceder, Joule, 2022, 6, 1654–1671 CrossRef CAS.
- Q. Ni, L. M. Zheng, Y. Bai, T. C. Liu, H. X. Ren, H. J. Xu, C. Wu and J. Lu, ACS Energy Lett., 2020, 5, 1763–1770 CrossRef CAS.
- C. F. Zhang, S. H. Park, S. E. O'Brien, A. Seral-Ascaso, M. Y. Liang, D. Hanlon, D. Krishnan, A. Crossley, N. McEvoy, J. N. Coleman and V. Nicolosi, Nano Energy, 2017, 39, 151–161 CrossRef CAS.
- Q. L. Wei, Q. Q. Wang, Q. D. Li, Q. Y. An, Y. L. Zhao, Z. Peng, Y. L. Jiang, S. S. Tan, M. Y. Yan and L. Q. Mai, Nano Energy, 2018, 47, 294–300 CrossRef CAS.
- H. Song, M. Luo and A. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 2875–2882 CrossRef CAS PubMed.
- Q. F. Fu, X. Z. Zhu, R. J. Li, G. S. Liang, L. J. Luo, Y. J. Chen, Y. L. Ding, C. F. Lin, K. K. Wang and X. S. Zhao, Energy Storage Mater., 2020, 30, 401–411 CrossRef.
- J. Jiang, W. Shi, J. Zheng, P. Zuo, J. Xiao, X. Chen, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2013, 161, A336–A341 CrossRef.
- A. Noori, M. F. El-Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Soc. Rev., 2019, 48, 1272–1341 RSC.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- G. Wang, X. Cui, J. Liu, Y. Wang, Y. Qiao, X. Shi, Y. Zhang, H. Liu and L. Li, Small, 2020, 16, 2003816 CrossRef CAS PubMed.
- M. Clites and E. Pomerantseva, Energy Storage Mater., 2018, 11, 30–37 CrossRef.
- C. K. Chan, H. Peng, R. D. Twesten, K. Jarausch, X. F. Zhang and Y. Cui, Nano Lett., 2007, 7, 490–495 CrossRef CAS PubMed.
- J. Zhu, H. Shen, X. Shi, F. Yang, X. Hu, W. Zhou, H. Yang and M. Gu, Anal. Chem., 2019, 91, 11055–11062 CrossRef CAS PubMed.
- Y.-H. Zhu, J.-Z. Wang, Q. Zhang, Y.-F. Cui, G. Huang, J.-M. Yan and X.-B. Zhang, Energy Environ. Sci., 2022, 15, 1529–1535 RSC.
- A. Kahn, Mater. Horiz., 2016, 3, 7–10 RSC.
- X. Shao, D. Li, J. Cai, H. Luo and C. Dong, Appl. Surf. Sci., 2016, 368, 477–482 CrossRef CAS.
- S. Gbadamasi, M. Mohiuddin, V. Krishnamurthi, R. Verma, M. W. Khan, S. Pathak, K. Kalantar-Zadeh and N. Mahmood, Chem. Soc. Rev., 2021, 50, 4684–4729 RSC.
- J. Mei, J. Shang, C. Zhang, D. Qi, L. Kou, B. Wijerathne, C. Hu, T. Liao, J. MacLeod and Z. Sun, Small Methods, 2022, 6, 2200658 CrossRef CAS PubMed.
- Z. Chen, V. Augustyn, J. Wen, Y. Zhang, M. Shen, B. Dunn and Y. Lu, Adv. Mater., 2011, 23, 791–795 CrossRef CAS PubMed.
- Y. Qian, X. Y. Cai, C. Y. Zhang, H. F. Jiang, L. J. Zhou, B. S. Li and L. F. Lai, Electrochim. Acta, 2017, 258, 1311–1319 CrossRef CAS.
- E. Lim, C. Jo, H. Kim, M. H. Kim, Y. Mun, J. Chun, Y. Ye, J. Hwang, K. S. Ha, K. C. Roh, K. Kang, S. Yoon and J. Lee, ACS Nano, 2015, 9, 7497–7505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02888c |
| This journal is © The Royal Society of Chemistry 2023 |