
Interacting resonances and antiresonances in conjugated hydrocarbons: exceptional points and bound states in the continuum†
Nikolay
Shubin
 a,
Aleksei
Emelianov
ab,
Yuriy
Uspenskii
a and
Alexander
Gorbatsevich
*a
a,
Aleksei
Emelianov
ab,
Yuriy
Uspenskii
a and
Alexander
Gorbatsevich
*a
aP.N. Lebedev Physical Institute of the Russian Academy of Sciences, 119991, Moscow, Russia. E-mail: gorbatsevichaa@lebedev.ru
bNational Research University of Electronic Technology, Zelenograd, 124498, Moscow, Russia
First published on 24th June 2021
Abstract
Quantum interference dramatically modulates electron transport that provides exciting prospects for molecular electronics. We develop a holistic picture of quantum interference phenomena in molecular conductors based on conjugated hydrocarbons taking into account the interaction of resonances and antiresonances (AR). This interaction can result in the coalescence of resonances and ARs accompanied by a significant quantum transparency change. As such a change results from a small variation of the system parameters, it is essential for reducing power consumption in electronics. We establish that the coalescence of ARs is intimately connected with the exceptional point of an underlying non-Hermitian Hamiltonian. The coalescence of ARs cannot be explained considering only the LUMO and HOMO without orbitals beyond them. Cyclobutadiene is discussed as an example. We show that the interaction of resonances and ARs can also result in the formation of a bound state in the continuum (BIC). Our formalism accounting for separate descriptions of resonances and ARs is especially suitable for describing BICs, which can be considered as either a resonance or an AR with zero width. In particular, we show that benzene in the para-configuration possesses BICs, which can be revealed as narrow Fano resonances (FRs) in the transmission spectrum by perturbing the molecule symmetry. Any BIC can be turned into an FR by a proper change of the system parameters, but the reverse is not true. We demonstrate that BICs are related to such chemical concepts as non-bonding orbitals, radicals, and diradicals. Our analytical results within the Hückel formalism are closely reproduced by ab initio simulations. Therefore, experimentally revealing these phenomena looks quite probable.
1 Introduction
Many applications of molecular conductors1,2 exploit the interference nature of quantum transport, which clearly manifests itself in the existence of resonances and antiresonances (ARs), i.e., peaks and dips in the transmission spectrum. The former are due to constructive quantum interference, while the latter appear as a result of destructive quantum interference (DQI)3–20 and are considered as convincing evidence of the wave nature of quantum transport in molecular conductors. Rapid progress in experimental techniques during the past years made it possible to observe DQI in various structures,3–7 providing results of great interest for molecular electronics,21,22 thermoelectrics,23,24 and sensor25 applications. These studies have revealed the physical mechanisms leading to formation of isolated ARs8–10 and have given a number of elegant rules describing DQI in either the atomic orbital (AO)11–13 or molecular orbital (MO)14,15,26 basis.Nevertheless, the intensive study of quantum interference in molecular conductors in the past years with deep insight into the relation between the molecular topology and electronic structure has not yet resulted even in conceptual understanding of how molecular switches could compete with modern semiconductor transistors. Molecular electronics offers ultimately small size and perfect reproducibility of individual elements. But the main problem of modern silicon electronics is the power consumption, which requires a decrease in the operating and switching voltages.27 Transport in small molecules is of resonant origin. However, as was stated in ref. 28, switching by a simple shift of resonances cannot provide small enough operating voltages. AR alone is not the solution for the same reason as well, because it possesses small contrast (on/off current ratio).29,30 Hence some mechanism of resonance suppression of an interference nature is required. A possible solution providing an extremely small switching voltage was proposed in ref. 31. It utilizes DQI combined with the coalescence of resonances near the exceptional point (EP)32–36 of an open quantum system formed by a molecule and electrodes.
In this paper, we present a unified picture of interference phenomena in molecular transport with a special emphasis on DQI and the interaction between the resonances and ARs as well as the interaction of ARs with each other. Why is such an interaction important? Because of two main reasons. At first, the interaction of resonances can result in coalescence of resonances and ARs accompanied by an essential change of quantum transparency resulting from a small change of system parameters.31 Therefore, studying interacting resonances and ARs in realistic molecular structures based on ab initio simulations is potentially of primary importance for the whole field of molecular electronics.
Secondly, the interaction of resonances and ARs results in new fundamental effects in molecules, an example of which is bound states in the continuum (BICs) – fully localized states within the energy range of continuous spectrum states.37 For a long time after BICs were introduced in quantum mechanics38 they have been considered as a pure mathematical trick. It was so until Friedrich and Wintgen39 recognized a BIC as a result of the interference interaction of (at least) two resonances. Soon after BICs became very popular objects of investigation in different fields.37,40–42 A BIC arises from destructive interference and in this sense is close to the Fano resonance (FR),43 but it is a rather more complicated physical phenomenon than FRs, which have been already studied in molecular transport.29,44 Here we expand the study of BICs to single-molecule quantum conductors.
In the present paper, we consider the interaction of ARs as a part of a general problem45 related to the unified description of resonances, ARs, asymmetric FRs,46 and BICs in molecular conductors. In Section 2, we describe our analytical and numerical methods for the analysis of resonant phenomena in quantum transport. In Section 3, we develop a general approach to a complicated picture of mutual influences, transformations, and coalescence of resonances, ARs, FRs, and BICs in single-molecule conductors and establish a direct relationship with the fundamental properties of open quantum systems near their EPs. In Section 4, we provide illustrative examples of the AR coalescence phenomenon in cyclobutadiene and BIC formation in benzene with electrodes in the para-configuration. In the latter case, the asymmetric perturbation can reveal the BIC as a narrow FR. In Section 5, we show that the obtained analytical results are in good agreement with our thorough ab initio simulations of electron transport in cyclobutadiene and benzene molecules. More examples could be found in the ESI.†
2 Methods
2.1 Unified analytical description of resonances, antiresonances, and BICs
We perform analytical calculations within the Hückel (tight-binding) formalism. The transport properties are studied via the non-equilibrium Green's function (NEGF) technique and utilizing the Landauer notation for the transmission coefficient.47 However, we use the transmission coefficient in a more convenient formulation for the analysis of interference phenomena45 (see the ESI,† Section S1.1, for details):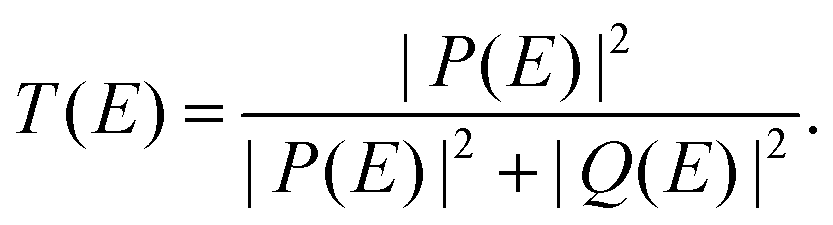 | (1) |
| P(E) = 2 det(ÎE − Ĥ0)u†r(ÎE − Ĥ0)−1us, | (2) |
| Q(E) = det(ÎE − Ĥaux). | (3) |
![[capital Gamma, Greek, circumflex]](https://www.rsc.org/images/entities/char_e132.gif) s − ir is the auxiliary Hamiltonian of the system.45 Coherent coupling to the electrodes16 is defined by coupling matrices (non-Hermitian parts of the electrode self-energies)48 s/r = us/ru†s/r with
s − ir is the auxiliary Hamiltonian of the system.45 Coherent coupling to the electrodes16 is defined by coupling matrices (non-Hermitian parts of the electrode self-energies)48 s/r = us/ru†s/r with| us = (γs1,γs2,…,γsN)T, ur = (γr1,γr2,…,γrN)T. | (4) |
![[Doublestruck R]](https://www.rsc.org/images/entities/char_e175.gif) governs the hopping between the i-th MO of the molecule and the corresponding electrode. In general, quantities γs/ri are energy-dependent, but here we use the wide-band limit (WBL) approximation49 and treat them as constants. Calculations beyond this assumption do not change the qualitative results obtained below. We assume that the couplings to the electrodes are large enough to prevent charge accumulation. In our analytic calculations, we also assume that the electrode Fermi energy coincides with the energy of the 2pz-orbitals of sp2 hybridized carbon atoms,11 which is set as the energy origin: EF = 0.
governs the hopping between the i-th MO of the molecule and the corresponding electrode. In general, quantities γs/ri are energy-dependent, but here we use the wide-band limit (WBL) approximation49 and treat them as constants. Calculations beyond this assumption do not change the qualitative results obtained below. We assume that the couplings to the electrodes are large enough to prevent charge accumulation. In our analytic calculations, we also assume that the electrode Fermi energy coincides with the energy of the 2pz-orbitals of sp2 hybridized carbon atoms,11 which is set as the energy origin: EF = 0.
Expression (1) is exact and provides a natural way to analyze transmission interference features: unity transmission (resonance) takes place only at energies equal to real roots of Q(E) and zero transmission (antiresonance) takes place only at real roots of P(E). If P(E) ≠ 0 at some resonance energy E0, where Q(E) turns to zero, then P(E0) defines the resonance width. Similarly, Q(E0) defines the width of the antiresonance if P(E0) = 0 and Q(E0) ≠ 0. A special situation takes place if the roots of P(E) and Q(E) coincide.45 This case can be referred to as a resonance or an antiresonance of zero width, which corresponds to the formation of a BIC.45
2.2 Numerical simulations
The DFTB+50 and Synopsys QuantumATK R-2020.0951 packages were used to calculate the optimal geometries, the density of states (DOS), MO wavefunctions, and transmission spectra. Details are presented in the ESI,† Section S1.2.3 Results and discussion
3.1 Crossing, avoided crossing and coalescence of antiresonances near the Fermi level
To describe a complicated picture of AR interactions, we consider a conjugated hydrocarbon molecule with N carbon atoms. The energies of ARs are determined by the zeroes of P(E). Following the definition of P(E) in eqn (2), one can get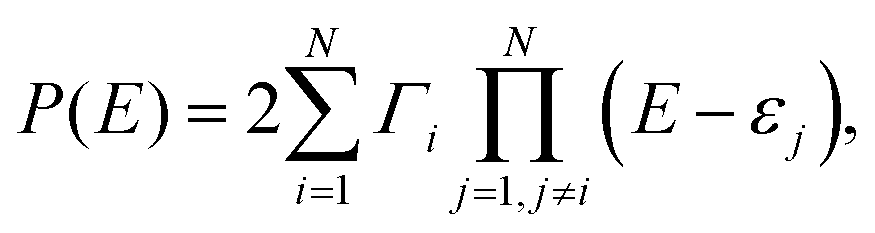 | (5) |
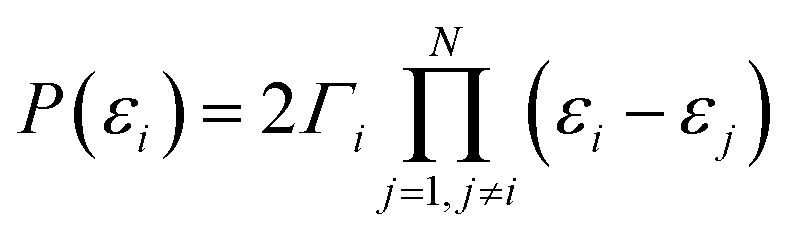 | (6) |
Moreover, if two MOs are degenerate (εi = εi+1), then P(εi) = 0 regardless of the sign of Γi or Γi+1, which sufficiently extends the conclusion of ref. 53. We recall that a real root of P(E) manifests itself as an AR if it does not coincide with a real root of Q(E). Otherwise, there is a BIC, and in this case the transmission value is defined by the multiplicity of the P(E) and Q(E) roots.45
In the vicinity of the Fermi energy EF = 0, contributions from the HOMO and LUMO states play the key role, and one can get the following approximation for P(E) from eqn (5):
| P(E) ≈ C1[ΓH(E − εL) + ΓL(E − εH) + (E − εH)(E − εL)C2], | (7) |
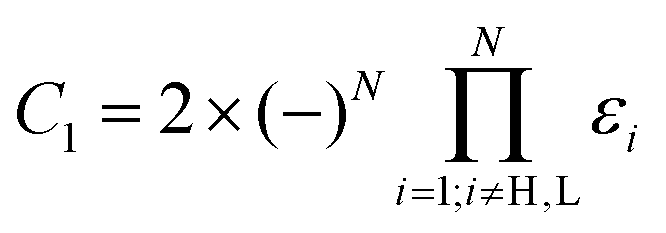 and
and 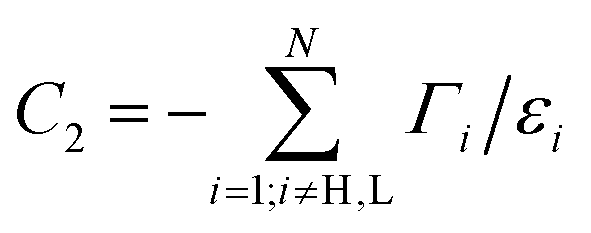 are constants and εH/L is the energy of the HOMO/LUMO states. This approximation takes into account distant MOs beyond the HOMO and LUMO by an energy-independent constant C2. Equating P(E) from (7) to zero, one gets an equation for determining DQI energies. A similar equation can be obtained from the well-known DQI condition for the Green's function u†rĜr(E)us = 0 [it arises directly from the standard definition of the transmission coefficient, see eqn (S1) in the ESI†], if one considers the eigenstate decomposition of the zero-order Green's function at the HOMO and LUMO energies and also takes distant MOs into account:
are constants and εH/L is the energy of the HOMO/LUMO states. This approximation takes into account distant MOs beyond the HOMO and LUMO by an energy-independent constant C2. Equating P(E) from (7) to zero, one gets an equation for determining DQI energies. A similar equation can be obtained from the well-known DQI condition for the Green's function u†rĜr(E)us = 0 [it arises directly from the standard definition of the transmission coefficient, see eqn (S1) in the ESI†], if one considers the eigenstate decomposition of the zero-order Green's function at the HOMO and LUMO energies and also takes distant MOs into account: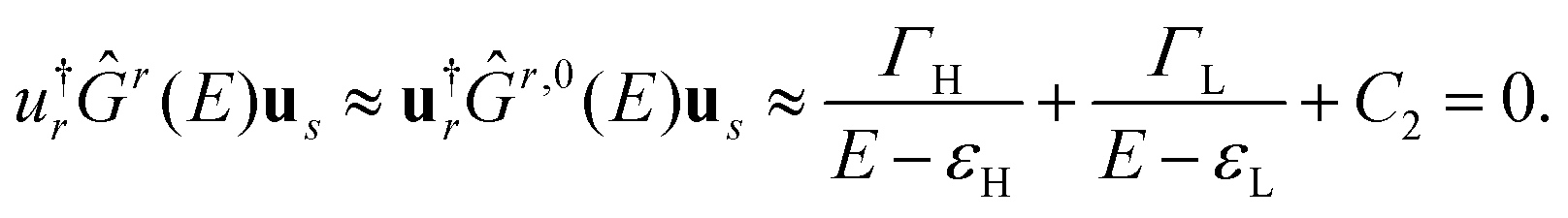 | (8) |
From eqn (7) we can see that P(E) is zero at
 | (9) |
| D = [C2(εL − εH) + ΓH − ΓL]2 + 4ΓHΓL. | (10) |
The second-order transmission supernode formed at the threshold D = 0 can be referred to as hard zero. For small positive D, the supernode splits into two ARs, and the gap between them behaves non-analytically as  . Negative D prevents P(E) in eqn (7) from turning to zero at real energies, which means the absence of zero transmission dips in this case.
. Negative D prevents P(E) in eqn (7) from turning to zero at real energies, which means the absence of zero transmission dips in this case.
If the hydrocarbon under consideration is alternant with an even number of carbon atoms N, and the leads are attached to atoms of different classes (starred and unstarred), then, using the Coulson–Rushbrooke theorem,55 we can conclude that γsL = γsH = γs and γrL = −γrH = −γr and the conditions for D = 0 simplify to
| εL = εH or εL − εH = −4γsγr/C2 | (11) |
It is instructive to describe different types of AR behavior graphically. Condition P(E) = 0 defines a hypersurface in the energy parameter space, and hence the behavior of ARs with a change of parameters can be described as a cross-section of this hypersurface. One can distinguish three types of such cross-sections, which correspond to qualitatively different AR position evolution patterns. Fig. 1a–c illustrate these cross-section types: (1) avoided crossing of ARs, when two zero-valued ARs always stay separate (Fig. 1a); (2) AR crossing, when zero-valued ARs intersect forming a supernode and energy splitting between them growing linearly with the parameter change (Fig. 1b); and (3) AR coalescence, when two zero-valued ARs coalesce into a supernode and then transform to a nonzero transmission dip (Fig. 1c). All three scenarios are illustrated in the next section for a generalized butadiene molecule model.
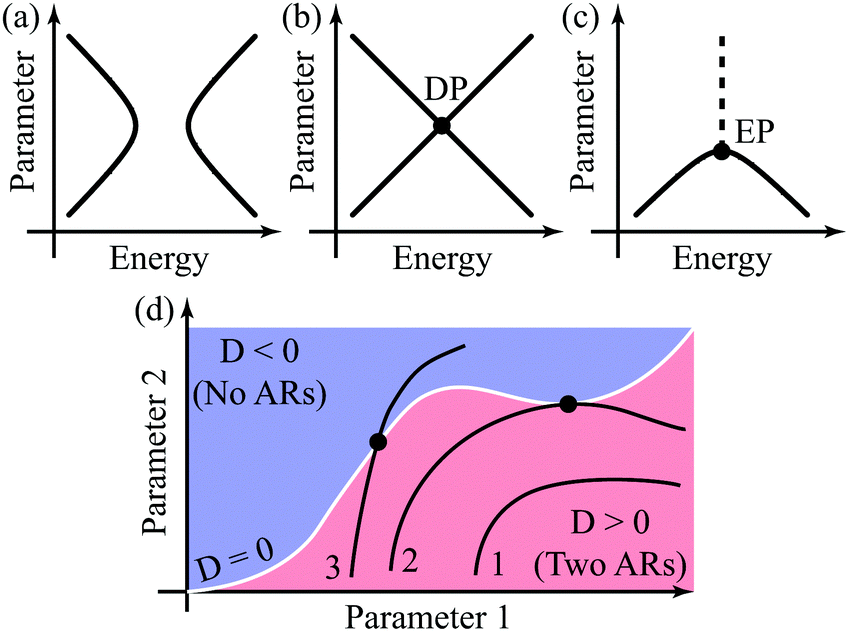 | ||
| Fig. 1 Schemes of different AR evolution types: (a) avoided crossing, (b) crossing, and (c) coalescence. The solid line represents zero-valued ARs, dashed line – non-zero transmission dips, and transmission supernode formation is marked by bold dots. (d) Different trajectories (black lines) in the parameter space and their position relative to the threshold line D = 0 (white line). Trajectory 1 corresponds to an AR avoided crossing, trajectory 2 – to AR crossing, and trajectory 3 – to AR coalescence. | ||
3.2 Exceptional points and coalescence of antiresonances
The AR coalescence illustrated by Fig. 1c resembles the behavior of the eigenvalues of some non-Hermitian operator near its EP.32–36 At the EP, two real eigenvalues of a non-Hermitian operator turn into two complex conjugate ones. We consider a molecular conductor comprised of a molecule and electrodes as an open quantum system (OQS). Such systems are characterized by non-Hermitian operators, which essentially complicates their description.56In contrast to the function Q(E),45 the function P(E) cannot be straightforwardly related to a characteristic polynomial of some Hamiltonian (regular eigenvalue problem). Depending on the positioning relative to the border surface D = 0 [see eqn (10)], the evolution trajectories can be classified into three types (1–3 in Fig. 1d), which results in different classes of Hamiltonians. Trajectory 1 can be related to a Hermitian Hamiltonian with distinct eigenvalues. Trajectory 2 can also be related to a Hermitian Hamiltonian having a diabolic point (DP) when the trajectory touches a surface D = 0. At the DP the operator has two orthogonal eigenvectors with degenerate eigenvalues, and hence it is diagonalizable. The energy splitting between its eigenvalues (AR positions) scales linearly with the perturbation. Trajectory 3 can be related only to a non-Hermitian Hamiltonian. In this case, the transmission supernodes (intersection with the D = 0 surface) correspond to the EPs of this Hamiltonian. At the EP, the operator has a form of a Jordan block with a single eigenvector, and hence it is non-diagonalizable.32,35,36 Non-analytical behavior of the energy splitting between ARs  is a characteristic of non-Hermitian Hamiltonian eigenvalues near the EP.57
is a characteristic of non-Hermitian Hamiltonian eigenvalues near the EP.57
A typical example of a non-Hermitian Hamiltonian possessing an EP is a ![[scr P, script letter P]](https://www.rsc.org/images/entities/char_e52f.gif)
![[scr T, script letter T]](https://www.rsc.org/images/entities/char_e533.gif) -symmetric Hamiltonian. Here stands for space inversion and – for time-reversal operations. Despite its non-Hermitian nature, it can have real eigenvalues, which may spontaneously coalesce into a complex conjugate pair with the variation of the system's parameters. This phenomenon is known as -symmetry breaking (PTSB)58,59 and takes place at the EP. Thus, the AR coalescence phenomenon can be associated with the PTSB of some underlying non-Hermitian -symmetric Hamiltonian. Indeed, one can write P(E) function (7) as P(E) ≈ 2C1C2
-symmetric Hamiltonian. Here stands for space inversion and – for time-reversal operations. Despite its non-Hermitian nature, it can have real eigenvalues, which may spontaneously coalesce into a complex conjugate pair with the variation of the system's parameters. This phenomenon is known as -symmetry breaking (PTSB)58,59 and takes place at the EP. Thus, the AR coalescence phenomenon can be associated with the PTSB of some underlying non-Hermitian -symmetric Hamiltonian. Indeed, one can write P(E) function (7) as P(E) ≈ 2C1C2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) det(ÎE − Ĥ) with
det(ÎE − Ĥ) with
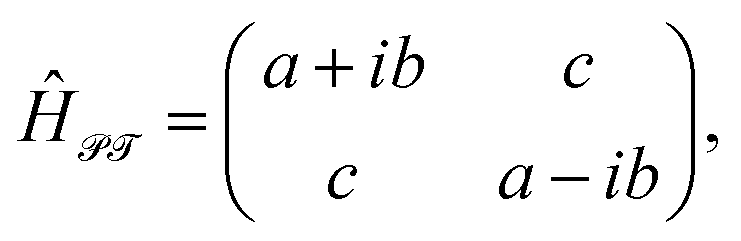 | (12) |
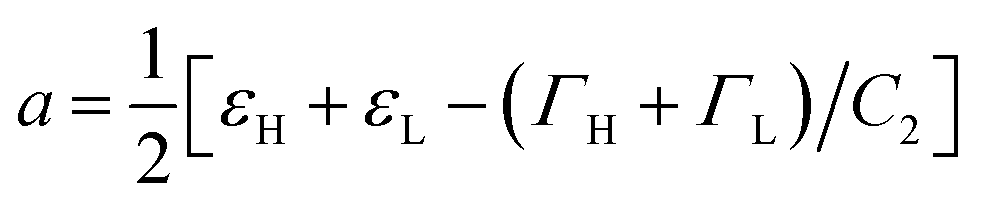 ,
,  , and
, and 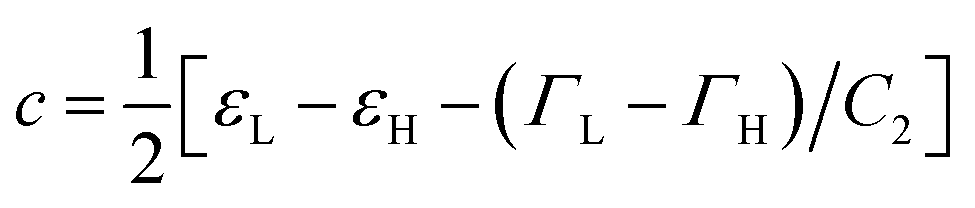 . Trajectory 3 can be realized only if ΓH and ΓL are of different sign. Thus, b in eqn (12) is real and operator Ĥ can be regarded as the Hamiltonian of some -symmetric model.‡ From eqn (12) one can conclude that the AR coalescence phenomenon can be always related to -symmetry breaking in some underlying -symmetric model.
. Trajectory 3 can be realized only if ΓH and ΓL are of different sign. Thus, b in eqn (12) is real and operator Ĥ can be regarded as the Hamiltonian of some -symmetric model.‡ From eqn (12) one can conclude that the AR coalescence phenomenon can be always related to -symmetry breaking in some underlying -symmetric model.
In real systems smooth tuning of the parameters to satisfy the condition D = 0 can be difficult. Thus, the very point of AR coalescence and hence the very EP may not be observable. Despite these difficulties, two different regimes related to the EP and manifesting its existence are distinguishable: one with two zero-valued ARs (D > 0) and another with a single non-zero transmission dip (D < 0). That is exactly what is observed in our ab initio simulations of cyclobutadiene (see Section 5), where the pristine molecule provides a regime with two ARs and the molecule with two –NH2 side groups provides a regime without DQI.
3.3 Bound states in the continuum in molecular conductors
Interference may lead to the formation of BICs37 – MOs which are effectively decoupled from the scattering channels (electrodes) but with an energy lying in the continuous spectrum range. While all electronic states of an isolated molecule are bound, most of them turn into delocalized ones when leads with a continuous electron spectrum are attached. This is not the case for BICs having no coupling with the lead states. Such MOs remain perfectly isolated from the electrodes (bound to the molecule) and do not contribute to transport through the system.BICs are being actively studied in different physical systems: acoustic cavities,41 various optical structures,42 quantum billiards,40etc. To the best of our knowledge, BICs in single-molecule conductors have not been described yet. However, taking BICs into account is crucial for understanding the resonant transport properties (either destructive or constructive interference) in molecular conductors. For instance, analysis of the numerator of the transmission coefficient [function P(E)] alone may give a spurious AR at an energy where a BIC is really formed.45
According to the formation mechanism, BICs can be divided into three main groups: symmetry-protected BICs, Fabry–Perot BICs, and Friedrich–Wintgen BICs.37 In a symmetry-protected BIC, the matrix element between the localized state and the continuum turns to zero due to the different symmetry of the states. A Fabry–Perot BIC is built up between two perfectly reflecting scatterers if the distance between them is appropriately tuned. The most intriguing and non-trivial is the Friedrich–Wintgen mechanism of BIC formation due to the destructive interference between two (nearly) degenerate localized states. Regardless of the particular formation mechanism, a BIC manifests as a real pole of the scattering matrix, which must coincide with its root to preserve finite values of the scattering matrix at real energies.60 Thus, from the point of view of our formalism, any BIC can be considered as a common real root of the P(E) and Q(E) functions.45
Being decoupled from the electrodes, BICs do not manifest themselves in transport. However, a slight perturbation of the system disturbing, for instance, the symmetry of the quantum system or its couplings to the leads can introduce a coupling between the BIC and the continuum in the leads. Typically, in this case, an asymmetric Fano resonance appears in the transmission spectrum.61,62 This fact can be easily understood if one appeals to the formalism of the P(E) and Q(E) functions. Indeed, suppose that there is a BIC at energy E = E0 and perturbation Δ shifts the roots of the P(E) and Q(E) functions with some ratio coefficients kP and kQ, respectively (kP ≠ kQ). Thus, one can estimate the transmission spectrum profile given by general eqn (1) in the vicinity of E0 and for small Δ as a standard FR profile43 with resonant energy Er = E0 + Δ(kP + kQ)/2, asymmetry parameter q = ±1 and width Γ = ±Δ(kQ − kP)/2. In fact, in ref. 63 this phenomenon was observed in the ab initio simulations of benzene and polyyne linear chains with different left and right electrodes. However, the authors did not study BICs in the context of the observed effect.
It must be noted that any BIC can be revealed as an FR, but the inverse is not true in general. Formation of a BIC in the standard model system with a simple Fano resonance (propagating channel with a continuous spectrum coupled to a single localized state)43 implies disruptive transformation to turn the matrix element between the continuum and localized state to zero. However, generally, as was shown by Friedrich and Wintgen in ref. 39, a more complicated picture without bond breaking takes place. BIC formation requires more degrees of freedom compared to a simple FR – at least two localized states coupled to the same continuum. In this configuration the Friedrich–Wintgen mechanism may allow for a BIC preserving non-zero coupling of both localized states with the continuum. Therefore, in this context, we admit that our approach provides a natural unified description of resonance phenomena, including any mechanism of BIC formation.
In molecular conductors any MO (the i-th one for definiteness) can be turned into a BIC if both coupling vectors us and ur(4) in the MO basis have zero entries in the i-th position. In the AO basis, the conditions for the i-th MO becoming a BIC imply a requirement of orthogonality: u†sci = u†rci = 0, where ci ∈ N is the wavefunction of the i-th MO in the AO basis. Thus, one can construct a BIC from any MO with appropriate adjustment of the electrode couplings. Condition u†s,rci = 0 for an arbitrary MO requires precise tuning of the tunneling matrix elements between the electrodes and each atom of the molecule, which is an arduous task in reality. However, according to the Coulson–Rushbrooke theorem,55 there is a special singly occupied MO – a non-bonding orbital (NBO) in alternant hydrocarbons with an odd number of carbon atoms. The NBO has the energy of sp2 hybridized carbon 2pz-orbitals, and it vanishes at inactive positions (unstarred atoms).64 Therefore, one can attach electrodes to these atoms and turn the NBO into a BIC. In other words, we conclude that, if a molecule is a radical with an NBO, then it possesses a BIC if coupled to electrodes in a particular configuration.
3.4 Close relation between BICs and the existence of diradicals
The close relation between DQI and the diradical existence was established in ref. 8. In a specific configuration, this correspondence can be extended to take BICs into account. Consider a molecular conductor with identical left and right leads attached to the same atom (Fig. 2). It is not obligatory for the electrodes to be coupled directly to the same atom, which is extremely tough in practice. Instead, one can use linear polyene spacers to extend each metal electrode. In this case, it can be regarded as a cross-conjugated molecule with an arbitrary side-group, which is known to possess DQI.65,66 For this configuration, we have ur = us = (γ,0,…,0)T in the AO basis (if the sites are numbered beginning from the one which is coupled to both leads). From the general formulas (2) and (3), we can derive the following:| P(E) = 2γ2det(ÎE − Ĥ0′), Q(E) = det(ÎE − Ĥ0). | (13) |
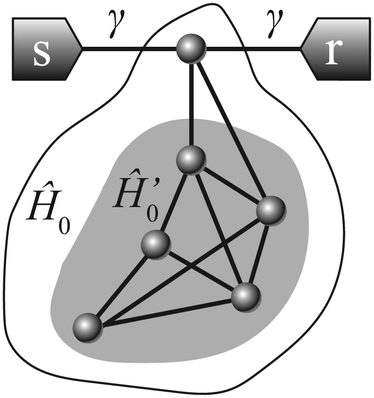 | ||
| Fig. 2 Schematic view of an example structure coupled to electrodes via a single atom. The eigenvalues of the bare Hamiltonian of the whole molecule Ĥ0 define perfect transmission resonances. The eigenvalues of the bare Hamiltonian of the reduced molecule Ĥ0′ define antiresonances. | ||
For brevity, we will refer to the initial molecule with one atom removed as a reduced molecule. Thus, unity-valued transmission resonances are located exactly at the MO energies of the whole molecule, and zero-valued ARs are exactly at the MO energies of the reduced molecule. This conclusion naturally generalizes the Fano–Anderson model (single pendant atom), and, in particular, the results of ref. 67–69. In the ESI† (Section S2) we consider a more general case of proportionate coupling: us = λur for any λ ∈ ![[Doublestruck C]](https://www.rsc.org/images/entities/char_e166.gif) .
.
If the molecule under consideration is a closed-shell alternant hydrocarbon with an even number of carbon atoms, it has no NBOs. The reduced molecule remains alternant but with an odd number of carbon atoms and hence appears to be radical with a single NBO. Therefore, DQI at zero energy is expected in this case due to P(0) = 0 and Q(0) ≠ 0. On the other hand, if the initial molecule is an odd atom alternant hydrocarbon, i.e., it is radical and has an NBO, then its reduced version with an even number of carbon atoms can be either a closed-shell molecule with no NBOs or a diradical with a doubly degenerate NBO. In the former case, there is a resonance at E = 0 (P(0) ≠ 0 and Q(0) = 0). In the latter case, both functions P(E) and Q(E) turn to zero at E = 0, which indicates the formation of a BIC.45 Function P(E) has a higher-order zero at E = 0 than Q(E) because of the two NBOs of the diradical, and hence, in this case, the BIC is accompanied by DQI. Thus, we show that if the alternant hydrocarbon can be represented as a diradical with one additional atom (attached to the electrodes), then there are DQI and a BIC at E = 0. The reverse is also true: if the electrodes are attached to the same atom of an alternant hydrocarbon molecule, and both DQI and a BIC are observed at E = 0, then the reduced molecule is a diradical. A specific illustrative example is presented in the ESI† (Section S3.1).
4 Illustrative examples of DQI and BICs within the Hückel formalism
4.1 Butadiene: a generic example
To illustrate all the types of AR interactions, we consider a butadiene molecule, in which 2pz-orbitals of carbon are described within the Hückel model (see Fig. 3a). The on-site energy of carbon 2pz-orbital ε0 is set as the energy origin. We take into account the hopping integrals for the nearest neighbors (τ and τ0), the next-nearest neighbors (τ2 < τ, τ0), and the next-next-nearest neighbors (τ3 < τ2). This generic model can describe either trans-butadiene with negligible τ3, or cis-butadiene with high τ3.16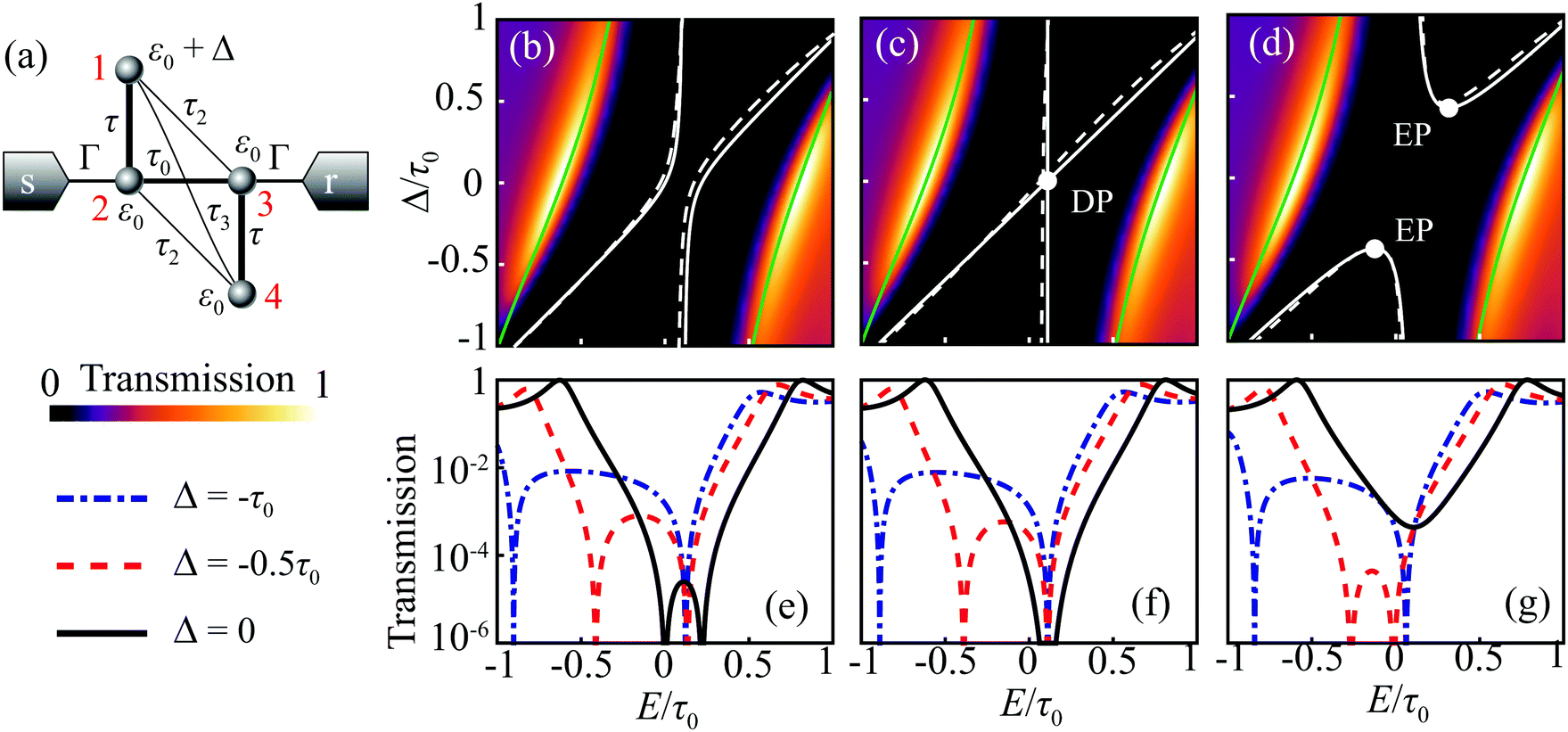 | ||
Fig. 3 (a) Carbon skeleton of a butadiene molecule as a graph for the Hückel model. (b)–(d) Evolution of the transmission spectrum with varying Δ for Γ = 0.3τ0, τ = 1.1τ0, and τ2 = 0.1τ0: 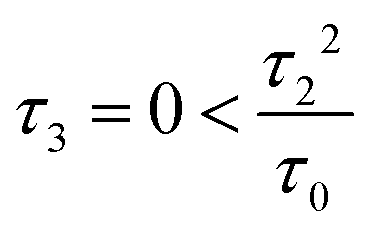 (b), (b), 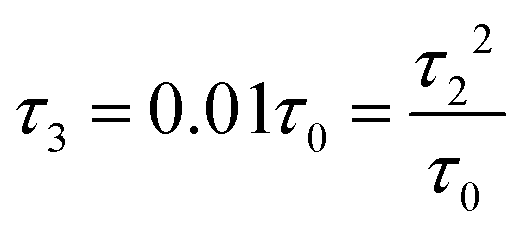 (c), and (c), and 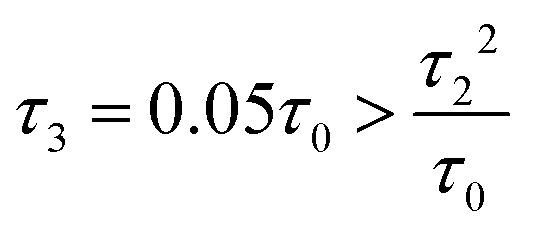 (d). The white solid lines show the exact position of the ARs (real roots of PB) and the white dashed lines depict the approximate position of the ARs from eqn (7). The solid green lines correspond to the energies of the butadiene HOMO and LUMO. (e)–(g) Transmission spectrum of the butadiene molecule in a configuration with τ3 = 0 (e), τ3 = 0.01τ0 (f), and τ3 = 0.05τ0 (g) for some particular value of Δ and Γ = 0.3τ0. (d). The white solid lines show the exact position of the ARs (real roots of PB) and the white dashed lines depict the approximate position of the ARs from eqn (7). The solid green lines correspond to the energies of the butadiene HOMO and LUMO. (e)–(g) Transmission spectrum of the butadiene molecule in a configuration with τ3 = 0 (e), τ3 = 0.01τ0 (f), and τ3 = 0.05τ0 (g) for some particular value of Δ and Γ = 0.3τ0. | ||
Suppose that an external perturbation (e.g. gating or mechanical stretching) shifts the energy of the 1st site by Δ (see Fig. 3a). Then, the transmission coefficient has the form (1) with
| PB(E) = 2Γ[τ0E2 − E(Δτ0 + 2ττ2) + Δττ2 + τ3(τ2 + τ22 − τ0τ3)]. | (14) |
The expression for QB(E) is quite cumbersome and is not of interest for the DQI description. PB(E) is a second-order polynomial and can be studied easily. Its discriminant is
| DB = 4Γ2[Δ2τ02+ 4(τ2 − τ0τ3)(τ22 − τ0τ3)]. | (15) |
For a large shift Δ, the discriminant is positive, which guarantees two separate ARs. However, if Δ is negligible, then the discriminant sign is defined by the second term in square brackets in eqn (15), and several scenarios are possible. For τ3 < τ22/τ0, discriminant DB is positive for any Δ, and there are two separate ARs. In this case, varying Δ qualitatively corresponds to trajectory 1 in Fig. 1d, which results in the avoided crossing of ARs. At τ3 = τ22/τ0, the discriminant is non-negative and turns to zero at Δ = 0. Thus, the regime of AR crossing takes place (trajectory 2 in Fig. 1d). Finally, for τ3 > τ22/τ0, discriminant DB becomes negative for 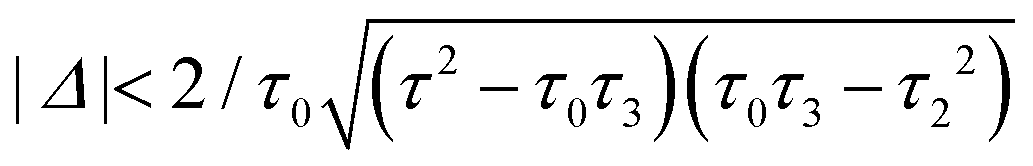 , which enables one to observe AR coalescence (trajectory 3 in Fig. 1d). All these regimes are illustrated in Fig. 3b–d. One can see good agreement between approximation (7) and the exact calculations.
, which enables one to observe AR coalescence (trajectory 3 in Fig. 1d). All these regimes are illustrated in Fig. 3b–d. One can see good agreement between approximation (7) and the exact calculations.
The discussed model reveals the already known in the literature phenomena of splitting ARs16,70 or the destruction of an AR16 in the butadiene molecule as a manifestation of two fundamentally different AR interaction regimes: avoided crossing and coalescence.
4.2 Cyclobutadiene: coalescence of antiresonances
The molecule of cyclobutadiene (CB) is very unstable;71 however, it represents an important structural block of many compounds, including diradicals,8 possessing interesting transport properties.31 Consider a CB molecule connected to two leads and describe its carbon 2pz-orbitals by the Hückel model (Fig. 4a). The ratio between the nearest-neighbor hopping integrals τ and τ0 is defined by the geometry of the molecule (Fig. 4b). | ||
| Fig. 4 (a) Carbon skeleton of CB as a graph for the Hückel model. (b) Transformation of the CB molecule geometry with varying τ/τ0 ratio. (c) Interchange of the HOMO and LUMO states of CB with increasing τ. For τ = τ0 CB becomes a disjoint diradical with two degenerate NBOs. The dashed line indicates the line of the HOMO/LUMO wavefunction nodes. | ||
The transmission coefficient of CB in the 1–4 configuration can be written in the general form (1) with
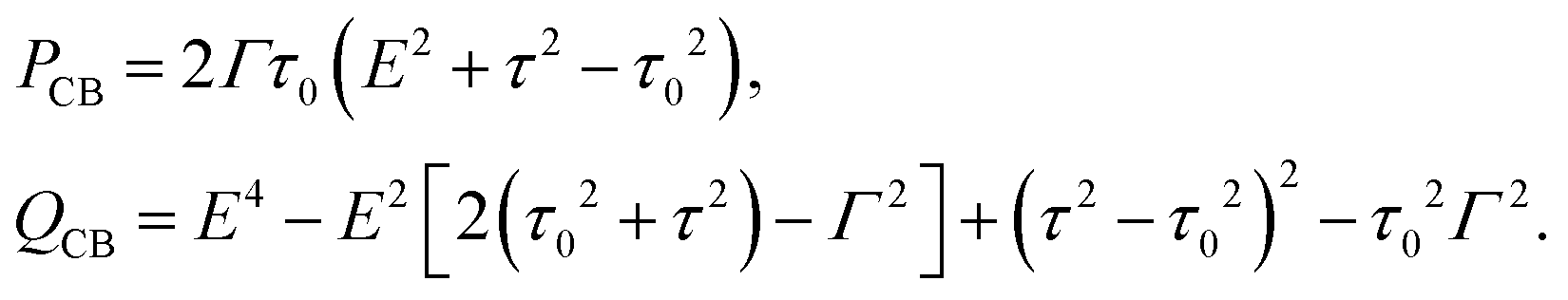 | (16) |
 , which coalesce for τ = τ0. These roots are real for τ < τ0 and become a complex conjugate for τ > τ0.
, which coalesce for τ = τ0. These roots are real for τ < τ0 and become a complex conjugate for τ > τ0.
For a small positive difference τ0 −τ, the energy splitting between ARs behaves non-analytically as  . This behavior can be described by an exact correspondence between energies of ARs and eigenvalues of a non-Hermitian Hamiltonian by rewriting PCB(E) as
. This behavior can be described by an exact correspondence between energies of ARs and eigenvalues of a non-Hermitian Hamiltonian by rewriting PCB(E) as
| PCB(E) = 2Γτ0det(EÎ − ĤCBzero), | (17) |
where
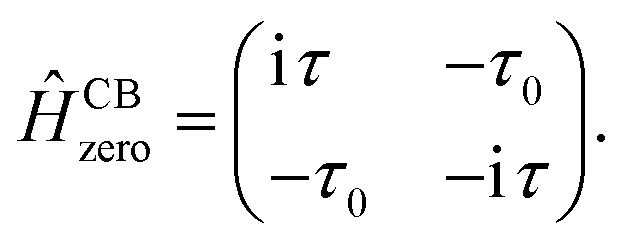 | (18) |
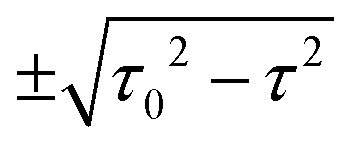 for τ < τ0 and two complex-conjugate eigenvalues
for τ < τ0 and two complex-conjugate eigenvalues 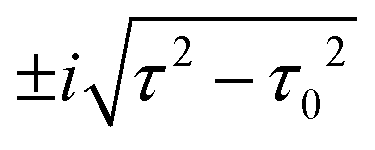 for τ > τ0. Therefore, there are two zero-valued antiresonances in the transmission spectrum of CB at
for τ > τ0. Therefore, there are two zero-valued antiresonances in the transmission spectrum of CB at  for τ < τ0, which coalesce at the EP of ĤCBzero (τ = τ0) and turn into a single transmission dip with a nonzero value at E = 0 for τ > τ0. Fig. 5 illustrates the transmission spectrum evolution by varying the τ to τ0 ratio.
for τ < τ0, which coalesce at the EP of ĤCBzero (τ = τ0) and turn into a single transmission dip with a nonzero value at E = 0 for τ > τ0. Fig. 5 illustrates the transmission spectrum evolution by varying the τ to τ0 ratio.
 | ||
| Fig. 5 (a) Density plot of the CB transmission coefficient evolution with varying τ/τ0 ratio for Γ = 0.3τ0. The black lines indicate perfect resonances (real roots of QCB), the white solid line shows the position of zero transmission (real roots of PCB), and the white dashed line depicts approximate AR positions (9). The solid red and dashed blue lines correspond to the energies of symmetric/antisymmetric MOs correspondingly. (b) Transmission spectrum for some particular value of the τ/τ0 ratio and Γ = 0.3τ0. | ||
AR coalescence in CB is related to the interchange of the HOMO and LUMO wavefunctions (Fig. 4c). Since the HOMO and LUMO can be characterized by a line (director) of wavefunction nodes, this switching can be considered as a reorientation transition rather than a truly symmetry breaking transition.
In the case of the CB molecule, two-level approximation (7) is not necessary, as the exact PCB(E) is already a characteristic determinant of 2 × 2 Hamiltonian (18). However, in general, there is no such exact formula for P(E), and approximation (7) does shed light on the AR coalescence phenomenon. In the ESI† (Section S3.2) we illustrate this for a more complicated molecule – benzocyclobutadiene.
4.3 Benzene: BICs and Fano resonances
Benzene is one of the most studied molecules in chemistry. In particular, benzene in the meta-configuration is a textbook example of quantum interference, which displays itself as AR at zero energy (at the LUMO–HOMO midgap).72 Here we show that in the para-configuration DQI is not less important and can be revealed as ARs at the LUMO/HOMO energies.The LUMO and HOMO states in benzene are doubly degenerate. According to eqn (5), the numerator of the transmission coefficient P(E) must turn to zero at the energies of these degenerate MOs E = ±τ (in the nearest-neighbor approximation with hopping integral τ) regardless of the configuration of the electrode attachment, which can be either ortho, meta, or para. It is well-known (see e.g.ref. 73 and 74) that for the meta- and ortho-configurations ARs of the transmission spectrum do take place at energies E = ±τ. However, for the para-configuration, DQI is absent. Such a fundamental difference arises from the fact that in the case of the para-configuration function Q(E) also turns to zero at E = ±τ, which indicates the formation of a BIC:
 | (19) |
BIC formation in benzene with electrodes in the para-configuration can be interpreted either as symmetry-protected or due to the Friedrich–Wintgen mechanism.39 Among two pairs of degenerate orbitals, there are two MOs (one LUMO and one HOMO) that vanish on two opposite carbon atoms, which we number as the 1st and the 4th ones (see the atom numbering in Fig. 6a). This vanishing takes place because the corresponding wavefunctions are antisymmetric with respect to the reflection in the plane normal to the cycle plane, which goes through the opposite 1st and 4th C atoms. The coupling to the leads at these sites (para-configuration) in the nearest neighbor approximation is also zero, and hence these wavefunctions are symmetry-protected BICs (Fig. 6c). At the same time, the possibility of “choosing” appropriate antisymmetric MOs arises from the fact of degeneracy, which is typical for a more general Friedrich–Wintgen mechanism.
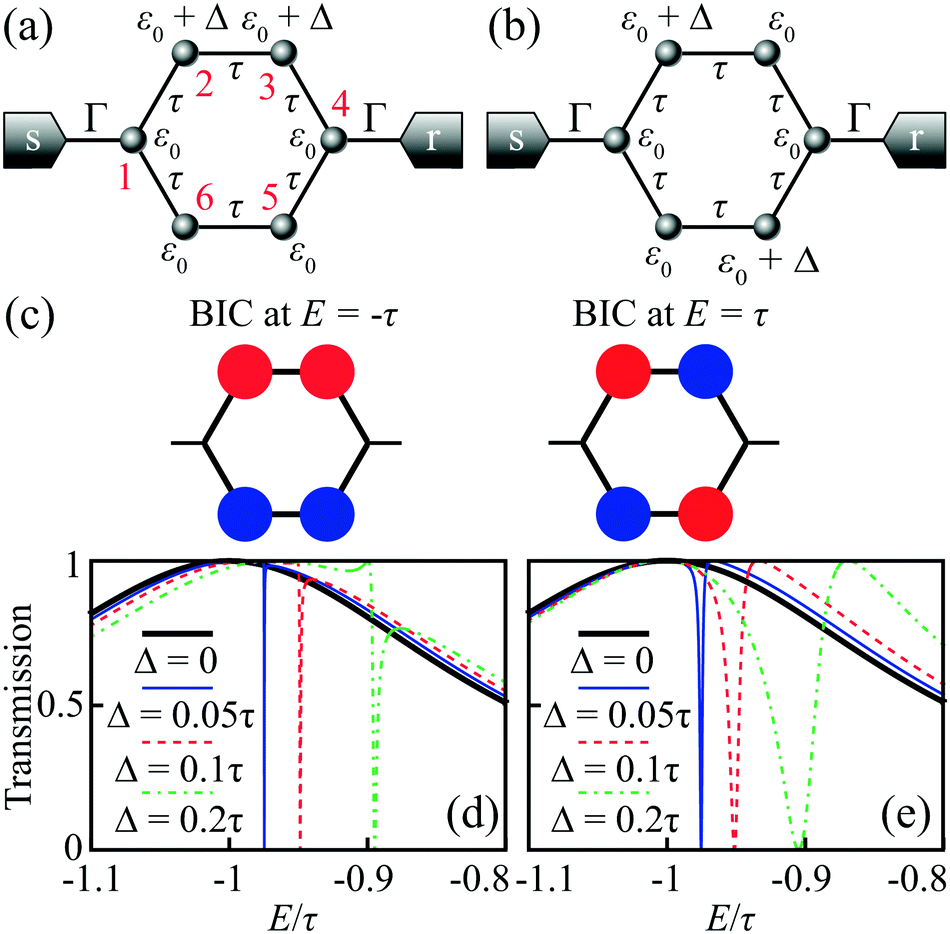 | ||
| Fig. 6 The carbon skeleton of para-benzene as a graph for the Hückel model with different configurations of the gate induced energy shift of atomic orbitals: (a) one-sided and (b) crossed. (c) BICs in benzene with the electrodes in the para-configuration. Transmission spectrum evolution with increasing Δ for (d) one-sided and (e) crossed configurations of the gate. We set Γ = 0.3τ. | ||
The presented here BIC in benzene in the para-configuration and BICs in cyclooctatetraene and anthracene discussed in the ESI† (Sections S3.3 and S3.4) can be treated as simple examples of such a mechanism. An example of a more general case is considered in the ESI† (Section S3.5). It should be noted that analysis of a BIC and the transmission coefficient features in its vicinity can be straightforwardly provided via the P(E) and Q(E) functions regardless of the particular mechanism of BIC formation.
A slight violation of the molecule symmetry with either an external field, asymmetric lead connection, or asymmetric addition of atomic groups makes these wavefunctions non-zero at the 1st and 4th atoms. This provides weak coupling between the bound states of a benzene molecule and the lead continuum. For instance, consider a gate induced energy shift Δ of the atomic orbital energy for carbon atoms from one side of the benzene cycle (Fig. 6a). By a direct calculation, one can check that ∂P/∂Δ ≠ ∂Q/∂Δ near BIC energy E = ±τ, and hence the roots of P(E) and Q(E) are shifted differently and the BIC is destroyed for Δ ≠ 0. Fig. 6d shows the transmission spectrum evolution near E = −τ with increasing Δ. For Δ = 0 the spectrum is smooth and for Δ ≠ 0 a narrow asymmetric Fano resonance profile43 appears.
Similarly, for a crossed configuration of gate induced energy shift (Fig. 6b), we also have ∂P/∂Δ ≠ ∂Q/∂Δ, and the BIC is destroyed for Δ ≠ 0. The corresponding evolution of the transmission spectrum near E = −τ is shown in Fig. 6e and also demonstrates the appearance of an FR for Δ ≠ 0. The crossed configuration of atoms affected by the gate gives P(E) = 4Γτ3[(E − Δ/2)2 − τ2 − Δ2/4] and shows an avoided crossing of ARs at Δ = 0, which corresponds to the smallest energy split between them.
5 Comparison with numerical simulations
5.1 Coalescence of antiresonances in cyclobutadiene
To verify our analytical results at a higher accuracy level, we perform ab initio (DFTB and DFT) simulations of some molecules considered above. Molecules comprising a CB cycle and polyene spacers were studied via the DFTB+ and QuantumATK packages (see the ESI,† Section S1.2, for details) in different geometries and with various side groups attached to the 2nd and 3rd carbon atoms in the CB cycle (Fig. 7a). Here we present the results for pristine CB and CB with –NH2 side groups, which shows the most prominent alteration of the electronic properties. Calculations with some more side groups and detailed atomic positions of the optimized geometry can be found in the ESI† (Sections S4 and S7). The interface between each gold electrode and molecule is realized through an –SCH3 group, which is relatively stable and provides reasonable transport and charge injection into the molecule from the gold contacts.75 The polyene spacers between the anchors and the CB cycle were attached to increase the whole length of the molecule to prevent significant direct gold–gold tunneling.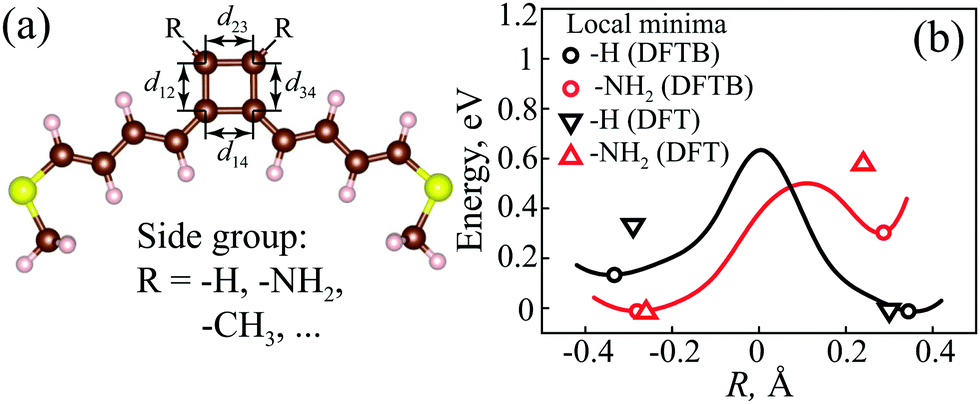 | ||
| Fig. 7 (a) Structure of a CB molecule with polyene spacers and –SCH3 anchor groups. (b) DFTB total energy calculations for different R values in the CB cycle with a fixed perimeter for –H and –NH2 groups attached to the 2nd and 3rd carbon atoms in the CB cycle. The black line corresponds to pristine CB, and the red line to CB with –NH2 side groups. In each case, the energy is measured from its global minimum. The open circles indicate local minima of the total energy. The open triangles show values of R and relative energies of local minima of the total energy calculated by the DFT method for CB with –H (black) and –NH2 (red) groups. | ||
For further description, it is illustrative to introduce a characteristic parameter R:
| R = d12 + d34 − d23 − d14. | (20) |
The isolated CB molecule has four 2pz MOs, two of which are degenerate in the perfectly square geometry.71 When coupled to the electrodes, this configuration corresponds exactly to the EP, i.e., to the AR coalescence point. However, the perfectly square configuration is unstable and due to the Jahn–Teller distortion the degeneracy breaks and two optimal geometries are possible. Fig. 7b depicts the relative total energy change of the whole molecular structure with tuning the ratio of the bond lengths inside the CB cycle (keeping the perimeter constant) with –H and –NH2 side groups. There are two local minima for both the –H and –NH2 side groups. Structural optimization via DFTB and DFT calculations shows that the global minimum in the case of the –H group (pristine CB) corresponds to R > 0, and one can expect the presence of two ARs in the transmission spectrum. On the other hand, for the –NH2 group, the optimal R is negative, and hence no ARs are expected.
Alternatively, polyene groups can be used to control the geometry (see the ESI,† Section S4, for details).
Slight variation of the geometry by adding a side group or applying a mechanical stretch to the CB molecule results in the swapping of the HOMO and LUMO orbitals, as has been discussed within the Hückel formalism in Section 4. When a mechanical stretch is applied to the CB molecule to shift the bond length by up to 0.01 Å from the perfectly square geometry (note that it is not the most stable geometry), the HOMO and LUMO levels swap, and other MOs stay in the same order (Fig. 8a). For the local and global energy minima, the order of the energy levels changes according to the R parameter sign. The DFT calculations also confirm the switching of the HOMO and LUMO, and the corresponding DOS spectra and orbitals are presented in the ESI† (Section S4).
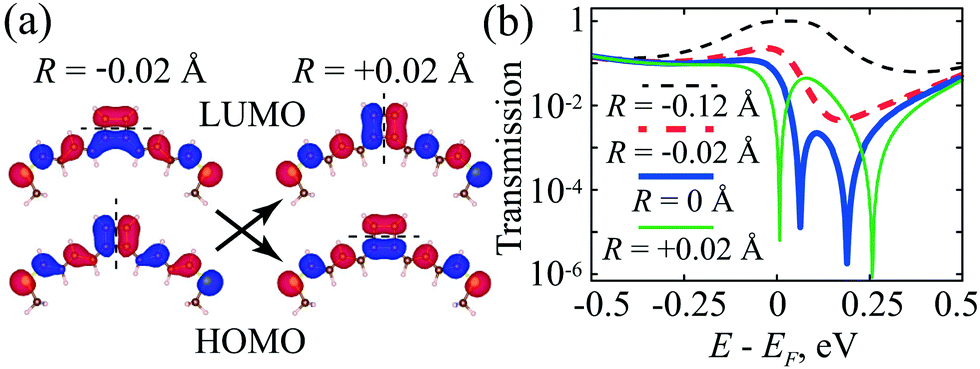 | ||
| Fig. 8 (a) Interchange of the HOMO and LUMO wavefunctions in the CB molecule with polyene spacers and –SCH3 anchor groups in the 1–4-configuration with the variation of the C–C distance in the cycle. The dashed line indicates the line of the HOMO/LUMO wavefunction nodes. (b) DFTB calculated transmission spectrum of CB for different values of R. | ||
Fig. 8b shows the DFTB calculated transmittance of the CB molecule for different R values, which we change by a slight mechanical stretch of the cycle in different directions keeping its perimeter constant. The AR coalescence takes place approximately when the HOMO and LUMO are degenerate (R = 0), similar to the results of ref. 76. As we have shown above in Section 3, this is a general property of alternant hydrocarbons with an even number of carbon atoms, where the electrodes are attached to atoms of different classes [see conditions (11)]. Variation of R by ±0.02 Å near this critical value R = 0 provides a three-order change of the minimum transmission value (compare the thin solid green and thick dashed red lines in Fig. 8b).
DFTB and DFT simulated transmission spectra of the CB molecule with –H and –NH2 side groups in two optimal geometries (corresponding to two energy minima) are presented in Fig. 9a–d. One can see that the transmission coefficient changes as expected for positive and negative values of R. The AR coalescence effect is very sensitive to parameter R. Indeed, according to the Hückel model, the splitting between ARs is proportional to  for small R, which is non-analytical at the threshold R = 0. Therefore, the ARs are far from the Fermi energy in almost all simulations. The Hückel model calculations shown as a dashed line as a comparison demonstrate good agreement with ab initio results.
for small R, which is non-analytical at the threshold R = 0. Therefore, the ARs are far from the Fermi energy in almost all simulations. The Hückel model calculations shown as a dashed line as a comparison demonstrate good agreement with ab initio results.
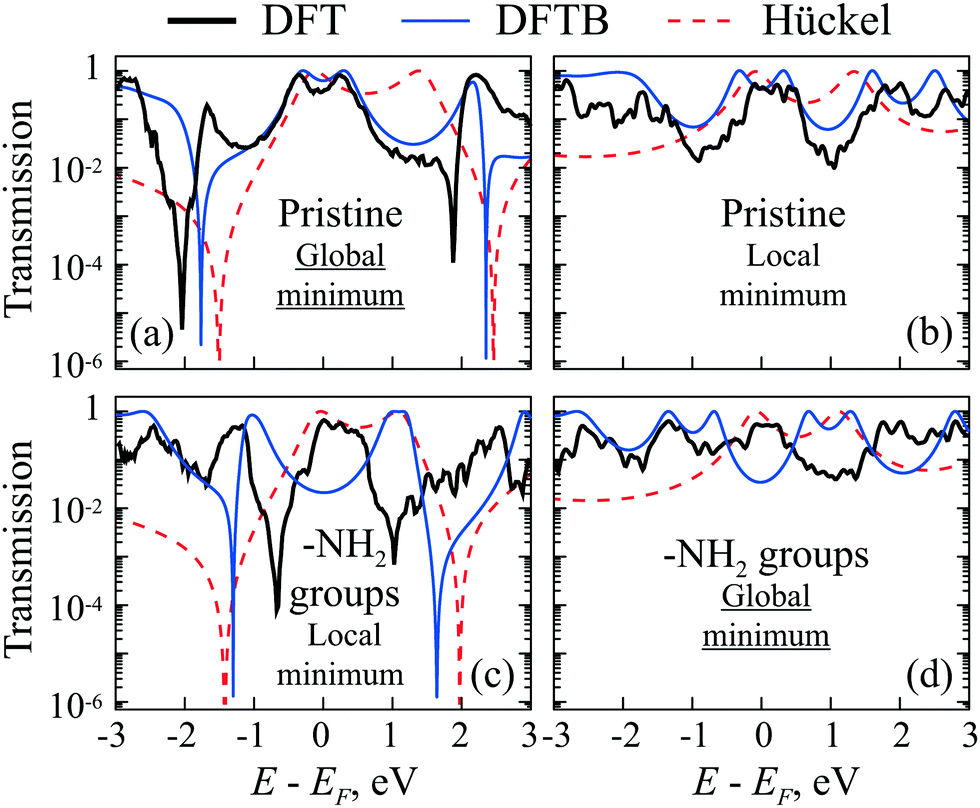 | ||
| Fig. 9 (a)–(d) Transmittance of pristine CB (with –H groups) (a) and (b) and with –NH2 groups (c) and (d) at two local minima of the total energy (see Fig. 7). The global minimum of CB with –H groups corresponds to the case (a) with positive R ≈ 0.33 Å for DFTB and R ≈ 0.3 Å for DFT, while case (b) shows pristine CB in a local minimum with negative R ≈ −0.34 Å for DFTB and R ≈ −0.29 Å for DFT. The global minimum of CB with –NH2 groups corresponds to the case (d) with negative R ≈ −0.28 Å for DFTB and R ≈ −0.26 Å for DFT, while case (c) refers to the local minimum with positive R ≈ 0.29 Å for DFTB and R ≈ 0.24 Å for DFT. The black thick solid lines correspond to DFT calculations, the blue thin solid lines to DFTB calculations, and the red dashed lines to a simple Hückel model (see the ESI,† Section S5, for details). | ||
5.2 Bound states in the continuum in benzene
We also use the DFTB and DFT methods to analyze the DOS, MOs, and transmission spectra of a benzene molecule with electrodes attached in a para-configuration. Here the –SH group was chosen as an anchor and a polyene chain as a spacer. The structures of the simulated molecular systems are shown in Fig. 10a. Detailed atomic positions of the optimized geometry are presented in the ESI† (Section S7). The DOS spectrum of pristine benzene connected to the leads in the para-configuration shows two very sharp peaks corresponding to two BICs (Fig. 10b and c).77,78 When the on-site energy is changed on one side or on diagonal atoms of the benzene ring via grafting of some side groups, the symmetry of the molecule becomes lower. As we have discussed in Section 4, this leads to the destruction of the BICs and the formation of FRs in the transmission. Indeed, two additional –NH2 groups on the same side of the benzene ring make the DOS peaks corresponding to the BICs decrease in amplitude and become wider (Fig. 10c).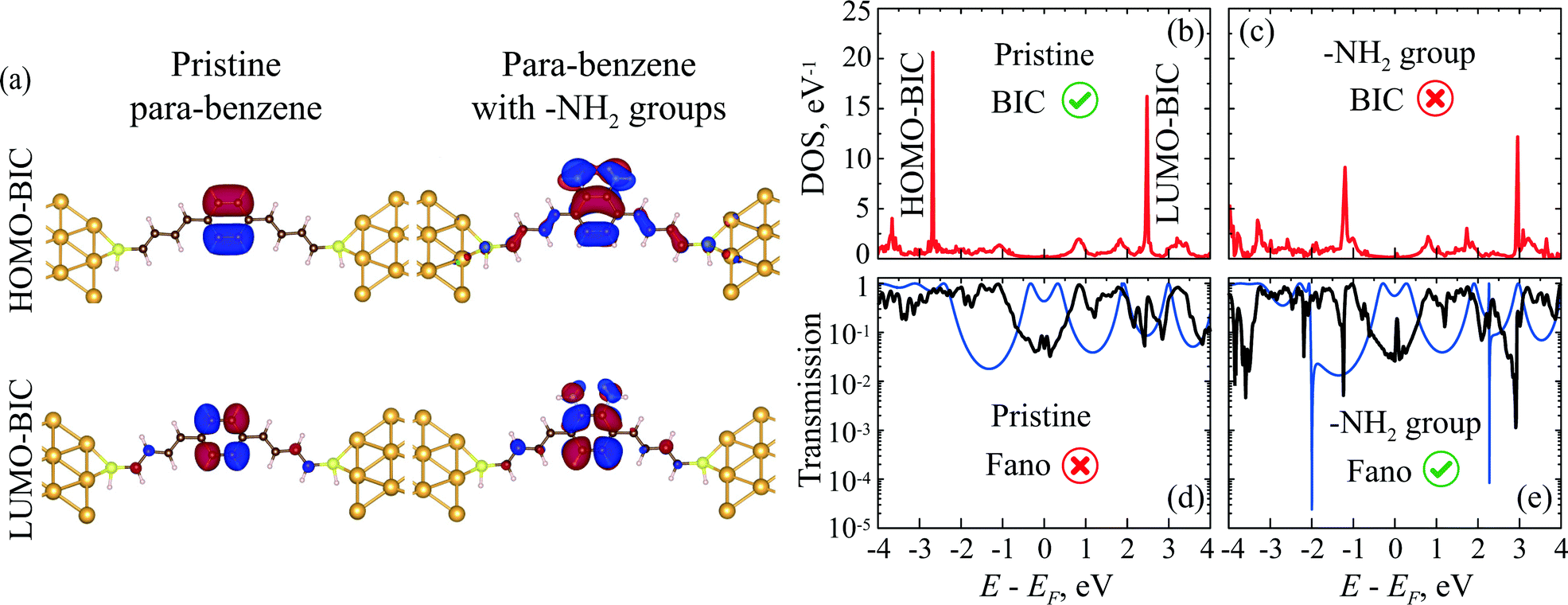 | ||
| Fig. 10 (a) DFT calculated localized MOs of BICs in pristine benzene and their transformation into delocalized MOs in benzene with –NH2 groups. DFT calculated DOS of (b) the pristine benzene molecule and (c) benzene with –NH2 groups with polyene spacers, anchor groups and electrodes in the para-configuration included in both cases. The DFT (thick black lines) and DFTB (thin blue lines) calculated transmission spectrum of (d) pristine benzene and (e) benzene with –NH2 groups. | ||
Fig. 10e illustrates localized BIC orbitals for pristine para-benzene between the gold electrodes, which become delocalized in para-benzene with –NH2 groups. The BIC arising from the HOMO orbital (HOMO-BIC in Fig. 10a) is fully localized on the benzene cycle as it should be (compare with the Hückel model in Fig. 6e). On the other hand, the MO of LUMO-BIC (Fig. 10a) is slightly delocalized even in the pristine molecule (compare with the Hückel model in Fig. 6e). This can be caused by a small displacement of the electron density across the molecule due to the nearest atoms from the polyene spacers, anchor groups, or electrodes. Introduction of –NH2 side groups makes the BIC orbitals become delocalized with the electron density spreading along the molecule into the electrodes. It is important to note that any other MO, which is not a BIC, is typically strongly delocalized regardless of any additional side groups (ESI,† Fig. S14).
The transmission coefficient of para-benzene after attaching the side groups does show two FRs (Fig. 10d and e). Grafting of other groups, such as –OH, SH, and –CH3, has a similar effect on the DOS and transmission spectra (ESI,† Section S6 and Fig. S15).
The results of this section show that the wavefunctions and transmission spectra obtained from the DFTB and DFT methods are in good agreement with the analytical calculations from Section 4.
6 Conclusion
Concluding, we made the next step towards a deep understanding of quantum transport in molecular conductors. The fundamental part of our investigation was done analytically in the context of the Hückel model in terms of functions P(E) and Q(E) [eqn (2) and (3)]. This approach is the most adequate for such studies as it provides a separate description of resonances and antiresonances (ARs), and, more importantly, permits simple consideration of their mutual interaction. In parallel, we performed ab initio simulations that validated the realistic nature of our analytical results.We focused on a group of interference phenomena caused by the interaction of transmission resonances and ARs, which remained practically unstudied in molecular electronics to the present day. The coalescence of resonances or ARs is most outstanding because it promises low-voltage switching of conductance, that is, a solution to modern electronics' central problem. Unlike the energy levels of isolated molecules, the resonances exhibit not only crossing and avoided crossing, but they can coalesce as well. Coalescence of resonances takes place at a point in the parameter space known as an exceptional point. The coalescence phenomenon can be crucial for molecular electronics applications because it changes the value of the integrated transparency (current) by orders of magnitude. At the same time, crossing or avoided crossing does not change it considerably. In the present paper, we have shown that ARs can exhibit similar behavior and studied all possible scenarios of interacting AR behavior: crossing, avoided crossing, and coalescence. For each scenario, a specific set of parameters was found, and conditions for the AR coalescence and exceptional point existence were formulated. The obtained results are promising for the development of new conductance switching mechanisms with low power consumption.
These findings were applied to butadiene, cyclobutadiene, and benzocyclobutadiene molecules connected to electrodes. It is noticeable that the transmission spectrum of cyclobutadiene is highly sensitive to the C–C interatomic distances, as is predicted by analytics. The variation of this distance by ±0.02 Å (less than 0.6%) is sufficient to change the transmission from a regime with two ARs to that without ARs (ARs coalesced) and to obtain by this means a three-order increase of the minimum transmission.
In our paper, we also studied bound states in the continuum (BICs) in molecular conductors. We demonstrated that BICs, being recognized as a widespread phenomenon of wave nature, are also ubiquitous in molecular electronics, where some correspondence with non-bonding orbitals, radicals, and diradicals can be revealed. The physical nature of BICs in molecular conductors is by no means trivial. A BIC can be considered simultaneously either as a resonance or an AR with zero width. Therefore, our description of BICs in benzene and other molecules was successful because of the original approach to quantum transport description in terms of the P(E) and Q(E) functions, which allow for separate analysis of resonances and ARs. Within this formalism, a BIC naturally appears as a common real root of P(E) and Q(E). A BIC can be destroyed by a small external influence (e.g., electrical or chemical gating) breaking this interference. In this case, it reveals itself as a very narrow FR or a number of FRs depending on the multiplicity of the common root of the P(E) and Q(E) functions in the unperturbed configuration. For this reason, the study of BICs is promising for molecular sensor applications.
We note that the highest sensitivity of devices (for conductance switching and sensing) will be obtained when a doubly degenerate molecular level falls in the vicinity of the Fermi energy, that is, for molecules of a diradical type. Similar materials are difficult for technology; therefore, future selection of molecule-candidates should be very careful. It will include the analysis of stability, Coulomb effects, etc. Our present results provide useful guidelines for such searching.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation under project #21-19-00808.Notes and references
- J. C. Cuevas and E. Scheer, Molecular electronics: an introduction to theory and experiment, World Scientific, 2010 Search PubMed.
- F. Evers, R. Korytár, S. Tewari and J. M. van Ruitenbeek, Rev. Mod. Phys., 2020, 92, 035001 CrossRef CAS.
- J. Bai, A. Daaoub, S. Sangtarash, X. Li, Y. Tang, Q. Zou, H. Sadeghi, S. Liu, X. Huang, Z. Tan, J. Liu, Y. Yang, J. Shi, G. Mészáros, W. Chen, C. Lambert and W. Hong, Nat. Mater., 2019, 18, 364–369 CrossRef CAS PubMed.
- M. H. Garner, H. Li, Y. Chen, T. A. Su, Z. Shangguan, D. W. Paley, T. Liu, F. Ng, H. Li, S. Xiao, C. Nuckolls, L. Venkataraman and G. C. Solomon, Nature, 2018, 558, 415–419 CrossRef CAS PubMed.
- Y. Li, M. Buerkle, G. Li, A. Rostamian, H. Wang, Z. Wang, D. R. Bowler, T. Miyazaki, L. Xiang, Y. Asai, G. Zhou and N. Tao, Nat. Mater., 2019, 18, 357–363 CrossRef CAS PubMed.
- C. Jia, M. Famili, M. Carlotti, Y. Liu, P. Wang, I. M. Grace, Z. Feng, Y. Wang, Z. Zhao, M. Ding, X. Xu, C. Wang, S.-J. Lee, Y. Huang, R. C. Chiechi, C. J. Lambert and X. Duan, Sci. Adv., 2018, 4, eaat8237 CrossRef CAS PubMed.
- K. Wang, A. Vezzoli, I. M. Grace, M. McLaughlin, R. J. Nichols, B. Xu, C. J. Lambert and S. J. Higgins, Chem. Sci., 2019, 10, 2396–2403 RSC.
- Y. Tsuji, R. Hoffmann, M. Strange and G. C. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E413–E419 CrossRef CAS PubMed.
- M. G. Reuter and T. Hansen, J. Chem. Phys., 2014, 141, 181103 CrossRef CAS PubMed.
- X. Zhao, V. Geskin and R. Stadler, J. Chem. Phys., 2017, 146, 092308 CrossRef.
- T. Markussen, R. Stadler and K. S. Thygesen, Nano Lett., 2010, 10, 4260–4265 CrossRef CAS PubMed.
- K. G. L. Pedersen, A. Borges, P. Hedegard, G. C. Solomon and M. Strange, J. Phys. Chem. C, 2015, 119, 26919–26924 CrossRef CAS.
- Y. Tsuji, E. Estrada, R. Movassagh and R. Hoffmann, Chem. Rev., 2018, 118, 4887–4911 CrossRef CAS PubMed.
- K. Yoshizawa, T. Tada and A. Staykov, J. Am. Chem. Soc., 2008, 130, 9406–9413 CrossRef CAS PubMed.
- Y. Tsuji and R. Hoffmann, Angew. Chem., Int. Ed., 2014, 53, 4093–4097 CrossRef CAS PubMed.
- Y. Tsuji, R. Hoffmann, R. Movassagh and S. Datta, J. Chem. Phys., 2014, 141, 224311 CrossRef PubMed.
- R. Movassagh, G. Strang, Y. Tsuji and R. Hoffmann, J. Math. Phys., 2017, 58, 033505 CrossRef.
- K. G. L. Pedersen, M. Strange, M. Leijnse, P. Hedegård, G. C. Solomon and J. Paaske, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125413 CrossRef.
- Y. Zhang, G. Ye, S. Soni, X. Qiu, T. L. Krijger, H. T. Jonkman, M. Carlotti, E. Sauter, M. Zharnikov and R. C. Chiechi, Chem. Sci., 2018, 9, 4414–4423 RSC.
- G. Yang, S. Sangtarash, Z. Liu, X. Li, H. Sadeghi, Z. Tan, R. Li, J. Zheng, X. Dong, J. Liu, Y. Yang, J. Shi, Z. Xiao, G. Zhang, C. Lambert, W. Hong and D. Zhang, Chem. Sci., 2017, 8, 7505–7509 RSC.
- C. M. Guédon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. van der Molen, Nat. Nanotechnol., 2012, 7, 305 CrossRef PubMed.
- B. Huang, X. Liu, Y. Yuan, Z.-W. Hong, J.-F. Zheng, L.-Q. Pei, Y. Shao, J.-F. Li, X.-S. Zhou, J.-Z. Chen, S. Jin and B.-W. Mao, J. Am. Chem. Soc., 2018, 140, 17685–17690 CrossRef CAS PubMed.
- J. P. Bergfield, M. A. Solis and C. A. Stafford, ACS Nano, 2010, 4, 5314–5320 CrossRef CAS PubMed.
- R. Miao, H. Xu, M. Skripnik, L. Cui, K. Wang, K. G. L. Pedersen, M. Leijnse, F. Pauly, K. Wärnmark, E. Meyhofer, P. Reddy and H. Linke, Nano Lett., 2018, 18, 5666–5672 CrossRef CAS PubMed.
- E.-C. Lee, Appl. Phys. Lett., 2012, 100, 153117 CrossRef.
- T. Stuyver, M. Perrin, P. Geerlings, F. De Proft and M. Alonso, J. Am. Chem. Soc., 2018, 140, 1313–1326 CrossRef CAS PubMed.
- C. Hu, Modern semiconductor devices for integrated circuits, Prentice Hall, Upper Saddle River, NJ, 2010, vol. 2 Search PubMed.
- Y. Li, J. A. Mol, S. C. Benjamin and G. A. D. Briggs, Sci. Rep., 2016, 6, 33686 CrossRef CAS.
- C. J. Lambert, Chem. Soc. Rev., 2015, 44, 875–888 RSC.
- A. A. Gorbatsevich and N. M. Shubin, Phys.-Usp., 2018, 61, 1100–1115 CrossRef CAS.
- A. A. Gorbatsevich, G. Y. Krasnikov and N. M. Shubin, Sci. Rep., 2018, 8, 15780 CrossRef PubMed.
- T. Kato, Perturbation Theory for Linear Operators, Springer-Verlag, Berlin, Heidelberg, 1995 Search PubMed.
- W. D. Heiss, J. Phys. A: Math. Gen., 2004, 37, 2455–2464 CrossRef.
- I. Rotter, J. Phys. A: Math. Theor., 2009, 42, 153001 CrossRef.
- I. Rotter and J. P. Bird, Rep. Prog. Phys., 2015, 78, 114001 CrossRef CAS PubMed.
- W. D. Heiss, J. Phys. A: Math. Theor., 2012, 45, 444016 CrossRef.
- C. W. Hsu, B. Zhen, A. D. Stone, J. D. Joannopoulos and M. Soljačić, Nat. Rev. Mater., 2016, 1, 16048 CrossRef CAS.
- J. von Neumann and E. Wigner, Phys. Z., 1929, 30, 465–467 Search PubMed.
- H. Friedrich and D. Wintgen, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 32, 3231–3242 CrossRef CAS PubMed.
- A. F. Sadreev, E. N. Bulgakov and I. Rotter, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235342 CrossRef.
- A. A. Maznev and A. G. Every, Phys. Rev. B, 2018, 97, 014108 CrossRef CAS.
- P. S. Pankin, B.-R. Wu, J.-H. Yang, K.-P. Chen, I. V. Timofeev and A. F. Sadreev, Commun. Phys., 2020, 3, 91 CrossRef.
- U. Fano, Phys. Rev., 1961, 124, 1866–1878 CrossRef CAS.
- T. A. Papadopoulos, I. M. Grace and C. J. Lambert, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 193306 CrossRef.
- A. A. Gorbatsevich and N. M. Shubin, Phys. Rev. B, 2017, 96, 205441 CrossRef.
- A. E. Miroshnichenko, S. Flach and Y. S. Kivshar, Rev. Mod. Phys., 2010, 82, 2257–2298 CrossRef CAS.
- S. Datta, Electronic Transport in Mesoscopic Systems, Cambridge University Press, Cambridge, 1997 Search PubMed.
- V. Sokolov and V. Zelevinsky, Ann. Phys., 1992, 216, 323–350 Search PubMed.
- D. Ryndyk, R. Gutiérrez, B. Song and G. Cuniberti, Energy Transfer Dynamics in Biomaterial Systems, Springer, Berlin, Heidelberg, 2009, pp. 213–335 Search PubMed.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafe, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrica, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Kohler, T. Kowalczyk, T. Kubar, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Rezac, C. G. Sanchez, M. Sternberg, M. Stohr, F. Stuckenberg, A. Tkatchenko, V. W.-Z. Yu and T. Frauenheim, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, S. T. Chill, F. Rasmussen, G. Penazzi, F. Corsetti, A. Ojanperä, K. Jensen, M. L. N. Palsgaard, U. Martinez, A. Blom, M. Brandbyge and K. Stokbro, J. Phys.: Condens. Matter, 2019, 32, 015901 CrossRef PubMed.
- E. Whittaker and G. Robinson, The Calculus of Observations: An Introduction to Numerical Analysis, Dover Publications, 1967 Search PubMed.
- Y. Tsuji and K. Yoshizawa, J. Phys. Chem. C, 2017, 121, 9621–9626 CrossRef CAS.
- K. Yoshizawa, Acc. Chem. Res., 2012, 45, 1612–1621 CrossRef CAS PubMed.
- C. A. Coulson, B. O'Leary and R. B. Mallion, Hückel theory for organic chemists, Academic Press, 1978 Search PubMed.
- N. Moiseyev, Non-Hermitian Quantum Mechanics, Cambridge University Press, Cambridge, 2011 Search PubMed.
- J. Wiersig, Phys. Rev. A, 2016, 93, 033809 CrossRef.
- C. M. Bender and S. Boettcher, Phys. Rev. Lett., 1998, 80, 5243–5246 CrossRef CAS.
- C. M. Bender, Rep. Prog. Phys., 2007, 70, 947 CrossRef.
- C. Blanchard, J.-P. Hugonin and C. Sauvan, Phys. Rev. B, 2016, 94, 155303 CrossRef.
- M. L. L. d. Guevara, F. Claro and P. A. Orellana, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 195335 CrossRef.
- M. L. Ladrón de Guevara and P. A. Orellana, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205303 CrossRef.
- C. Fang, D. Li, B. Cui, Y. Xu, G. Ji and D. Liu, Appl. Phys. Lett., 2012, 100, 023303 CrossRef.
- C. A. Ramsden, Advances in Heterocyclic Chemistry, Academic Press, 1980, vol. 26, pp. 1–113 Search PubMed.
- G. C. Solomon, D. Q. Andrews, R. H. Goldsmith, T. Hansen, M. R. Wasielewski, R. P. Van Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17301–17308 CrossRef CAS PubMed.
- J. Gu, W. Wu, T. Stuyver, D. Danovich, R. Hoffmann, Y. Tsuji and S. Shaik, J. Am. Chem. Soc., 2019, 141, 6030–6047 CrossRef CAS PubMed.
- A. E. Miroshnichenko and Y. S. Kivshar, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 056611 CrossRef PubMed.
- A. Chakrabarti, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 205315 CrossRef.
- T. A. Papadopoulos, I. M. Grace and C. J. Lambert, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 193306 CrossRef.
- G. C. Solomon, J. P. Bergfield, C. A. Stafford and M. A. Ratner, Beilstein J. Nanotechnol., 2011, 2, 862–871 CrossRef CAS PubMed.
- F. A. Carey and R. J. Sundberg, Advanced organic chemistry: part A: structure and mechanisms, Springer Science & Business Media, 2007 Search PubMed.
- K. Moth-Poulsen, Handbook of single-molecule electronics, CRC Press, 2016 Search PubMed.
- P. Sam-ang and M. G. Reuter, New J. Phys., 2017, 19, 053002 CrossRef.
- T. Hansen, G. C. Solomon, D. Q. Andrews and M. A. Ratner, J. Chem. Phys., 2009, 131, 194704 CrossRef PubMed.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- D. Nozaki, A. Lucke and W. G. Schmidt, J. Phys. Chem. Lett., 2017, 8, 727–732 CrossRef CAS PubMed.
- P. Sautet and C. Joachim, Chem. Phys. Lett., 1988, 153, 511–516 CrossRef CAS.
- R. Kucharczyk and S. G. Davison, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 195402 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the utilized analytical and numerical methods (including geometries of molecules for ab initio simulations); additional analytical examples: benzocyclobutadiene, cyclobutadiene, and octatetraene; additional numerical examples: cyclobutadiene and benzene with different side groups. See DOI: 10.1039/d1cp02504j |
‡ Mathematically this means that Ĥ = Ĥ. Typically, one takes complex conjugation as a time-reversal operation and for a particular Hamiltonian Ĥ one can take 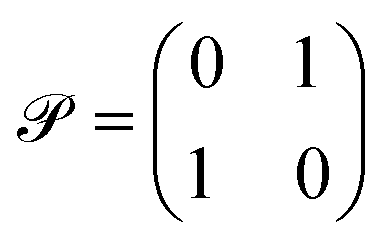 as a space inversion operation. as a space inversion operation. |
| This journal is © the Owner Societies 2021 |