
Molecular dynamics study on the aggregation behaviour of different positional isomers of sodium dodecyl benzenesulphonate†
Abstract
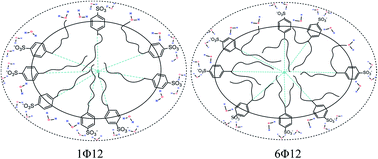
All-atom molecular dynamics (MD) simulations were performed to study the aggregation behaviour of different sodium dodecyl benzenesulphonate positional isomers (xΦ12) where x = 1, 2, 3, 4, 5 and 6. In the simulation, the solvent accessible surface area, carbon and sulphur distribution, angle possibility distribution, chain conformation, hydration numbers, distribution of polar heads on the micelle surface, and the interaction energy among the benzene rings were analyzed. The simulated results showed that these six isomer micelles are more elliptical than spherical and the micelle radius increases with the shifting of the benzenesulphonate group from one side to the middle of the alkyl chain. In the micellar aggregate, the short alkyl chains are located at the polar layer of the micelle while the long alkyl chains assemble in the central region of the micelle. In the six different isomers, 1Φ12 isomer shows some special structural features.
 Please wait while we load your content...
Please wait while we load your content...

