
In situ engineered ZnS–FeS heterostructures in N-doped carbon nanocages accelerating polysulfide redox kinetics for lithium sulfur batteries†
Wenda
Li
,
Zhijiang
Gong
,
Xiujuan
Yan
,
Dezhu
Wang
,
Jing
Liu
,
Xiaosong
Guo
,
Zhonghua
Zhang
 * and
Guicun
Li
*
* and
Guicun
Li
*
College of Materials Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: zhangzh@qust.edu.cn
First published on 22nd November 2019
Abstract
Electrode materials with efficient catalyzing capability for polysulfides in lithium sulfur batteries are currently receiving intensive research interest for next-generation portable electronic equipment. Herein, a novel hollow architecture composed of ZnS–FeS heterostructures encapsulated in N-doped carbon is designed for the first time as a high-efficiency catalyst to propel polysulfide redox kinetics, in which the ZnS–FeS heterostructures are mosaiced in the carbon framework through a simple in situ sulfuration process. Kinetic analyses and theoretical calculation verify that the abundant heterojunctions could facilitate electron and ion transfer, strengthen the combination with polysulfides and boost the polysulfide redox reaction kinetics. Ex situ electrochemical impedance spectroscopy (EIS) revealed the excellent interface solid–liquid–solid conversion reaction. Benefiting from the state-of-the-art design, the S@ZnS–FeS@NC electrode shows an outstanding rate capacity (718 mA h g−1 at 4.0C) and favorable cycling stability (822 mA h g−1 at 0.2C after 200 cycles). Our approach would be a proof-of-concept design of metal sulfide heterojunctions as an effective sulfur host in improving the polysulfide redox kinetics for lithium sulfur batteries.
1. Introduction
The development of lithium-ion batteries has been severely limited by the high cost and relatively low energy density.1,2 Compared with lithium-ion batteries, lithium–sulfur (Li–S) batteries are regarded as one of the most promising candidates for next-generation energy storage systems due to their high theoretical specific capacity (≈1675 mA h g−1) and earth-abundant sulfur distribution.3–7 However, their practical applications are hindered by the following problems: (1) the essentially insulating nature of sulfur, leading to a low active materials' utilization; (2) the large volume expansion of sulfur during the cycling process, causing an adverse impact on the cycling durability; (3) the shuttle effect of polysulfides, resulting in poor cycling stability and low coulombic efficiency; and (4) the sluggish redox kinetics of polysulfide conversion to Li2S2/Li2S, leading to a poor rate capacity.8–11To address these problems, tremendous efforts have been devoted towards designing the nanostructure of sulfur hosts.12–16 These attempts have made considerable progress in solving some aspects of these problems and accelerated the practical applications of Li–S batteries. Over the past few years, hollow carbon materials, including hollow carbon nanospheres, carbon nanotubes and carbon nanocages, as sulfur hosts have been investigated extensively to improve the conductivity of sulfur cathodes, which can also enhance physical entrapment of the polysulfides and ease volume expansion of sulfur simultaneously.17–20 Notwithstanding all these advantages, hollow structured carbon–sulfur cathodes usually suffer from rapid capacity decay due to the weak physical interaction between nonpolar carbon and polar polysulfides.21 These technical challenges unavoidably lead to insufficient sulfur utilization, sluggish reaction kinetics and low coulombic efficiency in Li–S batteries.
Recently, inorganic compound materials, such as metal oxides, metal sulfides and metal nitrides, have been employed as sulfur hosts for their strong polarity with polysulfides.22–29 Among them, metal sulfides have been widely applied as sulfur hosts for Li–S batteries because of their simple preparation process, high yield and strong chemical adsorption properties onto polysulfide species.30 Cui et al. systematically investigated the adsorption capability between various metal sulfides and polysulfides, which revealed that FeS possessed stronger adsorption than Li2S6 species.31 In addition, Wang et al. confirmed that ZnS can promote the fast conversion of polysulfides by combining first-principles calculations with experimental analysis.32 In order to enhance the redox kinetics of polysulfide conversion and obtain impressive rate performance, defect and heterojunction engineering is explored by many researchers as a novel research approach for Li–S batteries.33 Building heterostructures containing dissimilar coupling components with different bandgaps could promote interfacial reaction kinetics and accelerate charge carrier transport. Although VO2–VN, TiO2–TiN and MXene/1T-2H MoS2 heterostructures have been proved to exhibit preferable electrochemical performance as sulfur hosts for Li–S batteries,34 it is generally believed that building heterostructures containing dissimilar coupling components with different bandgaps could promote interfacial reaction kinetics and accelerate charge carrier transport.35 However, the heterostructures with smaller intrinsic energy bandgaps in previous reports might possess lower catalyzing capability to polysulfides due to the relatively weak built-in electric field. In addition, previous literature studies pertaining to heterostructures for sulfur hosts showed limited sulfur loading because of their irregular morphologies such as micronized particles, nanosheets and nanoflakes. It is still far from satisfactory owing to the large volume changes during discharge/charge processes and poor redox reaction kinetics of polysulfides.34 Therefore, engineering heterostructures with a strong intrinsic E-field accompanied by hollow structure design at the nano/microscopic level would be an effective path to high-performance Li–S batteries.
In this work, for the first time, we designed ZnS–FeS heterostructures encapsulated in N-doped carbon (ZnS–FeS/NC) as a multifunctional catalyst to propel the polysulfide redox kinetics, which significantly improved the electrochemical performance of the Li–S battery. The heterostructures are based on the different intrinsic energy bandgaps of ∼0.8 eV for FeS and ∼4.9 eV for ZnS. A strong E-field at the heterointerface is induced by the large energy bandgap between the integrated compounds, which would effectively promote polysulfide conversion. Furthermore, the hollow structures provide a large space for sulfur loading and buffer volume expansion during cycling. Kinetic analyses and first-principles calculation verified that the abundant heterostructures facilitated electron and ion transfer, strengthened the combination with polysulfides and boosted the surface redox reaction kinetics. Ex situ electrochemical impedance spectroscopy (EIS) revealed the excellent interface solid–liquid–solid conversion reaction. Importantly, owing to the ingenious designed and strong exclusive binding with polysulfides, the ZnS–FeS/NC hybrids show an outstanding rate capacity (718 mA h g−1 at 4.0C) and favorable cycling stability (822 mA h g−1 at 0.2C after 200 cycles).
2. Experimental
2.1 Materials preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) on the upstream side of a tube furnace and calcined at 650 °C for 2 hours in a flowing argon atmosphere. The FeS/NC nanocages were prepared by etching the ZnS–FeS/NC sample with NaOH solution.
:3 were mixed together and kept in a sealed vessel, which was heated to 155 °C for 12 h to produce the S@ZnS–FeS/NC (or S@ZnS/NC and S@FeS/NC) composites. The sulfur cathodes were prepared via a slurry coating method. In brief, the slurry was prepared by mixing the sulfur composites (S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC), Super P and polyvinylidene fluoride (PVDF) binder in a weight ratio of 7:2:1 in N-methyl-2-pyrrolidone (NMP) and was then uniformly compressed onto a carbon cloth current collector and dried in a vacuum oven at 60 °C overnight. The sulfur loading in all electrodes was controlled to be 1.02–3.42 mg cm−2.
:3) on the upstream side of a tube furnace and calcined at 650 °C for 2 hours in a flowing argon atmosphere. The FeS/NC nanocages were prepared by etching the ZnS–FeS/NC sample with NaOH solution.
:3 were mixed together and kept in a sealed vessel, which was heated to 155 °C for 12 h to produce the S@ZnS–FeS/NC (or S@ZnS/NC and S@FeS/NC) composites. The sulfur cathodes were prepared via a slurry coating method. In brief, the slurry was prepared by mixing the sulfur composites (S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC), Super P and polyvinylidene fluoride (PVDF) binder in a weight ratio of 7:2:1 in N-methyl-2-pyrrolidone (NMP) and was then uniformly compressed onto a carbon cloth current collector and dried in a vacuum oven at 60 °C overnight. The sulfur loading in all electrodes was controlled to be 1.02–3.42 mg cm−2.
2.2 Characterization of materials
The morphologies of all samples were characterized by field-emission scanning electron microscopy (FESEM, Hitachi S-4800, Japan). Energy dispersive spectroscopy (EDS) elemental mapping was performed on an Oxford Inca EDS detector on a Tecnai 2100. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were recorded using a JEOL-2100F. The crystal structures of all samples were examined by powder X-ray diffraction (XRD) with Cu Kα radiation (λ = 1.5 Å) over the range of 5–85° (2θ) at room temperature. X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI5300 instrument. Raman spectroscopy test was performed on a LabRAM HR Evolution in the range of 100–2000 cm−1. The specific surface area was measured by the multipoint Brunauer–Emmett–Teller (BET) method at 78 K with a Quantachrome NOVA-4200e system. Elemental analysis (EA, Vario EL III) and inductively coupled plasma (ICP, Thermo (iCAP Q)) were conducted to measure the elemental composition of the samples. The sulfur content of the cathode material was determined by thermogravimetric analysis (TGA, NETZSCH).2.3 Theoretical calculation
The calculations were performed using the Vienna Ab initio simulation package (VASP). The six outmost electrons for transition metals and chalcogens were treated as valence electrons. The generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE) was adopted for the exchange-correlation functional.36 Energy cut off for plane-wave expansion was set to 520 eV.37 Brillouin zone sampling was performed with Monkhorst–Pack (MP) special k point meshes. A supercell (2 2 1) with lattice constants a = b = 3.821 Å and c = 6.257 Å and a supercell (2 2 2) with lattice constants a = b = c = 5.419 Å were constructed for ZnS and FeS, respectively. A vacuum layer larger than 10 Å was added to avoid interaction between adjacent images. All atoms were allowed to relax until the calculated Hellmann–Feynman force on each atom was smaller than 0.01 eV Å−1.2.4 Electrochemical characterization
The electrochemical performances of the S@ZnS–FeS/NC cathode and comparison samples were examined by using CR-2032 coin-type cells with a lithium foil anode and a polypropylene (PP) separator. The electrolyte was prepared by dissolving lithium bis(trifluoromethanesulfonic)imide (1 M) in 1,3-dioxolane (DOL)/ethylene glycol dimethyl ether (DME) (1:1 v/v) containing lithium nitrate (1 wt%). All cells were assembled in a glovebox filled with argon with the concentration of oxygen below 1.0 ppm. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) tests were conducted on an Autolab electrochemical workstation. The charge–discharge measurements were performed using a LAND CT 2001A instrument at room temperature.
3. Results and discussion
The synthetic process of the S@ZnS–FeS/NC electrodes is shown in Fig. 1. First, the well-defined Fe/ZIF-8 polyhedra are facilely synthesized by a precipitation method in the presence of zinc nitrite, ferric acetylacetonate and 2-methylimidazole in methanol at room temperature. The ferric acetylacetonate molecule was well limited in the framework of ZIF-8 during the bridging process between Zn ions and 2-methylimidazole precursor. Then, the collected Fe/ZIF-8 polyhedra are etched with tannic acid (TA)38 and the detailed etching process is shown in Fig. S1.† The TA molecules can attach to the surface of Fe/ZIF-8 polyhedra and the free H+ ions can penetrate into polyhedra.39,40| C76H52O46 (TA) → C76H52O46 + H+ | (1) |
| Fe2(C8H4O4)3 + 6H+ → 2Fe3+ + 3C8H6O4 | (2) |
| Zn(C4H6N2)2 + 2H+ → Zn2++ 2C4H6N2 | (3) |
| Zn2++ C76H52O46 → Zn(C76H52O46)2 | (4) |
| C76H52O46 + 2Fe3+ → Fe2(C76H52O46)3 | (5) |
 | ||
| Fig. 1 Representative illustration of the preparation processes of the S@ZnS–FeS/NC electrodes. | ||
Consequently, the TA–Fe/Zn-ZIF nanocages with intrinsic crystalline frameworks are obtained. Third, in the following sulfuration process, the tannic acid molecules on the Fe/ZIF-8 rhombic surface and organic ligands transform into a highly conductive carbon matrix. Synchronously, the ferric acetylacetonate and zinc species are further in situ transformed into well-crystallized FeS and ZnS nanoparticles, respectively. The S@ZnS–FeS/NC electrodes were finally obtained by heating sulfur and ZnS–FeS/NC with a ratio of 7:3 in a sealed vessel at 155 °C for 12 h.
In order to investigate the morphology and nanostructure of the as-synthesized composites, the ZIF-8 and Fe/ZIF-8 polyhedra were characterized by SEM. As shown in Fig. 2a and b, both of them demonstrate a highly uniform dodecahedral structure with an average diameter of about 120 nm. In order to verify the critical role of ferric acetylacetonate in the synthesis process (Fig. S2†), the morphology of Fe/ZIF-8 polyhedra have also been investigated by adding varied contents of ferric acetylacetonate, demonstrating that the surface of Fe/ZIF-8 nanocrystals become rougher with the increase of ferric acetylacetonate. Besides, it has been revealed that the crystal phase of Fe/ZIF-8 polyhedra is consistent with ZIF-8 polyhedra by XRD analysis (Fig. S3†), which indicates that the introduction of ferric acetylacetonate does not destroy the original skeleton structure of ZIF-8 polyhedra. The TA–Fe/ZIF-8 nanocages obtained by etching with TA maintain a similar polyhedron shape with a smooth surface (Fig. 2c), indicating that tannic acid molecules are uniformly coated outside the Fe/ZIF-8 nanocages. The XRD analysis further verifies that the skeleton structure is still maintained well after being etched (Fig. S4†). The SEM image of ZnS–FeS/NC composites shown in Fig. 2d clearly demonstrates the typical hollow structure in a polyhedron.
 | ||
| Fig. 2 Typical SEM images of (a) ZIF-8 and (b) Fe/ZIF-8 polyhedra. (c) SEM images of the TA–Fe/ZIF-8 nanocages. (d) SEM images of the ZnS–FeS/NC nanocages. (e) SEM image of the ZnS–FeS/NC nanocages and the corresponding elemental mapping of S, Fe and Zn. (f) TEM images and (g–i) HRTEM images of the ZnS–FeS/NC nanocages. | ||
To illustrate the homogeneous distribution of elements in the ZnS–FeS/NC nanocages, energy dispersive spectroscopy (EDS) mapping has been performed as shown in Fig. 2e and S5,† which shows the uniform distribution of S, Zn, Fe, C and N throughout the samples. To further investigate the nanostructure of ZnS–FeS/NC nanocages, typical TEM images (Fig. 2f) of the obtained ZnS–FeS/NC nanocages show a uniform hollow architecture with the polyhedral shell. HRTEM images are also obtained and shown in Fig. 2g–i, which demonstrate that the ZnS–FeS/NC nanocage is assembled from ZnS and FeS nanoparticles with a particle size of 10–20 nm within a thin carbon wall. The lattice fringes of the ZnS–FeS heterostructure with a d-spacing of 0.31 and 0.26 nm correspond to the (111) and (200) crystal planes of ZnS and FeS, respectively.41,42 Moreover, the heterointerface between ZnS and FeS can be observed clearly, which acts as the highly active sites for catalyzing the conversion of polysulfides and enhancing the electron transfer kinetics.
The XRD patterns of the ZnS–FeS/NC, FeS/NC and ZnS/NC composites are presented in Fig. 3b and S6.† The XRD pattern for ZnS–FeS/NC exhibits major peaks at a 2θ of 26.9°, 28.5°, 30.5° and 39.6° corresponding to the (100), (002), (101), and (102) lattice planes of rhombohedra ZnS, which match well to JCPDS no. 36-1450 for ZnS.43 Besides the XRD peaks related to ZnS, additional peaks located at 28.5°, 47.4° and 56.2° can be observed, which can be indexed to the (111), (220) and (311) plane of FeS (PDF 23-1123), respectively.44 The XRD results suggest that hollow structured ZnS–FeS/NC composites are successfully obtained through sulfuration treatment. To investigate the Brunauer–Emmett–Teller (BET) specific surface area and porous structural characteristics of the ZnS–FeS/NC nanocages, the nitrogen adsorption/desorption measurement has been performed. The adsorption/desorption isotherms of the ZnS–FeS/NC nanocages show a type-IV adsorption branch at a relative pressure p/p0 of 0.1–1.0 (Fig. 3b).45 The BET surface area of the samples is calculated to be about 186.78 m2 g−1. In addition, the pore size distribution calculated by the Barrett–Joyner–Halenda (BJH) method is depicted in the inset of Fig. 3b. The pore structure of ZnS–FeS/NC nanocages is characterized by two kinds of pores. In detail, the pore sizes of the ZnS–FeS/NC nanocages can be divided into 5 nm and 80 nm. The mesopores of around 5 nm may originate from the vapor of organic species in the polyhedral shell, which could effectively reduce the Li-ion transfer barriers. The macropores of around 80 nm may be induced by TA etching, which could ease the volume expansion efficiently during the charge/discharge process.
 | ||
| Fig. 3 (a) XRD patterns of the ZnS–FeS/NC composites. (b) Nitrogen adsorption–desorption isotherms of the ZnS–FeS/NC composite (inset: the pore-size distributions curves). High-resolution (c) C 1s XPS spectra, (d) N 1s XPS spectra, (e) S 2p XPS spectra, (f) Zn 2p XPS spectra and (g) Fe 2p XPS spectra of the ZnS–FeS/NC composite. (h) Raman spectra of ZnS/NC, FeS/NC and ZnS–FeS/NC composites. | ||
The quantitative detection and chemical bonding state are described by ICP, EA and XPS characterization methods. For the ZnS–FeS/NC composite, the ratio of Zn and Fe elements was ≈2:1 based on the ICP spectroscopy result (Table S1†). In addition, the contents of C and N in the ZnS–FeS/NC composite were confirmed by EA (Table S2†). It showed that the C and N contents were 44.2 wt% and 6.4 wt%, respectively. As shown in Fig. S7,† it can be found that C 1s, N 1s, S 2p, Fe 2p and Zn 2p peaks appeared at about 284.8 eV, 400.1 eV, 164.2 eV, 722.5 eV and 1018.2 eV in the spectra, respectively. In the C 1s spectrum, three characteristic peaks centered at 284.6, 285.3, and 286.8 eV can be deconvoluted (Fig. 3c), which are assigned to the carbon atoms in C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, and C–O groups, respectively.46,47 The main peaks at 284.6 eV and 285.3 eV suggest that graphitic carbon and nitrogen doped carbon are the majority species in the samples. The N 1s spectra (Fig. 3d) of ZnS–FeS/NC can be deconvoluted into three peaks, namely the pyridinic N (398.4 eV), pyrrolic N (400.5 eV) and graphitic N (401.5 eV).48 The N content in ZnS–FeS/NC measured using XPS is 6.82%, which indicates a high N-element doping content in ZnS–FeS/NC samples. The nitrogen groups in the carbon matrix can provide strong polar interactions with polysulfides, thus leading to an effective confinement effect for sulfur species. The high-resolution XPS spectral peaks of the S 2p region are located at 162.4 and 163.6 eV (Fig. 3e) associated with the S 2p3/2 and S 2p1/2 of ZnS, respectively.49 A couple of peaks at 162.4 and 163.6 eV can be attributed to S 2p3/2 and S 2p1/2 of FeS, respectively. Another peak at 168.2 eV can be attributed to S–O species due to the exposure to an air atmosphere.50 The Zn 2p spectrum in the high-resolution XPS spectra is divided into one couple of peaks (Fig. 3f): the 2p3/2 (232.7 eV) and 2p1/2 (236.3 eV), which are attributed to ZnS.51 The Fe 2p spectrum in Fig. 4g shows two couples of peaks at 724.6 and 710.9 eV, which are attributed to Fe 2p1/2 and Fe 2p3/2, respectively.52–54 This suggests that Fe appears in the divalent state Fe2+. These XPS analyses are consistent with XRD and HRTEM results.
C, C–N, and C–O groups, respectively.46,47 The main peaks at 284.6 eV and 285.3 eV suggest that graphitic carbon and nitrogen doped carbon are the majority species in the samples. The N 1s spectra (Fig. 3d) of ZnS–FeS/NC can be deconvoluted into three peaks, namely the pyridinic N (398.4 eV), pyrrolic N (400.5 eV) and graphitic N (401.5 eV).48 The N content in ZnS–FeS/NC measured using XPS is 6.82%, which indicates a high N-element doping content in ZnS–FeS/NC samples. The nitrogen groups in the carbon matrix can provide strong polar interactions with polysulfides, thus leading to an effective confinement effect for sulfur species. The high-resolution XPS spectral peaks of the S 2p region are located at 162.4 and 163.6 eV (Fig. 3e) associated with the S 2p3/2 and S 2p1/2 of ZnS, respectively.49 A couple of peaks at 162.4 and 163.6 eV can be attributed to S 2p3/2 and S 2p1/2 of FeS, respectively. Another peak at 168.2 eV can be attributed to S–O species due to the exposure to an air atmosphere.50 The Zn 2p spectrum in the high-resolution XPS spectra is divided into one couple of peaks (Fig. 3f): the 2p3/2 (232.7 eV) and 2p1/2 (236.3 eV), which are attributed to ZnS.51 The Fe 2p spectrum in Fig. 4g shows two couples of peaks at 724.6 and 710.9 eV, which are attributed to Fe 2p1/2 and Fe 2p3/2, respectively.52–54 This suggests that Fe appears in the divalent state Fe2+. These XPS analyses are consistent with XRD and HRTEM results.
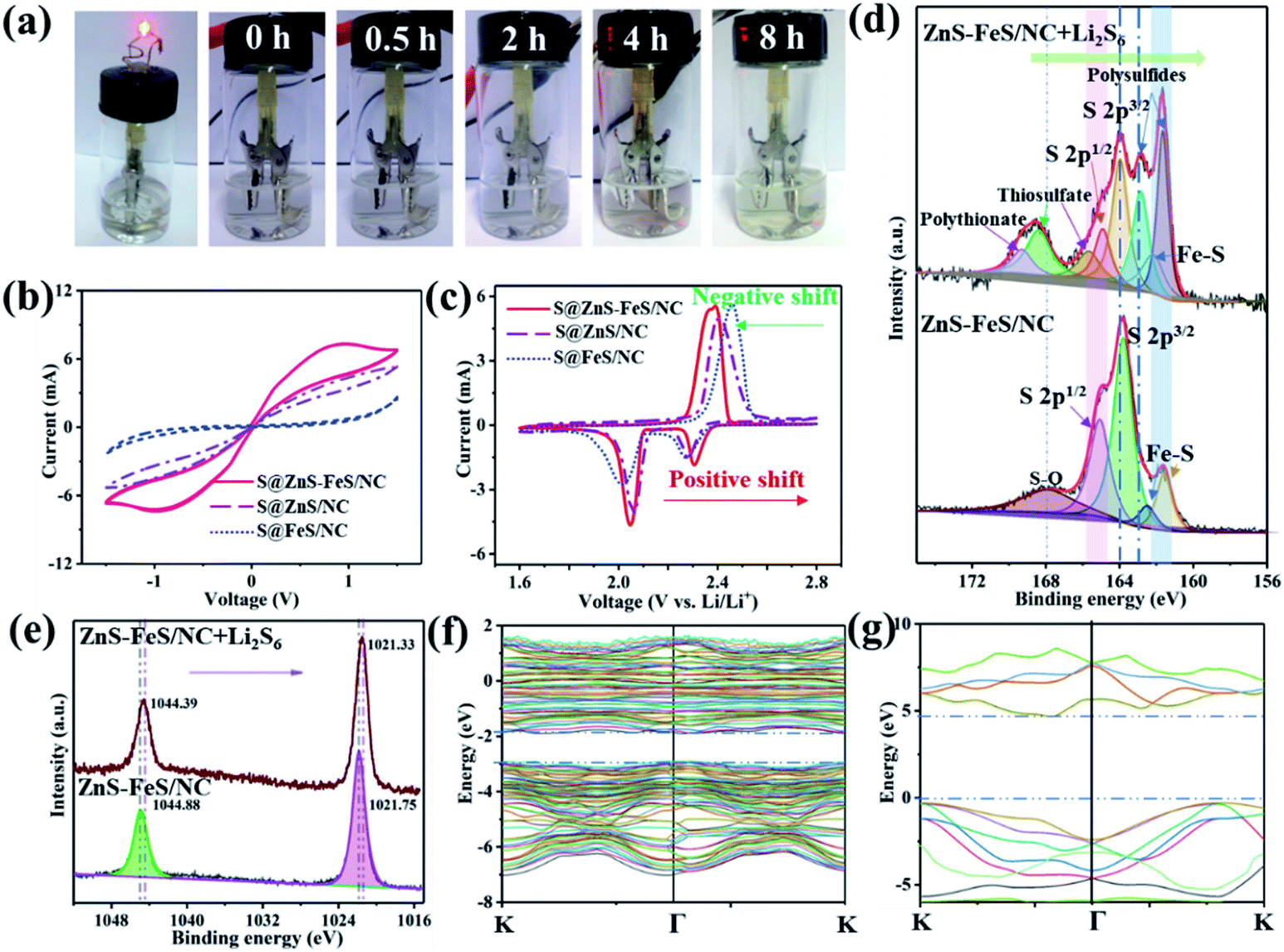 | ||
| Fig. 4 (a) Digital photos of the transparent Li–S cell at different discharge states of the S@ZnS–FeS/NC electrode. (b) CV curves of the Li2S6-containing symmetrical cell with working electrodes of the ZnS–FeS/NC, ZnS/NC and FeS/NC composites at a scan rate of 5 mV s−1. (c) The comparison of the CV curves of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC cathodes in the third cycle at a scan rate of 0.1 mV s−1. (d) XPS (S 2p spectra) of ZnS–FeS/NC and ZnS–FeS/NC + Li2S6. (e) XPS (Zn 2p spectra) of ZnS–FeS/NC and ZnS–FeS/NC + Li2S6. (f and g) The calculated band structure for ZnS and FeS, respectively. | ||
The Raman spectra (Fig. 3h) of the ZnS–FeS/NC, ZnS/NC and FeS/NC composites show two strong peaks at 1350 and 1580 cm−1, which are assigned to the defect-activated disordered D-band and the ordered G-band of graphitic layers of N-doped carbon nanocages, respectively. Moreover, several weak peaks can also be observed from around 200 to 500 cm−1 in ZnS–FeS/NC, which are related to the transverse mode of ZnS and FeS, respectively.50 These results further confirm the existence of ZnS–FeS heterostructures and are consistent with the TEM results.
The S@ZnS–FeS/NC composite maintains the polyhedral shape after sulfur is loaded (Fig. S8†). The XRD pattern was recorded to determine the crystalline phase of the S@ZnS–FeS/NC composite as shown in Fig. S9,† verifying that sulfur successfully penetrated into ZnS–FeS/NC. The content of sulfur in the S@ZnS–FeS/NC composite is estimated to be 69.9 wt% according to thermogravimetric analysis (TGA) (Fig. S10†). The morphology and distribution of Fe, Zn, S, and C elements in the S@ZnS–FeS/NC composite were investigated by TEM and EDS mapping (Fig. S11†). The darker contrast of the inside region of the S@ZnS–FeS/NC composite could be ascribed to the diffusion of sulfur into the inner space. According to the EDS-mapping images, sulfur has been homogeneously encapsulated within the ZnS–FeS/NC composite. The hollow structure could efficiently immobilize the polysulfides and ease volume expansion of sulfur simultaneously.
Polysulfide chemisorption is investigated using the transparent Li–S cells with the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC cathodes, respectively. As shown in Fig. 4a and S12,† the electrolyte color of Li–S cells using the S@ZnS–FeS/NC composite changes a little during the discharge/charge process, while the electrolyte color of Li–S cells with the S@ZnS/NC and S@FeS/NC cathodes changes from colorless to brown. These results evidently authenticate the stronger polysulfide chemisorption ability of the ZnS–FeS/NC composite towards soluble polysulfide species. To probe the contribution of the ZnS–FeS heterostructure to improving the reaction kinetics of polysulfide conversion in Li–S batteries, cyclic voltammetry (CV) tests have been conducted in the voltage range −1.5–1.5 V for Li2S6 symmetrical cells. The Li2S6 symmetrical cell with the ZnS–FeS/NC electrode shows much larger redox currents than the FeS/NC and ZnS/NC references (Fig. 4b), which can be identified from the distinct peaks (−0.8 V and 0.8 V). The higher current response verifies that ZnS–FeS heterostructures significantly enhance the lithiation/delithiation reaction kinetics of polysulfide conversion.55
The CV curves of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC composite cathodes for the selected first cycles at a scan rate of 0.1 mV s−1 are presented in Fig. 4c. Two reduction peaks are observed at around 2.31 and 2.03 V, corresponding to the reduction of S8 molecules to the long-chain polysulfides (Li2Sn) and further transformation into Li2S2/Li2S. In addition, one anodic peak at around 2.31 V corresponds to the conversion of Li2S2/Li2S to elemental S8. The profiles for the five cycles are well-overlapped, indicating the good electrochemical stability of the cells based on the S@ZnS–FeS/NC electrode (Fig. S13†). Specifically, the reduction peaks of the S@ZnS–FeS/NC electrode shift to higher potentials and the oxidation peak moves to a lower potential than those of the S@ZnS/NC and S@FeS/NC electrodes, resulting in a lower charge–discharge overpotential. This result demonstrates that the ZnS–FeS heterostructure is highly beneficial to enhance the redox reaction kinetics of polysulfides.
XPS analyses of the pristine ZnS–FeS/NC and ZnS–FeS/NC-Li2S6 composite after the Li2S6 adsorption experiment have been performed to provide additional evidence for the strong interaction between polysulfides and ZnS–FeS heterostructures. As shown in Fig. 4d, pristine ZnS–FeS/NC shows two couples of S 2p character peaks, attributed to ZnS and FeS, respectively. However, the S 2p spectra of the solid precipitates after the Li2S6 adsorption experiment from the Li2S6 solution display four predominant peaks, which can be attributed to short chain polysulfides with S–O groups (167.5 eV), polysulfides (161.7 eV), thiosulfate (165.2 eV) and polythiosulfate complexes (164.8 eV), respectively. The thiosulfate and polythiosulfate complexes may originate from the strong interactions between Li2S6 and the ZnS–FeS heterostructure.33 These new sulfur species possibly arise from the surface redox reaction between Li2S6 and ZnS–FeS/NC. Meanwhile, Zn 2p peaks of ZnS–FeS/NC-Li2S6 shift towards lower binding energy with regard to that of pristine ZnS–FeS/NC (Fig. 4e), demonstrating the increased electron density at the metal center.32 The relative intensity of Fe3+ significantly decreases from 64% to 48%, accompanied by the increase of Fe2+ from 36% to 52% (Fig. S14†). These results correspond to the changes of the S 2p spectra. Fig. S15† shows that N 1s spectrum of ZnS–FeS/NC consists of two main peaks. After adsorbing Li2S6, the peaks shift towards the higher binding energy region obviously, which is assigned to the strong interaction of N species in ZnS–FeS/NC with polysulfides.56 Therefore, XPS analyses strongly prove that there is chemisorption and surface redox reactions between polysulfides and ZnS–FeS/NC.
The first-principles calculation is utilized to reveal the band offset between ZnS and FeS. Both the conduction band and valence band of ZnS are higher than those of FeS as shown in the calculated band structures (Fig. 4f and g) and band alignment (Fig. S16†), which is in agreement with previous reports.57,58 ZnS possesses a wide energy bandgap of ∼4.90 eV (Eg = VB–CB) with a conduction band (CB) of ∼–0.04 eV and a valence band (VB) of 4.86 eV. In contrast, FeS serves as a good electron conductor with overlapping CB and VB, possessing a narrow energy bandgap of ∼0.80 eV. Therefore, the heterostructure consisting of ZnS and FeS could induce a strong built-in electric field and effectively promote the electronic conductivity of the composite. More importantly, the ZnS–FeS heterostructures could enhance polysulfide redox kinetics for lithium–sulfur batteries, benefitting from the strong build-in E-field that could effectively boost the charge-transfer processes during the electrochemical redox reaction occurring on the adsorption surface of ZnS–FeS heterostructures.
Based on the above experiments and theoretical analyses, the facilitated ion/electron migration, enhanced polysulfide adsorption and boosted polysulfide conversion process by employing the highly active ZnS–FeS heterostructures are schematically illustrated in Fig. 5. First, the heterostructures, consisting of an n-type semiconductor (ZnS) and a p-type semiconductor (FeS), possess abundant highly stable heterointerfaces that not only facilitate Li ion and electron migration, but also can effectively promote the reversible conversion of polysulfides to solid Li2S, leading to improved rate capabilities. Second, the strong chemical adsorption between polysulfides and polar FeS–ZnS can effectively suppress the diffusion of polysulfides into the electrolyte, which could be beneficial for the achievement of stable cycle performances. Third, the well-defined hollow structure could efficiently buffer volume expansion/shrinkage during the charge/discharge process.
 | ||
| Fig. 5 Illustration of the fast-redox conversion reaction of polysulfides catalyzed by ZnS–FeS heterostructures during the discharge/charge process. | ||
The electrochemical performances of ZnS–FeS/NC, ZnS/NC and FeS/NC as sulfur hosts for Li–S batteries are examined in CR2032-type coin cells. Fig. 6a shows the rate performances of the above three samples at different current densities. The discharge capacities of three sulfur cathodes decrease with the increase of current rate from 0.2 to 4.0C. During cycling at 0.2, 0.5, 1.0, 2.0 and 4.0C, the discharge specific capacities of S@ZnS–FeS/NC are around 1278, 1083, 911, 858 and 718 mA h g−1, respectively. The corresponding discharge capacities revert to 827, 864, 985, and 1136 mA h g−1 after decreasing the current density to 2C, 1C, 0.5C, and 0.2C, respectively. The S@ZnS–FeS/NC cathode exhibited the highest reversible capacity and superior rate properties compared to S@ZnS/NC and S@FeS/NC at the varying rates. The corresponding charge and discharge profiles at different current densities of the S@ZnS–FeS/NC cathode are depicted in Fig. 6b. Compared to S@ZnS/NC and S@FeS/NC (Fig. S17†), the S@ZnS–FeS/NC cathode retains the charge and discharge voltage plateaus at a high current density. In addition, the overpotentials (ΔE) of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC electrodes at different current rates are shown in Fig. 6c, which reveal that the S@ZnS–FeS/NC electrode delivers the lowest charge–discharge overpotentials, suggesting the excellent Li-ion diffusion kinetics and electron transportation in the ZnS–FeS/NC architecture.
 | ||
| Fig. 6 (a) The rate performance comparison of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC cathodes. (b) Voltage profiles and rate capacities of S@ZnS–FeS/NC at various current densities from 0.2 to 4.0C. (c) Overpotentials (ΔE) of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC electrodes at different current rates. (d) The comparison of cycling performance and coulombic efficiency of the S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC electrodes at 0.2C. (e) The cycling performance and coulombic efficiency of the S@ZnS–FeS/NC cathode at 1.0, 2.0 and 4.0C, respectively. (f) The cycling performance and coulombic efficiency of the S@ZnS–FeS/NC electrode with different S loadings at 0.2C. (g) Galvanostatic charge/discharge profiles of the S@ZnS–FeS/NC electrode with a S loading of 3.34 mg cm−2 for different cycles at 0.2C. | ||
Cycling performances of S@ZnS–FeS/NC, S@ZnS/NC and S@FeS/NC composites at 0.2C are shown in Fig. 6d. The S@ZnS–FeS/NC cathodes deliver a higher initial specific discharge capacity of ∼1232 mA h g−1 at 0.2C and retain ∼822 mA h g−1 after 200 cycles, which is superior to that of the S@ZnS/NC and S@FeS/NC electrodes. Fig. 6e shows the cycling performance of the S@ZnS–FeS/NC cathode at 1.0, 2.0 and 4.0C, respectively. These results demonstrate the excellent cycling stability at different rates. Fig. 6f shows the cycling performances at gradually increased sulfur loadings from 2.01 to 3.34 mg cm−2 at a current rate of 0.2C. After 200 cycles, the cathodes with a sulfur loading of 2.01, 2.40 and 3.34 mg cm−2 retain a capacity of 823, 811 and 796 mA h g−1, respectively. Fig. 6g shows the representative galvanostatic discharge/charge voltage profiles of the S@ZnS–FeS/NC cathodes with a sulfur loading of 3.34 mg cm−2. The discharge/charge voltage profiles can be well retained as those of the lower loading electrodes, which indicates that the well-designed ZnS–FeS heterostructure could effectively boost the cathode performance under high sulfur loadings. It is worth noting that the high specific capacity and preeminent rate properties achieved in S@ZnS–FeS/NC are superior to most of the literature reports (Table S3†).
The reduction between sulfur and Li ions is a multistep electrochemical process that introduces different intermediate species.59 The interface impedance of the cells sensitively depends on the solid–liquid–solid conversion processes, which is helpful for us to understand the detailed interface reduction of Li–S batteries. The ex situ EIS spectra of the S@ZnS–FeS/NC electrode are recorded as shown in Fig. 7a and b. Typically, all the Nyquist plots are composed of a depressed semicircle in the high-frequency region, a depressed semicircle in the middle-frequency region, and a sloping line in the low-frequency region.60 It is worth noting that there is an appreciable decrease in the interface resistance of the S@ZnS–FeS/NC electrode during the cell discharge process (I – V), verifying an optimized electron/ion conductivity and solid–liquid–solid conversion kinetics.61 During the charge process (V – VII), there are no significant changes in the interface resistance and the charge transfer resistance, exhibiting a stable interface reaction.62 In the final recharge state (VII), the interface resistance and the charge transfer resistance are lower than those in the origin state, indicating that the ZnS–FeS heterostructure optimizes the sulfur deposition. In addition, the S@ZnS–FeS/NC electrode after cycling is characterized by SEM as shown in Fig. 7c and d. As shown, the electrode shows a smooth surface and is characterized by a poriferous and lax structure. The high-resolution SEM image of the S@ZnS–FeS/NC electrode demonstrates the complete polyhedral structure, indicating that the hollow structure is stable during the charge/discharge process. Encouragingly, compared to the sulfur/carbon cathode, the S@ZnS–FeS/NC electrode demonstrates smaller changes after discharge (Fig. S18†), which present the ZnS–FeS/NC hollow structure can effectively buffer the volume expansion of sulfur upon lithiation. These results inevitably demonstrate the excellent structure and function design for Li–S batteries.
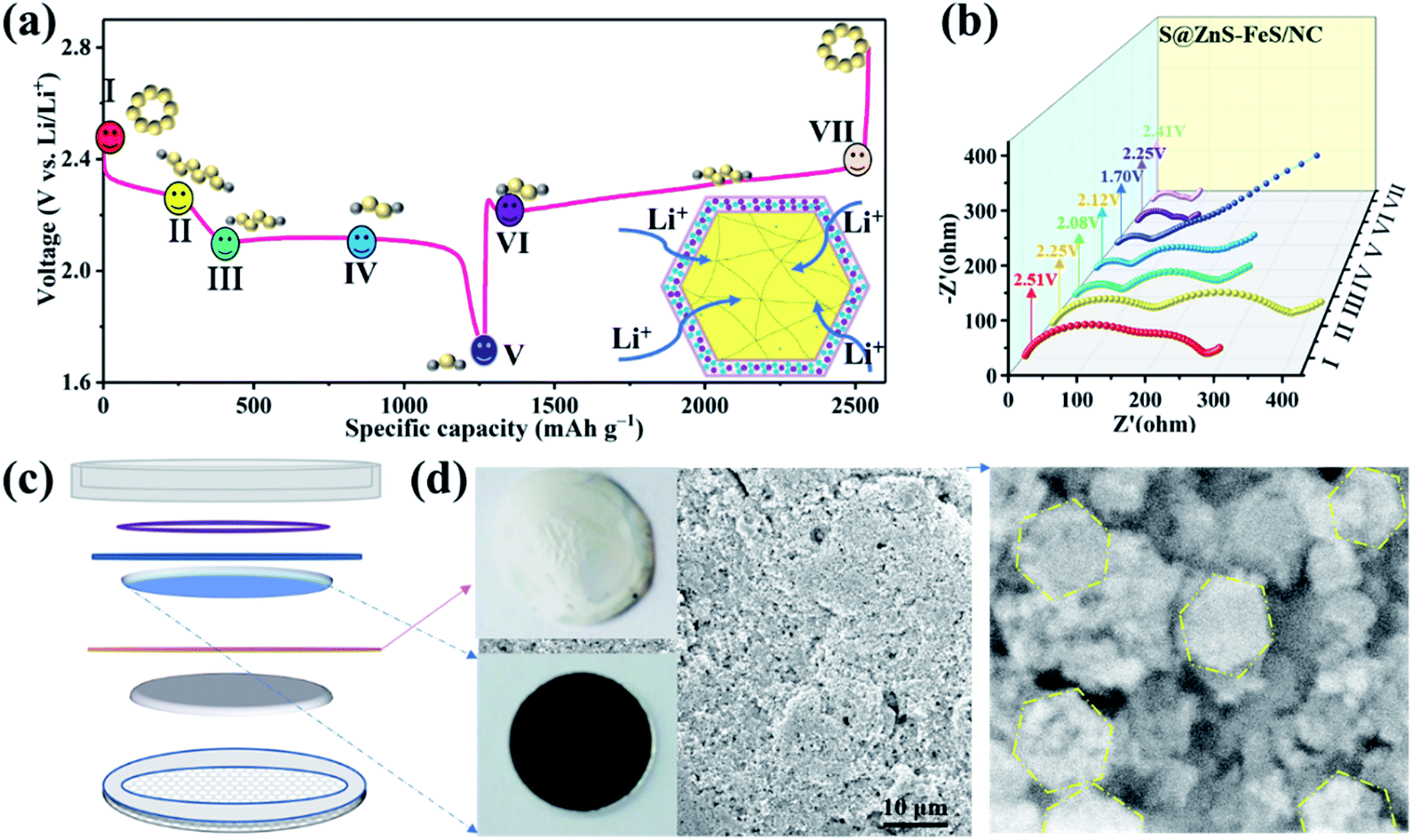 | ||
| Fig. 7 (a and b) Ex situ EIS spectra of the S@ZnS–FeS/NC electrode in the charge/discharge process. (c and d) SEM image of the S@ZnS–FeS/NC electrode after 100 cycles. | ||
4. Conclusions
In summary, a three-dimensional architecture composed of a ZnS–FeS heterostructure encapsulated in N-doped carbon nanocages is designed for the first time as a promising sulfur host for Li–S batteries. Such a binary sulfur host combines the merits of strong anchoring with polysulfides and high ultrafast electron/ion conduction. The highly efficient immobilization-catalyzing-conversion mechanism of polysulfides generated by the ZnS–FeS heterostructure greatly suppresses polysulfide shuttling, promotes the redox kinetics, and hence boosts the electrochemical performance of Li–S batteries. The assembled cathode based on the ZnS–FeS/NC hosts exhibits an impressive rate capability with 718 mA h g−1 at 4.0C and maintained long-term cyclability with a low capacity decay of 0.16% per cycle within 200 cycles at 0.2C. More remarkably, an areal capacity of 3.57 mA h cm−2 can be further attained with a high sulfur loading (3.34 mg cm−2) after 200 cycles. We believe that our work not only can provide conceptually novel opportunities for designing sulfur hosts, but also would hold great application potential in portable electronics and implantable devices for Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51672146, 21805157, 51972187), the Natural Science Foundation of Shandong Province (ZR2018BEM011, ZR2019MEM043 and ZR2019MB037), the Key R & D project of Shandong Province (2019GGX103034) and the Development Program in Science and Technology of Qingdao (19-6-2-12-cg).Notes and references
- H. Chu, H. Noh, Y. J. Kim, S. Yuk, J. H. Lee, J. Lee, H. Kwack, Y. Kim, D. K. Yang and H. T. Kim, Nat. Commun., 2019, 10, 188 CrossRef PubMed.
- H. Gao, T. Zhou, Y. Zheng, Q. Zhang, Y. Liu, J. Chen, H. Liu and Z. Guo, Adv. Funct. Mater., 2017, 27, 1702634 CrossRef.
- H. Pan, K. S. Han, M. H. Engelhard, R. Cao, J. Chen, J.-G. Zhang, K. T. Mueller, Y. Shao and J. Liu, Adv. Funct. Mater., 2018, 28, 1707234 CrossRef.
- N. Song, Z. Gao, Y. Zhang and X. Li, Nano Energy, 2019, 58, 30–39 CrossRef CAS.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- J. He and A. Manthiram, Energy Storage Mater., 2019, 20, 55–70 CrossRef.
- J. Zhang, H. Hu, Z. Li and X. W. Lou, Angew. Chem., Int. Ed. Engl., 2016, 55, 3982–3986 CrossRef CAS PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- G. Hu, C. Xu, Z. Sun, S. Wang, H. M. Cheng, F. Li and W. Ren, Adv. Mater., 2016, 28, 1603–1609 CrossRef CAS PubMed.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840–845 CrossRef CAS PubMed.
- J. He, G. Hartmann, M. Lee, G. S. Hwang, Y. Chen and A. Manthiram, Energy Environ. Sci., 2019, 12, 344–350 RSC.
- S. Wang, H. Chen, J. Liao, Q. Sun, F. Zhao, J. Luo, X. Lin, X. Niu, M. Wu, R. Li and X. Sun, ACS Energy Lett., 2019, 4, 755–762 CrossRef CAS.
- Y. Zhang, Z. Mu, C. Yang, Z. Xu, S. Zhang, X. Zhang, Y. Li, J. Lai, Z. Sun, Y. Yang, Y. Chao, C. Li, X. Ge, W. Yang and S. Guo, Adv. Funct. Mater., 2018, 28, 1707578 CrossRef.
- H. Yuan, W. Zhang, J.-g. Wang, G. Zhou, Z. Zhuang, J. Luo, H. Huang, Y. Gan, C. Liang, Y. Xia, J. Zhang and X. Tao, Energy Storage Mater., 2018, 10, 1–9 CrossRef.
- P. Geng, S. Cao, X. Guo, J. Ding, S. Zhang, M. Zheng and H. Pang, J. Mater. Chem. A, 2019, 7, 19465–19470 RSC.
- J. Zhang, Z. Li, Y. Chen, S. Gao and X. W. D. Lou, Angew. Chem., Int. Ed. Engl., 2018, 57, 10944–10948 CrossRef CAS PubMed.
- G. Xu, A. Kushima, J. Yuan, H. Dou, W. Xue, X. Zhang, X. Yan and J. Li, Energy Environ. Sci., 2017, 10, 2544–2551 RSC.
- D. H. Yang, H. Y. Zhou, H. Liu and B. H. Han, iScience, 2019, 13, 243–253 CrossRef CAS PubMed.
- D. Xiao, Q. Li, H. Zhang, Y. Ma, C. Lu, C. Chen, Y. Liu and S. Yuan, J. Mater. Chem. A, 2017, 5, 24901–24908 RSC.
- W. Zhou, C. Wang, Q. Zhang, H. D. Abruña, Y. He, J. Wang, S. X. Mao and X. Xiao, Adv. Energy Mater., 2015, 5, 1401752 CrossRef.
- Z. Chang, H. Dou, B. Ding, J. Wang, Y. Wang, X. Hao and D. R. MacFarlane, J. Mater. Chem. A, 2017, 5, 250–257 RSC.
- C. Zha, D. Wu, T. Zhang, J. Wu and H. Chen, Energy Storage Mater., 2019, 17, 118–125 CrossRef.
- Y. Tian, H. Huang, G. Liu, R. Bi and L. Zhang, Chem. Commun., 2019, 55, 3243–3246 RSC.
- Y. Zhong, D. Chao, S. Deng, J. Zhan, R. Fang, Y. Xia, Y. Wang, X. Wang, X. Xia and J. Tu, Adv. Funct. Mater., 2018, 28, 1706391 CrossRef.
- Y. An, Z. Zhang, H. Fei, S. Xiong, B. Ji and J. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 12400–12407 CrossRef CAS PubMed.
- M. Yu, S. Zhou, Z. Wang, Y. Wang, N. Zhang, S. Wang, J. Zhao and J. Qiu, Energy Storage Mater., 2019, 20, 98–107 CrossRef.
- N. Zhang, Y. Yang, X. Feng, S.-H. Yu, J. Seok, D. A. Muller and H. D. Abruña, J. Mater. Chem. A, 2019, 7, 21128–21139 RSC.
- B. He, Z. Zhou, P. Man, Q. Zhang, C. Li, L. Xie, X. Wang, Q. Li and Y. Yao, J. Mater. Chem. A, 2019, 7, 12979–12986 RSC.
- X.-T. Gao, X.-D. Zhu, L.-L. Gu, C. Wang, K.-N. Sun and Y.-L. Hou, Chem. Eng. J., 2019, 378, 122189 CrossRef CAS.
- Z. Xiao, Z. Yang, L. Zhang, H. Pan and R. Wang, ACS Nano, 2017, 11, 8488–8498 CrossRef CAS PubMed.
- D. S. Wu, F. Shi, G. Zhou, C. Zu, C. Liu, K. Liu, Y. Liu, J. Wang, Y. Peng and Y. Cui, Energy Storage Mater., 2018, 13, 241–246 CrossRef.
- J. Xu, W. Zhang, H. Fan, F. Cheng, D. Su and G. Wang, Nano Energy, 2018, 51, 73–82 CrossRef CAS.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- Y. Song, W. Zhao, L. Kong, L. Zhang, X. Zhu, Y. Shao, F. Ding, Q. Zhang, J. Sun and Z. Liu, Energy Environ. Sci., 2018, 11, 2620–2630 RSC.
- Y. Liu, T. Zhou, Y. Zheng, Z. He, C. Xiao, W. K. Pang, W. Tong, Y. Zou, B. Pan, Z. Guo and Y. Xie, ACS Nano, 2017, 11, 8519–8526 CrossRef CAS PubMed.
- J. Kang, S. Tongay, J. Zhou, J. Li and J. Wu, Appl. Phys. Lett., 2013, 102, 012111 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- M. Wu, C. Li, J. Zhao, Y. Ling and R. Liu, Dalton Trans., 2018, 47, 7812–7818 RSC.
- M. Hu, Y. Ju, K. Liang, T. Suma, J. Cui and F. Caruso, Adv. Funct. Mater., 2016, 26, 5827–5834 CrossRef CAS.
- Q. Li, M. Wu, J. Zhao, Q. Lü, L. Han and R. Liu, J. Electron. Mater., 2019, 48, 3050–3058 CrossRef CAS.
- W. Qin, D. Li, X. Zhang, D. Yan, B. Hu and L. Pan, Electrochim. Acta, 2016, 191, 435–443 CrossRef CAS.
- X. Wei, W. Li, J. A. Shi, L. Gu and Y. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27804–27809 CrossRef CAS PubMed.
- K. Yang, Q. Guo, H. Li, X. Hao, Y. Ma, M. Yang, T. Zhai, S. V. Savilov, V. V. Lunin and H. Xia, J. Power Sources, 2018, 402, 340–344 CrossRef CAS.
- C. Zhao, X. Shao, Z. Zhu, C. Zhao and X. Qian, Electrochim. Acta, 2017, 246, 497–506 CrossRef CAS.
- L. Liu, Y. Zeng, S. J. Tan, H. Xu, D. D. Do, D. Nicholson and J. Liu, Chem. Eng. J., 2019, 357, 358–366 CrossRef CAS.
- J. Wu, X. Li, H. Zeng, Y. Xue, F. Chen, Z. Xue, Y. Ye and X. Xie, J. Mater. Chem. A, 2019, 7, 7897–7906 RSC.
- S. Wang, W. Li, H. Song, C. Mao, Z. Zhang, H. Peng and G. Li, Inorg. Chem. Front., 2019, 6, 1275–1281 RSC.
- D. Li, X. Ren, Q. Ai, Q. Sun, L. Zhu, Y. Liu, Z. Liang, R. Peng, P. Si, J. Lou, J. Feng and L. Ci, Adv. Energy Mater., 2018, 8, 1802386 CrossRef.
- Y. Guo, X. Zhang, X. Zhang and T. You, J. Mater. Chem. A, 2015, 3, 15927–15934 RSC.
- C. Zhang, F. Han, J. Ma, Z. Li, F. Zhang, S. Xu, H. Liu, X. Li, J. Liu and A.-H. Lu, J. Mater. Chem. A, 2019, 7, 11771–11781 RSC.
- W. Sun, C. Cai, X. Tang, L.-P. Lv and Y. Wang, Chem. Eng. J., 2018, 351, 169–176 CrossRef CAS.
- F. Bu, P. Xiao, J. Chen, M. F. Aly Aboud, I. Shakir and Y. Xu, J. Mater. Chem. A, 2018, 6, 6414–6421 RSC.
- J. S. Cho, J.-S. Park and Y. C. Kang, Nano Res., 2016, 10, 897–907 CrossRef.
- Q. Ma, H. Song, Q. Zhuang, J. Liu, Z. Zhang, C. Mao, H. Peng, G. Li and K. Chen, Chem. Eng. J., 2018, 338, 726–733 CrossRef CAS.
- L. Kong, X. Chen, B. Q. Li, H. J. Peng, J. Q. Huang, J. Xie and Q. Zhang, Adv. Mater., 2018, 30, 1705219 CrossRef PubMed.
- H. Wang, W. Zhang, J. Xu and Z. Guo, Adv. Funct. Mater., 2018, 28, 1707520 CrossRef.
- L. D. Zhao, J. He, S. Hao, C. I. Wu, T. P. Hogan, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 16327–16336 CrossRef CAS PubMed.
- R. H. Misho and W. A. Murad, Sol. Energy Mater. Sol. Cells, 1992, 27, 335–345 CrossRef CAS.
- W. Li, Z. Chen, D. Wang, Z. Gong, C. Mao, J. Liu, H. Peng, Z. Zhang and G. Li, J. Power Sources, 2019, 435, 226778 CrossRef CAS.
- C. Ma, Z. Fu, C. Deng, X. Liao, Y. He, Z. Ma and H. Xiong, Chem. Commun., 2018, 54, 11348–11351 RSC.
- S. Niu, W. Lv, C. Zhang, Y. Shi, J. Zhao, B. Li, Q.-H. Yang and F. Kang, J. Power Sources, 2015, 295, 182–189 CrossRef CAS.
- C. Barchasz, J.-C. Leprêtre, F. Alloin and S. Patoux, J. Power Sources, 2012, 199, 322–330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta11451c |
| This journal is © The Royal Society of Chemistry 2020 |