
High-loaded sub-6 nm Pt1Co1 intermetallic compounds with highly efficient performance expression in PEMFCs†
Qingqing
Cheng‡
a,
Shuai
Yang‡
b,
Cehuang
Fu
c,
Liangliang
Zou
*a,
Zhiqing
Zou
a,
Zheng
Jiang
 b,
Junliang
Zhang
c and
Hui
Yang
*ad
b,
Junliang
Zhang
c and
Hui
Yang
*ad
aShanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201210, P. R. China. E-mail: zoull@sari.ac.cn; yangh@sari.ac.cn
bShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201204, China
cInstitute of Fuel Cells, key laboratory for Power Machinery and Engineering of MOE, Shanghai Jiao Tong University, 200240, China
dNingbo Cotron New Energy S&T Co., Ltd, Ningbo 315300, China
First published on 12th November 2021
Abstract
High-loaded oxygen reduction reaction (ORR) Pt intermetallic compounds with high performance expression under PEMFC operating conditions are prerequisite for practical application. Nevertheless, high metal-loading would lead to the severe agglomeration and nonuniformity of nanoparticles, imposing an enormous challenge on efficient synthesis. Herein we present a cobalt oxide aided structural evolution strategy to controllably synthesize high-loaded (44.7 wt%) sub-6 nm Pt1Co1 intermetallic compounds with a Pt-rich shell (Pt1Co1-IMC@Pt). Experiments and theory explicitly reveal that the ordered arrangement of Pt–Co atoms endows surface Pt with a lowered d band centre and enhanced oxidation-resistance of Pt/Co sites, thus simultaneously boosting the ORR activity and durability. The preeminent intrinsic ORR activity on an optimized catalyst reaches a mass activity as high as 0.53 A mg(Pt)−1@0.90 V RHE−1 (MA@0.9 V) in rotating disk electrode measurements. PEMFCs with such a catalyst deliver record-high power density (2.30/1.23 W cm−2 under H2–air/O2 conditions at 80 °C) and extraordinary stability, ranking the highest fuel cell performance among the Pt-based electrocatalysts. Significantly, the MA@0.9 V calculated from fuel cell attains 0.46 A mg(Pt)−1, exceeding the 2020 DOE target (0.44 A mg(Pt)−1) and very close to the intrinsic value, definitely confirming the superiority of high-loaded Pt1Co1-IMC@Pt/C in activity expression under fuel cell conditions. This study paves a new way for the future practical application of low-Pt catalysts in PEMFCs.
Broader contextCommercialization of fuel cell vehicles urgently requires the high-performance, long-life and low-Pt electrocatalysts towards the oxygen reduction reaction (ORR) to dramatically reduce the costly Pt usage. Pt-based intermetallic (IMC) catalysts have been regarded as the most promising alternative for the ORR due to the boosted activity and durability. Substantial progress has been achieved in the development of cost-effective IMC catalysts, but the advanced catalysts only meet the initial-activity target using the idealized rotating disk electrode (RDE) and rarely exhibit the desirable performance in the membrane electrode assembly (MEA) configuration. Here, we reported the high-loaded (44.7 wt%) and sub-6 nm Pt1Co1 intermetallic compound catalyst, which can be controllable synthesized through a cobalt oxide aided structural-evolution strategy. The resultant catalyst not only exhibits superb ORR activity and durability in RDE measurements, but also delivers the record-high power density and extraordinary stability in H2–air/O2 fuel cells, demonstrating that the excellent activity can be highly efficient expression under PEMFC operation conditions. This study opens a feasible path to bridge the performance gap between the model environment under half-cell and operating conditions in PEMFCs via constructing high-loaded intermetallic Pt1Co1 electrocatalyst with highly efficient activity expression. |
Introduction
Carbon-supported platinum nanoparticles (Pt-NPs/C), as the state-of-the-art oxygen reduction reaction (ORR) catalysts for PEMFCs,1,2 are still suffering from undesirable intrinsic activity, poor durability and high cost.3 Alloying Pt with a first-row transition metal (TM = Co, Fe, Ni, etc.)4–7 is current a feasible strategy to solve the above problems simultaneously by virtue of the unique geometric configuration and electronic structure.8,9 Pt intermetallic compounds (IMC), instead of the disordered PtM alloy,10,11 have been demonstrated to prominently improve the ORR intrinsic activity and stabilization of TM atoms through strong 3d–5d orbital coupling along the crystallographic c direction.12–15 For instance, Sun et al.16 reported an ordered L10-PtCo structure with greatly enhanced ORR activity. Meantime, the Co atoms remained stable after 24 h acid-leaching at 60 °C, indicative of an excellent structural stability. Abruña et al.17 designed an ordered Pt3Co alloys with a PtCo@Pt skin core–shell structure, enabling the superior long-term stability and significantly improved mass activity. Although the great progress have been achieved,15,18–20 the excellent ORR activities are always exclusively acquired from rotating disk electrode (RDE) measurements and rarely efficiently translated into a membrane electrode assembly (MEA) configuration.21–23 Therefore, how to obtain the preeminent performance for the PtM-IMC catalysts under fuel cell operating conditions is a key issue to be fixed urgently.High-loaded PtM-IMC/C catalysts with high-efficient performance expression have been regarded as the most promising candidates24 for the practical application in PEMFCs in comparison with its low-loaded (<20 wt%) counterparts.25–27 On the one hand, a high metal/carbon ratio would thin the catalytic layer (CL) to accelerate the mass transport, which in turn would significantly decrease the voltage loss,28 especially under high current densities (>1.0 A cm−2). On the other hand, high metal ratio might increase the accessibility of metal active sites and ionomer (e.g., Nafion resin), thereupon enlarging the triple-phase reaction interface29 and thus boosting the fuel cell performance. Last but not least, the low carbon support could weaken the risk of carbon corrosion30,31 under working environment (e.g., high potential, strong acid and high oxygen concentration), thereby effectively mitigating the Ostwald-ripening of nanoparticles (NPs) and prolonging the lifetime of fuel cells.32 Unfortunately, the formation of PtM-IMC structure usually involves the high-temperature annealing, which inevitably results in the serious IMC sintering with the NP sizes of ca. 8–10 nm,33,34 much larger than that of PtM alloys or Pt/C, consequently substantially lowering the Pt utilization and ORR activity. In particular, for the high-loaded PtM-IMC/C catalysts, the high-density NPs are extremely prone to agglomeration into large NPs, further increasing the difficulty of the controllable synthesis of small-sized PtM-IMC/C catalysts. To fulfil the above ambition, some sacrificial templates, such as SiO2,7 TiO2,35 MgO,36,37etc., were employed as the coating layer to restrict the coalescence of PtM-NPs under high-temperature annealing. Nevertheless, the coating and post-treatment processes make the synthetic procedures tedious and time-consuming. Even more seriously, the thicker coating layer may hinder the atomic inter-diffusion of Pt and M, thus leading to more difficult in phase-transformation from disordered alloys to the ordered IMC structure.38 In the meantime, the carbon residual derived from organic templates (e.g., PDA39,40) would contaminate the PtM–NP surface, and then degrading the ORR activity. Thus, innovative approaches are needed for facile, but efficient, synthesis of high-loaded and small-sized PtM-IMC/C catalyst for practical application in PEMFCs.
Herein we present a cobalt oxide aided thermal-migration strategy to ensure high-efficient structural evolution from an ultrafine Pt nanocrystal to a sub-6 nm high-loaded (ca. 44.7 wt%) Pt1Co1 intermetallic core–shell structure with 2–3 atomic layers of Pt shell (Pt1Co1-IMC@Pt/C). To be specific, the deposited Co3O4 not only facilitates the inter-diffusion of Pt/Co atoms due to the in situ formation of abundant oxygen-vacancies,12,36 but also prevents the NPs from sintering via space–barrier effect. Both X-ray adsorption spectroscopy (XAS) and DFT calculations highlighted the crucial role of the core–shell structure of Pt1Co1-IMC@Pt in tuning the lattice contraction,41 d band centre and oxidation-resistance of Pt/Co atoms. As a result, the optimized catalyst exhibited an excellent electrocatalytic ORR activity and stability both in RDE and MEA configurations. Impressively, the power densities reached the record-high values of 2.30/1.23 W cm−2 for H2–O2/air fuel cells at 80 °C, respectively, which were the highest performance to date. Meanwhile, the high ORR performance was faultlessly translated into MEA configuration with the MA@0.9 V of 0.48 A mg(Pt)−1, close to the intrinsic value (0.53 A mg(Pt)−1) in RDE measurements, which has rarely been reported to date. Thus, this high-loaded, small-sized Pt1Co1-IMC@Pt/C catalyst is highly suitable for the future practical application in low-Pt PEMFCs.
Results and discussion
Controllable structural evolution to construct Pt1Co1-IMC
The synthesized procedure is illustrated in Fig. 1a. Typically, the carbon (XC-72R) supported ultrafine Pt nanoparticles (Pt-NPs/C) were initially fabricated via the Pt-carbonyl method (Detailed experiments can be seen in the ESI†). H2PtCl6·6H2O was coordinated with the continuously purged CO gas to form the Pt(CO)x clusters accompanied by the change in solution colour from yellow to olive (Fig. S1, ESI†). Afterwards, the above solution was mixed with XC-72R/methanol slurry and evaporated, followed by slowly exposed to air at zero degree to trigger the decomposition of Pt(CO)x. The X-ray diffraction (XRD) pattern (Fig. S2a, ESI†) shows several diffraction peaks at ca. 39.8, 46.2, 67.5 and 81.3°, corresponding to the facets of (111), (200), (220) and (311) of face centre cubic crystalline Pt (JCPDS no. 04-0802). The TEM image (Fig. S2b, ESI†) presents the well-dispersed and uniformly sized Pt-NPs over the carbon matrix with the mean size of ca. 1.9 nm. Thermogravimetric (TG) curves combined with inductively coupled plasma atomic emission spectroscopy (ICP-AES) results (Fig. S3, ESI†) determine a Pt weight content of ca. 40 ± 1%. Notably, as the Pt(CO)x clusters are inclined to be oxidized in the presence of trace oxygen, the reaction system needs to be isolated from air, otherwise the resultant Pt-NPs will be larger in size (Fig. S4, ESI†). Thereafter, the Co ions (atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for Pt/Co) were deposited on the prepared Pt-NPs/C matrix as the form of Co3O4 precipitates through the ammonia (NH3)-evaporation method.42 The XRD pattern (Fig. S5a, ESI†) shows some new diffraction peaks, which are assigned to the cobalt oxide (Co3O4) phase (JCPDS no. 42-1467), confirming the formation of the Co3O4/Pt-NP hybrid complex. Moreover, TEM images combined with EDS-mapping (Fig S5b and c, ESI†) show that cobalt oxide nanoparticles are distributed around the Pt-NPs. Subsequently, the prepared Co3O4/Pt-NPs/C precursor was annealed under 90% Ar + 10% H2 at high temperature (T) for various times (t, t = 1, 2.5 and 4 h) to induce the structural evolution43 from the Pt nanocrystal to the Pt1Co1-IMC structure. The surface unstable Co atoms were then removed by immersing the annealed sample in warm 0.5 M H2SO4 solution to obtain the final samples for further study, denoted as T–PtCo/C-t.
:1 for Pt/Co) were deposited on the prepared Pt-NPs/C matrix as the form of Co3O4 precipitates through the ammonia (NH3)-evaporation method.42 The XRD pattern (Fig. S5a, ESI†) shows some new diffraction peaks, which are assigned to the cobalt oxide (Co3O4) phase (JCPDS no. 42-1467), confirming the formation of the Co3O4/Pt-NP hybrid complex. Moreover, TEM images combined with EDS-mapping (Fig S5b and c, ESI†) show that cobalt oxide nanoparticles are distributed around the Pt-NPs. Subsequently, the prepared Co3O4/Pt-NPs/C precursor was annealed under 90% Ar + 10% H2 at high temperature (T) for various times (t, t = 1, 2.5 and 4 h) to induce the structural evolution43 from the Pt nanocrystal to the Pt1Co1-IMC structure. The surface unstable Co atoms were then removed by immersing the annealed sample in warm 0.5 M H2SO4 solution to obtain the final samples for further study, denoted as T–PtCo/C-t.
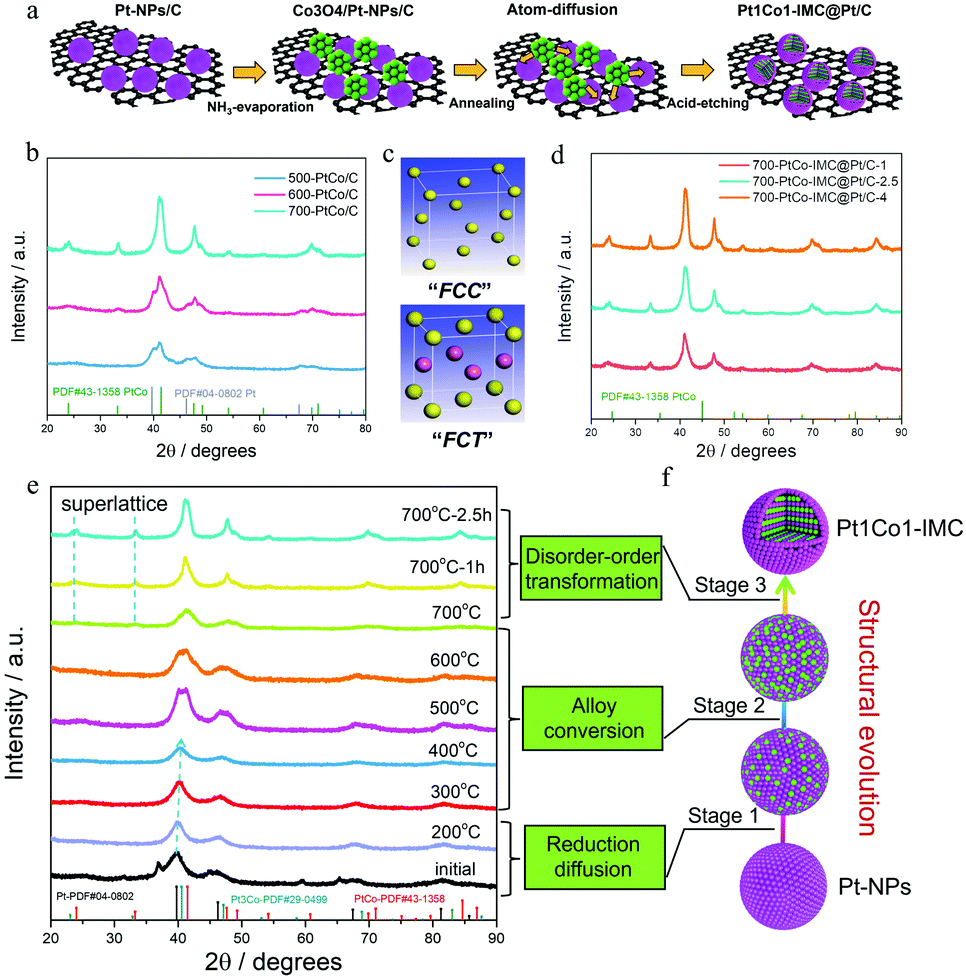 | ||
| Fig. 1 (a) Synthetic procedure for the Pt1Co1-IMC@Pt/C catalyst; (b) XRD patterns of the obtained PtCo/C samples under different annealing temperatures; (c) crystal structure for fcc Pt and fct Pt1Co1 intermetallic structure; (d) structural evolution of PtCo/C under different duration times at 700 °C; (e) temperature/time-evolution ex situ XRD patterns of Pt1Co1-IMC@Pt/C catalyst during the annealing process (f) schematic diagram of possible structural evolution mechanism. | ||
To explore the effect of annealing temperature on the structural evolution process, the Co3O4/Pt-NPs/C was then annealed at 500, 600 and 700 °C, respectively, to obtain the T-PtCo/C samples (T = 500, 600, and 700). XRD patterns (Fig. 1b) show that both 500 and 600-PtCo/C samples exhibit the hybrid phase structures, mainly including Pt crystalline and Pt1Co1 alloy, implying that Pt-NPs could be partially converted to alloys under relatively low temperatures. As temperature increases to 700 °C, no Pt nanocrystal but a Pt1Co1 alloy structure is observed, indicative of the accomplishment for structural evolution. Specifically, as for the 700-PtCo/C sample, all diffraction peaks shift to higher angles in comparison with the pristine Pt-NPs/C, suggesting the intensive lattice contraction. Moreover, some superlattice peaks (ca. 24.0° and 33.3°) corresponding to the (001) and (110) facets of ordered face-centred tetragonal (fct, JCPDS no. 43-1358) Pt1Co1 structure are clearly observed, illustrating that high-temperature is prerequisite for the structural evolution from fcc crystalline Pt to fct Pt1Co1-IMC (Fig. 1c). In addition, the duration time also plays a crucial role in the crystalline size and the ordering degree. The contrast sample annealed with short time (700-Pt1Co1-IMC@Pt/C-1) displays the small nanoparticle size while the superlattice peaks are not obvious (Fig. 1d), signifying a the partially ordered structure. A longer duration time (700-PtCo-IMC@Pt/C-4), in spite of the essentially improved ordering degree with distinct superlattice peaks, would readily result in the severe agglomeration of NPs, verified by sharp characteristic peaks in the XRD pattern. Hence, the moderate annealed time (i.e., t = 2.5 h) both guarantees the high ordering degree and prevents the NPs from further sintering.
Interestingly, the feeding ratio of Pt/Co atoms is another impact factor to control the structural evolution. To comprehensively investigate the regulation rule, the Co3O4/Pt-NPs/C precursors with various initial Pt/Co atomic ratios (Pt3Co1, Pt1Co3) were prepared and to form the corresponding PtCo/C contrast samples at 700 °C for 2.5 h, designated as PtCo/C (3:1) and PtCo/C (1:3), respectively. As expected, the PtCo/C (3:1) sample exhibits the partially ordered Pt3Co-IMC phase structure, as shown by the XRD pattern (Fig. S6, ESI†). Nevertheless, TEM images (Fig. S7, ESI†) show that the crystalline size of PtCo/C (3:1) is obviously larger than that of PtCo/C (1:1), which is probably attributed to the insufficient Co3O4 species as the space barriers to protect Pt-NPs against coalescence. To be surprised, the PtCo/C (1:3) sample displays the disordered Pt3Co1 phase structure (Fig. S6, ESI†) other than the ordered Pt3Co-IMC or Pt1Co3 alloy structure. The possible reason is that the excessive Co ions are deposited to form a thick Co3O4 protector, which seriously hinders the heat-conduction, thereby lowering the thermal-migration rate and leading to the incomplete structural evolution. It is noted that abundant Co3O4 can better prevent the NP aggregation, gaining relatively smaller Pt3Co-NPs compared to the other two samples. The above control experiments reveal that the annealing temperature, duration time as well as the feeding ratio of Pt/Co contribute significantly to the structural evolution from an ultrafine Pt nanocrystal to Pt1Co1-IMC with high-loaded sub-6 nm NPs.
To deeply investigate the structural evolution mechanism of Pt-NPs with Co3O4 to intermetallic compound, the temperature/time-dependence ex situ XRD were conducted to monitor the annealing process, as shown in Fig. 1e. It is observed that the Co3O4 phase disappears when the temperature increases to 200 °C while the diffraction peaks of fcc Pt positively shift to high angle compared to the initial Co3O4/Pt-NPs/C precursor, illustrating that Co3O4 is reduced to metallic Co atoms, which start to migrate and diffuse into the Pt lattice to form a PtxCo (x > 3) disordered alloy. When the temperature increases to 300 and 400 °C, more Co atoms diffuse into the Pt lattice to form a Pt3Co disorder alloy, verified by the continuous positive shift of diffraction peaks. When the annealing temperature reaches 500, 600 °C, the doublet peaks at around 40.5 and 41.4 are clearly observed on the XRD patterns, attesting the formation of alloy mixture (Pt3Co, Pt3Co/Pt1Co1) at this stage. Furthermore, the peak of the Pt3Co phase is gradually weakened and the Pt1Co1 phase is predominant with the temperature increasing to 700 °C, again demonstrating the incessant diffusion of Co atoms in the period of annealing. Thereafter, the superlattice peaks located at ca. 24.0, 33.2°, assigned to the fct Pt1Co1-IMC phase, gradually strengthen as prolonging the duration time (1, 2.5 h) at 700 °C, indicative of the occurrence of disorder–order phase transformation.
Based on ex situ XRD results, the structural-evolution mechanism is speculated and illustrated in Fig. 1f. initially, the Co3O4 is reduced to metallic Co atoms at a temperature below ca. 200 °C under a H2 atmosphere. The in situ formed Co atoms gradually diffuse into the Pt-NP lattice to form PtxCo (x > 3), Pt3Co, the Pt3Co/Pt1Co1 mixture and Pt1Co1 disorder alloys sequentially as the temperature uplifting from 300 to 700 °C. Subsequently, disorder–order transformation occurs and is accomplished after annealing at 700 °C for an appropriate time (i.e., 2.5 h). Finally, the core–shell Pt1Co1-IMC@Pt/C catalyst is obtained after warm-acid treatment.
As far as 700-Pt1Co1-IMC@Pt/C-2.5 is concerned, a low-magnification TEM image (Fig. 2a) shows that the NPs are highly dispersed on the carbon support. The NPs in the range of 4.5–6 nm are predominant with an average size of only ca. 5.3 nm (Fig. 2b). The d-spacing of ca. 0.217 nm for the adjacent lattice fringe, calculated from HR-TEM image (Fig. 2c) of a single NP, matches well with that of the (111) facet of Pt1Co1-IMC structure. The superlattice reflection, for instance (110), is clearly observed in the fast Fourier transform (FFT) pattern (inset in Fig. 2c), also evidencing the formation of ordered Pt1Co1-IMC at 700 °C. Aberration-corrected high-angle annual dark field scanning transmission spectroscopy (AC-HAADF-STEM) was carried out to acquire the detailed atomic arrangement in the nanocrystal. To be specific, Fig. 2d and Fig. S8 (ESI†) show the typical Z-contrast images for a single Pt1Co1-NP, displaying that the alternating brighter (Pt) and darker (Co) atoms stacking in one direction were clearly indicative of the Pt/Co atoms ordering, which can also be apparently observed in the enlarged image (Fig. 2e). The (001) facet is clearly recognized on a single NP, further proving the formation of the Pt1Co1 fct structure. Strikingly, two atomic Pt-layers are discerned at the margin of Pt1Co1-NP, confirming the formation of the Pt1Co1-IMC@Pt core–shell structure. Energy dispersive X-ray spectroscopy (EDS) elemental mapping images (Fig. 2f–h) depict that Pt and Co atoms are simultaneously distributed over one single nanoparticle in a highly ordered manner. Notably, the Pt map has a slightly larger area than the Co map, also indicates that a Pt-rich shell is formed at the surface of an ordered Pt1Co1-IMC core, which is in line with the EDS line-profile results (Fig. 2i). ICP measurements show that the Pt1Co1-IMC@Pt/C has a metal loading of ca. 44.7 wt% with an Pt/Co atom ratio of nearly 57:43 (Table S1, ESI†).
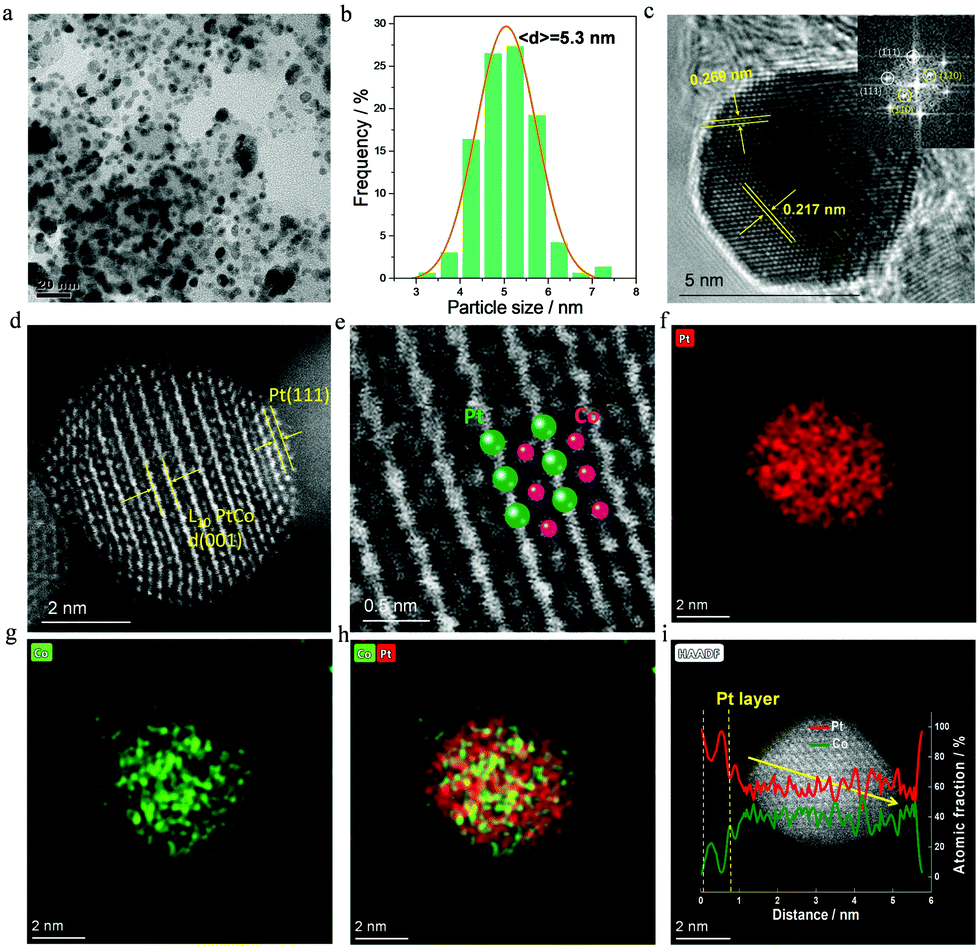 | ||
| Fig. 2 (a) Low-magnification TEM (b) size distribution histogram (c) HR-TEM, inset is the FFT image (d) atomic-resolution and (e) enlarged STEM images (f–h) EDS-mapping for the the 700-Pt1Co1-IMC@Pt/C-2.5 catalyst and (i) EDS line-profile. | ||
Electronic structure analysis
X-ray photoelectron spectroscopy (XPS) was initially carried out to identify the surface elemental compound and chemical state. High-resolution XPS of Pt 4f in 700-Pt1Co1-IMC@Pt/C-2.5 and Pt/C (Fig. 3a) show that the binding energy (BE) of Pt 4f shifts to higher value relative to the Pt/C one, which could be ascribed to the strong electronic interaction within the Pt1Co1-IMC structure, also suggesting a downshift of the d-band center.44 The deconvoluted peaks obviously show that the surface Pt in 700-Pt1Co1-IMC@Pt/C-2.5 is primary in the metallic Pt(0) state determined by its peak area percentage in comparison with that of commercial Pt/C, signifying that the Pt1Co1-IMC structure is conducive to improving the oxygen-species resistance for surface Pt, probably weakening the binding between O/OH and Pt sites, and achieving the fast H2O desorption and high activity.45 X-ray adsorption spectroscopies (XAS) were further conducted to elucidate the electronic state and the local coordination environment for the as-prepared catalyst. The X-ray adsorption near edge structure (XANES, Fig. 3b) spectra at the Pt L3-edge show that the white line (WL) peak is obviously lower than that of commercial Pt/C and PtO2, but very similar to that of Pt foil, signifying the less oxidized state of Pt in the 700-Pt1Co1-IMC@Pt/C-2.5 catalyst. As the zero valent Pt is more favorable for the ORR catalysis,46 the metallic Pt would be beneficial to accelerate the ORR kinetics. The XANES spectra at Co K-edge show (Fig. S9, ESI†) that the WL peak slightly shifts positively compared to Co foil, implying that Co atoms carried positive charges, confirming the intensive interaction between Pt and Co atom. In theory, the interaction in ordered intermetallic alloy includes the significant ionic bond and the electrons will transfer from low electronegative atom (Co, 1.8) to high one (Pt, 2.2), thus inducing the high valence of Co atoms in the Pt1Co1-IMC structure, which is thus in line with the above XANES results.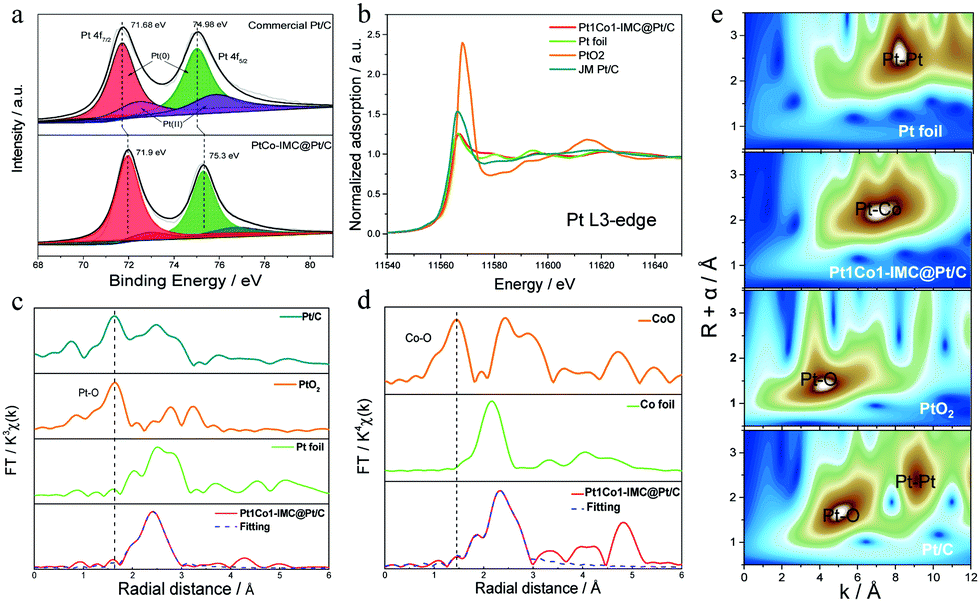 | ||
| Fig. 3 (a) XPS spectra of Pt 4f for commercial Pt/C and Pt1Co1-IMC@Pt/C catalyst; (b) XANES (c) EXAFS spectra at Pt L3-edge for Pt1Co1-IMC@Pt/C, commercial Pt/C, PtO2 and Pt foil; (d) Co K-edge EXAFS spectra for Pt1Co1-IMC@Pt/C, CoO and Co foil; (e) wavelet transform of Pt L3-edge EXAFS data for Pt foil, Pt1Co1-IMC@Pt/C, PtO2 and commercial Pt/C. | ||
Fourier transform extended X-ray adsorption fine structure (FT-EXAFS) spectroscopy and fitting results (fitting parameters were provided in Table S2, ESI†) at the Pt L3-edge (Fig. 3c) demonstrate that the 700-Pt1Co1-IMC@Pt/C-2.5 displays a significantly shortened Pt–Pt bond length (0.269 Å) compared to the Pt foil (0.276 Å), due to the incorporation of smaller Co atoms into the Pt crystal lattice. Noteworthily, the commercial Pt/C shows a distinct peak at nearly 1.66 Å, close to the Pt–O coordination of PtO2, demonstrating that the Pt atoms in Pt/C catalyst are prone to be oxidized. In contrast, almost no Pt–O bond could be observed in the Pt1Co1-IMC@Pt/C catalyst, confirming the oxidization-resistance featured for the ordered Pt1Co1-IMC@Pt structure, which is well consistent with the XPS results. Besides, Co K-edge experiment data and fitting results (Fig. 3d) show a noticeable increase of the Co–Co bond length from the bulk value of 2.49 Å to 2.61 Å, again demonstrating that the structural evolution from pure Pt to ordered Pt1Co1-IMC@Pt induces the strong electronic interaction between the Pt and Co layers. As reported,4,11,47 both Pt lattice contraction and ligand effect have been proved to facilitate the significant improvement of ORR activity. Moreover, the wavelet transform (WT) of Pt L3-edge EXAFS oscillation (Fig. 3e) displays that the intensity maxima of 700-Pt1Co1-IMC@Pt/C-2.5 is centered at ∼7.3 Å−1, lower than that of Pt foil (∼8.1 Å−1), which is associated with the Pt–Co attributions. Moreover, compared to the Pt/C and PtO2 references, there is only one intensity maximum and no Pt–O or Co–O attribution is observed, further confirming the oxygen-resistance property of the Pt1Co1-IMC@Pt structure.
Electrocatalytic ORR and mechanism study
The electrocatalytic ORR performance of the as-prepared 700-PtCo@Pt/C-t and reference Pt/C (Johnson Matthey) catalysts were evaluated by cyclic voltammetry (CV) and linear sweep voltammetry (LSV) in 0.1 M HClO4 solution at room temperature of ca. 25 °C. CV curves of 700-Pt1Co1-IMC@Pt/C-t (t = 1, 2.5 and 4 h) samples recorded in N2-saturated electrolyte at a scan rate of 50 mV s−1 are illustrated in Fig. 4a and Fig. S10 (ESI†). Clearly, the H2 underpotential deposition (HUPD) regions drop sharply when compared to commercial Pt/C one, demonstrating that the increased NP sizes restrict the high exposure of Pt atoms after high-temperature annealing. Accordingly, the electrochemical surface areas (ECSAs) of Pt1Co1-IMC@Pt/C-1, 2.5 and 4 samples are calculated as 52.6, 43.5 and 34.1 m2g(Pt)−1, which are much smaller than that of Pt/C one (65.6 m2g(Pt)−1). Noteworthily, the formation potential for the Pt–OHad and the reduction of Pt oxide significantly shift to being more positive for the 700-Pt1Co1-IMC@Pt/C-t samples with respect to that of Pt/C, indicative of the weakened oxophilicity on the Pt surface, which is also in good agreement with the XPS or EXAFS conclusion. On the basis of LSV curves towards ORR on various catalysts (Fig. 4b), 700-Pt1Co1-IMC@Pt/C-t samples show the obvious positive shift of both onset and half-wave potentials (Eonset, E1/2) in comparison with the commercial Pt/C, validating that the ordered P1Co1-IMC@Pt structure indeed boosts the ORR activity in spite of the largely decreased ECSA. In particular, the ORR activities of 700-Pt1Co1-IMC@Pt/C-t samples represent the “volcano-like” regularity with increasing duration time. In detail, the 700-Pt1Co1@Pt/C-1 shows the lowest activity among the 700-Pt1Co1-IMC@Pt/C-t samples, which stems mainly from its partially ordered structure whereas it still displayed superior ORR activity compared to Pt/C, attesting that the ORR activity is much more promoted after Co incorporation. On the other hand, even, though the 700-Pt1Co1-IMC@Pt/C-4 catalyst displays a highly ordered Pt1Co1-IMC@Pt structure, the activity shows an obvious decline in comparison with the 700-Pt1Co1-IMC@Pt/C-2.5 catalyst, also confirming the significance of the particle size in ORR activity.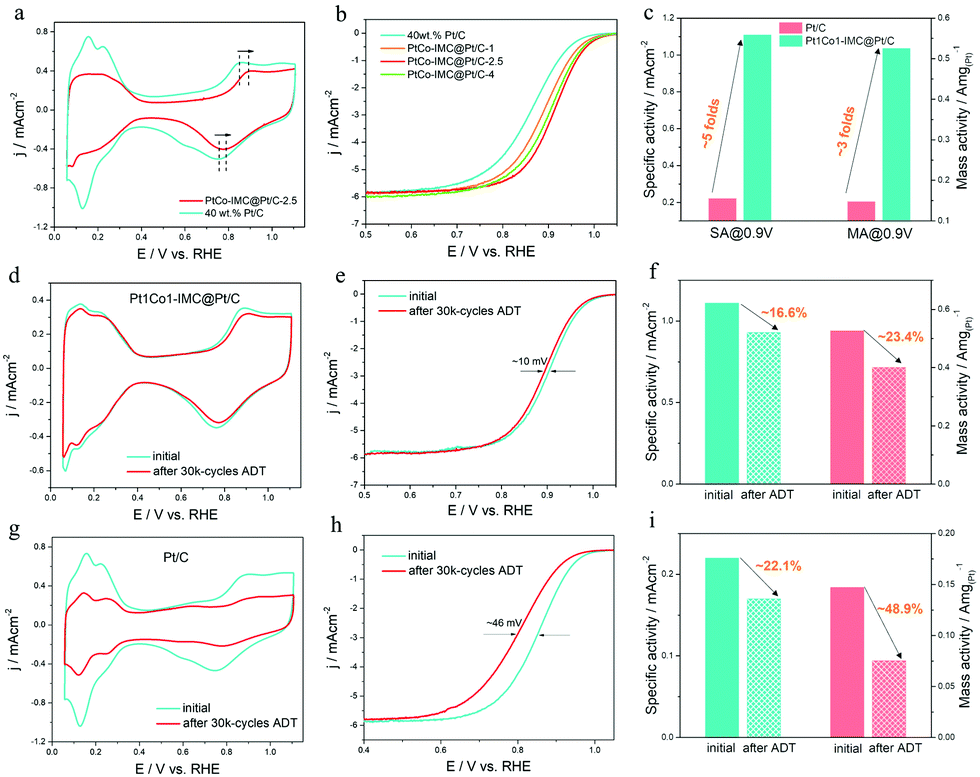 | ||
| Fig. 4 (a) Comparison of CV curves for Pt1Co1-IMC@Pt/C-2.5 and commercial Pt/C catalysts; (b) LSV curves of ORR on various Pt1Co1-IMC@Pt/C-t catalysts; (c) specific activities and mass activities at 0.9 V/RHE; (d and g) CV, (e and h) LSV (f and i) SAs and MAs on Pt1Co1-IMC@Pt/C-2.5 and Pt/C catalysts before and after 30k cycles ADT. | ||
To get further insight into the intrinsic activity for the Pt1Co1-IMC@Pt/C catalyst, the kinetic currents, calculated from the Koutecky–Levich equation, are generally normalized by ECSA and the mass of the loaded Pt to obtain the specific activity (SA) and mass activity (MA), respectively. Fig. 4c shows the SAs/MAs on 700-Pt1Co1-IMC@Pt/C-2.5 with the values of 1.11 mA cm−2/0.53 A mg(Pt)−1, being ∼5 times and 3.3 times as high as those (0.22 mA cm−2/0.15 A mg(Pt)−1) of the commercial Pt/C at 0.9 V/RHE, reflecting the highly boosted ORR intrinsic activity as well as the Pt utilization efficiency. Tafel plots (Fig S11, ESI†) exhibit the slopes of 60.7 and 78.3 mV dec−1 for the 700-Pt1Co1-IMC@Pt/C-2.5 and Pt/C, further assessing the greatly enhanced ORR kinetics by the construction of the ordered Pt1Co1-IMC@Pt structure.
Except for the electrocatalytic activity, the electrochemical durability of the ORR catalyst is also significantly critical from a practical perspective. The cycling stability was investigated by using the accelerated durability test (ADT) between 0.6 and 1.1 V RHE−1 in O2-saturated 0.1 M HClO4 at a scan rate of 100 mV s−1. As illustrated in Fig. 4d and e, after a 30k potential cycling, 700-Pt1Co1-IMC@Pt/C-2.5 exhibits a slight decrease of ECSA (∼7% loss) and a negative shift of only ∼10 mV in E1/2. Particularly, the specific activity and mass activity of the 700-Pt1Co1-IMC@Pt/C-2.5 catalyst display drops of 16.6% and 23.4% (Fig. 4f). On the contrary, the commercial Pt/C shows a clear decline of ECSA (Fig. 4g) and serious degradation of ORR activity with the E1/2 negative shift by ca. 46 mV (Fig. 4h). In the meantime, the Pt/C displays apparent losses 22.1% in SA, and 48.9% in MA (Fig. 4i), respectively, again confirming the poor durability of the commercial Pt/C catalyst. Furthermore, TEM images monitor the evolution of morphologies for 700-Pt1Co1-IMC@Pt/C-2.5 and Pt/C before and after ADT. The NPs in the commercial Pt/C catalyst shows an obvious coalescence with the mean size increasing from 2.2 to 5.5 nm (Fig. S12, ESI†), indicative of the extensive ripening of Pt-NPs during CV cycling. In contrast, only a slight growth of NPs could be discerned (Fig. S13, ESI†) for the 700-Pt1Co1-IMC@Pt/C-2.5 with the average size increasing from 5.3 to 6.2 nm, which explicitly interpret the maintenance in ECSA after potential cycling, also proving the antioxidation of Pt surface. Moreover, STEM images (Fig. S14a and b, ESI†) for the 700-Pt1Co1-IMC@Pt/C show that the ordered Pt1Co1 atomic arrangement is observation, certifying the superb structural stability. The EDS line-profile of a single NP (Fig. S14c and d, ESI†) displays the existence of the core–shell structure with a Pt/Co atomic ratio of ca. 56:44 and the ICP-AES result shows that almost no Co element is detected in the electrolyte after ADT, further revealing the positive effectiveness of the core–shell intermetallic structure in stabilization of Co atoms.
To get a deep insight into the enhanced mechanism for the ordered Pt1Co1-IMC@Pt structure in ORR activity and durability, DFT computations were conducted on the basis of pure-Pt and Pt1Co1-IMC@Pt models with 2 atomic layers of the Pt shell (Fig. 5a and b). As depicted in Fig. 5c, the Pt1Co1-IMC@Pt exhibits a lower d-band center with respect to the pure Pt, indicating that antibonding orbital is filled with more electrons and the oxygen adsorption is weakened, which matches well with the spectroscopic and CV results. The energy profiles of the ORR process on two models are displayed in Fig. 5d. It is noted that the adsorption energy for each reaction intermediate is obviously weakened in the Pt1Co1-IMC@Pt model in comparison with the pure-Pt structure, demonstrating that the ligand effect between Pt and Co atom that is triggered by the electron transfer from Co to Pt greatly influences the adsorption behavior of intermediates on Pt site. As a result, the onset potentials for pure Pt and Pt1Co1-IMC@Pt models are 0.397 V and 0.266 V, respectively, again confirming the greatly improved ORR kinetics after the formation of the Pt1Co1-IMC structure. Interestingly, to theoretically analyze the stabilization of the Co atom within the Pt1Co1-IMC@Pt structure, the disordered Pt1Co1 with randomly dispersed Co atoms model was also established (inset in Fig. 5e). Notably, the Co atom in ordered Pt1Co-IMC@Pt shows a much lower O adsorption energy than the disordered one (Fig. 5e), signifying that the periodical arrangement of the Pt1Co1 structure gives the Co atoms superb oxygen-resistance capacity, which thereby impedes the dissolution of Co atom from Pt1Co1 nanoparticles and improves the structural durability.
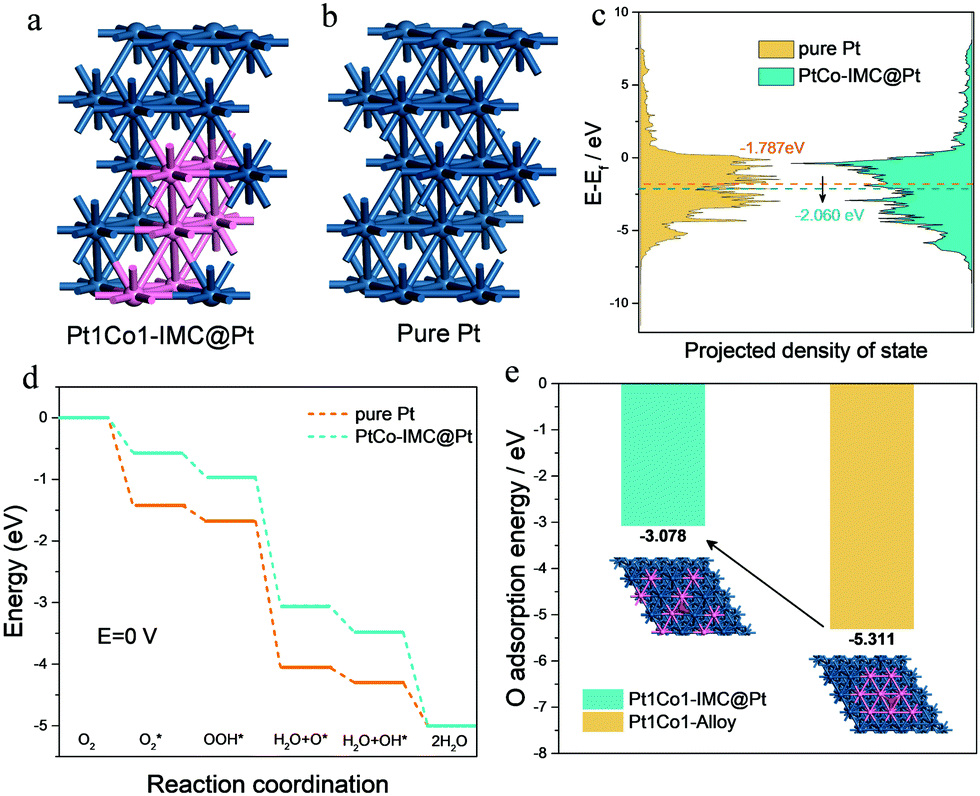 | ||
| Fig. 5 Calculation models for (a) Pt1Co1-IMC@Pt (b) pure Pt; (c) PDOS of Pt on two model structures; (d) energy profiles of ORR processes at U = 0 V; and (e) O adsorption energy of Co sites in the ordered and disordered Pt1Co1 alloy structure. | ||
Practical application in PEMFCs
Considering the high ORR activity and durability, the prepared Pt1Co1-IMC@Pt/C catalyst was used as the cathode of MEA under H2–air/O2 conditions to demonstrate the practical application in PEMFCs. For better comparison, the commercial 40 wt% Pt/C (Johnson Matthey) was used as the cathode catalyst to measure the performance under the same testing conditions. The steady-state polarization curves (Fig. 6a) show that the MEA the cathode of Pt1Co1-IMC@Pt/C exhibits obviously high cell voltages at various current densities compared to commercial Pt/C, indicative of the greatly enhanced ORR activity and mass transportation after the formation of high-loaded Pt1Co1-IMC@Pt/C structure. Noteworthily, at a voltage of 0.8 V, the current density is achieved of ∼0.28 A cm−2 (Fig. 6b), which nearly approaches that of the DOE target (0.3 A cm−2@0.8 V) and much higher than that of commercial Pt/C with a value of 0.2 A cm−2. In addition, at a practical working potential of 0.65 V, the Pt1Co1-IMC@Pt/C yields a current density of ca. 1.45 A cm−2. On the contrary, the MEA the commercial Pt/C cathode only delivers a low current density of 1.18 A cm−2. Compared to previously reported Pt-based catalysts27,37,48 (Table S3, ESI†), the Pt1Co1-IMC@Pt/C in this case is one of the best-performing cathode catalysts in fuel cells to date.The IR-free polarization curves (Fig. S15a, ESI†) were obtained by correcting the presented high-frequency resistance (HFR, Table S4, ESI†) to evaluate the intrinsic ORR activity operating in the PEMFC. The mass activities for Pt1Co1-IMC@Pt/C at various voltages (0.9 V, 0.8 V, and 0.7 V) can reach 0.18, 6.65 and 13 A mg(Pt)−1, respectively, which are much higher values than those of commercial Pt/C with the values of 0.05, 2 and 9.85 A mg(Pt)−1, respectively, as shown in Fig. S15b (ESI†). In particular, for lower Pt usage (0.05 mg cm−2 for the anode and 0.1 mg cm−2 for the cathode) MEA, the Pt1Co1-IMC@Pt/C displays high peak power density (1.13 W cm−2) and MA (0.24 A mg(Pt)−1, Fig. S16, ESI†) with respect to the commercial Pt/C (1.02 W cm−2), proving the greatly promoted Pt utilization in practical applications. Under H2–O2 fuel cell conditions (Fig. 6c), the Pt1Co1-IMC@Pt/C achieves a record-high power density of 2.30 W cm−2 at a current density of 4.0 A cm−2, which greatly surpasses that of the commercial Pt/C MEA with the value of 1.99 W cm−2, displaying an unprecedented discharging performance. The superior performance is also supported by an excellent MA@0.9 V (calculated from the IR-free currents, Table S5, ESI†) of 0.46 A mg(Pt)−1, which exceeds the DOE 2020 target (0.44 A mg(Pt)−1) and overwhelms the Pt/C one (Fig. S17, ESI†). Significantly, the MA@0.9 V obtained in MEA configuration is very close to the value in RDE, indicating the outstanding activity expression for the high-loaded Pt1Co1-IMC@Pt/C catalyst under fuel cell operating conditions.
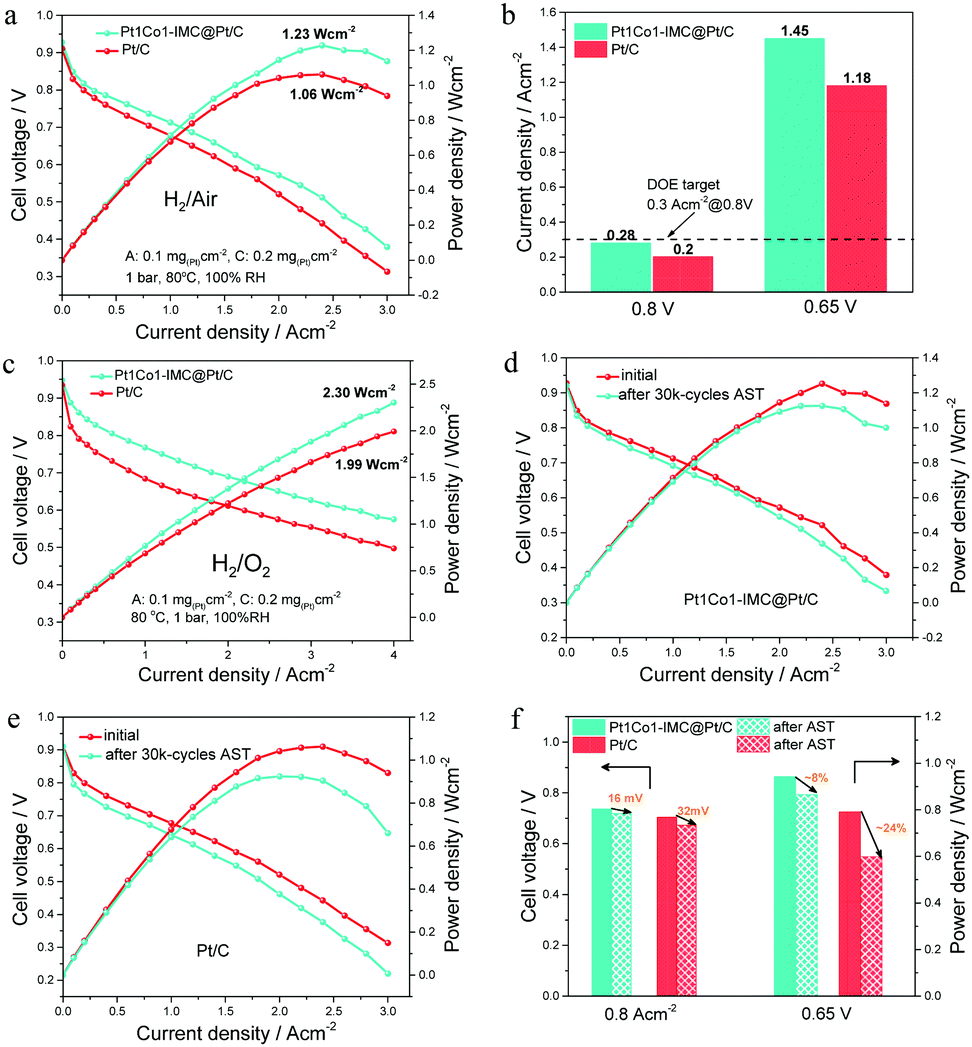 | ||
| Fig. 6 (a) Steady-state polarization curves of MEAs with Pt1Co1-IMC@Pt/C and commercial Pt/C as the cathode under H2–air conditions; (b) current densities for Pt1Co1-IMC@Pt/C and Pt/C at 0.8 and 0.65 V; (c) performance comparison of H2–O2 fuel cell prepared by Pt1Co1-IMC@Pt/C and Pt/C catalysts; polarization curves of H2–air fuel cell with (d) Pt1Co1-IMC@Pt/C and (e) commercial Pt/C before and after 30k-cycles AST; (f) voltage drops and power density losses after the AST test. | ||
The potential cycling on the MEA between 0.6 V and 0.95 V according to the DOE accelerate stability test (AST) protocol was carried out to evaluate the stability of H2–air PEMFC under the working conditions, as shown in Fig. 6d and e. After the 30k-cycles AST, the Pt1Co1-IMC@Pt/C exhibits the negligible voltage drops at various current densities compared to the commercial Pt/C (Fig. S18, ESI†). Particularly, at a current density of 0.8 A cm−2, the MEA with Pt1Co1-IMC@Pt/C shows a voltage drop of ∼16 mV (Fig. 6f), which is much lower than the loss of the DOE target of <30 mV. Meantime, at a practical voltage of 0.65 V, the power density is decreased by only ∼8% with the value from 0.943 W cm−2 to 0.865 W cm−2. In a contrast, commercial Pt/C exhibits a ∼32 mV voltage loss at 0.8 A cm−2 and the power density at 0.65 V decreases from 0.791 to 0.598 W cm−2 (∼24% loss). In addition, HFR-corrected MA at 0.8 V decreases by 24.8% from the value of 6.65 to 5.0 A mg(Pt)−1 (Fig. S19, ESI†), while the MA for Pt/C presents a 67.5% decay after AST. Moreover, the long-time stability test at 1 A cm−2 under H2–air conditions for Pt1Co1-IMC@Pt/C and commercial Pt/C catalyst was performed (Fig. S20, ESI†). The cell voltage of MEA prepared with Pt1Co1-IMC@Pt/C only shows ∼6.5% degradation even after 100 h discharging, while the commercial Pt/C one displays the rapid performance degradation with the voltage drop by ∼29.2%, also attesting the superb structural stability of core–shell Pt1Co1-IMC@Pt/C catalyst under the conditions of practical operation.
Conclusions
In summary, the sub-6 nm core–shell Pt1Co1-IMC@Pt/C catalyst with high metal loading (44.7 wt%) was successfully synthesized through a cobalt oxide aided structural evolution strategy. XPS and XAS measurements illuminated the modulation effect of periodic Pt1Co1 arrangement on the electronic structure of Pt/Co sites. As a result, the Pt1Co1-IMC@Pt/C catalyst exhibited extraordinary ORR activity and durability in the RDE configuration. DFT confirmed that the weakened adsorption of O*/OH* intermediates and improved oxidation-resistance of Pt/Co atoms were at the origin of enhanced activity and durability, respectively. PEMFCs with the Pt1Co1-IMC@Pt/C cathode achieved the record-high power densities of 2.30/1.23 W cm−2 under H2–O2/air conditions, demonstrating an unprecedented fuel cell performance to date. Particularly, the MA0.9V calculated from fuel cell attained ca. 0.46 A mg(Pt)−1, which surpassed that of the 2020 DOE target (0.44 A mg(Pt)−1) and was very close to the intrinsic value, indicating the superior performance expression under PEMFC operating conditions. This study provided an ideal strategy for the future development of high-loaded and efficient performance expression fuel cell electrocatalysts.Author contributions
Qingqing Cheng: conceptualization, methodology, validation, writing – original draft; Shuai Yang: formal analysis, methodology; Cehuang Fu: formal analysis, methodology; Liangliang Zou: investigation, data curation, resources; Zhiqing Zou: investigation, resources; Zheng Jiang: investigation, resources; Junliang Zhang: investigation, resources; Hui Yang: validation, data curation, supervision, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research Program (2017YFA0206500), the National Natural Science Foundation of China (22002184, 21673275 and 21573107), Shanghai Rising-Star Program (21QA1410100), the Science and Technology Service Network Initiative, CAS (KFJ-STS-QYZX-102), SARI Cutting Edge Projects and the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF).References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- R. D. Tilley and J. J. Gooding, Nat. Energy, 2016, 1, 16174 CrossRef CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- Z. Qi, C. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. Pei, X. Li, L. A. Curtiss and W. Huang, J. Am. Chem. Soc., 2017, 139, 4762–4768 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron and M. Mavrikakis, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- J. Li, Z. Xi, Y. T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- J. Liang, F. Ma, S. Hwang, X. Wang, J. Sokolowski, Q. Li, G. Wu and D. Su, Joule, 2019, 3, 956–991 CrossRef CAS.
- S. Zhang, S. Guo, H. Zhu, D. Su and S. Sun, J. Am. Chem. Soc., 2012, 134, 5060–5063 CrossRef CAS PubMed.
- X. X. Du, Y. He, X. X. Wang and J. N. Wang, Energy Environ. Sci., 2016, 9, 2623–2632 RSC.
- J. Li, S. Sharma, K. Wei, Z. Chen, D. Morris, H. Lin, C. Zeng, M. Chi, Z. Yin, M. Muzzio, M. Shen, P. Zhang, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2020, 142, 19209–19216 CrossRef CAS PubMed.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 CrossRef CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- D. Wang, Y. Yu, J. Zhu, S. Liu, D. A. Muller and H. D. Abruna, Nano Lett., 2015, 15, 1343–1348 CrossRef CAS PubMed.
- Z. Wang, X. Yao, Y. Kang, L. Miao, D. Xia and L. Gan, Adv. Funct. Mater., 2019, 29, 1902987 CrossRef.
- B. Zhang, G. Fu, Y. Li, L. Liang, N. S. Grundish, Y. Tang, J. B. Goodenough and Z. Cui, Angew. Chem., Int. Ed., 2020, 132, 7931–7937 CrossRef.
- Y. Hu, M. Zhu, X. Luo, G. Wu, T. Chao, Y. Qu, F. Zhou, R. Sun, X. Han, H. Li, B. Jiang, Y. Wu and X. Hong, Angew. Chem., Int. Ed., 2021, 60, 6533–6538 CrossRef CAS PubMed.
- X. X. Wang, S. Hwang, Y. T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 CrossRef CAS PubMed.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6, 475–486 CrossRef CAS.
- J. S. Lee, K. I. Han, S. O. Park, H. N. Kim and H. Kim, Electrochim. Acta, 2004, 50, 807–810 CrossRef CAS.
- M. Xie, Z. Lyu, R. Chen, M. Shen, Z. Cao and Y. Xia, J. Am. Chem. Soc., 2021, 143, 8509–8518 CrossRef CAS PubMed.
- Y. Ma, A. N. Kuhn, W. Gao, T. Al-Zoubi, H. Du, X. Pan and H. Yang, Nano Energy, 2021, 79, 105465 CrossRef CAS.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS PubMed.
- T. Suzuki, S. Tsushima and S. Hirai, J. Power Sources, 2013, 233, 269–276 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- J. Lee, J. M. Yoo, Y. Ye, Y. Mun, S. Lee, O.-H. Kim, H.-W. Rhee, H. I. Lee, Y.-E. Sung and J. Lee, Adv. Energy Mater., 2015, 5, 1402093 CrossRef.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16, 140–147 CrossRef CAS PubMed.
- P. Ren, P. Pei, Y. Li, Z. Wu, D. Chen and S. Huang, Prog. Energy Combust. Sci., 2020, 80, 100859 CrossRef.
- L. Zou, J. Fan, Y. Zhou, C. Wang, J. Li, Z. Zou and H. Yang, Nano Res., 2015, 8, 2777–2788 CrossRef CAS.
- L. Zou, J. Li, T. Yuan, Y. Zhou, X. Li and H. Yang, Nanoscale, 2014, 6, 10686–10692 RSC.
- J. Kim, S. Yang and H. Lee, Electrochem. Commun., 2016, 66, 66–70 CrossRef CAS.
- Q. Li, L. Wu, G. Wu, D. Su, H. Lv, S. Zhang, W. Zhu, A. Casimir, H. Zhu, A. Mendoza-Garcia and S. Sun, Nano Lett., 2015, 15, 2468–2473 CrossRef CAS PubMed.
- J. Liang, N. Li, Z. Zhao, L. Ma, X. Wang, S. Li, X. Liu, T. Wang, Y. Du, G. Lu, J. Han, Y. Huang, D. Su and Q. Li, Angew. Chem., Int. Ed., 2019, 58, 15471–15477 CrossRef CAS PubMed.
- C. Jung, C. Lee, K. Bang, J. Lim, H. Lee, H. J. Ryu, E. Cho and H. M. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 31806–31815 CrossRef CAS PubMed.
- D. Y. Chung, S. W. Jun, G. Yoon, S. G. Kwon, D. Y. Shin, P. Seo, J. M. Yoo, H. Shin, Y. H. Chung, H. Kim, B. S. Mun, K. S. Lee, N. S. Lee, S. J. Yoo, D. H. Lim, K. Kang, Y. E. Sung and T. Hyeon, J. Am. Chem. Soc., 2015, 137, 15478–15485 CrossRef CAS PubMed.
- Y. Zhao, C. Wang, J. Liu and F. Wang, Nanoscale, 2018, 10, 9038–9043 RSC.
- T. Zhao, G. Wang, M. Gong, D. Xiao, Y. Chen, T. Shen, Y. Lu, J. Zhang, H. Xin, Q. Li and D. Wang, ACS Catal., 2020, 10, 15207–15216 CrossRef CAS.
- Y. Li, B. Tan and Y. Wu, J. Am. Chem. Soc., 2006, 128, 14258–14259 CrossRef CAS PubMed.
- Q. Wang, S. Chen, F. Shi, K. Chen, Y. Nie, Y. Wang, R. Wu, J. Li, Y. Zhang, W. Ding, Y. Li, L. Li and Z. Wei, Adv. Mater., 2016, 28, 10673–10678 CrossRef CAS PubMed.
- B.-A. Lu, L.-F. Shen, J. Liu, Q. Zhang, L.-Y. Wan, D. J. Morris, R.-X. Wang, Z.-Y. Zhou, G. Li, T. Sheng, L. Gu, P. Zhang, N. Tian and S.-G. Sun, ACS Catal., 2020, 11, 355–363 CrossRef.
- J. Greeley, I. Stephens, A. Bondarenko, T. P. Johansson, H. A. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- J. Zhang, K. Sasaki, E. Sutter and R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- F. Dionigi, C. C. Weber, M. Primbs, M. Gocyla, A. M. Bonastre, C. Spori, H. Schmies, E. Hornberger, S. Kuhl, J. Drnec, M. Heggen, J. Sharman, R. E. Dunin-Borkowski and P. Strasser, Nano Lett., 2019, 19, 6876–6885 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02530a |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |