
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium alloys used as electrocatalysts for the oxygen reduction reaction
Tianlei
Wang
a,
Arunabhiram
Chutia
 b,
Dan J. L.
Brett
c,
Paul R.
Shearing
c,
Guanjie
He
*abc,
Guoliang
Chai
*d and
Ivan P.
Parkin
*a
b,
Dan J. L.
Brett
c,
Paul R.
Shearing
c,
Guanjie
He
*abc,
Guoliang
Chai
*d and
Ivan P.
Parkin
*a
aChristopher Ingold Laboratory, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk; i.p.parkin@ucl.ac.uk
bSchool of Chemistry, University of Lincoln, Brayford Pool, Lincoln, LN6 7TS, UK
cElectrochemical Innovation Lab (EIL), Department of Chemical Engineering, University College London (UCL), London WC1E 7JE, UK
dState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (CAS), Fuzhou, 350002 Fujian, P. R. China. E-mail: g.chai@fjirsm.ac.cn
First published on 15th March 2021
Abstract
Palladium-based alloy materials as cathodes for the reduction of oxygen are regarded as potential substitutes for platinum-based catalysts in fuel cells. In this work, we present a scientometric analysis and critically review the use of Pd alloys for the oxygen reduction reaction (ORR). Through scientometric analysis, publication information, research fronts and hotspots are identified. For the critical review, reaction mechanisms in different media are discussed, with the aid of volcano plots to show the general principles for catalyst modifications to maximise the ORR. Influencing factors, including alloying, structure, strain and ligands, particle size, crystal facets and dealloying are considered with a view to informing the theoretical feasibility to enhance the ORR activity. In addition, Pd-based alloys synthesized by different methods are presented and compared in terms of ORR activities. Future research directions are discussed and possible approaches to mass production for industrialization are also proposed.
 Tianlei Wang | Tianlei Wang received his Bacholar's degree in Chemistry from Lanzhou University and Master's degree in Chemical Engineering from Tianjin University in 2015 and 2018, respectively. He is currently a PhD candidate in the Department of Chemistry at University College London under the supervision of Prof. Ivan P. Parkin and Dr Guanjie He. His research interest is focused on electrocatalysts for the oxygen reduction reaction. |
 Guanjie He | Dr Guanjie He is a Senior Lecturer in Chemistry, Leader for Advanced Functional Materials Research Group and Programme Leader for MSc Battery Sciences, University of Lincoln, and an Honorary Lecturer at University College London (UCL). Dr He received a PhD degree from the Chemistry Department, UCL, under the supervision of Prof. Ivan P. Parkin. Dr He's research focuses on materials for electrochemical energy storage and conversion applications, especially electrode materials in aqueous electrolyte systems. |
 Guoliang Chai | Prof. Guo-Liang Chai has been a Professor at Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), since 2016. He received his PhD in 2012 from FJIRSM, CAS. After that, he worked as a Postdoc at Tokyo Institute of Technology from 2012 to 2014, and then joined University College London as a Research Associate from 2014 to 2016. His current research interests focus on functional materials for energy storage and conversion. |
 Ivan P. Parkin | Prof. Ivan P. Parkin is a Professor of Materials Chemistry, Dean of Faculty of Maths & Physical Sciences at University College London. He is a selected Fellow of the Royal Society of Chemistry and a Member of Academia Europaea. Prof. Parkin's work focuses on the development of functional inorganic materials and thin films for energy application, catalysis and wetting. |
Broader contextWith the rising global population and increasing energy demands, fuel cells are attracting considerable attention as alternative energy conversion devices for highly efficient direct chemical-to-electrical energy conversion. The oxygen reduction reaction (ORR) at the cathode is a multi-electron, multi-step reaction with sluggish kinetics. Platinum represents the most widely explored ORR electrocatalyst due to its excellent catalytic performance. Compared to Pt, the research on Pd as an ORR catalyst is relatively limited. However, with the advantages of lower cost, greater abundance and better methanol tolerance, Pd-based materials have promising prospects to replace Pt-based ones. Herein, we combine the scientometric analysis and critical review methods to present a deep and broad overview of Pd alloys for the ORR. Through scientometric analysis, the developments, fronts and hotspots in this field are identified. For the critical review, the mechanisms in different solutions, volcano plots of the ORR catalysts and the crucial elements to enhance the electrocatalytic performance are summarized. Additionally, various synthesis methods of Pd–metal alloy materials are concluded and compared. Finally, we propose perspectives and suggestions for the further development of Pd alloys used as ORR electrocatalysts. This work aims to provide comprehensive and critical insight and indicate a clear direction for future research. |
1. Introduction
The oxygen reduction reaction (ORR), one of the key chemical reactions in fuel cells and metal–air batteries, has been intensively investigated over the last several decades.1–3 Research on ORR catalysts can be traced back to the 1970s, where most attention was focused on platinum (Pt) based catalysts.4–6 Pt-based catalysts set the benchmark due to their excellent ORR performance and stability, and are widely used as cathode electrocatalysts in practical fuel cells.7 However, there are several obstacles for commercialization: (1) the high price and the scarcity of Pt materials; (2) the methanol crossover effect and poor methanol tolerance, which leads to low cell performance and efficiency when using Pt-based catalysts in direct methanol fuel cells (DMFCs),8 and (3) easy poisoning by impurities, such as CO and chloride ions in electrolytes. In recent years there have been attempts to reduce the amount of Pt catalyst but it has been found that under cathodic conditions Pt can easily migrate and aggregate on the carbon support. Additionally, if the particles are smaller in size then they can dissolve to form Pt cations, which then can deposit on other Pt particles, causing them to grow and deactivate, leading to the use of a large amount of Pt metal. Therefore, research effort has also been directed at the development of alternatives, such as other noble metals with relatively low-cost, non-noble metal materials and even metal-free catalysts. The emergence of these studies has greatly enriched the types of ORR electrocatalysts and paved several pathways for practical applications.Amongst these, palladium (Pd) based materials have attracted extensive attention as alternative electrocatalysts because of their comparable properties and slightly lower cost compared to Pt. For instance, the average prices of Pd and Pt in the last ten years (from 1 August 2010 to 1 August 2020) are $910.62 and $1230.96 per troy oz, respectively (http://www.platinum.matthey.com/prices). Specifically, both Pd and Pt are platinum group metals, and they are adjacent in the periodic table, resulting in similar chemical properties. For example, both are inactive and stable in air and humid environments at room temperature and easily form alloys with other metals. Also, the physical properties (such as colour, appearance, melting point, hardness and ductility) are similar. Pd also has very similar electronic properties to Pt.9 Catalysts based on Pd were claimed to have higher methanol tolerance than Pt-based catalysts,10 and be more stable than non-precious metal catalysts in acidic media and highly tolerant towards CO poisoning.11 Unlike Pt/C, Pd-based electrocatalysts did not show any methanol oxidation peak above 0.7 V.12 The methanol oxidation on Pt/C caused the onset of the net cathode current to shift negatively and resulted in a significant increase in overpotential.13 Thus, Pd-based electrocatalysts tend to exhibit better ORR selectivity than Pt-based ones in the presence of methanol.
The study of Pd-based catalysts can be traced back to the first half of the last century. In the early studies, researchers mostly investigated the catalytic oxidation performance, such as methanol oxidation,14 ethylene oxidation,15 carbon monoxide oxidation,16 and formate oxidation,17 as well as various physicochemical properties18–21 and structural characteristics.22,23 For a pure Pd catalyst, due to its intrinsic properties, it is difficult to achieve catalytic performance for the ORR and stability close to that of Pt. For example, the binding ability of Pd to oxygen is too strong, which hinders the reduction of oxygen. The ORR activity of poly-Pd is about five times lower than that of poly-Pt in HClO4 solution.24 In addition, by alloying with other elements (especially transition metals), the ORR activity of Pd based catalysts can be enhanced dramatically25 and the production cost of the electrocatalysts can be further reduced.
Overall, a large and growing number of studies have investigated the role of Pd alloys as ORR catalysts and strive to enhance the activity to match, or even exceed, Pt-based catalysts. Herein, scientometric analysis and traditional review methods are combined with an aim to provide comprehensive and critical insight and indicate a clear direction for future research in this area. In terms of scientometric analysis, based on the Bibliometrix/Biblioshiny R-package,26 Citespace27,28 and VOSviewer29 software, annual production, the top 10 sources and countries by publication, co-citation and keyword analysis, and co-keyword networks are discussed. In terms of the conventional review, this work elucidates the development of ORR mechanisms in different media, volcano plots of previously reported ORR electrocatalysts, the theoretical possibility of an enhancement in the ORR activity and the recent advances in Pd-based electrocatalysts.
2. Scientometric analysis on Pd alloys for the ORR
Before 1970, although there were several reports on the ORR of Pd alloys,24,30,31 most of them were not exhaustive and systematic. Based on the database in the Web of Science from 1970 to 2020, a total of 400 articles were obtained on Pd alloys for the ORR. As shown in Fig. 1, the first report appeared in 1997, and the studies in this field have grown significantly in the past decade. In this study, Pattabiraman et al. dispersed Pd catalysts on various carbon supports and reported their ORR activity in alkaline media for the first time.25 Also, amorphous PdP alloy catalysts for the ORR in alkaline media were reported by Podestá et al.32 In 2004, Savadogo et al. proposed active Pd based alloy catalysts for the ORR in acidic media for the first time and showed that the PdCo alloy (the atomic ratio of Pd to Co is 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28) may exhibit better performance for the ORR than Pt materials.33 Since then, the relevant research in this area has gradually increased.34–36 For example, Shao et al. synthesized PdFe nanoparticles for the ORR and found that the surface-specific activity of the PdFe alloys is related to the Pd–Pd bond distance: the shorter the bond distance, the higher the activity.36
:28) may exhibit better performance for the ORR than Pt materials.33 Since then, the relevant research in this area has gradually increased.34–36 For example, Shao et al. synthesized PdFe nanoparticles for the ORR and found that the surface-specific activity of the PdFe alloys is related to the Pd–Pd bond distance: the shorter the bond distance, the higher the activity.36
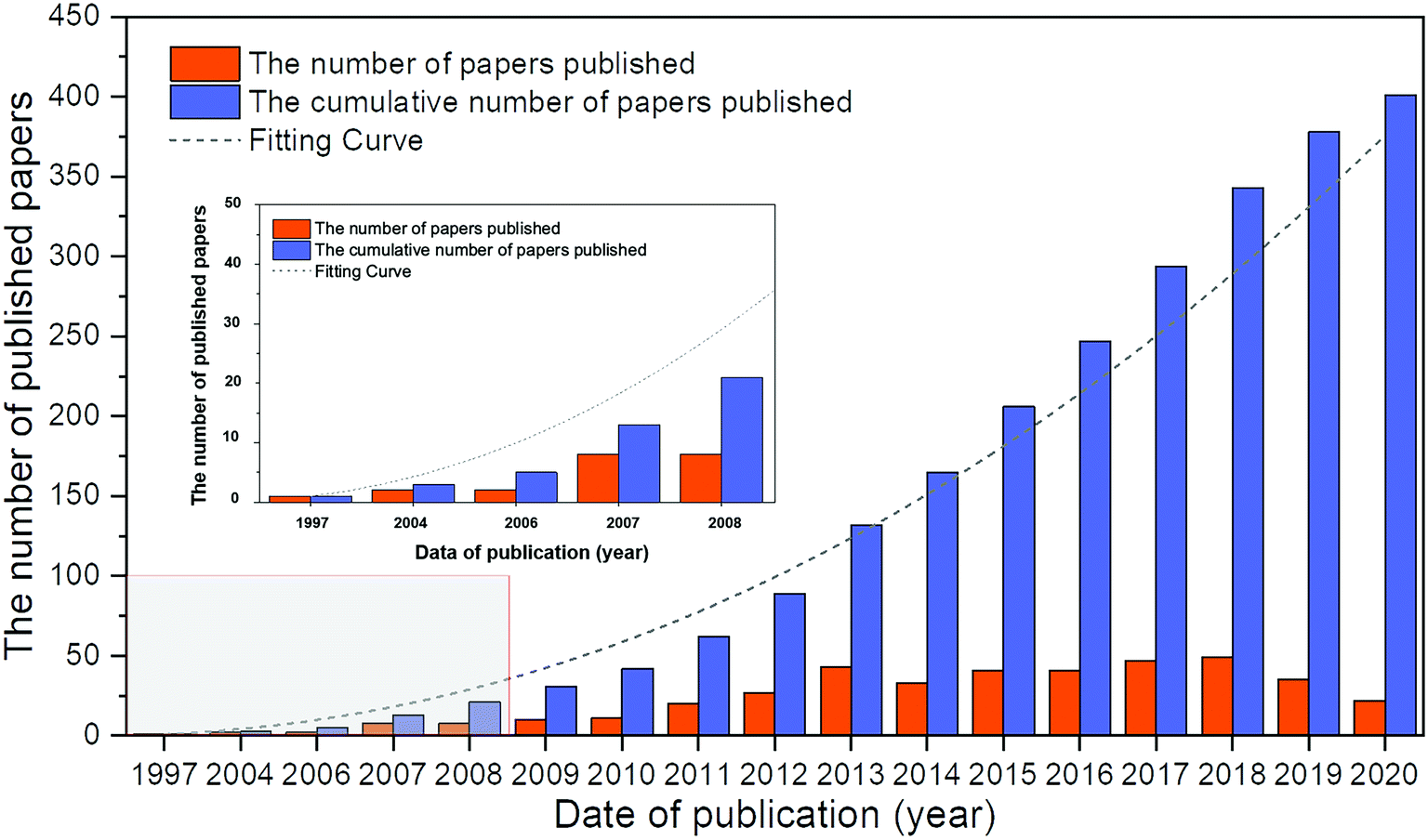 | ||
| Fig. 1 Number of Web of Science (WOS)-based research studies on Pd alloys for the ORR. Source: Literature statistics from the WOS platform. | ||
Based on the Citespace software, taking advantage of cluster analysis of title terms, emerging trends and new developments in this field can be easily identified, as shown in Fig. 2. Clusters (groupings) could be found and numbered in the size descending order of the cluster size. For example, the size of cluster #0 (Pd nano-particle) is the largest. Each point in the graph represents a node, and the node means keywords or references. For ‘tree ring’ shaped nodes, the number of citations the article received in a particular year affects the thickness of the nodes; a red ring shows that a particular year denotes a citation burst (a surge of citations). The network indicates the co-citation and co-occurrence of keywords. The network of different types of entities has different meanings. For instance, lines that connect nodes in a cited reference network are co-citation links. Table 1 presents the information about the largest four clusters by size. The silhouette value reflects the homogeneity and consistency of the cluster.
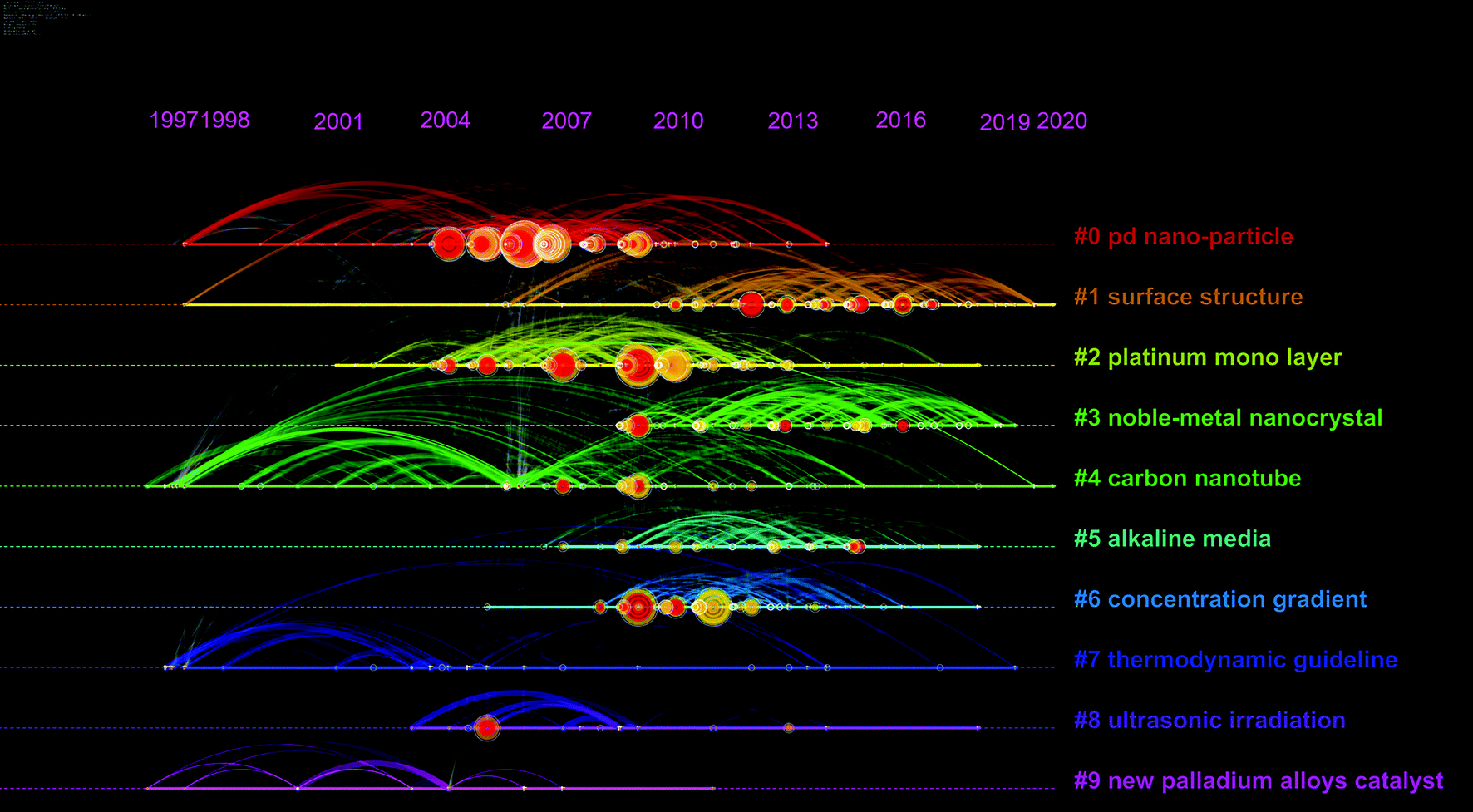 | ||
| Fig. 2 A timeline visualization of the network of co-citation and keyword co-occurrence of the literature about Pd alloys for the ORR between 1997 and 2020. | ||
| Cluster ID | Size | Silhouette | Mean cite year |
|---|---|---|---|
| #0 Pd nano-particle | 103 | 0.621 | 2007 |
| #1 surface structure | 93 | 0.578 | 2014 |
| #2 platinum mono layer | 88 | 0.502 | 2009 |
| #3 noble-metal nanocrystal | 82 | 0.655 | 2014 |
Pd nano-particle (cluster #0) is the focus of research in this field. The study of surface structure (cluster #1) exhibits a good trend for possible future development. Alloyed with platinum (cluster #2), carbon nanotube as the carrier (cluster #4), and applied in alkaline media (cluster #5) are also hotspots.
The keywords summarize the main purpose of the literature to a certain extent, and they can comprehensively interpret its content. Based on the VOSviewer software, co-keyword network visualization maps are shown in Fig. 3 to understand the hotspots and directions in this field. Although there is no specific quantitative data, the direct connection of keywords and the frequency and proportions of the research can be clearly identified from Fig. 3(a and b), respectively. For the topic, “Palladium (Pd) alloy(s) for ORR”, the keywords highlighted in the figure are: “nanoparticles”, “electrocatalysts”, “stability”, and “enhancement” and, through the co-keyword network, the top research directions and underlying problems in this field are revealed, which are namely:
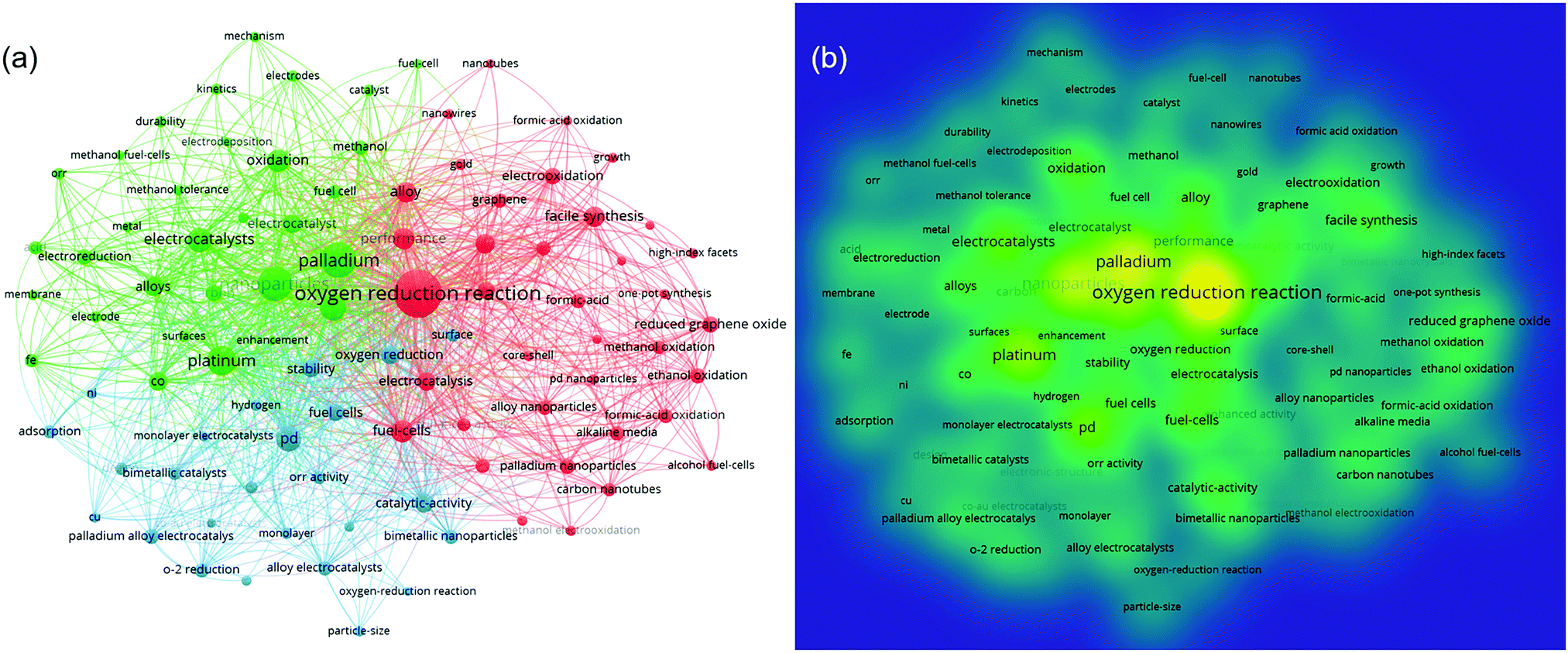 | ||
| Fig. 3 Co-keyword network visualization on Pd alloys for ORR research. Note: (a) network visualization based on co-keywords; and (b) density visualization map based on co-keywords. | ||
(1) Nanoparticles of palladium alloy(s) prepared by various methods are used for ORR catalysis. Catalysts with different morphologies such as nanowire, core–shell and monolayer are widely studied.
(2) The main metals alloyed with Pd are Pt, Co, Au, Fe, Ni and Cu.
(3) Stability and catalytic activity are still the focus in this field. Developing methods to realize comparable performance with Pt is one of the key difficulties in this research.
(4) The pursuit of simpler and more effective synthetic methods, such as facile synthesis and one-pot synthesis.
(5) The combination of carbon material supports, such as graphene, reduced graphene oxide, carbon nanotubes, etc.
(6) In addition to oxygen reduction, Pd alloy catalysts are widely used in formal acid oxidation, methanol oxidation, and ethanol oxidation. The preparation of dual-functional catalysts, which have both oxygen reduction activity and organic oxidation activity, is an interesting research topic.
(7) There is limited research on the mechanism and kinetics, and more in-depth work is needed.
3. ORR mechanisms
The ORR mechanisms on noble metal surfaces, such as Pd and Pt, have not been completely understood despite the intensive studies over the past few decades.37 In general, due to the similarities of Pd and Pt, the ORR processes on these two electrocatalysts are basically similar, that is, this reaction starts with the adsorption of molecular oxygen on the catalyst surface, and then two mechanisms have been generally accepted: one is a direct four-electron mechanism where O2 is reduced directly to water without the production of H2O2; the other is a multi-step four-electron mechanism where O2 is reduced to H2O2 and then further reduced to water. Depending on the pH value of the electrolyte, the electrochemical reduction of oxygen follows different pathways. Specifically, in acidic solution, the reaction is:| O2 + 4H+ + 4e− → 2H2O | (1) |
while in alkaline solution the overall reaction is:
| O2 + 2H2O + 4e− → 4OH− | (2) |
In acidic media, the ORR process undergoes the following steps:38
| O2(g) → O2(ads) | (3) |
Pathway a:
| O2(ads) → 2O(ads) | (4) |
| Oads + H+ + e− → OH(ads) | (5) |
| OHads + H+ + e− → H2O | (6) |
Pathway b:
| O2(ads) + H+ + e− → OOHads | (7) |
| OOHads → O + OHads | (8) |
| O + H+ + e− → OHads | (9) |
| OHads + H+ + e− → H2O | (10) |
| OOHads + H+ + e− → H2O2 | (11) |
| H2O2 → 2OHads | (12) |
For four-electron mechanisms, as shown in Fig. 4(a), after the oxygen gas is transferred to absorbed oxygen (eqn (3)), there are two common pathways. One is denoted as O2-diss-gas39 (Pathway a): the absorbed O is formed through the O2 dissociation step (eqn (4)), and then combines with a proton and an electron to produce absorbed OH (eqn (5)), followed by the reduction with protons and electrons to form H2O (eqn (6)).
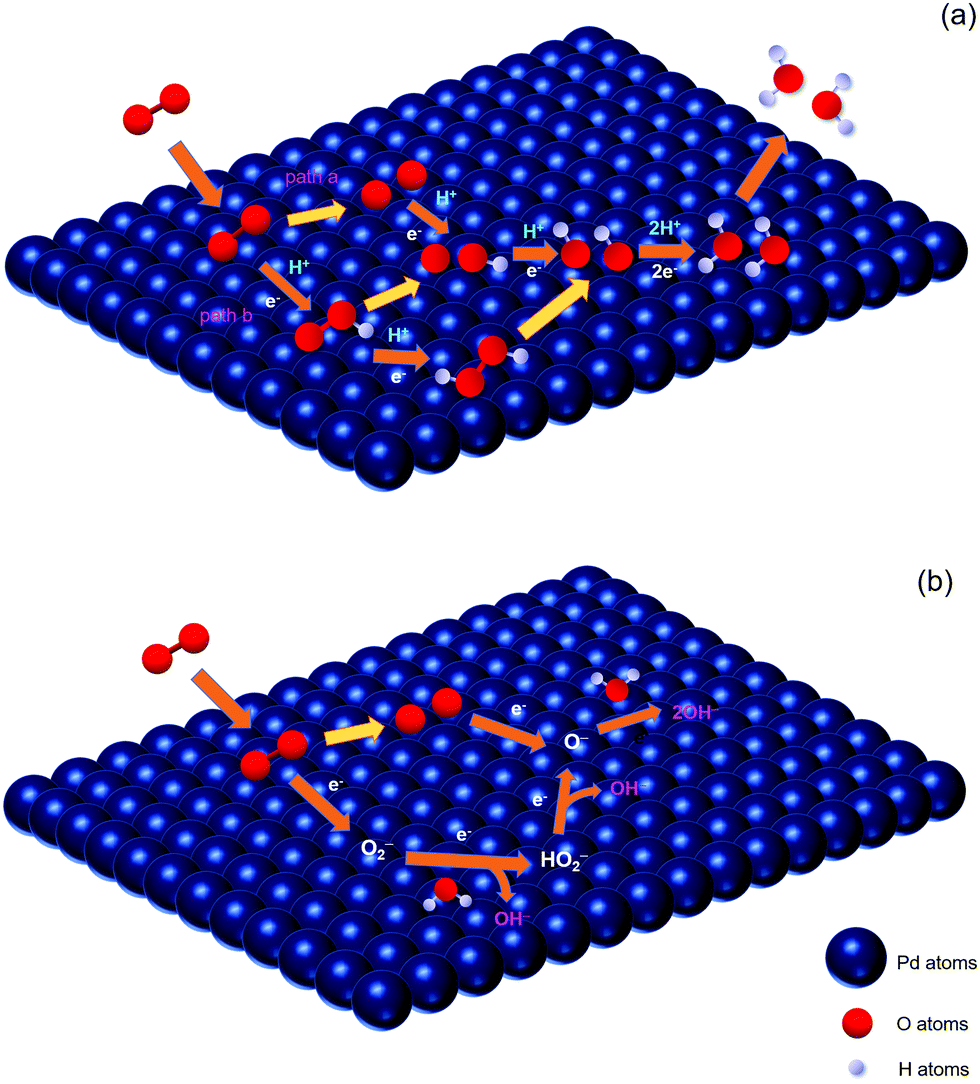 | ||
| Fig. 4 Schematic diagram of mechanisms for the ORR on Pd in acidic (a) and alkaline (b) media, respectively. | ||
The other one is called HOO-form-gas39 (Pathway b): the intermediate superoxide is formed by an oxygen molecule, a proton and an electron (eqn (7)). After the breaking of the O–O bond, the absorbed O and OH species are produced (eqn (8)) and then reduced to H2O by combining with protons and electrons (eqn (9) and (10)). Also, the intermediate superoxide combines with a proton and an electron to form hydrogen peroxide, and then becomes absorbed OH species by a cleavage reaction (eqn (11) and (12)).
According to the general ORR mechanism in alkaline solutions proposed by Anastasijevic et al.,40 it consists of the following steps:
| O2(ads) + e− → O2− | (13) |
| O2−+ H2O + e− → HO2− + OH− | (14) |
| HO2− + e− → O− + OH− | (15) |
| O− + H2O + e− → 2OH− | (16) |
| O2(ads) → 2O(ads) | (17) |
| O(ads) + e− → O− | (18) |
| O− + H2O + e− → 2OH− | (19) |
According to Kinoshita's work, a four-electron pathway appeared to be predominant for Pd electrocatalysts.7 However, Kim et al. researched oxygen reduction on bare Pd in 0.1 M LiOD solution and found that about one third of the O2 reacts at a bare Pd surface and is reduced directly to OD−via the four-electron pathway, while the rest is reduced to deuterium peroxide via the two-electron pathway.42 Therefore, ORR catalysts are expected to have an optimal balance between both the cleavage and the reduction of oxygen.
Furthermore, representative studies focused on the ORR process and other possible mechanisms were proposed, such as the oxygen hydration mechanism,43 peroxide mechanism,44 aquoxyl mechanism,44etc. While alloying with various elements, the mechanisms on Pd or Pt alloys for the ORR45–50 are more sophisticated and need further investigation. Advanced characterization techniques need to be applied to understand the mechanisms. For example, Wang et al. used shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) to illustrate the ORR processes that occur on the surface of bimetallic Pt3Co nanocatalyst structures.49 They found direct spectroscopic evidence of *OOH, which suggests that the ORR undergoes an associative mechanism on Pt3Co in both acidic and alkaline media.
With the aid of DFT calculations to study the ORR mechanism, there were more attempts in the field of Pt-based catalysts to study the ORR mechanism,50–55 and relatively fewer studies for Pd-based catalysts.55,56 Except for the similarities between the two, there are differences in the ORR mechanisms for Pd and Pt electrocatalysts. For instance, the enthalpies and barriers for each step are different and the ORR process varies under specific conditions. For example, Ford et al. indicted that, while the activation barriers for the O–O bond scission steps differ by ca. 0.1 eV on Pd and Pt, the hydrogenation steps (including O2 + H and OH + H) are kinetically less active on Pd than on Pt, suggesting that overall Pd is less effective than Pt.44 Sha et al. considered that, under conditions with high Had, another mechanism producing HOOH is possible, but this is unlikely under ordinary fuel cell operating conditions.43 Additionally, Ou et al. pointed out that, in the presence of hydrated protons, the mechanism of the ORR on the Pd(111) surface only involved the O2 molecule dissociation mechanism, whereas the mechanism of the ORR on the Pt(111) surface involved the dissociation mechanism of both O2 molecule and OOH species.55 As for the entire four-electron ORR, the protonation of adsorbed O atoms to form OH was the slowest step, and it was thus the rate-determining step (rds) for both the Pd(111) or Pt(111) surfaces. Such an rds finding of the ORR explained why Pt- and Pd-based electrocatalysts showing weaker bonding with atomic oxygen had higher ORR activities.
In addition, there is relatively limited research focusing on the ORR conducted in neutral media (pH = 7).57,58 However, microbial fuel cells (MFCs) operating under neutral conditions show promising application prospects. The low concentrations (10−7 M) of H+ and OH− have negative effects on the ORR kinetics, resulting in high overpotentials.59 Precious metal catalysts (Pt, Pd, etc.) tend not to be the first choice due to the high cost compared to the low power output produced.60 More breakthroughs are needed in this field. Hence, methods to enhance the catalytic activity and the factors affecting the ORR activity are described in detail below.
4. Volcano plots of ORR catalysts
The theoretical description of electrocatalytic phenomena is extremely challenging. In this regard, density functional theory (DFT) calculations have been extensively used to correlate the ORR activity with certain parameters of electrocatalysts. The experimental evidence suggests that the ORR activity is related to the strength of the metal–oxygen (M–O) bond, which corresponds to an adsorbed oxygen atom as the ORR intermediate.61 Metals with very negative oxygen binding energy easily cleave molecular oxygen but subsequently form strong inert oxides and have an inappropriate cathode potential. Metals with very positive oxygen binding energy, for example, noble metals such as Au and Ag, have a high barrier for the dissociative adsorption of oxygen, so they also tend to be poor ORR catalysts.62 With the deepening of our understanding in this area, researchers have linked the physical quantities representing the ORR activity of catalysts with some other factors and represent findings in so-called volcano plots. The development of volcano plots of the ORR activity against various factors is shown in Fig. 5. It is noted that these factors, including the d-band vacancy value, the incipient –O or –OH adsorption potential, the oxygen binding energy, the d-band center, the degree of alloying of the electrocatalysts and the adsorption strength for certain surface reactions, can account for various ORR catalytic performances. All these factors influence each other in determining the best conditions for efficient catalytic behavior for the ORR and, therefore, it can be noted that better catalytic activity can be achieved under the optimal conditions of these factors.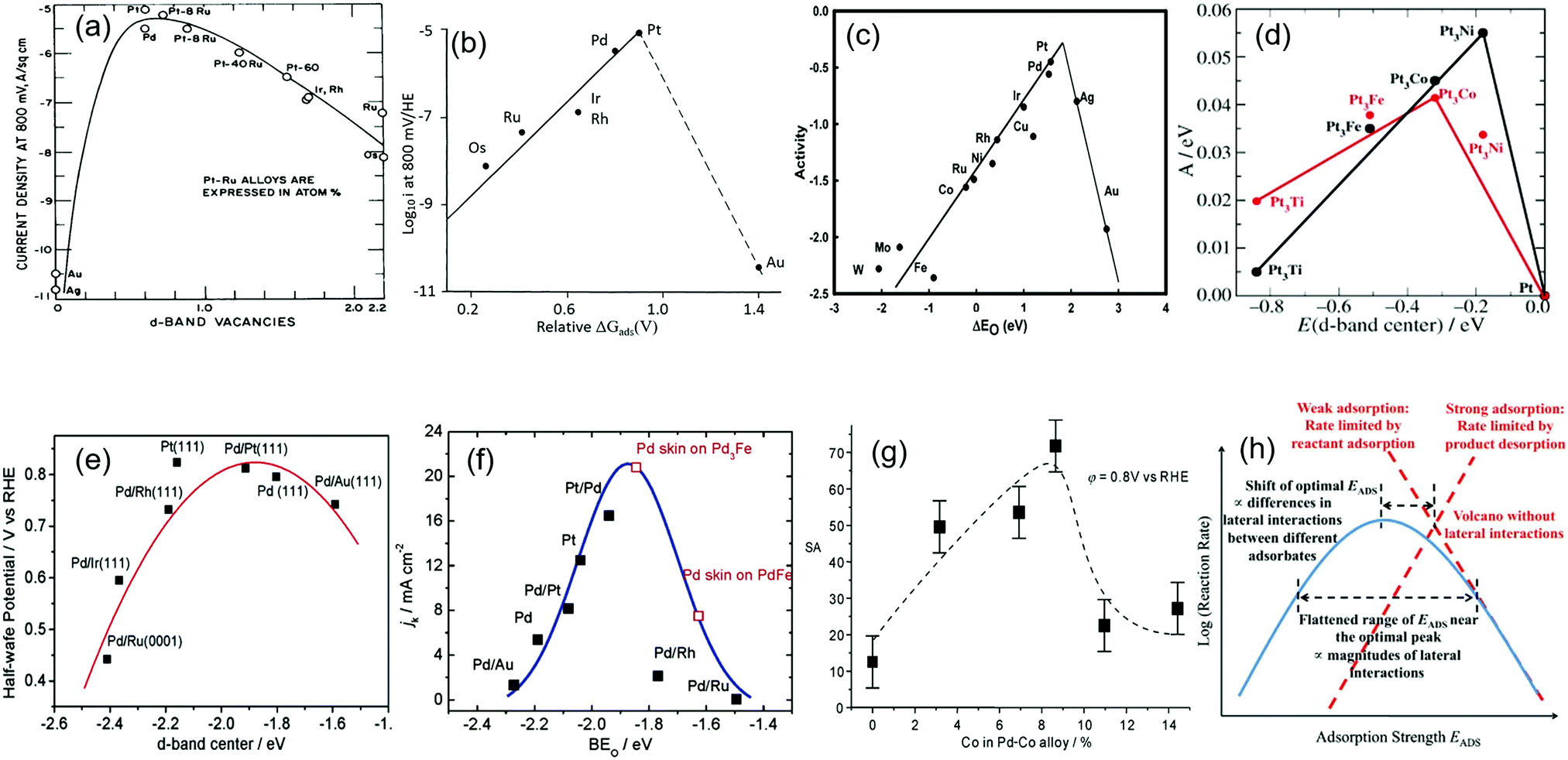 | ||
| Fig. 5 Volcano plots of ORR activity vs. various elements. (a) O2 reduction in 85% orthophosphoric acid: the plot of current density at q = −460 mV at 25 °C against the d-orbital vacancy value of the electrode metal. Reproduced with permission.63 Copyright 1971, Taylor & Francis. (b) Volcano plots of current density i in the ORR vs. relative ΔGads. Reproduced with permission.37 Copyright 1983, Springer. (c) Trends in ORR activity plotted as a function of the oxygen binding energy. Reproduced with permission.61 Copyright 2004, American Chemical Society. (d) The volcano plot of ORR activity vs. the experimentally measured d-band center. Reproduced with permission.64 Copyright 2006, Wiley-VCH. (e) The volcano plot of the half-wave potential of Pd-based alloys as a function of the calculated Pd d-band center (relative to the Fermi level). Reproduced with permission.65 Copyright 2006, American Chemical Society. (f) Volcano plots of the ORR activity (expressed as the kinetic current density) vs. the calculated oxygen-binding energy. Reproduced with permission.66 Copyright 2007, American Chemical Society. (g) The volcano-type relationship between the surface-specific activity (SA) of Pd–Co alloys and the degree of alloying. Reproduced with permission.67 Copyright 2007, Wiley-VCH. (h) The relationship between the catalytic activity and the adsorption strength for certain surface reactions. Reproduced with permission.68 Copyright 2012, Elsevier. | ||
In the 1970s, Appleby showed a volcano plot of the current density, i, in the ORR vs. d-band vacancy values of the electrode metal.63 Pt, Pd and PtRu alloys were at the peak position of this curve, as shown in Fig. 5(a). In 1983, Tarasevich et al. demonstrated the volcano plots of the current density, i, in the ORR vs. the relative ΔGads(V) (Fig. 5(b)).37 ΔGads is the incipient –O or –OH adsorption potential from anodic cyclic voltammetry. These similar volcano relationships can also be observed in the overpotential η–ΔGads plots at constant i for the ORR in acid solution for platinum group metals, Ag, Au and their alloys. In 2004, Nørskov et al. presented a method for calculating the stability of reaction intermediates of electrochemical processes based on electronic structure calculations.61 Trends in the ORR were plotted as a function of the oxygen binding energy. As shown in Fig. 5(c), in terms of pure metal, Pt has the best activity towards the ORR and Pd occupies the second place. The model explained the reason that Pt is the best elemental cathode material and alloying can be used to improve the activity of Pd.
Later, Stamenkovic et al. studied the Pt–metal system in detail and described that the variations in the electronic structure determine the trends in the catalytic activity of the ORR across the periodic table.64 The authors showed that Pt alloys involving 3d metals (such as Ti, V, Fe, Co, and Ni) are better catalysts than pure Pt because the electronic structure of the Pt atoms on the surface of these alloys has been modified slightly. At first, the catalytic activity is related to the adsorption energy of oxygen ΔEO. ΔEO should be moderate, not too high or too low (the Sabatier principle). It is well-established in a number of studies that surface bond energies correlate with the average energy of the d-states on the surface atoms to which the adsorbate binds (the d-band center).69–73 It is difficult to measure the oxygen bond energy; however, the d-band center is accessible by experiments such as synchrotron-based high-resolution photoemission spectroscopy.74 The measurement of the d-band center provides a powerful tool to directly correlate the variations in the ORR activity with the changes of the surface electronic structure. As shown in Fig. 5(d), the activity predicted from DFT simulations is shown in black, and the measured activity is shown in red. Although there is a certain deviation between the two, the overall trend is generally consistent.
Shao et al. reported a volcano-type dependence of activity on the energy of the d-band center of Pd monolayers, with Pd/Pt(111) at the top of the curve, by using DFT calculations, as presented in Fig. 5(e).65 The authors considered that, due to the strong surface segregation of Pd at high temperature, the downward shift of the d-band center of the Pd that constituted the alloy surface could lead to the high ORR activity. Later, Shao et al. established the volcano-type dependence of the ORR activity on the binding energy of oxygen (Fig. 5(f)) by combining experimental data and DFT calculations, and predicted that the Pd overlayer on a Pd3Fe(111) alloy might have the most active performance.66
In 2007, Suo et al. described the “volcano” relationship between the ORR activity and the degree of alloying, shown in Fig. 5(g), and noted that the catalytic activity correlates with the adsorption energy ΔEO, which in turn depends on the lattice strain due to alloying.67 In 2012, Qi et al. proposed a flattened Sabatier volcano plot in the reduction of oxygen as a result of the adsorbate interactions on surfaces, as shown in Fig. 5(h).68 Dashed lines are based on a simple Arrhenius relation; the solid line is obtained with the consideration of lateral interactions between adsorbed reaction intermediates. The finite interactions between different reaction intermediates may also shift the optimal adsorption energy and the corresponding maximum activity.
In Fig. 6, the development of volcano plots and their use to understand the ORR activity and the breakthroughs in the application of Pd (alloys) for the ORR are presented, which clearly shows that the combination of theories and experiments has greatly contributed to the rapid development of this field in recent years. Since 1997, when dispersed Pd-based catalysts on carbon for the ORR in alkaline media were first reported,32 there have been increasing numbers of novel Pd alloys with superior catalytic performance. From realizing comparable performance to Pt to far exceeding Pt towards ORR performances,75 research on Pd-based catalysts is constantly delivering breakthroughs. It is believed that Pd-based catalysts could deliver greater achievements in the near future.
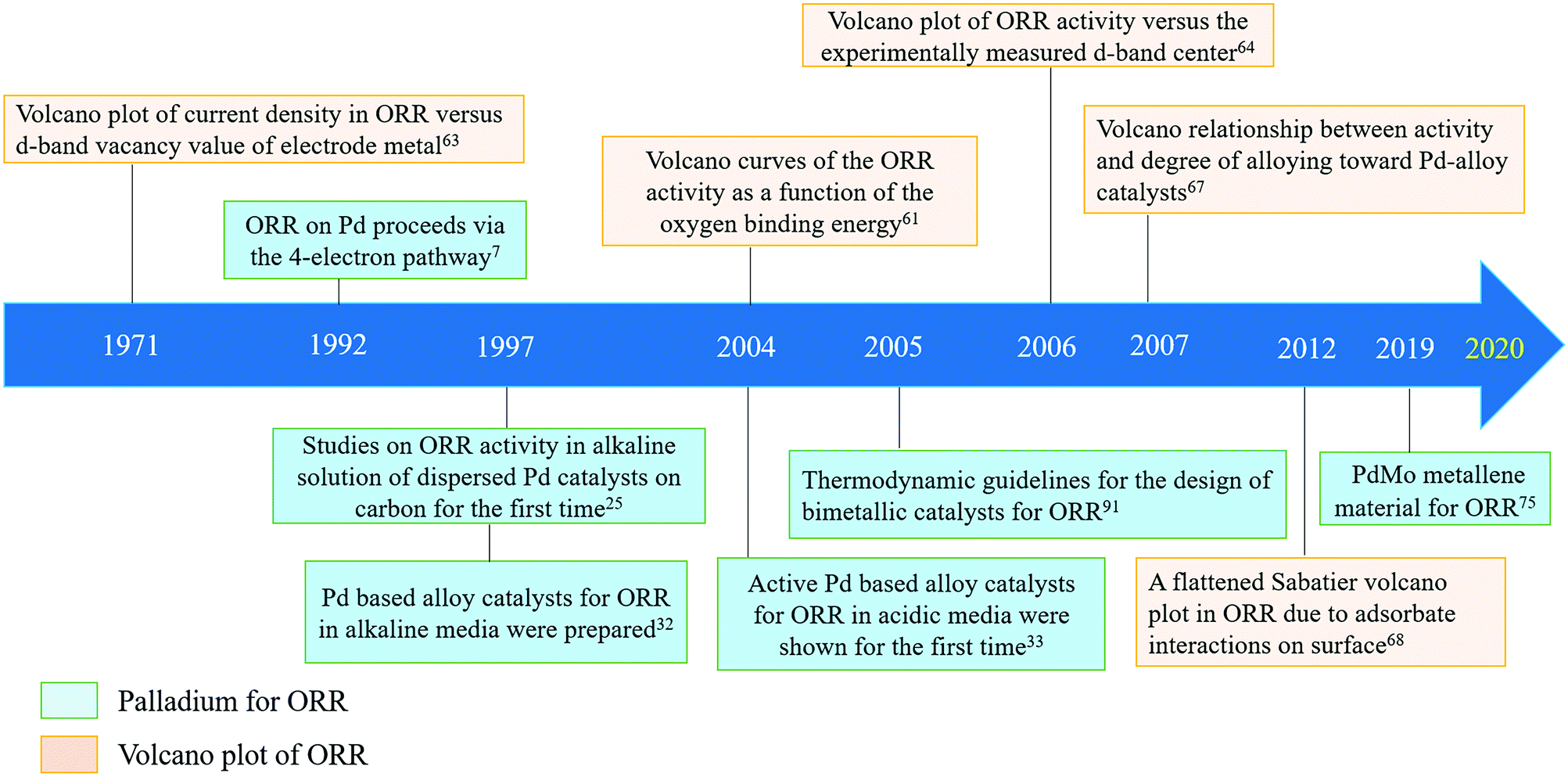 | ||
| Fig. 6 A timeline of palladium (alloys) for the ORR and volcano plots towards ORR activity development. | ||
5. Methods to enhance the ORR activity
There are many factors that contribute to the enhancement of the ORR activity. In this section, commonly used methods to enhance the ORR performance are concluded.5.1 Alloying
There is a growing recognition of enhancing ORR performances by the effect of the alloying process. The ORR activity of Pd can be dramatically enhanced by alloying with other metals, such as Pt,76 Co,33 Fe,36 Cr,77 Ni,78 Rh,79 Ag,80 Au,81 Cu,82 Mo,75 Ti,83 Sn,84 Y,85 Mn,86 W,87 Zn,88 Ru89 and Ir.90Fernundez et al. proposed a simple thermodynamic model to interpret the enhanced activity on alloying.91 This model involves the combination of one metal that will easily break the O–O bond of O2 (forming adsorbed atomic oxygen) with another metal that will easily reduce the adsorbed atomic oxygen. The analysis of the Gibbs free energies of these two reactions guides the selection of combinations of metals that can produce alloy surfaces with enhanced activity for the ORR. In their experiment, PdCo/C (10–30% Co) electrodes exhibited remarkable activities for the ORR, close to that of carbon-supported Pt. For Pd with fully occupied valence d-orbitals, Balbuena et al. argued that alloying with transition metals, such as Co with unoccupied valence d-orbitals, reduced significantly the Gibbs free energy of both the first charge-transfer step, and the steps involving the reduction of intermediates.92
In addition, intrinsic Pd surfaces bind oxygen too firmly to allow the efficient removal of the adsorbed reaction intermediates, which affects the ORR activity. For example, in Adzic et al.'s work, the volcano-type dependence of the ORR activity on the binding energy of the oxygen and the d-band center of the noble metal overlayers was established.66 They noted that alloying Pd with some transition metals could lower the d-band position of the noble-metal overlayers and therefore may improve the activity significantly by inducing strain and electron redistribution between the substrates and the overlayers. Henkelman et al. investigated the effect of the alloy composition in Pd/Cu nanoparticles on the ORR activity.93 The activity enhancement is due to the difference in how the two metals respond to a shift in their d-band centers. For instance, charge transfer from Cu to Pd raises the d band of Cu and lowers that of Pd, resulting in stronger binding between oxygen and Cu and weaker binding between oxygen and Pd. Henkelman et al. also pointed out the charge redistribution between the core and shell in Pd-based core–shell nanoparticles, which became an important factor for lowering the d-band center to promote oxygen reduction.94 Moreover, the binding energy of the dissociated O2 molecule is linearly related to the average d-band energy of electrons in the Pd shell. Onana et al. argued that the enhancement of PdCu alloys was attributed to an optimal d band property that makes OOH dissociative adsorption easier.95 Recently, Crooks and co-workers also demonstrated that the activity of the individual sites on the surface of the nanoparticles can influence the overall activity.96,97 For example, alloying Pd with Au weakens the O-binding energy on AuxPd(300−x) alloys, which results in increased ORR activity, but in PdxIr(100−x) the effect is opposite as the O-binding energy increases at the active sites containing Ir.96,97
Additionally, Ou revealed that an ideal Pd-based bimetallic alloy catalyst for the ORR should possess simultaneously negative alloy formation energy and negative surface segregation energy of Pd.98 The alloy formation energy of Pd with transition metals M can be mainly determined by their electron interaction, which could be the origin of the negative alloy formation energy for Pd–M alloys. The surface segregation energy of Pd is primarily determined by the surface energy and the atomic radius of M. M with a smaller atomic radius and higher surface energy would tend to favour the surface segregation of Pd in the corresponding Pd–M alloys.
Furthermore, recent studies found that the ORR activity was related to transition-metal dissolution. For example, Han et al. found that the amount of transition-metal dissolution from PtM (Pt–metal) nanoparticles increases when Pt is alloyed with more negative Vdissolve transition metals despite their strong alloy-formation energy, where Vdissolve or the dissolution potential is the thermodynamic potential of the dissolution of the transition metal (M ⇔ Mn+ + ne−) at pH = 0.99 Moreover, the increase of transition-metal dissolution and the decrease of Vdissolve were correlated with the ORR activity of PtM nanoparticles. Among all PtM nanoparticles examined, PtFe nanoparticles were found to have the highest ORR specific activities, roughly three times better than that of Pt nanoparticles before voltage cycling. Similar to PtM, PdM is expected to have an analogous effect.
5.2 Structure, strain and ligands
Many studies describe the role of the structures and morphologies of electrocatalysts in the enhancement of the ORR. There have been many efforts to manipulate the structure and shape of catalysts during the synthetic process to fabricate metallic nanoparticles. Structure or shape-controlled metallic catalysts have exhibited improved electrochemical activities because of the exposure of a particular surface, which is favourable for electrocatalytic reactions. Hong et al. synthesized PdPt alloy nanocrystals (NCs) with hollow structures such as nanocages with porous walls and dendritic hollow structures.76 They found that the type of surface facet plays a crucial role in determining the ORR activities of PdPt NCs. PdPt nanocages prepared from octahedral Pd NC templates exhibited the largest improvement of the ORR performance. Duan et al. prepared nanoporous PdCr alloys with uniform ligament dimensions and a controllable bimetallic ratio.77 Specifically, the nanoporous Pd75Cr25 alloy displayed the highest specific kinetic activity with a value of ∼0.24 mA cm−2 at 0.9 V, which is more than three times higher than that of Pd, and also higher than that of Pt/C (0.15 mA cm−2) catalysts. The Pd75Cr25 alloy exhibited a higher mass activity (0.16 A mg−1), which was nearly 1.4 times that of Pt/C, and 3.1 times that of nanoporous Pd. The improved overall ORR performances of the PdCr alloy could be ascribed to its excellent structural integrity and continuity as well as the appropriate changes in the Pd electronic structure induced by alloying with Cr. By DFT calculations, the authors clarified the reasons for the enhanced ORR performance. The decrease of electron back-donation from the Pd 4d orbital to the 2p* orbital of O usually results in the downshift of the d-band center of Pd, generating a weaker metal–O bond, which was proposed to be the possible cause for the higher ORR activity of PdCr alloys than Pt/C and Pd catalysts.100,101Core–shell nanoparticles often exhibit improved catalytic properties due to the lattice strain created in these core–shell particles and the substrate material provides an additional advantageous modification of the electronic structures of surface atoms. Chen et al. reported the synthesis of bimetallic PdAu nanoparticles with a core–shell construction.102 The sufficient lattice strain imposed by the Au core and Ag removal, which could tailor the d-band center of the Pd shell, accounted for the enhanced ORR performance of the core–shell Au@Pd nanoparticles. Also, Suo et al. noted that the key to improving Pd-based catalysts for the ORR is alloying Pd with elements of smaller atomic size to form a “Pd-shell/alloy-core” structure to take advantage of the lattice-strain effect and to prevent the disadvantageous surface-ligand effect.67
Xiong et al. also studied the surface structure and the strain in PdPt core–shell nanocrystals.103 From a geometrical phase analysis, they found that most of the Pt shells in the icosahedral Pd@Pt nanocrystals are dominated by compressive strain at a specific size, while compressive and tensile strains co-exist in the Pt shells of the octahedral Pd@Pt nanocrystals, which might be responsible for their different ORR properties. Furthermore, Yang et al. recently prepared three bimetallic PdZn nanoparticles for the ORR and found that the catalytic performance followed the order of Pd@Zn_core–shell > PdZn_ordered ≫ PdZn_disordered, which can be attributed to the effects of different surface structures of the catalysts.88 Firstly, from the structural perspective, the well-defined core–shell structure and ordered structure can provide more predictable control over geometric and structural effects for catalysis optimization. Secondly, stronger electronic interactions exist in Pd@Zn_core–shell over PdZn_ordered and over PdZn_disordered. Such electronic interactions could make the d-band center of Pd downshift, weakening the adsorption of the oxygenated intermediates.
He et al. prepared AuM (M = Pt/Pd) alloyed flowerlike-assembly nanochains (FANs) for an enhanced ORR.104 The authors discovered that the enhanced performances of AuM FANs are mainly ascribed to the interconnected porous bimetallic-alloyed structures, which provide an enlarged electrochemical surface area (ECSA) and more available active sites for the ORR, promote O2 diffusion and electron transport, and suppress Ostwald ripening. Furthermore, the highly ordered structures and tight interconnection of AuM FANs facilitate the mass transportation of reactant molecules and increase the electrochemically utilized Pt/Pd atoms.
Conformal deposition of Pt as ultrathin shells on facet-controlled Pd nanocrystals offers a great opportunity to enhance the catalytic performance while reducing the mass loading. Wang et al. proposed such a system by depositing the active metal as shells of only a few atomic layers on nanocrystals made of another metal, together with an optimized surface structure.105 Owing to lateral confinement imposed by twin boundaries and thus vertical relaxation only, the platinum overlayers evolve into a corrugated structure under the compressive strain. Brandiele et al. described the facile synthesis of Pd3Y alloy nanoparticles and confirmed that a strong ligand effect due to the introduction of Y in the Pd lattice affords better catalytic activity through electrochemical characterization.85 Wang et al. describe a facile method for the preparation of Pd-rich PdxCo alloy nanoparticles supported on carbon, using an adsorbate-induced surface segregation effect.106 The electronic properties of Pd were modulated by alloying with different amounts of Co, which affects the ORR activity. Xiao et al.'s work offered compelling evidence that the surface strain in Pd-based alloyed nanoparticles can be readily tuned by adding Co and Fe elements to achieve optimal electrocatalytic performance.107 Moreover, Ham et al. showed that the electrochemical activity of Pd3Co alloy catalysts towards the ORR can be enhanced by adding a small amount of Ir.90 Their study highlighted that the enhancing effect is attributed to the synergetic interplay between the surface electronic structure modification due to underlying Ir atoms and the compressive strain caused by the Pd3Co substrate. DFT calculations showed that the PdIrCo ternary alloying leads to a noticeable reduction in the DOS peak intensity near the Fermi level and a downshift in the d-valence band center, compared to the monometallic Pd(111) surface. They further noted that the addition of Ir to the Pd3Co alloy causes a slight increase in the activation energy for O–O bond breaking but it significantly decreases for O and OH hydrogenation, leading to improved ORR activity. The ORR activities of typical Pd-based electrocatalysts with different structures mentioned in this section are compared in Table 2.
| Materials | Mass activity (mA mgPGM−1) | Specific activity (μA cmPGM−2) | Half-wave potential E1/2 (V vs. RHE) | ECSA (m2 g−1) | Measured potential (V vs. RHE) | Electrolyte |
|---|---|---|---|---|---|---|
| a mA mgPt−1. b μA cmPt−2. | ||||||
| PdPt nanocages76 | 764.7 | N/A | N/A | 40.3 | 0.85 | 0.1 M HClO4 |
| Pd75Cr25 alloys77 | 160 | 240 | N/A | N/A | 0.90 | 0.1 M HClO4 |
| Pd@Pt icosahedra103 | 3490a | 3020b | N/A | N/A | 0.90 | 0.1 M HClO4 |
| Pd@Zn core–shell88 | 44.05 | 48.83 | 0.82 | 90.22 | 0.85 | 0.1 M KOH |
| AuPd FANs104 | 142.21 | 480 | N/A | N/A | 0.90 | 0.1 M KOH |
| PdY NPs85 | 146 | 575 | 0.851 | 24.83 | 0.90 | 0.1 M H2SO4 |
| PdY NPs85 | 213 | 174 | 0.883 | 24 | 0.90 | 0.1 M KOH |
| Pd2FeCO@Pt/C107 | 2500a | 128b | N/A | N/A | 0.90 | 0.1 M HClO4 |
5.3 Particle size and crystal facets
The particle size of catalysts is also a complicated element in determining the ORR activity and many studies focused on the relationship between the size of Pd nanomaterials and the catalytic performance. For example, Zhou et al. noted that the Pd nanoparticle size has a strong impact on the ORR in acidic solutions and an optimal average Pd particle size is in the range from 5.0 to 6.0 nm.108 Meanwhile, Jiang et al. conducted similar studies in alkaline solutions and pointed out that the ORR activity of the catalysts varies with the Pd particle size and carbon-supported Pd catalysts with average Pd particle sizes from 3 to 16.7 nm are highly active for the ORR.109 It is noted that the stronger adsorption of OH on smaller particles would block the active reaction sites, so the particle sizes are not as small as possible.In regards to the particle size effect of PdM alloys, the number of related studies is relatively limited. Castegnaro et al. revealed that, in alkaline electrolyte, for bimetallic nanoparticles consisting of highly crystalline nanoalloys with a size of about 5 nm, the charge transfer involving Pd and M atoms affects the activity of the catalysts.110 Furthermore, they noted that different compositions may induce different valence band structure and the materials whose d-band center is closer to the Fermi level will have higher ORR activity.
Crystal facets are also crucial aspects, since relevant research found that the ORR activity depends strongly on the orientation of the Pd surface.10 For instance, Kondo et al. studied catalytic behaviours for the ORR in acidic electrolytes on low index planes, the n(100)–(111) and n(100)–(110) series of Pd, and concluded that the (100) lattice plane is a possible active site for Pd and the ORR activity does not depend on the step structure.111 Specifically, the specific activity has the following order on the low index planes of single crystal Pd at 0.90 V vs. RHE in 0.1 M HClO4: Pd(110) < Pd(111) < Pd(100). This order is completely opposite to that of Pt in 0.1 M HClO4: Pt(100) < Pt(111) ≤ Pt(110). Later, Hitotsuyanagi et al. extended the study to the ORR on stepped surfaces of the n(111)–(100) series of Pd in HClO4 and revealed that the activity increases with increasing terrace atom density, showing that the (111) terrace is the active site for the ORR on the n(111)–(100) series of Pd.112
In contradiction to the above results, the high catalytic activity of Pd nanorods as compared to spherical Pd nanoparticles has been attributed to the prevalence of Pd(110) facets.113 Related oxygen reduction studies on Pd nanocubes with a preferential (100) surface orientation have been published.114,115 For example, Shao et al. indicated that the ORR activity of Pd nanocubes enclosed by (100) facets was one order of magnitude higher than that of Pd octahedra enriched with (111) facets.114 They have demonstrated that the ORR activity was strongly dependent on the surface structure of Pd nanocatalysts with exposed (100) facets being much more active than (111) facets.
5.4 Dealloying
Dealloying has generally been known as the selective removal of a less noble component for a given bimetallic alloy. Dealloying is an effective and crucial strategy to control and modify the surface electronic structure and the chemical composition of an alloyed electrocatalyst, thus enhancing the activity and stability. Much of the related research has been focused on Pt-based catalysts synthesized by dealloying towards the ORR.116,117 For Pd based catalysts, relatively limited investigations have been conducted. These Pd based ORR catalysts include PdCo,118 PdCu,119 PdZr120 and a dealloyed PdAg core Pt monolayer shell electrocatalyst.121Yang et al. investigated the ORR activity of electrochemically dealloyed PdCu3 thin films and noted that the ORR activity enhancement is due to the compressive strain in the Pd overlayer in the dealloyed Pd–Cu films.122 Also, the researchers found that the dealloyed structures and the ORR activity are dependent on the nature of the noble component of the alloy. Gunji et al. synthesized an electrochemically dealloyed PdCu3 intermetallic compound for the ORR.119 They indicated that, after electrochemical dealloying, the oxygen binding energy of PdCu3 was lower than that of the Pd(111) structure, and the longer distance between oxygen and the catalyst surface explained the weaker binding of oxygen, which led to the superior ORR activity.
In Lu et al.'s research, the electrocatalytic activity was enhanced by the dealloying process, which partially leached out the inactive transition metal species (Mn and Ni) on the surface and generated Pd-rich surfaces on the nanoparticles.86 The Pd-based dealloyed catalysts exhibited only a slight degradation in ORR activity in alkaline media, which could be reversed by repeating the dealloying process. Mondal et al. demonstrated electrochemical dealloying-assisted Pd-based catalysts (CoxCuyPdz) for the ORR and indicated that the elemental composition and dealloying-induced lattice strain and the change in the electronic structure due to the downshift in the d-band center of Pd control the overall performance of the alloy electrocatalysts.123 However, the dealloying effect of electrocatalysts for the ORR can be controversial. Lee et al. reported a comparison of alloyed and dealloyed AgPdPt nanoframes for catalyzing the ORR and concluded that the mass activity of the ternary catalysts was higher than that of the dealloyed ones with less Ag.124
Thus, to understand the dealloying effect of PdM, more studies including both experimental and theoretical approaches need to be conducted. For example, addressing questions of how to conduct systematic research in this area and to provide a general design for these systems is required.
The enhancement of the ORR activity can be attributed to many components, like alloying, structure, strain, ligands, particle size, crystal facets and dealloying. Most previous studies use theoretical methods to verify their correctness by establishing simple models, and the surface atomic structure of the alloy could be a very important factor for predicting the activity. However, bonding between metal atoms may also result in more complicated alloy properties. Catalytic reactions can also take place at surface defects, which is difficult to simulate and verify. The formation of intermediates (such as hydroxyl) could also dominate the kinetics, or these could bind irreversibly to cleaved oxygen. These factors need to be further examined.
6. PdM (Pd–metal) nanoparticles obtained by different preparation methods
There are many methods reported in the literature to prepare PdM alloys and they could be divided into chemical reduction reactions, electrochemical methods, dealloying and so on. The synthesis methods play a vital role in the morphology of catalysts, which significantly affects the performance of the catalysts. In the following chapter, the preparation methods of PdM nanoparticles for the ORR will be discussed in detail.6.1 Chemical reduction reactions
The chemical reduction method is the most common method for synthesizing alloys. In general, metal precursors are mixed with surfactants, reducing agents and/or capping agents in a solvent (aqueous phase or organic phase), and, by controlling the reaction conditions, alloy catalysts are prepared.Ramanathan et al. synthesized Pd3Ni/C catalysts by a co-precipitation method from a mixture of Pd(NO3)2·2H2O and Ni(NO3)2·6H2O salts in an aqueous solution.125 Pd–Ni hydroxides were formed by slowly adding sodium hydroxide (NaOH) solution and hydrogen was bubbled as a reducing agent at 80 °C, which was further annealed in a reducing atmosphere (10% H2 + 90% Ar) to reduce the hydroxides to the metallic form at about 300 °C for 2 hours. Similarly, Wang et al. prepared PdCu alloys by a reduction reaction in a hydrogen atmosphere.126 They pointed out that the catalyst with the best ORR performance was the alloy nanoparticles with a Pd–Cu molar ratio of approximately 1:1.
Dai et al. reported PdW alloy catalysts for the ORR by reducing palladium(II) chloride and ammonium tungstate with H2 in a tube furnace,87 wherein Pd19W/C had the best catalytic activity for the ORR. The mass activity of the PdW alloys was two-fold that of Pd/C; however, it was lower than that of commercial Pt/C.
In addition, according to recent reports, PdCoMo catalysts,34 carbon-supported PdPtFe alloys,127 core–shell Pd–Co@Pd/C nanoparticles,106 Pd3M (M = Fe, Ni, Cu, Co) alloys,128,129 bimetallic Pt surface-enriched PtPd(x) nanoparticles,130 and core–shell fct-PdFe@Pd nanoparticles131 were prepared by using H2 to reduce metal precursors.
Yang et al. synthesized three types of PdZn with different surface architectures by using NaBH4 as the reducing agent.88 Through heat treatment under a 0.1 MPa Ar/H2 (10%) atmosphere at 600 °C for 5 h, the disordered PdZn alloys turned into ordered PdZn alloys, and the mass activity (MA) and specific activity (SA) of ordered PdZn were markedly higher than those of disordered PdZn. Core–shell PdZn was also obtained through a slightly different reaction process and had better ORR performance. The TEM images of these disordered PdZn, ordered PdZn and core–shell Pd@Zn are presented in Fig. 7(a–c).
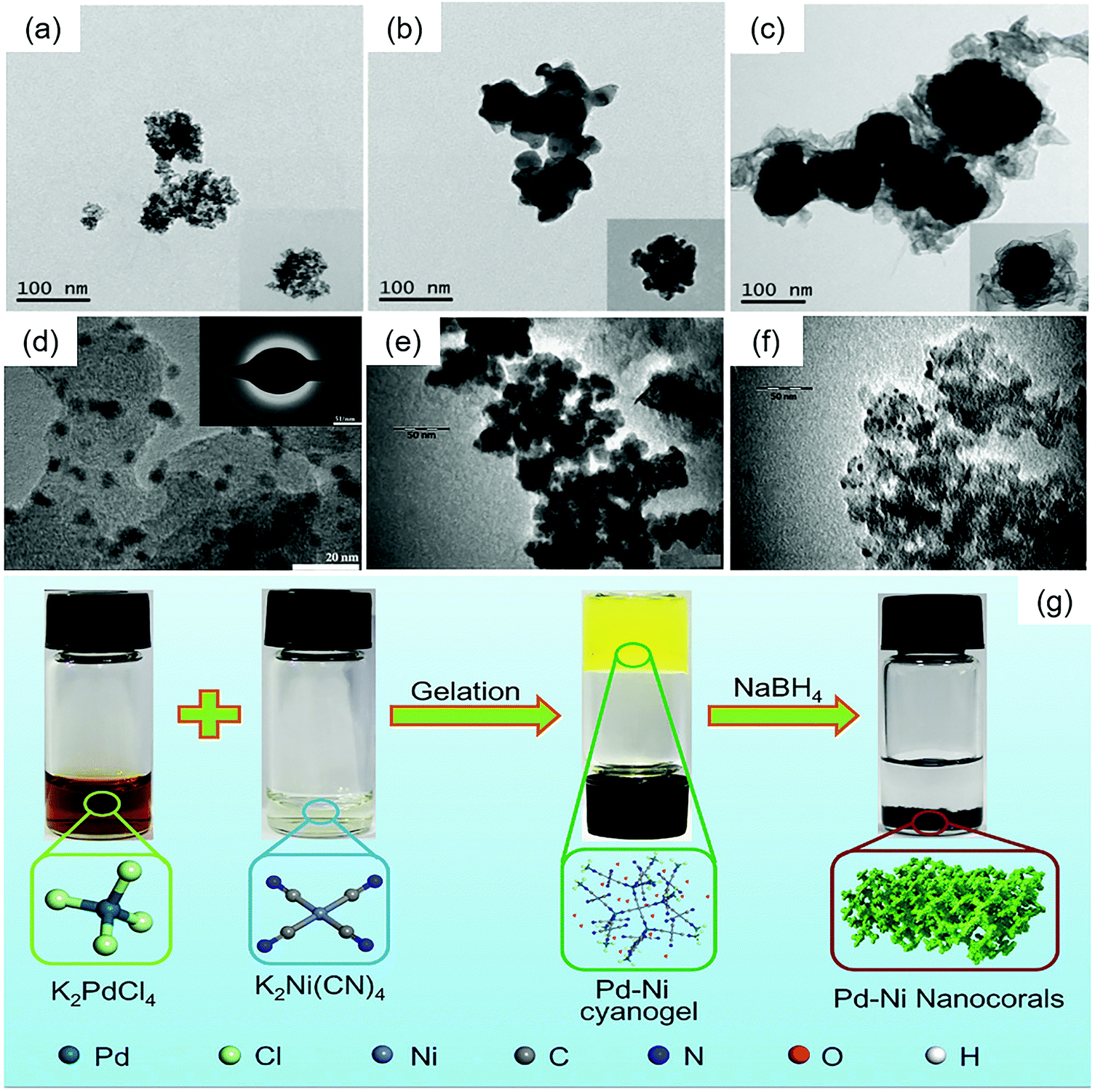 | ||
| Fig. 7 Transmission electron microscopy (TEM) images of (a) disordered PdZn, (b) ordered PdZn, and (c) core–shell Pd@Zn. Reproduced with permission.88 Copyright 2020, Elsevier. (d) 20 wt% Pd3Fe/C. The inset shows selected area diffraction patterns of Pd3Fe/C. Reproduced with permission.12 Copyright 2011, Elsevier. (e) PdNi2/VC and (f) PdNi2/MC. Reproduced with permission.133 Copyright 2012, Elsevier. (g) Schematic illustration of the synthesis of Pd–Ni nanocorals through a cyanogel-reduction method. Reproduced with permission.134 Copyright 2018, Wiley-VCH. | ||
Fernández et al. prepared carbon supported PdCoAu electrocatalysts by a reverse microemulsion method using sodium dioctylsulfosuccinate (AOT) as the surfactant, heptane as the oil phase and sodium borohydride as the reducing reagent.83 Remona et al. synthesized PdPt alloys by a microemulsion method using NaBH4 as the reductant at room temperature.132
Neergat et al. prepared carbon-supported PdFe electrocatalysts (Pd to Fe ratios = 1:1, 2:1, and 3:1) by a co-reduction method at 80 °C in alkaline media (pH = 10) with sodium borohydride (NaBH4) as a reducing agent and without any stabilizing agents,12 as shown in Fig. 7(d). Similarly, Ramos-Sánchez et al. reported that mesoporous carbon-supported nanoparticulated PdNi2 (Fig. 7(e and f)) exhibited higher catalytic activity for the ORR, which was synthesized by the reduction of metal chlorides with NaBH4 in aqueous media.133
The preparation of carbon-supported PdCo alloys,34,135 PdV alloys,136 PdFe alloys,12 and PdAu nanoparticles137 also used the NaBH4 reduction method.
Recently, some PdM alloys were fabricated via the cyanogel-reduction method with NaBH4 as the reducing agent. A schematic illustration is presented in Fig. 7(g). Firstly, a cyanogel of two metals is formed, and then reduced with NaBH4. Liu et al. reported the preparation of PdNi nanocorals with hierarchical porosity materials through the formation of a Pd–Ni cyanogel followed by chemical reduction with NaBH4 solution.134 Xu et al. prepared polyallylamine (PAA)-functionalized PdCo alloy nanonetworks by a functional molecule assisted cyanogel-reduction method using a NaBH4/PAA mixture as the reductant.138 A similar strategy was used to prepare polyethyleneimine (PEI)-functionalized PdNi alloy nanostructures,139 and reduced graphene oxide supported PdNi alloy nanocrystals.140
Zhao et al. reported a polyol reduction method using ethylene glycol as the solvent as well as reducing reagents to synthesize carbon-supported PdNi nanoalloy electrocatalysts.78 After the heat treatment at 500 °C, the degree of alloying (atomic percentage of Ni in the alloy) was increased from 0 to 13.3%. They found that, with increasing the heat-treatment temperature (or Ni content), more Ni atoms get into the Pd lattice, resulting in higher lattice strain, weaker M–Oads bonding, and higher specific activity. However, the ECSA decreases significantly with temperature, and thus the mass activity shows a volcano relationship with temperature. PdPt catalysts were also synthesized by using a modified polyol process and ethylene glycol (EG) was used as both a solvent and a reducing agent.141–143 A similar process was used to synthesize PdIr alloy networks.144
Jang et al. presented an ultrasound-assisted polyol process without any added surfactant, pH adjuster, or stabilizer to prepare PdCo nanoparticles with two different structures (core–shell structure and bimetallic alloy nanoparticles, as shown in Fig. 8(a and b)).145 The difference between the preparation methods of the two structures is whether the Co precursor is firstly added to prepare cobalt seeds or Pd and Co precursors are added to the reaction at the same time. The Pd4Co core–shell structure shows dramatically enhanced ORR activity (its mass activity was 173 mA mg−1 and its specific activity was about 480 μA cm−2).
 | ||
| Fig. 8 TEM images of (a) as-synthesized Pd80Ni20 and Pd80Ni20 after heat treatment at (b) 500 °C. Reproduced with permission.78 Copyright 2010, Elsevier. (c) PdMo bimetallene. Reproduced with permission.75 Copyright 2019, Springer Nature. (d) Pd6Ni icosahedra.150 (e) RhPd alloy cubes and (f) RhPd alloy octahedra. Reproduced with permission.79 Copyright 2015, Royal Society of Chemistry. (g) Truncated cubic, (h) cuboctahedral, and (i) branched (tetrapod-like) CuPd nanocrystals. Reproduced with permission.152 Copyright 2012, Royal Society of Chemistry. (j) Schematic diagrams and TEM images of the tree-like Pd3Ag nanocrystals. Reproduced with permission.153 Copyright 2019, Royal Society of Chemistry. Insets: (c) HRTEM image of PdMo bimetallene; (e and f) TEM images of individual nanocrystals at a higher magnification; (g) HRTEM image of a truncated nanocube and (h) high resolution scanning electron microscopy (HRSEM) image of the cuboctahedra. | ||
In addition, PdM (M = Ni, Fe, Co) nanoparticles were obtained through the reduction of the metal precursors, and 1,2-hexadecanediol was used as a reducing agent and oleic acid and oleylamine as capping agents.146 Octahedral Pt–Pd nanoparticles were synthesized by reducing metal precursors with glycerol as a reducing agent and showed good ORR activity in buffer solution.58
Luo et al. reported PdMo bimetallene (Fig. 8(c)) for the reduction of oxygen with the highest mass activity (16.37A mgPGM−1) so far in 0.1 M KOH solution at 0.90 V.75 This activity was 77.9 and 327.4 times higher than that of commercial Pt/C and Pd/C catalysts, respectively. The value of the specific activity was 11.64 mA cmPGM−2. They used ascorbic acid (AA) to reduce [Pd(acac)2] and Mo(CO)6 in oleylamine at 80°C for 12 h to obtain this material. In acid electrolytes, PdMo bimetallene also exhibited better ORR activity than Pt/C, but its stability was poor for practical applications. Zuo et al. prepared PdCuCo anisotropic structure catalysts by using ascorbic acid (AA) to reduce metal ions.148 At 0.8 V vs. RHE, the SA of this catalyst could reach 1.61 mA cm−2, and the MA was 1.135 A mg−1 in 0.1 M HClO4 solution.
Wang et al. prepared PdCuNi nanocrystals (NCs) by reducing palladium acetylacetonate ([Pd(acac)2]), copper acetylacetonate ([Cu(acac)2]), and nickel acetylacetonate ([Ni(acac)2]) in oleylamine (OAm) with ascorbic acid (AA) or benzoic acid (BA) serving as the reducing agents.149 Through the surface treatment protocol by adding a mixed solution of H2O2 and sulfuric acid to remove the surface Ni and Cu atoms, the treated catalysts (PdCuNi-AB-t/C) exhibited a mass activity of 0.45 A mg−1 Pd in alkaline medium at 0.90 V vs. RHE. It is noted that, before the acid treatment, the MA of the catalysts was 0.10 A mg−1 Pd under the same conditions. Besides, different reducing agents resulted in various catalytic activities; the combination of BA and AA was better than sole BA or sole AA in this research. Feng et al. prepared PdNi icosahedra (Fig. 8(d)) for the ORR with a mass activity of 0.22 A mg−1 Pd and a specific activity of 0.66 mA cm−2 Pd at 0.9 V vs. RHE in 0.1 M KOH solution.150 Pd(acac)2 and Ni(HCO2)2·2H2O were used as precursors, ascorbic acid (AA) was selected as a reducing agent, and oleylamine (OAm) and 1-octadecene (ODE) were applied as solvents and stabilizers. PdPt alloy nanodendrites151 were also prepared by using AA to reduce metal ions. Yan et al. reported the synthesis of RhPd alloy nanocrystals for the ORR by using an extremely slow injection method79 (Fig. 8(e and f)). This synthesis involved the simultaneous injection of Na3RhCl6 and Na2PdCl4 using a syringe pump at a rate of 2 mL h−1 into ethylene glycol (EG) with ascorbic acid (AA) and KBr serving as reducing and capping agents, respectively. The Rh8Pd92 alloy octahedra exhibited high mass activity with a value of 0.18 mA μg−1 in terms of the equivalent Pt cost.
The hierarchical Pd4Fe nanoflowers (NFs) (Fig. 9(a)) in Lian et al.'s work were prepared by reducing palladium acetylacetonate with Fe(CO)5 and oleylamine at 120 °C.155 The MA of the Pd4Fe NFs was 521 mA mgPd−1 at 0.85 V, and the SA of this catalyst was 1.57 mA cm−2 in a 0.1 M HClO4 aqueous solution. Meanwhile, in a 0.1 M KOH aqueous solution at 0.85 V, the MA and SA were 4.07 A mgPd−1 and 11.6 mA cm−2, respectively. Regardless of acidic or alkaline electrolytes, the Pd4Fe NF catalyst exhibited excellent catalytic activity towards the ORR. Cui et al. prepared a Pd3Pb intermetallic compound (Fig. 9(b)) for the ORR by a modified impregnation–reduction approach.156 It included a mixture of Pd and Pb precursors with carbon black in THF, co-reduction of the Pd and Pb precursors, and heat treatment. Potassium triethylborohydride (KEt3BH) and lithium triethylborohydride (LiEt3BH) were used as reducing agents because they both had fast reduction kinetics. The MA of the Pd3Pb catalysts was 168.9 mA mgPd−1 at 0.9 V in KOH solution. He et al. synthesized alloyed PdAu nanochain networks (NCNs) (Fig. 9(c)) by using freshly-prepared hydrazine (80%) to reduce an allantoin solution of HAuCl4 and H2PdCl4.157 At 0.85 V vs. RHE in 0.1 M KOH solution, the SA and MA of the PdAu NCNs were 0.59 mA cmPd−2 and 86.01 mA mg−1, respectively. The presence of allantoin suggested an increased degree of alloying of Au with Pd, which was conducive to the improvement of the catalytic activity.
 | ||
| Fig. 9 Representative TEM images of (a) Pd4Fe NFs. Reproduced with permission.155 Copyright 2018, Royal Society of Chemistry. (b) Ordered Pd3Pb/C. Reproduced with permission.156 Copyright 2016, American Chemical Society. (c) Alloyed PdAu nanochain networks. Reproduced with permission.157 Copyright 2015, Elsevier. (d) Pt1Pd1/C.161 (e) Pd31Bi12/C nanoparticles and the corresponding selected area electron diffraction (SAED) pattern. Reproduced with permission.172 Copyright 2019, American Chemical Society. (f) SEM images of NP-Pd75Cu25, and (g) alloys by dealloying the source alloys in 0.2 M H2SO4 solution for 48 h at room temperature. The inset of (g) is the TEM image of NP-Pd75Cu25. Reproduced with permission.82 Copyright 2013, Elsevier. SEM (h) and TEM (i) images of the resulting samples by dealloying the PdNiAl alloy in 0.5 M NaOH solution for 48 h at room temperature. Reproduced with permission.173 Copyright 2013, Royal Society of Chemistry. | ||
Kuai et al. prepared PdAu core–shell nanoparticles by using polyvinylpyrrolidone (PVP) to reduce a solution of HAuCl4 and H2PdCl4.158 Moreover, if cetyltrimethylammonium bromide (CTAB) was added, polycrystalline PdAu alloys were prepared. PdPt popcorn-shaped catalysts were obtained via a hydrothermal method involving the co-reduction of K2PtCl4 and PdCl2 in an aqueous solution containing PVP and NaI.159 The authors found that if NaI was absent, octahedra (≈20 nm) and very small particles (≈3 nm) were generated for Pt and Pd. The bio-synthesis of palladium nanocubes (PdNCs) was realised using pine needle extract as the reducing agent and CTAB as the capping agent. As eco-friendly and readily available biomass, pine needle extract avoided the use of highly polluting chemical reducing agents.160 1-Naphthol ethanol solution as the reductant and structure-directing agent was applied to fabricate tree-like PdxAgy nanocrystals153 (as shown in Fig. 8(j)). Chemical reduction reactions can be used to prepare metal alloy catalysts with various morphologies and properties, and a reasonable selection of reducing agents cannot be ignored. However, the reaction temperature ranges from room temperature to several hundred degrees Celsius, and the types of reagents and experimental methods used in the reaction vary greatly. To obtain a catalyst with excellent performance, it is very important to explore the experimental conditions.
6.2 Electrochemical methods
Electrochemical methods are another way to prepare alloys. Compared with traditional chemical reduction methods, electrochemical methods are environmentally friendly without organic reducing agents, surfactants or high temperature, and provide a rapid and effective route to prepare hollow nanostructures. However, to accurately control the structure, these methods often require complicated multistep operations and specific equipment, such as pulsed electrodeposition, which may not be available in many laboratories.Betancourt et al. reported a strategy to prepare PdAg alloys for the ORR using a UPD method.80 They have used underpotentially deposited Cu and subsequent galvanic displacement to deposit atomically dispersed Pd to achieve precise tuning of the electronic properties. The Pd@Ag/C catalysts had a higher normalized Pd mass activity with a value of 878 mA mg−1 in 0.1 M NaOH at −0.1 V vs. Hg|HgO. In addition, Gobal et al. synthesized PdCu alloys by an electrodeposition method.167
Xu et al. prepared nanotubular mesoporous PdCu catalysts through a galvanic replacement reaction using dealloyed nanoporous Cu as both the template and reductant.170 This material exhibited a half-wave potential of 0.840 V for the ORR, superior to the commercial Pt/C (0.825 V) in 0.1 M HClO4 solution. PdFe@PdPt/C was also prepared by the galvanic replacement reaction between PdFe/C alloy nanoparticles and PtCl42−.127
Wang et al. presented a pulsed electrodeposition process to prepare PdBi alloy electrocatalysts for the ORR.172 Pd31Bi12 ordered alloys (Fig. 9(e)) were grown directly onto carbon supports by deposition from an aqueous electrolyte containing ethylenediammenetetracetic acid (EDTA), Bi(C2H3O2)3 and Pd(NO3)2 at 30 °C. The SA and MA of Pd31Bi12 were 2.42 ± 0.2 mA cmPd−2 and 0.95 ± 0.18 A mgPd−1 at 0.9 V vs. RHE in 0.1 M KOH electrolyte.
6.3 Dealloying
Nanoporous materials obtained by the dealloying method have been proved to represent a particular class of multifunctional catalytic nanoarchitectures with interconnected nanoscale skeletons and voids,174 which are especially beneficial for electron and mass transport during electrocatalytic processes. The advantages of dealloying are simple and high-yielding preparation methods without any assistance from organic solvents.Zhang et al. showed that nanoporous (NP) PdCu alloys (as presented in Fig. 9(f and g)) synthesized by dealloying from ternary PdCuAl source alloys exhibited superior ORR activity and higher catalytic durability compared with the Pt/C catalyst.82 PdCuAl alloy foils were made by refining pure Pd, Cu, and Al at high temperature in an arc furnace, followed by melt-spinning under an Ar atmosphere. The nanoporous metals were prepared by dealloying the alloy foils in 0.2 M H2SO4 solution. The mass activity of NP-Pd50Cu50 is 0.15 A mgPd−1, and the specific activity is 0.22 mA cm−2. Similarly, Xu et al. presented NP-PdNi alloys by etching PdNiAl alloy foils in 0.5 M NaOH solution,173 as exhibited in Fig. 9(h and i). NP-PdNi showed higher specific activity with a value of about 0.21 mA cm−2 at 0.9 V and the mass activity of NP-PdNi is about 0.15 A mgPd−1. PdTi alloys175 and PdCe176 were also prepared by a similar method. In Begum et al.'s study, Pd nanonetworks (Pd-Net) without any support material were prepared by using 0.1 M HCl etching of PdZn alloys.177 The introduction of CTAB plays a crucial role in the structure and morphology of the catalysts. Pt-Net had a higher ECSA and ORR performance than those of commercial Pt/C and homemade Pd nanoparticles in alkaline media.
Sun et al. reported a process for converting colloidally synthesized ordered intermetallic PdBi2 to ordered intermetallic Pd3Bi nanoparticles under ambient conditions by electrochemical dealloying to significantly enhance the ORR activity,180 as shown in Fig. 10(a and b). The SA and MA of converted Pd3Bi were 2.3 ± 0.19 mA cmPd−2 and 1.2 ± 0.08 A mgPd−1 at 0.9 V in alkaline media.
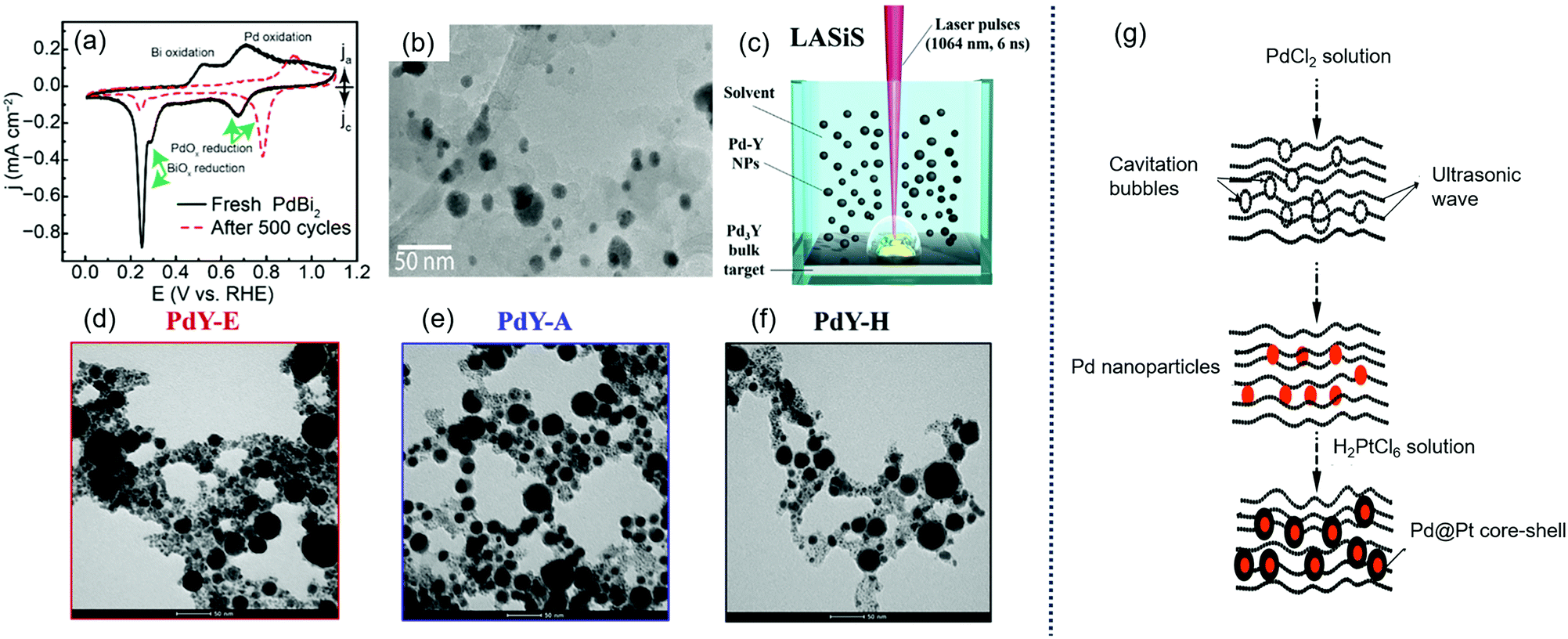 | ||
| Fig. 10 (a) Cyclic voltammograms (CVs) of PdBi2 in Ar-saturated 0.1 M KOH before and after the electrochemical dealloying; and (b) TEM image of dealloyed PdBi2 supported on Vulcan carbon. Reproduced with permission.180 Copyright 2019, American Chemical Society. (c) Sketch of PdY NPs preparation by laser ablation synthesis in solution (LASiS); and representative TEM images of PdY NPs prepared in (d) ethanol (PdY–E), (e) acetone (PdY–A) and (f) n-hexane (PdY–H). Reproduced with permission.85 Copyright 2019, Elsevier. (g) Schematic illustration of the synthesis of Pd@Pt core shell using ultrasonication. Reproduced with permission.184 Copyright 2019, Elsevier. | ||
Gunji et al. synthesized PdM (M = Fe, Co, Ni) alloy catalysts and then electrochemically dealloyed these alloys to enhance the ORR activity.129 The dealloyed Pd–M nanoparticles had a core–shell structure with a Pd3M-core and a Pd-shell.
6.4 Other methods
In addition to the above synthesis methods, other methods can also be used for the preparation of PdM alloys. These methods are not widely used in the synthesis of Pd-based alloys, although they are extensively applied in the preparation of other materials, such as carbon materials, other metal nanoparticles (Au181 and Ag182), etc. Due to the interoperability of synthetic methods, learning from the preparation of other materials can facilitate the study of Pd-based alloy materials.:1)/rGO alloy was 1.01 mA mgPd−1, and the specific activity was 806 μA cmPd−2. Zheng et al. prepared Pd@Pt/C core–shell nanoparticles via sonochemical synthesis,184 as Fig. 10(g) demonstrates.
In short, various synthetic methods of PdM alloys are summarized in Fig. 11. Among the synthetic methods of Pd-based alloys, the most commonly used method is chemical reduction, while electrochemical methods are regarded as simple and environmentally friendly routes to prepare nanomaterials with a tunable size distribution and chemical composition and desired morphology. Moreover, the number of other methods has gradually increased, and various new methods have also appeared to be used in the synthesis of Pd-based alloys with excellent ORR performance. In addition, during the synthesis of Pd-based alloys, the following concerns need to be noted.
 | ||
| Fig. 11 Comparison of different synthetic methods to obtain PdM alloys. | ||
(1) The effect of heat treatment. (a) Change the structure of alloys. In Yang et al.'s work, the disordered type is changed to the ordered type via heat treatment, which increases the ORR activity.88 Also, Maiti et al. mentioned that, through high-temperature treatment at ∼500 °C in an Ar/H2 atmosphere, phase transformation of face-centered cubic (fcc) to face-centered tetragonal (fct) PdFe alloys was achieved, which is due to the movement of core Pd atoms to the surface of the alloy as Pd has higher adsorption enthalpy of H than Fe, resulting in the formation of fct-PdFe@Pd structures.131
(b) For core–shell structures, heat treatment at a low temperature can facilitate core–shell structure formation, which plays a key role in enhancing the ORR activity of PdCo alloy catalysts.190
(c) For PdFe and PdCu alloys, annealing at elevated temperatures enabled both Pd enrichment on the surface and an increase in the ECSA, thus enhancing the ORR performance.128
(d) The degree of alloying can be increased to a certain extent, which is conducive to the improvement of the catalytic activity, but the post-treatment temperature should not be too high, which may lead to an increase in particle size and a consequent decrease in the electrochemically active surface area.78,83,135,191 But there are some exceptions; in the case of the Pd60V40 sample, heat treatment at 300 °C decreased the ORR performance of the as-prepared sample.136
(e) Remove the organic reagents (such as surfactants, capping agents, etc.).192
Therefore, these issues are clearly complicated and further experimental and theoretical work is required to fully understand the effect of heat treatment on the electrocatalytic activity.
(2) The effect of the CO reagent. Through the comparison of Fig. 12, it is noted that a CO reagent is used in the synthesis process of many Pd-based alloy catalysts with extremely high catalytic activity, such as PdMo bimetallene75 and Pd4Fe nanoflowers.155 Besides, Ahmad et al. used W(CO)6 to synthesize W-doped Pd nanocubes.193 At 0.9 VRHE, the specific activity of 1.2% W-doped Pd nanocubes/C was 1.18 mA cm−2, which surpassed most reported Pd-based catalysts for the ORR in alkaline media.
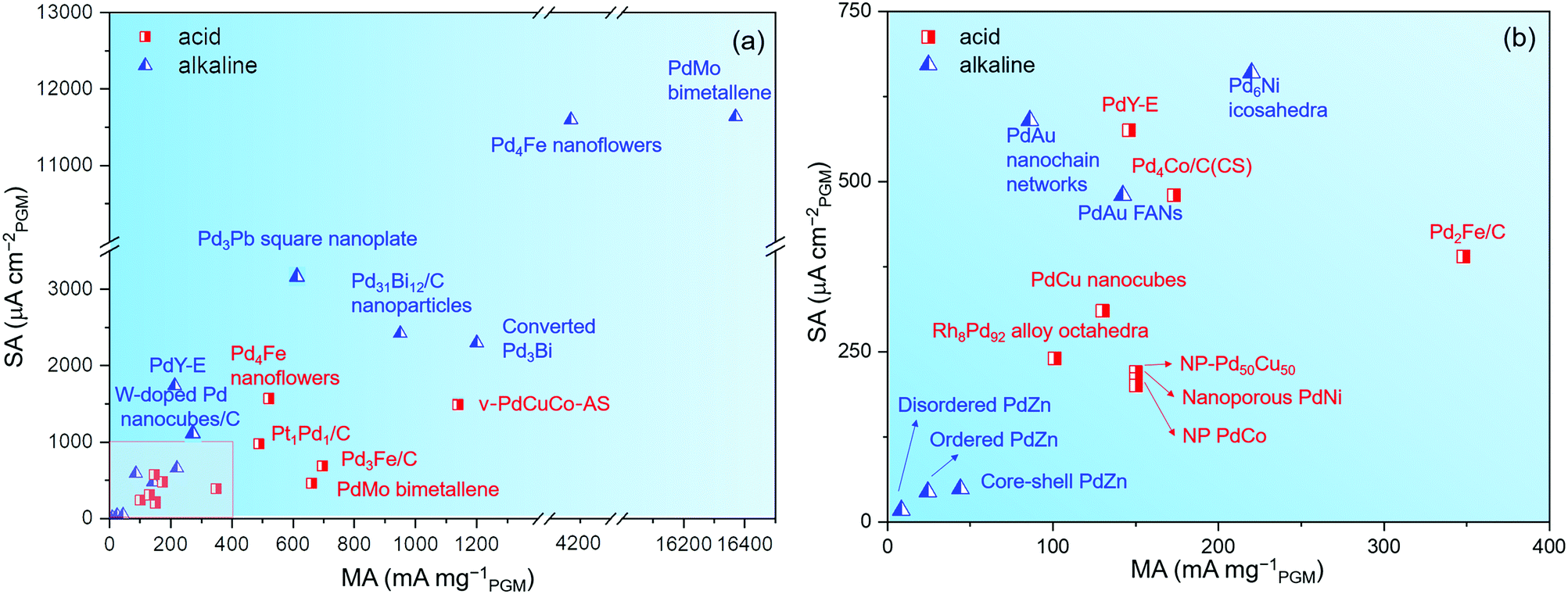 | ||
| Fig. 12 (a) MAs and SAs for different Pd-based catalysts in alkaline and acid media in recent years. (b) Partial enlarged view of (a). | ||
Some researchers have specifically studied the role of the CO reagent. For example, Wei et al. reported the effect of CO treatment for PdCoAu catalysts.194 The authors found that the CO treatment can modify the PdAu and PdCo alloy ratios in the bulk structure and result in the formation of Pd–Au alloy enriched on the outer shell, thereby increasing the ORR performance. The PdCu/C catalysts heat-treated in CO have a higher alloying degree and electrochemical activity towards the ethanol oxidation reaction compared with those treated under O2 and H2.195 The CO-confinement effect, which is the strong binding of CO on the metal (111) facets, is used to induce the formation of 2D nanomaterials.196 Therefore, more in-depth research in this aspect needs to be done.
(3) The effect of solvents. In chemical reactions, the role of the solvents is often less noticeable, but its impact cannot be ignored. The solvent will affect the morphology and structure of Pd-based catalysts.159 Zheng et al. found that, during the synthesis of both Pt and Pd nanoparticles using H2 as the reducing agent at room temperature, ethylene glycol (rather than the commonly used water) used as the media can minimize the aggregation of particles during their formation due to the high viscosity, thereby controlling the particle size.130 Also, it is found that the solvent played significant roles in controlling the synthesis of intermetallic Pd3Pb square nanoplates.154
(4) The effect of doping.
(a) To improve the selectivity. Pd-based alloys also have high catalytic activity for the methanol/ethanol oxidation reaction. To avoid the poisoning problem induced by the alcohol crossover effect, achieving the selectivity of Pd-based alloys towards the ORR is highly desired. The surface chemical functionalization of metal nanocrystals was used in Pd-based alloys to enhance the ORR selectivity, such as polyallylamine (PAA)-functionalized PdCo alloy nanonetworks.138
(b) To improve the methanol tolerance. For instance, the essential role of Rh in obtaining high methanol tolerance capacity was reported in Rh-doped PdAg nanoparticles.192
(c) To improve the ORR activity. The surface decoration induced structural change could directly influence the electronic structure, thereby improving the catalytic activity, such as for Pt submonolayer decorated PdAu/C nanocatalysts.137 Besides, polyethyleneimine (PEI) as a dopant was reported.139 Specifically, –NH2 groups on the modified PEI polymer can been protonated into –NH3+ in an acidic medium, which will increase the interfacial H+ concentration on the Pd surface and consequently accelerate proton-coupled electrocatalytic reactions (such as the ORR and HER).197
(5) The effect of reductants. Calderon et al. studied the effect of five different reductants (methanol, ethanol, n-propanol, formaldehyde and ascorbic acid) on the ORR activity of PdPt catalysts.198 Different reductants can tune the size and surface Pt content, thus influencing the properties of the catalysts. In addition to several common reducing agents, the introduction of other new reducing agents may bring surprises, such as pine needle extract as the reducing agent,160 1-naphthol ethanol solution,153 urea slowly degraded to NH3 during pyrolysis199 and a borane tert-butylamine complex.200
(6) The effect of additives. The introduction of some additives can affect the morphology and properties of the catalyst, such as EDTA,158,177 arginine,201 glycine,202 NaI,159 amines,196etc.
(7) The effect of carriers. In this review, we did not conduct a detailed analysis of the role of the carriers. But a suitable carrier will greatly enhance the catalytic performance of Pd-based alloy catalysts, such as ordered mesoporous carbon (OMC),143 defective mesoporous carbon,203 nitrogen-doped graphene,199 a CoFe2O4–Vulcan composite,200 carbide-derived carbons (TiC, Mo2C and SiC),204etc. The issue that carbon-support corrosion leads to a decrease in the stability of catalysts is worthy of further investigation.205
(8) Other considerations. For Pd-based alloys prepared with different metals M or different synthesis methods, the ratio of Pd to M at the optimal activity is uncertain. For example, the maximum activity for the ORR was observed at an alloy composition of ∼60 at% Pd for three types of PdM alloys (M = Co, Ni, Cr).186 Meanwhile, in Liu et al.'s investigation, Pd70Co30/C alloys had the highest catalytic activity.206 As for PdCu alloys, a molar ratio of approximately 1
:1 showed the optimal alloy composition.95,126 Using a combination of multiple synthesis methods to synthesize highly active ORR catalysts is a method to control the structures of the targeted materials. For instance, in You et al.'s work, three catalysts with PdNi, PdNiCu and PdCu cores and a PdIr shell were fabricated by a polyol method and galvanic replacement.207 Chen et al. reported a process to prepare nanoporous PdCe (NP-PdCe) nanocubes by melt spinning combined with chemical dealloying.176 Carbon supported PdCu alloyed catalysts were prepared by using a two-step process involving the synthesis of Cu nanoparticles, followed by galvanic substitution of Pd on Cu nanoparticles.208 Focusing on efficient bifunctional catalysts, for example, PdIr alloys144 as efficient bifunctional catalysts for oxygen reduction and oxygen evolution reactions, tree-like PdAg nanocrystals towards anodic formic acid oxidation and cathodic oxygen reduction153 were reported. Additionally, Pd alloyed with novel elements is worthy of attention, such as PdTe,209 PdCe,176etc.
Finding suitable ORR catalysts used in proton exchange membrane fuel cells (PEMFC) is a key concern, and ideal catalysts should meet the requirements of good reliability, durability and stability in acidic electrolytes.210 One of the main issues for Pd-based catalysts is Pd dissolution under the operating conditions including low pH, high temperature, high potential and frequent start–stop cycling.10 Compared with Pt, Pd is much more easily dissolved.
In acid solution, the main pathways for Pd dissolution include direct dissolution (eqn (20)) or oxide film formation (eqn (21)) and a subsequent chemical reaction (eqn (22)).211
| Pd → Pd2+ + 2e− | (20) |
| Pd + H2O → PdO + 2H+ +2e− | (21) |
| PdO + 2H+ → Pd2+ + H2O | (22) |
In the direct dissolution pathway, the equilibrium potential of Pd dissolution is 0.2 V lower than that of Pt. In the chemical dissolution pathway, the Pd2+ equilibrium concentration is ∼5 orders of magnitude higher than that of Pt2+. Thus, Pd is much less stable than Pt in an acid medium.212
It is noted that Pd/C was not regarded as an ideal catalyst for the ORR in acid solution due to the gradual degradation during potential cycling.213 However, some Pd-based alloys which had good stability in acid media were reported.148,173
In addition, Ou et al. compared the ORR mechanisms on the Pd(111) and Pt(111) surfaces in acidic solution by DFT,55 and revealed that the adsorption and dissociation processes of O2 molecules more easily occurred on the Pt(111) surface and that the serial protonation of the dissociative product to form H2O molecules could also occur more easily on the Pt(111) surface than on the Pd(111) surface, indicating that Pt can serve as a better ORR electrocatalyst than Pd.
In general, Pd-based catalysts are still not comparable with Pt-based ones in terms of ORR activity and stability in acidic electrolytes. More efforts need to be made to achieve new breakthroughs.
7. Electrochemical analysis
Electrochemical analysis determines the specific performance of electrocatalysts, which is particularly important for Pd-based electrocatalyst optimization. Accordingly, typical methods of electrochemical analysis, including determination of the electron transfer number (n) and electrochemically active surface area (ECSA), durability evaluation protocols and standardization of electrochemical analysis processes are concluded in this section, as well as the performance of representative Pd-based ORR electrocatalysts.7.1 Electron transfer number (n)
The electron transfer number per O2 molecule (n) is one of the key parameters for the ORR, which can provide information on both the oxygen conversion efficiency and the mechanisms, thus helping to evaluate the performance of electrocatalysts. The rotating ring-disk electrode (RRDE) method214 and the Koutecky–Levich (KL) method215 are the two most common experimental methods to calculate the value of n. The n values of PdPt (0.5 M H2SO4,161 0.1 M HClO4216), PdCo (0.1 M HClO4),217 PdRu (0.1 M KOH),89 PdAu (0.1 M KOH)218 alloys, etc. were investigated by the RRDE method. Based on the KL equation, the n values of Pd nanoparticles (both 0.05 M H2SO4 and 0.1 M KOH),219 PdPt (0.1 M KOH,220 0.5 M H2SO4221), PdPb (0.1 M KOH),154 PdAg (0.1 M KOH)222 and PdRh (0.1 M KOH)223 alloys were reported. Furthermore, some studies verified the n values in both ways, including PdCo (0.5 M H2SO4,135,224 0.1 M HClO4225), PdNi (0.1 M KOH,226 both 0.1 M HClO4 and 0.1 M KOH227) and PdZn (0.1 M KOH)88 alloys. These studies showed that the n values are near to four, indicating excellent performance with limited amounts of H2O2 generated in the ORR process for these Pd-based electrocatalysts.However, according to recent studies, the n values obtained by the RRDE (nRRDE) and KL (nKL) methods have discrepancies,42,224,228,229 and the n values from the RRDE method are believed to be more accurate. The KL plots are often not linear, and the nKL values sometimes exceed theoretical limits. Masa et al. proved that the KL plots are affected by the coverage of electrocatalysts on the working electrode and demonstrated that the surface area ratio (total electroactive surface area to geometric area of the rotating disc electrode (RDE) surface) should be taken into account when inferring electrocatalytic effects on the basis of KL analysis of RDE data.230 Zhou et al. pointed out that, from a theoretical viewpoint, the ORR is neither a single-step nor a one-way reaction, and therefore does not fulfil the assumptions from the KL method.231 From an experimental viewpoint, the n values are significantly dependent on the angular velocity (ω) of the RDE, contradicting the assumption of constant n in KL theory. Issues with the RRDE method were also presented in detail by Zhou et al.'s study.231 For example, the collection efficiency (NC) of the RRDE decreases significantly with the catalyst loading. Moreover, when the electrode surface is rough, NC also decreases dramatically with ω. In addition, the widely applied RRDE method with a Pt ring is not suitable for H2O2 collection in alkaline electrolytes because the oxidation of H2O2 on Pt is not a mass-transfer-limited process. A properly biased Au ring rather than a Pt ring is more suitable. As a result, the RRDE method with a properly biased Au ring is recommended to determine n values for the ORR in alkaline electrolytes, supplemented by the calibration of the collection efficiency. Therefore, as for Pd-based electrocatalysts, the best possible way is that the RRDE with the relevant calibration is the major approach, and the KL method is an auxiliary verification.
7.2 Electrochemically active surface area (ECSA)
The ECSA of ORR electrocatalysts is a key parameter to evaluate mass activities and specific activities. It can be determined by CO-stripping (oxidation of pre-adsorbed CO monolayers) and the Hupd method (integrating the underpotentially deposited hydrogen (Hupd) regions from cyclic voltammetry).232 In previous work, the ECSAs of PdZn88 and PdAu157 alloys were estimated by CO-stripping measurements and the ECSAs of PdFe155 and PdPt161 alloys were calculated by the Hupd method, respectively.It is noted that, unlike Pt, Pd could absorb a certain amount of H2, resulting in the fact that it is impossible to quantitatively differentiate between Hupd and absorbed hydrogen (Habs). Therefore, it is hard to obtain the exact ECSA by the Hupd method. Recent studies reported that the ECSA of a Pd-based catalyst is measured by Pd oxide reduction analysis.233,234 The Coulomb charge of Pd-based catalysts is calculated by using the chemically adsorbed oxygen reduction peak instead of the hydrogen adsorption/desorption peak in Pt-based catalysts. The ECSAs of PdCo,224,235 PdPb,154 and PdAg233 alloys have also been measured by this approach. In addition, the use of underpotentially deposited copper (CuUPD) is feasible;236 however, the major disadvantage of this method is that the metal ions might largely affect catalytic reactions. The ECSAs of PdFe alloys12 were determined by this method.
Shao et al. studied the ECSA measurements of Pt- and Pd-based nanoparticles in detail and demonstrated that the ECSAs follow HUPD < CO stripping < CuUPD and CuUPD is the most accurate method.237 In Luo et al.'s work, the ECSAs of PdMo catalysts were determined by Hupd, CO stripping, Cuupd and Pd oxide reduction, and the ECSAs determined from the Cuupd method were used for further analysis.75
Some research pointed out that one of the most suitable techniques to determine ECSAs is differential electrochemical mass spectrometry (DEMS),238 which allows in situ measurements of supported catalysts under controlled mass flow conditions, but this technique is not available in most labs.
7.3 Durability test
Durability is regarded as a key factor for ORR electrocatalysts in practical applications. Durability tests in different studies of Pd-based electrocatalysts varied greatly, and even a small proportion of studies did not provide specific durability performances. Standard durability test protocols are summarized.| Item | Test conditions and protocols | Diagnostic test/criteria | |
|---|---|---|---|
| Half cell (RDE) | MEA | ||
| Start/stop cycle | 1.0–1.5 V vs. RHE, triangular-wave potential cycle | ||
| 25 °C, 0.1 M HClO4, N2 saturated | 80 °C, 100% relative humidity, H2/N2 | • Cyclic voltammetry for ECSA measurement, and ORR activity measurement at every 10 to 1k-cycles. | |
|
|
• If ECSA is less than 50% vs. initial values, finish the cycle test. Otherwise, continue up to 60k-cycles. | |
| Load cycle | 0.6–1.0 V vs. RHE, rectangular-wave potential cycle | ||
| 25 °C, 0.1 M HClO4, N2 saturated | 80 °C, 100% relative humidity, H2/N2 | • Cyclic voltammetry for ECSA measurement, and ORR activity measurement at every 10 to 1k-cycles. | |
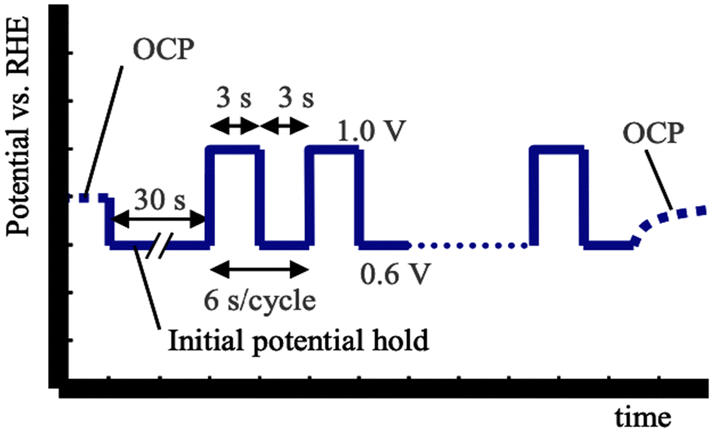
|
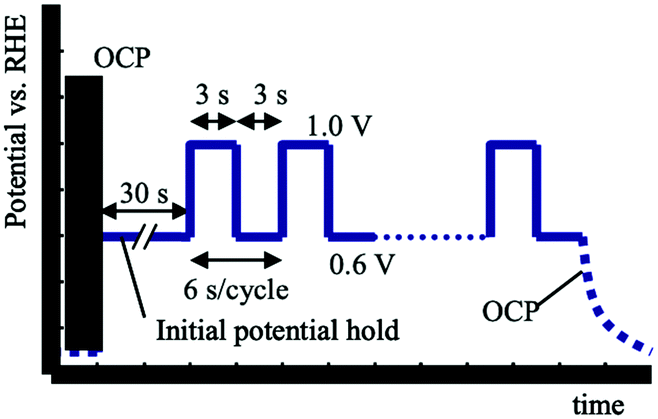
|
• If ECSA is less than 50% vs. initial values, finish the cycle test. Otherwise, continue up to 400k-cycles. | |
| Cycle | Step change: 30 s at 0.7 V and 30 s at 0.9 V. Single cell 25–50 cm2 |
| Cycle number | 30k |
| Cycle time | 60 s |
| Temperature | 80 °C |
| Relative humidity | Anode/cathode 100/100% |
| Fuel/oxidant | H2/N2 |
| Pressure | 150 kPa |
| Metrics | Frequency | Target |
|---|---|---|
| Catalytic activity | Start and end of life | ≤60% loss of initial activity |
| Polarization curve from 0 to ≥ 1.5 A cm−2 | After 0, 1k, 5k, 10k, and 30k cycles | ≤30 mV loss at 0.8 A cm−2 |
| ECSA/cyclic voltammetry | After 1, 10, 30, 100, 300, 1k, 3k cycles and thereafter every 5k cycles | ≤ 40% loss of initial area |
7.4 Standardization of electrochemical analysis
Cyclic voltammograms (CV) and linear sweep voltammograms (LSV) on RDEs are extensively collected to evaluate the ORR performance of Pd-based catalysts. These methods allow studying the material in a large potential and time-scale domain, presenting information on the thermodynamic and kinetic behaviors of the electrocatalysts. However, due to the differences in test methods and conditions, it is hard to compare the ORR performances of electrocatalysts using MA or SA values. To better compare the ORR activities of different electrocatalysts, more standardized and reasonable guidance needs to be proposed.Mayrhofer et al. scrutinized the thin-film rotating disc electrode (TF-RDE) method for investigating the activities of high surface area electrocatalysts.244 They pointed out: (a) Normalized activities have to be determined. The normalization can be based on the noble metal loading (mass activity) or surface area (specific activity). (b) The active surface area has to be evaluated by applying a proper method that also considers the capacity of the support. (c) The electrode thickness, the diffusion limited current of the ORR and the current under the specific potential need to be limited.
Garsany et al. studied the impact of film drying procedures on RDE characterization and found that films prepared with the rotational drying method are more reproducible than films prepared with the stationary method.245 Takahashi et al. emphasized the importance of ink preparation for the RDE method.246
In different acidic electrolytes, the ORR activity varies. The catalytic activity of Pt/C followed the trend HClO4 > H2SO4 > H3PO4 and the durability of Pt/C in H2SO4 was found to be lower than in HClO4.246 Pd is less studied in this aspect, but it can be predicted that Pd may exhibit similar trends to Pt. Therefore, it is necessary to unify the types and concentrations of the electrolyte during the test.
Moreover, the ultimate estimation of the electrocatalyst catalytic activity towards the ORR should be performed in MEAs. To a certain extent, it can be seen whether the catalyst is suitable for practical applications rather than the laboratory level. For example, Luo et al. evaluated the performances of PdMo bimetallene catalysts in Zn–air and Li–air batteries.75 Hence, the MEA test is strongly recommended for Pd-based electrocatalysts in future work.
7.5 Summary of the electrocatalytic performance of representative Pd-based catalysts
As Tables 5 and 6 and Fig. 12 show, regarding the ORR activity, Pd-based alloy materials had better performance in alkaline electrolytes than in acidic electrolytes, and some of them, such as PdMo bimetallene and Pd4Fe nanoflower materials, even exceeded that of Pt-based catalysts. They exhibited excellent ORR activity in alkaline solutions and also had good activity in acidic solutions. However, in acid solution, the stability of Pd alloy catalysts was often not as good as that of Pt-based materials. Indeed, Pd is more susceptible to oxidation at more negative potentials than Pt and is more easily poisoned by anions such as ClO4− and other oxygen-containing species as compared to Pt.10 Pd-based catalysts with different element compositions and morphologies and structures had obvious activity differences. Although previous work has studied the relationship between morphology and activity, there is still no exact direct relationship between material composition, structure and performance. Methods to obtain the expected structure and electrocatalytic performance through experimental preparation are still a challenge.| Materials | Mass activity (mA mgPGM−1) | Specific activity (μA cmPGM−2) | ECSA (m2 g−1) | E 1/2 (V vs. RHE) | Onset potential (V vs. RHE) | Measured potential (V vs. RHE) | Stability |
|---|---|---|---|---|---|---|---|
| a 0.5 M KOH. | |||||||
| Disordered PdZn88 | 8.37 | 17.36 | 48.2 | 0.74 | 0.93 | 0.85 | After continuous operation of about 8 h, the current dropped to 78.30% of its initial value at 0.5 V and 900 rpm. |
| Ordered PdZn88 | 24.44 | 44.41 | 55.04 | 0.81 | 0.97 | 0.85 | After continuous operation of about 8 h, the current dropped to 91.58% of its initial value at 0.5 V and 900 rpm. |
| Core–shell PdZn88 | 44.05 | 48.83 | 90.22 | 0.82 | 0.98 | 0.85 | After continuous operation of about 8 h, the current dropped to 94.30% of its initial value at 0.5 V and 900 rpm. |
| Pd4Fe nanoflowers155 | 4070 | 11600 |
33 | 0.903 | N/A | 0.85 | After 5k cycles, the loss of ECSA and MA was 19% and 17%, respectively, by cycling the potential between 0.6 and 1.0 V. |
| Pd6Ni icosahedra150 | 220 | 660 | N/A | 0.89 | 1.04 | 0.90 | After 10k cycles, the catalyst exhibited a 5.6% loss of its initial mass activity by cycling the potential between 0.4 and 1.0 V at 100 mV s−1. |
| PdAu nanochain networks157 | 86.01 | 590 | 45.24 | 0.848 | 0.986 | 0.85 | After 1k cycles, the half-wave potentials only negatively shift about 5 mV by sweeping between 0.2 and 1.1 V at 5 mV s−1. |
| Pd31Bi12/C nanoparticles172 | 950 | 2420 | N/A | 0.92 | 0.97 | 0.90 | After 10k cycles, retention of ∼60% of the initial activity by cycling the voltage repeatedly between 0.6 and 1.0 V at a sweep rate of 100 mV s−1. |
| Converted Pd3Bi180 | 1200 | 2300 | N/A | N/A | 0.97 | 0.90 | After 10k cycles, the catalyst retained 86% and 69% of the initial SA and MA, respectively, whereas the ECSA decreased by 20%, by cycling the samples from 0.6 to 1.0 V at a sweep rate of 100 mV s−1. |
| PdY-E85 | 213a | 1740 | 24 | N/A | N/A | 0.90 | N/A |
| PdAu FANs104 | 142.21 | 480 | 34.5 | N/A | N/A | 0.90 | After 1k cycles, negative shifts of about 7 mV in the half-wave potentials of AuPd FANs, by applying continuous potential sweeps between 0.21 and 1.21 V at a scan rate of 5 mV s−1. |
| PdMo bimetallene/C75 | 16370 |
11640 |
139.7 | 0.95 | N/A | 0.90 | After 30k cycles, the catalyst retained over 60% of the initial mass activity, by cycling the potential between 0.6 and 1.0 V at 50 mV s−1. |
| W-Doped Pd | |||||||
| Nanocubes/C193 | 250 | 1180 | 21.1 | N/A | N/A | 0.90 | After 10k cycles, the catalyst showed a decrease of 14.8% with respect to the initial mass activity. |
| Pd3Pb square nanoplate154 | 620 | 3590 | 17.3 | N/A | N/A | 0.90 | After 10k cycles, the catalyst showed a 21% loss of its initial mass activity. |
| Materials | Mass activity (mA mgPGM−1) | Specific activity (μA cmPGM−2) | ECSA (m2 g−1) | E 1/2 (V vs. RHE) | Measured potential (V vs. RHE) | Stability |
|---|---|---|---|---|---|---|
| a 0.5 M H2SO4. b 0.1 M H2SO4. c V vs. NHE. | ||||||
| Pd3Fe/C12 | 696 | 690 | 100 | 0.832 | 0.80 | N/A |
| Pd2Fe/C12 | 348 | 390 | 90 | N/A | 0.80 | N/A |
| Pd4Co/C(CS)145 | 173 | 480 | ∼35 | N/A | 0.75 | N/A |
| Pd4Fe nanoflowers155 | 521 | 1570 | 33 | 0.833 | 0.85 | N/A |
| Pt1Pd1/C161 | 488a | 976 | 107.7 | N/A | 0.85 | After 10k cycles, the catalyst showed a mass activity loss of only 3.5% by applying linear potential sweeps between 0.5 and 1.0 V at 50 mV s−1. |
| NP-Pd50Cu5082 | 150 | 220 | N/A | N/A | 0.90 | A retention of ∼30% of the initial specific activity by using the chronoamperometry method for 3000 s at 0.9 V. |
| Nanoporous PdNi173 | 150 | 210 | N/A | N/A | 0.90 | After 5k cycles, the catalyst underwent a loss of 12% relative to the initial ECSA with a negative half-wave potential shift by 14 mV from 0.6 to 1.0 V. |
| Rh8Pd92 alloy octahedra79 | 101 | 240 | 42.7 | N/A | 0.90 | After 30k cycles, the catalyst showed ∼75% of the initial activity between 0.6 and 1.0 V. |
| NP PdCo247 | 150 | 200 | N/A | N/A | 0.90 | After 5k cycles, the catalyst showed degradation with a negative half-wave shift of 19 mV that corresponded to an ∼17% loss of the ECSA between 0.6 and 1.0 V |
| PdCu nanocubes152 | 130b | 310 | 41.8 | 0.812 | 0.85c | N/A |
| PdY–E85 | 146b | 575 | 24.83 | 0.851 | 0.90 | N/A |
| PdMo bimetallene/C75 | 660 | ∼460 | 139.7 | N/A | 0.90 | Its stability is not sufficient for practical applications |
| v-PdCuCo-AS148 | 1135 | 1610 | N/A | N/A | 0.80 | After 4k cycles between 0.6 and 1.1 V, the catalyst showed a 0.26% loss of MA at 0.9 V. |
Other factors, including the electrochemically active surface area (ECSA), half wave potential (E1/2), Tafel slope, onset potential (Eonset), etc., are also factors that affect the performance of electrocatalysts and could not be ignored. Many catalysts reach high mass activities (MAs) by increasing the specific activity (SA) at the expense of a reduced ECSA. In the potential region of kinetic control, low ECSA is counterbalanced by high specific activities. At higher overpotentials (usually used in actual systems), the reaction rate is not limited by kinetics, but by mass transfer.248 Therefore, it is necessary to obtain materials which combine a high mass activity with a high ECSA. In addition, E1/2 is another common parameter that allows comparing the ORR activity of catalysts, especially between platinum group metal (PGM) catalysts and PGM-free catalysts. A positive shift in E1/2 indicates a high ORR activity.249 However, there is an issue that E1/2 increases with RDE catalyst loading. Thus, comparisons between catalysts by E1/2 are biased towards studies that used higher loading, and the loading dependence complicates the comparison between studies of different loadings.250 The Tafel slope is derived from the microkinetic model, and thus represents the reaction mechanism and surface kinetics.251 Different electrocatalysts show distinct rate-determining steps, such as the dissociation of O2 or desorption of OH.251 In general, the smaller the Tafel slope, the faster the ORR kinetics. In the case of cathode Pd-based catalysts, the decrease in onset potential and the increase in current density detected during the ORR process are considered to be key parameters for evaluating the catalyst performance.252 How to achieve it is the goal that researchers are striving for.
8. Conclusions and perspectives
Recent studies on Pd-based alloy catalysts for the oxygen reduction reaction are summarized in this review. There is a trend of rapid development and electrocatalysts are being developed that show excellent activities for the ORR, which often even exceed the performance of Pt-based catalysts, especially in alkaline media and in the presence of methanol. We note that theoretical research often only focuses on one or a few factors, while actual experiments are very complicated. Therefore, the development of related theories and the combination of theory with experiments can help us to predict development trends and prepare high-performance materials more efficiently. The alloys formed from palladium and various elements may have good catalytic activity. In addition, Pd alloys with different morphologies and structures can be obtained by different synthesis methods, and their catalytic performance is quite different. Regarding the mass activity and specific activity towards the ORR, PdMo bimetallene, PdBi alloys and Pd4Fe nanoflower materials seem to be the most promising cathode electrocatalysts; however, other parameters, like stability, and catalyst activity under actual industrial conditions rather than laboratory conditions, are more important factors in determining whether commercialization can be achieved. In addition, some other issues for mass production need more attention.(1) In terms of reaction thermodynamics and kinetics, more in-depth research is needed to pave the way for future industrialization. The establishment of universal reaction kinetics and thermodynamic models could be achieved through more advanced machine learning techniques for building up correlations to produce high-performance Pd alloy catalysts.
(2) Regarding catalyst deactivation, studies need to focus on deactivation mechanisms, especially for precious metal catalysts. The study of the deactivation mechanism can better understand the catalytic process, thereby helping to extend the service life and understand how to regenerate the catalyst.
(3) As for recycling, it needs to be determined if there is a facile way to regenerate deactivated catalysts. If so, how can this be achieved with efficient circulation and still maintaining high activity and stability. If not, whether the abandoned catalysts can be processed for other applications needs to be studied.
(4) At present, little research has been reported on the life cycle assessment (LCA) of ORR catalysts (not just the Pd or Pt series), so it is difficult to evaluate the environmental impact of these electrocatalysts during the production process. For example, the production of a certain catalyst using low-cost raw materials under harsher reaction conditions is a dilemma. Through LCA, the advantages and disadvantages can be obtained, thus deciding whether this catalyst is worth producing.
In short, the research on Pd-based electrocatalysts towards the ORR is moving in the direction of low cost, high activity, stable performance and long lifespan. Researchers need to analyze and solve the above problems to promote the industrialization of this material set, as demonstrated in Fig. 13.
 | ||
| Fig. 13 Schematic illustration of the ideal properties and future perspectives on Pd-based alloy electrocatalysts for the ORR. | ||
Conflicts of interest
There are no conflicts to declare.Appendix A. Supplementary data
1. Methodology
1.1. Data collection
The data was obtained from the Web of Science Core Collection (WoSCC) using the advanced search “TS = (“Palladium alloy*” or “Pd* alloy*” or “Palladium catalysts” or “Pd* catalysts”) AND TS= (“oxygen reduction reaction” or “ORR”)”. English was the only chosen language, and the type of documents was restricted to the article. The timespan was from 1970 to 2020. For citation indexes, the data was only selected from Science Citation Index Expanded (SCI-EXPANDED) – 1970–present. As a result, 400 documents were identified on 6 July 2020.1.2. Scientometric analysis methods
1.3 Other scientometric analysis on Pd-based alloys for the ORR
Fig. 14(a) describes the top 10 sources of these 400 articles. Amongst them, Electrochimica Acta was the most productive source with 37 articles, which is intended for studies in the field of electrochemistry. In terms of average citations, Journal of the Electrochemical Society (J ELECTROCHEM SOC) was the highest journal, because three highly cited articles (Lee K, 2006;35 Jiang L, 2009;253 and Wang X, 2008126) were published in this journal. Besides, Journal of Electroanalytical Chemistry (J ELECTROANAL CHEM), Journal of Power Sources (J POWER SOURCES) and Journal of Physical Chemistry C (J PHYS CHEM C) had higher average citations.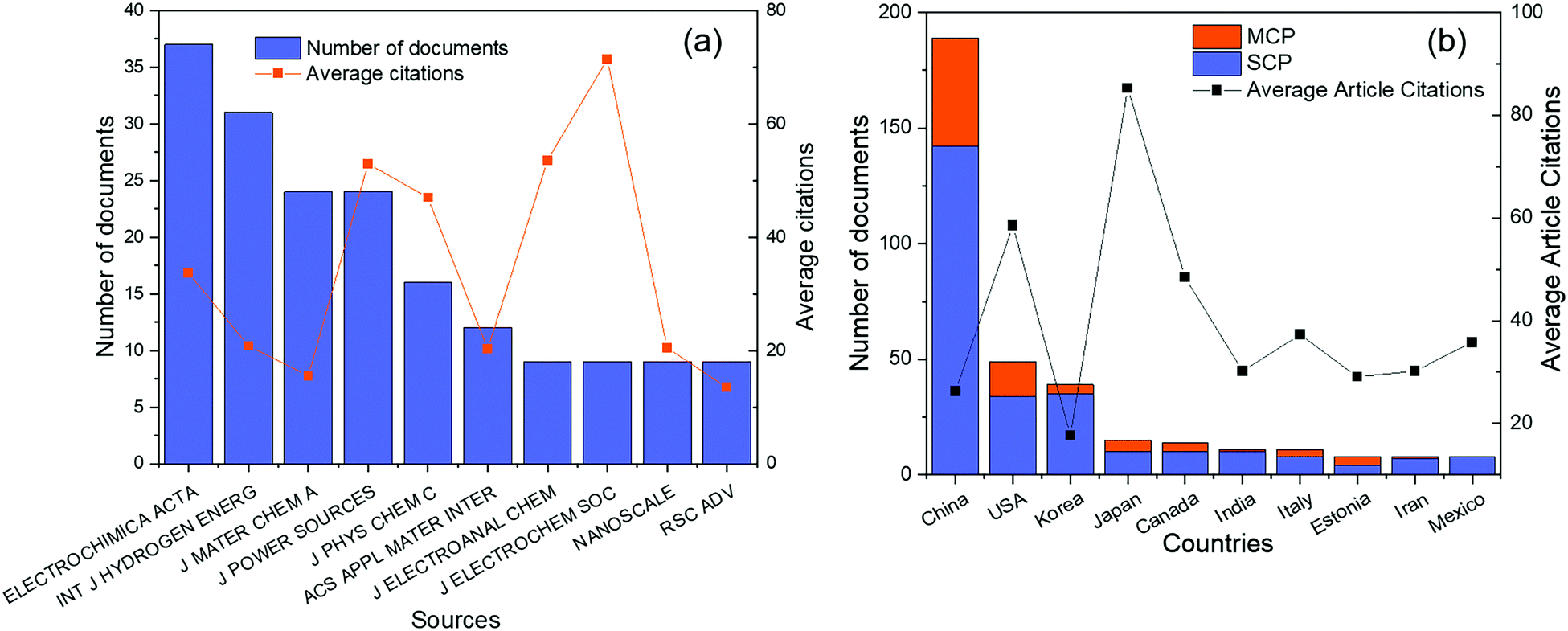 | ||
| Fig. 14 (a) Top 10 impactful sources and (b) top 10 countries by publications and their average citations on Pd alloys for the oxygen reduction reaction. | ||
Fig. 14(b) depicted the top 10 countries by publication number. According to all author's addresses, the publications were divided into Single Country Publication (SCP) and Multiple Country Publication (MCP). China published the most articles (189), which was almost four times that of the USA (ranking in second place). But the average citation number was less than that from the USA. Although the number of publications is not high, Japan had the largest average citations. The number of MCP articles to the total number of articles (so-called MCP ratio) could reflect the extent of inter-country collaborations to a certain degree. Estonia and Japan had higher MCP ratios, while Mexico and India had lower ones.
Table 7 displays the top 10 references with the strongest citation bursts sorted by strength. The centrality value indicates the importance of the position in the network. High values of centrality identify potentially revolutionary publications.256 Sigma is a hybrid metric, used to measure the structural centrality and citation burstness of a cited reference, suggesting the scientific novelty of a document.257 From Table 7, 8 in 10 are from cluster #0 (Pd nano-particle).
| References | Strength | Centrality | Sigma | Cluster | Duration 1997–2020 |
|---|---|---|---|---|---|
| O. Savadogo, Electrochem. Commun., 2004, 6, 10533 | 17.22 | 0.02 | 1.43 | #0 |

|
| M. H. Shao, J. Am. Chem. Soc., 2006, 128, 352636 | 13.59 | 0.02 | 1.35 | #0 |

|
| K. Lee, J. Electrochem. Soc., 2006, 153, 0186 | 13.39 | 0.01 | 1.20 | #0 |

|
| J. L. Fernandez, J. Am Chem. Soc., 2005, 127, 35791 | 13.09 | 0.02 | 1.22 | #0 |

|
| M. H. Shao, Langmuir, 2006, 22, 1040965 | 12.64 | 0.03 | 1.40 | #0 |

|
| J. L. Fernandez, J. Am. Chem. Soc., 2005, 127, 1310083 | 11.78 | 0.01 | 1.15 | #0 |

|
| M. H. Shao, Chem. Rev., 2016, 116, 3594254 | 10.58 | 0.02 | 1.20 | #1 |

|
| H. A. Gasteiger, Appl. Catal B-Environ., 2005, 56, 9255 | 9.19 | 0.04 | 1.40 | #8 |

|
| V. Raghuveer, J. Phys. Chem. B, 2005, 109, 2290934 | 8.97 | 0.01 | 1.12 | #0 |

|
| W. M. Wang, J. Power Sources, 2007, 167, 243217 | 7.98 | 0.02 | 1.17 | #0 |

|
Among these articles, a paper published by Savadogo33 in 2004 had the strongest burst strength (17.22), largest Sigma value (1.43), longest burst duration (ranging from 2004 to 2012) and longest citation duration (2004–2020). In this work, extremely active palladium-based alloy catalysts without platinum for the ORR in an acid solution were shown for the first time. It opened the curtain for the follow-up study of Pd alloy catalysts. In addition, in Gasteiger's article,255 the authors quantified the activities and voltage loss modes for MEAs (membrane electrode assemblies), and provided benchmark oxygen reduction activities for electrocatalysts by using two different testing procedures to establish the relative merit of candidate catalysts. Its centrality was the largest (0.04) and the Sigma value was the second largest (1.40). Besides, it is noted that Shao MH was the first author of three references in Table 7, suggesting that he/she has made outstanding contributions in this field.
Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council (EPSRC, EP/V027433/1; EP/L015862/1; EP/533581/1), The Royal Society (RGS\R1\211080; IEC\NSFC\201261), STFC Batteries Network (ST/R006873/1), RSC Mobility Grant (M19-7656), Faraday Institution (EP/S003053/1) Degradation project (FIRG001) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000) for financial support.References
- J.-P. Randin, Electrochim. Acta, 1974, 19, 83–85 CrossRef CAS.
- R. Zurilla, R. Sen and E. Yeager, J. Electrochem. Soc., 1978, 125, 1103–1109 CrossRef CAS.
- L. Kreja and R. Dabrowski, J. Power Sources, 1981, 6, 35–46 CrossRef.
- G. Gruver, L. Bregoli, R. Pascoe and H. Kunz, J. Electrochem. Soc., 1978, 125, 366 CrossRef.
- S. Mukerjee, S. Srinivasan and A. J. Appleby, Electrochim. Acta, 1993, 38, 1661–1669 CrossRef CAS.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201–224 CrossRef CAS.
- K. Kinoshita, Electrochemical Oxygen Technology, John Wiley & Sons, New Jersey, 1992 Search PubMed.
- Q. Li, T. Wang, D. Havas, H. Zhang, P. Xu, J. Han, J. Cho and G. Wu, Adv. Sci., 2016, 3, 1600140 CrossRef PubMed.
- M. Rao, A. Damjanovic and J. O. M. Bockris, J. Phys. Chem., 1963, 67, 2508–2509 CrossRef CAS.
- M. Shao, J. Power Sources, 2011, 196, 2433–2444 CrossRef CAS.
- S. T. Nguyen, D. S. L. Tan, J.-M. Lee, S. H. Chan, J. Y. Wang and X. Wang, Int. J. Hydrogen Energy, 2011, 36, 9645–9652 CrossRef CAS.
- M. Neergat, V. Gunasekar and R. Rahul, J. Electroanal. Chem., 2011, 658, 25–32 CrossRef CAS.
- H. Yang, N. Alonso-Vante, C. Lamy and D. L. Akins, J. Electrochem. Soc., 2005, 152, A704 CrossRef CAS.
- F. Kadirgan, B. Beden, J. Leger and C. Lamy, J. Electroanal. Chem. Interfacial Electrochem., 1981, 125, 89–103 CrossRef CAS.
- R. Moss, H. Gibbens and D. Thomas, J. Catal., 1970, 16, 117–125 CrossRef CAS.
- A. Baiker, D. Gasser and J. Lenzner, J. Chem. Soc., Chem. Commun., 1987, 1750–1751 RSC.
- B. Beden, C. Lamy and J. Leger, Electrochim. Acta, 1979, 24, 1157–1166 CrossRef CAS.
- A. Bryant, W. Bugden and J. Pratt, Acta Metall., 1970, 18, 101–107 CrossRef CAS.
- P. Chinh, S. Skalski and J. Budnick, J. Appl. Phys., 1970, 41, 1080 CrossRef.
- G. L. Holleck, J. Phys. Chem., 1970, 74, 503–511 CrossRef CAS.
- T. Skoskiewicz, A. Szafranski and B. Baranowski, Phys. Status Solidi B, 1973, 59, 135–136 CrossRef.
- J. Hedman, M. Klasson, R. Nilsson, C. Nordling, M. Sorokina, O. Kljushnikov, S. Nemnonov, V. Trapeznikov and V. Zyryanov, Phys. Scr., 1971, 4, 195 CrossRef CAS.
- E. Allison and G. Bond, Catal. Rev., 1972, 7, 233–289 CrossRef CAS.
- A. Damjanovic and V. Brusić, Electrochim. Acta, 1967, 12, 1171–1184 CrossRef CAS.
- R. Pattabiraman, Appl. Catal., A, 1997, 153, 9–20 CrossRef CAS.
- M. Aria and C. Cuccurullo, J. informetr., 2017, 11, 959–975 CrossRef.
- M. B. Synnestvedt, C. Chen and J. H. Holmes, AMIA Annu. Symp. Proc., 2005, 724–728 Search PubMed.
- C. Chen, J. Am. Soc. Inf. Sci. Technol., 2006, 57, 359–377 CrossRef.
- N. J. Van Eck and L. Waltman, Scientometrics, 2010, 84, 523–538 CrossRef PubMed.
- A. Damjanovic and V. Brusić, J. Electroanal. Chem. Interfacial Electrochem., 1967, 15, 29–33 CrossRef CAS.
- J. Fishman and M. Yarish, Electrochim. Acta, 1967, 12, 579–581 CrossRef CAS.
- J. Podestá and R. Piatti, Int. J. Hydrogen Energy, 1997, 22, 753–758 CrossRef.
- O. Savadogo, K. Lee, K. Oishi, S. Mitsushima, N. Kamiya and K.-I. Ota, Electrochem. Commun., 2004, 6, 105–109 CrossRef CAS.
- V. Raghuveer, A. Manthiram and A. J. Bard, J. Phys. Chem. B, 2005, 109, 22909–22912 CrossRef CAS PubMed.
- K. Lee, O. Savadogo, A. Ishihara, S. Mitsushima, N. Kamiya and K.-I. Ota, J. Electrochem. Soc., 2006, 153, A20–A24 CrossRef CAS.
- M.-H. Shao, K. Sasaki and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 3526–3527 CrossRef CAS PubMed.
- M. Tarasevich, A. Sadkowski and E. Yeager, in Comprehensive Treatise of Electrochemistry, ed. B. E. Conway, J. O. Bockris, E. Yeager, S. U. M. Khan and R. E. White, Springer, Boston, MA, 1983, pp. 301–398 Search PubMed.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- T. Jacob and W. A. Goddard III, ChemPhysChem, 2006, 7, 992–1005 CrossRef CAS PubMed.
- N. Anastasijević, V. Vesović and R. Adžić, J. Electroanal. Chem. Interfacial Electrochem., 1987, 229, 317–325 CrossRef.
- M.-h. Shao, P. Liu and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 7408–7409 CrossRef CAS PubMed.
- J.-D. Kim, S.-I. Pyun, T.-H. Yang and J.-B. Ju, J. Electroanal. Chem., 1995, 383, 161–166 CrossRef.
- Y. Sha, T. H. Yu, B. V. Merinov, P. Shirvanian and W. A. Goddard III, J. Phys. Chem. Lett., 2011, 2, 572–576 CrossRef CAS.
- D. C. Ford, A. U. Nilekar, Y. Xu and M. Mavrikakis, Surf. Sci., 2010, 604, 1565–1575 CrossRef CAS.
- E. López-Chávez, A. Garcia-Quiroz, G. González-García, Y. A. Peña-Castañeda, J. A. Díaz-Góngora and F. de Landa Castillo-Alvarado, Int. J. Hydrogen Energy, 2016, 41, 23281–23286 CrossRef.
- T. T. Vo Doan, J. Wang, K. C. Poon, D. C. Tan, B. Khezri, R. D. Webster, H. Su and H. Sato, Angew. Chem., Int. Ed., 2016, 55, 6842–6847 CrossRef PubMed.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. T. Goh, T. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- R. Rahul, R. Singh, B. Bera, R. Devivaraprasad and M. Neergat, Phys. Chem. Chem. Phys., 2015, 17, 15146–15155 RSC.
- Y. H. Wang, J. B. Le, W. Q. Li, J. Wei, P. M. Radjenovic, H. Zhang, X. S. Zhou, J. Cheng, Z. Q. Tian and J. F. Li, Angew. Chem., 2019, 131, 16208–16212 CrossRef.
- Q. Jia, K. Caldwell, J. M. Ziegelbauer, A. Kongkanand, F. T. Wagner, S. Mukerjee and D. E. Ramaker, J. Electrochem. Soc., 2014, 161, F1323 CrossRef CAS PubMed.
- S. Sharma, C. Zeng and A. A. Peterson, J. Chem. Phys., 2019, 150, 041704 CrossRef PubMed.
- Q. Zhang and A. Asthagiri, Catal. Today, 2019, 323, 35–43 CrossRef CAS.
- H.-C. Tsai, Y.-C. Hsieh, T. H. Yu, Y.-J. Lee, Y.-H. Wu, B. V. Merinov, P.-W. Wu, S.-Y. Chen, R. R. Adzic and W. A. Goddard III, ACS Catal., 2015, 5, 1568–1580 CrossRef CAS.
- X. Wang, X. Li, S. Liao and B. Li, Comput. Mater. Sci., 2018, 149, 107–114 CrossRef CAS.
- L. Ou and S. Chen, J. Phys. Chem. C, 2013, 117, 1342–1349 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov and W. A. Goddard III, ACS Catal., 2014, 4, 1189–1197 CrossRef CAS.
- I. Roche and K. Scott, J. Appl. Electrochem., 2009, 39, 197–204 CrossRef CAS.
- Y.-W. Lee, S.-E. Oh and K.-W. Park, Electrochem. Commun., 2011, 13, 1300–1303 CrossRef CAS.
- F. Harnisch and U. Schröder, Chem. Soc. Rev., 2010, 39, 4433–4448 RSC.
- S. Rojas-Carbonell, C. Santoro, A. Serov and P. Atanassov, Electrochem. Commun., 2017, 75, 38–42 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. L. Fernández, J. M. White, Y. Sun, W. Tang, G. Henkelman and A. J. Bard, Langmuir, 2006, 22, 10426–10431 CrossRef PubMed.
- A. Appleby, Catal. Rev., 1971, 4, 221–244 CrossRef.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- M. Shao, T. Huang, P. Liu, J. Zhang, K. Sasaki, M. Vukmirovic and R. Adzic, Langmuir, 2006, 22, 10409–10415 CrossRef CAS PubMed.
- M. Shao, P. Liu, J. Zhang and R. Adzic, J. Phys. Chem. B, 2007, 111, 6772–6775 CrossRef CAS PubMed.
- Y. Suo, L. Zhuang and J. Lu, Angew. Chem., Int. Ed., 2007, 46, 2862–2864 CrossRef CAS PubMed.
- L. Qi and J. Li, J. Catal., 2012, 295, 59–69 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141 CrossRef CAS PubMed.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- B. Hammer and J. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- L. A. Kibler, A. M. El-Aziz, R. Hoyer and D. M. Kolb, Angew. Chem., Int. Ed., 2005, 44, 2080–2084 CrossRef CAS PubMed.
- B. S. Mun, C. Lee, V. Stamenkovic, N. M. Markovic and P. N. Ross Jr, Phys. Rev. B, 2005, 71, 115420 CrossRef.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang and Y. Qin, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410–2419 CrossRef CAS PubMed.
- H. Duan and C. Xu, Phys. Chem. Chem. Phys., 2016, 18, 4166–4173 RSC.
- J. Zhao, A. Sarkar and A. Manthiram, Electrochim. Acta, 2010, 55, 1756–1765 CrossRef CAS.
- Y. Yan, F. Zhan, J. Du, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Nanoscale, 2015, 7, 301–307 RSC.
- L. E. Betancourt, A. Rojas-Pérez, I. Orozco, A. I. Frenkel, Y. Li, K. Sasaki, S. D. Senanayake and C. R. Cabrera, ACS Appl. Energy Mater., 2020, 3, 2342–2349 CrossRef CAS.
- J.-J. Lv, S.-S. Li, A.-J. Wang, L.-P. Mei, J.-R. Chen and J.-J. Feng, Electrochim. Acta, 2014, 136, 521–528 CrossRef CAS.
- H. Zhang, Q. Hao, H. Geng and C. Xu, Int. J. Hydrogen Energy, 2013, 38, 10029–10038 CrossRef CAS.
- J. L. Fernández, V. Raghuveer, A. Manthiram and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 13100–13101 CrossRef PubMed.
- S. Salomé, A. Ferraria, A. B. Do Rego, F. Alcaide, O. Savadogo and R. Rego, Electrochim. Acta, 2016, 192, 268–282 CrossRef.
- R. Brandiele, V. Amendola, A. Guadagnini, G. A. Rizzi, D. Badocco, P. Pastore, A. A. Isse, C. Durante and A. Gennaro, Electrochim. Acta, 2019, 320, 134563 CrossRef CAS.
- X. Lu, M. Ahmadi, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 5891–5898 CrossRef CAS.
- Y. Dai, P. Yu, Q. Huang and K. Sun, Fuel Cells, 2016, 16, 165–169 CrossRef CAS.
- H. Yang, K. Wang, Z. Tang, Z. Liu and S. Chen, J. Catal., 2020, 382, 181–191 CrossRef CAS.
- J. Tian, W. Wu, Z. Tang, Y. Wu, R. Burns, B. Tichnell, Z. Liu and S. Chen, Catalysts, 2018, 8, 329 CrossRef.
- H. C. Ham, D. Manogaran, K. H. Lee, K. Kwon, S.-A. Jin, D. J. You, C. Pak and G. S. Hwang, J. Chem. Phys., 2013, 201104 CrossRef PubMed.
- J. L. Fernández, D. A. Walsh and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 357–365 CrossRef PubMed.
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2005, 109, 18902–18906 CrossRef CAS PubMed.
- W. Tang, L. Zhang and G. Henkelman, J. Phys. Chem. Lett., 2011, 2, 1328–1331 CrossRef CAS PubMed.
- W. Tang and G. Henkelman, J. Chem. Phys., 2009, 130, 194504 CrossRef PubMed.
- F. Fouda-Onana, S. Bah and O. Savadogo, J. Electroanal. Chem., 2009, 636, 1–9 CrossRef CAS.
- H. Guo, J. A. Trindell, H. Li, D. Fernandez, S. M. Humphrey, G. Henkelman and R. M. Crooks, J. Mater. Chem. A, 2020, 8, 8421–8429 RSC.
- J. A. Trindell, Z. Duan, G. Henkelman and R. M. Crooks, ChemElectroChem, 2020, 7, 3824–3831 CrossRef CAS.
- L. Ou, J. Chem., 2015, 2015, 932616 Search PubMed.
- B. Han, C. E. Carlton, J. Suntivich, Z. Xu and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 3971–3978 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., 2006, 118, 2963–2967 CrossRef.
- S. Zuluaga and S. Stolbov, J. Chem. Phys., 2011, 135, 134702 CrossRef PubMed.
- D. Chen, C. Li, H. Liu, F. Ye and J. Yang, Sci. Rep., 2015, 5, 11949 CrossRef PubMed.
- Y. Xiong, H. Shan, Z. Zhou, Y. Yan, W. Chen, Y. Yang, Y. Liu, H. Tian, J. Wu and H. Zhang, Small, 2017, 13, 1603423 CrossRef PubMed.
- L.-L. He, P. Song, A.-J. Wang, J.-N. Zheng, L.-P. Mei and J.-J. Feng, J. Mater. Chem. A, 2015, 3, 5352–5359 RSC.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie and J. A. Herron, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- D. Wang, H. L. Xin, H. Wang, Y. Yu, E. Rus, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Chem. Mater., 2012, 24, 2274–2281 CrossRef CAS.
- W. Xiao, M. A. L. Cordeiro, M. Gong, L. Han, J. Wang, C. Bian, J. Zhu, H. L. Xin and D. Wang, J. Mater. Chem. A, 2017, 5, 9867–9872 RSC.
- W. Zhou, M. Li, O. L. Ding, S. H. Chan, L. Zhang and Y. Xue, Int. J. Hydrogen Energy, 2014, 39, 6433–6442 CrossRef CAS.
- L. Jiang, A. Hsu, D. Chu and R. Chen, J. Electrochem. Soc., 2009, 156, B643–B649 CrossRef CAS.
- M. V. Castegnaro, W. J. Paschoalino, M. R. Fernandes, B. Balke, M. C. M. Alves, E. A. Ticianelli and J. Morais, Langmuir, 2017, 33, 2734–2743 CrossRef CAS PubMed.
- S. Kondo, M. Nakamura, N. Maki and N. Hoshi, J. Phys. Chem. C, 2009, 113, 12625–12628 CrossRef CAS.
- A. Hitotsuyanagi, S. Kondo, M. Nakamura and N. Hoshi, J. Electroanal. Chem., 2011, 657, 123–127 CrossRef CAS.
- L. Xiao, L. Zhuang, Y. Liu and J. Lu, J. Am. Chem. Soc., 2009, 131, 602–608 CrossRef CAS PubMed.
- M. Shao, T. Yu, J. H. Odell, M. Jin and Y. Xia, Chem. Commun., 2011, 47, 6566–6568 RSC.
- H. Erikson, A. Sarapuu, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochem. Commun., 2011, 13, 734–737 CrossRef CAS.
- P. Strasser and S. Kühl, Nano Energy, 2016, 29, 166–177 CrossRef CAS.
- P. Strasser, in Handbook of Fuel Cells – Fundamentals, Technology and Applications, ed. W. Vielstich, H. A. Gasteiger, A. Lamm and H. Yokokawa, John Wiley & Sons, New Jersey, 2010 Search PubMed.
- S. Tominaka, T. Hayashi, Y. Nakamura and T. Osaka, J. Mater. Chem., 2010, 20, 7175–7182 RSC.
- T. Gunji, S. H. Noh, F. Ando, T. Tanabe, B. Han, T. Ohsaka and F. Matsumoto, J. Mater. Chem. A, 2018, 6, 14828–14837 RSC.
- H. Duan and C. Xu, J. Power Sources, 2016, 316, 106–113 CrossRef CAS.
- H.-F. Yang, Y.-Y. Feng, L.-X. Du, Z.-H. Liu and D.-S. Kong, RSC Adv., 2016, 6, 16904–16910 RSC.
- R. Yang, W. Bian, P. Strasser and M. F. Toney, J. Power Sources, 2013, 222, 169–176 CrossRef CAS.
- S. Mondal and C. R. Raj, ACS Appl. Mater. Interfaces, 2019, 11, 14110–14119 CrossRef CAS PubMed.
- C.-L. Lee, K.-L. Huang, Y.-L. Tsai and Y.-J. Chao, Electrochem. Commun., 2013, 34, 282–285 CrossRef CAS.
- M. Ramanathan, B. Li, J. Greeley and J. Prakash, ECS Trans., 2010, 33, 181 CrossRef CAS.
- X. Wang, N. Kariuki, J. T. Vaughey, J. Goodpaster, R. Kumar and D. J. Myers, J. Electrochem. Soc., 2008, 155, B602 CrossRef CAS.
- J. Yang, W. Zhou, C. H. Cheng, J. Y. Lee and Z. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 119–126 CrossRef CAS PubMed.
- M. Ramanathan, V. Ramani and J. Prakash, Electrochim. Acta, 2012, 75, 254–261 CrossRef CAS.
- T. Gunji, R. H. Wakabayashi, S. H. Noh, B. Han, F. Matsumoto, F. J. DiSalvo and H. D. Abruña, Electrochim. Acta, 2018, 283, 1045–1052 CrossRef CAS.
- Y. Zheng, L. Zhang, P. He, D. Dang, Q. Zeng, J. Zeng and M. Liu, Electrocatalysis, 2018, 9, 495–504 CrossRef CAS.
- K. Maiti, J. Balamurugan, S. G. Peera, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 18734–18745 CrossRef CAS PubMed.
- A. M. Remona and K. Phani, J. Fuel Cell Sci. Technol., 2011, 8, 011001 CrossRef.
- G. Ramos-Sánchez, M. M. Bruno, Y. R. Thomas, H. R. Corti and O. Solorza-Feria, Int. J. Hydrogen Energy, 2012, 37, 31–40 CrossRef.
- Z. Liu, X. Yang, L. Cui, Z. Shi, B. Lu, X. Guo, J. Zhang, L. Xu, Y. Tang and Y. Xiang, Part. Part. Syst. Charact., 2018, 35, 1700366 CrossRef.
- L. Zhang, K. Lee and J. Zhang, Electrochim. Acta, 2007, 52, 3088–3094 CrossRef CAS.
- S.-Y. Ang and D. A. Walsh, Appl. Catal., B, 2010, 98, 49–56 CrossRef CAS.
- W. Li, Z. Le, T. Zhou, M. Liao, H. Liu, B. Na, Y. Yu, B. Wang, H. Zhou and Z. Liao, Int. J. Electrochem. Sci., 2018, 13, 9292–9301 CrossRef CAS.
- G. R. Xu, C. C. Han, Y. Y. Zhu, J. H. Zeng, J. X. Jiang and Y. Chen, Adv. Mater. Interfaces, 2018, 5, 1701322 CrossRef.
- Y. Zhao, Y. Ding, B. Qiao, K. Zheng, P. Liu, F. Li, S. Li and Y. Chen, J. Mater. Chem. A, 2018, 6, 17771–17777 RSC.
- H.-M. Liu, S.-H. Han, Y.-Y. Zhu, P. Chen and Y. Chen, Green Energy Environ., 2018, 3, 375–383 CrossRef.
- S. An, J.-H. Park, C.-H. Shin, J. Joo, E. Ramasamy, J. Hwang and J. Lee, Carbon, 2011, 49, 1108–1117 CrossRef CAS.
- N. V. Long, T. D. Hien, T. Asaka, M. Ohtaki and M. Nogami, Int. J. Hydrogen Energy, 2011, 36, 8478–8491 CrossRef.
- J. Liu, J. Yin, B. Feng, F. Li and F. Wang, Appl. Surf. Sci., 2019, 473, 318–325 CrossRef CAS.
- A. T. N. Nguyen and J. H. Shim, J. Electroanal. Chem., 2018, 827, 120–127 CrossRef CAS.
- J.-H. Jang, C. Pak and Y.-U. Kwon, J. Power Sources, 2012, 201, 179–183 CrossRef CAS.
- F. Pires and H. Villullas, Int. J. Hydrogen Energy, 2012, 37, 17052–17059 CrossRef CAS.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma and D. Su, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- Y. Zuo, D. Rao, S. Li, T. Li, G. Zhu, S. Chen, L. Song, Y. Chai and H. Han, Adv. Mater., 2018, 30, 1704171 CrossRef PubMed.
- H. Wang, W. Luo, L. Zhu, Z. Zhao, B. E. W. Tu, X. Ke, M. Sui, C. Chen and Q. Chen, Adv. Funct. Mater., 2018, 28, 1707219 CrossRef.
- Y. Feng, Q. Shao, Y. Ji, X. Cui, Y. Li, X. Zhu and X. Huang, Sci. Adv., 2018, 4, eaap8817 CrossRef PubMed.
- L. B. Venarusso, C. V. Boone, J. Bettini and G. Maia, J. Mater. Chem. A, 2018, 6, 1714–1726 RSC.
- L. Zhang, F. Hou and Y. Tan, Chem. Commun., 2012, 48, 7152–7154 RSC.
- X. Jiang, Y. Xiong, Y. Wang, J. Wang, N. Li, J. Zhou, G. Fu, D. Sun and Y. Tang, J. Mater. Chem. A, 2019, 7, 5248–5257 RSC.
- S. Luo, M. Tang, X. Wu, Y. Ou, Z. Wang, N. Jian, X. Li, Y. Lin, Y. Yan and J. Huang, CrystEngComm, 2019, 21, 290–296 RSC.
- C. Lian, Y. Cheng, L. Chen, X. Han, X. Lei, Y. Liu and Y. Wang, Chem. Commun., 2018, 54, 7058–7061 RSC.
- Z. Cui, H. Chen, M. Zhao and F. J. DiSalvo, Nano Lett., 2016, 16, 2560–2566 CrossRef CAS PubMed.
- L.-L. He, P. Song, J.-J. Feng, W.-H. Huang, Q.-L. Wang and A.-J. Wang, Electrochim. Acta, 2015, 176, 86–95 CrossRef CAS.
- L. Kuai, X. Yu, S. Wang, Y. Sang and B. Geng, Langmuir, 2012, 28, 7168–7173 CrossRef CAS PubMed.
- Y. Ma, L. Yin, G. Cao, Q. Huang, M. He, W. Wei, H. Zhao, D. Zhang, M. Wang and T. Yang, Small, 2018, 14, 1703613 CrossRef PubMed.
- X. Peng, Z. Cui, X. Bai and H. Lv, IET Nanobiotechnol., 2018, 12, 1031–1036 CrossRef PubMed.
- W. Wang, Z. Wang, J. Wang, C. J. Zhong and C. J. Liu, Adv. Sci., 2017, 4, 1600486 CrossRef PubMed.
- M. Yang, Z. Wang, W. Wang and C.-J. Liu, Nanoscale Res. Lett., 2014, 9, 405 CrossRef PubMed.
- G. Zangari, Coatings, 2015, 5, 195–218 CrossRef CAS.
- E. Herrero, L. J. Buller and H. D. Abruña, Chem. Rev., 2001, 101, 1897–1930 CrossRef CAS.
- S. R. Brankovic and G. Zangari, Advances in Electrochemical Science and Engineering: Electrochemical Engineering Across Scales: from Molecules to Processes, Wiley-VCH, Weinheim, 2015 Search PubMed.
- A. Brenner, Electrodeposition of alloys: principles and practice, Elsevier, Amsterdam, 2013 Search PubMed.
- F. Gobal and R. Arab, J. Electroanal. Chem., 2010, 647, 66–73 CrossRef CAS.
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- C. Xu, Y. Zhang, L. Wang, L. Xu, X. Bian, H. Ma and Y. Ding, Chem. Mater., 2009, 21, 3110–3116 CrossRef CAS.
- V. Richoux, S. Diliberto, C. Boulanger and J. Lecuire, Electrochim. Acta, 2007, 52, 3053–3060 CrossRef CAS.
- Y. Wang and A. S. Hall, ACS Energy Lett., 2019, 5, 17–22 CrossRef.
- C. Xu, Y. Liu, Q. Hao and H. Duan, J. Mater. Chem. A, 2013, 1, 13542–13548 RSC.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund and H. Ogasawara, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- Y. Liu and C. Xu, ChemSusChem, 2013, 6, 78–84 CrossRef CAS PubMed.
- J. Chen, Y. Li, N. Lu, C. Tian, Z. Han, L. Zhang, Y. Fang, B. Qian, X. Jiang and R. Cui, J. Mater. Chem. A, 2018, 6, 23560–23568 RSC.
- H. Begum, M. S. Ahmed, S. Cho and S. Jeon, Int. J. Hydrogen Energy, 2018, 43, 229–238 CrossRef CAS.
- L. Sun, C.-L. Chien and P. C. Searson, Chem. Mater., 2004, 16, 3125–3129 CrossRef CAS.
- Q. Zhang and Z. Zhang, Phys. Chem. Chem. Phys., 2010, 12, 1453–1472 RSC.
- D. Sun, Y. Wang, K. J. Livi, C. Wang, R. Luo, Z. Zhang, H. Alghamdi, C. Li, F. An and B. Gaskey, ACS Nano, 2019, 13, 10818–10825 CrossRef CAS PubMed.
- T. Tsuji, Y. Higashi, M. Tsuji, Y. Ishikawa and N. Koshizaki, Appl. Surf. Sci., 2015, 348, 10–15 CrossRef CAS.
- W.-L. Chan, R. S. Averback, D. G. Cahill and A. Lagoutchev, Phys. Rev. B, 2008, 78, 214107 CrossRef.
- Y. Han, S. Wu, E. Dai, Y. Ye, J. Liu, Z. Tian, Y. Cai, X. Zhu and C. Liang, ChemPhysChem, 2017, 18, 1133–1139 CrossRef CAS PubMed.
- H. Zheng, M. S. Matseke and T. S. Munonde, Ultrason. Sonochem., 2019, 57, 166–171 CrossRef CAS PubMed.
- Ş. Korkmaz, B. Gecici, S. D. Korkmaz, R. Mohammadigharehbagh, S. Pat, S. Özen, V. Şenay and H. H. Yudar, Vacuum, 2016, 131, 142–146 CrossRef.
- K. Lee, O. Savadogo, A. Ishihara, S. Mitsushima, N. Kamiya and K.-I. Ota, J. Electrochem. Soc., 2005, 153, A20 CrossRef.
- M. Tarasevich, G. Zhutaeva, V. Bogdanovskaya, M. Radina, M. Ehrenburg and A. Chalykh, Electrochim. Acta, 2007, 52, 5108–5118 CrossRef CAS.
- N. Pokhrel, P. K. Vabbina and N. Pala, Ultrason. Sonochem., 2016, 29, 104–128 CrossRef CAS PubMed.
- L. Karuppasamy, S. Anandan, C.-Y. Chen and J. J. Wu, Electrocatalysis, 2017, 8, 430–441 CrossRef CAS.
- D.-S. Kim, J.-H. Kim, I.-K. Jeong, J. K. Choi and Y.-T. Kim, J. Catal., 2012, 290, 65–78 CrossRef CAS.
- Q. Hu, W. Zhan, Y. Guo, L. Luo, R. Zhang, D. Chen and X. Zhou, J. Energy Chem., 2020, 40, 217–223 CrossRef.
- Y. Sun, B. Huang, N. Xu, Y. Li, M. Luo, C. Li, Y. Qin, L. Wang and S. Guo, Sci. Bull., 2019, 64, 54–62 CrossRef CAS.
- F. Ahmad, L. Luo, X. Li, H. Huang and J. Zeng, Chin. J. Catal., 2018, 39, 1202–1209 CrossRef CAS.
- Y.-C. Wei, C.-W. Liu and K.-W. Wang, Chem. Commun., 2011, 47, 11927–11929 RSC.
- W.-D. Kang, Y.-C. Wei, C.-W. Liu and K.-W. Wang, Electrochem. Commun., 2011, 13, 162–165 CrossRef CAS.
- Q. Yang, L. Shi, B. Yu, J. Xu, C. Wei, Y. Wang and H. Chen, J. Mater. Chem. A, 2019, 7, 18846–18851 RSC.
- J.-X. Feng, S.-Y. Tong, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2018, 140, 5118–5126 CrossRef CAS PubMed.
- J. C. Calderón, L. Ndzuzo, B. J. Bladergroen and S. Pasupathi, Int. J. Hydrogen Energy, 2018, 43, 16881–16896 CrossRef.
- M. Thi, T. Tran, P. H. Anh, H.-T. Nhac-Vu and Q. Bui, J. Alloys Compd., 2019, 797, 314–324 CrossRef CAS.
- M. Martins, Ö. Metin, B. Šljukić, M. Sevim, C. Sequeira and D. Santos, Int. J. Hydrogen Energy, 2019, 44, 14193–14200 CrossRef CAS.
- Y. Chen, X. Jiang, Y. Li, P. Li, Q. Liu, G. Fu, L. Xu, D. Sun and Y. Tang, Adv. Mater. Interfaces, 2018, 5, 1701015 CrossRef.
- J. Ju, X. Wang and W. Chen, Int. J. Hydrogen Energy, 2020, 45, 6437–6446 CrossRef CAS.
- G. Li, B. Shi, Y. Gong, Y. Zhang, X. Wang, M. Guo and X. Lyu, Mater. Chem. Phys., 2020, 243, 122570 CrossRef CAS.
- M. Lüsi, H. Erikson, A. Sarapuu, M. Merisalu, M. Rähn, A. Treshchalov, P. Paiste, M. Käärik, J. Leis and V. Sammelselg, ChemElectroChem, 2020, 7, 546–554 CrossRef.
- Nancy Garland, Thomas Benjamin and John Kopasz, ECS Trans., 2007, 11, 923 CrossRef CAS.
- H. Liu, W. Li and A. Manthiram, Appl. Catal., B, 2009, 90, 184–194 CrossRef CAS.
- D. J. You, D. H. Kim, J. R. De Lile, C. Li, S. G. Lee, J. M. Kim and C. Pak, Appl. Catal., A, 2018, 562, 250–257 CrossRef CAS.
- Q. Gong, S. Gong, T. Zhang, X. Cheng and H. Li, J. Electrochem. Soc., 2019, 166, F906 CrossRef CAS.
- Y. Zhang, B. Huang, G. Luo, T. Sun, Y. Feng, Y. Wang, Y. Ma, Q. Shao, Y. Li and Z. Zhou, Sci. Adv., 2020, 6, eaba9731 CrossRef CAS PubMed.
- A. Albarbar and M. Alrweq, Proton Exch. Membr. Fuel Cells, 2018, 9–29 Search PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solution, National Association of Corrosion Engineers, Houston, 1974 Search PubMed.
- J. Solla-Gullon, V. Montiel, A. Aldaz and J. Clavilier, Electrochem. Commun., 2002, 4, 716–721 CrossRef CAS.
- P. Wells, E. Crabb, C. King, R. Wiltshire, B. Billsborrow, D. Thompsett and A. Russell, Phys. Chem. Chem. Phys., 2009, 11, 5773–5781 RSC.
- U. Paulus, T. Schmidt, H. Gasteiger and R. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- O. El Mouahid, C. Coutanceau, E. Belgsir, P. Crouigneau, J. Léger and C. Lamy, J. Electroanal. Chem., 1997, 426, 117–123 CrossRef CAS.
- W. Wang, Q. Huang, J. Liu, Z. Zou, Z. Li and H. Yang, Electrochem. Commun., 2008, 10, 1396–1399 CrossRef CAS.
- W. Wang, D. Zheng, C. Du, Z. Zou, X. Zhang, B. Xia, H. Yang and D. L. Akins, J. Power Sources, 2007, 167, 243–249 CrossRef CAS.
- W. Yan, Z. Tang, L. Wang, Q. Wang, H. Yang and S. Chen, Int. J. Hydrogen Energy, 2017, 42, 218–227 CrossRef CAS.
- H. Erikson, A. Sarapuu, N. Alexeyeva, K. Tammeveski, J. Solla-Gullón and J. Feliu, Electrochim. Acta, 2012, 59, 329–335 CrossRef CAS.
- X. Zhong, Y. Qin, X. Chen, W. Xu, G. Zhuang, X. Li and J. Wang, Carbon, 2017, 114, 740–748 CrossRef CAS.
- K. Jukk, N. Kongi, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochem. Commun., 2015, 56, 11–15 CrossRef CAS.
- L. Xu, Z. Luo, Z. Fan, X. Zhang, C. Tan, H. Li, H. Zhang and C. Xue, Nanoscale, 2014, 6, 11738–11743 RSC.
- Y. Qi, J. Wu, H. Zhang, Y. Jiang, C. Jin, M. Fu, H. Yang and D. Yang, Nanoscale, 2014, 6, 7012–7018 RSC.
- P. Chandran, A. Ghosh and S. Ramaprabhu, Sci. Rep., 2018, 8, 1–11 CAS.
- X. Li, Q. Huang, Z. Zou, B. Xia and H. Yang, Electrochim. Acta, 2008, 53, 6662–6667 CrossRef CAS.
- B. Li and J. Prakash, Electrochem. Commun., 2009, 11, 1162–1165 CrossRef CAS.
- J. Jiang, H. Gao, S. Lu, X. Zhang, C.-Y. Wang, W.-K. Wang and H.-Q. Yu, J. Mater. Chem. A, 2017, 5, 9233–9240 RSC.
- Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., 2013, 125, 3192–3198 CrossRef.
- J. Duan, Y. Zheng, S. Chen, Y. Tang, M. Jaroniec and S. Qiao, Chem. Commun., 2013, 49, 7705–7707 RSC.
- J. Masa, C. Batchelor-McAuley, W. Schuhmann and R. G. Compton, Nano Res., 2014, 7, 71–78 CrossRef CAS.
- R. Zhou, Y. Zheng, M. Jaroniec and S.-Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- K. Kinoshita and P. Stonehart, in Modern Aspects of Electrochemistry, ed. J. O. Bockris and B. E. Conway, Springer, Boston, MA, 1977, pp. 183–266 Search PubMed.
- K. Kakaei and M. Dorraji, Electrochim. Acta, 2014, 143, 207–215 CrossRef CAS.
- X. Wang, W. Wang, Z. Qi, C. Zhao, H. Ji and Z. Zhang, Int. J. Hydrogen Energy, 2012, 37, 2579–2587 CrossRef CAS.
- R. N. Singh and C. S. Sharma, Eng. Technol. Appl. Sci. Res., 2012, 2, 295–301 CrossRef.
- T. Nagel, N. Bogolowski and H. Baltruschat, J. Appl. Electrochem., 2006, 36, 1297–1306 CrossRef CAS.
- M. Shao, J. H. Odell, S.-I. Choi and Y. Xia, Electrochem. Commun., 2013, 31, 46–48 CrossRef CAS.
- Z. Jusys, J. Kaiser and R. J. Behm, Phys. Chem. Chem. Phys., 2001, 3, 4650–4660 RSC.
- A. Iiyama, S. Iguchi, A. Daimaru and K. Shinohara, Presented in part at Fuel cell commercialization conference of Japan, 2007.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775 CrossRef CAS.
- Y. Hashimasa and T. Numata, Int. J. Hydrogen Energy, 2015, 40, 11543–11549 CrossRef CAS.
- C. Takei, K. Kakinuma, K. Kawashima, K. Tashiro, M. Watanabe and M. Uchida, J. Power Sources, 2016, 324, 729–737 CrossRef CAS.
- K. Kakinuma, M. Hayashi, T. Hashimoto, A. Iiyama and M. Uchida, ACS Appl. Energy Mater., 2020, 3, 6922–6928 CrossRef CAS.
- K. Mayrhofer, D. Strmcnik, B. Blizanac, V. Stamenkovic, M. Arenz and N. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS.
- Y. Garsany, I. L. Singer and K. E. Swider-Lyons, J. Electroanal. Chem., 2011, 662, 396–406 CrossRef CAS.
- I. Takahashi and S. S. Kocha, J. Power Sources, 2010, 195, 6312–6322 CrossRef CAS.
- C. Xu, Y. Liu, H. Zhang and H. Geng, Chem. – Asian J., 2013, 8, 2721–2728 CrossRef CAS PubMed.
- G. W. Sievers, A. W. Jensen, J. Quinson, A. Zana, F. Bizzotto, M. Oezaslan, A. Dworzak, J. J. Kirkensgaard, T. E. Smitshuysen and S. Kadkhodazadeh, Nat. Mater., 2020, 1–6 Search PubMed.
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- D. E. Beltrán and S. Litster, ACS Energy Lett., 2019, 4, 1158–1161 CrossRef.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- J. C. Calderón Gómez, R. Moliner and M. J. Lázaro, Catalysts, 2016, 6, 130 CrossRef.
- L. Jiang, A. Hsu, D. Chu and R. Chen, J. Electrochem. Soc., 2009, 156, B643 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- C. Chen, Proceedings of the 10th international conference on Intelligent user interfaces, 2005, 98-105.
- C. Chen, F. Ibekwe-SanJuan and J. Hou, J. Am. Soc. Inf. Sci. Technol., 2010, 61, 1386–1409 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |