
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-atom cluster catalysts for efficient electrocatalysis
Libo
Sun
ab,
Vikas
Reddu
 a and
Xin
Wang
*ab
a and
Xin
Wang
*ab
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459, Singapore. E-mail: WangXin@ntu.edu.sg
bCambridge Centre for Advanced Research and Education in Singapore Ltd (Cambridge CARES), CREATE Tower, Singapore 138602, Singapore
First published on 5th October 2022
Abstract
Multi-atom cluster catalysts have turned out to be novel heterogeneous catalysts with atomic dispersion for electrochemical energy applications. Beyond a simple combination of single-atom catalysts, they could offer boosted activity as a result of the synergistic effects between adjacent atoms. Meanwhile, the multiple active sites in the catalytic center may render them versatile binding modes toward adsorbates and provide an opportunity for catalyzing complex reactions with diverse products. Herein, a comprehensive review of the recent development of multi-atom cluster catalysts for electrochemical energy applications is provided. Specifically, the origin of synergistic effects in multi-atom cluster catalysts and related modulation methods are illustrated and summarized. The introduction of multi-atom cluster catalysts to circumvent the scaling relationships as well as their potential for developing new descriptors is then discussed. Subsequently, the methods for fabricating multi-atom cluster catalysts and related characterization techniques are reviewed. This is followed by the discussion of their application in key electrochemical reactions such as water splitting, oxygen reduction, and carbon dioxide/monoxide reduction, as well as the real-time techniques for their mechanistic study. Finally, the future challenges and opportunities concerning the improvement of multi-atom cluster catalysts are outlined, which are essential to make such electrocatalysts viable for electrochemical energy conversion.
 Libo Sun | Libo Sun received his bachelor's degree in chemistry (2009) from Jilin University. Then, he received his PhD in inorganic chemistry (2014) under the supervision of Prof. Jihong Yu from the State Key Laboratory of Inorganic Synthesis & Preparative Chemistry, Jilin University. He then started his further research in The University of Hong Kong and Nanyang Technological University. In 2017, he joined Prof. Xin Wang's group and worked in the Cambridge Centre for Advanced Research and Education in Singapore. His current research focuses on heterogeneous electrocatalytic CO2 reduction and nitrate reduction with advanced molecular and cluster catalysts. |
 Vikas Reddu | Vikas Reddu received his BE (Hons.) degree in chemical engineering from Birla Institute of Technology and Science (India) in 2013, and his PhD in 2022 from Nanyang Technological University, under the supervision of Prof. Xin Wang. He has been working on electrocatalytic reduction of carbon dioxide to fuels and feedstocks. His research interests include electrochemical energy conversion and storage technologies. |
 Xin Wang | Prof. Xin Wang is Cheng Tsang Man Chair Professor in Energy at Nanyang Technological University, Singapore. He received his bachelor's (1994) and master's (1997) degrees in chemical engineering from Zhejiang University, and PhD (2002) in chemical engineering from Hong Kong University of Science and Technology. He has research interest in nanostructure control, surface functionalization and interface tuning for selective electrocatalysis towards key electrochemical reactions for energy and environmental applications. He is a Fellow of the Academy of Engineering, Singapore, and a Fellow of the Royal Society of Chemistry. |
1. Introduction
Practical application of electrocatalytic energy conversion relies on the availability of electrocatalysts with high activity, selectivity, and durability. Noble metal (Pt, Au, Ru, Ir, etc.) based electrocatalysts have been used for electrocatalytic CO2 reduction reaction (CO2RR), hydrogen evolution reaction (HER), oxygen evolution/reduction reaction (OER/ORR), nitrogen reduction reaction (NRR), etc.1–5 However, the high cost and natural scarcity of these materials accompanied by insufficient activity hamper their large-scale applications. An alternative choice is to develop either non-precious metal electrocatalysts or noble metal-based electrocatalysts with highly efficient atom utilization and activity, which can tackle the above-mentioned problems while maintaining excellent performance.Recently, catalysts dispersed at an atomic scale have drawn tremendous research interest for being good candidates for various applications.6 It is widely accepted that the catalysts dispersed at an atomic scale can exhibit better performance with improved atom utilization than the bulk ones due to the unsaturated metal environment and unique electronic structure.7,8 Recent studies have confirmed that single-atom catalysts (SACs) demonstrate a boost in catalytic performance in many electrocatalytic applications.9,10 The strategy of using SACs could further facilitate their intrinsic performance by careful tuning of their coordination and electronic configurations.11 Although great breakthroughs have been achieved by using SACs, a major shortcoming of these catalysts is the lack of structure complexity, which makes them unsuitable when faced with complex reactions. As such, complex reactions follow various reaction pathways via diverse intermediates to form multiple products with the assistance of multiple active sites on a catalyst.12–15 Besides, the single active site of SAC materials makes it unlikely for them to be able to regulate the binding modes of adsorbed intermediates during reactions due to the lack of diverse synergistic interactions,16–18 thus limiting their ability to surpass the intrinsic performance by circumventing the scaling relationship.19 This calls for further development of suitable electrocatalysts.
The introduction of additional metal atoms in the vicinity of single active sites of SACs to form multi-atom cluster catalysts, including dual atom catalysts (DACs), triple atom catalysts (TACs), etc., could allow for the inheritance of the atomic dispersity, high atomic utilization, high activity and selectivity of SACs, and break the intrinsic limits as well.20–26 To date, research on multi-atom cluster catalysts has revealed the opportunities in various applications, such as CO oxidation,27–30 NO oxidation,31,32 CO2 hydrogenation,33–35 glycerol hydrogenolysis,36 alkene epoxidation,37 oxidation of alcohols,38 semi-hydrogenation of alkynes, and Suzuki coupling reaction along with other applications.39–43 Although their development is still at an early stage, they already exhibit fascinating activity.44 Notably, Fe–Co hetero-DAC exhibited superior oxygen reduction activity compared to Pt/C and its counterpart SACs.45 The efficient activation of the binding of O2 allows it to perform stably in H2/O2 fuel cells. The Ni/Fe–N–C hetero-DAC is highly selective (>90%) for CO2RR to produce CO over a wide potential range (−0.5 to −0.9 V vs. reversible hydrogen electrode (RHE)), wherein 98% selectivity could be obtained at −0.7 V vs. RHE with good long-term stability for at least 30 h.46 Pt–Ru hetero-DAC also exhibited greatly improved hydrogen evolution activity and stability when compared to that of commercial Pt/C.47 These studies proved that the multi-atom cluster catalysts could exhibit excellent electrocatalytic activity. One advantage of using these catalysts is that such materials can provide multiple active sites to bind with the adsorbent and optimize the binding mode through C-, O- and N-binding sites, which favor complex reaction paths and formation of complex products.48 Moreover, through the synthesis of either homo- or hetero- multi-atom clusters, their electronic structure can be systematically tuned, thus providing a desired synergistic effect altering their intrinsic catalytic ability, which in turn renders the possibility to tune a reaction.49,50
Considering that multi-atom cluster catalysts are still in their infancy for electrocatalytic applications, we hope that through a thorough compilation of published work on multi-atom cluster electrocatalysts, a clear fundamental research map could be established (Fig. 1). Different from SACs, the synergistic effect that exists in multi-atom cluster catalysts (either homo- or hetero-atom clusters) is one of their main characteristics and contributes the most to boosted activity. The synergistic effect may affect the electronic structure of active centers and the binding behaviour of intermediates, and thus, the necessary parameters behind it and the methods of modulating these parameters are discussed first (including inter-atomic interaction, coordination environment, and substrate interactions). Subsequently, the theoretical understanding of the influence on the scaling relationships arising from the multi-atom cluster catalysts and the descriptors that may affect the intrinsic activity of such electrocatalysts are discussed. Then, as precisely controlled fabrication of multi-atom cluster catalysts remains a challenge, recently developed methods and related characterization techniques are reviewed to inspire more advanced preparation strategies. Furthermore, the experimental applications in CO2RR, OER/ORR, HER, and NRR as well as the real-time techniques for mechanism investigation are summarized for a better understanding of the advantages of using multi-atom cluster catalysts. We hope this review could inspire interested readers in the further development of multi-atom cluster electrocatalysts.
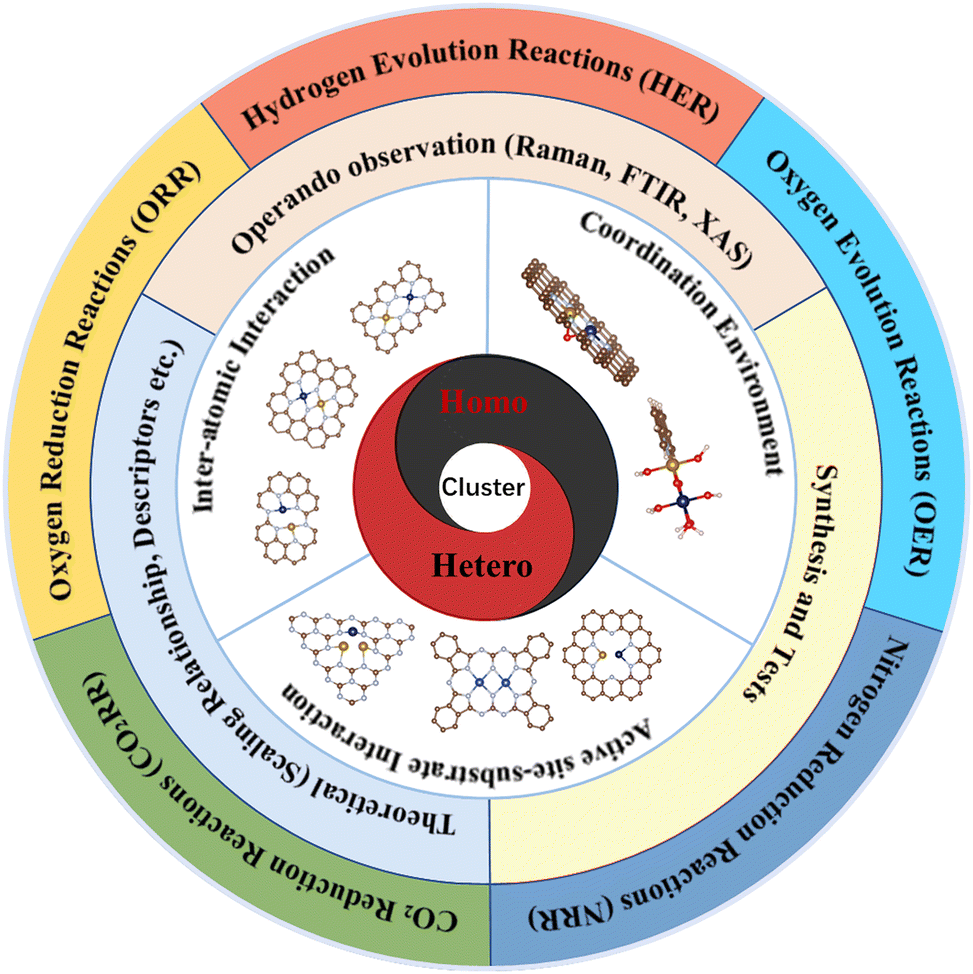 | ||
| Fig. 1 Schematic view of topics covered in the review for the advancement of multi-atom cluster catalysts. | ||
2. Synergistic effects in multi-atom cluster catalysts
2.1 Synergistic effects
The synergistic effect plays a key role in boosting the performance of an electrocatalyst. It is believed that such an effect may be derived from the combined benefit from the components A and B forming synergistic (A + B)syn, which should render higher activity outperforming the individual components A and B, so that (A + B)syn > A + B. As the electrocatalytic reactions usually take place on a surface, any modification of the surface may affect its ability to interact with adsorbates.51–53 Evidence suggests that either the electronic or geometric modifications of a surface can lead to a synergistic effect, resulting in improved activity, selectivity, or stability of a catalyst.54–56 The synergistic effects are common in nanostructures, where the electronic and geometric modifications can be achieved by various techniques such as alloying, segregation-driven interphase formation, core–shell modification, etc.57,582.2 The origin of synergistic effects in multi-atom cluster catalysts
It is necessary to specify the scope of multi-atom cluster catalysts discussed in this work (Fig. 2a). According to the equation by Kubo,59 the band gap (δ) can be approximately expressed as follows: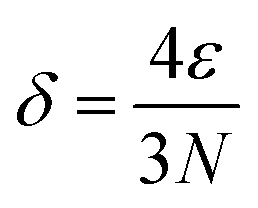 | (1) |
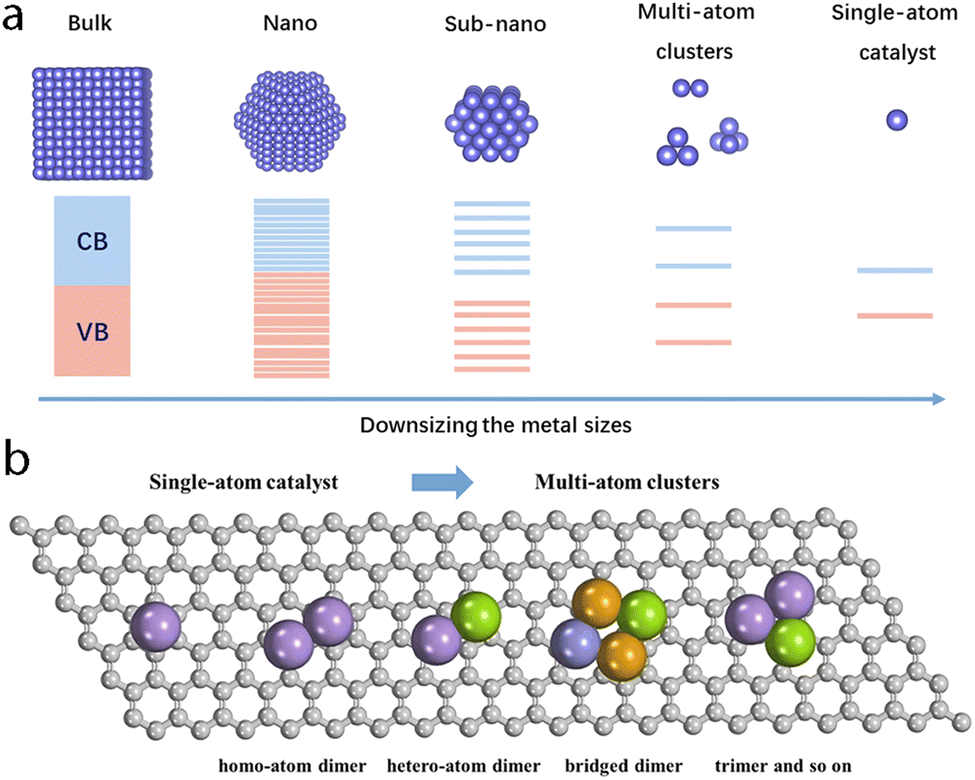 | ||
| Fig. 2 (a) Representative geometric and electronic differences of materials with diverse sizes. (b) Possible multi-atom cluster catalysts. | ||
As shown in Fig. 2b, multi-atom cluster catalysts may possess diverse configurations in electrocatalysis, including homo- and hetero-atom clusters such as homo-atom dimers, hetero-atom dimers, bridged dimers, homo- or hetero-atom trimers, and so on. The presence of various configurations in multi-atom cluster catalysts suggests that diverse interactions exist between various metal atom sites, and they cannot simply be regarded as separated entities.64–66 Therefore, the construction of multi-atom cluster catalysts usually results in significantly different catalytic activity than that of single-atom catalysts.67 The main factor leading to this observed difference is usually attributed to the synergistic effects after the introduction of additional atoms in the active centers. However, further explanation behind the synergistic effect is necessary. Through tuning the local geometric and electronic configuration of active sites,68–72 various parameters, such as the degree of d-band filling, the spin state, charge difference, electronegativity, and so on, could be systematically regulated, which may be used to explain the origin of synergistic effects of multi-atom cluster catalysts (Fig. 4a–c).73–78 When such modified active centers are involved in the interaction with an adsorbate, they could optimize the binding mode, leading to effective activation of the adsorbate and optimized reaction energies, eventually tuning the activity of multi-atom cluster catalysts (Fig. 3a).
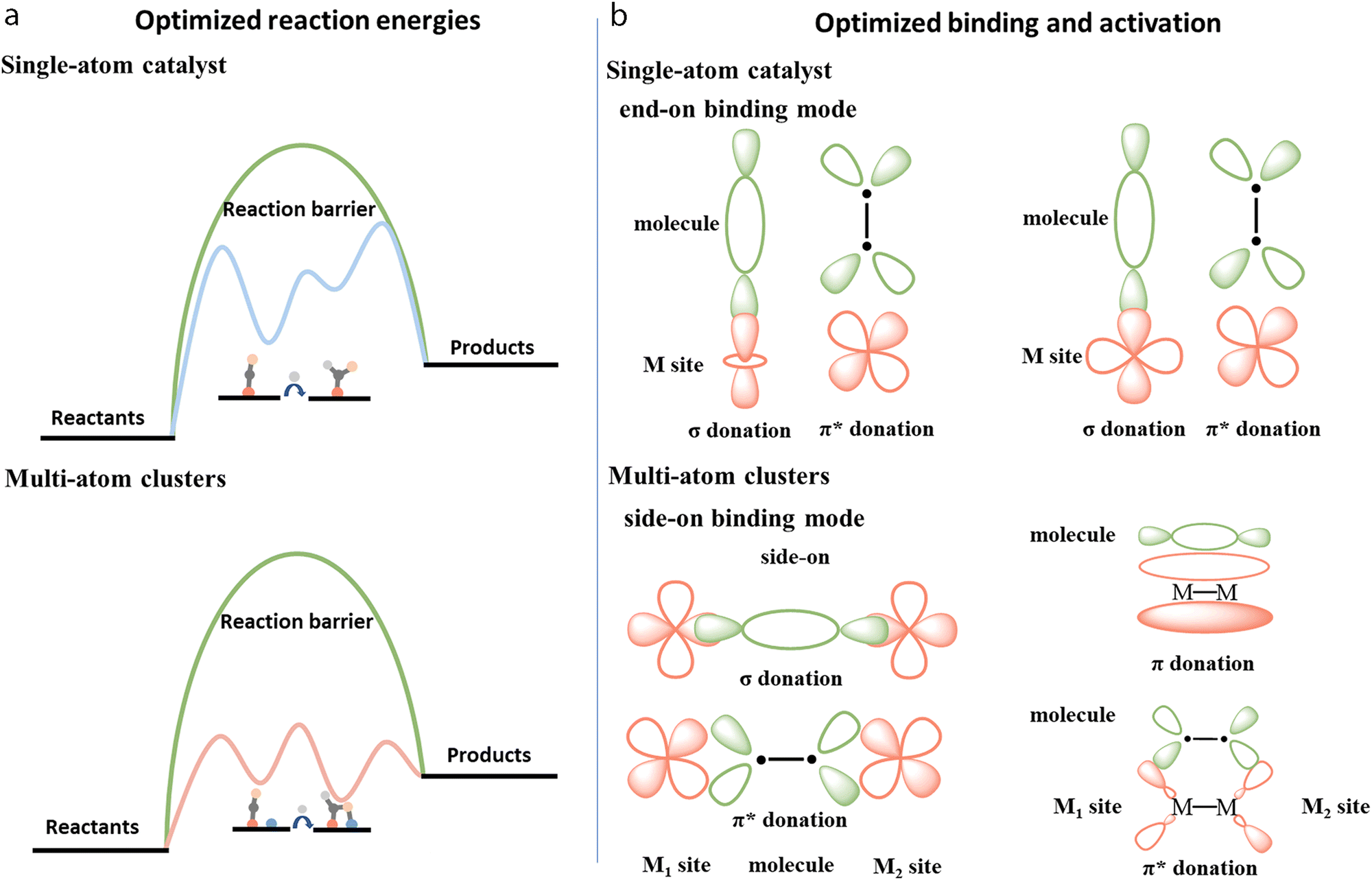 | ||
| Fig. 3 Illustration of (a) optimized reaction energies, and (b) binding and activation of multi-atom cluster catalysts when compared with single-atom catalysts. | ||
Specifically, after the incorporation of multi-atoms, the orbitals of metals and adsorbates could overlap with each other through more efficient orbital interactions with optimized σ or π donation, and π* back-donation bonds, leading to better activation of the adsorbates (Fig. 3b).79–86 As such, Jiang et al. established TM2@C2N (M = Pt, Co, Ni, Cu) dual atom models and compared them with their counterpart TM@C2N for ORR.87 Taking Co2@C2N as an example, Co2@C2N presented remarkably higher adsorption energy (2.39 eV) for the O2 molecule with side-on binding configuration than Co@C2N (1.18 eV) with end-on binding configuration. Therefore, the O–O bond length of 1.22 Å in the free O2 molecule was elongated to 1.47 Å with Co2@C2N, which was longer than 1.33 Å in Co@C2N. This could be ascribed to a shifted d-band center to the Fermi level, which increased orbital coupling between Co 3d orbital and O2 2p orbital. This may have increased charge transfer to O2 from 0.51 to 0.76e−, thus leading to better dissociation of O2 for ORR with dual atom configuration of metal atoms.
The spin polarization of multi-atom cluster catalysts also plays a role in electrocatalytic reactions.88,89 For instance, as oxygen is a paramagnetic molecule, the ferromagnetic half-metal (HM, with one spin-orbital crossing the Fermi level) of [(Co and/or Fe)-Nx-gra, x = 1–6] dual sites with spin-polarized electrons could create a moderate non-uniform magnetic field to couple and activate O2 in ORR.90 Meanwhile, the formation of the O–O intermediate in OER also requires spin conservation to yield the paramagnetic triplet state of oxygen by interaction with active centers. Thus, the spin-polarization of Co–Fe dual atoms could guarantee the parallel spin alignment of oxygen molecules during the reaction, to facilitate either the activation of O2 in ORR or the formation of O–O in OER. Therefore, the spin polarization was capable of describing the activity of ORR and OER, wherein the spin moment of Fe should be between 0.4 and 1.5μB for an ideal dual atom Fe–Co bifunctional electrocatalyst. Recently, Zhang et al. reported the incorporation of Mn in the vicinity of Fe to regulate it from a low spin state of t2g5eg0 to an intermediate spin state of t2g4eg1.88 This one eg electron (dz2 orbital) may couple with the anti-bonding π-orbital of oxygen and activate it to improve the ORR.
In general, the surface polarized active sites may also indicate the existence of strong interactions with the intermediates during reactions. For instance, Chen et al. calculated and found that Cu2@C2N may synergistically exhibit a large charge density of +0.62e− than that of Cu-bonded N atoms (−0.09e−) during CO2RR.91 Later, Zapol et al. found that for bimetallic dimers, the charge difference (ΔQ(N–M)) was linearly correlated with the electronegativity difference (ΔχM–N).76 Accordingly, the total electron transfer from the metal dimers to the substrate could be described using χMN = 0.69χN + 0.31χM. This equation suggested that the total charge transferred between the cluster and the substrate is related to the electronegativity difference of metals. This charge transfer may affect the subsequent binding strength of intermediates during reactions. Later, Wu et al. were able to establish an equation of η = 5.67Δχ + 0.13 to describe such relationships of activity with the electronegativity difference.92 Du et al. found that in the Mo–Ru model for NRR, the Mo-end could transfer 0.35e− and the Ru-end could transfer 0.18e− to the N2 molecule.93 Thus, the electrons in the d orbitals of Mo and Ru atoms may be donated to the empty π* orbital of N atoms to activate the triple bond in N2. In contrast, the N2 molecule on the SAC may have one N atom gaining electrons and the other one losing electrons. The charge difference of N atoms adsorbed onto Mo and Ru could also polarize the N2 molecule to further weaken the triple bond. Besides, through the bond order analysis of projected crystal orbital Hamilton popular (pCOHP), the integrated pCOHP value (ICOHP) of −7.19 for the adsorbed N–N bond on Mo–Ru was also closer to the Fermi level than that of −10.15 on the Ru SAC, suggesting a stronger N–N bond for the latter when interacting with the N2 molecules.
2.3 Techniques in modulating key parameters
In the above section, we mentioned that the parameters such as the degree of d-band filling, the magnetic moment, charge difference, and electronegativity could result in the synergistic effects of multi-atom cluster catalysts. As these parameters are physical variables experimentally adjustable, the understanding of such parameters from an experimental point of view could help better modulate them.94–96 From the atomic level, the introduction of one or more kinds of atoms in the vicinity of active sites can form new atomic interfaces. Such an atomic interface may tune the electronic configuration of the active sites and lead to new binding sites, modes, and different binding strengths with the intermediates. Besides, the interaction of catalysts with the substrate cannot be neglected when considering factors influencing catalytic activity. Previous research has disclosed that the selection of diverse substrates can affect selectivity through effective (but varied) interactions between the catalytic sites and the substrate.76,77 The substrate can act as an electron sponge and help tune the valence state of the active sites, may stabilize the metal atoms and can facilitate the electron transfer to the catalytic moieties.27 Research also indicates that the coordination environment of the active sites can affect the activity of catalysts.90 Therefore, we will elaborate on the modulation of the atomic interface interactions from these three aspects: inter-atomic interface, coordination environment, and substrate (Fig. 4d–f).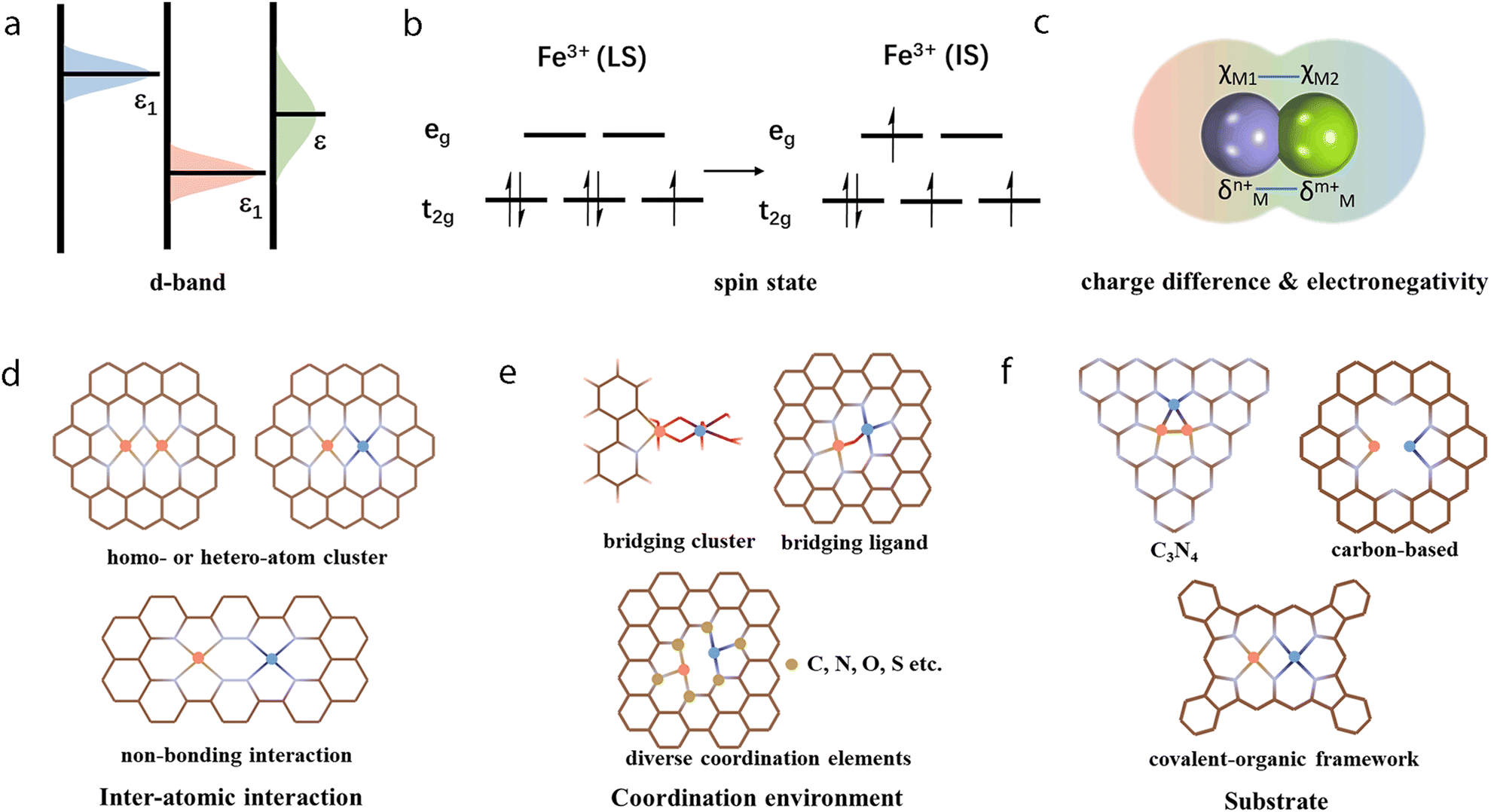 | ||
| Fig. 4 The origin of synergistic effects in multi-atom cluster catalysts, including (a) d-band filling, (b) spin state, (c) charge difference and electronegativity. Modulation techniques, including (d) inter-atomic interaction, (e) coordination environment, (f) substrate. | ||
For instance, Sun's group identified with the help of DFT models that the use of metal dimers (Cu2, MnCu, and NiCu) for CO2RR could offer different selectivity. Cu2 was found to be active for CO production, MnCu was promising for CH4 generation, while NiCu yielded methanol.97 Furthermore, a detailed inspection of metal dimers and single-atom catalysts for ORR by Hankel et al. found that CoPt@N8V4 was more promising for ORR than the others.89 Besides, the DFT calculations disclosed a volcano correlation of onset potential with the free energy of the adsorbed hydroxyl intermediate, where the -OH group can be related to the spin moment change of cobalt atoms through d-orbital splitting. Thus, the introduction of the additional atoms could result in the tuning of d-orbitals and the reaction diversity of electrocatalysts. In addition, researchers recognized that the d-band center could also be used to evaluate the activity of multi-atom cluster catalysts. A systematic investigation of heteroatom metal dimers anchored on graphdiyne monolayers (FeM@ and NiM@GDY, M = Ti, V, Cr, Mn, Fe, Co, Ni, and Cu) for NRR suggested that FeCo@ and NiCo@GDY were suitable for NRR with HER suppression ability. This might have been due to the modulated d-band center by effective cooperation of the additional hetero-atom.98
It is worth pointing out that the above-mentioned modulation of the materials should be regarded as a complex process of tuning the electrocatalysts. In most systems, electrocatalysts are not affected merely by one kind of effect, and therefore, multi-effects should be considered. One example that has been reported is for NRR, with an iron-based metal dimer anchored on graphene (Fe–TMDA/GS, TM = Ti, V, Cr, Mn, Fe, Co, Ni, and Cu), where Fe–TiDA/GS had the highest NRR activity among them, with a limiting energy of 0.88 eV.113 The DFT calculation results showed that an effective electron transfer might have resulted in moderate nitrogen adsorption for Fe–TiDA/GS, and the introduction of a second metal atom might have promoted the electron transfer between N2 and the substrate, finally improving the catalytic activity of NRR. Another example is one using Fe–Co dual metal-doped N-coordinated graphene (FeCoN6, FeCoN7, and FeCoN8) for ORR/OER.43 It was reported that FeCoN7 and FeCoN8 were more promising for ORR/OER than FeCoN6, and Co was found to be more active than Fe. However, the presence of Fe was also required as it could modify the electronic structure of the Co atom and helped improve the binding behavior to the *O intermediate. Besides, the d-band center of the Co atom could be correlated with the ΔGO* of the intermediates during reactions for predicting the overpotential of such Fe–Co dual metal catalysts for ORR/OER.
3. The scaling relationship
Multi-atom cluster catalysts represent a new class of catalysts that combine the concept of single-atom catalysts and multiple atom clusters through the introduction of additional atoms to single-atom catalysts.114–116 Either homo-nuclear or hetero-nuclear moieties can be constructed as multi-atom cluster catalysts by the selection of additional atoms.117,118 Multiple active sites are beneficial for electrochemical reactions with a complex reaction path and intermediates possessing several binding sites. For example, in the electrochemical CO2 reduction, several kinds of intermediates (*CHO, *COOH, *COH, etc.) including C-affinity and O-affinity sites can bind to multiple active sites of an electrocatalyst at the same time and lead to a different binding mode than that of SACs, which may help either lower the reaction barrier or change the reaction path, resulting in diverse selectivity and enhanced activity.48,119,120 Such decoupling of the original scaling relationship also requires new descriptors, all of which have further been discussed below.3.1 The origin of scaling relationship
In order to explore the most suitable materials for a certain reaction, one has to minimize the overpotentials (η) during reactions.121 Researchers are able to predict η in electrocatalytic reactions by calculating the relative stability of adsorbed intermediates using the computational hydrogen electrode (CHE) model.119,122 The rate-determining step energy, namely limiting potential (![[U with combining low line]](https://www.rsc.org/images/entities/i_char_0055_0332.gif) lim), calculated from the energy difference of adsorbed intermediates, is used as per the below equation to obtain η:
lim), calculated from the energy difference of adsorbed intermediates, is used as per the below equation to obtain η:| η = Ueq − Ulim | (2) |
Furthermore, the classical Sabatier principle suggests that the best catalysts should possess an optimum bond strength between the adsorbed species and surfaces, neither too weak to disfavor activation nor too strong to prevent the desorption of the adsorbed species.123 Previous research has successfully established thermodynamic energy scaling relationships, generally linearly related, which can be used to correlate different adsorbed species across the reaction path for a series of catalyst surfaces.124–126 One of the early examples was reported by Abild-Pedersen et al., who suggested that the linear scaling relationships may exist for the intermediates of CHx (x = 0, 1, 2, 3), NHx (x = 0, 1, 2), OHx (x = 0, 1), and SHx (x = 0, 1) to that of C, O, N, and S atoms, respectively, during the reaction process.127 In general, considering the adsorbates AHx and the relevant A atom as a common expression, they assumed that A has an optimal electron density of n0 with the contribution of both x hydrogen atoms and surface. From the bond order conservation view,128 the surface contribution of electron density (nsurf) to A can be written as
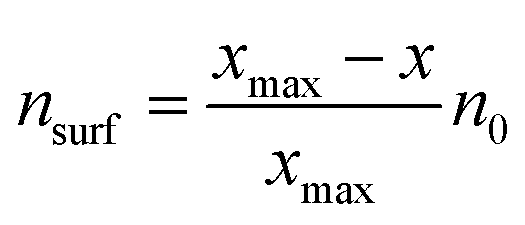 | (3) |
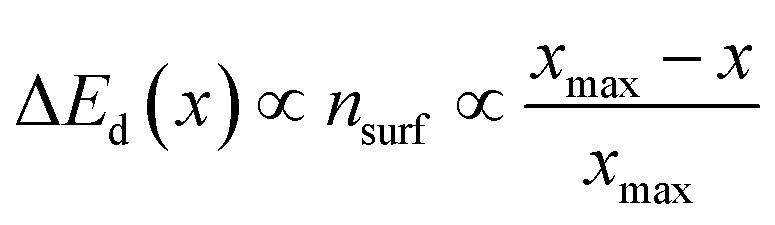 | (4) |
| ΔEAHx = γΔEA + ξ | (5) |
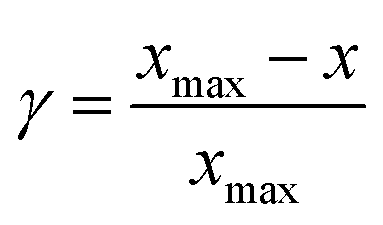 | (6) |
Eqn (5) suggests that a linear relation of adsorption energy exists between adsorbates AHx and A (Fig. 5), where the slope γ is constant and can be obtained with the bond-counting rules and the intercept ξ is sensitive to different coordination environments. According to eqn (6), the slope between adsorbate OH and O is 0.5 when considering that xmax for the O atom is 2, while the slope between CH3, CH2, CH, and C is 0.25, 0.5, and 0.75, respectively, when considering that xmax for C is 4. This can explain well the trend found by Abild-Pedersen et al. The concept of scaling relationships can be further extended to other systems with complex intermediates.129–131 For instance, the scaling relation of ΔG(*OOH) = ΔG(*OH) + 3.2 eV was found to explain the OER/ORR trend for the surface of low index metals, alloys, metal oxides, etc.132 The scaling relationships also exist in electrocatalytic CO2 reduction, where the adsorption energy of Eb(*CHO) = 0.88Eb(*CO) + 2.03 eV, Eb(*COOH) = 0.73Eb(*CO) + 1.82 eV, Eb(*CH2O) = 0.54 Eb(*CO) + 0.25 eV, etc. can be found.45 Furthermore, the combination of the linear relations with the Brønsted–Evans–Polanyi (BEP) correlations of activation energy and reaction energy for surface reactions can be used to establish the full potential energy diagram for a certain reaction, which can predict the maximum reaction rates and screen promising catalysts.133
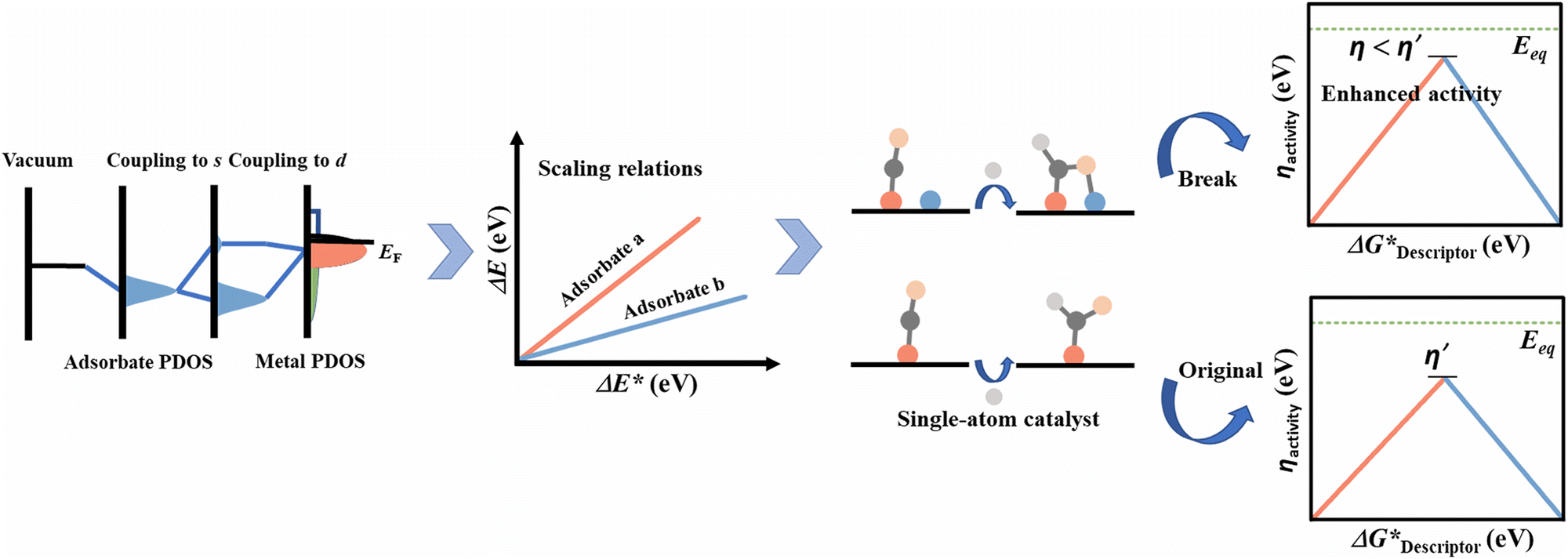 | ||
| Fig. 5 Illustration of the interaction between the adsorbate and the surface of the electrocatalyst to form the linear relations and to break such relations during a reaction (the left figure was replotted with permission,148 Copyright 2008 Elsevier). | ||
3.2 The limitation and breaking of the scaling relationship
The combination of scaling relationships and CHE models can facilitate the prediction of optimal electrocatalysts for certain electrocatalytic reactions with fewer independent variables to be considered. However, these relationships inevitably impose a thermodynamic limitation on the catalysts to be predicted. For reactions with complex reaction intermediates of the same central atom, like that in CO2RR, it is hard to circumvent the relationship by strengthening the adsorption energy of one kind of intermediate. For example, the strengthening of adsorption energy for *CHO would weaken it for the other (e.g. *CO) due to the linear relationship between them.134,135 Similarly, in the ORR, the scaling relationships exist between intermediates *O, *OOH, and *OH. The apex points of the volcano curves represent a maximum limiting potential of around 0.86 V. As the equilibrium potential for each elementary step is 1.23 V with a linear relation of ΔG(*OOH) = ΔG(*OH) + 3.2 eV, the minimum η is calculated to be 0.37 V from equation 2.121 However, if the adsorbing species could interact with a surface carrying various kinds of active sites, it may allow circumventing the scaling relationships.5,134,136–140 Jiang et al. established a new relation of ΔG(*OOH) = ΔG(*OH) + 2.41 eV with dual active site models, which is very close to the ideal linear relation of ΔG(*OOH) = ΔG(*OH) + 2.46 eV.70 The new relation could provide a maximum limiting potential of around 1.04 V and proved the possibility of using multi-atom clusters to break the scaling relationships.For multi-atom cluster catalysts, the introduction of additional atoms to form multi-active sites may decouple the inherent scaling relationships.73,141–143 This strategy is quite useful for reactions that involve complex reaction paths.70,109,144 An example was reported by Wang et al. in the context of CO2RR, wherein they established models containing twenty-one kinds of heteronuclear transition metal dimers embedded in a C2N substrate.48 Through the calculations, they found that both the metal atoms could bind with the adsorbed species through oxygen and carbon sites. This resulted in the decoupling of Eb(*CO) versus Eb(*CHO) and Eb(*CO) versus Eb(*COOH) from the original scaling relationship. Therefore, the free energy profiles obtained showed that both CuCr/C2N and CuMn/C2N could produce CH4 with limiting potentials as low as −0.37 V and −0.32 V, respectively. These results proved the efficiency improvement as a result of introducing multi-active sites which helped in circumventing the original scaling relationships to obtain complex products with high activity.
3.3 Descriptor
The electrochemical reactions are usually multi-step processes accompanied by multiple electron–proton transfers. During a reaction, the interaction of adsorbed species with the surface of a catalyst may lead to diverse binding strength on different catalysts, which results in complex reaction pathways and the production of diverse products. To selectively control the production of desired products through a certain reaction pathway with high activity, one has to screen for the most suitable catalysts, which is usually tedious and laborious. Thus, it will be helpful if key parameters could be identified and help reduce the complexity of grouping the reactions and guide a rational catalyst design strategy with the assistance of techniques such as DFT calculations, and also with the newly emerging machine learning technique.132,145 In this regard, the descriptors mentioned in the previous section could help provide a catalyst design strategy by elucidating the activity trends and hence unraveling the most promising catalysts.So far, the successful application of a descriptor for this purpose has been exemplified in the use of the binding energy of adsorbed species *O and *OH as a descriptor 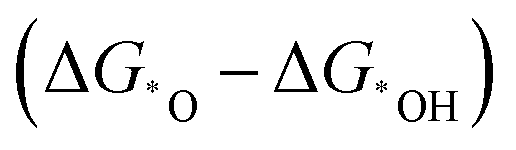 for disclosing the activity trend of OER/ORR reactions.132,146,147 However, it is hard to regard it as a universal principle to predict new series of catalysts as it is still ambiguous about the intrinsic factors of the catalysts that govern the binding strength of adsorbates in a simple adsorption–activity relationship. Therefore, it is necessary to identify the real intrinsic characteristics of the catalysts to help establish a more universal descriptor that can help adjust the adsorption behavior of adsorbates, in turn affecting the activity.148 Some progress has been achieved for electrocatalysts, where the d-band center,149–152 p-band center,153 eg occupancy,125,154 surface distortion,155 coordinative unsaturated metal cation,156 electronegativity,126etc., have been adopted to elaborate on these relationships. Nevertheless, none of these descriptors hold promise to represent a universal descriptor to disclose the intrinsic feature of diverse catalysts. Therefore, in the development of multi-atom cluster catalysts, specific descriptors should be built for a better adjustment of the adsorption energies of intermediates and the activity of such catalysts.98,157
for disclosing the activity trend of OER/ORR reactions.132,146,147 However, it is hard to regard it as a universal principle to predict new series of catalysts as it is still ambiguous about the intrinsic factors of the catalysts that govern the binding strength of adsorbates in a simple adsorption–activity relationship. Therefore, it is necessary to identify the real intrinsic characteristics of the catalysts to help establish a more universal descriptor that can help adjust the adsorption behavior of adsorbates, in turn affecting the activity.148 Some progress has been achieved for electrocatalysts, where the d-band center,149–152 p-band center,153 eg occupancy,125,154 surface distortion,155 coordinative unsaturated metal cation,156 electronegativity,126etc., have been adopted to elaborate on these relationships. Nevertheless, none of these descriptors hold promise to represent a universal descriptor to disclose the intrinsic feature of diverse catalysts. Therefore, in the development of multi-atom cluster catalysts, specific descriptors should be built for a better adjustment of the adsorption energies of intermediates and the activity of such catalysts.98,157
Wu's group developed a series of homonuclear (M2, M = Mn–Cu) and heteronuclear (FeM, M = Mn–Cu) nitrogen co-doped binuclear transition metal models for ORR, apart from developing a linear relationship between the overpotential and electronegativity difference. They also developed descriptors based on free energies (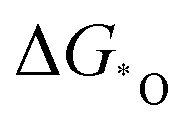 and
and 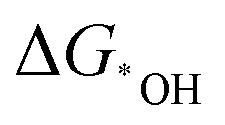 ) and d band center to screen these catalysts. FeCoN6-Gra turned out to be the most promising one, which coincided with the experimental results.92 Similar descriptors can also be used for the NRR with the use of two-dimensional bi-atom catalysts, where the adsorption energy of *H and *N2H can be used as a suitable descriptor.158 The above descriptor is commonly used in diverse electrocatalytic systems, yet it has the limitation to cover all kinds of catalysts.159 Specific descriptors considering the parameters mentioned in the above section to screen efficient multi-atom cluster catalysts effectively are still waiting to be developed. These descriptors, including the ones based on the N2 polarization charge for NRR by Zhao's group and the dissociative chemisorption energy of water to depict HER by Qiao's group, could help the rational design of multi-atom cluster catalysts.160,161 However, as those descriptors can help correlate the intrinsic feature of catalysts to the activity and selectivity among a series of catalysts, the use of more accurate and universal descriptors is crucial in assisting the experimental screening of optimized multi-atom cluster catalysts and is worth more endeavors.162
) and d band center to screen these catalysts. FeCoN6-Gra turned out to be the most promising one, which coincided with the experimental results.92 Similar descriptors can also be used for the NRR with the use of two-dimensional bi-atom catalysts, where the adsorption energy of *H and *N2H can be used as a suitable descriptor.158 The above descriptor is commonly used in diverse electrocatalytic systems, yet it has the limitation to cover all kinds of catalysts.159 Specific descriptors considering the parameters mentioned in the above section to screen efficient multi-atom cluster catalysts effectively are still waiting to be developed. These descriptors, including the ones based on the N2 polarization charge for NRR by Zhao's group and the dissociative chemisorption energy of water to depict HER by Qiao's group, could help the rational design of multi-atom cluster catalysts.160,161 However, as those descriptors can help correlate the intrinsic feature of catalysts to the activity and selectivity among a series of catalysts, the use of more accurate and universal descriptors is crucial in assisting the experimental screening of optimized multi-atom cluster catalysts and is worth more endeavors.162
4. Preparation method of multi-atom cluster catalysts
To better utilize the resulted synergistic effects in electrocatalysis, it is necessary to prepare multi-atom cluster catalysts with precise structural characteristics, and this requires researchers to develop novel and rational strategies. However, accurate control over the synthesis of a given homo- or hetero-composition for multi-atom cluster catalysts remains more challenging than the well-developed top-down and bottom-up synthesis strategies of SACs. The metal atoms also tend to aggregate to form nanoparticles or decompose to single atoms during the synthesis process. It would be of utmost importance to find out rational ways that facilitate the formation of multi-atom cluster catalysts without any unwanted structures. To date, various strategies, such as precursor preselection, spatial confinement–pyrolysis, coordination stabilization, and electrochemical strategies, atomic layer deposition method, and some other methods (electrospinning, ball-milling, mass-selected soft landing, etc.) have been reported to prepare multi-atom cluster catalysts and are discussed below (Fig. 6).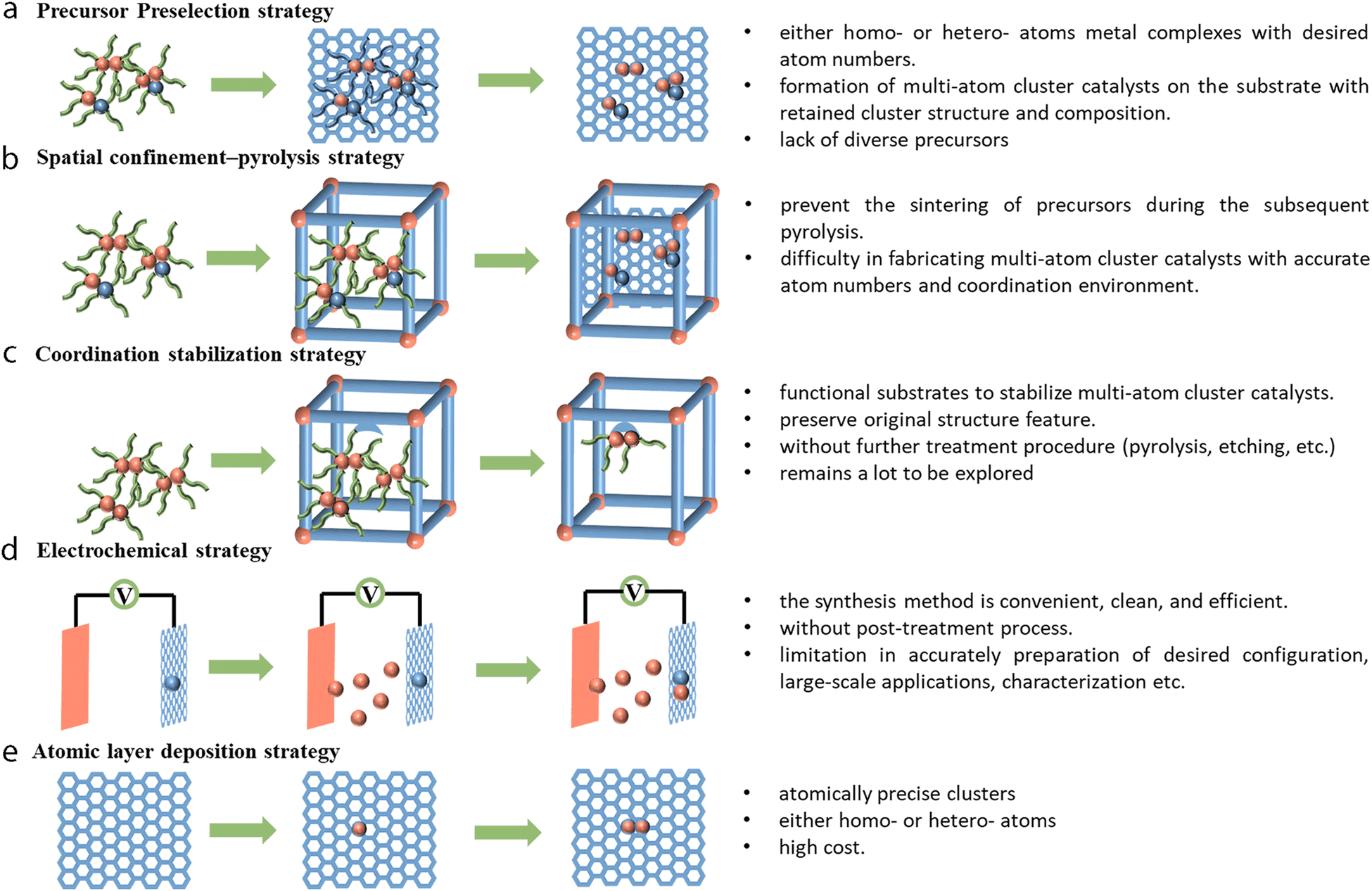 | ||
| Fig. 6 (a) Precursor preselection strategy. (b) Spatial confinement-pyrolysis strategy. (c) Coordination stabilization strategy. (d) Electrochemical route strategy. (e) Atomic layer deposition strategy. | ||
4.1 Precursor preselection strategy
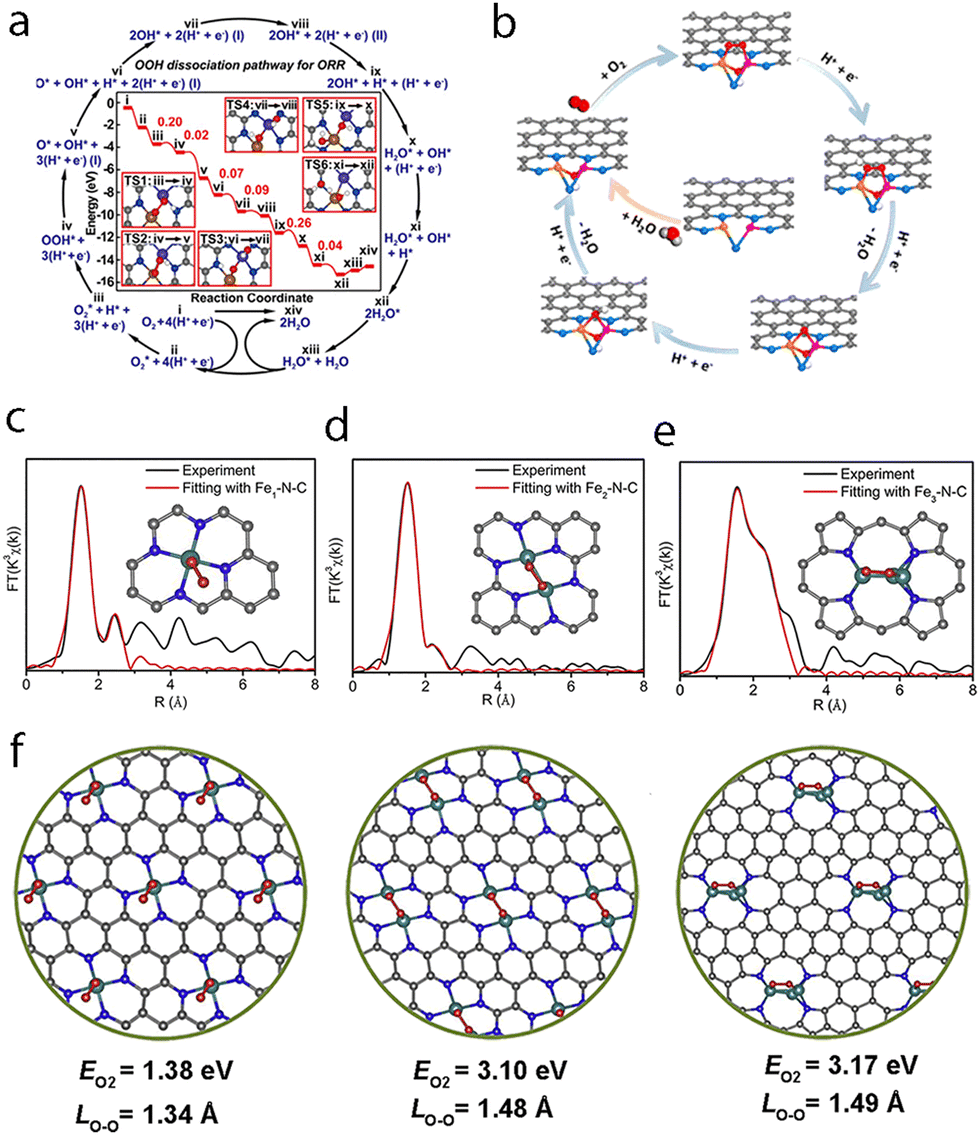
The precursor preselection method can anchor metal clusters (such as metal complexes) of either homo- or hetero-atoms with desired atom numbers onto a substrate (oxides, graphene, etc.).163 Such precursors possess characteristic metal clusters protected by a chelating ligand, which can help hamper the aggregation or decomposition of the metal clusters. Subsequent pyrolysis ensures the formation of multi-atom cluster catalysts on the substrate with retained cluster structure and composition. Using this strategy, Li et al. reported the use of bis(dicarbonyl-cyclopentadienyl iron) (Fe2O4C14H10) as a precursor, which was then dispersed on mpg-C3N4.37 Careful pyrolysis under a 5% H2/Ar atmosphere at 300 °C finally gave Fe2/mpg-C3N4. The precursor preselection method was also used to prepare a series of tunable Fe clusters using Fe(acac)2, Fe2(CO)9, and Fe3(CO)12, which were encapsulated in ZIF-8 to prepare Fe–N–C, Fe2–N–C, and Fe3–N–C respectively.164 Later, trinuclear Fe3, Fe2Zn, and Fe2Co were reported to be post-impregnated into [Zn(BPB)]·guest (AIST-1) for the preparation of Fe3-NPs/GNCL, Fe2-GNCL, and Fe2/Co1-GNCL with isolated Fe2 and Co sites on the substrate.165 Recently, Yao et al. reported that using Ni2(dppm)2Cl3 (dppm = bis(diphenylphosphino)methane) as the precursor, further encapsulation and pyrolysis in MOFs could produce a Ni2/NC dual-site material.166 The precursor preselection method turned out to be a convenient method in terms of both the availability of precursors and the ease of operation for the preparation of multi-atom cluster catalysts. Although there are already some examples available for preparing the homo-clusters with such a method, the demand for the synthesis of hetero-clusters still calls for more endeavors due to the lack of suitable precursors.4.2 Spatial confinement–pyrolysis strategy
The spatial confinement–pyrolysis method is widely used as an effective tool for the synthesis of atomic-scale materials. Considering that mere impregnation may cause the formation of isolated SACs or give rise to nanoparticle impurities rather than multi-atom cluster catalysts, the encapsulation of metal precursors in spatially confined porous materials such as metal–organic frameworks (MOFs) or covalent–organic frameworks (COFs) can prevent the sintering of precursors during the subsequent pyrolysis.160,161 Several paths of using the spatial confinement method are listed. In the first example, Wang et al. developed a Zn/Co bimetallic MOF by using metal-organic frameworks as a confined space. Then, such a porous material could be used as a host to capture the second element of iron salt in the cavities.45 During the pyrolysis process, the Zn2+ ions get reduced by the as-generated carbon and are vaporized due to the relatively low boiling point of Zn, while the Fe3+ moieties are reduced and get connected with the cobalt atoms in their vicinity to form the DAC Fe–Co structure. This atomic phase transformation could be traced through HAADF-STEM images and was proved with EXAFS fitting. Another example is the use of as-prepared CoNi-MOFs to fabricate a Co–Ni DAC.167 The CoNi-MOFs with two different metal elements could be prepared from chemical co-precipitation of Ni salt and Co(CN)63− during the synthesis. After being coated with dopamine during the self-polymerization process, the material was carbonized with NH3 at 500 °C to obtain CoNi–NPs/NC. The nanoparticles were then removed by acid leaching to obtain CoNi-SAs/NC. It is worth noting that the above two paths still lack precise control in obtaining multi-atom custers with designated structures, and the spatial confinement method to encapsulate precursors within well-defined structures could potentially offer better control of the composition. One example was reported by Ji et al., where Ru3(CO)12 was dispersed in the cages of ZIF-8 through one-pot synthesis wherein the as-prepared Ru3(CO)12@ZIF-8 decomposed to uniformly dispersed Ru3 clusters upon heat treatment.38 The method of spatial confinement is much easier to adapt to fabricate multi-atom cluster catalysts. However, as mentioned, an arbitrary combination of different elements to form clusters with an accurate multi-atom cluster structure and coordination environment remains difficult for such techniques; thus it is better if smart precursors with well-defined structures could be utilized.4.3 Coordination stabilization strategy
In the spatial confinement–pyrolysis strategy, it is necessary to overcome the high surface energy and anchor the clusters to the substrate by pyrolysis treatment. However, multi-atom clusters can also be stabilized directly by strong surrounding coordination sites to overcome their high surface energy.168,169 Therefore, substrates possessing coordination sites, such as MOFs, COFs, organic ligands, functional carbon substrates, etc., could be used to prepare multi-atom cluster catalysts.170 For instance, a series of crystalline trinuclear copper clusters were prepared by Lan et al. through solvothermal reaction using copper halides, imidazole, and pyrazole, where their precise structural features were verified from the single-crystal diffraction analysis.170 From the result, the Cu3(μ3-OH) core was stabilized by equatorial bridging pyrazole ligands, and the remaining square-planar vacancy of copper was coordinated by the imidazole ligand. Another example was reported by Duan et al. with the use of graphdiyne (GDY) to stabilize copper nanoclusters.171 The triple bond of graphdiyne made it behave as an electron-donating substrate for immobilized copper atoms. The uniform pore of graphdiyne rendered it the ability to confine the size of metal clusters and enhance its interaction with electrolytes. The use of ligands to stabilize the metal clusters was reported by Li et al.,172 where the co-facial dual palladium complex, {cis-[(2,2′-bipyridine)palladium(II)]2(μ-1,3-NO3)2} core, could be precipitated onto acetylene black to prepare the Pd2 catalysts. Here, each palladium atom was coordinated by a 2,2′-bipyridine ligand. The bidentate nitrate in the complex helps to chelate and bridge two palladium atoms to form the co-facial arrangement. In summary, the method of coordination stabilization strategy could prepare multi-atom clusters without further treatment procedures (pyrolysis, etching, etc.) and thus can preserve the original structure feature. This may help to build a precise moiety model to investigate the reaction mechanism and such a strategy remains to be explored a lot.4.4 Electrochemical strategy
The electrochemical route could be regarded as convenient, clean, and efficient for producing multi-atom cluster catalysts without the post-treatment process.173 For instance, through the electrochemical leaching, segregation, and deposition process, the atoms may be etched and dealloyed gradually from one electrode and migrate through the electrolyte to deposit onto the other electrode under an applied voltage. The electrochemical potential for leaching an element from the bulk materials can be overcome by operating at a suitably applied voltage. Experimentally, such a method was reported for the synthesis of SACs and can be extended for the synthesis of multi-atom clusters.174 Theoretically, Liu et al. investigated the feasibility and universality of such a strategy to prepare a series of materials using oxides, graphene, and graphdiyne as substrates with a different atomic scale of the catalysts (ranging from SACs to MACs).175,176 Zhang et al. prepared a Co–Pt DAC (A–CoPt–NC) with this method to prepare multi-atom cluster catalysts.177,178 The as-prepared Co-MOF was first pyrolyzed to form cobalt nanoparticles (Co-NC), and then the Co-NC were drop-cast onto a glassy carbon electrode and with the subsequent long-term cyclic voltammetry activation (0.1 to 1.1 V vs. RHE, 8000 cycles) of the sample with Pt wire as a counter electrode, A–CoPt–NC could be prepared by slow segregation and deposition of Pt on to the glassy carbon electrode. Cyclic voltammetry scanning can also be used to deposit metal ions directly from the electrolyte or form clusters. One such example has been reported by Wang et al., where the sulfur atom decorated graphene oxide (HG) was used to chelate Ni(acac)2 to form HG–Ni.179 Then the as-prepared HG–Ni was drop-cast onto a glassy carbon electrode (GCE) followed by the cyclic voltammetry scanning of pristine HG–Ni/GCE with FeCl3 in the electrolyte to form a pristine HG–NiFe dual-site. All these electrochemical methods can be extended to fabricate materials with different substrates, but limitations still exist when faced with the accurate synthesis of desired configuration, large-scale synthesis, characterization, etc.4.5 Atomic layer deposition strategy
Atomic layer deposition (ALD) can be used for the deposition of noble metals or metal oxides to form atomically precise clusters with the use of gas-phase precursors, and this method allows the preparation of atomically precise clusters of either homo- or hetero-atoms. Yan et al. reported the preparation of oxidized Pt2 dimers on graphene for hydrolytic dehydrogenation of ammonia borane.180 A first dose of precursor trimethyl(methylcyclopentadienyl)-platinum(IV) (MeCpPtMe3) was introduced. Then the self-limiting surface reaction between graphene and MeCpPtMe3 followed by subsequent exposure to O2 at 250 °C helped to form Pt1/graphene. The next reaction of Pt1 single atoms as nucleation sites with MeCpPtMe3 at 150 °C helped to form Pt2/graphene without any metal aggregation. The subsequent exposure of samples to ozone successfully removed the ligand to form oxidized Pt2Ox, which exhibited a much higher specific rate of 2800 molH2 molPt−1 min−1 for hydrolytic dehydrogenation of ammonia borane. This was around 17- and 45-fold higher than that of the Pt SAC and nanoparticles on graphene, respectively. The ALD technique calls for a stable interaction between the substrates and the precursors and the precursors should be easy to evaporate and anchor onto the substrates. The subsequent removal of protective ligands to obtain catalysts requires researchers to find relatively cheap sacrificial precursors to reduce the cost.4.5 Other strategies
The techniques of electrospinning,181 ball-milling,182 soft template-directed interlayer confinement,183 mass-selected soft landing,33etc. are also promising for preparing multi-atom cluster catalysts. For instance, Fu et al. reported using Fe, Co-Phen, polyacrylonitrile (PAN), and ZnO nanoparticles as precursors for the preparation of nanofibers by the electrospinning technique.184 The ZnO introduced could be reduced and evaporated during the subsequent pyrolysis process to create numerous hollow pores. These hollow pores could combine with the nanofibers prepared through the electrospinning technique to increase the total metal loading to about 9.8 wt%. The ball-milling method is capable of kilogram-scale synthesis of materials,182,185 and as such, M2/N-C (M2 = Ni, Co) and Fe1M1/N-C (M1 = Pd, Ni) were prepared and used for the reverse water–gas shift (r-WGS) reactions with the ball-milling method.163 The technique of using soft and reactive landing of mass-selected ions to deposit them onto the surfaces turns out to be a powerful tool due to its ability to accurately control the synthesis of catalysts with selected element species, cluster size, and total charge. This technique can be used to build more accurate and reliable catalytic models, which can help gain insights into the nature of the catalytic reactions.33 For instance, Let et al. reported that Agn (n = 2 to 4) clusters go through a quadrupole mass filter and deflector assembly and are then deposited on the oxide supports.186 Similarly, as-generated Cu4 clusters in Al2O3 thin film exhibited CO2 hydrogenation to methanol at both high and low CO2 partial pressures.34 However, this method is of both high cost and low yield, thus limiting its practical application.The preparation methods summarized above can complement each other in the fabrication of multi-atom cluster catalysts. So far, most of the methods developed are not universal and there is a high demand for the development of new strategies that can be used to prepare arbitrary and precise homo- or hetero-clusters. This can be regarded as a vital and fundamental requirement for the further study of the structure–activity relationship.187
5. Advanced characterization methods of multi-atom cluster catalysts
The successful preparation of multi-atom cluster catalysts should be confirmed using advanced characterization methods.25 Besides, verifying the existence of effective synergistic effects through observing their physical and chemical properties, such as the electronic state, coordination environment, morphology, the possible single-crystal structure, etc., is also necessary.188 Furthermore, monitoring the reaction kinetics (coordination environment, geometric structure, valence state, electronic structure, intermediate, adsorbate, etc.) during the catalytic process with multi-atom cluster catalysts with advanced characterization techniques is also necessary.189 This may deepen the comprehensive understanding of intrinsic features of these materials, facilitate their mechanistic investigation, and thus help to develop advanced electrocatalysts.5.1 Structure characterization
Considering that the amount of multi-atom cluster catalysts loaded on the substrate is usually quite small, only highly sensitive techniques can be used for their characterization. To date, the following characterization techniques such as scanning transmission electron microscopy (STEM),190,191 X-ray absorption spectroscopy (XAS),192,193 single-crystal X-ray diffraction (SC-XRD),194 X-ray photoelectron spectroscopy (XPS),195etc., are commonly used to probe the structure information of multi-atom cluster catalysts.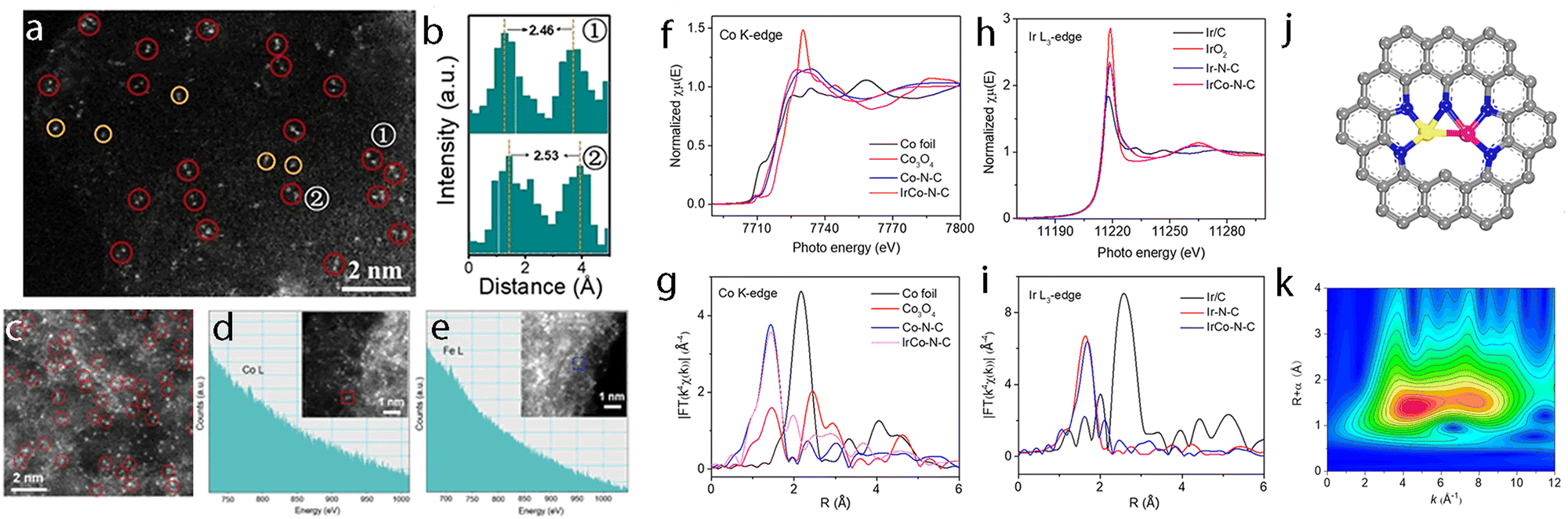 | ||
| Fig. 7 (a) HAADF-STEM image and (b) the corresponding intensity profile of Ni-CNC-1000. Reproduced with permission.181 Copyright 2021 John Wiley & Sons. (c) Aberration-corrected HAADF-STEM image. The dashed circles highlight the paired bright spots. (d) Cobalt element and (e) Iron element identification with EELS analysis of the selected paired spot and single spot in the inset HAADF-STEM images of Co2/Fe–N@CHC. Reproduced with permission.191 Copyright 2022 John Wiley & Sons. (f) Co K-edge XANES spectra. (g) Fourier transform of extended X-ray adsorption fine structure (FT-EXAFS) spectra of Co K-edge. (h) Ir L3-edge XANES spectra. (i) Fourier transform of extended X-ray adsorption fine structure (FT-EXAFS) spectra of Ir L3-edge. (j) Illustration of the structure of the IrCo–N–C catalyst. (k) WT of the Ir L3-edge for IrCo–N–C. Reproduced with permission.193 Copyright 2021 American Chemical Society. | ||
5.2 Electrocatalytic process evolution
The real-time characterization techniques are also indispensable for observing catalytic processes on multi-atom cluster catalysts.188,189 So far, several in situ/operando techniques have been explored, including Raman spectroscopy, Fourier transform infrared spectroscopy, XAS (XANEs and EXAFS), etc.198 By observing the coordination environment and geometric structure evolution, valence state and electronic structure comparison, and intermediate and adsorbate identification, opportunities could be identified to observe the local active sites and the reaction mechanisms. A brief introduction will be given in this section and a further discussion is provided in Section 7 (Tables 1–5).| Catalysts | Metal content (ICP/XPS, wt%) | M–M distance (STEM/XAS, Å) | Electrolyte | Overpotential at 10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|---|
| Pt–Ru dimer on NCNTs | Pt 0.9, Ru 0.31/— | 2.4 ± 0.4/2.34 ± 0.04 | 0.5 M H2SO4 | ∼20 | 28.9 | 47 |
| A–CoPt–NC | — | 3.4/— | 0.5 M H2SO4/1 M KOH | 27/50 | 31/48 | 178 |
| Pd/Cu–Pt NRs | — | —/2.64 | 0.5 M H2SO4 | 22.8 | 25 | 210 |
| Pt1@Fe−N−C | Pt 2.1, Fe 1.0/— | 3.0/— | 0.5 M H2SO4 | 60 | 42 | 211 |
| Fe–Rh SAs | —/Rh 1.1, Fe 1.8 | —/2.59 ± 0.02Fe–Fe, 2.59 ± 0.04Rh–Fe | 0.5 M H2SO4 | 36 | 26 | 212 |
| NiCo–SAD–NC | Ni 3.719, Co 3.331/— | 2.41 ± 0.24/2.55 | 0.5 M H2SO4/1 M KOH | 54.7/61 | 31.5/55 | 213 |
| W1Mo1–NG DAC | W 6.82, Mo 1.02/— | 3.0–3.6/3.29 | 0.5 M H2SO4/1 M KOH | 24/67 | 30/45 | 214 |
| Catalysts | Metal content (ICP/XPS, wt%) | M–M distance (STEM/XAS, Å) | Electrolyte | Half-wave potential (V vs. RHE) | Onset potential (V vs. RHE) | Limiting current density (mA cm−2)1600 rpm | Tafel slope (mV dec−1) | Specific activity (mA cm−2) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| (Fe,Co)/N-C | Fe 0.93, Co 1.17/— | —/2.25 | 0.1 M HClO4 | 0.863 | 1.06 | ∼5.7 | 66 | 2.842 (0.90 V) | 45 |
| Ni66Fe34–NC | Fe 0.29, Ni 0.14/— | —/2.35–2.51 | 0.1 M KOH | 0.85 | — | ∼5.6 | 107 | — | 49 |
| Co2N5/Co NP | Co 4.3/Co 1.06 | 2.11–2.12/2.12 | 0.1 M HClO4 | 0.79 | 0.92 | ∼6.0 | 55.8 | — | 64 |
| FeNi–N6–C | Fe 1.448, Ni 1.472/— | — | 0.1 M HClO4 | ∼0.8 | — | ∼5.2 | — | — | 66 |
| Fe–NiNC-50 | Fe 0.33, Ni 0.57/— | 1.8/2.43 | 0.1 M KOH | 0.85 | ∼1.0 | ∼7.0 | 55 | — | 67 |
| FeCo-IA/NC (X60-900) | Fe 0.26, Co 1.06/— | — | 0.1 M KOH | 0.88 | 0.98 | ∼6.5 | 97 | — | 70 |
| (Zn,Co)/NSC | —/Zn 0.13, Co 0.26 | 2.64 ± 0.2/— | 0.1 M KOH | 0.893 | 1.07 | ∼5.3 | 50 | — | 101 |
| FeCoNx/C | —/Fe 1.06, Co 1.12 | 2.2–2.3/2.1–2.2 | 0.1 M HClO4 | 0.86 | 1.02 | ∼5.7 | 47.9 | 16.07 (0.80 V) | 105 |
| Fe2–N–C | Fe 0.38/— | —/2.60 | 0.5 M H2SO4/0.1 M KOH | 0.78/0.905 | — | ∼5.7/ | 83/— | 16.4 (0.75 V) | 164 |
| Fe2/Co1-GNCL | —/Fe 0.48, Co 0.19 | 2.6Fe–Fe/2.6896Fe–Fe | 0.1 M KOH | 0.846 | — | ∼5.8 | — | — | 165 |
| CoNi-SAs/NC | — | —/2.22 | 0.1 M KOH | 0.76 | 0.88 | 4.95 | — | 23.2 (0.50 V) | 167 |
| (Fe,Co)/CNT | Fe 1.21, Co 1.13/— | —/2.22 | 0.1 M KOH | 0.954 | 1.15 | 5.5 | — | 17.51 (0.90 V) | 175 |
| a(Co–Pt)@N8V4 | Co 1.72, Pt 0.16/— | —/2.5–2.9 | 0.1 M KOH | 0.96 | — | 5.7 | — | 1.41(0.90 V) | 177 |
| Fe,Co SAs-PNCF | Co 4.32, Fe 5.48/— | ∼2.30/2.37–2.55 | 0.1 M HClO4/0.1 M KOH | 0.78/0.93 | 0.94/1.04 | ∼5.8/∼6.2 | 126/77 | — | 184 |
| Co2/Fe–N@CHC | Co 2.13, Fe 0.98/Co 1.94, Fe 0.85 | 6.3–7.6Fe–Co/5.3–8.8Fe–Co, 2.41Co–Co | 0.1 M HClO4/0.1 M KOH | 0.812/0.915 | — | ∼5.8 | 73/62 | 8.36/9.44 (0.80 V) | 191 |
| IrCo−N−C | Ir 0.18, Co 1.02/— | ∼2.30/∼2.30 | 0.1 M KOH | 0.911 | 1.00 | ∼4.4 | 54 | 29.73 (0.85 V) | 193 |
| Pt1@Fe−N−C | Pt 2.1, Fe 1.0/— | 3.0/— | 0.5 M H2SO4 | 0.80 | 0.93 | ∼6.3 | — | — | 211 |
| M/FeCo-SAs-N-C | Fe 3.32, Co 3.33/Fe 5.12, Co 4.39 | —/2.52Fe–Fe | 0.1 M HClO4 | 0.851 | 0.981 | ∼4.8 | 83 | 32.04 (0.80 V) | 215 |
| AC@f-FeCoNC900 | —/Fe 0.53, Co 0.54 | —/2.55 | 0.1 M KOH | 0.89 | 1.05 | 5.75 | — | — | 216 |
| (Au-Co) DP-NPAs | Au 1.18, Co 3.48/Au 1.55, Co 3.35 | 2.05/1.93 | 0.1 M HClO4 | 0.820 | 0.918 | ∼4.7 | 69 | — | 218 |
| Zn/CoN-C | Zn 0.33, Co 0.14/— | 2.2 ± 0.4/2.1 | 0.1 M HClO4/0.1 M KOH | 0.796/0.861 | 0.97/1.004 | /6.1 | /67 | — | 219 |
| Meso/micro-FeCo-Nx-CN-30 | Fe 0.40, Co 0.12/— | — | 0.1 M KOH | 0.886 | 954 | 6.3 | — | — | 220 |
| Co/Zn–NCNF | Co 4.89, Zn 4.04/— | 2.3/2.49 | 0.1 M HClO4 | 0.797 | 0.997 | ∼5.4 | — | — | 221 |
| NCAG/Fe–Co | Fe 0.7, Co 1.1/Fe 0.9, Co 1.0 | —/2.787 | 0.1 M KOH | 0.89 | 1.04 | ∼5.6 | — | 8.5 (0.85 V) | 222 |
| Fe-Zn-SA/NC | Fe 0.22, Zn 0.1/— | —/2.45 | 0.1 M HClO4/0.1 PBS/0.1 KOH | 0.78/0.72/0.85 | 0.87/0.94/0.94 | 4.72/∼4.2/∼4.8 | 54.3/112.6/41.7 | — | 224 |
| Fe1Co1-CNF | Fe 0.62, Co 0.54/— | —/2.43–2.55 | 0.1 M KOH | 0.87 | 0.99 | 4.3 | 88 | — | 225 |
| FeCo-DACs/NC | Co 0.80, Fe 0.76/— | ∼4/— | 0.1 M KOH | 0.877 | 0.984 | 5.7 | 85 | 11.05 (0.85 V) | 228 |
| Ni7Fe3-CNG | Ni 2.2, Fe 5.3/Ni 3.4, Fe 2.2 | /∼3.0 | 0.1 M KOH | 0.82 | — | 5.6 | — | — | 257 |
| Catalysts | Metal content (ICP/XPS, wt%) | M–M distance (STEM/XAS, Å) | Electrolyte | Onset potential (V vs. RHE) | E (V) at 10 mA cm−2 | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|---|---|
| Ni66Fe34-NC | Fe 0.29, Ni 0.14/— | —/2.35–2.51 | 0.1 M KOH | 1.51 | 1.697 | 120 | 49 |
| Fe–NiNC-50 | Fe 0.33, Ni 0.57/— | 1.8/2.43 | 1 M KOH | 1.50 | 1.57 | 54 | 67 |
| NiFe@PCN | Ni 15.36, Fe 8.82/— | — | 1 M KOH | — | 1.54 | 38 | 69 |
| Fe2/Co1-GNCL | —/Fe 0.48, Co 0.19 | 2.6Fe–Fe/2.6896Fe–Fe | 1 M KOH | — | 1.58 | 70 | 165 |
| CoNi-SAs/NC | —/2.22 | 0.1 M KOH | — | 1.57 | 58.7 | 167 | |
| Co−Fe−N−C (Fe activated) | Co 9.4 (± 1.4) × 10−8, Fe 8.6 (± 1.6) × 10−9 mol cm2/— | —/2.51–2.71 | 1 M KOH (10 ppm Fe(III)) | — | 1.539 | 37 | 173 |
| HG–NiFe | — | —/2.7 | 1 M KOH (12 μM Fe(III)) | — | — | 39 | 179 |
| IrCo−N−C | Ir 0.18, Co 1.02/— | ∼2.30/∼2.30 | 0.1 M KOH | — | 1.53 | 79 | 193 |
| Pt1@Fe−N−C | Pt 2.1, Fe 1.0/— | 3.0/— | 0.1 M KOH | 1.33 | 1.54 | 62 | 211 |
| Meso/micro-FeCo-Nx-CN-30 | Fe 0.40, Co 0.12/— | — | 1 M KOH | 1.60 | 1.67 | 57 | 220 |
| NCAG/Fe–Co | Fe 0.7, Co 1.1/Fe 0.9, Co 1.0 | —/2.787 | 1 M KOH | — | 1.523 | 70 | 222 |
| Fe1Co1-CNF | Fe 0.62, Co 0.54/— | —/2.43–2.55 | 0.1 M KOH | 1.56 | 1.73 | 102 | 225 |
| FeCo-DACs/NC | Co 0.80, Fe 0.76/— | ∼4/— | 1 M KOH | — | 1.60 | 82.7 | 228 |
| Ni7Fe3-CNG | Ni 2.2, Fe 5.3/Ni 3.4, Fe 2.2 | —/∼3.0 | — | 1.50 | 74 | 257 |
| Catalysts | Metal content (ICP/XPS, wt%) | M–M distance (STEM/XAS, Å) | Electrolyte | Potential (V vs. RHE) | Partial current density (mA cm−2) | Faradaic efficiency (%) | Tafel slope (mV dec−1) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Ni/Fe–N–C | Ni 0.97, Fe 0.34/— | —/2.06 | 0.5 M KHCO3 | −0.70 | 7.4 (CO) | 98 (CO) | 96 | 2.13 | 46 |
| CuFe/N–C | Fe 0.52, Cu 1.74/— | —/2.53 | 0.5 M KHCO3 | −0.4 | ∼0.5 (CO) | 95.5 (CO) | 104 | 1.08 (−0.90 V) | 71 |
| Ni2−N6 | Ni 0.18/— | 2.9/2.83–2.94 | 1.0 M KHCO3 | — | 150 (CO) | 94.3 (CO) | 111 | — | 166 |
| Cu/C-0.4 | —/Cu 0.32 | —/∼2.5 | 0.1 M KHCO3 | −0.7 | 1.23total | ∼91 (ethanol) | 164 | 0.0145 | 168 |
| Cu/C-0.8 | —/Cu 0.65 | —/∼2.7 | 0.1 M KHCO3 | −0.7 | 1.38total | ∼90 (ethanol) | 178 | 0.0087 | 168 |
| Cu10-Cu1x+/Pd10Te3 nanowires | Cu 0.10/— | 2.11–2.12/2.49–2.51 | 0.2 M NaHCO3 | −0.78 | 8.6 (CO) | 92 (CO) | — | — | 169 |
| Cu3Br | — | — | 0.5 M KOH | −0.7 | 73.24 (C2H4) | 55.01 (C2H4) | — | — | 170 |
| Cu3Cl | — | — | 0.5 M KOH | −0.9 | 205.35 (C2H4) | 44.68 (C2H4) | — | — | 170 |
| Cu3I | — | — | 0.5 M KOH | −0.8 | 109.42 (C2H4) | 31.9 (C2H4) | — | — | 170 |
| Cu nanoclusters | — | — | 1 M KOH | −1.0 | 312(C2+) | 93.9 (C2+) | 171 | ||
| Pd2 DAC | Pd 0.37/— | 1.96/2.64 | 0.5 M KHCO3 | −0.85 | 6.76 (CO) | 98.2 (CO) | 148.9 | — | 172 |
| Ni2–N4–C2 | Ni 1.7/Ni 0.53 | ∼2.5/∼2.5 | 0.5 M KHCO3 | −0.8 | 17.9 (CO) | 96.6 (CO) | 124.6 | 1.25 (−1.0 V) | 181 |
| Ni 1.7/Ni 0.53 | ∼2.5/∼2.5 | 1.0 M KHCO3 (flow cell) | −0.8/−1.0 | —/56.1total | 96.8 (CO)/∼92(CO) | — | — | 181 | |
| Ni 1.7/Ni 0.53 | ∼2.5/∼2.5 | 1.0 M KOH (flow cell) | −0.5/−0.9 | ∼33total/∼100total | 97.8 (CO)/81.3 (CO) | — | — | 181 | |
| Ag2-G | Ag 0.10/— | ∼3.5/2.9 | 0.5 M KHCO3 | −0.7 | 11.87 (CO) | 93.4 (CO) | 265 | — | 229 |
| Cu–Fe–N6–C | Fe 0.83, Cu 0.91/— | —/2.60 | 0.1 M KHCO3 | −0.7 | 3.8 (CO) | 98 (CO) | 244.74 | — | 230 |
| N4–Ni–Sn–N4 | —/Sn 0.30, Ni 0.28 | — | 0.5 M KHCO3 | −0.82 | 20.8 (formate) | 86.1 (formate) | 120 | 1.32 | 231 |
| CoPc@Fe–N–C | Fe 1.04, Co 0.65/— | — | 0.5 M KOH | −0.84 | 275.6 (CO) | 93 (CO) | — | — | 232 |
| Ni/Cu–N–C | Ni 0.27, Cu 0.31/— | 2.6/— | 0.5 M KHCO3 (flow cell) | −0.49/−0.59/−0.69 | 6.6/14.5/22.4 (CO) | 98/99/99 (CO) | 86 | 1.10 | 233 |
| Ni/Cu–N–C | Ni 1954 ppm, Cu 1878 ppm/Ni 0.3, Cu 0.33 | 2.38–2.41/2.4 | 0.5 M KHCO3 (flow cell) | −0.6 | 95.21(CO) | 97.7 (CO) | 175 | 5.75 | 234 |
| Ag@Ag8@Ag6 | — | 3.0–3.2/ | 0.5 M KHCO3 | −0.6 | ∼2.5 (CO) | ∼95 (CO) | — | 6.37 (−1.1 V) | 235 |
| Fe1−Ni1−N−C | Fe 0.45, Ni 0.42/— | ∼4.1/∼4.1 | 0.5 M KHCO3 | −0.5 | 2.4 (CO) | 96.2 (CO) | 83 | — | 237 |
| Cu8-2 | — | — | 0.5 M KHCO3 | −1.0 | ∼19.3(HCOOH) | 92(HCOOH) | — | — | 239 |
| Ni–Zn–N–C | Ni 0.84, Zn 1.32/— | 1.99–2.28/2.45–2.52 | 0.5 M KHCO3 | −0.8 | 20.23 (CO, −0.90 V) | 99 (CO) | 89.7 | 0.33 (−0.90 V) | 240 |
| Catalysts | Metal content (ICP/XPS, wt%) | M–M distance (STEM/XAS, Å) | Electrolyte | Potential (V vs. RHE) | NH3 yield rate (μg h−1 mgcat.−1) | Faradaic efficiency (FE, %) | Ref. |
|---|---|---|---|---|---|---|---|
| Au4Pd2(SR)8 | — | — | 0.1 M HCl | −0.2 | 27.1 | 12.3 | 250 |
| Au4Pt2(SR)8 | 0.1 M HCl | −0.1 | 23.6 | 9.7 | 250 | ||
| CNT@C3N4–Fe&Cu | Fe 7.0, Cu 7.81/— | 1.8–2.5/— | 0.25 M LiClO4 | −0.8/−1.2(Ag/AgCl) | —/10.27 | 34/— | 251 |
| PdCu/NC | Pd 2.23, Cu 2.32/— | —/∼2.3–2.5 | 0.05 H2SO4 | −0.45 | 69.2 ± 2.5 | 24.8 ± 0.8 | 252 |
6. Electrocatalytic applications of multi-atom cluster catalysts
6.1 H2 production
Hydrogen is one of the most attractive and alternative fuels to replace traditional fossil fuels due to its high energy density and the advantage of yielding non-polluting products. Therefore, many techniques have been explored for the production of hydrogen wherein the electrochemical hydrogen production with the direct use of water as a source has been regarded as a promising one to produce high-purity hydrogen. Although the precious platinum catalyst has proven to be the most effective for the HER, it is expensive and scarce, which hamper its scale-up and practical application. Therefore, great efforts have been devoted to developing other promising alternatives with high activity. Besides, some non-noble metal-based electrocatalysts such as single-atom catalysts emerged as excellent candidates with promising performance for HER, but the simplicity of the active sites limited the further regulation of their electronic structure to boost the activity of active centers. However, the synergistic interaction of multi-atom cluster catalysts provides a new opportunity to regard them as some of the best HER electrocatalysts.210,211It has been found that the charge polarization between metals in adjacent layers is essential to help boost the activity towards HER. Zhang et al. reported the preparation of dispersed A–CoPt–NC by the electrochemical activation method.178 In their method, the Co-MOF was used to prepare atomically dispersed Pt, where Pt and Co atoms were well-trapped on the defective sites of the hollow graphite spheres. The researchers suggested based on the comprehensive understanding of the mechanism that a charge polarization may exist between the Pt and Co atoms, where a charge polarization derived from the inner layer of A–CoPt–NC may affect and alter the charge distribution of the outmost layer (Fig. 8a and b). Therefore, the adsorption strength towards *H could be tuned by the charge polarization effect around the Co atom, which leads to minimized Gibbs free energy change during the reaction. Such an optimized electronic structure could be observed from the experimental results, where the HER activity of A–CoPt–NC outperformed that of the commercial Pt/C (20 wt% Pt) in both acidic and alkaline electrolytes. Finally, the overpotential at 10 mA cm−2 was reported to be 27 mV for A–CoPt–NC whereas it was 59 mV for Pt/C, in an acidic solution, while the overpotential was 50 mV and 65 mV, respectively, in an alkaline solution (Fig. 8c and d).
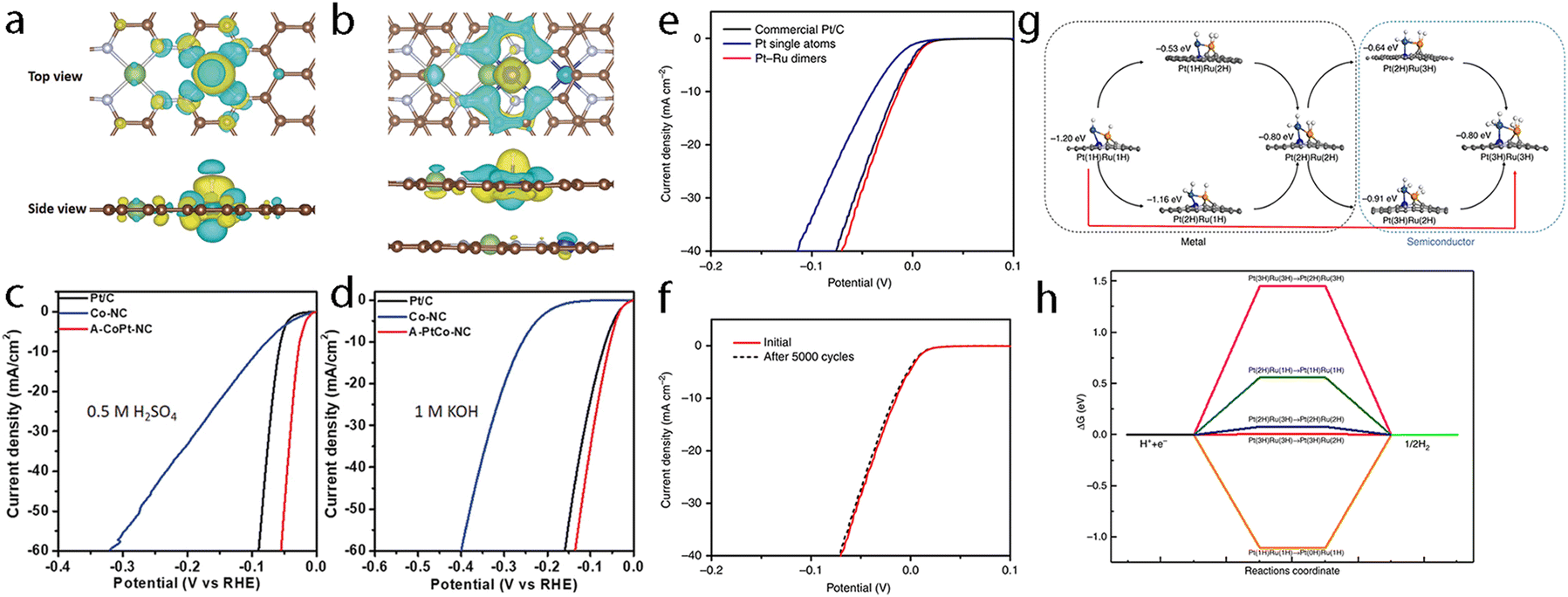 | ||
| Fig. 8 (a) Side view and top view of the charge distribution of single-layered model a(Co–Pt)@N8V4. (b) Side view and top view of the charge distribution of double layered model 2(a(Co–Pt)@N8V4). C brown, N green, Pt white, and Co blue. Yellow and cyan iso-surfaces represent electron accumulation and electron depletion. (c) LSV curves of Co-NC, A-CoPt-NC and Pt/C for HER in a 0.5 M H2SO4 electrolyte. (d) LSV curves of Co–NC, A–CoPt–NC and Pt/C for HER in a 1 M KOH electrolyte. Reproduced with permission.178 Copyright 2019 John Wiley & Sons. (e) The HER polarization curves recorded on Pt–Ru dimers, Pt single atoms and Pt/C catalysts. (f) Durability measurement of Pt–Ru dimer catalysts. (g) The atom structures of different H adsorption along with the adsorption energies. (h) The schematic of ΔGH for the different hydrogen coverages of typical configurations. For each coverage, all the possible pathways were considered, and the lowest ΔGH for each coverage is typically shown. Here, Pt(nH)Ru(mH) represents n H atoms adsorbed on Pt and m H atoms adsorbed on Ru. The orange, dark blue, light blue and gray balls represent Ru, Pt, N and C atoms, respectively. Reproduced with permission.47 Copyright 2019 Springer Nature. | ||
Later, Sun et al. reported the preparation of a hetero-dimer structure (Pt–Ru dimers) with the use of trimethyl(methylcyclopentadienyl)-platinum(IV) (MeCpPtMe3) and bis(ethylcyclopentadienyl)ruthenium(II) through an atomic layer deposition (ALD) method.47 The electrocatalytic HER tests of the Pt–Ru dimer were compared with that of Pt/C, which showed a much-improved mass activity of 23.1 A mg−1vs. 0.43 A mg−1 at an overpotential of 0.05 V (Fig. 8e). Good stability was also obtained for this catalyst as concluded after long-term stability tests (Fig. 8f). With the assistance of DFT calculations (Fig. 8g), it was found that the interaction between Ru atoms and the hydrogen intermediate could be modulated by Pt atoms and results in orbital rearrangement, where the dxz orbital of Ru became unoccupied due to sequential addition of hydrogen, thus leading to minimized Gibbs free energy towards H*, which finally improved the HER activity (Fig. 8h).
Precise synthesis of atomic-scale dual-metal electrocatalysts without the formation of nanoparticles still remains a challenge due to the inevitable aggregation during the fabrication process. Zhou et al. reported a top-down strategy using a dual-metal interbond as a chemical facilitator to spontaneously convert Fe nanoparticles to SACs.212 The formation of the Rh–Fe bond may help enhance the thermodynamic stability of the dual-metal, hindering the formation of nanoparticles, thus helping to strip the Fe atom to form Rh–Fe dual metal clusters from the Fe nanoparticles (Fig. 9a). The formation of the Rh–Fe bond may endow such a catalyst with good performance towards electrocatalytic HER. This electrocatalyst outperformed commercial Pt/C, requiring an overpotential 36 mV at 10 mA cm−2, with good stability in an acidic solution. From the DFT calculations, it was proved that the Rh moiety could capture one hydrogen atom spontaneously from water and act as the active site (Fig. 9b and c). This can also be observed through the projected density of state (pDOS), where the density of state of Rh is one order of magnitude larger than that of Fe below the Fermi level, thus assuring Rh is active for hydrogen reduction. Meanwhile, the Fe moiety could help tune the charge distribution and the electronic structure of Rh through the Rh–Fe bond (Fig. 9d and e), releasing extra electrons from Rh for reduction. Such Rh–Fe interaction was favorable for hydrogen atom adsorption and reduction and leads to increased activity towards HER.
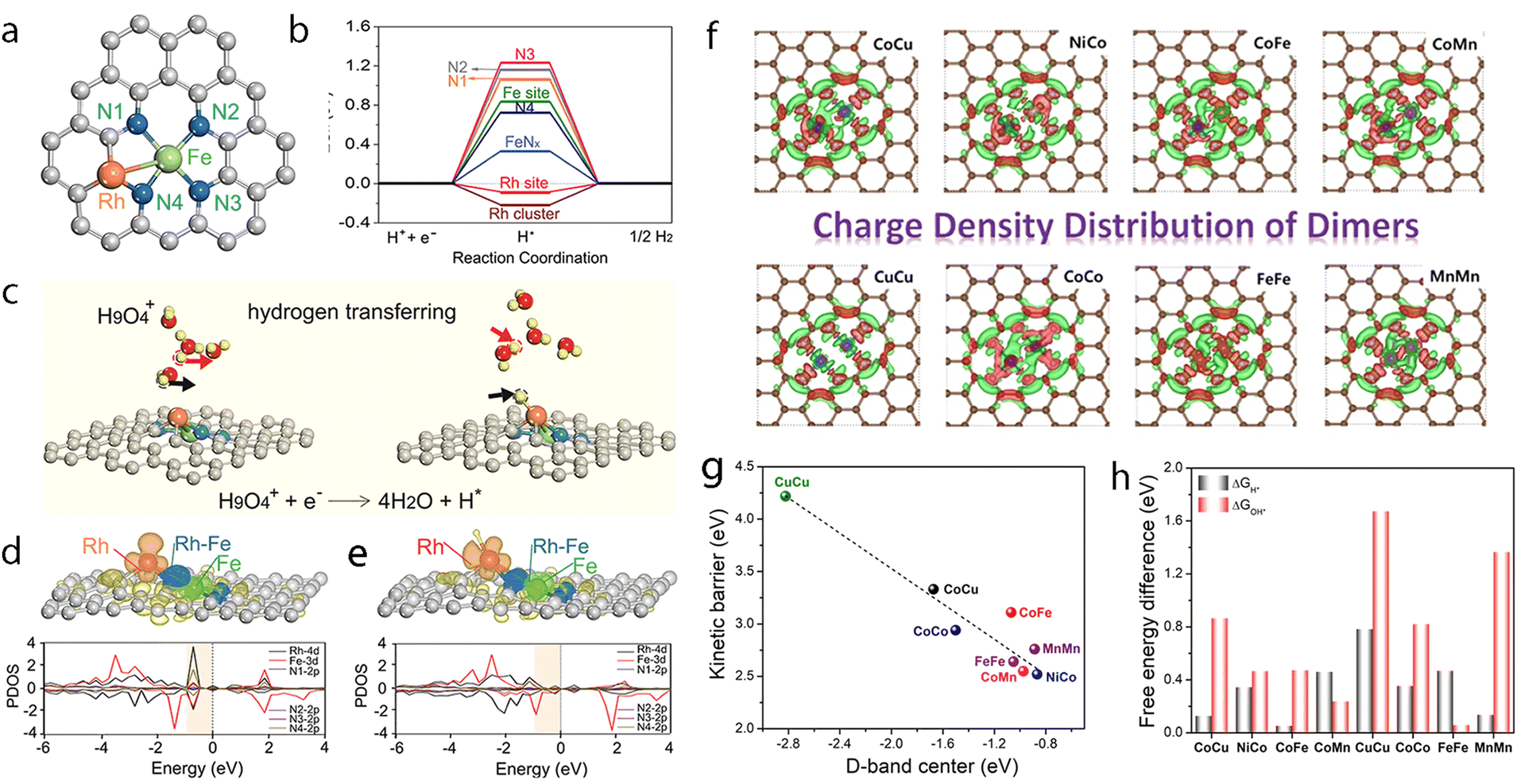 | ||
Fig. 9 (a) Possible sites to adsorb H in FR-NCS. (b) Hydrogen adsorption free energies (ΔGH) of different sites in FR-NCS and the single Fe metal atom in NCS (Fe–Nx) and the Rh7 cluster in NCS (Rh cluster). The Rh site in FR-NCS shows the closest value of ΔGH to 0 eV, identifying it as the active site to bond with H. (c) Schematic illustration of the hydrogen transfer process during HER on the Rh site in FR-NCS with H9O4+ in an acidic electrolyte. Projected density of state (pDOS) before (d) and after (e) H being adsorbed on the Rh atoms of FR-NCS with the corresponding charge distribution changes in the upper panels of each pDOS. Atomically dispersed Fe via the Rh–Fe bond retrieves some electrons in Rh to maintain a suitable number of active electrons in the Rh active site, which facilitates a moderate value of ΔGH. The blue, red, and green areas correspond to the electron distribution in Rh, Rh–Fe bonding, and Fe atom, respectively. Reproduced with permission.212 Copyright 2020 Wiley-VCH GmbH. (f) Different charge density distribution of TM-SAD-N6C. The charge depletion and accumulation are denoted by green and red colors, respectively. (g) Linear correlation between the d-band center and the kinetic energy barrier. (h) Free energy changes of the hydronium 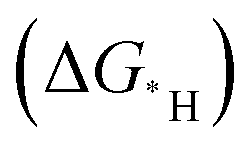 and hydroxide and hydroxide 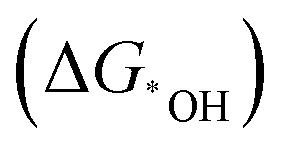 desorption step for TM-SAD-N6C. Reprinted with permission.213 Copyright 2021 Springer Nature. desorption step for TM-SAD-N6C. Reprinted with permission.213 Copyright 2021 Springer Nature. | ||
Recently, Kumar et al. reported the development of a pH-universal electrocatalyst, namely NiCo-SAD-N6C.213 The models of CoCu-SAD-N6C, NiCo-SAD-N6C, CoFe-SAD-N6C, CoMn-SAD-N6C, CuCu-SAD-N6C, NiNi-SAD-N6C, CoCo-SAD-N6C, FeFe-SAD-N6C, and MnMn-SAD-N6C were considered and established to screen and guide the experimental synthesis (Fig. 9f). They found that the kinetic dissociation energy of water molecules for these models was linearly correlated with the optimized d-band centers of these electrocatalysts, where NiCo-SAD-N6C possessed the minimum kinetic energy barrier (Fig. 9g). The calculations and comparison of the reaction profiles and the limiting step potentials also proved NiCo-SAD-N6C to be superior to the others for HER (Fig. 9h). In total, NiCo-SAD-N6C selected could exhibit an upshifted d-band center due to the synergistic interaction of the Ni–Co dual-site and may accelerate water dissociation and enhance HER activity, as shown by DFT calculations. Inspired by the DFT results, a precisely controlled NiCo-SAD-N6C was obtained by co-trapping of nickel and cobalt into polydopamine, followed by pyrolysis treatment. For the HER tests in alkaline electrolytes, it required about 61 and 189 mV overpotentials to achieve −10 and −100 mA cm−2, whereas for Pt/C (20%), it was about 51.5 and 227 mV, respectively. When assessed in an acidic electrolyte, the overpotentials needed were 54.7 and 116.8 mV for NiCo-SAD-N6C to reach −10 and −100 mA cm−2, while the state-of-the-art Pt/C (20%) needed 54.7 and 116.8 mV, respectively. Such an electrocatalyst also exhibited high stability with no obvious degradation during long-term stability tests, where at least 15 h runtime at −100 mA cm−2 and 50 h runtime at −50 mA cm−2 could be achieved. This work is a good example of DFT-assisted experimental screening of potential electrocatalysts for HER and is worth further endeavors.
The above studies proved that the resulted synergistic effects based on N-bridged hetero dimers, including precious metals, could help improve the HER performance. In a different approach, Yang et al. developed an O-bridged W-Mo non-precious transition metal hetero-dimer capable of pH-universal electrocatalytic hydrogen evolution.214 The EXAFS fitting disclosed that such dimers exhibited a W–O–Mo–O–C configuration, where an oxygen-bridged tetrahedron [WO4] and a distorted octahedron [MoO6] unit coexisted in the structure. With the assistance of theoretical calculations, they found that the non-metal atom around the metal atoms provided the active sites that were responsible for binding the hydrogen atoms with an optimized Gibbs free energy during the HER tests. The electrons from the metal centers were redistributed to the oxygen sites due to the synergistic interaction and enhanced the binding strength of the hydrogen atom, leading to an improved HER activity throughout the entire pH range.
6.2 Oxygen electrocatalysis
Catalysts capable of both ORR and OER are highly desirable for water splitting, fuel cells, metal-air batteries, etc. Noble metal catalysts such as Pt-based alloys and Ir/Ru-oxides are recognized as state-of-the-art ORR and OER electrocatalysts, respectively. But as rare metals, their limited availability in nature and high price hamper their scale-up application from economic considerations. Further, the multi-electron reactions of OER and ORR need large thermodynamic overpotentials, while sluggish reaction kinetics are also encountered due to their high reaction barriers. Thus, the development of novel electrocatalysts for the rapid OER and ORR remains a big challenge and the exploration of such earth-abundant element-based catalysts is urgent. Although the SACs already proved their outstanding or even better performance than the Pt-based nanocatalysts in ORR, some of the SACs tend to lose their stability during the reaction. For instance, the Fe SAC, which has been proved to be the most active one towards ORR in alkaline electrolytes, may experience the Fenton reaction during the ORR, which may destroy the catalyst, thus affecting its durability. The capability of SACs for catalyzing the OER is still unsatisfactory. However, multi-atom cluster catalysts can help enhance the catalytic activity of the active sites by reducing the reaction barriers for *OOH, *OH, and *O through synergistic effects. They can also increase the activity by increasing the loading of metals, providing a more economic use of metal elements with the maximum utilization of active metal sites.The ORR activity of non-precious metal-based electrocatalysts is usually not satisfactory due to their sluggish reaction rates in acidic media. Despite improvement by SACs, the expectations for better activation of O2 with lowered reaction barrier, proven reaction mechanism, smaller overpotential, and good long-term stability still call for better electrocatalysts.215–217 In 2017, Li's group reported a kind of Fe–Co dual-site embedded into N-doped porous carbon (Fig. 10a).45 Such a catalyst was synthesized through a host–guest strategy, where a Zn/Co bimetallic MOF was used to encapsulate the FeCl3 salts in the cavity of the MOF. Subsequent pyrolysis and graphitization resulted in the production of (Fe,Co)/N–C, which was confirmed by HAADF-STEM, XAFS, and Mössbauer spectra. The (Fe,Co)/N–C catalyst exhibited an onset potential of 1.06 V vs. RHE and a half-wave potential of 0.863 V vs. RHE compared to 1.03 V vs. RHE and 0.858 V vs. RHE in acid solution, respectively, of commercial Pt/C. Long-term cyclic voltammetry testing for 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times and H2/air fuel cell testing for 100 h further established the excellent stability of this dual-site catalyst. From the DFT calculations, it was suggested that the efficient activation of O2 from the synergistic influence of Fe–Co dual sites contributed the most towards the improved activity. The free energies for the conversion of O2 and *OOH into *O and *OH were greatly decreased for (Fe,Co)/N–C compared to that for the Fe SAs/N–C and Co SAs/N–C catalysts. Further, similar Fe–Co dual sites were investigated for ORR in alkaline media, and a better onset potential of 1.15 V vs. RHE and a half-wave potential of 0.954 V vs. RHE were exhibited, compared with 1.05 V vs. RHE and 0.842 V vs. RHE of commercial Pt/C.218 The use of such Fe–Co catalysts was also extended to Zn–air batteries, where a high voltage of 1.31 V and 1.23 V was obtained at discharge current densities of 20 mA cm−2 and 50 mA cm−2, respectively. In their model for theoretical calculations, it is noted that the terminal -OH groups were used to bridge dual atoms during the reaction path to help tune the reaction energies. Later, Xiao et al. explained in detail how a -OH coordinated Fe–Co dual-site could affect the ORR activity through the optimization of adsorption–desorption behavior of oxygen species through the electronic and geometric regulation of such catalysts (Fig. 10b).105 As -OH is an electron-withdrawing functional group, the coordination with -OH could upshift the Eredox of Fe(III)/Fe(II), which could be used as an effective M–O binding energy indicator. The higher Eredox may indicate a weaker Fe–O binding strength and lead to easy intermediate desorption, thus leading to promoted activity.
000 times and H2/air fuel cell testing for 100 h further established the excellent stability of this dual-site catalyst. From the DFT calculations, it was suggested that the efficient activation of O2 from the synergistic influence of Fe–Co dual sites contributed the most towards the improved activity. The free energies for the conversion of O2 and *OOH into *O and *OH were greatly decreased for (Fe,Co)/N–C compared to that for the Fe SAs/N–C and Co SAs/N–C catalysts. Further, similar Fe–Co dual sites were investigated for ORR in alkaline media, and a better onset potential of 1.15 V vs. RHE and a half-wave potential of 0.954 V vs. RHE were exhibited, compared with 1.05 V vs. RHE and 0.842 V vs. RHE of commercial Pt/C.218 The use of such Fe–Co catalysts was also extended to Zn–air batteries, where a high voltage of 1.31 V and 1.23 V was obtained at discharge current densities of 20 mA cm−2 and 50 mA cm−2, respectively. In their model for theoretical calculations, it is noted that the terminal -OH groups were used to bridge dual atoms during the reaction path to help tune the reaction energies. Later, Xiao et al. explained in detail how a -OH coordinated Fe–Co dual-site could affect the ORR activity through the optimization of adsorption–desorption behavior of oxygen species through the electronic and geometric regulation of such catalysts (Fig. 10b).105 As -OH is an electron-withdrawing functional group, the coordination with -OH could upshift the Eredox of Fe(III)/Fe(II), which could be used as an effective M–O binding energy indicator. The higher Eredox may indicate a weaker Fe–O binding strength and lead to easy intermediate desorption, thus leading to promoted activity.
 | ||
| Fig. 10 (a) Energies of intermediates and transition states in the mechanism of ORR at (Fe,Co)/N-C from DFT. Reprinted with permission.45 Copyright 2017 American Chemical Society. (b) Proposed ORR mechanism on the FeCoN5−OH site. Reprinted with permission.105 Copyright 2019 American Chemical Society. The k3-weighted Fourier-transform experimental Fe K-edge EXAFS spectrum (black line) and the fitting curve (red line) of (c) Fe1−N−C, (d) Fe2−N−C, and (e) Fe3−N−C. The insets of (c), (d), and (e) show the optimized structural model of Fe2−N−C, Fe1−N−C, and Fe3−N−C (Fe green, N blue, C gray, and O red) through theoretical calculations, respectively. (f) Superoxo-like adsorption at Fe1−N−C, peroxo-like adsorption at Fe2−N−C and peroxo-like adsorption at Fe3−N−C. Reprinted with permission.164 Copyright 2019 Elsevier. | ||
As discussed above, previous research suggests that the Fe SAC is an excellent catalyst for the ORR. The practical demand for performing the ORR under suitably applied bias still requires catalysts that could help with better oxygen activation and accelerate the reaction kinetics. Besides, the inevitable Fenton reaction between the Fe active sites and the by-product of H2O2 may produce hydroxyl and oxygen radicals, which may affect the active sites, leading to reduced durability. Ye et al. prepared a series of catalysts through the precursor preselection method and obtained tunable Fe1–N–C, Fe2–N–C, and Fe3–N–C electrocatalysts (Fig. 10c–e).164 They found that these three moieties could exhibit different O2 adsorption, as concluded through the observations made from low-temperature Fourier-transform infrared spectroscopy, superoxo-like adsorption configuration (end-on adsorption mode) for Fe1–N–C, and peroxo-like adsorption (side-on adsorption mode) for Fe2–N–C and Fe3–N–C (Fig. 10f). Also, the Fe clusters could promote the tailoring of N-species towards higher activity and electron transport, which were embedded in the carbon matrix during the pyrolysis process. The adsorption configuration of O2 onto Fex–N–C may contribute to the activation of O2 molecules. In detail, in the side-on mode, more electrons from the multi-active sites could be donated into the empty orbitals of the adsorbed oxygen and improve the activation of this molecule. Besides, Fe2–N–C could provide more catalytically active sites than Fe3–N–C when they both have comparable Fe content. Furthermore, Fe2–N–C could provide more pyridinic N species than Fe1–N–C and was more graphitized than Fe3–N–C. As a result, Fe2–N–C possessed an excellent performance in the acidic media than Fe3–N–C and the poorest one to perform was Fe1–N–C. Fe2–N–C with a half-wave potential of 0.78 V vs. RHE was the best among the three materials and was almost comparable to the commercial Pt/C with only −20 mV and −79 mV disparity in 0.5 H2SO4 and 0.1 M HClO4, respectively. A slight −20 mV shift was observed after the accelerated test of 20000 cycles, which indicated the remarkable durability of Fe2–N–C. Further measurements of these catalysts in an alkaline solution also suggested that Fe2–N–C could be operated in a universal pH range.
All the examples listed above used either the homo-atom or hetero-atom Fe-based clusters for their potential application for ORR.191 The use of other hetero-atom clusters is also favorable for ORR due to the inherent electronegativity difference between the atoms, which may lead to different adsorption strengths towards the intermediates and thus activate the adsorbate. For instance, Lu et al. reported the preparation of Zn/CoN-C with the dispersion of zinc and cobalt salts in chitosan followed by pyrolysis and a subsequent etching process.219 As the electronegativity of zinc (1.65) is slightly smaller than that of cobalt (1.88), it makes it easier for zinc to donate the electrons from the outermost orbital due to the synergistic interaction. Experimentally, the XANES spectrum (Fig. 11a and b) of Co atoms in Zn/CoN-C suggested a valence state between that of cobalt foil and CoN-C, while the XANES spectrum of Zn atoms in Zn/CoN-C indicated a valence state higher than that of ZnN–C. This coincided with the electronegativity trend mentioned above and allowed zinc to donate electrons and helped tune the electronic structure of Zn/CoN-C. Therefore, the O–O bond in the adsorbed *O2 could be elongated from 1.23 Å to 1.43 Å by the ZnCoN6(OH) moiety (Fig. 11c). It could be shown through the DFT calculations that this may lead to decreased dissociation energy for *O2. For comparison, the length of the O–O bond for ZnN4 and CoN4 moieties was only 1.29 Å and 1.30 Å, respectively (Fig. 11c). Furthermore, the higher activation ability for Zn/CoN-C towards the O–O bond led to greatly improved activity with a high half-wave potential of 0.861 V vs. RHE, which is more positive than that of Pt/C (by 35 mV) with good stability (Fig. 11d).
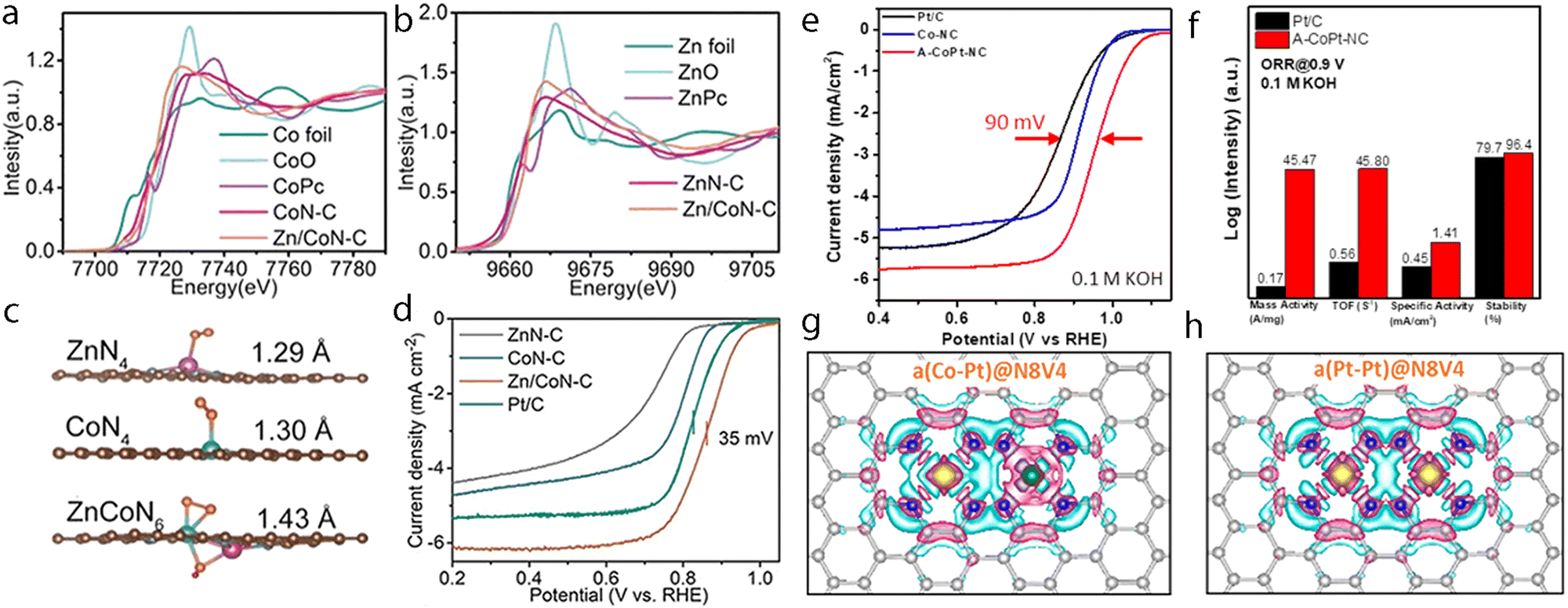 | ||
| Fig. 11 (a) Co K-edge XANES. (b) Zn K-edge XANES. (c) Optimized geometry of O2 adsorption configuration on the ZnN4, CoN4, and ZnCoN6(OH) systems. The brown, blue, yellow, green, purple, and red balls are C, N, O, Zn, Co, and H atoms, respectively. (d) ORR linear scan voltammogram (LSV) curves for different catalysts in an O2 saturated 0.1 M KOH solution. Reprinted with permission.219 Copyright 2019 John Wiley & Sons. (d) ORR linear sweep voltammetry curves of Co–NC, A–CoPt–NC, and Pt/C in a 0.1 M KOH electrolyte. (e) Comparison of mass activity, TOF, specific activity, and stability of A–CoPt–NC and Pt/C for ORR. (f) ORR free energy profiles of a(Co–Pt)@N8V4 at the equilibrium potential (U = 1.23 V), onset potential, and zero potential. (g and h) Local densities of state of a(Co–Pt)@N8V4 and a(Pt–Pt)@N8V4. (i and j) Top view of the charge densities of a(Co–Pt)@N8V4 and a(Pt–Pt)@N8V4. The pink and aqua isosurfaces with an isosurface level of 0.0025e/a0 represent electron accumulation and depletion areas, respectively. Reprinted with permission.177 Copyright 2018 American Chemical Society. | ||
It has been known for SACs that the selection of the substrate has a great impact on their performance towards ORR. A similar conclusion could also be made for multi-atom cluster catalysts. In addition, interaction with other atoms could tune their electronic structures. The electronic structure and the substrate can exert a combined effect on the ORR. Zhang et al. prepared a Pt–Co nitrogen–carbon-based catalyst (A–CoPt–NC).177 The local coordination environment of the metal pair was directly observed using the HAADF imaging, which suggested that the two metals were trapped in the defect sites of the substrate. The ORR measurements revealed a half-wave potential of 0.96 V vs. RHE, which was 90 mV superior to that of Pt/C in alkaline media (Fig. 11e and f). Both a(Co–Pt)@N8V4 and a(Pt–Pt)@N8V4 models were designed to investigate the reaction mechanism. The free energy reaction profiles suggested that the first protonation (O2 to *OOH) was the rate-determining step, where the change in free energy was 0.3 eV for a(Co–Pt)@N8V4. This value was smaller than that of a(Pt–Pt)@N8V4 (1.07 eV) and suggested a strong binding effect between a(Co–Pt)@N8V4 and O2. Further electronic structure analysis suggested that 3d orbitals of Co in the a(Co–Pt)@N8V4 moiety were up-shifted and might enhance the binding strength between the a(Co–Pt)@N8V4 moiety and O2 and other intermediates. A charge accumulation on Co and a charge depletion on Pt, leading to an asymmetric deployment of the surface charge, ensured the effective reduction of O2 on Co (Fig. 11g and h). Thus, the synergistic combination of defect coordination and electronic structure could enhance the ORR activity of a(Co–Pt)@N8V4.
The multi-atom site catalysts are usually more active than their respective single-atom counterparts for the OER in an alkaline medium. Wang et al. proved this using molecular dual atom-doped graphene (Fig. 12a–c).179 In detail, as-synthesized pristine Ni sites exhibited poor activity towards the OER in an alkaline solution. However, the introduction of Fe salts into the electrolyte and the subsequent cyclic voltammetry scanning led to the in situ generation of the Ni–Fe dual atom moiety, which could exhibit remarkably enhanced OER activity. They concluded that the adsorption of OH− to form a bridge geometry with Ni–Fe dual-sites helped improve the activity of such Ni–Fe catalysts. Furthermore, Hu's group reported the in situ formation of Co–Fe–N–C, where the Co–Fe dual sites were reconstructed from the single atom Co catalysts (Co–N–C) through cyclic voltammetry scanning of an Fe(III) containing alkaline electrolyte, and explained how the dual atom sites could be formed through operando XAS techniques (Fig. 17d–g).173 Recently, they provided further insights into the mechanism of OER with such dual sites, with a combination of operando XAS and electrokinetic methods.176
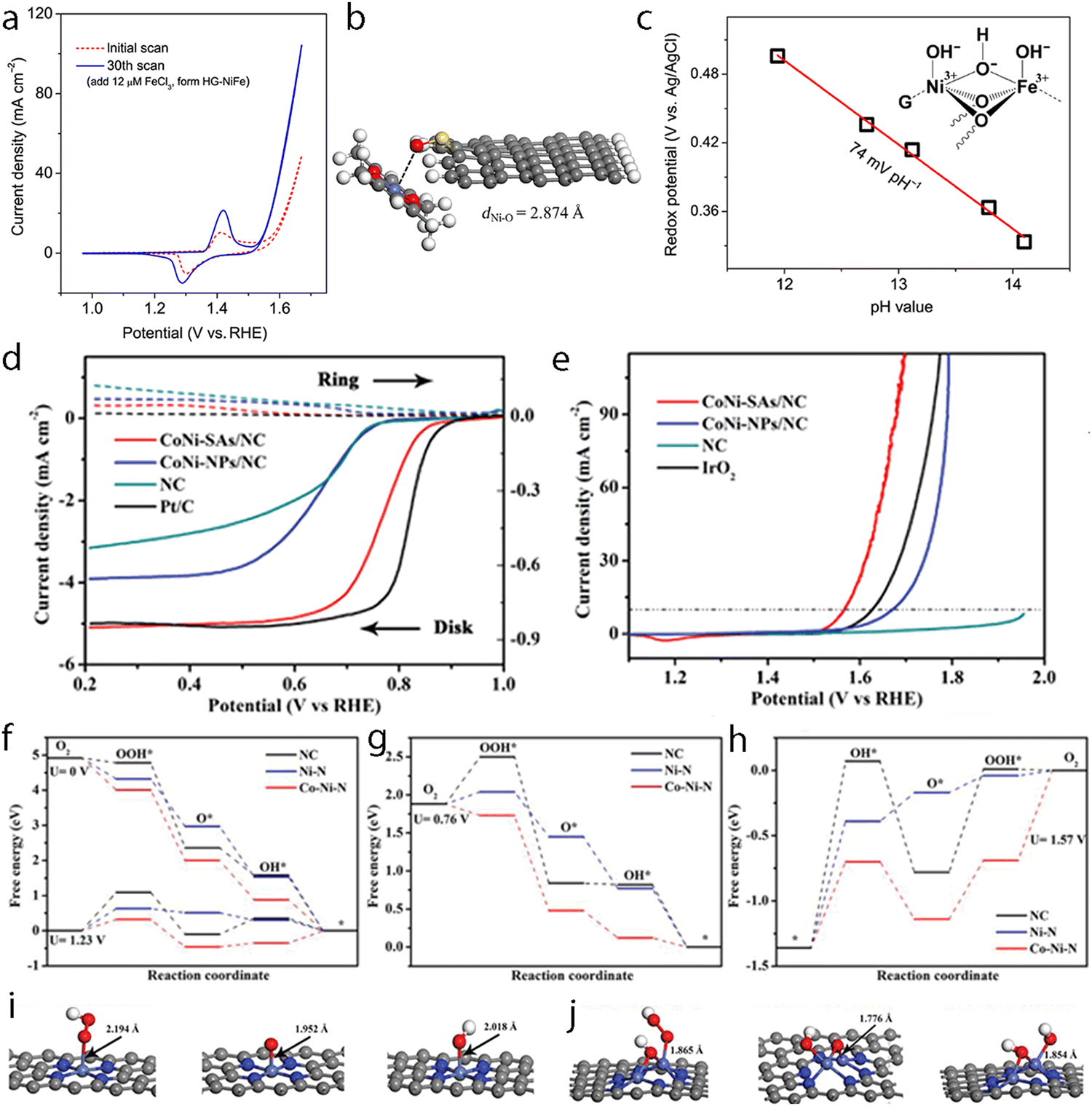 | ||
| Fig. 12 (a) Comparison of the initial and steady cyclic voltammetry curves of HG–Ni conducted in a glass cell containing 1 M KOH + 12 μM FeCl3. Scan rate, 50 mV s−1; rotation rate, 2000 rpm. (b) A DFT calculation-derived model to indicate the conjugation of Ni(acac)2 onto HG. The dopant of HG was a sulfoxide group as one representative interacting site. The gray, white, red, and yellow spheres represent C, H, O, and S atoms, respectively. The Ni atom is represented by a dark blue sphere. (c) Pourbaix diagram, illustrating the formal potentials of redox versus pH values of KOH solutions. The inset shows the proposed configuration for the adsorption of HO− ions onto Ni–Fe sites before OER. Reprinted with permission from AAAS.179 Copyright 2018. (d) ORR polarization curves at 1600 rpm. (e) OER polarization curves. Free energy diagram of (f) Ni–N and Co–Ni–N models at 0 and 1.23 V, (g) ORR, and (h) OER processes on NC, Ni–N, and Co–Ni–N. Reprinted with permission. Proposed reaction path on Ni–N (i) and Co–Ni–N (j). Reprinted with permission.167 Copyright 2019 John Wiley and Sons. | ||
The development of a bifunctional ORR/OER catalyst is necessary for its use in rechargeable metal–air batteries.193,220–228 Multi-atom cluster catalysts are also promising as bifunctional ORR/OER catalysts. Han et al. reported an atomically dispersed binary Co–Ni catalyst prepared by the pyrolysis of dopamine-coated metal–organic frameworks.167 This DAC was embedded in N-doped hollow carbon nanotubes (denoted as CoNi-SAs/NC). The remarkable activity of CoNi-SAs/NC for both ORR and OER in alkaline media is reflected by an exceptional Egap of 0.81 V between the half-wave potential of ORR and that of OER at a current density of 10 mA cm−2 (Fig. 12d and e). This performance was close to that of Pt/C-IrO2 catalysts and outperformed most of the other bifunctional nonprecious metal electrodes. Through theoretical calculations, they found that Co–Ni–N could change the rate-determining step (RDS) from hydrogenation of molecular O2 in NC and Ni–N to protonation of OH* with reduced energy difference for the ORR step, where a similarly reduced RDS energy difference was also found in the OER for Co–Ni–N compared to the others (Fig. 12f–j). Inspired by these results, this catalyst was further assembled to evaluate its activity in the Zn–air battery, where a better charging and discharging polarization curve could be observed for CoNi-SAs/NC, with 1.16 V (charging) and 2.05 V (discharging) at 10 mA cm−2, and 0.97 V (charging) and 2.22 V (discharging) at 50 mA cm−2, when compared with that of Pt/C-IrO2 as the cathode (1.17 V (charging) and 2.12 V (discharging) at 10 mA cm−2, 1.05 V (charging) and 2.28 V (discharging) at 50 mA cm−2). The maximum power density was 101.4 mW cm−2 and the discharge-specific capacity was 750.9 mA h g−1 for CoNi-SAs/NC-based cells, while these were 112 mW cm−2 and 719.2 mA h g−1, respectively, for Pt/C. Besides, such a catalyst also exhibited a good cycling rechargeability under 5 mA cm−2, where negligible voltage fading was found after 95 cycles. This was much better than the obvious decay observed for Pt/C-IrO2, just after 46 cycles. The further assembly of flexible solid-state Zn–air batteries and related measurements at diverse angles also proved the stable discharge and charge performance of such catalysts.
6.3 Electrocatalytic CO2 reduction reaction
The exploitation of fossil fuels has triggered an excessive emission of greenhouse CO2 gas, which causes severe environmental problems. Thus, there is a great demand for developing powerful techniques that can reduce CO2 to value-added products. The electrochemical CO2 reduction technique can convert CO2 into different kinds of energetic carbon products with the use of renewable electric energy. Thus, it can be regarded as a promising strategy to realize carbon utilization. However, the difficulty in overcoming the high thermodynamic reaction energy barrier and improving the reaction rate kinetically with high selectivity makes it imperative to develop highly active catalysts. Although the recent research on single-atom catalysts has already proven their advantage in CO2 reduction to CO, they are normally inefficient in catalyzing CO2 to more complex products. Thus, the incorporation of more atoms to form multi-atom cluster catalysts may hold promise to either boost the performance of CO2RR or provide more catalytic reaction sites to facilitate complex reactions and produce CO, HCOOH, CH4, C2H4, etc., through effective interactions.172,229–239It is well known that the transition metals can exhibit varied interaction strength with the intermediates due to their electronic features. Thus, the inclusion of a hetero-atom can help tune the activity through the tuning of the interaction strength with intermediates.46,230,231,233,240 As mentioned in previous sections, the difference in electronegativity can lead to diverse binding strength towards the adsorbate, leading to a possible decrease in free energy change to certain intermediates, thus tuning the activity during reactions. For instance, the Ni SAC could exhibit high selectivity for CO production, but a large overpotential was required for the first step of *COOH formation and thus suffered from sluggish reaction kinetics. Other catalysts such as the Fe SAC and Co SAC showed a high reaction rate for the formation of *COOH, yet the strong interaction of *CO with the metal made it difficult for CO to desorb from the catalyst's surface. Accordingly, Ren et al. reported the synthesis of a uniform Ni–Fe DAC, which exhibited a high selectivity of above 90% for CO production over a wide potential range from −0.5 to −0.9 V vs. RHE and also retained high stability even after the long-term electrolysis testing (30 hours) (Fig. 13a).46 Their conclusion from the DFT calculations was that the Ni–Fe dual-site could synergistically decrease the reaction barrier of the first step (*COOH formation), while the CO-adsorbed moiety could facilitate the release of CO (Fig. 13b and c). Ni–Fe dual sites also gave an opportunity for their use as bifunctional electrocatalysts of CO2RR and OER for Zn–CO2 batteries.241 Recently, Chen et al. reported that the introduction of Cu atoms to form a hetero Ni/Cu–N–C material could decrease the energy barrier for intermediate adsorption and regulate the d-band center to obtain optimized binding affinity towards intermediates. Thus, NiCuN6 displays excellent selectivity for CO production than its counterpart Ni–N-C and Cu–N–C with large current density in flow cell tests (Fig. 13d–g).234 Later, other kinds of similar hetero-atom electrocatalysts were also reported, such as Ni–Fe, Ni–Cu, Ni–Zn, Ni–Sn, Cu–Fe, etc.,230,231,233,234,236,237,250 and proved the existence of effective synergistic effects between them during CO2RR.
 | ||
| Fig. 13 (a) FECO for catalysts obtained in a CO2-saturated 0.5 M KHCO3 solution. (b) Calculated free energy diagrams for CO2RR yielding CO on different catalysts. (c) The catalytic mechanism on a diatomic metal–nitrogen site based on the optimized structures of adsorbed intermediates COOH* and CO*. Reprinted with permission.46 Copyright 2019 John Wiley and Sons. (d) faradaic efficiency for CO production at various applied potentials. (e) Tafel plots for CO production on various electrodes. (F) pDOS of Ni in NiN4 and NiCuN6. (f) Schematic illustration explaining the change of metal–adsorbate interaction by altering the metal d band center (Ed). Reprinted with permission. Copyright 2022 American Chemical Society.234 | ||
In the above example, hetero-atom configurations were used for the synergistic benefit of decreasing the reaction barrier. By optimizing the electronic structures, homo-atom-based electrocatalysts also hold potential as promising candidates for CO2RR. Jiao et al. found that the Cu-doped Pd10Te3 nanowires featured high activity for CO2 conversion to CO, as the active sites in this catalyst comprised of Cu10–Cu1x+ pairs, which were stabilized by the Te surface defects from Pd10Te3 nanowires.169 Later, Yao reported using Ni2(dppm)2Cl3 (dppm = bis(diphenylphosphino)methane) as a precursor to be pyrolyzed and prepared N2/NC electrocatalysts.166 They found, through the operando XAS (Fig. 17h–i), that the oxygen bridged O–N2–N6 configuration could be formed during reactions, which significantly lowered the free energy change of *COOH intermediate (Fig. 14b and c) formation and led to >94% faradaic efficiency during CO2RR (Fig. 14a). Recently, Zhang et al. used the electrospun-pyrolysis method to prepare Ni2–N4–C2 with Ni(acac)2. The samples pyrolyzed at 1000 °C could form such dual-site materials.181 Theoretical calculation showed that the modulated d-band center could exhibit optimized adsorption towards intermediates, leading to boosted activity for CO2RR.
 | ||
| Fig. 14 (a) CO FE in the current density range 50–200 mA cm−2. (b) Calculated Gibbs free energy diagrams for CO2 electroreduction to CO on various catalysts. (c) Proposed reaction pathways on O–Ni2–N6. Reprinted with permission.166 Copyright 2021 American Chemical Society. (d) The molecular structure characteristics of Cu3-X. (e) faradaic efficiency (FE) (H2, CH4, CO and C2H4) of Cu3–Br at different potentials (−0.6 V to −1.1 V). (f) The contrast of FE for CO2RR catalyzed by Cu3–Cl, Cu3–Br and Cu3–NO3 at different applied potentials (−0.6 V to −1.1 V). Reprinted with permission.170 Copyright 2021 John Wiley and Sons. | ||
Multi-atom cluster catalysts have proved their outstanding performance towards CO2 reduction to CO. Yet, the further reduction of CO2 to more valued-added multi-carbon chemicals is challenging.170,171,242 Copper-based catalysts are promising as diverse carbon-based fuels could be produced by them due to their moderate strength with various intermediates during the reaction. The copper-based multi-atom clusters could provide multi-active sites for the complex reaction pathways while inheriting the advantage of high activity and selectivity from SACs. The previous research based on copper complexes successfully observed the dynamic and reversible transformation of copper to form large nanoclusters, which are necessary for CO2 conversion to methane.243 A similar reversible process was also reported by Karapinar et al. for CO2RR to ethanol with the Cu–N–C SAC.244 Further, Xu et al. reported a carbon-supported, atomically dispersed Cu SAC, fabricated from an amalgamated Cu–Li method to produce ethanol.168 Recently, Lan et al. also reported using crystalline trinuclear copper clusters for the conversion of CO2 to C2H4 (Fig. 14d–f).170 In detail, Cu3–X (X = Cl, Br, NO3) complexes containing three different counter anions were fabricated (Fig. 14d). Among them, Cu3–Br could deliver the highest C2H4 production performance with considerable stability. It exhibited good selectivity from −0.7 V to −1.1 V vs. RHE. For instance, the faradaic efficiency of Cu3–Br at −0.7 V vs. RHE was about 55% (Fig. 14e and f). The theoretical calculations also suggested that two adjacent Cu centers played a key role in the C–C coupling of *CHO intermediates and promoted the conversion of CO2 to C2H4.
6.4 Electrocatalytic N2 reduction reaction
Ammonia contributes the most to modern agriculture and is essential for the synthesis of a great variety of pharmaceutical products. Yet the industrial production of ammonia mainly relies on the Haber–Bosch process. Harsh production conditions, high global energy consumption, and high CO2 emissions call for the development of new techniques to achieve the fixation of nitrogen under mild conditions. Thus, the electrocatalytic nitrogen reduction reaction (NRR) is emerging as an attractive strategy due to its favorable economics and environmentally benign fixation of nitrogen under ambient conditions. Since the emergence of multi-atom cluster catalysts, theoretical researchers have put great efforts to guide the development of NRR from the calculation aspect.245–247 Very recently, there have been some reports on the use of multi-atom cluster catalysts for NRR from the experimental research standpoint as well.248,249The precise synthesis by anchoring metal precursors to the substrate can be used to obtain the precious metal multi-atom cluster catalysts for NRR. Yao et al. developed the synthesis of bimetallic Au4Pt2(SR)8 clusters.250 After anchoring these clusters onto defective graphene, a hetero metal electrocatalyst of Au4Pt2/G with precise structural information could be obtained (Fig. 15a–d). The electrochemical NRR (Fig. 15c and d) using Au4Pt2/G demonstrated that this material could generate a maximum NH3 yield of up to 23.6 μg mg−1 h−1 at −0.1 V vs. RHE with a faradaic efficiency of ca. 9%. The yield of NH3 was better than that from the Au4Pt2(SR)8 electrocatalyst, where a maximum NH3 yield rate of 7.9 μg mg−1 h−1 could be obtained at −0.1 V with a faradaic efficiency of 9.7%. Furthermore, Au4Pd2/G obtained by the replacement of the Pt atom with Pd could provide a maximum NH3 yield rate of 27 μg mg−1 h−1 at −0.2 V with a faradaic efficiency of ca. 12%, which was higher than that of Au4Pt2/G.
 | ||
| Fig. 15 (a and b) Atomic structures of the clusters. (c) NH3 production rate. (d) faradaic efficiency of ammonia production at different potentials. Reprinted with permission.250 Copyright 2020 Springer Nature. | ||
The confinement effect can help enhance the interaction between the reactant and the active sites from the catalysts. The unique microenvironment formed due to such a confinement effect can also promote an effective interaction between multiple active sites, which is beneficial for the complex reaction processes of NRR. Therefore, the concept of “sub-nano reactors” was adopted by Wang et al. with the use of graphitic carbon nitride to confine multiple Fe and Cu atoms with the surrounding nitrogen atoms.251 The obtained CNT@C3N4-Fe&Cu could achieve an NH3 yield of 9.86 μg mg−1 h−1 with a faradaic efficiency of 34% (Fig. 16a and b). This was a nearly double yield and a 54% increase in the faradaic efficiency when compared with its single metal counterparts. From the theoretical understanding obtained with DFT calculations, they found that the formation of Fe–Cu could help optimize the reaction process of N2 reduction. The rate-determining step could be altered from the 5th hydrogenation step (NH* → NH2*, 0.71 eV) for CNT@C3N4-Fe and the N2 adsorption step (0.86 eV) for CNT@C3N4-Cu to the initial hydrogenation step (N2* → N2H*, −0.58 eV) for CNT@C3N4-Fe&Cu (Fig. 16c). The introduction of Cu could also provide an electron reservoir to supply electrons to the Fe atom. The existence of Fe–Cu also improved the electron transfer when compared to single-atom catalysts. All these combined helped to boost the activity of CNT@C3N4-Fe&Cu towards NRR.
 | ||
| Fig. 16 (a) The faradaic efficiencies of the materials at different potentials. (b) The ammonia yield rate for 3 and 10 h tests at −1.2 V vs. Ag/AgCl in 0.25 M LiClO4. Free energy diagrams of NRR on Fe2Cu (c) clusters in the cavity of g-C3N4. Reprinted with permission.251 Copyright 2020 Wiley-VCH GmbH. (d) NH3 yield rates and (e) FEs of PdCu/NC, Pd/NC, and Cu/NC at each given potential in N2-saturated 0.05 M H2SO4. (f) Partial density of states (pDOS) of N2 adsorption on Pd/NC, Cu/NC, and PdCu/NC. Blue lines: 2p orbital of N, orange lines: 3d orbital of Cu, and cyan lines: 4d orbital of Pd. (g) Projected crystal orbital Hamilton population (–pCOHP) and its integrated value (ICOHP) of N2 adsorption on Pd/NC, Cu/NC, and PdCu/NC. Orange and cyan lines indicate the bondings between N–Cu and N–Pd. The energy value is with reference to the Fermi level. Calculated free energy diagrams of NRR and HER on (h) PdCu/NC systems. The alternating pathway is favored on Pd/NC and Cu/NC, whereas the distal pathway is the most energetically preferred on PdCu/NC. The asterisk (*) represents a reactive intermediate or a vacant surface site. Insets: Structural models. (i) Difference between the limiting potentials for NRR and HER of Pd/NC, Cu/NC, and PdCu/NC. Reprinted with permission.252 Copyright 2020 Wiley-VCH GmbH. | ||
Based on the above-mentioned examples, effective interaction and activation of N2 molecules from a heteroatom configuration can help enhance the intrinsic performance of an electrocatalyst. However, the NRR process is inevitably accompanied by the HER, and therefore, the development of catalysts capable of suppressing the HER and improving the NRR simultaneously is important. A Pd-hydride can help promote the hydrogenation of the intermediate *NN in the NRR process.252 But the strong interaction between Pd and hydrogen may also block the effective reaction sites. The introduction of Cu atoms can improve the hydrogenation process and enhance the electron transfer by strong d–d orbital interactions with Pd. Along this line, the introduction of a secondary Cu to Pd was reported by Han et al. to obtain dual atom PdCu/NC.252 Such a catalyst could achieve an NH3 yield rate of ca. 69 μg mg−1 h−1 at −0.45 V with a high faradaic efficiency of ca. 24.8% in the acidic electrolyte, which outperformed the yield rate of Pd/NC by a factor of 4.2 and faradaic efficiency by 14.6 (Fig. 16d and e). Theoretical analysis disclosed that the free energy change needed for the rate-determining step of N2 chemisorption and the following hydrogenation step could be decreased by using PdCu/NC (Fig. 16h–i). They further proved that the introduction of Cu to form PdCu/NC could not only tune the PDOS of Pd to the Fermi level (Fig. 16f), but also promote the activation of the N2 molecule through back-donation bond formation between Pd and N2 with an effective d-2π* coupling (Fig. 16g).
7. In situ/operando characterization for mechanistic investigation
As mentioned above, a better understanding of the fundamental reaction mechanism and dynamic adsorption of intermediates and catalytic site evolution is critical for catalyst design. Real-time techniques such as in situ Raman spectroscopy, in situ Fourier transform infrared spectroscopy (FTIR), on-line mass spectroscopy, operando XAS, etc., could facilitate the understanding of possible phase transformation, structure reconstruction, valence state change, reaction pathways, adsorbed intermediates on active sites, etc. during reactions.197,242,253–256 When combined with both theoretical and experimental results, they can provide opportunities for the mechanistic understanding of certain reactions, thus guiding the further design of electrocatalysts.For example, through in situ FTIR, Lu et al. reported the direct observation of adsorbed intermediates on NiZn–N6–C during CO2RR.240 This electrocatalyst could exhibit an earlier signal of adsorbed *CO species than that from the Ni–N4–C and Zn–N4–C counterparts, which indicated weaker adsorption and difficulty in producing CO for the latter ones (Fig. 17a–c). Yao et al. demonstrated that during CO2RR the Ni2–N6 configuration could be transformed into an oxygen bridged O–Ni2–N6 moiety under open circuit potential and this transformation could be observed by operando XAS (Fig. 17h–i).166 The shortened distance between Ni atoms could lead to an enhanced Ni–Ni interaction. When combined with theoretical calculations, a significantly lowered Gibbs free energy change for *COOH formation could be obtained, which explained the boosted CO2RR activity. The structure reconstruction of active sites during reactions was also reported by Patzke et al. for ORR/OER (Fig. 17i and j), where the single Ni sites were easily transformed to form Ni–O–Ni and Ni–O–Fe configurations, while Fe–O–Fe was not observed during operando XAS.257 Hu et al. reported another example of operando XAS measurement, used to observe the in situ formation of Co–Fe dual atom catalysts during the OER process (Fig. 17d–g).173 Recently, they further disclosed the OER reaction mechanisms of a series of dual-atom electrocatalysts based on Fe, Co, and Ni SACs through a combination of operando XAS and electrokinetic measurements.176 Take the formation of Ni–Fe–N–C as an example: the fitting result from operando Ni K-edge XAS data of the original Ni–N–C suggested a single Ni configuration coordinated with 3N, 1C, and 1O. After immersion in a 1 M KOH electrolyte, two N donors were replaced by three O donors (OH−/H2O). After five cyclic voltammetry scans, a new single-scattering path was found which was due to the formation of Ni–Fe dual-atoms via oxygen bridges. The XANES data also suggested that the oxidation state of Ni was around the range of +2 to +3 and changed little with extended electrolysis. Meanwhile, operando Fe K-edge XAS data were also collected, where the signal was only observed after one hour of electrolysis. The oxidation state of Fe XANES data also suggested the existence of Fe(IV) species during the electrolysis. With the assistance of electrokinetic measurements, they successfully explained how hydroxyl groups, protons, and electron transfers affect the catalytic cycles of OER and proposed a possible reaction mechanism.
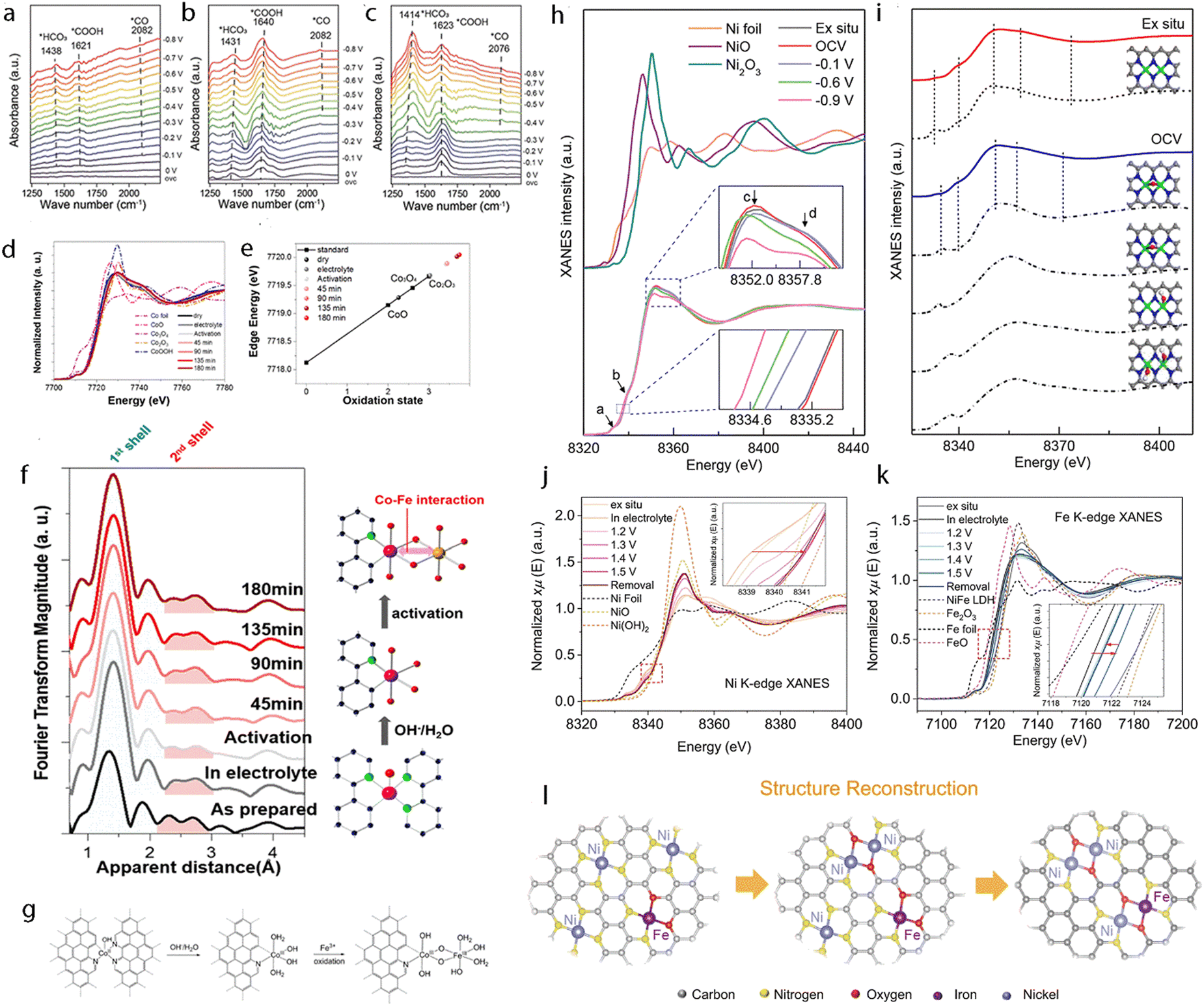 | ||
| Fig. 17 (a–c) In situ FTIR spectra for the co-adsorption of NiZn–N6–C (a), Ni–N4–C (b), and Zn–N4–C (c) in a 0.5 M KHCO3 aqueous solution. Reprinted with permission.240 Copyright 2021 Wiley-VCH GmbH. (d) XANES spectra and (e) K-edge energies (at 50% level) of XANES spectra in panel a and cobalt reference compounds containing Co(0), Co(II), or Co(III). (f) Fourier transform of Co K-edge EXAFS spectra without phase correction for as-prepared Co−N−C and the catalyst after activation as well as under OER for various durations. (g) Proposed model for the formation of a Co–Fe double-atom catalyst. After the precatalyst is immersed in the electrolyte solution, either two N or one N and one C were replaced by O. For simplicity, only the model where two N were replaced is shown. Reprinted with permission.173 Copyright 2019 American Chemical Society. (h) Operando XANES spectra recorded at the Ni K-edge of Ni DSC, at different applied potentials from the open-circuit voltage to −0.9 V RHE during the electrocatalytic CO2RR, and the XANES data of the referenced standards of NiO, Ni2O3, and Ni foil. Inset: Magnified absorption edge and white-line peak of the XANES region. (i) Comparison between the Ni K-edge XANES experimental spectra (solid lines) and the theoretical spectra (dashed lines) calculated with the depicted structures (inset). Reprinted with permission.166 Copyright 2021 American Chemical Society. (j and k) Ni, Fe K-edge. (i) Structural reconstruction of NiFe-CNG during OER from separate Ni and Fe centers to Ni–O–Fe/Ni moieties. Reprinted with permission.257 Copyright 2021 Springer Nature. | ||
8. Conclusions and outlook
In this review, great emphasis has been laid on multi-atom cluster catalysts, where the origin of synergistic effects, such as optimized d-band filling, the spin state, charge difference, electronegativity, etc., was discussed. The methods of modulating these parameters through effective interactions, such as inter-atomic interface, coordination environment, and substrate, were summarized. Their ability to circumvent scaling relationships and the need to develop advanced descriptors during electrocatalysis were also depicted with further discussions around the latest and well-developed synthesis methods. Following this, the recent experimental developments of such kinds of catalysts in various electrocatalytic applications (HER, OER, ORR, CO2RR, and NRR) were reviewed, where the boosted activity compared to their SAC counterparts proved the opportunity offered by multi-atom cluster catalysts as an excellent class of electrocatalysts. Despite all the progress made, this area remains challenging to the researchers.8.1 Maximizing the mass utilization
One of the obstacles is the relatively low mass loading of multi-atom cluster catalysts onto the substrates, which is also a pressing problem in the preparation of SACs, hampering their device-level activity. Simply increasing metal loading may not work, as most of the synthesis processes involve high-temperature pyrolysis, which may cause a severe aggregation of atomic-scale catalysts to form sub-nano clusters or nanoparticles. One solution is to enhance the interaction between the metals and the substrate to stabilize the catalysts and avoid aggregation.245,258–260 Therefore, the use of substrates possessing plenty of functional anchoring sites to tightly bind with metal sites is likely to avoid possible aggregation. For instance, Jiang et al. computed that a TAC catalyst with loading as high as 35.8% could be achieved when a graphdiyne and graphene composite was introduced as the substrate.187 The strong hybridization between the Fe-3d orbitals from the metal and the C-2p orbital from the ethynyl of the skeleton may help to stabilize the catalyst.8.2 Precisely controlled synthesis techniques
The technique for precisely controlled synthesis of multi-atom cluster catalysts is still not mature, and therefore, their synthesis is much more challenging than that of SACs. The synthesis of catalysts with a precise structure is both meaningful and necessary. Theoretically, researchers have built plenty of models to investigate the properties of multi-atom cluster catalysts in diverse applications, but due to the limitations in the controlled synthesis of these materials, it is not easy to verify the theoretical prediction. Although the degree of atomic dispersion of the catalysts can be directly observed by HAADF-STEM and their coordination environment can be obtained from XAF spectra, their synthesis (homo- or hetero-moieties) onto substrates without introducing impurities such as SACs or nanoparticles is difficult. Some techniques such as the mass-selected deposition method have turned out to be effective to prepare such materials, but they are not easy for large-scale applications. The precursor preselection method also has its limitations and calls for a judicious selection of suitable precursors. The subsequent pyrolysis to anchor the metal catalysts onto the substrate is necessary, which makes the whole process a bit cumbersome and energy-consuming. Therefore, other reliable techniques capable of precise synthesis of multi-atom cluster catalysts are still needed.8.3 Advanced theoretical method to simulate the real catalytic environment
The theoretical computation method is beneficial and indispensable for developing new kinds of catalysts.261 It aids in a better understanding of the reaction mechanisms, correlates activity to the intrinsic electronic structure, accelerates the screening of effective catalysts with effective descriptors, etc.250 As discussed in the review, researchers have put great efforts into the understanding of multi-atom cluster catalysts from the theoretical aspect and some credible conclusions have already been made. However, the electrochemical reactions are rather complicated. First, a given reaction may occur in between the electrode and electrolyte interface with the electrical double layer. So, the inclusion of more external conditions into the calculation system and a precise depiction of the real reaction environment are difficult but worth pursuing. Besides, although most calculations for an electrocatalytic reaction pathway rely solely on the thermodynamic states during the process, the kinetics associated with the dissociation and formation of some key intermediates are required to help the researchers understand the reactions. Moreover, even though it will be better if a model built from a computational aspect can be large enough to reflect a real reaction system as closely as possible, the calculations would still be both time and resource-consuming. So new techniques allowing researchers to study relatively large model systems should be developed.8.4 Development of efficient descriptors and screening electrocatalysts with high activity
Finally, theoretical calculations already play an important role nowadays. They can not only help explain the reactions but also guide the design of experiments. Therefore, the universal descriptors to guide the fabrication of different catalysts are needed to better assist the screening of the optimized catalysts. Besides, the use of machine learning has also been a hot topic recently, and it will be interesting to direct some efforts in combining the concept of multi-atom cluster catalysts with machine learning to achieve more interesting progress.262 In that case, the combination of geometrical and electronic characteristics of electrocatalysts, such as coordination atoms, fine-tuned ligands, details about bond length, atom electronegativity, d-band center, magnetic moment, etc. could help build a database, which can be trained with the machine learning-based programs and screen the effective electrocatalysts.8.5 Exploring real-time characterization techniques for mechanism understanding
The identification of catalytically active sites, the dynamic evolution behaviours, and observable reaction intermediates on multi-atom cluster catalysts during reactions with various characterization methods may help deepen the mechanistic understanding of such electrocatalysts. However, a low loading of such catalysts poses a challenge to the subsequent characterization. Therefore, the atomic scale dispersion of these catalysts requires measurements with instruments that offer high resolutions. Recently developed real-time techniques such as in situ FTIR, in situ Raman, on-line mass spectroscopy, operando XAS, etc., could facilitate the observation of possible structure evolution and the real active centers, help identify valence state change, etc., during reactions. Those techniques render researchers the ability to get information pertaining to the geometric and electronic characteristics of the active sites in a specific reaction, which would subsequently help guide the designing of novel electrocatalysts. While capturing the related information with an in situ setup to fit into diverse measuring equipment is still in its infancy, it is worth more effort.8.6 Optimized reaction pathways and widespread applications
Multi-atom cluster catalysts can provide multiple active sites for interaction with the intermediates and help circumvent the scaling relationship with the formation of complex products, thus helping to tune the reaction pathways for certain reactions. They have demonstrated their potential in various reactions, such as glycerol hydrogenolysis,36 oxidation of alcohols,38 CO2 conversions to methanol,34etc., which require multiple reaction sites to act synergistically. While the report of multi-atom clusters towards the electrochemical catalysis to produce complex products is limited, there is one study showing the highly selective synthesis of ethanol from CO2 on Cun clusters (n = 3 and 4) by involving the participation of C–C coupling from the cluster.168 It echoes the claim made in the previous sections that multi-atom clusters may unlock the selective catalysis to obtain complex and diverse products due to the presence of multiple active sites.In addition, more interest has been drawn toward the engineering applications of electrocatalysts. For example, in CO2RR, significant achievements have been made with a flow cell setup. This could also provide good opportunities for the further techno-economic application of multi-atom cluster catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Research Foundation (NRF), Prime Minister's Office, Singapore, under its Campus for Research Excellence and Technological Enterprise (CREATE) program. We also acknowledge financial support from the academic research fund AcRF tier 1 (M4012076 RG118/18), Ministry of Education, Singapore.References
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef
.
-
Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, NY, 2008, pp. 89–189 Search PubMed
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed
- J. Li, Y. Li and T. Zhang, Sci. China: Mater., 2020, 63, 889–891 Search PubMed
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS
- H. Rong, S. Ji, J. Zhang, D. Wang and Y. Li, Nat. Commun., 2020, 11, 5884 CrossRef CAS PubMed
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC
- Y. Pan, C. Zhang, Y. Lin, Z. Liu, M. Wang and C. Chen, Sci. China Mater., 2020, 63, 921–948 CrossRef CAS
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS
- J. Zhang, Q.-A. Huang, J. Wang, J. Wang, J. Zhang and Y. Zhao, Chin. J. Catal., 2020, 41, 783–798 CrossRef CAS
- S. Chen, M. Cui, Z. Yin, J. Xiong, L. Mi and Y. Li, ChemSusChem, 2021, 14, 73–93 CrossRef CAS PubMed
- S. Zhang, Y. Wu, Y.-X. Zhang and Z. Niu, Sci. China: Chem., 2021, 64, 1908–1922 CrossRef CAS
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2022, 12, 2102715 CrossRef CAS
- J. Wang, C. Liu, S. Li, Y. Li, Q. Zhang, Q. Peng, J. S. Tse and Z. Wu, Chem. Eng. J., 2022, 428, 132558 CrossRef CAS
- C. Cheng, X. Zhang, Z. Yang and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 32903–32912 CrossRef CAS
- F. Li and Z. Chen, Nanoscale, 2018, 10, 15696–15705 RSC
- F. Li, X. Liu and Z. Chen, Small Methods, 2019, 3, 1800480 CrossRef
- P. Zhou, X. Hou, Y. Chao, W. Yang, W. Zhang, Z. Mu, J. Lai, F. Lv, K. Yang, Y. Liu, J. Li, J. Ma, J. Luo and S. Guo, Chem. Sci., 2019, 10, 5898–5905 RSC
- N. Biswakarma, P. J. Sarma, S. D. Baruah, N. K. Gour and R. C. Deka, J. Phys. Chem. C, 2020, 124, 3059–3068 CrossRef CAS
- Z. Gao, X. Liu, A. Li, X. Li, X. Ding and W. Yang, Mol. Catal., 2019, 470, 56–66 CrossRef
- B. Yang, X. Yu, A. Halder, X. Zhang, X. Zhou, G. J. A. Mannie, E. Tyo, M. J. Pellin, S. Seifert, D. Su and S. Vajda, ACS Sustainable Chem. Eng., 2019, 7, 14435–14442 CrossRef CAS
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed
- X. Zhang, G. Cui, H. Feng, L. Chen, H. Wang, B. Wang, X. Zhang, L. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W.-C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9, 2353 CrossRef
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. Duan, D. Wang, C. Draxl and Y. Li, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS
- E. Vorobyeva, E. Fako, Z. Chen, S. M. Collins, D. Johnstone, P. A. Midgley, R. Hauert, O. V. Safonova, G. Vilé, N. López, S. Mitchell and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2019, 58, 8724–8729 CrossRef CAS PubMed
- D.-H. Xing, C.-Q. Xu, Y.-G. Wang and J. Li, J. Phys. Chem. C, 2019, 123, 10494–10500 CrossRef CAS
- C. Cui, Z. Luo and J. Yao, CCS Chem., 2019, 1, 215–225 CAS
- J. Fu, J. Dong, R. Si, K. Sun, J. Zhang, M. Li, N. Yu, B. Zhang, M. G. Humphrey, Q. Fu and J. Huang, ACS Catal., 2021, 11, 1952–1961 CrossRef CAS
- C. Dong, Y. Li, D. Cheng, M. Zhang, J. Liu, Y.-G. Wang, D. Xiao and D. Ma, ACS Catal., 2020, 10, 11011–11045 CrossRef CAS
- Z. W. Chen, J.-M. Yan and Q. Jiang, Small Methods, 2019, 3, 1800291 CrossRef
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC
- M. Ma, A. Kumar, D. Wang, Y. Wang, Y. Jia, Y. Zhang, G. Zhang, Z. Yan and X. Sun, Appl. Catal., B, 2020, 274, 119091 CrossRef CAS
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Small, 2020, 16, 1906782 CrossRef CAS PubMed
-
J. K. Nørskov, F. Studt, F. Abild-Pedersen and T. Bligaard, Fundamental Concepts in Heterogeneous Catalysis, 2014, pp. 155–174 Search PubMed
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS
- B. Garlyyev, J. Fichtner, O. Piqué, O. Schneider, A. S. Bandarenka and F. Calle-Vallejo, Chem. Sci., 2019, 10, 8060–8075 RSC
- X. Zhang, A. Chen, Z. Zhang and Z. Zhou, J. Mater. Chem. A, 2018, 6, 18599–18604 RSC
- H.-L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC
- L. Jasin Arachchige, Y. Xu, Z. Dai, X. Zhang, F. Wang and C. Sun, J. Phys. Chem. C, 2020, 124, 15295–15301 CrossRef CAS
- W. Huang and W.-X. Li, Phys. Chem. Chem. Phys., 2019, 21, 523–536 RSC
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC
- R. Kubo, J. Phys. Soc. Jpn., 1962, 17, 975–986 CrossRef CAS
- D. Buceta, Y. Piñeiro, C. Vázquez-Vázquez, J. Rivas and M. A. López-Quintela, Catalysts, 2014, 4, 356–374 CrossRef
- J. A. Alonso, Chem. Rev., 2000, 100, 637–678 CrossRef CAS PubMed
- V. N. Popok, I. Barke, E. E. Campbell and K. H. Meiwes-Broer, Surf. Sci. Rep., 2011, 66, 347–377 CrossRef CAS
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, Nano Energy, 2018, 46, 396–403 CrossRef CAS
- Y. Zhou, W. Yang, W. Utetiwabo, Y.-M. Lian, X. Yin, L. Zhou, P. Yu, R. Chen and S. Sun, J. Phys. Chem. Lett., 2020, 11, 1404–1410 CrossRef CAS PubMed
- J. Shan, C. Ye, Y. Jiang, M. Jaroniec, Y. Zheng and S. Z. Qiao, Sci. Adv., 2022, 8, eabo0762 CrossRef CAS PubMed
- X. Zhu, D. Zhang, C.-J. Chen, Q. Zhang, R.-S. Liu, Z. Xia, L. Dai, R. Amal and X. Lu, Nano Energy, 2020, 71, 104597 CrossRef CAS
- Y. Xu, W. Zhang, Y. Li, P. Lu, Y. Wang and Z.-S. Wu, Front. Mater., 2019, 6 CAS
- C. Wu, X. Zhang, Z. Xia, M. Shu, H. Li, X. Xu, R. Si, A. I. Rykov, J. Wang, S. Yu, S. Wang and G. Sun, J. Mater. Chem. A, 2019, 7, 14001–14010 RSC
- L. Chen, Y. Zhang, L. Dong, W. Yang, X. Liu, L. Long, C. Liu, S. Dong and J. Jia, J. Mater. Chem. A, 2020, 8, 4369–4375 RSC
- F. Wang, H. Xie, T. Liu, Y. Wu and B. Chen, Appl. Energy, 2020, 269, 115029 CrossRef CAS
- R. Ku, G. Yu, J. Gao, X. Huang and W. Chen, Phys. Chem. Chem. Phys., 2020, 22, 3254–3263 RSC
- X. Gao, L. Mei, Y. Zhou and Z. Shen, Nanoscale, 2020, 12, 7814–7821 RSC
- Y. Yang, H. Zhang, Z. Liang, Y. Yin, B. Mei, F. Song, F. Sun, S. Gu, Z. Jiang, Y. Wu and Z. Zhu, J. Energy Chem., 2020, 44, 131–137 CrossRef
- F. He, Z. Wang, S. Wei and J. Zhao, Appl. Surf. Sci., 2020, 506, 144943 CrossRef CAS
- H. He, C. Morrissey, L. A. Curtiss and P. Zapol, J. Phys. Chem. C, 2018, 122, 28629–28636 CrossRef CAS
- T. Yang, S. Tang, X. Li, E. Sharman, J. Jiang and Y. Luo, J. Phys. Chem. C, 2018, 122, 25441–25446 CrossRef CAS
- G. Zhu, M. Kan, Q. Sun and P. Jena, J. Phys. Chem. A, 2014, 118, 304–307 CrossRef CAS PubMed
- G. Zheng, L. Li, Z. Tian, X. Zhang and L. Chen, J. Energy Chem., 2021, 54, 612–619 CrossRef
- H. Shen, Y. Li and Q. Sun, J. Phys. Chem. C, 2017, 121, 3963–3969 CrossRef CAS
- T. Deng, C. Cen, H. Shen, S. Wang, J. Guo, S. Cai and M. Deng, J. Phys. Chem. Lett., 2020, 11, 6320–6329 CrossRef CAS PubMed
- Y. Zhao, S. Zhou and J. Zhao, iScience, 2020, 23, 101051 CrossRef CAS PubMed
- R. Hu, Y. Li, Q. Zeng, F. Wang and J. Shang, ChemSusChem, 2020, 13, 3636–3644 CrossRef CAS PubMed
- W. Pei, S. Zhou, J. Zhao, X. Xu, Y. Du and S. X. Dou, Nano Energy, 2020, 76, 105049 CrossRef CAS
- R. Guo, M. Hu, W. Zhang and J. He, Molecules, 2019, 24, 1777 CrossRef CAS
- C. Hu, Y. Wang, J. Chen, H. F. Wang, K. Shen, K. Tang, L. Chen and Y. Li, Small, 2022, 2201391 CrossRef CAS PubMed
- X. Li, W. Zhong, P. Cui, J. Li and J. Jiang, J. Phys. Chem. Lett., 2016, 7, 1750–1755 CrossRef CAS PubMed
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed
- M. A. Hunter, J. M. T. A. Fischer, Q. Yuan, M. Hankel and D. J. Searles, ACS Catal., 2019, 9, 7660–7667 CrossRef CAS
- Y. Sun, J. Wang, Q. Liu, M. Xia, Y. Tang, F. Gao, Y. Hou, J. Tse and Y. Zhao, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS
- Y. Meng, C. Yin, K. Li, H. Tang, Y. Wang and Z. Wu, ACS Sustainable Chem. Eng., 2019, 7, 17273–17281 CrossRef CAS
- T. He, A. R. Puente Santiago and A. Du, J. Catal., 2020, 388, 77–83 CrossRef CAS
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2020, 10, 1902844 CrossRef CAS
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed
- Y. Pan, C. Zhang, Z. Liu, C. Chen and Y. Li, Matter, 2020, 2, 78–110 CrossRef
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS
- D. Ma, Z. Zeng, L. Liu, X. Huang and Y. Jia, J. Phys. Chem. C, 2019, 123, 19066–19076 CrossRef CAS
- E. F. Holby and C. D. Taylor, Sci. Rep., 2015, 5, 9286 CrossRef PubMed
- J. Wang, S. Dou and X. Wang, Sci. Adv., 2021, 13, eabf3989 CrossRef
- D. Liu, B. Wang, H. Li, S. Huang, M. Liu, J. Wang, Q. Wang, J. Zhang and Y. Zhao, Nano Energy, 2019, 58, 277–283 CrossRef CAS
- B. Wang, J. Zou, X. Shen, Y. Yang, G. Hu, W. Li, Z. Peng, D. Banham, A. Dong and D. Zhao, Nano Energy, 2019, 63, 103851 CrossRef CAS
- M. A. Hunter, J. M. T. A. Fischer, M. Hankel, Q. Yuan and D. J. Searles, J. Chem. Inf. Model., 2019, 59, 2242–2247 CrossRef CAS PubMed
- X. Zhao, X. Liu, B. Huang, P. Wang and Y. Pei, J. Mater. Chem. A, 2019, 7, 24583–24593 RSC
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, J. Am. Chem. Soc., 2019, 141, 17763–17770 CrossRef CAS PubMed
- Q. Huang, H. Liu, W. An, Y. Wang, Y. Feng and Y. Men, ACS Sustainable Chem. Eng., 2019, 7, 19113–19121 CrossRef CAS
- Y. Meng, K. Li, D. Xiao, Y. Yuan, Y. Wang and Z. Wu, Int. J. Hydrogen Energy, 2020, 45, 14311–14319 CrossRef CAS
- X. Bai, Q. Li, L. Shi, X. Niu, C. Ling and J. Wang, Small, 2020, 16, 1901981 CrossRef CAS PubMed
- Z. Liang, M. Luo, M. Chen, C. Liu, S. G. Peera, X. Qi, J. Liu, U. P. Kumar and T. L. T. Liang, J. Colloid Interface Sci., 2020, 568, 54–62 CrossRef CAS PubMed
- Y. Meng, C. Yin, K. Li, H. Tang, Y. Wang and Z. Wu, Appl. Surf. Sci., 2019, 480, 905–911 CrossRef CAS
- H. Zhang, C. Cui and Z. Luo, J. Phys. Chem. C, 2020, 124, 6260–6266 CrossRef CAS
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS
- W. Yang, H. Huang, X. Ding, Z. Ding, C. Wu, I. D. Gates and Z. Gao, Electrochim. Acta, 2020, 335, 135667 CrossRef CAS
- J. Liu, D. Cao, H. Xu and D. Cheng, Nano Sel., 2021, 2, 251–270 CrossRef CAS
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC
- E. Guan, J. Ciston, S. R. Bare, R. C. Runnebaum, A. Katz, A. Kulkarni, C. X. Kronawitter and B. C. Gates, ACS Catal., 2020, 10, 9065–9085 CrossRef CAS
- Z. He, K. He, A. W. Robertson, A. I. Kirkland, D. Kim, J. Ihm, E. Yoon, G.-D. Lee and J. H. Warner, Nano Lett., 2014, 14, 3766–3772 CrossRef CAS
- J.-C. Liu, X.-L. Ma, Y. Li, Y.-G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610 CrossRef PubMed
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS
- M. T. M. Koper, Chem. Sci., 2013, 4, 2710–2723 RSC
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed
- G. Lendvay, J. Phys. Chem., 1989, 93, 4422–4429 CrossRef CAS
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS
- Z. Xu, J. Rossmeisl and J. R. Kitchin, J. Phys. Chem. C, 2015, 119, 4827–4833 CrossRef CAS
- E. M. Fernández, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martínez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4683–4686 CrossRef PubMed
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, J. Catal., 2004, 224, 206–217 CrossRef CAS
- A. Vojvodic and J. K. Nørskov, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS
- J. Greeley, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 605–635 CrossRef PubMed
- M. T. Darby, M. Stamatakis, A. Michaelides and E. C. H. Sykes, J. Phys. Chem. Lett., 2018, 9, 5636–5646 CrossRef CAS PubMed
- M. Andersen, A. J. Medford, J. K. Nørskov and K. Reuter, ACS Catal., 2017, 7, 3960–3967 CrossRef CAS
- M. J. Craig, G. Coulter, E. Dolan, J. Soriano-López, E. Mates-Torres, W. Schmitt and M. García-Melchor, Nat. Commun., 2019, 10, 4993 CrossRef PubMed
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef
- T. Sours, A. Patel, J. Nørskov, S. Siahrostami and A. Kulkarni, J. Phys. Chem. Lett., 2020, 11, 10029–10036 CrossRef CAS PubMed
- G. Luo, Y. Jing and Y. Li, J. Mater. Chem. A, 2020, 8, 15809–15815 RSC
- Y. Qian, Y. Liu, Y. Zhao, X. Zhang and G. Yu, EcoMat, 2020, 2, e12014 CrossRef CAS
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS
- L. Gong, D. Zhang, Y. Shen, X. Wang, J. Zhang, X. Han, L. Zhang and Z. Xia, J. Catal., 2020, 390, 126–134 CrossRef CAS
- L. Wu, T. Guo and T. Li, Adv. Funct. Mater., 2022, 2203439 CrossRef CAS
- Z. F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1494–1519 CrossRef
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS
-
A. Nilsson and L. G. M. Pettersson, in Chemical Bonding at Surfaces and Interfaces, ed. A. Nilsson, L. G. M. Pettersson and J. K. Nørskov, Elsevier, Amsterdam, 2008, pp. 57–142 Search PubMed
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed
- J. H. Montoya, A. D. Doyle, J. K. Nørskov and A. Vojvodic, Phys. Chem. Chem. Phys., 2018, 20, 3813–3818 RSC
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- R. Chattot, O. Le Bacq, V. Beermann, S. Kühl, J. Herranz, S. Henning, L. Kühn, T. Asset, L. Guétaz, G. Renou, J. Drnec, P. Bordet, A. Pasturel, A. Eychmüller, T. J. Schmidt, P. Strasser, L. Dubau and F. Maillard, Nat. Mater., 2018, 17, 827–833 CrossRef CAS PubMed
- H. B. Tao, L. Fang, J. Chen, H. B. Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed
- X. Guan, W. Gao and Q. Jiang, J. Mater. Chem. A, 2021, 9, 4770–4780 RSC
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS
- C. Jia, Q. Wang, J. Yang, K. Ye, X. Li, W. Zhong, H. Shen, E. Sharman, Y. Luo and J. Jiang, ACS Catal., 2022, 12, 3420–3429 CrossRef CAS
- H. Li, Z. Zhao, Q. Cai, L. Yin and J. Zhao, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC
- X. Liu, Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 3648–3654 RSC
- C. Zhu, M. Wang, C. Wen, M. Zhang, Y. Geng, G. Zhu and Z. Su, Adv. Sci., 2022, 9, 2105697 CrossRef CAS PubMed
- H. Chen, Y. Zhang, Q. He, H. Zhang, S. Xu, X. He and H. Ji, J. Mater. Chem. A, 2020, 8, 2364–2368 RSC
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865–2878 CAS
- Y.-S. Wei, L. Sun, M. Wang, J. Hong, L. Zou, H. Liu, Y. Wang, M. Zhang, Z. Liu, Y. Li, S. Horike, K. Suenaga and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 16013–16022 CrossRef CAS PubMed
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS PubMed
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed
- Y.-F. Lu, L.-Z. Dong, J. Liu, R.-X. Yang, J.-J. Liu, Y. Zhang, L. Zhang, Y.-R. Wang, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 26210–26217 CrossRef CAS PubMed
- W. Rong, H. Zou, W. Zang, S. Xi, S. Wei, B. Long, J. Hu, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 466–472 CrossRef CAS PubMed
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS PubMed
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed
- M. Li, K. Duanmu, C. Wan, T. Cheng, L. Zhang, S. Dai, W. Chen, Z. Zhao, P. Li, H. Fei, Y. Zhu, R. Yu, J. Luo, K. Zang, Z. Lin, M. Ding, J. Huang, H. Sun, J. Guo, X. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS
- J.-C. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2020, 142, 3375–3383 CrossRef CAS
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, Nat. Energy, 2021, 6, 1054–1066 CrossRef CAS
- L. Zhang, J. M. T. A. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757–10763 CrossRef CAS
- L. Zhang, Y. Jia, H. Liu, L. Zhuang, X. Yan, C. Lang, X. Wang, D. Yang, K. Huang, S. Feng and X. Yao, Angew. Chem., Int. Ed., 2019, 58, 9404–9408 CrossRef CAS
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, eaap7970 CrossRef PubMed
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li, X. Huang, T. Yao, J. Yang, S. Wei and J. Lu, Nat. Commun., 2017, 8, 1070 CrossRef PubMed
- X. Cao, L. Zhao, B. Wulan, D. Tan, Q. Chen, J. Ma and J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202113918 CAS
- H. Wang, X. Wang, J. Pan, L. Zhang, M. Zhao, J. Xu, B. Liu, W. Shi, S. Song and H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 23154–23158 CrossRef CAS PubMed
- Y. Wu, C. Ye, L. Yu, Y. Liu, J. Huang, J. Bi, L. Xue, J. Sun, J. Yang, W. Zhang, X. Wang, P. Xiong and J. Zhu, Energy Storage Mater., 2022, 45, 805–813 CrossRef
- M. Jiang, F. Wang, F. Yang, H. He, J. Yang, W. Zhang, J. Luo, J. Zhang and C. Fu, Nano Energy, 2022, 93, 106793 CrossRef CAS
- X. He, Y. Deng, Y. Zhang, Q. He, D. Xiao, M. Peng, Y. Zhao, H. Zhang, R. Luo, T. Gan, H. Ji and D. Ma, Cell Rep. Phys. Sci., 2020, 1, 100004 CrossRef
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed
- Z. W. Chen, L. Chen, M. Jiang, D. Chen, Z. Wang, X. Yao, C. V. Singh and Q. Jiang, J. Mater. Chem. A, 2020, 8, 15086–15093 RSC
- X. Li, X. Yang, J. Zhang, Y. Huang and B. Liu, ACS Catal., 2019, 9, 2521–2531 CrossRef CAS
- J. Li and J. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC
- D. S. Su, B. Zhang and R. Schlögl, Chem. Rev., 2015, 115, 2818–2882 CrossRef CAS PubMed
- Z. Wang, X. Jin, C. Zhu, Y. Liu, H. Tan, R. Ku, Y. Zhang, L. Zhou, Z. Liu, S.-J. Hwang and H. J. Fan, Adv. Mater., 2021, 33, 2104718 CrossRef CAS PubMed
- J. Timoshenko and B. Roldan Cuenya, Chem. Rev., 2021, 121, 882–961 CrossRef CAS PubMed
- M. Xiao, J. Zhu, S. Li, G. Li, W. Liu, Y.-P. Deng, Z. Bai, L. Ma, M. Feng, T. Wu, D. Su, J. Lu, A. Yu and Z. Chen, ACS Catal., 2021, 11, 8837–8846 CrossRef CAS
- W. L. Bragg, Proc. Math. Phys., 1913, 89, 248–277 CAS
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., 2020, 38, 063204 CrossRef CAS
- X. Peng, H.-X. Liu, Y. Zhang, Z.-Q. Huang, L. Yang, Y. Jiang, X. Wang, L. Zheng, C. Chang, C.-T. Au, L. Jiang and J. Li, Chem. Sci., 2021, 12, 7125–7137 RSC
- X. Li, H.-Y. Wang, H. Yang, W. Cai, S. Liu and B. Liu, Small Methods, 2018, 2, 1700395 CrossRef
- G. Gourdin and V. Doan-Nguyen, Cell Rep. Phys. Sci., 2021, 2, 100660 CrossRef CAS
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X. F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista and Y. Liang, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed
- D. H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C. T. Dinh, F. P. García de Arquer, Y. Wang, Z. Liang, A. H. Proppe and C. S. Tan, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang and Y. Huang, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed
- E. Martínez-Periñán, M. Revenga-Parra, J. Pastore, F. Pariente, F. Zamora, O. Castillo, E. Lorenzo and H. D. Abruña, J. Phys. Chem. C, 2018, 122, 12377–12383 CrossRef
- C. J. Chang, S. F. Hung, C. S. Hsu, H. C. Chen, S. C. Lin, Y. F. Liao and H. M. Chen, ACS Cent. Sci., 2019, 5, 1998–2009 CrossRef CAS PubMed
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S. F. Hung and B. Chen, Nat. Catal., 2020, 3, 75–82 CrossRef CAS
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed
- Z. Chen, S. Jiang, G. Kang, D. Nguyen, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2019, 141, 15684–15692 CrossRef CAS PubMed
- K. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed
- S. Zhu, T. Li, W. B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS
- T. Chao, X. Luo, W. Chen, B. Jiang, J. Ge, Y. Lin, G. Wu, X. Wang, Y. Hu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16047–16051 CrossRef CAS
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef
- Y. Zhou, E. Song, W. Chen, C. U. Segre, J. Zhou, Y.-C. Lin, C. Zhu, R. Ma, P. Liu, S. Chu, T. Thomas, M. Yang, Q. Liu, K. Suenaga, Z. Liu, J. Liu and J. Wang, Adv. Mater., 2020, 32, 2003484 CrossRef CAS PubMed
- A. Kumar, V. Q. Bui, J. Lee, L. Wang, A. R. Jadhav, X. Liu, X. Shao, Y. Liu, J. Yu, Y. Hwang, H. T. D. Bui, S. Ajmal, M. G. Kim, S.-G. Kim, G.-S. Park, Y. Kawazoe and H. Lee, Nat. Commun., 2021, 12, 6766 CrossRef CAS PubMed
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed
- S.-H. Yin, J. Yang, Y. Han, G. Li, L.-Y. Wan, Y.-H. Chen, C. Chen, X.-M. Qu, Y.-X. Jiang and S.-G. Sun, Angew. Chem., Int. Ed., 2020, 59, 22160–22163 CrossRef
- G. Zhang, Y. Jia, C. Zhang, X. Xiong, K. Sun, R. Chen, W. Chen, Y. Kuang, L. Zheng, H. Tang, W. Liu, J. Liu, X. Sun, W.-F. Lin and H. Dai, Energy Environ. Sci., 2019, 12, 1317–1325 RSC
- F. Kong, R. Si, N. Chen, Q. Wang, J. Li, G. Yin, M. Gu, J. Wang, L.-M. Liu and X. Sun, Appl. Catal., B, 2022, 301, 120782 CrossRef CAS
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 131, 2648–2652 CrossRef
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed
- J. Zang, F. Wang, Q. Cheng, G. Wang, L. Ma, C. Chen, L. Yang, Z. Zou, D. Xie and H. Yang, J. Mater. Chem. A, 2020, 8, 3686–3691 RSC
- Y. Chen, S. Hu, F. Nichols, F. Bridges, S. Kan, T. He, Y. Zhang and S. Chen, J. Mater. Chem. A, 2020, 8, 11649–11655 RSC
- Y.-j Wu, X.-h Wu, T.-x Tu, P.-f Zhang, J.-t Li, Y. Zhou, L. Huang and S.-g Sun, Appl. Catal., B, 2020, 278, 119259 CrossRef CAS
- J. Xu, S. Lai, D. Qi, M. Hu, X. Peng, Y. Liu, W. Liu, G. Hu, H. Xu, F. Li, C. Li, J. He, L. Zhuo, J. Sun, Y. Qiu, S. Zhang, J. Luo and X. Liu, Nano Res., 2021, 14, 1374–1381 CrossRef CAS
- Y. Wang, Z. Li, P. Zhang, Y. Pan, Y. Zhang, Q. Cai, S. R. P. Silva, J. Liu, G. Zhang, X. Sun and Z. Yan, Nano Energy, 2021, 87, 106147 CrossRef CAS
- T. Cui, Y.-P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CAS
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS
- M. Liu, N. Li, S. Cao, X. Wang, X. Lu, L. Kong, Y. Xu and X.-H. Bu, Adv. Mater., 2022, 34, 2107421 CrossRef CAS PubMed
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef CAS
- R. Yun, F. Zhan, X. Wang, B. Zhang, T. Sheng, Z. Xin, J. Mao, S. Liu and B. Zheng, Small, 2021, 17, 2006951 CrossRef CAS
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS PubMed
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed
- L. Qin, F. Sun, X. Ma, G. Ma, Y. Tang, L. Wang, Q. Tang, R. Jin and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26136–26141 CrossRef CAS PubMed
- J. Hao, Z. Zhuang, J. Hao, C. Wang, S. Lu, F. Duan, F. Xu, M. Du and H. Zhu, Adv. Energy Mater., 2022, 2200579 CrossRef CAS
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC
- L. J. Liu, Z. Y. Wang, Z. Y. Wang, R. Wang, S. Q. Zang and T. C. Mak, Angew. Chem., Int. Ed., 2022, 61, e2022056 Search PubMed
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Nat. Commun., 2022, 13, 1322 CrossRef CAS PubMed
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed
- Y. Wu, C. He and W. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 47520–47529 CrossRef CAS
- J.-C. Chen, H. Cao, J.-W. Chen, S.-J. Qian, G.-J. Xia, Y.-G. Wang and J. Li, J. Phys. Chem. C, 2021, 125, 19821–19830 CrossRef CAS
- Y. Wu, C. He and W. Zhang, J. Am. Chem. Soc., 2022, 144, 9344–9353 CrossRef CAS PubMed
- Y. Li, Q. Zhang, C. Li, H.-N. Fan, W.-B. Luo, H.-K. Liu and S.-X. Dou, J. Mater. Chem. A, 2019, 7, 22242–22247 RSC
- P. Saha, S. Amanullah and A. Dey, J. Am. Chem. Soc., 2020, 142, 17312–17317 CrossRef CAS PubMed
- C. Yao, N. Guo, S. Xi, C.-Q. Xu, W. Liu, X. Zhao, J. Li, H. Fang, J. Su, Z. Chen, H. Yan, Z. Qiu, P. Lyu, C. Chen, H. Xu, X. Peng, X. Li, B. Liu, C. Su, S. J. Pennycook, C.-J. Sun, J. Li, C. Zhang, Y. Du and J. Lu, Nat. Commun., 2020, 11, 4389 CrossRef CAS
- X. Wang, S. Qiu, J. Feng, Y. Tong, F. Zhou, Q. Li, L. Song, S. Chen, K.-H. Wu, P. Su, S. Ye, F. Hou, S. X. Dou, H. K. Liu, G. Q. Lu, C. Sun, J. Liu and J. Liang, Adv. Mater., 2020, 32, 2004382 CrossRef CAS PubMed
- L. Han, Z. Ren, P. Ou, H. Cheng, N. Rui, L. Lin, X. Liu, L. Zhuo, J. Song, J. Sun, J. Luo and H. L. Xin, Angew. Chem., Int. Ed., 2020, 60, 345–350 CrossRef PubMed
- K. S. Adarsh, N. Chandrasekaran and V. Chakrapani, Front. Chem., 2020, 8, 137 CrossRef CAS
- L. Jin and A. Seifitokaldani, Catalysts, 2020, 10, 481 CrossRef CAS
- Z. Chen, Y. Xu, D. Ding, G. Song, X. Gan, H. Li, W. Wei, J. Chen, Z. Li, Z. Gong, X. Dong, C. Zhu, N. Yang, J. Ma, R. Gao, D. Luo, S. Cong, L. Wang, Z. Zhao and Y. Cui, Nat. Commun., 2022, 13, 763 CrossRef CAS
- L. Han, P. Ou, W. Liu, X. Wang, H. T. Wang, R. Zhang, C. W. Pao, X. Liu, W. F. Pong, J. Song and Z. Zhuang, Sci. Adv., 2022, 8, eabm3779 CrossRef CAS
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS PubMed
- B. Hu, A. Huang, X. Zhang, Z. Chen, R. Tu, W. Zhu, Z. Zhuang, C. Chen, Q. Peng and Y. Li, Nano Res., 2021, 14, 3482–3488 CrossRef CAS
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 17, 2104445 CrossRef CAS PubMed
- Y. Wang, X. Wan, J. Liu, W. Li, Y. Li, X. Guo, X. Liu, J. Shang and J. Shui, Nano Res., 2021, 1–8 Search PubMed
- H. Li, S. Di, P. Niu, S. Wang, J. Wang and L. Li, Energy Environ. Sci., 2022, 15, 1601–1610 RSC
- J. Liu, W. Luo, L. Wang, J. Zhang, X.-Z. Fu and J.-L. Luo, Adv. Funct. Mater., 2022, 32, 2110748 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |