
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular layer deposition of photoactive metal-naphthalene hybrid thin films†
Melania
Rogowska
 *a,
Per-Anders
Hansen
a,
Henrik Hovde
Sønsteby
a,
Joanna
Dziadkowiec
b,
Håkon
Valen
c and
Ola
Nilsen
*a
*a,
Per-Anders
Hansen
a,
Henrik Hovde
Sønsteby
a,
Joanna
Dziadkowiec
b,
Håkon
Valen
c and
Ola
Nilsen
*a
aCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, 0315 Oslo, Norway. E-mail: ola.nilsen@kjemi.uio.no
bThe NJORD Centre, Department of Physics, University of Oslo, 0371 Oslo, Norway
cNordic Institute of Dental Materials, 0855 Oslo, Norway
First published on 26th August 2021
Abstract
We here report on photoactive organic–inorganic hybrid thin films prepared by the molecular layer deposition (MLD) method. The new series of hybrid films deposited using 2,6-naphthalenedicarboxylic acid (2,6-NDC) and either hafnium chloride (HfCl4), yttrium tetramethylheptanedionate (Y(thd)3) or titanium chloride (TiCl4) were compared with the known zirconium chloride (ZrCl4) based system. All metal-naphthalene films are amorphous as-deposited and show self-saturating growth as expected for an ideal MLD process with varied growth rates depending on the choice of metal precursor. The growth was studied in situ using quartz crystal microbalance (QCM) and the films were further characterised using spectroscopic ellipsometry (SE), Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), and UV-Vis and photoluminescence (PL) spectroscopy to obtain information on their physicochemical properties. The hybrid thin films display intense blue photoluminescence, except for the Ti-organic complex in which titanium clusters were found to be an effective PL quencher for the organic linker. We demonstrate how the optical properties of the films depend on the choice of metal component to make a foundation for further studies on these types of organic–inorganic hybrid materials for applications as photoactive agents.
Introduction
Organic–inorganic hybrids emerge as a creative alternative for designing functional materials with unique features. Combinations of the physical and chemical properties (e.g., flexibility, electrical conductivity and catalytic activity) of organic and inorganic components offer the opportunity to build materials with new or improved properties.1 These functional hybrids can be used in a wide range of fields like optics and photoluminescence,2,3 electronics,4,5 biology,6,7 protective coatings,8 and sensors.9,10 However, most existing micro- and nanotechnologies require ultrathin film materials with uniform and conformal coverage, preferably at the atomic/molecular level. Design of such hybrid thin film materials can be achieved through gas-phase deposition techniques like atomic/molecular layer deposition (ALD/MLD).ALD has become an essential tool for deposition of thin films of inorganic materials with precise thickness control.11 The MLD approach extends on the ALD technique by using larger building blocks as precursors, and can be used to grow both hybrid organic–inorganic and entirely organic films.12–16 Many of the existing MLD processes are based on aromatic carboxylic acids as organic building blocks since they often provide uniform films that are stable in air. In addition, the rigid structure of the aromatic ring reduces the possibility of dual reactions with the substrate, and the high reactivity of carboxylic acids allows for a wide range of metal precursors to be used. Furthermore, the conjugated structure enables absorption bands in the UV and visible light region, which makes them attractive as photoactive agents. A high diversity of aromatic dicarboxylic acids have been extensively applied as organic building blocks in designing materials by MLD.17–20
The attractive optical properties of such hybrid thin films have recently drawn significant attention. Khayyami et al.21,22 reported reversibly photoresponsive Zn-, and Fe-azobenzene (4,4′-azobenzenedicarboxylic acid) structures. Giedraityte et al.23,24 describe a series of Eu-based hybrid materials with different aromatic dicarboxylic acids that show intense red photoluminescence involving the energy transfer between the organic component and Eu3+ cations. In a related work, they demonstrate blue and green photoluminescence by combining nucleobase like uracil and adenine with Na, Ba and La metals.25 Furthermore, photoactive organic molecules can be combined with biocompatible metals like Zr or Ti to create antimicrobial coatings. These photoactive components can utilize light energy to form reactive oxygen species and induce a phototoxic effect upon living microorganisms. Lausund et al.26 successfully deposited photoactive hybrid materials based on Zr-clusters and bi-aromatic linkers with promising luminescent and antimicrobial properties.
The physicochemical properties, including optical properties of the organic–inorganic hybrids are determined by the interaction between the organic ligand and the metal ion. Hybrids containing transition metal ions, exhibit photoabsorption and emission that is typically centered on the organic ligand rather than on the metal, but can also involve charge and energy transfer between these components.27 Importantly, the electronic configuration of the transition metal, but also the orientation of the organic ligand and the bonding geometry has a significant effect on the optical properties. Nasalevich et al.28 reported that variations on the d0-metal component in crystalline metal–organic frameworks (MOFs) change the electronic properties of these materials. They demonstrated that Ti-based MOF promotes a ligand-to-metal charge transfer (LMCT) upon photoexcitation due to the formation of Ti3+, whereas the d-orbitals of Zr4+ and Hf4+ cannot overlap with the π* orbital of the ligand and thus these MOFs show nearly identical performance with linker-based activity.
Until now most systematic studies on MLD hybrid coatings (also with MOF-like structures) have focused on variations on the organic linkers.16,24,29 Our approach has been to investigate the impact of various isoelectronic metals on the photophysical properties of the hybrid organic–inorganic thin films. We foresee that this type of new hybrid materials could be exciting in applications as photoactive coatings on flexible and/or rigid nanostructured surfaces. In this work, we use 2,6-naphthalenedicarboxylic acid (abbreviated here as 2,6-NDC) as the organic building block and d0 transition metals of different valences such as Ti, Zr, Hf or Y as the connector. Tetravalent Ti, Zr and Hf were selected because of their comparatively similar electronic and optical properties. It has been assumed that these metals can form M3+ species with charge transfer to oxygen to affect their optical properties. Whereas trivalent Y cannot participate in a similar charge transfer state, and thus it does not show any photochemical activity. Furthermore, the corresponding element oxides like ZrO2, HfO2 and Y2O3 exhibit relatively large optical bandgap in the range of 5.8–6.0 eV and rather negligible absorption from near-UV to visible light, while TiO2 with Eg = ∼3.3 eV shows strong UV and some visible-light photocatalytic activity.30
The model π-conjugated organic linker used in this work has a delocalized aromatic structure that controls its chemical and physical properties, like near-UV absorbance. Furthermore, this bulky ligand with high structural rigidity offers many advantages in the construction of metal–organic frameworks. We recently demonstrated that 2,6-NDC precursor readily form hybrid with Zr,26 however, to the best of our knowledge no attempts have been reported so far with other transition metals such as Ti, Hf and Y.
Experimental methods
The hybrid Ti-, Zr-, Hf-, and Y-naphthalene thin films were deposited with an F-120 Sat ALD reactor (ASM Microchemistry Ltd) at a reactor pressure of 6.6 mbar. Nitrogen (99.999%, Schmidlin Sirocco 5 V1 N2 generator) was used as a carrier and purging gas with a total flow of ca. 400 sccm (cm3 min−1) throughout the experiments (inner chamber flow 300 sccm and outer chamber flow 100 sccm). The precursors used in this study were ZrCl4 (Strem Chemicals, >99.9%), HfCl4 (Strem Chemicals, >99.9%), Y(thd)3 (thd: 2,2,6,6-tetramethyl-3,5-heptanedionate) (Strem Chemicals, >98%), TiCl4 (Fluka, >99%) and 2,6-naphthalenedicarboxylic acid (Sigma-Aldrich, 99%). The sublimation temperatures for powder precursors were 165 °C for ZrCl4 and HfCl4, 135 °C for Y(thd)3 and 225 °C for 2,6-NDC. The liquid TiCl4 precursor was kept in an external bubbler at room temperature. The films were deposited on Si(100) substrates precleaned with ethanol (99.96%, VWR). The growth rate of the films was measured as a function of deposition temperature and number of cycles.The thickness and the refractive index of the films were determined with an alpha-SE spectroscopic ellipsometer (J. A. Woollam) in the wavelength range of 390–900 nm at an incident angle of 70°. The data were fitted to a Cauchy model using the CompleteEASE software. The thickness of the native oxide layer was determined before the deposition process and added as an individual layer in the interpretation of film thickness.
The growth dynamics of the films were monitored by an in situ quartz crystal microbalance (QCM) using two 6 MHz gold coated quartz crystals (Inficon) placed ∼5 cm apart in a home-made holder. The resonance frequency of the crystals during the deposition is proportional to the total oscillating mass of the sensor. The QCM data were processed by averaging 16 consecutive cycles. The changes in resonance frequency were converted to mass per area (ng cm−2) by using the thickness and density of the film measured by XRR on films deposited separately from QCM.
Information on the structural parameters of the thin films such as thickness, density and surface roughness was obtained from X-ray reflectivity (XRR) measurements. XRR analysis was performed using an Empyrean diffractometer (Panalytical) equipped with a Cu Kα (λ = 1.5406 Å) source powered at 45 kV and 40 mA, and a parallel beam mirror. The XRR results were analysed using the X'Pert Reflectivity software provided by Panalytical.
Fourier transform infrared spectroscopy (FTIR) measurements of the films were performed using Bruker Vertex 70 spectrometer equipped with Pike VeeMAX III specular reflection accessory with an angle of incidence at 75°. All data were acquired in the reflection mode using 64 scans in the wavenumber range of 4000–370 cm−1 and 2 cm−1 resolution. Films for FTIR measurements were deposited on electropolished steel substrates. A spectrum obtained from the uncoated electropolished steel substrate was used as the background during the measurements. FTIR measurement of the organic compound was performed using PerkinElmer Spectrum One instrument in the transmission mode in the wavenumber range of 4000–400 cm−1 and 1 cm−1 resolution. The pellet was prepared by mixing ∼1 mg of the sample with 100 mg KBr and pressed into a pellet in a hydraulic press using a force of approximately 10 tons.
X-ray photoelectron spectroscopy (XPS) was performed with a Theta Probe Angle-Resolved XPS (Thermo Scientific) using a standard monochromated Al Kα source (hv = 1486.6 eV) and the analysis chamber was maintained at ∼10−8 mbar during analysis. The spectra were acquired using a pass energy of 200 eV for survey spectra and 100 eV for detailed peak scans with an analysis spot of roughly 400 μm. The binding energy scale was calibrated by setting the main line of the adventitious C 1s, C–C peak, at 284.8 eV.
UV-Vis transmittance spectra of the films were collected with Shimadzu UV-3600 UV-Vis-NIR spectrophotometer in the wavelength range of 200–850 nm with 1 nm resolution. Photoluminescence (PL) measurements were performed using 280 nm excitation diode and a USB2000+ spectrometer (OceanOptics) in the wavelength range of 187–1035 nm at room temperature. Photobleaching effects were determined using the same setup. Films for UV-Vis and PL measurements were deposited on 1 × 1 cm2 fused silica substrates precleaned with ethanol (99.96%, VWR).
Results and discussion
Film growth and deposition parameters
In this work, we created four possible hybrid complexes Ti-NDC, Zr-NDC, Hf-NDC and Y-NDC (Fig. 1). The growth dynamics of all systems were initially investigated using in situ QCM. The standard pulsing sequences and reaction temperatures used throughout the QCM experiments for all systems were chosen after some preliminary experiments and are presented in Table 1. Note that for the Zr-NDC process we used the deposition parameters proposed by Lausund et al.,26 except for the deposition temperature, which was fixed in this work to 260 °C. The QCM experiments were performed by gradually increasing pulse and purge length one at a time while keeping the other parameters constant based on the standard pulsing sequence. The growth rate as a function of inorganic and organic precursor pulse length is shown in Fig. 2a and b. All investigated systems show self-limiting growth for both precursors. The relatively low density of the films measured by XRR was determined to 2.1 g cm−3 for Hf-NDC and ∼1.7 g cm−3 for Ti-, Zr-, and Y-NDC (Fig. S1†).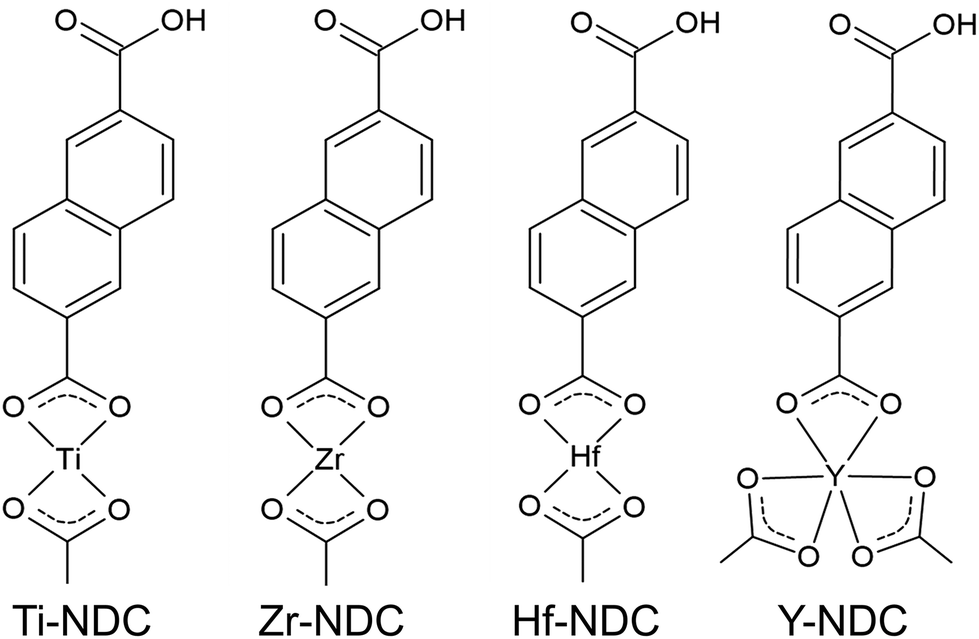 | ||
| Fig. 1 Possible structures of metal-naphthalene hybrids deposited in this work. | ||
 | ||
| Fig. 2 The growth rate as a function of (a) inorganic and (b) organic precursors pulse lengths as measured by the QCM method. The standard pulse/purge lengths for all systems were fixed to: Zr-NDC (3–2–20–2 s), Hf-NDC (12–2–10–2 s), Ti-NDC (1–5–15–2 s) and Y-NDC (4–3–8–3 s). (c) Growth per cycle (GPC) as a function of deposition temperature and (d) film thickness versus number of deposition cycles. The dashed lines show a fitted linear regression. The error bars in (c–d) indicate the standard deviation from measuring a total of nine points on the substrates from each deposition. In some cases the error bars are within the sizes of the marks. | ||
| Inorganic precursor | Pulse/purge (s) | 2,6-NDC pulse/purge (s) | Deposition temperature (°C) |
|---|---|---|---|
| ZrCl4 | 3/2 | 20/2 | 260 |
| TiCl4 | 1/5 | 15/2 | 260 |
| HfCl4 | 12/2 | 10/2 | 260 |
| Y(thd)3 | 4/3 | 8/3 | 260 |
The inorganic precursors saturate the surface faster (ZrCl4 3 s, TiCl4 1 s, and Y(thd)3 4 s) than the organic precursor, except for HfCl4, which requires roughly 12 s to saturate the surface. This significant delay in the nucleation and growth when using HfCl4 was unexpected and we are uncertain about its origin, particularly since according to thermodynamics, the vapour pressure of HfCl4 is more than twice that of ZrCl4. Although the sublimation taking place during delivery is not an equilibrium process and hence also influenced by kinetics and effective surface area of the precursor. It may also be related to variations in the structure of the material during growth, leading to different amounts of reactive sites on the surface. The growth was not improved, however, by elevating the sublimation temperature of HfCl4 (from 165 °C to 175 °C), but only by increasing its pulse length to 12 s. The delay in the growth of Hf-layer can also be seen in Fig. 3, where we plot the mass evolution over time for Ti-, Zr-, Hf-, and Y-NDC systems. Moreover, this graph proves a self-saturating growth for all precursors. In order to understand the rather complex growth mechanism of Hf-NDC, a more thorough investigation of this system should be performed.
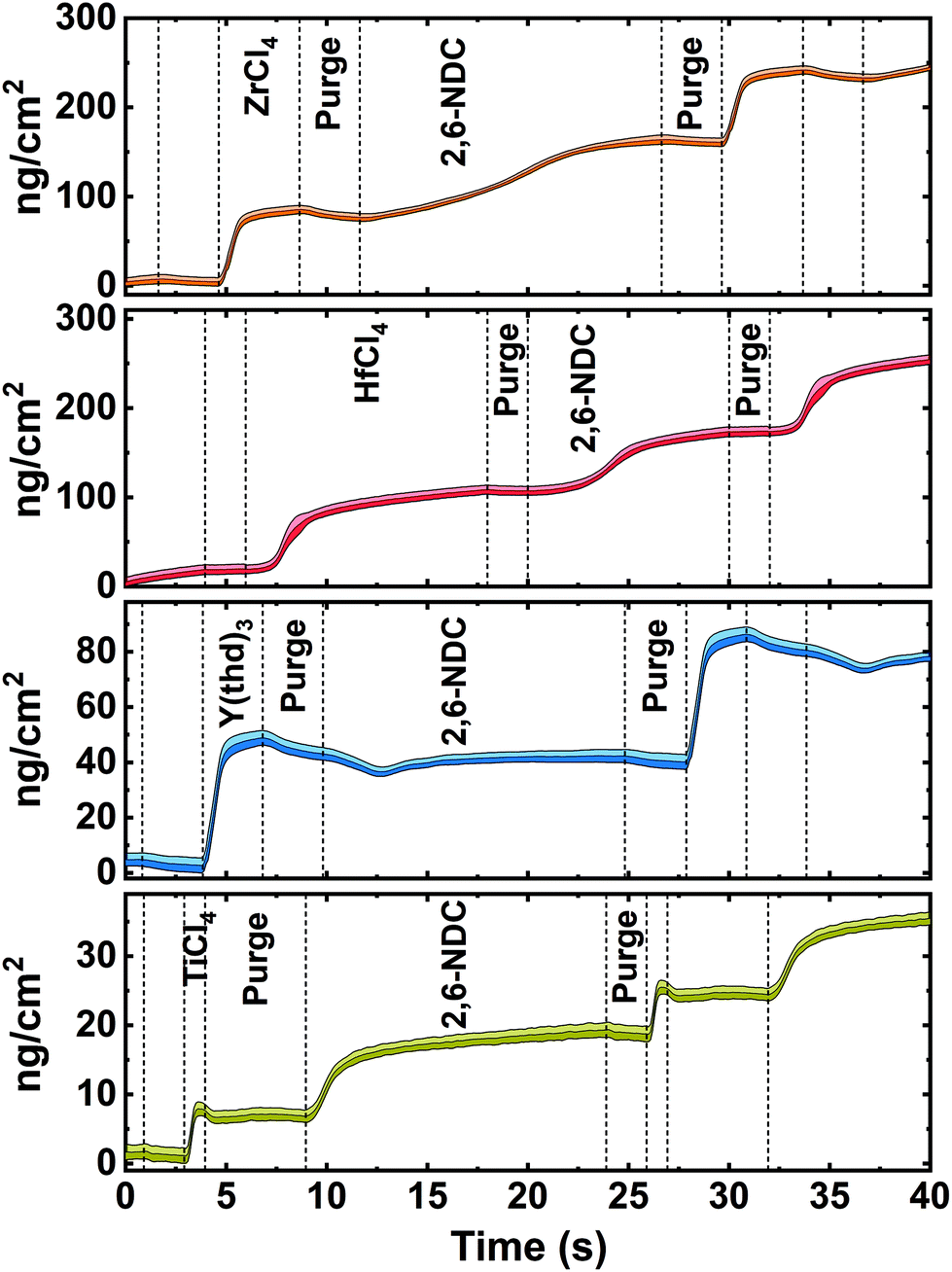 | ||
| Fig. 3 Mass increase as a function of time measured in situ with QCM for Zr-NDC, Hf-NDC, Y-NDC and Ti-NDC. Inorganic precursor pulses and 2,6-NDC pulses are separated by inert gas (N2) purges. The shaded area represents the statistical variation during 16 cycles. | ||
The growth rates of the different systems were also investigated as a function of deposition temperature based on the pulsing parameters initially chosen from the QCM analysis (Fig. 2c). The GPC value decreases with increasing reaction temperature for Hf-, and Y-NDC systems, especially above 280 °C. This reduction in growth rate is probably caused by increased thermal motion and disorder of the organic molecules at higher temperatures that prevent the adsorbed molecules from self-organizing on the surface.17 Importantly, however, no changes in the type of the coordination mode nor possible decomposition of the film was observed in the FTIR spectra of the Y-NDC film deposited at different temperatures (Fig. S2†). For Ti-NDC, the GPC remains nearly constant throughout the temperature range, which is similar to what was found for the ZrCl4 + 2,6-NDC (Zr-NDC) process.26 A relatively constant growth rate is seen for all processes between 260–280 °C.
Among the three deposited systems, the highest growth rate is for the Hf-NDC process (∼7.1 Å per cycle), which is comparable with Zr-NDC (∼8 Å per cycle),26 while the lowest GPC is for the Ti-NDC process (0.8 Å per cycle). Surprisingly, the growth rate for Ti-NDC is much lower than anticipated. Previous studies of the growth of pure metal oxides have shown similar GPC values of around 0.5 Å per cycle for TiCl4/H2O (TiO2),31 ZrCl4/H2O (ZrO2),32 and HfCl4/H2O (HfO2)33 processes, and thus it can be assumed that the size of the halide precursors does not impact the growth rate of our systems. For the Y-NDC process, on the other hand, the low growth rate (2 Å per cycle) is possibly caused by the bulky metal thd-precursor. This β-diketonate reactant can reduce surface coverage due to steric hindrance and can exhibit low reactivity leading to low growth rates. Finally, in Fig. 2d, we show that the film thickness increases linearly with the increasing number of deposition cycles for Ti-NDC, Hf-NDC and Y-NDC systems, which is an expected characteristic for an ideal ALD/MLD process of amorphous films.
Bonding characteristics
The organic moiety of the deposited films and the coordination towards the metal were investigated with FTIR spectroscopy, see Fig. 4. For comparison, the spectrum of the corresponding carboxylic acid precursor is included as reference. Firstly, the absorptions at around 770 cm−1 and 1500 cm−1 are related to the C–H and C–C stretching vibrations in the aromatic ring of the 2,6-NDC molecule.34,35 Furthermore, the characteristic absorption peaks for symmetric and asymmetric stretching vibrations of carboxylate groups are seen at 1400–1600 cm−1.17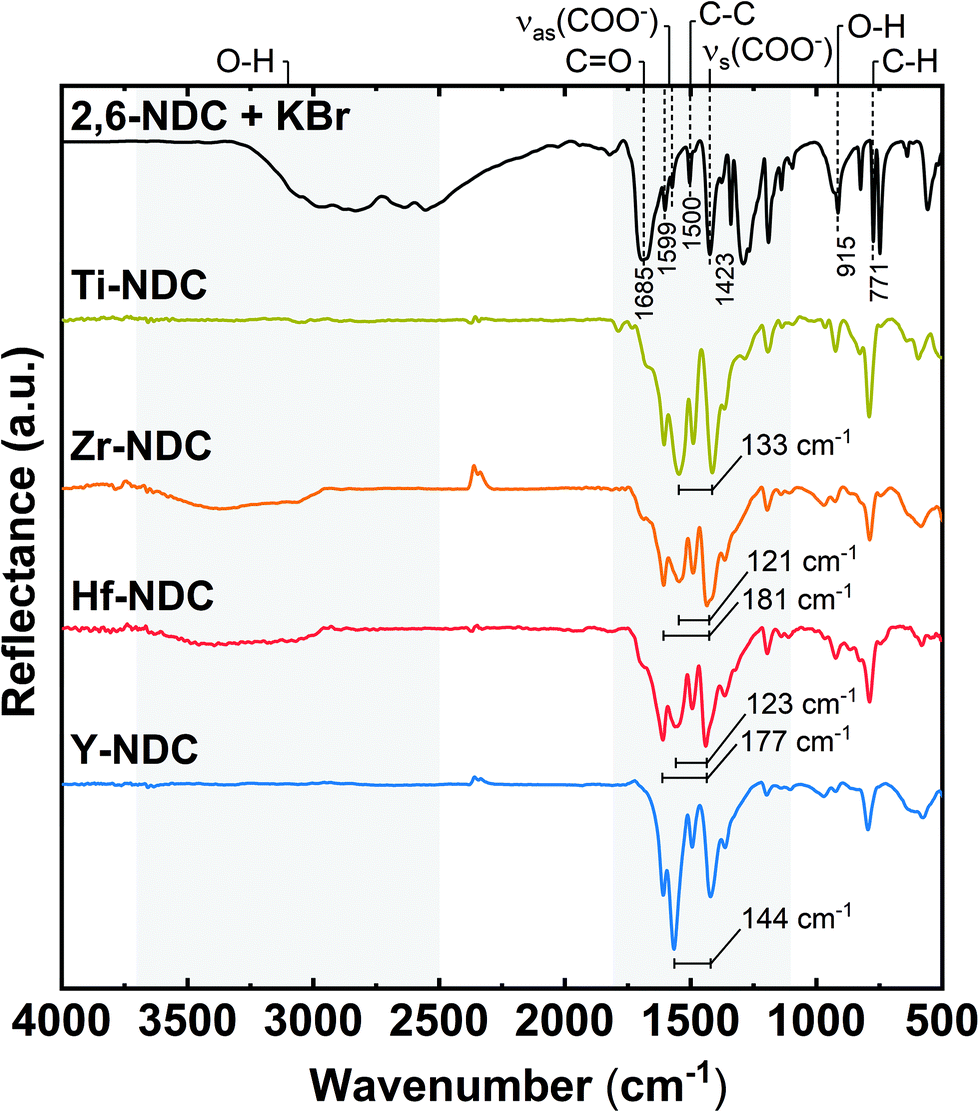 | ||
| Fig. 4 FTIR reflection spectra of representative hybrid thin films: Ti-NDC (100 nm), Zr-NDC (109 nm), Hf-NDC (108 nm), Y-NDC (99 nm), and the corresponding organic precursor. | ||
A significant reduction or disappearance of the sharp signal around 1685 cm−1 from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations indicates that it coordinates to the metal atom during formation of the hybrid structure.21 Similarly, the absence of the broad absorption for OH groups around 2900–3700 cm−1 in the spectra of Ti-NDC and Y-NDC films, suggest that the oxygen is deprotonated and bonded to the metal. In the Zr-NDC and Hf-NDC spectra, on the other hand, the weak absorption due to OH groups indicates that the deposited films absorbed water upon short storage in air (24 hours). In addition, we observe that this weak absorption band increased in the Zr-NDC sample after much longer (5 months) exposure to air, which confirms that the OH stretching band most likely comes from the absorbed water (Fig. S3†). However, it cannot be excluded that the OH bands present in the as-deposited Zr-NDC and Hf-NDC samples are the result of unreacted OH groups present in the films. Moreover, a reduction of another sharp absorption band of OH groups at around 915 cm−1 in all spectra supports the assumption that the metals bond to the 2,6-NDC molecule via two carboxylic oxygen atoms.36
O stretching vibrations indicates that it coordinates to the metal atom during formation of the hybrid structure.21 Similarly, the absence of the broad absorption for OH groups around 2900–3700 cm−1 in the spectra of Ti-NDC and Y-NDC films, suggest that the oxygen is deprotonated and bonded to the metal. In the Zr-NDC and Hf-NDC spectra, on the other hand, the weak absorption due to OH groups indicates that the deposited films absorbed water upon short storage in air (24 hours). In addition, we observe that this weak absorption band increased in the Zr-NDC sample after much longer (5 months) exposure to air, which confirms that the OH stretching band most likely comes from the absorbed water (Fig. S3†). However, it cannot be excluded that the OH bands present in the as-deposited Zr-NDC and Hf-NDC samples are the result of unreacted OH groups present in the films. Moreover, a reduction of another sharp absorption band of OH groups at around 915 cm−1 in all spectra supports the assumption that the metals bond to the 2,6-NDC molecule via two carboxylic oxygen atoms.36
According to Verpoort et al.,37 the separation between two dominant absorption peaks from the symmetric and asymmetric stretching O–C–O vibrations varies depending on how the carboxylate group coordinates to a metal atom (Fig. S4†). The splitting values Δ between these peaks are marked in all spectra of deposited films and are summarized in Table 2. Bidentate (chelating) complexes exhibit a splitting in the range between 50 and 150 cm−1, while monodentate complexes show a separation larger than 200 cm−1. For bridging coordination, a splitting is greater than in bidentate complexes with Δ values between 130 and 200 cm−1. The Δ value for Ti-NDC film indicates a bidentate type of coordination. Whereas the doublet band with the second intensive absorption peak in the higher wavenumber values seen in the spectra of Zr-NDC and Hf-NDC samples suggests a possible twofold nature of these systems with neighbouring bidentate and bridging complexes. For the Y-NDC sample a border-line value (Δ = 144 cm−1) points in the direction of bidentate or bridging coordination of the carboxylate group to yttrium atoms. Hence, we may conclude that in all deposited films the metal atoms bond to the organic precursor through both carboxylic oxygen atoms from the carboxylate unit, however, the bonding scheme varies some.
| Sample | ν as(COO−) | ν s(COO−) | Δν (COO−) [νas(COO−) − νs(COO−)] |
|---|---|---|---|
| Ti-NDC | 1548 | 1415 | 133 |
| Zr-NDC | 1608, 1548 | 1427 | 181, 121 |
| Hf-NDC | 1613, 1559 | 1436 | 177, 123 |
| Y-NDC | 1565 | 1421 | 144 |
XPS analysis was carried out for qualitative and quantitative investigation of the composition and chemistry of the structures and to further support the FTIR results. The survey spectra confirm a hybrid nature of the films (Fig. S5† and Table 3). For the samples where chlorine containing precursors were used, i.e. for Ti-, Zr-, and Hf-NDC, Cl 2p (at ∼200 eV) is barely detectable (not seen here in the wide energy range spectra) and the calculated Cl concentration is below 0.1%, if any.
| Sample | M | C | O | Cl | C/M ratio | C/O ratio |
|---|---|---|---|---|---|---|
| Ti-NDC | 3.8 | 71 | 25 | ∼0.1 | 18.7 | 2.84 |
| Zr-NDC | 3.0 | 71 | 26 | — | 23.7 | 2.73 |
| Hf-NDC | 3.0 | 71 | 26 | ∼0.1 | 23.7 | 2.73 |
| Y-NDC | 3.8 | 70 | 26 | — | 18.4 | 2.69 |
The core level peaks of Ti 2p, Zr 3d, Hf 4f and Y 3d are shown in Fig. 5, and fit well with Ti4+, Zr4+, Hf4+ and Y3+, respectively. The Ti 2p peak has spin orbit splitting of 6 eV and binding energy of 2p3/2 and 2p1/2 peaks at 458.4 eV and 464.4 eV, which is characteristic for Ti4+ oxidation state in TiO2 (assigned to Ti–O bonds).38 Two components at 183.1 eV (3d5/2) and 185.5 eV (3d3/2) in the Zr 3d spectrum can be attributed to Zr–O bonds, however, the position of these components is slightly shifted to higher energies as compared to pure ZrO2 film by ALD.39 The Hf 4f core level peak shows two contributions at 17.7 eV (4f7/2) and 19.3 eV (4f5/2) which are typical for Hf–O bonds, while two components at 157.6 eV (3d5/2) and 159.6 eV (3d3/2) on Y 3d spectrum correspond to Y–O bonds.40,41 These peaks, however, are also slightly shifted to higher BE as compared to the pure metal oxides prepared by ALD suggesting some possible metal–OH bonds. The existence of metal–OH bonds on the surfaces could be explained by adsorbed water species due to the contact of the surfaces with ambient conditions.
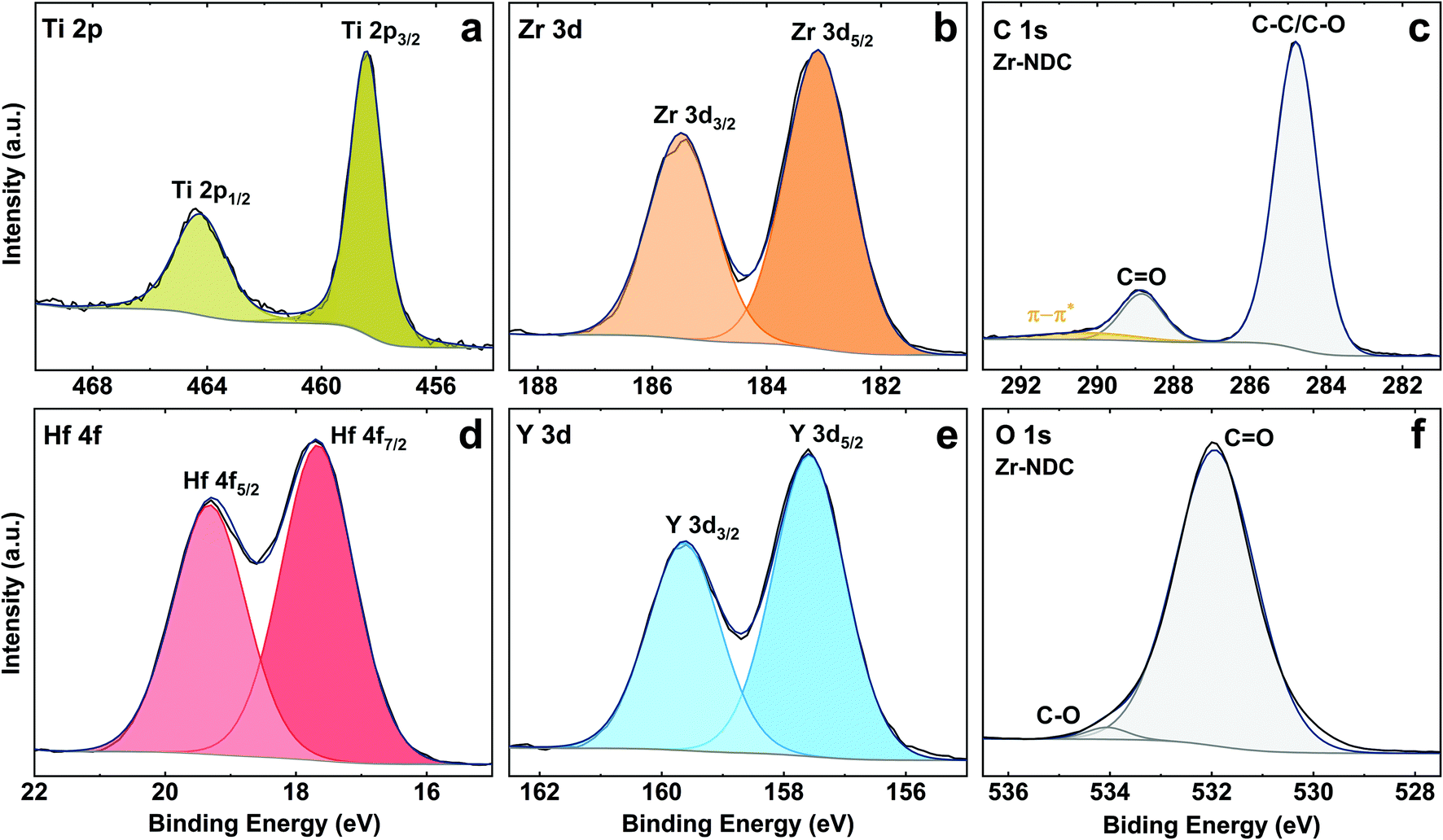 | ||
| Fig. 5 XPS spectra of the hybrid thin films. (a) Ti 2p region of Ti-NDC, (b) Zr 3d region of Zr-NDC, (d) Hf 4f region of Hf-NDC and (e) Y 3d region of Y-NDC. (c) Representative C 1s region of Zr-NDC sample. (f) Representative O 1s region of Zr-NDC sample. Black curve: measured data. Dark blue curve: total fit. Colour region: fitted regions for different core contributions. | ||
The detailed C 1s spectra for all four samples indicates two main components at 284.8 eV (C–C/C–O) and 288.8 eV (CO) from the aromatic ring and carboxyl group of the ligand with the ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 between these species,42 see Fig. 5c. The weak peak from the carbonyl group CO in these spectra confirms that the organic precursor bonds to the metal atoms via both OH and CO active sites. Furthermore, the π–π* satellite structure seen around 6 eV from the main C 1s peak is attributed to delocalised electrons in the aromatic ring of the 2,6-NDC molecule. The O 1s spectra for all samples show the main peak seen at 531.9 eV for oxygen in carbonyl group (Fig. 5f).24 Note that due to the air storage and carriage of the samples, physically adsorbed carbon and oxygen (e.g., CO2, H2O) may also be present at the surface. The C:O atomic ratio differs from the expected 3:1 and indicates a slight oxygen surplus in all deposited samples.
:1 between these species,42 see Fig. 5c. The weak peak from the carbonyl group CO in these spectra confirms that the organic precursor bonds to the metal atoms via both OH and CO active sites. Furthermore, the π–π* satellite structure seen around 6 eV from the main C 1s peak is attributed to delocalised electrons in the aromatic ring of the 2,6-NDC molecule. The O 1s spectra for all samples show the main peak seen at 531.9 eV for oxygen in carbonyl group (Fig. 5f).24 Note that due to the air storage and carriage of the samples, physically adsorbed carbon and oxygen (e.g., CO2, H2O) may also be present at the surface. The C:O atomic ratio differs from the expected 3:1 and indicates a slight oxygen surplus in all deposited samples.
Photoabsorption and luminescence characteristics
The deposited hybrid films can be utilized as photoactive materials, thus we investigated their optical properties and thereby the effect of different metal clusters on these properties by performing UV-Vis spectroscopy. The transmission spectra for the four thin films and the solution spectrum of the corresponding 2,6-NDC organic precursor are depicted in Fig. 6. The prominent doublet peak of 2,6-NDC precursor located between 325 and 360 nm is attributed to π–π* intraligand transition in the UVA region associated with the aromatic rings of the ligand. For the absorption spectra of deposited films, the π–π* transition is redshifted which reflects the interaction between the naphthalene and metal atoms.43 Similar shift to longer wavelengths was seen on the spectra of deposited films for the absorption peak located between 270 and 300 nm also assigned to the π–π* transition.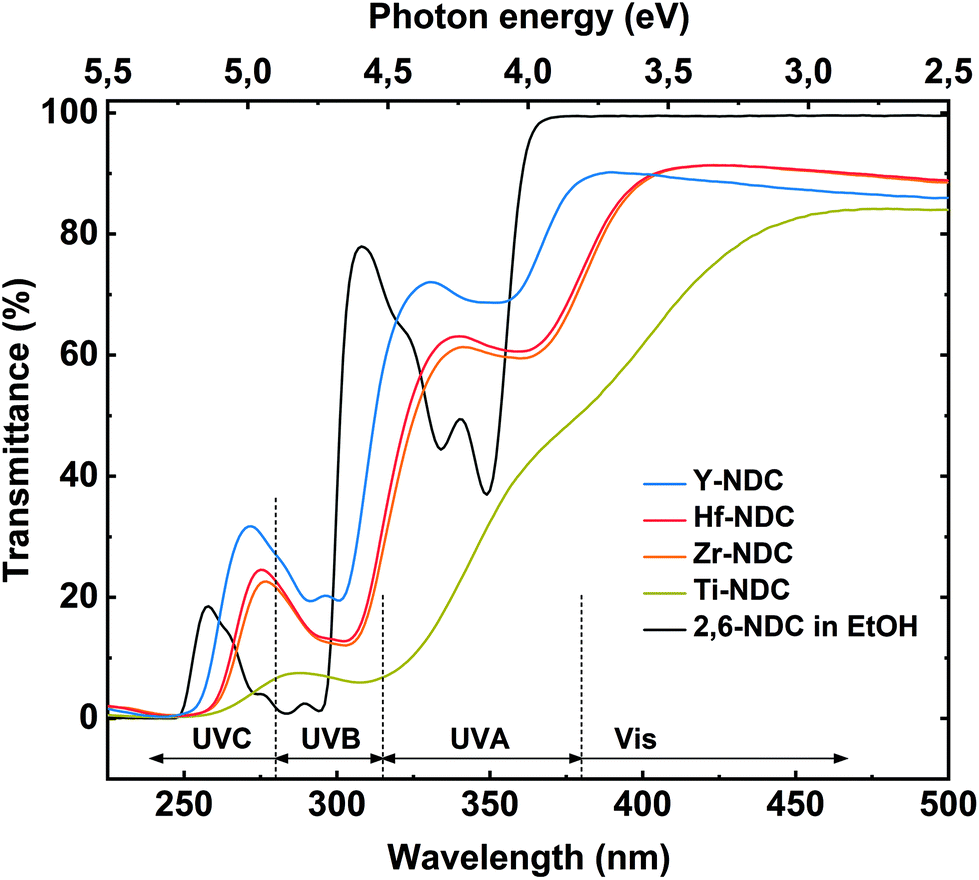 | ||
| Fig. 6 UV-Vis transmission spectra of the four hybrid thin films: Ti-NDC (100 nm), Zr-NDC (109 nm), Hf-NDC (108 nm), Y-NDC (99 nm), and the corresponding organic precursor dissolved in ethanol. Note that the dotted line separating the UVC and UVB regions at 280 nm also shows the light emission of the UV diode used in the PL measurements. | ||
The absorption properties of the films strongly depend on the metal component. All deposited films absorb in the near UV region where the π–π* adsorption edge of the aromatic ring is expected, however, the main absorption edge differs within the samples. This effect can be correlated with the different electronic characteristic and charge density of the metal constituents. The Y-NDC film is transparent throughout the visible region with the main absorption edge at around 380 nm, whereas Zr-, and Hf-NDC samples show a weak absorption in the visible region with the main absorption edge at ∼400 nm. The spectrum for Ti-NDC film differs compared to the other deposited systems by a much stronger absorption in the visible region with the absorption edge at around 450 nm. Also, when comparing the Ti-NDC film with pure TiO2 film (Fig. S6†), it is evident that by combining conjugated organic linker with transition metal cluster we can shift and increase the absorption throughout the visible region due to electronic coupling between the ligand and d orbitals on the metal. Furthermore, the difference between the absorption properties of deposited films and the corresponding organic linker indicates that there is a strong influence of the metal clusters on the electronic structure of the linker. It may be assumed that the Ti4+ cation with its relatively small ionic radius of 0.068 nm and a high charge decreases the electron density of the naphthalene ring, which in consequence decrease the HOMO–LUMO gap and redshift the material absorption. The tetravalent Zr4+ and Hf4+ cations have a comparably larger ionic radius of 0.079 nm and 0.078 nm, respectively, and therefore a weaker effect on the cation–aromatic interaction. Whereas Y3+ with an ionic radius of 0.092 nm provides the lowest redshift of the absorption, which is consistent with the theoretical expectations, the trend is therefore Ti4+ > Zr4+ ≥ Hf4+ > Y3+. The ionic radius data were obtained from Ahrens.44
Photoluminescence emission spectra of the four deposited systems are shown in Fig. 7a. The emission of the Zr-NDC, Hf-NDC and Y-NDC are similar in shape with the strong doublet peak. The maximum emission peaks for these samples are respectively: Zr-NDC: 446/485 nm, Hf-NDC: 443/483 nm and Y-NDC: 435/481 nm. For the Ti-NDC complex, Ti-clusters in the hybrid structure quench the ligand-based luminescence. This effect can be attributed to the interfacial electron transfer through the partially filled d-orbitals of the Ti atom and the π orbitals of the aromatic ring.45 Note that the absorption of the TiO2 film (Fig. S6†) overlaps in some extent with the characteristic emission region of the metal-naphthalene hybrids. Moreover, Ti-NDC complex absorbs wavelengths slightly longer than the 280 nm emission light from the diode used in this study. However, no visible luminescence for this complex was detected when using 254 nm and 365 nm UV lamp.
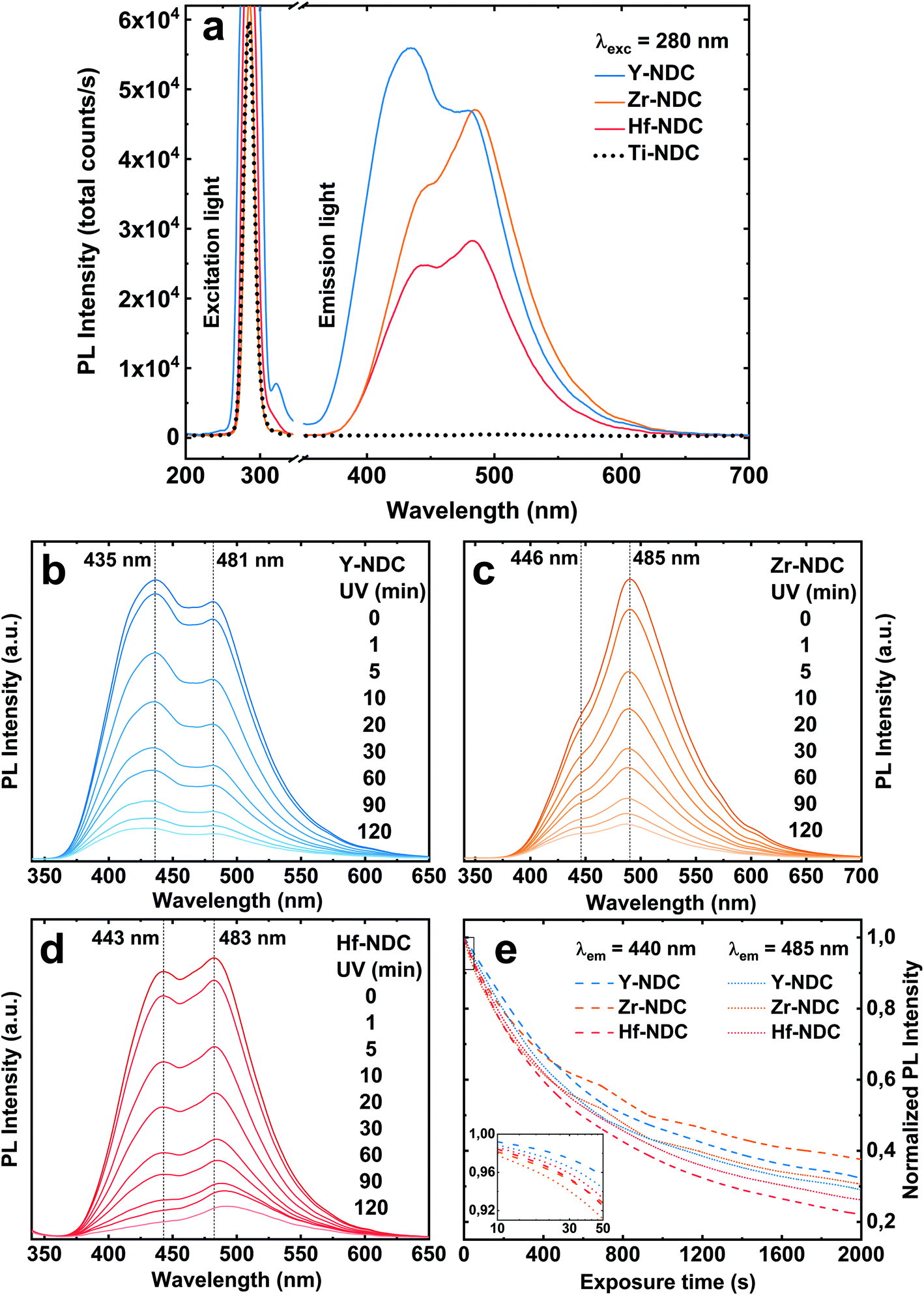 | ||
| Fig. 7 (a) Photoluminescence emission spectra of the hybrid films: Ti-NDC (100 nm), Zr-NDC (109 nm), Hf-NDC (108 nm) and Y-NDC (99 nm). PL was recorded using a 280 nm diode. (b–d) The time evolution of the PL spectra upon UV exposure. (e) Normalized PL intensity of the two emission peaks at 440 nm and 485 nm vs. UV exposure time. The inset shows a zoomed-in view of the first 50 s on a logarithmic scale. | ||
The emission spectra for luminescent samples are somewhat a mirror image of the absorption of pure 2,6-NDC molecules and thus it can be assumed that the luminescence is centred on the organic linker with π-conjugated backbone, rather than on the metal. However, the metal ions can influence the emission characteristics, depending on the electronic configuration and ionic radius of the metal and relative metal and linker orbital energies.27 The noticeable differences in the emission spectra of these three systems can be hence triggered by the spacing and orientation between the organic linkers and the bonding configuration depending on the system. It was reported that the emission intensity of pure 2,6-NDC ligand can be significantly enhanced when combined with metal clusters such as Zr due to the strong electronic coupling between the neighbouring ligands through metal clusters.46
The total emission intensity of all samples with the comparable thickness (∼100 nm) vary within the materials depending on the metal component in order of Ti ≪ Hf < Zr < Y, which confirms the effect of the ionic radius on the fluorescent properties. The intensity for yttrium-based material is almost 2 times enhanced when compared to that with hafnium. The variations in the photoluminescence intensity can also be related to the differences in the absorption properties of these complexes. It should be noted that the absorption of the films overlaps in different magnitude with the light emission at 280 nm from the diode used in this study. Furthermore, the emission spectra exhibit the characteristic tail at around 550 nm which can be assigned to the formation of excimers (excited state dimers) due to the high concentration of the naphthalenes.47 When the aromatic molecules are close enough to each other to form aggregates, they can transform to excimers if one of the partners is excited.
The blue emission of the samples decreases gradually with time upon UV exposure (Fig. 7b–e), suggesting a photobleaching effect. This process could be explained by the formation of highly oxidative radicals (OH˙ and O2˙−) during the photoexcitation of the sample, which can attack the naphthalene molecule leading to the aromatic ring opening and loss of luminescence.48,49 Although there is a significant reduction of both emission peaks under long time UV exposure, no apparent shift in the peak positions was observed. The low and high energy emission peaks decrease relatively at the same rate, see Fig. 7e, which is in contradiction with our earlier findings.26 It was suggested that the high energy peak originates only from the aromatic molecule, whereas the peak in lower energy arises from the interaction between the metal and the carboxylate group of the organic molecule. Our current results show that the peak at low emission energy has a similar position for all samples, while the one at higher energy varies within the samples, especially for the Y-NDC system. Furthermore, although the emission intensity decreases with a similar rate, the final photobleaching effect is slightly higher for the Hf-NDC sample in the order of Hf-NDC > Y-NDC > Zr-NDC.
The photostability of photoactive materials can sometimes be recovered by tempering.50 However, the luminesce of the Zr-, Hf-, and Y-NDC samples were not improved after tempering the films at 300 °C for 1 h, and also not improved by keeping the samples in the dark for 24 h (data not shown). Furthermore, it should be noted that FTIR analysis of the films after 3 hours of irradiation by 365 nm UV light did not show any significant changes in the reflection spectra as compared to the as-deposited samples, proving no chemical photodegradation of the samples (Fig. S8†).
Finally, all our MLD processes yielded relatively smooth and amorphous thin films (Fig. S9 and S10†). Although the Atomic Force Microscopy (AFM) images of the samples based on the chlorine precursors show very small and regular islands on the otherwise almost flat surface, these islands do not produce any diffraction pattern when measured by XRD. The root-mean-square (RMS) surface roughness in islands-free regions estimated by AFM (as described in ESI†) was close to 0.4 nm for ca. 100 nm thick films, which is in good agreement with XRR data that gave roughness value of 0.5 nm for Y-NDC (50 nm), Ti-NDC (45 nm), Zr-NDC (120 nm) and Hf-NDC (100 nm). However, the line profile across the islands on the surface from the AFM images show height variations of around 6 nm for Hf-NDC, 8 nm for Zr-NDC and 12 nm for Ti-NDC. Additionally, all films exhibit rather good stability when exposed to DI water, except the Y-NDC sample, which completely dissolved after 24 hours of water treatment (Table S1†). The wettability of the surfaces was thus investigated by contact angle measurements, see Fig. S12,† showing that Y-NDC has a more hydrophilic nature than the other deposited systems (Ti-NDC = 80°, Zr-NDC/Hf-NDC = 78° and Y-NDC = 68°). Furthermore, sample-tip adhesion measured with AFM was significantly higher for Y-NDC (95.1 ± 4.1 nN) than for Hf-NDC (2.3 ± 0.1 nN) (Fig. S11†). Such high adhesion measured in air at ambient humidity conditions results from capillary condensation and formation of water bridges between an AFM tip and the sample, and points to more hydrophilic surface properties of Y-NDC films in comparison with Hf-NDC.51
Conclusions
In this work, we investigated a group of hybrid metal-naphthalene thin film materials made by the MLD technique. We selected d0-metals, Ti4+, Zr4+, Hf4+ and Y3+ as the metal centres and 2,6-naphthalenedicarboxylic acid as the organic unit. All deposited systems show self-saturating growth, possess apparent ALD windows and variations in growth rates depending on the metal constituents in the order of Ti < Y ≪ Hf < Zr. The hybrid nature of the films and the fact that the organic precursor reacts with metal through both the OH and the CO groups from the carboxylate unit was proved by XPS and FTIR analysis. Moreover, the Ti-NDC complex was formed by bidentate coordination, whereas Zr-, Hf-, and Y-NDC complexes have a bidentate or bridging type of coordination. The films are amorphous, smooth and hydrophobic, except for the Y-NDC system, which exhibits a more hydrophilic nature. Incorporation of different transition metal clusters in the structure with the same organic linker results in changes in absorption and luminescence properties. Furthermore, we can tune the light absorption towards the visible part of the spectrum by using metals with a high charge density. The films containing Zr, Hf and Y were found to show intense blue photoluminescence when exposed to UV and the emission intensity and energy depends on the metal component. In particular, the presence of Y clusters enhances the luminescence intensity, whereas the Zr and Hf components shift the emission towards the higher wavelengths. The emission intensity decreases under long time UV exposure, suggesting photodegradation of the organic ligand in the hybrid structure. Furthermore, the luminescence from Ti-NDC system was completely quenched by Ti ions in the structure. All data collected in this study indicate that our uniform hybrid metal-naphthalene thin film materials with interesting optical properties could be beneficial for many potential applications such as antimicrobial coatings in photodynamic therapy or optical sensors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No. 765378. The authors would like to also acknowledge the Research Council of Norway through the TRALALALA project No. 244087 within the ENERGIX program.References
- P. Gomez-Romero, Adv. Mater., 2001, 13, 163–174 CrossRef CAS.
- S. Parola, B. Julián-López, L. D. Carlos and C. Sanchez, Adv. Funct. Mater., 2016, 26, 6506–6544 CrossRef CAS.
- P. Escribano, B. Julián-López, J. Planelles-Aragó, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23–40 RSC.
- C. R. Kagan, D. B. Mitzi and C. D. Dimitrakopoulos, Science, 1999, 286, 945–947 CrossRef CAS.
- D. B. Mitzi, K. Chondroudis and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 29–45 CAS.
- A. Li, H. Shen, H. Ren, C. Wang, D. Wu, R. Martin and D. Qiu, J. Mater. Chem. B, 2015, 3, 1379–1390 RSC.
- L. Momtazi, H. H. Sønsteby, D. A. Dartt, J. R. Eidet and O. Nilsen, RSC Adv., 2017, 7, 20900–20907 RSC.
- W. A. Zoubi, J. H. Min and Y. G. Ko, Sci. Rep., 2017, 7, 7063 CrossRef PubMed.
- E. Bescher and J. D. Mackenzie, Mater. Sci. Eng., C, 1998, 6, 145–154 CrossRef.
- S. Wang, Y. Kang, L. Wang, H. Zhang, Y. Wang and Y. Wang, Sens. Actuators, B, 2013, 182, 467–481 CrossRef CAS.
- M. Leskelä and M. Ritala, Thin Solid Films, 2002, 409, 138–146 CrossRef.
- S. M. George, B. Yoon and A. A. Dameron, Acc. Chem. Res., 2009, 42, 498–508 CrossRef CAS PubMed.
- Q. Peng, B. Gong, R. M. VanGundy and G. N. Parsons, Chem. Mater., 2009, 21, 820–830 CrossRef CAS.
- B. H. Lee, B. Yoon, I. Abdulagatov, R. A. Hall and S. M. George, Adv. Funct. Mater., 2013, 23, 532–546 CrossRef CAS.
- M. Karppinen and P. Sundberg, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- D. J. Hagen, L. Mai, A. Devi, J. Sainio and M. Karppinen, Dalton Trans., 2018, 47, 1579–15800 RSC.
- K. B. Klepper, O. Nilsen and H. Fjellvåg, Dalton Trans., 2010, 39, 11628–11635 RSC.
- K. B. Lausund and O. Nilsen, Nat. Commun., 2016, 7, 13578 CrossRef CAS PubMed.
- K. B. Lausund, V. Petrovic and O. Nilsen, Dalton Trans., 2017, 46, 16983–16992 RSC.
- A. Tanskanen and M. Karppinen, Sci. Rep., 2018, 8, 8976 CrossRef CAS PubMed.
- A. Khayyami and M. Karppinen, Chem. Mater., 2018, 30, 5904–5911 CrossRef CAS PubMed.
- A. Khayyami, A. Philip and M. Karppinen, Angew. Chem., Int. Ed., 2019, 58, 13400–13404 CrossRef CAS PubMed.
- Z. Giedraityte, P. Sundberg and M. Karppinen, J. Mater. Chem. C, 2015, 3, 12316–12321 RSC.
- Z. Giedraityte, L.-S. Johansson and M. Karppinen, RSC Adv., 2016, 6, 103412–103417 RSC.
- Z. Giedraityte, J. Sainio, D. Hagen and M. Karppinen, J. Phys. Chem. C, 2017, 121, 17538–17545 CrossRef CAS.
- K. B. Lausund, M. S. Olsen, P.-A. Hansen, H. Valen and O. Nilsen, J. Mater. Chem. A, 2020, 8, 2539–2548 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed.
- M. Safdar, A. Ghazy, M. Tuomisto, M. Lastusaari and M. Karppinen, J. Mater. Sci., 2021, 56, 12634–12642 CrossRef CAS.
- J. Roberts, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2000, 18, 1785–1791 CrossRef.
- J.-P. Niemelä, G. Marin and M. Karppinen, Semicond. Sci. Technol., 2017, 32, 093005 CrossRef.
- M. Ritala and M. Leskelä, Appl. Surf. Sci., 1994, 75, 333–340 CrossRef CAS.
- K. Kukli, M. Ritala, T. Sajavaara, J. Keinonen and M. Leskelä, Thin Solid Films, 2002, 416, 72–79 CrossRef CAS.
- T. Suthan, N. P. Rajesh, P. V. Dhanaraj and C. K. Mahadevan, Spectrochim. Acta, Part A, 2010, 75, 69–73 CrossRef CAS PubMed.
- N. Mukwevho, R. Gusain, E. Fosso-Kankeu, N. Kumar, F. Waanders and S. S. Ray, J. Ind. Eng. Chem., 2020, 81, 393–404 CrossRef CAS.
- B. C. Smith, Spectroscopy, 2018, 33(1), 14–20 CAS.
- F. Verpoort, T. Haemers, P. Roose and J.-P. Maes, Appl. Spectrosc., 1999, 53, 1528–1534 CrossRef CAS.
- A. C. Bronneberg, C. Höhn and R. van de Krol, J. Phys. Chem. C, 2017, 121, 5531–5538 CrossRef CAS.
- L. Sygellou, V. Gianneta, N. Xanthopoulos, D. Skarlatos, S. Georga, C. Krontiras, S. Ladas and S. Kennou, Surf. Sci. Spectra, 2011, 18, 58–67 CrossRef CAS.
- Q. Tao, G. Jursich and C. Takoudis, Appl. Phys. Lett., 2010, 96, 192105 CrossRef.
- I.-S. Park, Y. C. Jung, S. Seong, J. Ahn, J. Kang, W. Noh and C. L. Matras, J. Mater. Chem. C, 2014, 2, 9240–9247 RSC.
- A. Schöll, Y. Zou, M. Jung, Th. Schmidt, R. Fink and E. Umbach, J. Chem. Phys., 2004, 121, 10260–10267 CrossRef.
- M. Gutiérrez, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2016, 18, 5112–5120 RSC.
- L. H. Ahrens, Geochim. Cosmochim. Acta, 1952, 2, 155–159 CrossRef CAS.
- J. Fujisawa, R. Muroga and M. Hanaya, Phys. Chem. Chem. Phys., 2015, 17, 29867 RSC.
- W. Zhang, H. Huang, D. Liu, Q. Yang, Y. Xiao, Q. Ma and C. Zhong, Microporous Mesoporous Mater., 2013, 171, 118–124 CrossRef CAS.
- C. Spies and R. Gehrke, J. Phys. Chem. A, 2002, 106, 5348–5352 CrossRef CAS.
- J. Zhang, S. Fan, B. Lu, Q. Cai, J. Zhao and S. Zhang, R. Soc. Open Sci., 2018, 6, 181779 CrossRef PubMed.
- J. P. Hoogenboom, M. H. P. van Dijk, N. F. van Hulst and M. F. García-Parajó, Phys. Rev. Lett., 2005, 95, 097401 CrossRef PubMed.
- M. N. Getz, P.-A. Hansen, H. Fjellvåg and O. Nilsen, RSC Adv., 2017, 7, 8051–8059 RSC.
- D. L. Sedin and K. L. Rowlen, Anal. Chem., 2000, 72, 2183–2189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02201f |
| This journal is © The Royal Society of Chemistry 2021 |