
An efficient and durable anode for ammonia protonic ceramic fuel cells†
Hua
Zhang‡
a,
Yucun
Zhou‡
 b,
Kai
Pei
a,
Yuxin
Pan
a,
Kang
Xu
a,
Yong
Ding
b,
Bote
Zhao
a,
Kotaro
Sasaki
c,
YongMan
Choi
*d,
Yu
Chen
*a and
Meilin
Liu
*b
b,
Kai
Pei
a,
Yuxin
Pan
a,
Kang
Xu
a,
Yong
Ding
b,
Bote
Zhao
a,
Kotaro
Sasaki
c,
YongMan
Choi
*d,
Yu
Chen
*a and
Meilin
Liu
*b
aSchool of Environment and Energy, South China University of Technology, Guangzhou, 510006, China. E-mail: eschenyu@scut.edu.cn
bSchool of Material Science and Engineering, Georgia Institute of Technology, Atlanta, GA 30309, USA. E-mail: Meilin.liu@mse.gatech.edu
cChemistry Department, Brookhaven National Laboratory, Upton, NY 11973, USA
dCollege of Photonics, National Yang Ming Chiao Tung University, Tainan 71150, Taiwan. E-mail: ymchoi@nctu.edu.tw
First published on 19th November 2021
Abstract
Ammonia protonic ceramic fuel cells (PCFCs) have the potential to be a highly efficient power source with high energy density. However, the inadequate catalytic activity of the existing anodes for utilization of ammonia greatly limits the performance of PCFCs. Here we report an Fe-modified state-of-the-art Ni cermet anode with greatly enhanced activity and durability toward utilization of ammonia. Cells with an Fe-decorated Ni-BaZr0.1Ce0.7Y0.1Yb0.1O3 (Ni–BZCYYb) anode demonstrate an excellent performance, achieving peak power densities of 0.360, 0.723, 1.257, and 1.609 W cm−2 at 550, 600, 650, and 700 °C, respectively, which reveal the highest performance of solid oxide fuel cells fueled on ammonia. In addition, the cells show an excellent durability when operated at a constant current density of 0.5 A cm−2 (or a power density of ∼0.435 W cm−2) at 650 °C. The superior activity and durability of the Fe-modified Ni/BZCYYb anode are attributed to the alternation of NH3 adsorption strength and N2 desorption barrier heights, as confirmed by first-principles based mechanistic and microkinetic modeling. Our research provides a valuable guidance for the development of efficient electro-catalysts for ammonia PCFCs.
Broader contextAmmonia protonic ceramic fuel cells (PCFCs) have the potential to be a highly efficient power source with high energy density. Unfortunately, the limited catalytic activity of the existing anodes for the direct utilization of ammonia greatly inhibits the activity and durability of PCFCs. In particular, the development of materials or structures with high electrocatalytic activity for ammonia decomposition, and excellent durability is imperative to the deployment of commercially viable fuel cells to be operated with ammonia. One of the most effective strategies is to create a unique anode surface that offers high activity and durability toward ammonia utilization at low cost. In this article, we report our findings in the design and development of an Fe-modified state-of-the-art Ni cermet anode with greatly enhanced activity and durability toward the direct utilization of ammonia. Our research provides a valuable guidance for the development of efficient electro-catalysts for ammonia PCFCs. |
Introduction
Ammonia has been identified as one of the most promising hydrogen carriers for protonic ceramic fuel cells (PCFCs) because of its unique properties, such as the high hydrogen content (17.7 wt%), high energy density (4 kW h kg−1), zero-carbon emission, low cost, facile liquefaction under mild conditions (−33.4 °C at atmospheric pressure or 10 atm at ambient temperature), easy transportation and storage.1–4 The ammonia decomposition reaction is an endothermic process, and the full conversion of ammonia can be achieved at 500 °C and atmospheric pressure.5–7 Hence, it is greatly feasible to utilize ammonia as a fuel for PCFCs at relatively lower temperatures (500–700 °C) than oxygen ion-based SOFCs (O-SOFCs).8–10 Steam is produced in the cathode in PCFCs, enabling high fuel utilization.8,9,11 In addition, since no oxygen ion is incorporated in the anode or electrolyte, no undesirable NOx is formed.12,13 When ammonia is supplied to PCFCs, ammonia utilization in the cell involves thermal- and electro-catalytic processes that can be described as follows: adsorption of NH3 on the anode surface, decomposition of NH3 into H2 and N2, splitting of H2 to monoatomic hydrogen, and electrochemical oxidation of hydrogen to a proton, and transport of the proton from the anode to the cathode, where H2O is formed, as shown schematically in Fig. 1a. | ||
| Fig. 1 Microstructure of a fuel cell. (a) Schematic of a single PCFC fueled by ammonia (the orange and gray grains represent the BZCYYb and Ni phase, respectively); (b) a typical scanning electron microscope (SEM) image of an anode supported cell, including a porous cathode, a dense electrolyte, an AFL and an ASL; (c) a detailed SEM image of the cathode; (d) a detailed SEM image of the anode; (e) a detailed TEM image and (f) a HRTEM of Ni grains covered by Fe nanoparticles; and (g) a STEM-EDX mapping of an Fe-modified Ni–BZCYYb anode. | ||
However, many challenges still remain to significantly advance the PCFCs operated in NH3, including the poor catalytic activity and stability of the state-of-the-art fuel electrode materials toward decomposition of ammonia, a lack of suitable cathodes designed for PCFC operation (typically between ∼400 and 650 °C) and fabrication, inadequate proton conductivity of the electrolytes, instability of the interfaces, as well as variations in cell fabrication and testing. Furthermore, rapid degradation under typical operating conditions is a significant challenge for the development of fuel cells in NH3.14,15 Recently, considerable efforts have been made to improve the performance of NH3 PCFCs by applying thin film electrolytes and developing more reactive anode materials/structures, which is of great significance to the enhancement of activity and durability for ammonia PCFCs.14,16,17 For instance, Yoo et al. fabricated a Ba0.1Ce0.8Y0.2O3−δ (BCY) electrolyte with a thickness of 10–15 μm by using a wet colloidal spray method. As a result, when fueled by NH3, the peak power density (Pmax) of the Ni–BCY cells with a Ba0.5Sr0.5Co0.8 Fe0.2O3−δ (BSCF) cathode has been increased to 0.08 W cm−2 at 600 °C.18,19 Xie et al. fabricated a dense BaCe0.8Sm0.2O3−δ electrolyte layer with a thickness of only 10 μm on porous anode supports by using a modified suspension spray process; a Pmax of 0.53 W cm−2 was obtained at 700 °C when fueled by ammonia, much higher than that those (0.35 W cm−2 at 700 °C) of cells with a 50 μm thick electrolyte via co-pressing.20–22 However, a thin and dense proton-conducting electrolyte fabricated by a wet colloidal spray method or a suspension spray process may be unsuitable for large scale fabrication. Thus, it is necessary to choose a cost-effective method for the fabrication of dense electrolyte materials with high proton conductivity and suitable thickness before the practical application of ammonia power generation by PCFC technologies. Another effective method to improve the anode performance is the surface modification with nano-structured catalysts via a solution infiltration method, providing more active sites and more choices for catalyst development.23 It was confirmed that tailoring the capabilities of NH3 adsorption and N2 desorption by forming alloys with a strong NH3 adsorption and a weak nitrogen binding energy is an effective strategy to improve the cell performance.24 For example, an anode-based La0.55Sr0.30TiO3−δ (LST) perovskite substrate with NiCo alloy nanoparticles (NPs) on its surface demonstrated a superior performance for NH3 decomposition because of the rich active sites and balanced NH3 adsorption and N2 desorption capability. With such an anode, a Pmax of 0.361 W cm−2 was obtained at 800 °C using ammonia. Inaba et al. found that Ni–Fe/Sm-doped CeO2 (SDC) and Ni–Mo/SDC anodes showed improved performance for ammonia oxidation due to the higher nitrogen adsorption enthalpy of Fe and Mo than Ni.25,26 Shao et al. reported that a PCFC with the Ni–BZCYYbPd anode and the BZCYYbPd electrolyte (∼17 μm) can achieve a Pmax of 0.724 W cm−2 (vs. 0.45 W cm−2 for the cell without Pd incorporation) at 650 °C operated in NH3. The improved power output for the incorporation of a small amount of Pd boosts the catalytic activity of the anode for NH3 decomposition and increases proton conductivity from the creation of B-site cation deficiency and electrolyte sintering.27 However, Pd is not feasible for practical application in SOFCs due to its high cost. Moreover, the high basicity of supports and high tolerance to the water/hydrogen poisoning effect on NH3 decomposition are of crucial importance to improve the catalytic activity and durability for ammonia decomposition over the anode.25,28–34 For example, Eguchi's group found that the catalytic activity of Ni/BaCe0.4Zr0.4Y0.2O3−δ (Ni/BCZY, 100% at 600 °C), Ni/BaCe0.75Y0.25O3−δ (Ni/BCY25, 98.6% at 600 °C) and Ni/BaCe0.9Y0.1O3−δ (Ni/BCY10, 100% at 607 °C) for ammonia decomposition was remarkably higher than that of Ni/GDC (62% at 600 °C), Ni/SDC (57% at 607 °C), or Ni/YSZ (44% at 607 °C).28,31,33,35,36 Among these anode materials, Ni/BZCY exhibited the highest peak power density of 0.32 W cm−2 at 700 °C using ammonia as the fuel.36 Wang et al. recently reported a Ni/YSZ anode surface-decorated with Ba, displaying an enhanced performance since a small amount of Ba species alleviated the negative effect of the water/hydrogen on the active sites of Ni/YSZ catalysts.34 While surface modification is effective, the selection of suitable catalysts is critical to the performance and durability enhancement. To be effective in designing better materials for NH3 utilization, it is necessary to gain deeper understanding of the reaction mechanism and elementary steps. To the best of our knowledge, none of the reported electrode materials have sufficient durability under realistic operating conditions. In addition, the reaction mechanisms of NH3 conversion on anode surfaces are poorly understood. Accordingly, mechanistic understanding may be vital to achieving rational designs of novel better materials and electrode structures for high-performance and durable PCFCs.
Here we reported a re-structured anode surface for NH3-fueled PCFC via a surface modification of Fe on the Ni–BZCYYb anode. The catalytic decomposition of ammonia on various metallic catalysts (e.g., Pt, Cu, and Ni) has been widely studied, with more attention on Fe and Ru. In particular, Ru is the most active one, and has been extensively studied.37,38 However, Ru is not feasible for practical application due to its high cost. In this study, Fe is chosen as the reforming catalyst for a PCFC fueled with NH3 since Fe has similar activity to Ru. Interestingly, Fe tends to stay on Ni surfaces due most likely to their similar crystal structures. The cells with such anodes demonstrate a superior catalytic activity and durability when fueled with ammonia under typical PCFC operation conditions. An anode supported single cell with FeNi–BZCYYb anodes achieved a peak power density of 1.257 W cm−2, and a power density of 0.435 W cm−2 at a current density of 0.5 A cm−2 at 650 °C for 100 h, when fueled by NH3. Characterization and analyses indicate that the surface of the anode is much more active for the NH3 conversion reaction, suggesting that Fe modified anode surfaces are a highly promising electrocatalyst material for low-cost, high-performance, and durable PCFCs in NH3. Moreover, the mechanism of the remarkable performance under realistic operation conditions has been unraveled using electrochemical impedance spectroscopy (EIS) and density functional theory (DFT) calculations, providing vital information for the rational design of more efficient and durable catalysts for NH3 PCFCs.
Results and discussion
A double perovskite cathode PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF)39,40 exhibiting high activity for the oxygen reduction reaction (ORR) is printed onto a thin BZCYYb electrolyte, which is supported on a multi-layered Ni–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (Ni–BZCYYb) anode. BZCYYb has been applied as an electrolyte because of its high ionic conductivity and relatively low electronic conductivity.41 The upper layer of the anode is a fine Ni–BZCYYb anode functional layer (AFL), designed to maximize the triple phase boundary (TPB) length. The surface of the Ni–BZCYYb anode supporting layer (ASL) is also decorated with Fe NPs for enhancing the NH3 conversion (Fig. 1a). Fig. 1b shows the typical cross-sectional view of an anode supported single cell with a porous anode supporting layer (ASL, Ni–BZCYYb, ∼800 μm thick), an anode functional layer (AFL, Ni–BZCYYb, ∼20 μm thick), a dense electrolyte (BZCYYb, ∼7 μm thick) and a porous cathode (PBSCF, ∼50 μm thick). More details about the cathode and anode can be found in Fig. 1c and d, respectively. Briefly, a porous and fine PBSCF cathode (with a grain size of ∼500 nm) is expected to provide more reaction sites for the ORR, while the porous Ni–BZCYYb anode is able to facilitate ammonia decomposition and mass transport (Note S1 and Fig. S1, ESI†). Ammonia is first decomposed over the anode surface to N2 and H2. H2 was then split into protons, which then transport through a proton conducting electrolyte to the cathode.42 The Ni–BZCYYb anode showed a higher catalytic activity for ammonia decomposition than the conventional Ni/YSZ anode (Note S2 and Fig. S2, ESI†). Fe(NO3)3 water solution was dispersed on the sintered NiO–BZCYYb surface followed by firing at 700 °C in air for 3 h and then in H2 for 3 h. The typical SEM images of anode surfaces before (Fig. S3a–c, ESI†) and after (Fig. S3d–f, ESI†) modification with catalysts reveal that the nanoparticles are preferentially deposited on the surface of the Ni grain rather than on the BZCYYb surface (Note S3 and Fig. S3, ESI†). Fe and Ni are close to each other in the periodic table. Accordingly, they follow the Hume-Rothery rule very well; a solid solution of Fe–Ni (alloy) can be readily formed. Under our experimental conditions, an alloy with a possible composition of FeNi3 was formed, which is supported by the XRD patterns (Note S4 and Fig. S4, ESI†) and XPS analysis (Note S5 and Fig. S5, S6, ESI†) of anodes after testing. Fig. 1e and f show the transmission electron microscopy (TEM) images of the Ni–BZCYYb grains coated with nanoparticles. It is observed that Ni surfaces are partially covered by nanoparticles (Fig. 1e, Note S4 and Fig. S5, ESI†). The interplanar spacing of the surface coating (Fig. S5, ESI†) is ∼0.177 nm, corresponding likely to the (200) plane of the FeNi3 alloy (PDF#38-0419), whereas the main grain spacing of ∼0.203 nm is the Ni(111) plane (Fig. 1f). Fig. 1g and Fig. S3g (ESI†) show the scanning transmission electron microscopy (STEM) image of the anode functional layer with Fe modification, and the energy-dispersive X-ray (EDX) mapping of Ni, Fe, Ba, Zr, Ce, and Y. It is also shown that Fe tends to deposit on the surface of Ni grains due most likely to the similar lattice constant of Ni and Fe. According to this observation, an FeNi(111) surface model was constructed to examine the enhanced Fe-modified Ni anode using DFT calculations.The electrochemical performance of the cells with such a unique microstructure was further evaluated using dry ammonia or wet H2 (as a reference) as the fuel and ambient air as the oxidant at 550–700 °C. Fig. 2a and b show the typical current density–voltage–power density (IVP) curves and electrochemical impedance spectra (EIS) of the single cells with a bare anode (without Fe modification). When H2 was used as the fuel, the cell shows a Pmax of 1.86 W cm−2, a ROhm of 0.068 Ω cm2 and a Rp of 0.048 Ω cm2 at 700 °C (see the black line in Fig. 2a and b). After switching to NH3, the Pmax dropped to 1.398 W cm−2 at 700 °C (see the blue squares) under the same conditions. With the decrease of the operating temperatures, the Pmax of cells in NH3 is abruptly dropped to 1.020, 0.691, and 0.332 W cm−2 at 650, 600, and 550 °C, respectively. Apparently, the peak power outputs (Pmax values) of our cell at each operating temperature (550–700 °C) are higher than those reported by others (see Fig. 2e). It is noted that the OCV of the cells in NH3 increased with the decrease in operation temperature, like hydrogen fuel cells, with the values of 0.99, 1.01, 1.029, and 1.015 V at 700, 650, 600, and 550 °C, respectively. The oxidation of ammonia in PCFCs may follow a two-step process: ammonia is initially decomposed to hydrogen and nitrogen at the anode, and hydrogen molecules are then dissociated and oxidized to protons, which transport through the electrolyte to the cathode and react with oxygen to produce water.42 Also, it is noted that both the ROhm and Rp of the cell operating in NH3 are higher than that in hydrogen. The relatively higher resistance in ammonia might be also attributed to the presence of nitrogen at the active sites for fuel oxidation, which will dilute the H2 in the vicinity of the active sites on the anode (more analysis can be found in Note S7 and Fig. S7, S8, ESI†).42,43
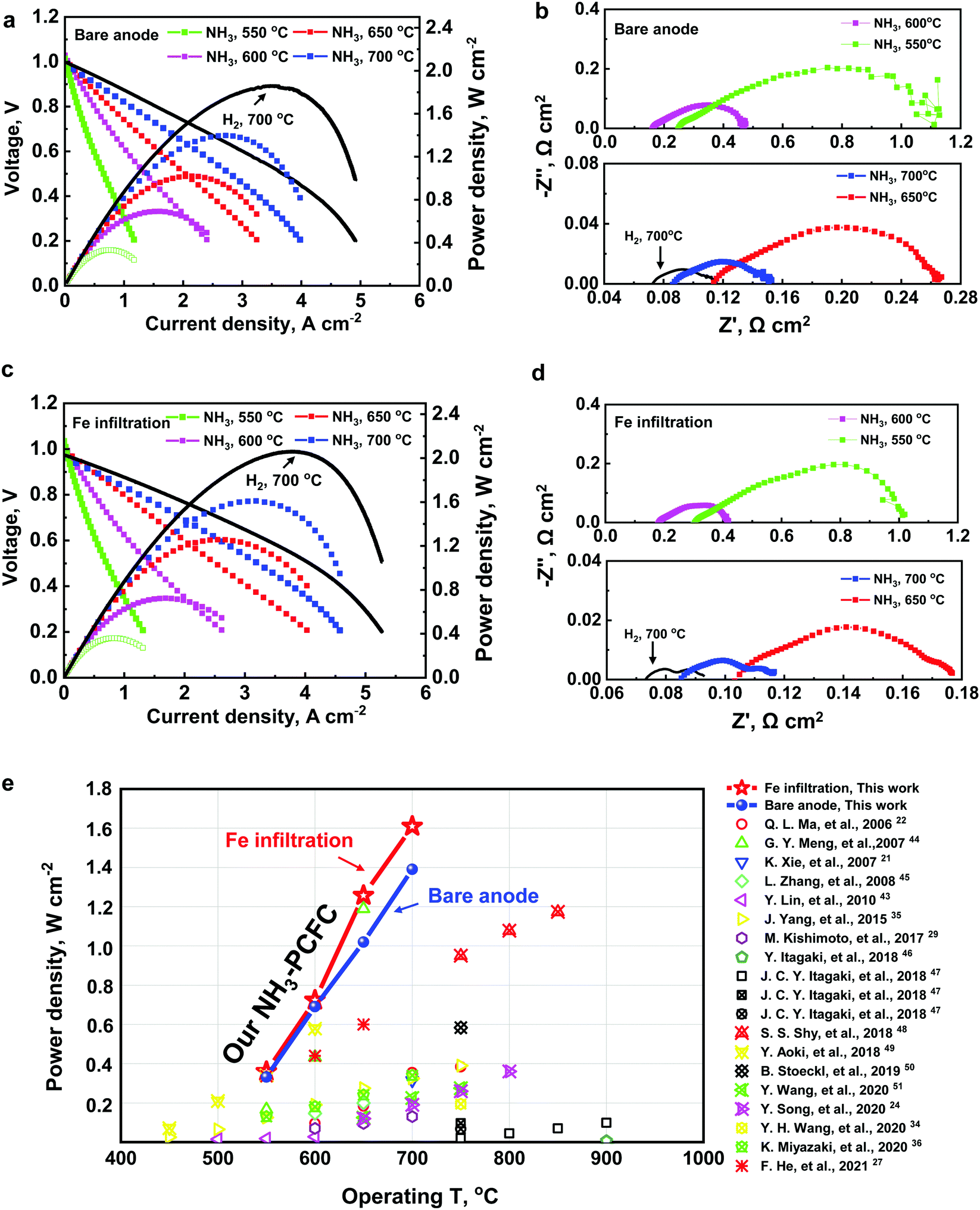 | ||
| Fig. 2 Electrochemical performance of a fuel cell using H2 or NH3 as the fuel. Typical IVP curves (a), and EIS (b) of a single cell tested with the bare Ni–BZCYYb anode at 550–700 °C using wet H2 (black lines) (3% humidity) and dry ammonia as the fuel and ambient air as the oxidant; typical IVP curves (c), and EIS (d) of a single cell with the Fe-modified Ni–BZCYYb anode, tested at 550–700 °C using wet H2 (black lines) (3% humidity) and ammonia as the fuel and ambient air as the oxidant; and (e) comparison of Pmax of the NH3-fueled fuel cells (with electrolytes of either oxygen ion or proton conductor) at different temperatures. | ||
With the Fe modification on the anode surface, the electrochemical performance was enhanced when the cells were operated with both H2 and NH3, suggesting a good reproducibility of our cells (Note S6, Fig. S7 and Table S1, ESI†). The performance improvement can be attributed to the Fe modification of the anode. As shown in Fig. 2c, with Fe modification, the Pmax of the cells increases from 1.86 W cm−2 to 2.062 W cm−2 in H2, and from 1.398 W cm−2 to 1.609 W cm−2 in NH3 at 700 °C. In comparison with the cells operated in H2, the Pmax values of cells in NH3 fuel were reduced by 21.9% and 24.8% for the cells with Ni–BZCYYb with and without Fe modification, respectively. The enhanced performance may indicate that Fe-modified Ni/BZCYYb could improve the decomposition of NH3 under operating conditions since the FeNi alloy formed (Fig. 1f) on the anode surfaces would effectively modify the NH3 adsorption phenomenon and the N2 desorption process. More detailed computational results will be discussed in the following. Fig. 2d shows the typical EIS results of the cell with the Ni–BZCYYb anode coated with Fe at various temperatures at open circuit voltage (OCV). Rp is larger than ROhm at all testing temperatures for the bare Ni–BZCYYb anode, suggesting that the electrode polarization (Rp) makes significant contribution to the total resistance of the PCFC. For the cell with the Fe–Ni/BZCYYb anode, the Rp decreased because of the Fe modification. The ohmic resistance (ROhm) makes dominant contribution to the total cell resistance at 700 and 650 °C, suggesting that the cell performance could be effectively improved by developing a more conductive electrolyte or reducing its thickness. As shown, the Rp values dropped to 0.031, 0.072, 0.237, and 0.716 Ω cm2 in NH3 at 700, 650, 600, and 550 °C, respectively (vs. 0.064, 0.151, 0.303, and 0.872 Ω cm2 for the bare Ni–BZCYYb anode, seen in Fig. 2b, Note S8 and Fig. S9, ESI†). The results further indicated that Fe modification can largely accelerate anodic reactions and decrease the Rp, thus improving the cell performance. A more detailed comparison of Pmax (see Fig. 2e) of cells based on protonic or oxygen ionic electrolytes manifests that our performance is among the best.21,22,24,27,28,35,36,43–51 More details of cell configurations, and electrolyte/electrode materials can be found in Table S2, ESI.† For example, when using NH3 as the fuel, a Pmax of ∼0.723 W cm−2 at 600 °C was achieved in our study, higher than the one reported with a Pmax of 0.63 W cm−2 at 600 °C based on a PCFC with BZY20 electrolyte and Ni/BZY cermet anode2. Our performance is also higher than the one reported by Meng et al. from an Sm-doped ceria electrolyte-based cell: 0.434 W cm−2 at 600 °C and 1.19 W cm−2 at 650 °C, fueled by NH3.44 However, the intrinsic electronic conduction (the electronic transference number of our cell is ∼0.09, while the one in Meng's report is ∼0.35 at 650 °C) of the ceria-based electrolyte under typical fuel cell operation conditions reduces the OCV (a value of ∼0.768 V at 650 °C) and hence the energy (voltage) efficiency,44 more so with reduced electrolyte thickness at higher temperatures.
Fig. 3a shows the voltages of the three single cells, with bare- and Fe-modified anodes, operated at 0.5 A cm−2 and 650 °C using wet H2 (3% H2O) or dry ammonia as the fuel and ambient air as the oxidant. It is noted that there is a slight degradation in performance of the cells fueled with ammonia than the one fueled with wet H2. However, the cell with the Fe decorated Ni/BZCYYb anode showed better performance and a slower deterioration rate (around 0.0022 V h−1) than the one with the bare anode (around 0.008 V h−1). The performance degradation rate of the cells in NH3 is close to that of the cell in H2. To gain more insight into the degradation of the cells in H2 or NH3, the IVP curves and the Rp of the three cells were collected at the beginning of the test (0 h) and after 100 h of continuous operation in H2 or NH3 at 650 °C for 100 h, as demonstrated in Fig. 3b–g. The 100 h exposure to NH3 at 650 °C for the cell with the bare anode resulted in an apparent performance degradation, especially at lower terminal voltage, as shown in Fig. 3d. Furthermore, the Rp values significantly increased (Fig. 3e, Note S9 and Fig. S10, ESI†), which could be attributed to the Ni coarsening and agglomeration after the treatment in NH3, as revealed by SEM images (Note S10 and Fig. S11c, ESI†). However, the current density decreased slightly at the low terminal voltage region for the cell with the Fe–Ni–BZCYYb anode after being tested in NH3 at 650 °C for 100 h (Fig. 3f). Moreover, the Rp values increased slightly from 0.077 Ω cm2 to 0.11 Ω cm2 (see Fig. 3g) (vs. the Rp values increased from 0.105 Ω cm2 to 0.21 Ω cm2 for the bare Ni–BZCYYb anode after being tested in NH3, see Fig. 3e), which may be due to the strong interaction of NH3 and anode surfaces: the surface could provide a good thermo-mechanical stability and a superior anti-sintering capability, as revealed by SEM images (Note S10 and Fig. S11d, ESI†). No significant change of Ni particle sizes was observed after running in NH3 for 100 h. Moreover, careful characterization of the anodes after long-term testing using XRD indicates that no nitrides were formed on either the bare anode or the Fe–Ni/BZCYYb anode (Note S11 and Fig. S12, ESI†). In addition, the stability testing of the single cells in NH3 with various Fe loading anodes at 0.5 A cm−2 at 650 °C is displayed in Note S12 and Fig. S13, ESI.† The deterioration rate decreased with the increase in Fe content at first and reached the minimum deterioration rate (around 0.0022 V h−1) at a loading amount of 10 μL (∼0.36 mg cm−2). A further increase in the Fe loading resulted in a poor performance and a much faster deterioration rate. A more detailed comparison of stability results (see Table S3, ESI†) of the O-SOFC and PCFCs in NH3 indicates that our cell stability is superior. More discussion can be found in Note S13 and Table S3, ESI.† To gain more insight into the mechanism of this degradation in performance, we resorted to DFT-based calculations.
 | ||
| Fig. 3 Durability of PCFCs in H2 or NH3. (a) An operation stability of the single cells at a constant current density of 0.5 A cm−2 for a period of ∼100 h at 650 °C in wet H2 (with 3% H2O) or dry NH3; (b), (d) and (f) typical IVP curves of cells operated at 650 °C before and after the stability test; and (c), (e) and (g) EIS of cells operated at 650 °C under OCV conditions before and after the stability test. Black, blue and red represent the performance of cells with the bare Ni/BZCYYb anode in wet H2 (with 3% H2O) fuel, the bare Ni/BZCYYb anode in dry NH3 fuel, and the Fe-modified Ni–BZCYYb anode in dry NH3 fuel, respectively. | ||
To theoretically describe the surface chemical reactions for NH3 decomposition in detail and to accurately understand the enhanced activity of the Fe-modified Ni/BZCYYb anode (Note S14), we resorted to DFT calculations in conjunction with microkinetic modeling. We examined the dehydrogenation of NH3 and N2 desorption on FeNi(111) and Ni(111) surfaces (Fig. 4a and b) to elucidate the reaction mechanisms on the Ni/BZCYYb and the Fe-modified Ni/BZCYYb anode. We assumed that FeNi(111) and Ni(111) surfaces could reasonably represent catalytically active sites for characterizing the surface reactions.52,53 As compiled in Table S4, ESI,†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 54 it is reasonably anticipated that the adsorption energy of intermediate species on Ni surfaces may be different from that on Ni–Fe alloy surfaces. To investigate the enhancement of FeNi–BZCYYb, one of the Ni atoms on the top layer of a (3 × 3) Ni(111) surface model (36 Ni) was replaced by an Fe atom (FeNi alloy with 2.7 mol% Fe). The most stable adsorption site for each surface species was first located as summarized in Table S5, ESI.† Briefly, H3N*, N2H*, and HN* adsorb at the top, bridge, and fcc hollow sites, respectively, while N* and H* sit at fcc and hcp hollow sites, respectively. For estimating the production rates of hydrogen and nitrogen, the zero-point energy (ZPE) correction was included. Then the detailed reaction pathways on FeNi(111) and Ni(111) were mapped out using the CI-NEB method.55 As displayed in Fig. 4a, the adsorption process of NH3 occurs without a reaction barrier. The adsorption energy of NH3 on FeNi(111) becomes slightly stronger than Ni(111) (−0.60 eV versus −0.57 eV, respectively). Then it follows the dehydrogenation taking place with a well-defined reaction barrier (H3N* → H2N* + H* → HN* + 2H* → N* + 3H*). It denotes that the rate-determining step on FeNi(111) is changed to the dehydrogenation of HN* rather than that of H3N* on Ni(111) (1.15 eV versus 1.23 eV, respectively). After the dehydrogenation processes, since the surface poisoning by nitrogen species is crucial, the effective removal of nitrogen molecules was carefully taken into account. We located the transition state of the associative desorption of N2, resulting in a lower reaction height by adding Fe in Ni (1.80 eV versus 1.92 eV, respectively). Based on the mechanistic details for modification of the NH3 adsorption energy and the associative desorption barrier height of nitrogen, one may expect that the Fe addition can effectively improve the fuel cell performance as discussed in the experimental section.24 In addition, the d-band centers of Ni and FeNi surfaces were calculated to examine the electronic effect. It leads to a linear correlation with the adsorption energies N* and H3N* (Fig. S14, ESI†). As the concentration of Fe increases from 6.3% to 25%, their adsorption becomes stronger along with an upward shift to the Fermi level (−1.2 eV to −0.73 eV, respectively).56 Furthermore, microkinetic modeling (Note S15) was carried out to estimate production rates of hydrogen and nitrogen using the MKMCXX software package57 at T = 700 °C in which the superiority of Fe-modified Ni–BZCYYb to Ni–BZCYYb is remarkable (Fig. 2e). To determine the parameters in the expression for the rate constant, the statistical-theory calculations were performed.58 As summarized in Table S6, ESI,† the NH3 decomposition is composed of four elementary steps of dehydrogenation. The associative desorption of nitrogen and hydrogen is also considered to take into account the fast transfer of hydrogen species into the bulk BZCYYb electrolyte after a rapid surface diffusion by overcoming its barrier of ∼0.1 eV (Table S6, ESI†). Fig. S15 (ESI†) shows a representative incorporation process of hydrogen species into the bulk electrolyte with oxygen vacancies. Rate constants (ki) were precisely calculated using the pre-exponential factors (Ai) at 700 °C and ZPE-corrected reaction barriers (Ea) (Table S7, ESI†). The first-principles microkinetic simulation was first executed to examine the pressure effect on Ni(111), clarifying that as the pressure increases, the production rates decrease (Note S15 and Fig. S16, ESI†). Then we observed that the production rate on Fe-modified Ni(111) is ∼5 times as high as that on Ni(111) at 700 °C (Note S14, Fig. S17 and Table S8, ESI†). In summary, first-principles based mechanistic study in conjunction with microkinetic modeling supports the enhanced activities of FeNi–BZCYYb compared to Ni–BZCYYb by increasing the NH3 adsorption energy and lowering the associative N2 desorption reaction barrier. This was also verified that the d-band center model can be used as a descriptor for the rational design of novel NH3-fueled PCFC anode materials.
54 it is reasonably anticipated that the adsorption energy of intermediate species on Ni surfaces may be different from that on Ni–Fe alloy surfaces. To investigate the enhancement of FeNi–BZCYYb, one of the Ni atoms on the top layer of a (3 × 3) Ni(111) surface model (36 Ni) was replaced by an Fe atom (FeNi alloy with 2.7 mol% Fe). The most stable adsorption site for each surface species was first located as summarized in Table S5, ESI.† Briefly, H3N*, N2H*, and HN* adsorb at the top, bridge, and fcc hollow sites, respectively, while N* and H* sit at fcc and hcp hollow sites, respectively. For estimating the production rates of hydrogen and nitrogen, the zero-point energy (ZPE) correction was included. Then the detailed reaction pathways on FeNi(111) and Ni(111) were mapped out using the CI-NEB method.55 As displayed in Fig. 4a, the adsorption process of NH3 occurs without a reaction barrier. The adsorption energy of NH3 on FeNi(111) becomes slightly stronger than Ni(111) (−0.60 eV versus −0.57 eV, respectively). Then it follows the dehydrogenation taking place with a well-defined reaction barrier (H3N* → H2N* + H* → HN* + 2H* → N* + 3H*). It denotes that the rate-determining step on FeNi(111) is changed to the dehydrogenation of HN* rather than that of H3N* on Ni(111) (1.15 eV versus 1.23 eV, respectively). After the dehydrogenation processes, since the surface poisoning by nitrogen species is crucial, the effective removal of nitrogen molecules was carefully taken into account. We located the transition state of the associative desorption of N2, resulting in a lower reaction height by adding Fe in Ni (1.80 eV versus 1.92 eV, respectively). Based on the mechanistic details for modification of the NH3 adsorption energy and the associative desorption barrier height of nitrogen, one may expect that the Fe addition can effectively improve the fuel cell performance as discussed in the experimental section.24 In addition, the d-band centers of Ni and FeNi surfaces were calculated to examine the electronic effect. It leads to a linear correlation with the adsorption energies N* and H3N* (Fig. S14, ESI†). As the concentration of Fe increases from 6.3% to 25%, their adsorption becomes stronger along with an upward shift to the Fermi level (−1.2 eV to −0.73 eV, respectively).56 Furthermore, microkinetic modeling (Note S15) was carried out to estimate production rates of hydrogen and nitrogen using the MKMCXX software package57 at T = 700 °C in which the superiority of Fe-modified Ni–BZCYYb to Ni–BZCYYb is remarkable (Fig. 2e). To determine the parameters in the expression for the rate constant, the statistical-theory calculations were performed.58 As summarized in Table S6, ESI,† the NH3 decomposition is composed of four elementary steps of dehydrogenation. The associative desorption of nitrogen and hydrogen is also considered to take into account the fast transfer of hydrogen species into the bulk BZCYYb electrolyte after a rapid surface diffusion by overcoming its barrier of ∼0.1 eV (Table S6, ESI†). Fig. S15 (ESI†) shows a representative incorporation process of hydrogen species into the bulk electrolyte with oxygen vacancies. Rate constants (ki) were precisely calculated using the pre-exponential factors (Ai) at 700 °C and ZPE-corrected reaction barriers (Ea) (Table S7, ESI†). The first-principles microkinetic simulation was first executed to examine the pressure effect on Ni(111), clarifying that as the pressure increases, the production rates decrease (Note S15 and Fig. S16, ESI†). Then we observed that the production rate on Fe-modified Ni(111) is ∼5 times as high as that on Ni(111) at 700 °C (Note S14, Fig. S17 and Table S8, ESI†). In summary, first-principles based mechanistic study in conjunction with microkinetic modeling supports the enhanced activities of FeNi–BZCYYb compared to Ni–BZCYYb by increasing the NH3 adsorption energy and lowering the associative N2 desorption reaction barrier. This was also verified that the d-band center model can be used as a descriptor for the rational design of novel NH3-fueled PCFC anode materials.
 | ||
| Fig. 4 Mechanistic studies of NH3 decomposition using DFT calculations. (a) Minimum energy pathways of the NH3 decomposition on FeNi(111) and Ni(111). The energies are relative to that of gas-phase NH3 and a bare surface of FeNi(111) or Ni(111). Those in parentheses denote the NH3 decomposition on Ni(111). The top view of each process shows the most stable configuration of surface intermediates on FeNi(111). (b) Scheme for microkinetic modeling of the NH3 decomposition on FeNi(111) and Ni(111). “ads” and “des” are adsorption and desorption, respectively. For simplicity, the associative desorption barriers (FeNi: 1.80 eV versus Ni: 1.92 eV) represent N* + N* → N2(g) + 2*. | ||
Conclusions
In summary, a re-structured anode surface via a surface modification of Fe on the Ni–BZCYYb anode greatly enhances the activity and durability toward ammonia utilization. The cells with such anodes achieved a peak power density of 1.257 W cm−2, and a power density of 0.435 W cm−2 at a current density of 0.5 A cm−2 at 650 °C for 100 h, when fueled by NH3. First-principles based calculations suggest that the enhanced activities of FeNi–BZCYYb compared to Ni–BZCYYb are due to the modification of the NH3 adsorption strength on the surface and the associative desorption barrier of nitrogen. The demonstrated surface restructuring of anodes could dramatically improve the performance and stability of PCFCs in ammonia for promising transportation and other applications. Compared to a system consisting of a separate ammonia reformer and a fuel cell, our PCFCs with a suitable catalyst integrated with the anode can eliminate the need for an ammonia cracking reactor or a reformer and a heat exchanger, greatly simplifying the design of the entire system, reducing the cost, and increasing the energy efficiency.Author contributions
Yu C. and M. L. conceived, designed and supervised the project. H. Z., Y. Z., and K. P. carried out the fabrication of the powders, cells and conducted data analysis of all electrochemical experiments. Y. P. and K. X. collected the data of X-ray diffraction, and SEM. K. S. and YongMan C. conducted the DFT calculation. Y. D. and B. Z. conducted TEM analysis. H. Z., Yu C., YongMan C., and M. L. contributed to writing the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (22005105 and 22179039), the Natural Science Foundation of Guangdong Province (2021A1515010395), and the Pearl River Talent Recruitment Program (2019QN01C693 and 2021ZT09L392). Computational studies were supported by the Ministry of Science and Technology (MOST Grant No. 110-2221-E-A49-017-MY3), the National Center for High-performance Computing (NCHC), and the Higher Education Sprout Project of the National Yang Ming Chiao Tung University and Ministry of Education (MOE), Taiwan. DFT calculations were performed using the resources of the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- Z. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. Zhan and S. A. Barnett, Nature, 2005, 435, 795–798 CrossRef CAS PubMed.
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O'Hayre, Nature, 2018, 557, 217–222 CrossRef CAS PubMed.
- F. Jiao and B. Xu, Adv. Mater., 2019, 31, 1805173 CrossRef.
- Y. Li, H. S. Pillai, T. Wang, S. Hwang, Y. Zhao, Z. Qiao, Q. M. Mu, S. Karakalos, M. J. Chen, J. Yang, D. Su, H. L. Xin, Y. S. Yan and G. Wu, Energy Environ. Sci., 2021, 14, 1449–1460 RSC.
- F. Ishak, I. Dincer and C. Zamfirescu, J. Power Sources, 2012, 212, 73–85 CrossRef CAS.
- F. Chang, W. Gao, J. Guo and P. Chen, Adv. Mater., 2021, 2005721, DOI:10.1002/adma.202005721.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef CAS.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Adv. Mater., 2012, 24, 195–208 CrossRef CAS.
- Y. Zhang, R. Knibbe, J. Sunarso, Y. Zhong, W. Zhou, Z. Shao and Z. Zhu, Adv. Mater., 2017, 29, 1700132 CrossRef PubMed.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- Y. Matsuzaki, Y. Tachikawa, T. Somekawa, T. Hatae, H. Matsumoto, S. Taniguchi and K. Sasaki, Sci. Rep., 2015, 5, 12640 CrossRef CAS.
- M. Ni, M. K. H. Leung and Y. C. Leung, Int. J. Hydrogen Energy, 2010, 33, 943–959 Search PubMed.
- G. Jeerh, M. F. Zhang and S. W. Tao, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- M. Li, M. Zhao, F. Li, W. Zhou, V. K. Peterson, X. Xu, Z. Shao, I. Gentle and Z. Zhu, Nat. Commun., 2017, 8, 13990 CrossRef CAS.
- C. Xia, Y. Mi, B. Wang, B. Lin, G. Chen and B. Zhu, Nat. Commun., 2019, 10, 1707 CrossRef PubMed.
- C. Ding and T. Hashida, Energy Environ. Sci., 2010, 3, 1729–1731 RSC.
- B. Hua, N. Yan, M. Li, Y. F. Sun, Y. Q. Zhang, J. Li, T. Etsell, P. Sarkar and J. L. Luo, Adv. Mater., 2016, 28, 8922–8926 CrossRef CAS.
- Y. Yoo, N. Lim, M. Phongaksorn, A. McFarlan and N. Maffei, Fuel Cells, 2008, 12, 691 CAS.
- Y. Yoo, M. Tuck, N. Lim, A. McFarlan and N. Maffei, ECS Trans., 2007, 7, 2305 CrossRef CAS.
- K. Xie, R. Q. Yan, G. Y. Meng and X. Q. Liu, Ionics, 2009, 15, 115–119 CrossRef CAS.
- K. Xie, Q. Ma, B. Lin, Y. Jiang, J. Gao, X. Liu and G. Meng, J. Power Sources, 2007, 170, 38–41 CrossRef CAS.
- Q. L. Ma, R. R. Peng, Y. J. Lin, H. F. Gao and G. Y. Meng, J. Power Sources, 2006, 161, 95–98 CrossRef CAS.
- S. P. Jiang, Int. J. Hydrogen Energy, 2012, 37, 449–470 CrossRef CAS.
- Y. Song, H. Li, M. Xu, G. Yang, W. Wang, R. Ran, W. Zhou and Z. Shao, Small, 2020, 16, 2001859 CrossRef CAS.
- W. Akimoto, T. Fujimoto, M. Saito, M. Inaba, H. Yoshida and T. Inagaki, Solid State Ionics, 2014, 256, 1–4 CrossRef CAS.
- M. Hashinokuchi, R. Yokochi, W. Akimoto, T. Doi, M. Inaba and J. Kugai, Solid State Ionics, 2016, 285, 222–226 CrossRef CAS.
- F. He, Q. N. Gao, Z. Q. Liu, M. T. Yang, R. Ran, G. M. Yang, W. Wang, W. Zhou and Z. P. Shao, Adv. Energy Mater., 2021, 2003916, DOI:10.1002/aenm.202003916.
- K. Miyazaki, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2017, 365, 148–154 CrossRef CAS.
- M. Kishimoto, N. Furukawa, T. Kume, H. Iwai and H. Yoshida, Int. J. Hydrogen Energy, 2017, 42, 2370–2380 CrossRef CAS.
- Y. Wang, J. Yang, J. Wang, W. Guan and J. Chen, ECS Trans., 2019, 91, 1611–1619 CrossRef CAS.
- A. F. S. Molouk, J. Yang, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Power Sources, 2016, 305, 72–79 CrossRef CAS.
- A. F. S. Molouk, J. Yang, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, J. Electrochem. Soc., 2015, 162, X27 CrossRef CAS.
- J. Yang, A. F. S. Molouk, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, ACS Appl. Mater. Interfaces, 2015, 7, 7406–7412 CrossRef CAS PubMed.
- Y. H. Wang, J. Yang, J. X. Wang, W. B. Guan, B. Chi, L. C. Jia, J. Y. Chen, H. Muroyama, T. Matsui and K. Eguchi, J. Electrochem. Soc., 2020, 167, 064501 CrossRef CAS.
- J. Yang, T. Akagi, T. Okanishi, H. Muroyama and T. Matsui, Fuel Cells, 2015, 15, 390–397 CrossRef CAS.
- K. Miyazaki, H. Muroyama, T. Matsui and K. Eguchi, Sustainable Energy Fuels, 2020, 4, 5238–5246 RSC.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- S. B. Simonsen, D. Chakraborty, I. Chorkendorff and S. Dahl, Appl. Catal., A, 2012, 447, 22–31 CrossRef.
- S. Choi, S. Yoo, J. Kim, S. Park, A. Jun, S. Sengodan, J. Kim, J. Shin, H. Y. Jeong, Y. Choi, G. Kim and M. Liu, Sci. Rep., 2013, 3, 2426 CrossRef.
- S. Choi, C. J. Kucharczyk, Y. G. Liang, X. H. Zhang, I. Takeuchi, H. I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef PubMed.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, J. Power Sources, 2008, 183, 682–686 CrossRef.
- Y. Lin, R. Ran, Y. M. Guo, W. Zhou, R. Cai, J. Wang and Z. P. Shao, Int. J. Hydrogen Energy, 2010, 35, 2637–2642 CrossRef.
- G. Y. Meng, C. R. Jiang, J. J. Ma, Q. L. Ma and X. Q. Liu, J. Power Sources, 2007, 173, 189–193 CrossRef CAS.
- L. Zhang and W. Yang, J. Power Sources, 2008, 179, 92–95 CrossRef CAS.
- Y. Itagaki, J. Cui, N. Ito, H. Aono and H. Yahiro, J. Ceram. Soc. Jpn., 2018, 126, 870–876 CrossRef CAS.
- J. C. Y. Itagaki, N. Ito, H. Aono and H. Yahiro, ECS Trans., 2018, 85, 779–786 CrossRef.
- S. S. Shy, S. C. Hsieh and H. Y. Chang, J. Power Sources, 2018, 396, 80–87 CrossRef CAS.
- Y. Aoki, T. Yamaguchi, S. Kobayashi, D. Kowalski, C. Zhu and H. Habazaki, Global Challen., 2018, 2, 1700088 CrossRef PubMed.
- B. Stoeckl, V. Subotic, M. Preininger, M. Schwaiger, N. Evic, H. Schroettner and C. Hochenauer, Electrochim. Acta, 2019, 298, 874–883 CrossRef.
- Y. Wang, Y. Gu, H. Zhang, J. Yang, J. Wang, W. Guan, J. Chen, B. Chi, L. Jia, H. Muroyama, T. Matsui, K. Eguchi and Z. Zhong, Appl. Energy, 2020, 270, 115185 CrossRef.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, Nat. Catal., 2019, 2, 659–670 CrossRef.
- X. Z. Duan, J. Ji, G. Qian, C. Fan, Y. Zhu, X. G. Zhou, D. Chen and W. K. Yuan, J. Mol. Catal. A: Chem., 2012, 357, 81–86 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef.
- S. Bhattacharjee, U. V. Waghmare and S. C. Lee, Sci. Rep., 2016, 6, 35916 CrossRef CAS PubMed.
- I. A. Filot, R. A. van Santen and E. J. Hensen, Angew. Chem., Int. Ed., 2014, 53, 12746–12750 CrossRef CAS PubMed.
- K. J. Laidler, Chemical Kinetics, Harper and Row, New York, 3rd edn, 1987 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02158c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |