
Non-aqueous electrolytes for sodium-ion batteries
A.
Ponrouch
ab,
D.
Monti
abc,
A.
Boschin
bc,
B.
Steen
d,
P.
Johansson
*bc and
M. R.
Palacín
*ab
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus UAB, 08193 Bellaterra, Catalonia, Spain. E-mail: rosa.palacin@icmab.es
bALISTORE-ERI European Research Institute, 33 rue Saint Leu, 80039 Amiens, France
cDepartment of Applied Physics, Chalmers University of Technology, SE-412 96 Göteborg, Sweden. E-mail: patrik.johansson@chalmers.se
dDepartment of Energy and Environment, Chalmers University of Technology, SE-412 96 Göteborg, Sweden
First published on 16th October 2014
Abstract
The first review of the various electrolytes currently used and developed for sodium-ion batteries (SIBs), both in terms of materials and concepts, is presented. In contrast to the Li-ion battery (LIB), which is a mature technology for which a more or less unanimously accepted “standard electrolyte” exists: 1 M LiPF6 in EC/DMC, the electrolyte of choice for SIBs has not yet fully conformed to a standard. This is true for both materials: salts, solvents, or additives, and concept, using the main track of organic solvents or aiming for other concepts. SIB research currently prospers, benefitting from using know-how gained from 30 years of LIB R&D. Here the currently employed electrolytes are emphasized and their effects on practical SIB performance are outlined, scrutinizing the rationale for specific choices made, salts, solvents, additives, concentrations, etc. for each specific cell set-up and usage conditions.
 A. Ponrouch | Dr Alexandre Ponrouch received his Masters Degree from Paul Sabatier University (Toulouse, France) and his PhD from the Institut National de la Recherche Scientifique (INRS-EMT, Canada) in 2010 working on electrodeposition of metals, alloys and oxides for application in fuel cells and supercapacitors. He is currently working as a postdoctoral fellow in the Institut de Ciència de Materials de Barcelona (ICMAB-CSIC, Spain). His current research is mainly focused on electrolyte formulation for rechargeable batteries. |
 D. Monti | Damien Monti received his B.Sc. (2009) and M.Sc. (2011) from the Université de Provence of Marseille where he majored in materials science. He carried out a six months internship in the MADIREL laboratory on polymer electrolytes and a second one in the Institut de Ciència de Materials de Barcelona (ICMAB-CSIC) on the microwave synthesis of titanate anodes. At present, he is performing a shared PhD project between Chalmers University of Technology, Sweden and ICMAB-CSIC on ionic liquid based electrolytes for SIBs. |
 A. Boschin | Andrea Boschin received his B.Sc. (2004) and M.Sc. (2010) in Science and Technology of Materials at the Ca'Foscari University of Venice, Italy. While studying for his M.Sc., he worked in the field of protective organic coatings for wood and metals. He acquired experience in the electrocatalysis field working at the Vrije Universiteit, Brussels, Belgium, and the University of Gothenburg, Sweden (2010–2012). Currently he is a PhD student at Chalmers University of Technology, Sweden. His research focuses on solid polymer electrolytes for application in sodium batteries. |
 B. Steen | Prof. Bengt Steen got his PhD in chemical engineering at Chalmers University of Technology in 1975. He was employed by the Swedish Environmental Research Institute in 1968 as an environmental scientist and held various positions there until 1996. In 1996 he joined Chalmers again as an adjunct professor. His research was initially focused on measurement techniques, but during the 1980s his research was mostly on mechanisms of generation and impacts of air pollutants. Since 1990 his research has been focused on Environmental Life Cycle Assessments (LCA). In 2012 he got the SETAC Europe Life Time Award for Outstanding Achievements in LCA. |
 P. Johansson | Prof. Patrik Johansson (Chalmers University of Technology) runs several scientific activities within the area of materials for Li-ion and next generation batteries: Na-ion, Mg, Li–S, Li–air, etc., often with Swedish and European industries involved. He has a group of 10 PhD students and postdocs, leads the theoretical activities within the ALISTORE-ERI, and has >100 scientific publications. After a PhD from Uppsala University, Sweden, and a postdoc at Northwestern University, Evanston, IL, he joined Chalmers in 1999. His main interest is to increase the understanding of new electrolyte materials by combining computational methods and vibrational spectroscopy with macroscopic property assessments. |
 M. R. Palacín | Dr M. Rosa Palacín has been a staff researcher at the Institut de Ciència de Materials de Barcelona (ICMAB-CSIC) since 1999. After her PhD in Chemistry (UAB), she entered battery research in 1996 through a post-doctoral stay with Prof. Jean-Marie Tarascon. Her activities have since been fully focused on rechargeable battery materials, either nickel-, lithium- and, more recently, sodium-based. She has led many research projects with public or industrial funding dealing with different aspects of battery materials, both electrodes and electrolytes, and is actively involved in the ALISTORE-ERI, which she has been jointly leading with Prof. Patrice Simon since 2010. |
1 Introduction and scope
1.1 Introduction
The electrolytes for any electrochemical energy storage device – such as batteries – are typically given much less attention than the active materials (electrodes). The rationale behind this is that the properties of the latter define the energy density (gravimetric and volumetric) of the system and are thus most eye-catching. The role of the electrolyte should, however, not be neglected as it is in large part responsible for the life-length and the realistically possible performance in terms of practically accessible capacity, rate capability, safety, etc. This notion is now being realized by a wider part of the battery research and development (R&D) community,1,2 coupled also to the development of new analysis techniques revealing the role of the electrolytes in obtaining the best possible interfaces to the electrodes and thus full cell performance.3–7 Amongst the various important energy storage figures of merit, the total energy throughput of a cell, until end-of-life (EOL), is perhaps one of the most difficult tasks to obtain, but also the one of particular interest for all large cell or larger-scale applications. It is directly related to the extent to which parasitic side reactions take place and is ultimately dependent on the choice of electrolyte. From an application perspective EOL deserves as much attention as the more often emphasized gravimetric and volumetric energy density at beginning-of-life (BOL).The electrolyte is sometimes called “passive”, “only” being responsible for containing and shuttling the charge carrier ion of choice (H+, Li+, Na+, Mg2+, etc.) between the electrodes. This is for the vast majority of applications made possible simply by using a source of the very same charge carriers, a salt, dissolved in significant amounts in one or more solvents. The energy separation between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) of the electrolyte components determines the theoretical upper limit of each electrolyte's thermodynamic electrochemical stability window (ESW). Quite stringent requirements exist for the electrolytes, as they should exhibit the widest possible ESW and this is in contact with strongly reducing/oxidizing electrode active materials. Degradation reactions often take place in the case of liquid electrolytes, which result in insoluble products adhering to the negative electrode surface and forming a protective solid passivation layer, the solid electrolyte interphase, SEI.8 An interphase is also formed at the positive electrode, sometimes denoted the surface layer (SL) to differentiate it from the SEI. Hence, cell operation is in practice most often made possible through chemical passivation of both electrode surfaces – extending the thermodynamic limits imposed by the HOMO and LUMO levels of the electrolyte by the SL and SEI, respectively. Finally, additives are often used in the electrolyte formulations to tune the composition of the SEI and enhance its stability,9,10 and for many other purposes to altogether create a functional electrolyte, e.g. reduce flammability, prevent overcharging events, assist the wetting of electrodes, etc.11–13
Here we review the choices of electrolyte components for the sodium-ion battery (SIB) both covering the main path of R&D and giving some hints at emerging alternative concepts. Many of the observations are in general valid also for the very similar and today dominant Li-ion battery (LIB). Hence, we will make extensive use of comparisons between the two batteries and carefully note the important exceptions to the rule.
1.2 Why sodium-ion batteries?
To develop a new battery technology, as an alternative or complement to the prevailing LIB based on other charge carrying ions or concepts is a major undertaking. The main motivation stems from concerns of a future limited lithium supply. This is related to a likelihood of enlarged generalized implementation of the LIB technology from portable electronics to larger scale applications, such as different kinds of XEVs (BEVs, PHEVs, HEVs) and also grid storage to enable integration of renewable energy sources. Although probably too alarmist, it should be remembered that Li is widely used for glasses and ceramics, is foreseen to be extensively used in the next generation of nuclear power plants, and is already an important ingredient in the pharmaceutical drug industry – a sector growing at a rapid pace.14 It is of uttermost importance to acknowledge that a limited supply issue is not anywhere nearly the same thing as simply looking at the resources such as elemental abundance in the crust of the Earth. The measure of the reserves is much more important and is a complex interplay of availability, production, recycling, geographical/political constraints, and not the least cost issues – Li might be “the new gold”.14–16 Thus, the actual Li reserves are dynamic, with the resources being the absolute maximum.
One of the most appealing alternatives is to use sodium instead of lithium. Indeed, sodium mineral resources are “unlimited”, attainable at low cost, and geographically distributed. Given the similar chemistry of these alkali ions a transfer of LIB know-how can, in general, be foreseen. Historically, back in the 1980s and early 1990s,17,18 the progress in SIBs paralleled that of LIBs and several electrochemical cells were assembled using first TiS2 (ref. 19) and later on Na
x
CoO2 (ref. 20 and 21) as positive electrode materials using either liquid or polymer electrolytes, and some first attempts to build Na-ion cells with Na15Pb3 negative electrodes (as opposed to using Na metal) were also reported.22,23 Unfortunately, this research path was largely abandoned due to the advent of the LIB technology for portable electronics and its prospects of a much more generalized field of application. There was one remarkable exception: the patent of a full SIB by Valence Technologies (US) in 2002,24,25 consisting of a carbonaceous negative electrode coupled to a sodium transition metal phosphate positive electrode using 1 M NaClO4 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC (2:1) as an electrolyte. However, the topic of SIBs started to re-emerge by 2010 and is now progressing at rapid pace (more than 250 scientific papers published in 2013). SIBs offer prospects of the very same performance as LIBs with a much better long-term sustainability perspective. The obtainable performance at the cell level depends on the electrochemical capacities and operating potentials of the negative and positive electrodes. Thus, and this is most important, such figures are not in any way related to the capacity values of Na metal anodes and there is no reason to beforehand assume that SIBs will exhibit lower energy densities than LIBs. The only issue to consider is that the lowest negative electrode potential limit is set by the standard redox potential of the metal itself, which is only somewhat higher for sodium (by ∼330 mV). From a practical point of view, an added benefit for SIBs is that low weight and less expensive aluminium current collectors can be used for both positive and negative electrodes (as sodium, in contrast to lithium, does not form alloys with aluminium).
:DMC (2:1) as an electrolyte. However, the topic of SIBs started to re-emerge by 2010 and is now progressing at rapid pace (more than 250 scientific papers published in 2013). SIBs offer prospects of the very same performance as LIBs with a much better long-term sustainability perspective. The obtainable performance at the cell level depends on the electrochemical capacities and operating potentials of the negative and positive electrodes. Thus, and this is most important, such figures are not in any way related to the capacity values of Na metal anodes and there is no reason to beforehand assume that SIBs will exhibit lower energy densities than LIBs. The only issue to consider is that the lowest negative electrode potential limit is set by the standard redox potential of the metal itself, which is only somewhat higher for sodium (by ∼330 mV). From a practical point of view, an added benefit for SIBs is that low weight and less expensive aluminium current collectors can be used for both positive and negative electrodes (as sodium, in contrast to lithium, does not form alloys with aluminium).
We foresee that within the next 10 years SIBs will have major attention from the scientific community, while their commercial competitiveness vs. LIBs ultimately will depend on the final performance shown and also on the application niches aimed at, for which the role of the electrolyte is instrumental.
Even if the statements in the section above on limited supply of Li and how Na can be a replacer/reliever all seem plausible, there is a need for more solid scientific support for sustainability. In order to substantiate the sustainability of SIBs vs. LIBs, the full cell configuration resulting from our laboratory studies27 was compared with three different LIB cells, using different chemistries and sizes (7 sizes in the range of ca. 60–120 W h kg−1), using the BatCap software tool.28 This software enables calculation of full battery parameters considering not only active materials but also inactive materials (separator and electrolyte, current collectors, casing, etc.). As the use of an aluminium negative electrode current collector is considered to be a major advantage for SIBs, not the least as the commodity market price of Cu (6755 US$ per tonne) is >3 times higher than for Al (1920 US$ per tonne),29 we did also include in the comparison an LIB configuration using an Al current collector with a Li4Ti5O12 (LTO) negative electrode that operates at a potential significantly higher than that of Li–Al alloy formation. For each LIB and SIB configuration all the components were evaluated and the exact contained mass ratios of each chemical element taken into account in order to evaluate its sustainability.
There are many different sustainability indicators suggested in the literature.30 In order to facilitate trade-offs with other battery performance features, we here chose to use a monetary sustainability indicator, the natural capital cost, developed for the EPS 2000d method.31 In this method the sustainability indicator is equal to people's willingness to pay (WTP) to restore changes in five safeguard subjects: human health, ecosystem production capacity, biodiversity, abiotic resources, and recreational and cultural values. WTP values for present OECD inhabitants are used, and the impacts value integrated over time and space, where it occurs. For abiotic resources, all future generations are influenced, and their WTP is estimated by their cost to produce a concentrate similar to the present resources. For copper and nine other metals, the cost of producing ore-like concentrates from the corresponding average metal concentration in the earth's crust was determined by Steen and Borg.32 The cost for extracting abiotic resources from mineral reserves is regarded as a natural capital cost.
The natural capital costs for the four different chemistries are shown in Table 1 for a medium sized cell (270 cm3), three out of the seven simulated in BatCap, with the qualitative results valid for all cell sizes considered.
| Calculated natural capital cost [€ per kW h capacity] | ||||
|---|---|---|---|---|
| SIBs | LIBs | |||
|
a
1 M NaPF6 in EC:PC:DMC HC = hard carbon, NVPF = Na3V2(PO4)2F3, C6 = graphite, LP30 = LIB standard electrolyte, LFP = LiFePO4, and LMO = LiMn2O4.
|
||||
| Elem. | HC|eleca |NVPF | C6|LP30|LFP | C6|LP30|LMO | LTO|LP30|LMO |
| Al | 0.36 | 0.18 | 0.11 | 0.51 |
| C | 0.073 | 0.08 | 0.057 | 0.068 |
| Cu | — | 185 | 110 | — |
| F | 2.17 | 1.09 | 0.8 | 2.42 |
| H | 0 | 0 | 0 | 0 |
| Fe | — | 0.6 | — | — |
| Li | — | 0.0085 | 0.0078 | 0.04 |
| Mn | — | — | 6.56 | 15.1 |
| O | 0 | 0 | 0 | 0 |
| P | 1.43 | 0.15 | 0.081 | 0.24 |
| Na | 0 | — | — | — |
| Ti | — | — | — | 1.56 |
| V | 27.5 | — | — | — |
| Sum | 31.5 | 187 | 117 | 19.9 |
Including the natural capital cost in the battery price would thus be highly problematic for C6|LP30|LFP and C6|LP30|LMO LIB cells – both popular chemistries for XEVs. Very efficient recycling of Cu would be needed. We also find that the SIB costs much better than these two major LIBs, and it is the lack of a Cu current collector and vanadium that makes the LTO|LP30|LMO cell the winner. A SIB without V in the cathode, and of similar capacity, would thus be even more sustainable. From an elemental point of view the three less sustainable elements are: Cu ≫ V > Mn. From an electrolyte perspective, it is re-assuring that the contribution from this part of the cell is hardly noticeable compared to the active materials and current collectors (as the contributions from the elements H, Li, O, and C are either 0 or very small – the only significant electrolyte element is F due to the LiPF6 salt used).
1.3 Scope of this review
The present paper covers non-aqueous electrolytes for SIBs operating close to room temperature. Even if some elevated temperature electrolyte concepts will be noted in passing, no attempt will be made to cover electrolytes for the following battery concepts:• Batteries with aqueous electrolytes.
• Na–air or Na–O2 batteries.
• Na–S batteries.
• Na–NiCl2 (ZEBRA) batteries.
Hereby we keep the scope to the mainstream of electrolytes for rechargeable batteries aimed for the consumer electronics market, the electromobility revolution (XEVs), and the large-scale storage applications for renewable energy sources (though batteries with aqueous electrolytes are gaining attention for the latter application – today dominated by Na–S and ZEBRA batteries).33,34 These limitations are chosen partly due to the SIB research focus overall globally, but also with the purpose to remain within the area where the corresponding knowledge of the LIB technology and its electrolytes – materials used, cells employed, properties, etc., is vast, to allow for extensive comparisons. In addition, special emphasis will be placed on the electrolytes used and developed after the recent upsurge in SIB research, rather than including all old studies made in the 1980–90s prior or concomitant to the commercialization of LIBs.
Examples of how the electrolytes limit and/or make possible the final cell performance of different SIBs will be given. However, the present review will not directly deal with the SIBs overall or on the outcome of the continuous exploration of different electrode materials. For this we kindly point to a number of recent reviews.35–42 None of these, however, takes a stance in the need of better SIB electrolytes and their development.
2 Electrolyte basics and materials
2.1 Electrolyte basics
While it is the active electrode materials used that set the energy output possible for any battery and as such attract the more attention, the electrolyte has other, and as we will argue, equally important roles to play in the cell. The property most common to first mention is the ionic conductivity, which in a more true sense should for LIBs and SIBs focus on the species that is to be intercalated at the electrodes – Li+ or Na+. We would, however, rather like to stress that placed between the two electrodes and interacting with both of them to create interfaces often dictating the true performance, it is the stability or meta-stability of the electrolyte that is its prime property. There should ideally be no chemical reactions or changes involving the electrolyte during the SIB cell operation, something that is cumbersome to attain while pushing the low (negative) and high (positive) limits of the cell voltage window further and further apart in an attempt to enhance the energy density as much as possible. If a meta-stable solution is chosen, then the reactions should be as controlled and predictable as possible.A generic list of properties needed for a SIB electrolyte totally complies with those usually compiled for LIB oriented electrolytes:43
(i) chemically stable – no chemical reactions during cell operation including both within itself, with the separator and electrodes used, and with the current collectors and packaging materials employed,
(ii) electrochemically stable – large separation of high and low onset potentials for decomposition by oxidation or reduction, respectively,
(iii) thermally stable – a wide liquidus range; both the melting and boiling points should be well outside the (internal) operation temperatures, and
(iv) ionically conductive and electronically insulating – to sustain cell operation by facile ion (here Na+) transport and to minimize self-discharge of the cell, respectively.
In addition to these operational requirements the SIB electrolyte of choice needs to meet other practical criteria:
(v) it must have low toxicity and successfully meet also other measures of limited environmental hazard,
(vi) it must be based on sustainable chemistries – abundant elements and as low impact synthesis process (energy, pollution, etc.) as possible, and
(vii) it must carry as low total cost, materials and production, as possible.
While the above requirements are general and not given in terms of specific optimal or even lower threshold values, there is of course a need to optimize them. This can be made both separately and in conjunction with each specific cell chemistry and application e.g. low temperature operation calls for an extended temperature window as compared to a cell designed and controlled to operate at room temperature. Target or threshold values are indeed often set, the most commonly known being perhaps the threshold of >1 mS cm−1 for the ionic conductivity. A single measure can, however, never tell the full story e.g. the crucial measure is a fast enough Na+ transport compared to the usage conditions.
Some of the SIB electrolyte properties mainly originate in the salts employed, others in the solvents used, and yet another set are more “holistic” – the latter being difficult to predict prior to actual testing in full cells. This is true for both SIB and LIB electrolytes, and we again stress that the know-how gained from more than 30 years of LIB electrolyte development can and should be extensively applied to SIBs. There are though some features that differ – both from fundamental physical aspects when changing from Li+ to Na+ chemistry (e.g. the different charge/radius ratio, the lower Lewis acidity, etc.) and in terms of application. Indeed, as the electrodes used are not the same the interfaces and/or interphases will not be the same e.g. the SIB negative electrodes do not contain graphite – as graphite cannot intercalate Na+ ions – but often various different carbonaceous materials such as hard carbon. Very recently Na+ co-intercalation into graphite was shown using diglyme (G2) as the electrolyte solvent, creating a complex [Na(G2)2]+ possible to intercalate, resulting in a ternary compound with an estimated formula of Na(G2)2C20.44 While fundamentally quite interesting, it is thus still not a bare Na+ ion that is intercalated.
The electrolyte optimisation is currently primarily made by varying the constituents of the electrolyte, Na-salt(s), solvent(s), and additive(s), and their respective ratios. Each component affects the above requirements to a quite different degree. The choice of Na-salt, that is changing the counter-anion to Na+, affects both the chemical and electrochemical stability, as well as the ionic conductivity. The anions are most often the electrolyte component that is oxidized first, setting the upper voltage limit for the ESW (while the lower limit is more often dictated by solvent reduction). At the same time, the strength of the ion–ion interactions determines the amount of charge carriers available – as extensive ion-pairing can occur – and thereby affects the ionic conductivity. The strength of the cation–anion interactions is reduced down to ca. 80%, moving from Li+ to Na+ as the cation.45 There is a strong temperature dependence of the ion-pairing and thus an optimal salt concentration for a maximum ionic conductivity – and this will thus also likely differ for SIB electrolytes when compared to LIB electrolytes.
The ion transport in total is, however, much more affected by the choice of solvent(s) – the conductivity, or more correctly the mobility, is as a rule of thumb approximately inversely proportional to the viscosity (thus proportional to the fluidity), basically based on the Stokes–Einstein relationship (while not straightforward totally valid for molecular sized diffusing species). Most often the total ionic conductivity is expressed as the sum over all species i of the electrolyte for the 3-part product of: the number of charge carriers, n
i
, their mobility, μ
i
, and their charge, z
i
. The problematic point is that while the total ionic conductivity is easily measured, the ion transport itself, and more importantly, the part carried by the species of interest during migration, here Na+, is cumbersome to attain. The sodium transference number (tNa) that quantifies the Na transport is defined as:
| tNa = μNa/∑μ i |
The fact that the anions are more weakly solvated by any solvents used, by virtue of having smaller charge/radius ratios, largely determines the mobility – and in a negative manner. As the cation, Na+ as well as Li+, carries a more stable solvation shell than the anions, the cation transference numbers are always inferior (often ca. 0.2–0.4 for the cation – and thus 0.6–0.8 for the anion). Thus we almost always have more efficient anion conducting electrolytes – the exception is when the anions are tethered to the matrix e.g. in single-ion conducting polymer electrolytes – where ideally tNa = 1 can be obtained. Overall, the weaker solvation shell for the sodium ion is, however, an advantage compared to the lithium ion (see below).
A rapid enough ion transport across the electrolyte is one of the fundamental requirements for operation at high powers (C-rates). This is dependent both on a fast enough ion transport in the bulk, to reduce polarisation resistance, and on the delivery and kinetics of the ion intercalation processes at the electrodes (charge transfer) including the transfer across the interfaces. The bulk ion transport properties will somewhat differ from LIBs to SIBs by virtue of different solvation shells, with Na+ often exhibiting larger coordination numbers in solution than Li+. Overall the nature of solvent coordination is, however, very similar for the two alkali cations, most of the solvent complexes having similar structures for both central ions – only a few differ by changing from mono-dentate coordination to Li+ to bi-dentate for Na+ (made possible due to its larger radius).46 Energetically the smaller charge/radius ratio of Na+ results in total binding energies reduced by ca. 20% as compared to the analogous Li+ complexes.45
Turning to the charge transfer at the electrolyte–electrode interface, SIB electrolytes seem to possibly have an upper hand compared to LIB electrolytes. For LIBs the Li+ transfer at the electrolyte–electrode interface can indeed be the rate determining step. The activation barriers show excellent correlation with the de-solvation energetics of the last solvent molecule for a wide range of battery solvents.47–50 Okoshi et al.46 used this notion and evaluated the de-solvation energies for various cations, including Li+ and Na+, with a range of organic solvents (27!) using DFT calculations. A trend to smaller de-solvation energies for Na+ as compared to Li+ was shown – attributed to the former's weaker Lewis acidity – by up to 40–70 kJ mol−1 (or ca. 25–30% lower). This thermodynamic approach correlates nicely both with recent DFT computational studies45 as well as the experimental observations of Sagane et al.51 and Mizuno52 of lower activation barriers for Na-ion insertion, which all taken together imply faster charge–discharge kinetics to be possible for SIBs as compared to LIBs.
The solvent(s), or more generally the solvent concept employed, has profound effects on the thermal stability of the electrolytes. The low viscosity organic solvents most often used for LIBs or SIBs often have rather high vapour pressures, creating flammable vapours upon elevated temperatures, while polymers or ionic liquids have no vapour pressure at all. At the same time, both the latter concepts have too high viscosities to allow for low temperature or even room-temperature operation. The above two properties are thus seemingly almost orthogonal to each other and must be properly considered with the specific application and operation conditions in mind – there is no unique solution. A prominent example is the standard LIB electrolytes with problems for operation at temperatures that are too high (above 50 °C) or too low (below −20 °C) – originating in the combination of solvents (EC/DMC) and the thermally sensitive LiPF6 salt.42
When each of the SIB electrolyte requirements and important properties has been optimised as far as seen possible and there are still some obstacles remaining for optimal cell operation; then the need has come for electrolyte additives to bridge those. The additives can be targeting safety oriented properties, originating in the electrolyte not reaching enough chemical and electrochemical stability, or be more performance oriented, like a need to stretch the operating window in terms of voltage or increasing the C-rates by reducing the viscosity. In our opinion, the number and amount of additives should truly be kept to a minimum – as they most often create a more complex and unpredictable chemistry (in terms of possible side reactions) and carries cost issues. They might, however, be unavoidable to finally arrive at a functional electrolyte.
The various methods to study the basic properties of SIB electrolytes range from spectroscopic analysis of the local structures, solvation shells and electrode–electrolyte interfaces, to basic macroscopic level properties such as density, viscosity, and ionic conductivities. For assessment of the ion transport diffusion NMR is especially valuable, together with proper impedance spectroscopy analysed via equivalent circuits. The electrochemical stabilities are evaluated via cyclic voltammetry either vs. model inert electrodes or the “real” electrodes that are to be used in the SIB cell. For safety assessment, the vapour pressures, the flash points, the ignition temperature and times, and the self-extinguishing times are all important measures used. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) or accelerating rate calorimetry (ARC) are the appropriate methods needed to study both the safe and/or possible operation temperature ranges and long-term stabilities at elevated temperatures including reactions vs. electrodes, current collectors, and packaging materials.
The electrochemistry approaches including cell testing have up to now mainly focussed on the properties of the electrolytes themselves separately. This is exactly how progress was made historically for LIBs and much of it relied on large empirical efforts and sometimes serendipity rather than rational design and development. A change in methodology, where a development towards better SIB electrolytes is more based on a holistic evaluation of the SIB cell properties rather than e.g. maximising a single property such as the ionic conductivity, has recently emerged.27,53 This also acknowledges the electrolyte to be equally important as the suitable choice of electrodes to make an operational SIB.
We are convinced that the best path forward to rapidly obtain better SIB electrolytes relies on a proper understanding of all the electrolyte basics, aspects, and requirements above. Especially important is to know how (and when) these are similar or different to those of LIBs, and then apply this knowledge to the most modern materials together with extensive testing in operational SIB cells. There are, however, many reasons to start from the materials available and to review the state-of-the-art for salts and solvents as well as additives, before scrutinizing the promises and problems of different SIB electrolyte concepts and compositions. One reason is that we by an Occam's razor approach might be able to deduce the origin of problems to a single material prior to the complexity arising from creating an SIB electrolyte or even the full SIB cell.
2.2 Sodium salts
As outlined above the salt used is one of two major components of any SIB electrolyte – with profound effects on the final performance. Amongst the properties directly affecting the salt choice one can highlight: (i) the solubility in the solvent(s) used – in order to create an electrolyte with enough charge carriers present, (ii) the stability vs. reduction as well as oxidation – possibly setting the limits of the electrochemical stability window (ESW), (iii) the chemical stability vs. the other materials of the electrolyte, the electrodes, and the current collectors, and (iv) non-toxicity and other safety related aspects.Features (i) and (ii) do unfortunately reduce the number of salt candidates to a very small number. The classic approach is to look for inorganic anions based on a central atom with ligands withdrawing electron density to create delocalized negative charge and thereby weakly coordinating anions (WCAs).54 Such anions are also more likely to be stable vs. oxidation (by their suitably large HOMO energies). For SIB electrolytes we mainly find the same prospective anions as applied in the field of LIB electrolytes for many years; ClO4−, BF4−, PF6−, CF3SO3− (Tf), and [N(CF3SO2)2]− (TFSI). As many resulting properties are more often dependent on the anion than on the cation, also the promises and drawbacks are more or less the very same. To briefly summarize, all the above anions have some problems – as already observed in LIB cells;42 ClO4− is a strong oxidant and therefore more or less banned for any practical cell development; BF4− produces less conductive electrolytes by virtue of a stronger interaction with the cation and thus less charge carriers present; PF6− – while being the anion of choice (the best compromise candidate) for LIBs – has severe safety issues, especially at elevated temperatures and in the presence of moisture, suffering hydrolysis to yield PF5, POF3, and HF; Tf has the same main problem as BF4− (less conductive electrolytes), and is also corroding the aluminium current collectors – also the main problem for the academically popular TFSI anion. The corrosion has an onset potential at ca. 2.7 V vs. Li+/Li for Tf in a LIB electrolyte.55
Scrutinizing the problematic features above, some can be altered by moving from SIBs to LIBs (e.g. solubility), while others are more anion inherent properties (ESW and Al corrosion).
Sodium salts in general have higher melting points than lithium salts – which contribute to making them easier to dry than their Li equivalents, with the larger thermal stability also being expected to bring in advantages in terms of safety. In Table 2 some physico-chemical properties of the most used sodium salts for SIB electrolytes are summarized.
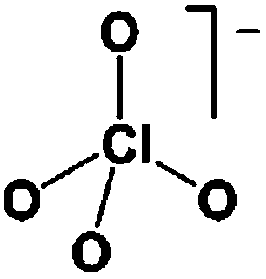
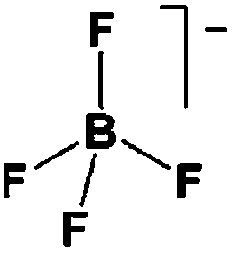
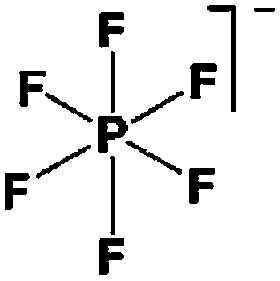
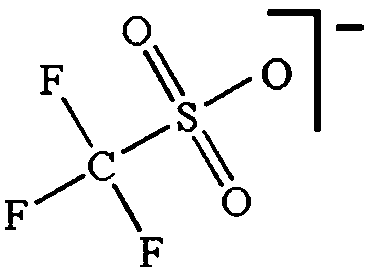
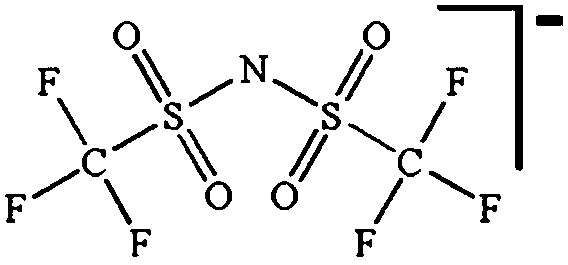
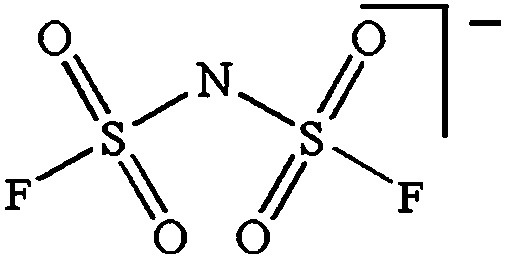
While being only of academic interest (see above), the most commonly used salt (in ca. 2/3 of the published SIB papers) is NaClO4, likely due to a combination of historical and cost reasons. An especially problematic issue, apart from the safety aspects, is that these salts are notoriously difficult to dry56 (although less hygroscopic). While the water content of the electrolytes is seldom reported in the literature, even after drying the powder at 80 °C under vacuum overnight NaClO4 based electrolytes do usually exhibit higher water contents (>40 ppm) as compared to e.g. NaPF6 based ones (<10 ppm). The second most popular salt is indeed NaPF6, which enables comparisons with many LIB studies. Emerging as strong Na-salt of choice candidates (despite the Al corrosion issues) are NaTFSI and NaFSI, partly due to the roles of these anions in creating suitable ionic liquid matrices. While NaTFSI and NaFSI perhaps cannot be applied as single salts for SIB electrolytes (Al corrosion), their non-toxicity, higher thermal stabilities than both NaPF6 and NaBF4, and the resulting higher conductivities than when using NaTf are clearly promising. It is worth pointing out that FSI has yet an unclear status when it comes to corrosion issues. Indeed, it was early on reported to corrode Al, which was later attributed to Cl impurities remaining from the salt synthesis, but there is an additional recent paper reporting on SS corrosion.57
Comparative studies across different Na-salts are rare. Bhide et al.58 compared NaPF6, NaClO4, NaTf, and NaTFSI using a common EC:DMC matrix and measured the ionic conductivity as a function of the salt concentration. For both NaTf and NaClO4 the dependence was markedly more obvious than for NaPF6. The maximum conductivities obtained were 6.8 mS cm−1 for 0.6 M (NaPF6), 5.0 mS cm−1 for 1 M (NaClO4), while 0.8 M NaTf showed a too low value.
New anions only very slowly enter the field of electrolyte studies and just a few academic groups focus on the development of new salts for battery electrolytes (SIBs and LIBs alike). This is largely due to the number of requirements that has to be met – and this with only a few atoms to choose from and combine to a small enough anion. At the same time, however, compared to the field of electrode materials, it is usually straightforward to make the Na-salt once the Li-salt has been made (or vice versa). A few examples of new anions/Na-salts are given below – often these at present exist only on an academic or in the best case semi-commercial scale and thus it is difficult to assess their full set of promises and problems.
One path recently explored is the use of heterocyclic rings as the framework structure for anions.59,60 Within this scope the NaTDI salt, as well as the analogous NaPDI salt, was very recently reported.61 Both salts show conductivities of ca. 4 mS cm−1 (in 1 M concentration in PC at room temperature) and similar electrochemical stability promise vs. oxidation, >4 V vs. Na/Na+, as other heterocyclic anions such as DCTA (TADC)62 and similar [N5C n ]− anions.58 The NaTDI and NaPDI salts also offer very high stability vs. moisture – which is promising both for handling and practical application. In addition, the melting point of NaTDI is above 330 °C (as compared to the mere 160 °C observed for LiTDI).63
The replacement of the electro-withdrawing F atom by the pseudo-halogen –CN group has been another recurring theme in recent LIB salt development. For example the BF4− anion analogue [B(CN)4]− was recently employed to create totally F-free electrolytes.64 Interestingly, the salts of this anion show very low solubilities in regular organic solvents like PC, but are soluble in PEGDME. The electrochemistry is complicated and using above ca. 4 V seems problematic.
Yet another example of routes to new Na-salts without the presence of any F atom was suggested in a purely computational study by Jónsson et al.65 targeting pseudo-delocalized anions i.e. anions with a central positive core and negatively charged extremities – thus not complying with the standard WCA recipe of total delocalization of the negative charge. Some of the anions suggested seem to be competitive with those used today in terms of the weakness of ion–ion interaction and the anion oxidation potential, but experimental proof is urged for.
We finally note that Na-salt properties may have an impact beyond the development of the SIB electrolyte of choice. The higher solubility of the Na2CO3 salt as compared to the Li2CO3 salt in the electrolytes can alter the nature of the SEI formed during cell operation quite drastically between the two alkali-ion battery concepts.
As one example of how complex these issues can be, the ion–ion interaction is, as noted above, reduced when moving from LIBs to SIBs and thus the number of charge carriers could at a first instance be expected to increase, also for less WCAs. However, as also the ion–solvent interactions become weaker (and possibly by the same amount) – we might arrive at a status quo.45 This might be one reason why almost all studies use a 1 M concentration for SIBs – just as found optimal for LIBs. There are though little hard facts supporting this choice at present as any detailed concentration studies are lacking.
2.3 Solvents
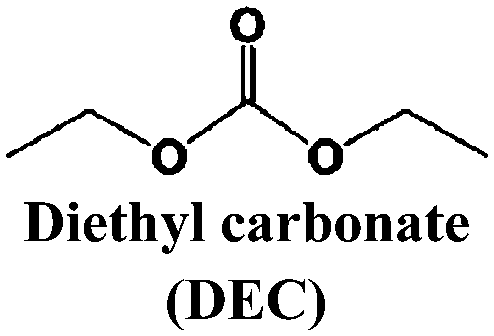
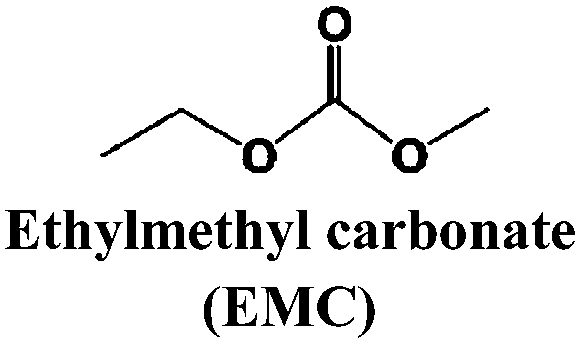
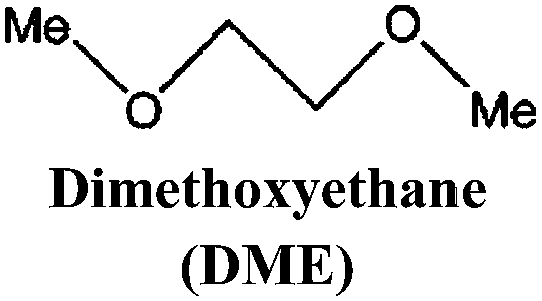
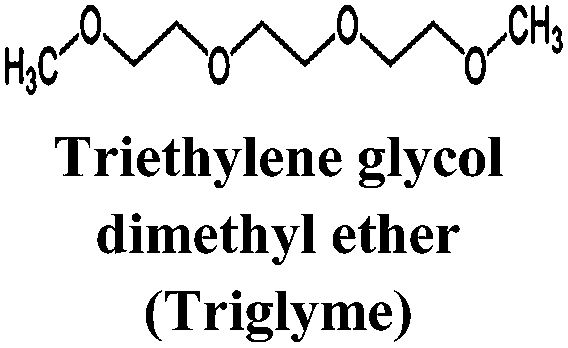
Just as for LIBs, the strongly reducing negative electrodes and strongly oxidizing positive electrodes in SIBs require electrolyte solvents with large ESWs, which turns into the stringent and absolute demand that solvents with active protons cannot be used (hence non-aqueous should indeed more correctly be aprotic). The solvents also need to comply with most of the prerequisites already set for the Na-salts: stable, non-toxic, inexpensive, etc. In addition, the presence of polar groups to dissolve sufficient amounts of Na-salt is a compulsory feature. This total set of properties can, however, be accomplished in many different ways by diverse types of solvent concepts, each having their own set of promises and problems, which we outline in separate sections below. While the combinations of salt and solvent may differ between LIBs and SIBs, there is yet no specific solvent developed specifically for SIBs and thus most information below is generic to both.The families of organic solvents investigated are much the same as those used for LIBs:42 organic carbonates (linear and cyclic), esters and ethers, for which the most significant properties are listed in Table 3. The Lewis acidity/basicity concept (i.e. electron acceptor/donor ability) of a solvent is of primary importance as it will influence the ESW. The acceptor (AN) and donor (DN) numbers of a solvent correlate with its HOMO/LUMO levels. Moreover, the acidity (basicity) of solvents will also determine their solvation properties; strong (low) acidity/basicity results in easy (hard) solvation of anions/cations.66 According to the hard and soft acids and bases (HSAB) concept67 this will determine the solvent–solvent and ion–solvent interactions. Li+ being a stronger acid than Na+ should impact the ion–solvent interactions, i.e. the nature of the solvation shell and solvation energy, as also shown in a recent DFT study.45
| Solvent | T m (°C) | T b (°C) | T f (°C) | η (cP) 25 °C | ε 25 °C | AN (DN) |
|---|---|---|---|---|---|---|
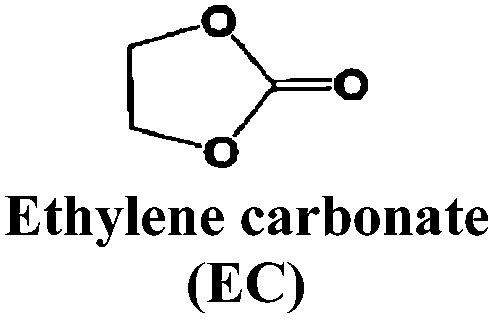
|
36.4 | 248 | 160 | 1.9 (40 °C) | 89.78 | (16.4) |

|
−48.8 | 242 | 132 | 2.53 | 64.92 | 18.3 (15.1) |
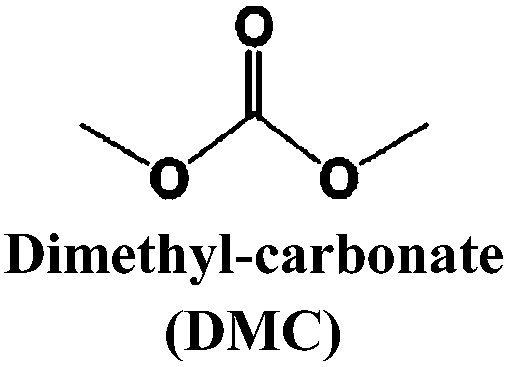
|
4.6 | 91 | 18 | 0.59 | 3.107 | |
|
−74.3 | 126 | 31 | 0.75 | 2.805 | (16.0) |
|
−53 | 110 | 0.65 | 2.958 | ||
|
−58 | 84 | 0 | 0.46 | 7.18 | 10.9 (18.6) |

|
−64 | 162 | 57 | 1.06 | 7.4 | 9.9 (19.2) |
|
−46 | 216 | 111 | 3.39 | 7.53 | 10.5 (14) |
 | ||
| Fig. 1 The two most common IL cations employed in electrolytes: Pyr13 or BMP (left) and C2mim or EMIm (right). | ||
As IL anions are chosen amongst WCAs in Na-salts (most often TFSI or FSI for RTILs), it is the choice of IL cation that can impose further restrictions on the electrolyte. As an example, the imidazolium based C n mim (also known as EMIm (n = 2), BMIm (n = 4), etc.) cations have problems with the cathodic limit of the ESW, while the pyrrolidinium based Pyr 1x family of cations has better electrochemical stability.
There are a few main drawbacks of using ILs for SIB electrolytes. The first, and most important one, is that almost all ILs have rather high viscosities, in the order of tens of cP at room temperature, and furthermore the viscosity often increases further upon doping with the charge carrier i.e. Na-salt. This is due to the formation of even stronger ion–ion interactions, which turns the mobility of the Na-ions being a complex, un-resolved issue (it has been suggested that they are moving as doubly negatively charged species).70 The second main drawback is the price of ILs. The price is limited by the anion and as the WCA is often the same as for the salts employed the total material cost will be high, sometimes believed to be excessively high for any practical purposes. The use of e.g. FSI based ILs rather than those based on TFSI can reduce this problem somewhat – as FSI has a less cumbersome and thus expensive synthesis path. As an additional drawback we must add the number of studies made with not dry enough or impure ILs, which make some results dubious and somewhat hindering the best development of the field.
Polymers are typically (very) bad solvents for ions – they have very high viscosities and low dielectric constants – often in the range 3–5. Therefore, the polymers employed should have strongly solvating groups like etheric oxygen atoms, carbonyl groups, or nitrile groups, spaced in an appropriate manner to fit the preferred solvation shell structure of the cations. The anions are usually not solvated by the polymer directly, but directed to the free volume of the matrix. The archetype of the polymer applied for solid polymer electrolytes (SPEs) or dry polymer electrolytes is poly(ethylene oxide), PEO. PEO was the first polymer used in 1973,71 launched as an electrolyte for lithium batteries in 1978,72 and still remains at the centre-court of SPE research and development. Quite many different polymers have been suggested as solvents, but as of today almost all rely on the solvating power of oligo(ethylene)oxide fragments – in the shape of block or graft co-polymers, etc.
The main criticism of the concept of using polymers as solvents is that the interfacial contact with the electrodes will always suffer and that the fabrication into thin films (needed due to low conductivities – see 3.3.1) counteracts their safety advantage by pin-hole formation and/or internal short-circuits.73 The polymer may also have limited stability at high voltages.
Some of the drawbacks of SPEs based on a polymer as the only solvent can be counteracted by the conception of gel polymer electrolytes (GPEs). In a GPE the polymer acts to provide the mechanical support and is swollen by an organic solvent (often the same as those in 2.3.1) which handles the ion transport in a liquid manner. In addition, the cations of the solute often contribute to dynamic cross-linking to different coordination sites of both the solvent and the polymer. The solvent can be one of the organic solvents mentioned in Section 2.3.1 or an IL. GPEs are thus closely related to an organic solvent based electrolyte contained in a polyolefin (or other) separator. As such they are not suitable vs. metallic anodes, they have lower mechanical strength than a SPE, but at the same time they offer high ionic conductivities and better contact with the electrodes. The GPE concept allows for the application of many different polymers, PEO, poly(acrylonitrile) (PAN), poly(methyl methacrylate) (PMMA) and poly(vinylidene fluoride) (PVdF) being the more commonly used. All are preferentially used with the same salts and solvents that are applied in liquid electrolytes – for example the LiTFSI has no upper hand here (as compared to the case for SPEs, where it increases the conductivity by enhancing the chain dynamics). It is worth mentioning that the “Bellcore LIB concept”74 is partially based on a co-polymer of PVdF with hexafluoropropylene (HFP) – PVdF-HFP.
2.4 Additives
As outlined in the Introduction, the third major component, often needed to create a functional electrolyte, is additives. An additive is basically the remedy used to address shortcomings of the original electrolyte recipe by addition of a new chemical in small amounts – usually less than 5 wt%. Such a limited amount needed originates from the preferred actions taking place at the electrolyte–electrode interfaces rather than in the bulk of the electrolyte. Typical interface/surface actions by an additive are to modify the SEI, increase the wetting of the surface, and protect towards overcharging events by redox shuttles taking on the extra charge. There are also additives with actions that are more global to the electrolyte, flame-retardants, fluidity enhancers/viscosity decreasers, impurity or radical scavengers, etc.The emphasis has for long been primarily on additives acting to create as beneficial SEIs as possible, with respect to both performance and safety (C-rates, life-length, and thermal and mechanical stability). Such additives should have LUMO energies lower than the electrolyte solvents (and salt anions) used, in order to be reduced first at the negative electrode – regardless of its nature and potential. Ideally the additives decompose in a controlled manner to create a thin SEI film (ion conducting, electronically insulating) which is insoluble in the electrolyte. Here the difference between SIBs and LIBs is clear-cut – we have already stressed that the different solubilities of the Li2CO3 and Na2CO3 carbonates as well as LiF and NaF may affect the nature of the SEI formed.
Currently, but this is a field with possibility of fast changes, it is the singly fluorinated EC, FEC, that is the most common SEI enhancer for SIBs. In a paper published in 2011,75 several additives were for the first time screened for SIB electrolytes including FEC, VC, ethylene sulphite (ES) and the doubly fluorinated EC (DFEC), and results pointed to a clear advantage of FEC with respect to the others. Interestingly, VC, very popular in the field of LIBs, was not found to act the same way for SIBs. The beneficial effect of FEC with hard carbon (HC) anodes (exhibiting a plateau at very low potential that cannot fully be reached if cell polarization is too high) remains controversial. A beneficial effect was reported when using 1 M NaClO4 in PC, which is known to build a poor SEI on HC electrodes.53 In contrast, when tested in electrolytes allowing the building of a high quality SEI per se (such as EC:PC, see Section 4.1) the addition of FEC was found to be detrimental.76 It is important to stress that such differences are not nested in the different content of water impurities in both electrolytes, as these have been proven to be <40 ppm, but on the more resistive SEI build in the presence of FEC than in EC:PC which caused larger cell polarization. The benefits of FEC are, however, clearly proven for electrode materials displaying significant volumetric expansion, such as SnO2, Sn, Sb, SnSb, and red phosphorus, all operating at higher potential values than hard carbon.77–79 Indeed, the use of FEC is in such cases mandatory to achieve decent capacity retention and it allows cells with sodium counter electrodes to even outperform their lithium analogues.
2.5 Brief summary
In all, there is a range of salts, solvents and solvent concepts that today compete to become part of the golden standard electrolyte of SIBs for the future. We would, however, like to stress that the path forward for SIBs will likely not be a single one leading to a single concept choice and not even to a single choice in any of the components. This is due to the fact that we now look at energy storage in a much more diversified and global manner. One large difference compared to when LIBs emerged on the market is that the safety aspect of the electrolyte is, if possible, even more important today. This is due to the interest in larger cells, for XEVs and large-scale energy storage, and to the fact that the consequences of an abuse event are somewhat proportional to the size of the cell.3 Electrolytes and performance
3.1 Organic liquid based electrolytes
Historically, propylene carbonate (PC) was among the first solvents used in Li and Na cells, due to its high dielectric constant and large liquid range. However, it was soon replaced by ethylene carbonate (EC) for Li-ion cells as EC passivates the surface of graphitic electrodes while PC co-intercalates between the graphene layers causing exfoliation.80 As hard carbon (HC) is currently the only viable carbonaceous negative electrode material for SIBs, PC has remained the main solvent used and is the base of about 60% of the electrolyte formulations in the SIB literature. Nonetheless, HC tested in pure PC based electrolytes exhibits strong capacity fading and low Coulombic efficiencies, which has been attributed to the continuous growth of the SEI.53
Alcántara et al.81 tested disordered carbonaceous electrodes in different electrolyte solvent mixtures and NaClO4 salt already in 2005, and concluded that the performance was improved with the use of THF, which unfortunately has a too narrow ESW to be applied in practical cells. More recently (2011), Komaba and co-workers showed (using a 1 M NaClO4 concentration) that EC:DEC or PC outperformed EC:DMC, or EC:EMC.82 Along a similar line, Vidal-Abarca et al.83 noted how moving from PC to a EC:DEC mixture led to an improved performance for a sodium fluorophosphate cathode. Decomposition at low voltages was observed for the 1 M NaPF6 in PC electrolyte, while for the corresponding EC:DEC electrolyte the upper voltage set the limit.
Systematic studies dealing with different electrolyte compositions were published soon after these recent but seminal papers. These include studies of thermal behaviour, ionic conductivity, viscosity and the ESW, both using inert and “real” HC electrodes (see Fig. 2 and 3). The results indicate that using a mixture of EC and PC as solvent (with either 1 M NaClO4 or NaPF6) resulted in a much better capacity retention for HC than using PC alone, which was correlated with EC inducing the formation of a more stable SEI (see Section 4.1) with lower polarization thus enabling the achievement of full capacity on the low potential plateau of HC.76
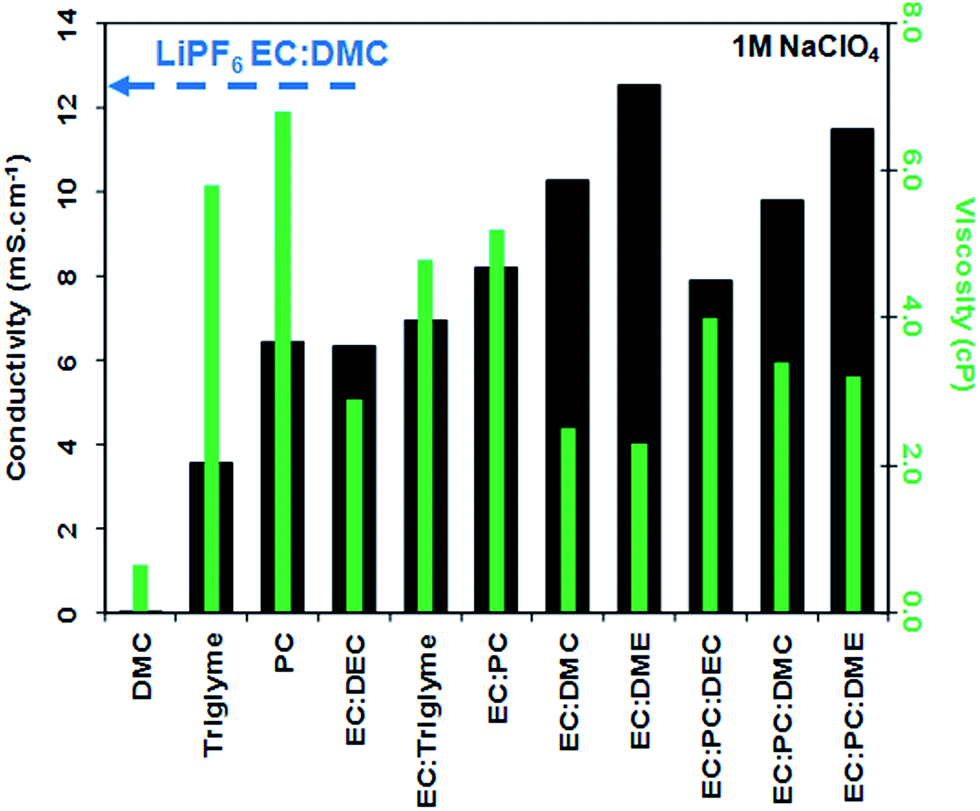 | ||
| Fig. 2
Conductivity (black bars and left hand side y axis) and viscosity (green bars and right hand side y axis) values of electrolytes consisting of 1 M NaClO4 dissolved in various solvents and solvent mixtures (reproduced with permission from The Royal Society of Chemistry).27,53 The ionic conductivity of 1 M LiPF6 in EC:DMC is also given for comparison.
| ||
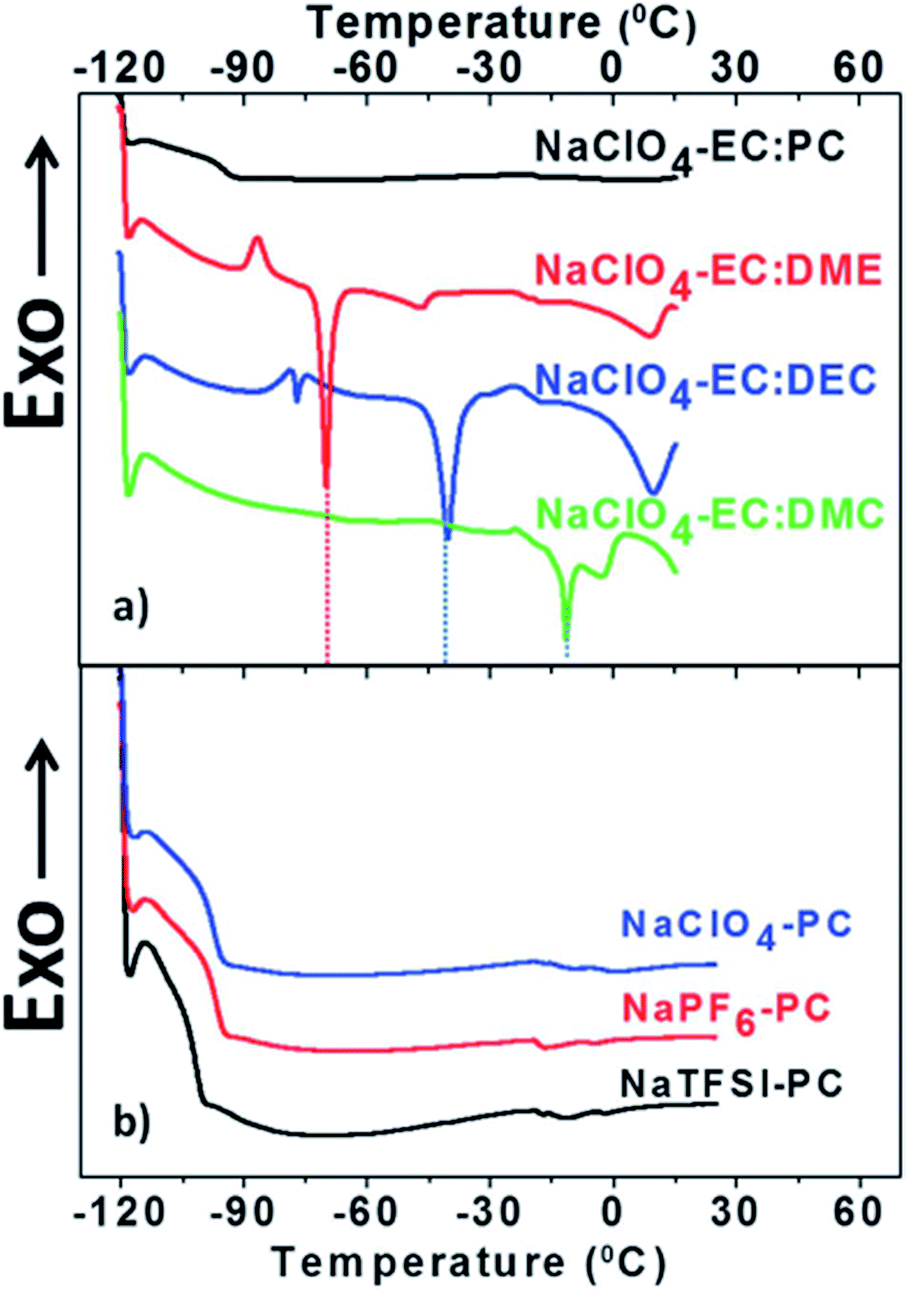 | ||
| Fig. 3 DSC heating curves of electrolytes after cooling the sample down to 120 °C. (a) Electrolytes based on 1 M NaClO4 dissolved in various solvent mixtures and (b) PC based electrolytes with 1 M of various Na salts. Reproduced with permission from The Royal Society of Chemistry.53 | ||
These results are not too surprising as LIBs also enlist mixed solvents in their electrolytes, as the diverse and sometimes contradictory requirements can hardly be met by an individual compound (use of EC alone is for instance prevented by its high melting point). Along this path of development, addition of 10% DMC to an EC:PC mixture resulted in decreased viscosity and enhanced ionic conductivity. The DMC does not seem to participate in the sodium solvation shell, but rather acts in the bulk of the electrolyte (Fig. 4).27
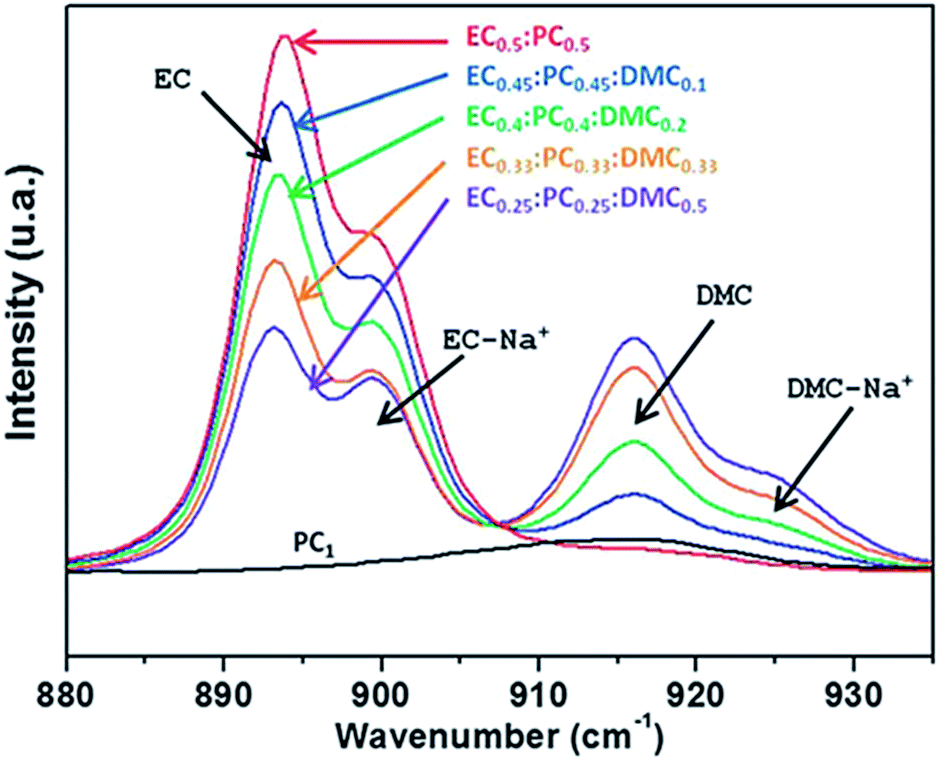 | ||
| Fig. 4 Raman spectra of electrolytes of 1 M NaTFSI in various solvent mixtures. The ratio between the bands corresponding to the unperturbed solvent and the same solvent coordinated by a sodium ion, respectively, provides an indication of the composition of the sodium ion solvation shell. Reproduced with permission from The Royal Society of Chemistry.27 Note that compositions are given in weight ratios and black arrows indicate peak positions. | ||
In another systematic study, a set of sodium salts (NaPF6, NaTf, and NaClO4) were dissolved in EC:DMC (3:7) and the influence on the conductivity was determined as a function of salt concentration and temperature.58 The best results were achieved with 0.6 M NaPF6 both in terms of ionic conductivity (6.8 mS cm−1) and the ESW against inert and Na0.7CoO2 electrodes, in the latter case the ESW improvement was attributed to the formation of an electrochemically stable surface film.
In terms of thermal stability, various electrolytes show interesting and sometimes somewhat puzzling results by DSC i.e. only a glass transition near −95 °C was observed for PC based electrolytes (see Fig. 2). In addition, an EC:PC solvent mixture showed a single exothermic peak around 250 °C resulting in an impressive liquidus range.53 Moreover, most of the Na salts used in the literature (NaClO4, NaPF6 and NaBF4) are thermally more stable than their Li analogues.
3.2 Ionic liquid based electrolytes
Ionic liquid (IL) based electrolytes have only attracted the attention of the SIB community during the very last few years – in stark contrast to, for example, polymer based electrolytes. A similarity, however, is the initial heavy focus on the electrolyte material properties prior to any real SIB studies being made. The reason for IL based electrolytes at all being considered originates from the wish to create safer electrolytes, but to still maintain the main properties of liquid electrolytes – not the least keeping the operating temperature of the SIB to ambient levels (still though, the room temperature performance of IL based electrolytes is often less than encouraging and often much reduced as compared to organic electrolytes).As mentioned in Section 2.3.2 IL based electrolytes consist of a Na-salt dissolved in an RTIL of the same WCA. It is of uttermost importance to highlight that these electrolytes are composed only of ions – thus the ionic conductivities reported will always be appealing, but that either assessment of the Na charge carrier transport features or electrochemical tests/experiments involving cycling of “real” electrode materials are compulsory for a true appraisal of their potential as SIB electrolytes (the same omission is true for IL based LIB studies).
Starting with Pyr based ILs Ding et al.84 reported NaFSI in Pyr13FSI (2:8) to have conductivities of 3.2 and 15.6 mS cm−1 at 298 and 353 K, respectively, and an anodic ESW limit of 5.2 V vs. Na+/Na. A recent follow-up85 used the same system and aimed to optimise the salt/IL ratio for a specific operating temperature, measured as capacity and rate capability of a Na/NaFSI in Pyr13FSI/NaCrO2 cells.
Noor et al.86 conducted a systematic study using NaTFSI in Pyr14TFSI, and reported ionic conductivities of 1–2 mS cm−1 at room temperature. They did not observe any deposition of Na metal on the Cu working electrode until −0.2 V vs. Na+/Na. This is most probably due to the high viscosity of the system (ca. 100 cP) which is common for ILs87 and impacts the mass transport properties. A more recent study by the same group88 deals with the basic properties and specific ion transport of NaTFSI in Pyr13FSI via23Na NMR spectroscopy. Of most interest is, however, the stable cycling vs. Na metal observed even at elevated temperatures (up to 100 °C) with a stable CV of plating/stripping on the Ni working electrode – dismissing doubts about this set-up made by Ding et al.85via a careful assessment of the role of water contamination at the ppm level. This also stresses one problem of ILs in general – purity and dryness – which, as already mentioned above, can obstruct progress.
Another study dealing with NaTFSI in Pyr14TFSI89 reports an ionic conductivity of ca. 1 mS cm−1 together with DSC analysis showing melting at −30 °C. In addition, a promising new IL, Pyr24TFSI, is advertised for the near future.
Very recently,90 0.45 M NaTFSI in Pyr14FSI was shown to outperform an 0.5 M NaPF6 in PC electrolyte in terms of cyclability with respect to a Na0.45Ni0.22Co0.11Mn0.66O2 electrode in half cells against sodium metal electrodes – which was attributed to the higher stability of the FSI anion at higher potentials (see above) and the formation of a suitable SEI. Another recent91 study on the effect of concentration (0.1–1 M of NaTFSI in Pyr14TFSI) and rate capability (0.05–5 C) revealed a promising capacity retention after 100 cycles at 0.3 C and 50 °C (87%) as compared to 1 M NaClO4 in EC:DEC (62%) with the same cell set-up (Na/electrolyte/NaFePO4) and conditions (note though the Na-salt difference in this comparison). Interestingly, the authors also argue for a lower salt concentration (0.5 M) to provide the best capacity at this temperature – which can be due to a change in the charge carrier nature/balance.
The other major class of ILs was used by some of us70 in a study focusing on the basic physico-chemical properties comparing EMImTFSI and BMImTFSI doped with NaTFSI and LiTFSI. By combining Raman spectroscopy and DFT calculations the stable Na+ charge carrying species in these IL systems was shown to, in general, be a doubly negatively charged [Na(TFSI)3]2− complex, as compared to [Li(TFSI)2]− for Li systems.92 This can be expected to hold true for most IL based Na conducting electrolytes – with implications for the conduction mechanism. In a short recent communication93 the equivalent FSI system, NaFSI-EMImFSI, was used for both basic properties evaluation and some cycling tests vs. Na metal. The latter again confirmed the wide ESW for systems using the FSI anion (here 5.1 V vs. Na+/Na) previously noted for the Pyr 1x systems.
While not directly along the main track of IL based electrolyte development, a proof-of-concept SIB symmetric cell assembly by Yamaki and co-workers, using Na3V2(PO4)3 as both the positive and negative electrodes with an 1 M NaBF4 in EMImBF4 electrolyte, was published as early as 2010.94 The cell did, however, not even remotely access the ESW of ILs – a cell voltage of 1.8 V resulted from the difference between the potentials observed for the two redox couples: V4+/V3+ (3.4 V) and V3+/V2+ (1.6 V) vs. Na+/Na.
A clear deviation from the standard IL concept is the use of small oligomers of PEO, “glymes”, and a salt to create “solvate ILs” by [M(glyme)]+ cationic charge carriers being formed.95 For Na, pentaglyme, G5, in an equimolar mixture with NaTFSI, was recently96 shown to form an electrolyte with an ionic conductivity of 0.61 mS cm−1 (30 °C) and with an upper ESW limit compatible with 4 V class positive electrodes. Subsequently, a proof of concept was shown (100 mA h g−1 for 50 cycles) for a Na/[Na(G5)]TFSI/Na0.44MnO2 cell, operated at 60 °C.
Yet another deviation from the main IL track was made by Egashira et al.97 using a ternary electrolyte composed of NaBF4 salt in PEGDME and using the IL DEMEBF4 as a co-solvent. The aim was to improve the cell safety and the conductivities, viscosities, and thermal properties were studied. For the 8:1:2 (PEG:NaBF4:DEMEBF4) molar ratio, ca. 1.2 mS cm−1 was measured, which is higher than the conductivity exhibited by the analogous lithium based electrolyte.
Along the track of ILs, Forsyth et al.98 recently investigated a solid state cousin of the ILs, an organic ionic plastic crystal (OIPC), made from Na x C2mpyr (1−x) TFSI. The OIPC displays an ionic conductivity of 0.3 mS cm−1 at 60 °C for x = 0.4. By XRD and NMR analysis the presence of two phases rich in NaTFSI and C2mpyrTFSI, respectively, was ascertained, with the volume and distribution being modified with temperature.
3.3 Polymer based electrolytes
The status of SPEs for sodium batteries can be exemplified by the recent study on the NaTFSI–PEO system103 (its Li analogue reported almost 20 years ago).104 The ionic conductivity reached 10−3 S cm−1 above 70 °C and an addition of SiO2 ceramic nano-fillers (7 nm) was needed/made to improve the mechanical properties and enhance the capacity retention for a Na/NaTFSI(PEO)20/Na symmetric cell. The use of NaTFSI as the salt is instrumental as the TFSI anion by its internal flexibility and large volume acts as a plasticizing agent.105 This reduces the rate of increase in glass transition temperature concomitant with salt addition to the polymer host and hence allows for softer, and more conductive, electrolytes than those based on other salts. Moreover, TFSI acts as a crystallization inhibitor for Li+ based systems,106 the parallel effect for Na+ based systems remaining to be verified. Interestingly, the use of both Na and TFSI seems to render the SPEs even “too” soft as ceramic particles are needed to create films with mechanical integrity.
A more “modern” approach is the single-ion conductor (SIC) developed by Villaluenga et al.107 The main advantage of SICs compared to SPEs is the suppression of concentration polarisation, as there are no mobile counter-anions. The developed polymer–SiO2 hybrid electrolytes (SiO2–PEG–anion or SiO2–anion) consist of 18 nm or 14 nm particles dispersed in a PEO matrix (for easy handling), with conductivities of ca. 2 × 10−5 S cm−1 at RT and an anodic ESW limit higher (4.4 V) than usually found for polyether based SPEs (3.9 V). 23Na and 19F NMR line-widths were used to measure and differentiate between the cation and anion contributions to the overall transport, respectively. The anion shows a broadening three times that usually observed in SPEs, and also in some instances ca. three times that of Na – thus moving significantly slower and confirming the materials as SICs.
3.4 Eutectic salt mixtures as electrolytes
While the use of “traditional” ILs as matrices is currently the most trodden path to create safer electrolytes, there are alternative approaches explored such as some of the SPEs and GPEs outlined above. Yet another more tempting possibility for SIBs is the use of eutectic salt mixtures as electrolytes, most simply binary NaX–MX systems (M being an alternative alkali ion). In 2010 the group of Hagiwara presented the thermal properties of several alkali MTFSI salts and their binary mixtures.110 Indeed, the melting points of the pure salts (Li–Cs, 403–368 K) can be lowered down to 325 K for the lowest melting Na system (Na0.47Cs0.53TFSI) and in the range 325–349 K for all choices of second M to Na. Such eutectics, while not applicable at room temperature, were later111 studied at 423 K and exhibit an ionic conductivity of ca. 12.1 mS cm−1 with an anodic ESW limit of 4.9 V vs. Na+/Na, thus extremely appealing. As a further proof of concept, also a NaFSI–KFSI eutectic electrolyte exhibiting a conductivity of 3.3 mS cm−1 was used in cells with NaCrO2 and Na metal as positive and negative electrodes, respectively, at 353 K.112,113 Being stable up to 5.2 V vs. Na+/Na and also showing no corrosion of the aluminium current collector, this type of electrolyte is clearly interesting, especially for high temperature operation. We note in passing, however, that the NaK alloy is a liquid at room-temperature for a wide range of compositions (ca. 40–90 wt% K), possibly causing problems for this electrolyte/negative electrode combination. The same electrolyte was also tested vs. a negative alloy electrode (Sn–Na)114 (at 363 K) and on a Na2FeP2O7 positive electrode in another study from the same group.115 Also ternary systems like NaFSI–KFSI–CsFSI, with a melting point of ca. 323 K and stable up to 4.9 V vs. Na+/Na, have been presented.110An inherent advantage with eutectic electrolytes is their fully inorganic nature – likely to reduce the number of side-reactions with the electrodes (including less drastic consequences in the case of abuse and thermal runaway). The main drawbacks are likely cost, just as for IL based electrolytes, and the operating temperature range, yet virtually impossible at regular ambient temperatures and thus even more so at lower temperatures.
3.5 Brief summary
Other concepts, starting with IL based electrolytes, are highly interesting as they might enable a safe(r) use of HC electrodes and also better capacity retention at only slightly elevated temperatures. This alongside with the prospects of creating almost unlimited combinations of salts and ILs give room to believe in further optimisation to be accomplishable. To this, however, must be added that the ion transport details and mechanisms in these systems are yet unexplored and is a venue for new scientific endeavours. The main drawbacks for implementation in SIBs as we see it are the current cost issues of ILs and the associated problems of purity and dryness, the uncertainty of performance at lower than ambient temperatures as a consequence of high viscosities, and the yet uncertainty on safety aspects – is a very high first exothermic reaction temperature more difficult to handle once a thermal runaway occurs? The eutectics have the same basic promises, but the need to find systems working at lower (room) temperature determines the prospects of these materials for SIBs.
For both organic solvent and IL based electrolytes there is also a need for a separator, a microporous film, to contain the electrolyte and separate the electrodes – as the electrolyte itself has no appreciable mechanical strength, while it is not necessary in cells using SPEs or GPEs. Even though the type of separators used in SIBs could affect the power/energy densities, capacity retention, and safety as observed for LIBs,116 no studies on the influence of the separators are available. Most SIB studies are made using glass microfiber or polypropylene film separators, as is the case for LIBs. Finally, while the SPE approach fundamentally holds large promise in almost all electrolyte aspects, we fail to find any convincing evidence of current progress towards a standard (not thin-film) SIB operating at room-temperature using an SPE. The GPEs are extremely much mirroring the Li systems in terms of materials, and also in application and scientific interest. However, we find the current interest in Na based GPEs very limited.
4 SIB cell aspects and tests
Having set the stage with the different electrolyte materials and concepts, we now turn to how these perform in real cells. As the interfaces/interphases with the electrodes are of uttermost importance, the SEI and SL are in focus together with the thermal stability prior to putting the few practical SIB attempts in perspective.4.1 SEI and SL
:EMC (3:7) electrolyte, was studied by FTIR and sodium alkoxides and alkyl carbonates were identified. Alternative XPS studies,120–124 using alloys as negative electrode materials (Sn, Mo3Sb7, Cu2Sb, Sb and In) and 1 M NaClO4 in PC as an electrolyte, indicate that thick layers (>5 nm) rich in carbonates (Na2CO3 and NaCO3R) fully cover the electrode after full discharge (0 V vs. Na+/Na), which are either thinner or cracked after charging the electrodes up to 2 V vs. Na+/Na.
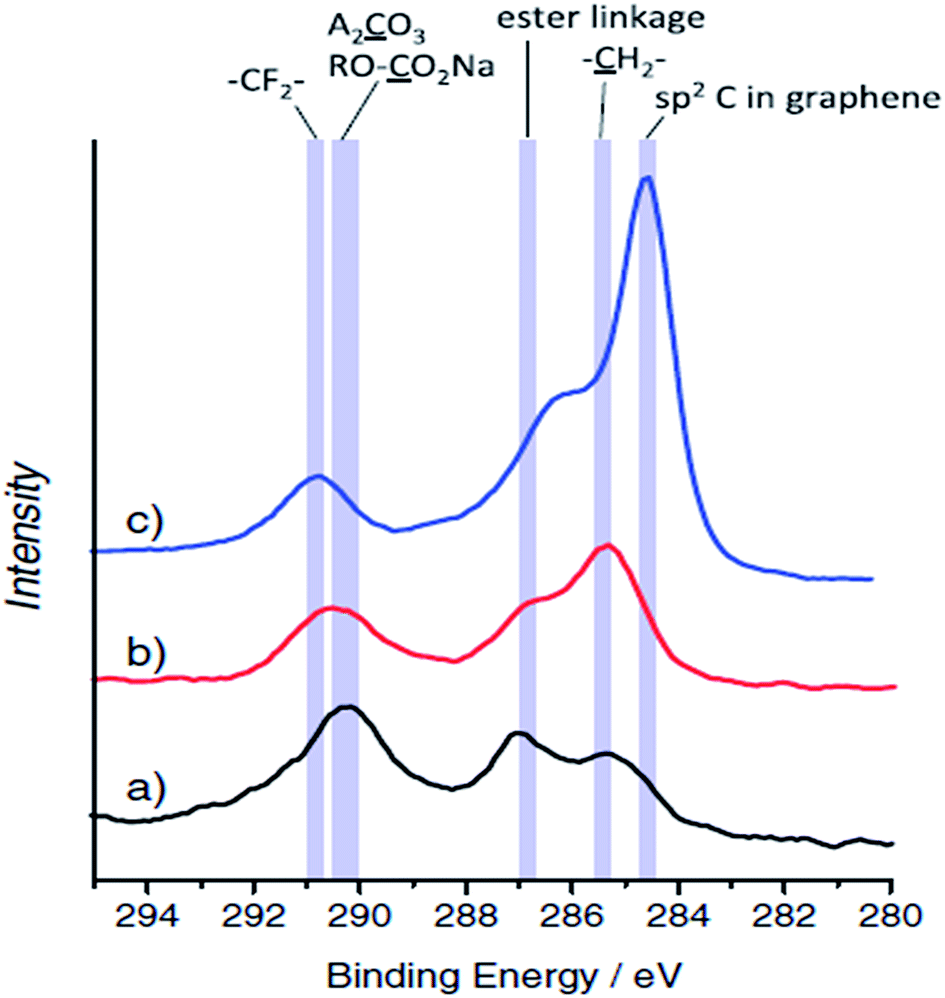 | ||
| Fig. 5 XPS C1s spectra for HC electrodes tested in: (a) sodium and (b) lithium cells after the first cycle, and (c) pristine electrode. Reproduced with permission,82 Copyright 2011, Wiley-VCH. | ||
 | ||
| Fig. 6 XPS C1s spectra of pristine HC powder and of HC electrodes after discharge of Na//HC cells down to 3 mV vs. Na+/Na in various electrolytes. Reproduced with permission from The Royal Society of Chemistry.27 | ||
Very recently Philippe et al. reported on comparative depth sensitive XPS analysis of the SEI layers formed onto Fe2O3 electrodes by varying the photon energy125 using either NaClO4 or LiClO4 in EC:DEC as electrolytes and sodium or lithium, respectively, as counter electrodes. The differences observed are similar to those reported for HC negative electrodes,82 with the SEI grown in the sodium cell being thicker and richer in inorganic species. The depth profile revealed that the lithium containing SEI exhibits a layered structure while a more homogeneous distribution of the components was found in the case of sodium.125
A significant difference in the morphology of the SEI formed on sodiated or lithiated HC electrodes using NaClO4 and LiClO4 in PC electrolytes was observed by HRTEM.82 For the former, the SEI surface was rough and non-uniform, while smooth and slightly thicker for the latter. Such findings have prompted re-introduction of EC as a co-solvent for SIB electrolytes as it promotes a more stable SEI, which is likely related to the formation of ether functionalities upon reduction.27
Very few studies report on the electrolyte stability vs. the positive electrode, and thus the surface layers (SLs) created have not been given much attention so far – which is true also for LIBs.126,127 A very thin SL was detected at the surface of Na3V2(PO4)2F3 (ref. 27) after the first full oxidation to 4.3 V vs. Na+/Na and further studies are underway to elucidate the electrode/electrolyte reactivity at high potentials. Recent studies report enhanced electrochemical stability vs. oxidation for both EC:PC and ethylmethyl sulfone (EMS) based electrolytes,128 although in the latter case the low stability upon reduction is still an issue. The use of NaClO4 or NaBF4 in PC was found to result in low Coulombic efficiency and poor capacity retention for Na3V2(PO4)3, while much better results were achieved using NaFSI in PC or EC:DEC and NaPF6 in EC:DEC electrolytes.129 Such results seem to point to a more general trend, as better Coulombic efficiencies and capacity retentions are often achieved for NaPF6 in EC containing electrolyte than for NaClO4 in PC based electrolytes.
Nonetheless, care has to be taken when interpreting results derived from tests in half cells, as the strong reactivity of the sodium counter electrode with the electrolyte can also have a major detrimental impact on the Coulombic efficiency.130,131
Finally, the mechanical properties (Young's modulus) of the SEI built on a Cu electrode cycled in a 1 M NaPF6 in EC:DEC electrolyte were determined by Weadock et al.132 by colloidal probe AFM. The observed heterogeneous SEI had a Young's modulus varying by over an order of magnitude (from 50 to 500 MPa) over a 25 × 25 μm surface area. It is clear at this stage that a deeper understanding of SEI properties, for SIBs as well as LIBs, will emerge from this kind of local analysis, that should serve to complement results from the most commonly used techniques for SEI characterization (e.g. XPS, EIS, FTIR, etc.), which merely provide averaged information.
The present consensus is that the SEI breaks down/cracks upon heating, and exothermic reactions subsequently occur between the now SEI-free electrode and the electrolyte, leading to the formation of a fresh SEI. In the next round, thermal decomposition of the re-formed SEI occurs upon further heating, together with reactions with the binder, which in turn generate additional heat.133 On the positive electrode, thermal decomposition of the active material itself with concomitant oxygen evolution and exothermic reactions with the solvents are the main causes for heat generation.134
All the exothermic processes described above are possible sources for disastrous thermal runaway of the battery pack. A safety figure of merit is therefore the magnitude of energy liberated by the reaction between fully charged electrodes (most reactive state) and the electrolyte at elevated temperatures. In view of the similarities of the electrolytes used, the present knowledge as well as the preferred ways to evaluate the thermal stability have been directly transferred from LIBs to SIBs. The present level of direct knowledge for SIBs is though limited; only a few electrolyte/electrode combinations have been evaluated either by DSC or by ARC. DSC enables the thermal response of individual and selected combinations of cell components to be measured over a broad temperature range, scanned at a fixed rate, while ARC tests are conducted on full cells and cell components under adiabatic conditions. The cell heating rate is a function of the intrinsic heat generating reactions in the cell and the thermal heat capacitance of the cell components.135 For HC electrodes DSC traces indicate that 1 M NaPF6 in EC:PC exhibits the highest exothermic peak onset temperature and lowest enthalpy of reaction amongst a variety of electrolytes. The first feature correlates with the higher thermal stability of the SEI, which is found to be higher for NaPF6 than for NaClO4 based electrolytes, with also some minor effect of the solvent used, EC0.5:DEC0.5 < PC < EC:PC.53 This points to a higher thermal stability of the SEI built in the presence of EC:PC. Fully sodiated HC in 1 M NaPF6 in EC:PC seems to compare very well with lithiated graphite in terms of safety: (i) a similar total heat generated and (ii) a higher exothermic peak onset temperature. Comparative DSC studies for fully lithiated and sodiated HC electrodes in various electrolytes by different authors fully confirm this trend.136 In contrast, ARC studies137,138 seem to indicate that graphite, lithiated in 1 M LiPF6 in EC:DEC, is thermally more stable than HC sodiated using 1 M NaPF6 in EC:DEC, which is attributed to the lower thermal stability of LiPF6 as compared to NaPF6. Moving to the positive electrodes, a range of SIB layered positive electrode materials (Na
x
FeO2, Na
x
CoO2, Na
x
CrO2, Na
x
Ni0.5Mn0.5O2)139–142 has been studied using DSC or ARC, with the main exothermic process being in all cases attributed to the active material decomposition involving oxygen release together with solvent combustion, in analogy with LiCoO2 in LIBs.143 However, no systematic study on the impact of the electrolyte or the effect of the SL on the total heat generated for SIB positive electrode materials has been carried out to date.
4.2 Practical SIBs
Besides the examples given in the Introduction for early proof of concept,21,23 which exhibited rather poor performance in terms of capacity retention, no other reports dealing with full cell assembly appeared until a decade later when Barker et al. at Valence Technologies built a 3.7 V sodium ion cell using NaVPO4F and HC as electrode materials, which was certainly a side result from their intense research on phosphates for lithium batteries at the time.24,25 The electrolyte used was 1 M NaClO4 in EC:DMC and the cell, tested at room temperature at C/10, showed a reversible capacity of 80 mA h g−1 on the second cycle, which faded to 50% on the 30th cycle. Another 8–9 years later on, Komaba et al.81 reported on the cycling performance of a HC|Na[Ni0.5Mn0.5]O2 cell using 1 M PC solutions of different salts (NaClO4, NaPF6, or NaTFSI) as the electrolytes. Although all cells showed initial capacities above 200 mA h per (g carbon) at 1 C rate with an operation voltage of 3 V, drastic capacity fading was observed for the cell with NaClO4, while those containing NaPF6 or NaTFSI retained 70% of the initial capacity after 50 cycles. Remarkably, Johnson and co-workers144 achieved a stable capacity of 100 mA h per (g cathode active material) for 150 cycles at 0.5 C rate for a Na
y
C|Na
1−y
[Ni1/3Fe1/3Mn1/3]O2 (y = 0.46) cell with an operating voltage of 2.75 V using 1 M NaClO4 in PC as the electrolyte. Rate capability tests performed between 0.1 and 1 C yielded reversible capacities ranging from 130 to 94 mA h g−1 (see Fig. 7). Oh et al. reported on a full SIB based on a carbon-coated Fe3O4 anode, Na[Ni0.25Fe0.5Mn0.25]O2 layered cathode, and NaClO4 in FEC/EMS as the electrolyte. This battery operated reversibly around 2.4 V and delivered a capacity of about 130 mAh per (g cathode active material) with 76.1% capacity retention after 150 cycles and a Coulombic efficiency approaching 100%.128 Full SIBs were also recently reported using organic negative electrodes (mono- or di-sodium terephthalate) and Na0.75Mn0.7Ni0.23O2 as the positive electrode using 1 M NaPF6 in EC:EMC as the electrolyte. This SIB exhibits an operating voltage of 3.6 V and delivered ca. 268 mA h per (g cathode active material) discharge capacity after 50 cycles.119
 | ||
| Fig. 7
Voltage versus capacity profiles for HC‖NVPF full Na-ion cells cycled in 1 M NaPF6 in EC0.45:PC0.45:DMC0.1 (reproduced with permission from The Royal Society of Chemistry27) and Na
y
C|Na
1−y
[Ni1/3Fe1/3Mn1/3]O2 (y = 0.46) cell using 1 M NaClO4 in PC as the electrolyte recorded at different rates (reproduced with permission,144 Copyright 2012, Elsevier).
| ||
Some of us reported on full HC|Na3V2(PO4)2F3 (NVPF) cells27 using 1 M NaClO4 or NaPF6 in EC0.45:PC0.45:DMC0.10 as the electrolyte, which exhibits an average potential of 3.75 V and a theoretical energy density comparable to that of graphite|LFP LIB cells. A stable capacity of 97 mA h per (g positive active material) for 120 cycles at C/5 and a Coulombic efficiency >98.5% and good performance at high rates was shown (see Fig. 7).
Besides the openly disclosed academic development described above, industrial R&D has also been disclosed to some extent. In addition to press releases, from which the true performance is always difficult to evaluate, some reports are available on the fabrication by Sumitomo of HC|NaFe0.4Mn0.3Ni0.3O2 coin cells and laminated SIBs using 1 M NaPF6 in PC as the electrolyte, without any FEC additive included.145 These SIBs both exhibit good cycle life and rate capability, although metrics to compare with similar LIB cells are missing. Comparative heating and overcharging tests seem to indicate better results for SIBs, as 200% overcharge only resulted in swelling, without burst or ignition. Cell performance has also recently been disclosed by Faradion for their 3Ah SIB pouch cells using a HC negative electrode and a layered positive electrode material and an electrolyte based on NaPF6 dissolved in a carbonate solvent mixture – a cell which seems to be comparable to the LIB state of the art.146
5 Conclusions
The field of SIBs has boomed during the last five years as a result of a growing sustainability concern and the emerging interest in large scale energy storage applications (XEVs, grids, etc.) for which it could become competitive with respect to LIBs. The performance of SIBs is progressing at a quick pace, not least as it takes substantial advantage of the chemical analogies between lithium and sodium and the widely accumulated know-how for LIBs over the years. Besides electrode materials research, electrolytes are crucial in determining the battery performance. Moreover, the electrolyte formulation is the key to decrease as much as possible the parasitic side reactions and foremost to delay battery EOL – which even more stresses sustainability, but also cost issues as they are strongly connected with EOL and the total energy throughput. This “hidden” role of the electrolyte which has been gradually “discovered” for LIBs is luckily present from the very beginning in the development of SIBs – and should therefore enable both faster and more reliable progress.In order to practically assess the viability of the SIB technology, optimization of each individual component is of course still urgently needed, and comprehensive studies enabling the building of laboratory-scale prototypes are compulsory. Already now studies devoted to full cell SIBs show these to exhibit performance comparable to the current state of the art LIBs at the laboratory scale and reports are starting to be disclosed on larger prototypes, which seem to sustain that trend. In all, this confirms that the SIB technology is well placed in the quest for alternative energy storage technologies, while emphasizing the need to pursue research to turn such prospects into a commercial reality.
Acknowledgements
We all would like to sincerely acknowledge the ALISTORE-ERI community at large and especially the sodium battery brainstorm members for sharing many fruitful discussions. PJ also would like to acknowledge the continuous financial support by the Swedish Energy Agency, the Swedish Science Council (VR) and the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS) as well as the support by several Chalmers Area of Advance, Materials, Energy, and Transport. RP and AP are grateful to Ministerio de Ciencia e Innovación (Spain, grant MAT2011-24757) for financial support. AB is grateful for the financial support by the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS); DM is grateful for the financial support by the Swedish Science Council (VR), Stiftelsen J. Gust. Richert, and Chalmers Energy Area of Advance.Notes and references
- J. C. Burns, A. Kassam, N. N. Sinha, L. E. Downie, L. Solnikova, B. M. Way and J. R. Dahn, J. Electrochem. Soc., 2013, 160, A1451 CrossRef CAS PubMed.
- L. J. Krause, L. D. Jensen and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A1451 CrossRef PubMed.
- Y. Akita, M. Segawa, H. Munakata and K. Kanamura, J. Power Sources, 2013, 239, 175 CrossRef CAS.
- S. Péres-Villar, P. Lanz, H. Schneider and P. Novák, Electrochim. Acta, 2013, 106, 506 CrossRef.
- D. Takamatsu, Y. Koyama, Y. Orikasa, S. Mori, T. Nakatsutsumi, T. Hirano, H. Tanida, H. Arai, Y. Uchimoto and Z. Ogumi, Angew. Chem., Int. Ed., 2012, 51, 11597 CrossRef CAS.
- P. Mukherjee, A. Lagutchev and D. D. Dlott, J. Electrochem. Soc., 2012, 159, A244 CrossRef CAS.
- J. E. Owejan, J. P. Owejan, S. C. DeCaluwe and J. A. Dura, Chem. Mater., 2012, 24, 2133 CrossRef CAS.
- K. Xu and A. von Cresce, J. Mater. Chem., 2011, 21, 9849 RSC.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379 CrossRef CAS.
- D. Y. Wang, N. N. Sinha, R. Petibon, J. C. Burns and J. R. Dahn, J. Power Sources, 2014, 251, 311 CrossRef CAS.
- M. Yoshio, H. Yoshitake and K. Abe, The Electrochemical Society Extended Abstracts, 2003, vol. 2003–2 Search PubMed.
- K. Abe and H. Yoshitake, Electrochemistry, 2004, 72, 520 Search PubMed.
- S. Wilken, P. Johansson and P. Jacobsson, in Lithium Batteries: Advanced Technologies and Applications, ISBN: 978-1-118-18365-6, ed. B. Scrosati, K. M. Abraham, W. Schalkwijk and J. Hassoun, John Wiley & Sons, 2013, 39–70 Search PubMed.
- J. M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- H. Vikström, S. Davidsson and M. Höök, Appl. Energy, 2013, 110, 252 CrossRef.
- C. Grosjean, P. H. Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735 CrossRef.
- M. S. Whittingham, Prog. Solid State Chem., 1978, 12, 41 CrossRef CAS.
- K. M. Abraham, Solid State Ionics, 1982, 7, 199 CrossRef CAS.
- A. S. Nagelberg and W. L. Worrell, J. Solid State Chem., 1979, 29, 345 CrossRef CAS.
- C. Delmas, J. J. Braconnier, C. Fouassier and P. Hagenmuller, Solid State Ionics, 1981, 4, 165 CrossRef.
- L. W. Shacklette, T. R. Jow and L. Townsend, J. Electrochem. Soc., 1985, 135, 2669 CrossRef.
- T. R. Jow, L. W. Shacklette, M. Maxfield and D. Vernick, J. Electrochem. Soc., 1987, 134, 1730 CrossRef CAS.
- Y. Ma, M. M. Doeff, S. J. Visco and L. J. De Jonghe, J. Electrochem. Soc., 1993, 140, 2726 CrossRef CAS.
- J. Barker, Y. Saidy and J. L. Swoyer, US Patent 2002/0192553 .
- J. Barker, Y. Saidy and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, 6, A1 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS.
- A. Ponrouch, R. Dedryvere, D. Monti, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Energy Environ. Sci., 2013, 6, 2361 CAS.
- P. A. Nelson, K. G. Gallagher, I. Bloom and D. W. Dees, Modeling the Performance and Cost of Lithium-ion Batteries for Electric Vehicles Chemical Sciences and Engineering Division, Argonne National Laboratory, ANL-11/32, Argonne, IL USA, 2011 Search PubMed.
- London Metal Exchange, accessed 2014-10-08.
- U. Palme, The Role of Indicators in Developing Sustainable Urban Water Systems, PhD thesis, Environmental System Analysis, Department of Energy and Environment, Chalmers University of Technology, Göteborg, Sweden 2007 Search PubMed.
- B. Steen, A Systematic Approach to Environmental Priority Strategies in In Product Development (EPS), version 2000 – Models and Data, Chalmers University of Technology, Centre for Environmental Assessment of Products and Material Systems (CPM) Report, 1999:5, Gothenburg, 1999 Search PubMed.
- B. Steen and G. Borg, Ecol. Econ., 2002, 42, 401 CrossRef.
- X. Lu, G. Xia, J. P. Lemmon and Z. Yang, J. Power Sources, 2010, 195, 2431 CrossRef CAS.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734 CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- S. W. Kim, D. H. Seo, S. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710 CrossRef CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- B. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168 CrossRef CAS PubMed.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312 CAS.
- H. Pan, Y. S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15029 RSC.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N. S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067 CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169 CrossRef CAS PubMed.
- E. Jónsson and P. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 10774 RSC.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160 CrossRef CAS.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120 CrossRef CAS.
- T. Abe, F. Sagane, M. Othsuka, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2151 CrossRef.
- F. Sagane, T. Abe, Y. Iriyama and Z. Ogumi, J. Power Sources, 2005, 146, 749 CrossRef CAS.
- Y. Yamada, F. Sagane, Y. Iriyama, T. Abe and Z. Ogumi, J. Phys. Chem. C, 2009, 113, 14528 CAS.
- F. Sagane, T. Abe and Z. Ogumi, J. Power Sources, 2010, 195, 7466 CrossRef CAS.
- H. Mizuno, Electrochim. Acta, 2012, 63, 139 CrossRef.
- A. Ponrouch, E. Marchante, M. Courty, J. M. Tarascon and M. R. Palacín, Energy Environ. Sci., 2012, 5, 8572 CAS.
- S. H. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320 CrossRef CAS.
- D. J. Vevlin and P. Herley, React. Solids, 1987, 3, 75 CrossRef.
- T. Evans, J. Olson, V. Bhat and S.-H. Lee, J. Power Sources, 2014, 269, 616 CrossRef CAS PubMed.
- A. Bhide, J. Hofmann, A. K. Dürr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987 RSC.
- P. Johansson, H. Markusson, P. Jacobsson and M. Armand, Phys. Chem. Chem. Phys., 2004, 6, 895 RSC.
- J. Scheers, P. Johansson, P. Szczecinski, W. Wieczorek, M. Armand and P. Jacobsson, J. Power Sources, 2010, 195, 6081 CrossRef CAS PubMed.
- A. Plewa-Marczewska, T. Trzeciak, A. Bitner, L. Niedzicki, M. Dranka, G. Z. Zukowska, M. Marcinek and W. Wieczorek, Chem. Mater., 2014, 26, 4908 CrossRef CAS.
- M. Egashira, B. Scrosati, M. Armand, S. Béranger and C. Michot, Electrochem. Solid-State Lett., 2003, 6, A71 CrossRef CAS PubMed.
- L. Niedzicki, G. Z. Zukowska, M. Bukowska, P. Szczecinski, S. Grugeon, S. Laruelle, M. Armand, S. Panero, B. Scrosati, M. Marcinek and W. Wieczorek, Electrochim. Acta, 2010, 55, 1450 CrossRef CAS PubMed.
- J. Scheers, D.-H. Lim, J.-K. Kim, E. Paillard, W. A. Henderson, P. Johansson, J.-H. Ahn and P. Jacobsson, J. Power Sources, 2014, 251, 451 CrossRef CAS PubMed.
- E. Jónsson, M. Armand and P. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 6021 RSC.
- K. Izutsu, in Electrochemistry in Nonaqueous Solutions, ISBN: 3-527-30516-5, WILEY-VCH Verlag GmbH, 2002 Search PubMed.
- R. G. Pearsons, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef.
- Thematic issue MRS Bull., 2013, 38, 7. The whole issue deals with ionic liquids Search PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS.
- D. Monti, E. Jónsson, M. R. Palacin and P. Johansson, J. Power Sources, 2014, 245, 630 CrossRef CAS PubMed.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- M. B. Armand, J. M. Chabagno and M. Duclot, Proceedings of the 2nd Int. Conf. On Solid Electrolytes, St Andrews, Scotland, 1978, Abstract 65 Search PubMed.
- M. Anderman, Solid State Ionics, 1994, 69, 336 CrossRef CAS.
- J. M. Tarascon, A. S. Gozdz, C. Schmutz, F. Shokoohi and P. C. Warren, Solid State Ionics, 1996, 86–88, 49 CrossRef CAS.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165 CAS.
- A. Ponrouch, A. R. Goñi and M. R. Palacín, Electrochem. Commun., 2013, 27, 85 CrossRef CAS PubMed.
- Y.-X. Wang, Y.-G. Lim, M.-S. Park, S.-L. Chou, J. H. Kim, H.-K. Liu, S.-X. Dou and Y.-J. Kim, J. Mater. Chem. A, 2014, 2, 529 CAS.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633 CrossRef CAS PubMed.
- R. Fong, U. von Sacken and J. R. Dahn, J. Electrochem. Soc., 1990, 137, 2009 CrossRef CAS PubMed.
- R. Alcantara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222 CrossRef CAS PubMed.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859 CrossRef CAS.
- C. Vidal-Abarca, P. Lavela, J. L. Tirado, A. V. Chadwick, M. Alfredsson and E. Kelder, J. Power Sources, 2012, 197, 314 CrossRef CAS PubMed.
- C. S. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 238, 296 CrossRef CAS PubMed.
- C. S. Ding, T. Nohira, R. Hagiwara, K. Matsumoto, Y. Okamoto, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2014, 269, 124 CrossRef CAS PubMed.
- S. A. M. Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766 CrossRef.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238 CrossRef CAS.
- H. Yoon, H. Zhu, A. Hervault, M. Armand, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2014, 16, 12350 RSC.
- J. Serra Moreno, G. Maresca, S. Panero, B. Scrosati and G. B. Appetecchi, Electrochem. Commun., 2014, 43, 1 CrossRef CAS PubMed.
- L. G. Chagas, D. Buchholz, L. M. Wu, B. Vortmann and S. Passerini, J. Power Sources, 2014, 247, 377 CrossRef CAS PubMed.
- N. Wongittharom, T. C. Lee, C. H. Wang, Y. C. Wang and J. K. Chang, J. Mater. Chem. A, 2014, 2, 5655 CAS.
- J. C. Lassègues, J. Grondin, C. Aupetit and P. Johansson, J. Phys. Chem. A, 2009, 113, 305 CrossRef PubMed.
- K. Matsumoto, T. Hosokawa, T. Nohira, R. Hagiwara, A. Fukunaga, K. Numata, E. Itani, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 265, 36 CrossRef PubMed.
- L. S. Plashnitsa, E. Kobayashi, Y. Noguchi, S. Okada and J. I. Yamaki, J. Electrochem. Soc., 2010, 157, A536 CrossRef CAS PubMed.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323 CrossRef CAS PubMed.
- S. Terada, T. Mandai, R. Nozawa, K. Yoshida, K. Ueno, S. Tsuzuki, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 11737 RSC.
- M. Egashira, T. Asai, N. Yoshimoto and M. Morita, Electrochim. Acta, 2011, 58, 95 CrossRef CAS PubMed.
- M. Forsyth, T. Chimdi, A. Seeber, D. Gunzelmann and P. C. Howlett, J. Mater. Chem. A, 2014, 2, 3993 CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Atlung, J. Electrochem. Soc., 1985, 132, 3061 CrossRef CAS PubMed.
- Y. Ma, M. M. Doeff, S. J. Visco and L. C De Jonghe, J. Electrochem. Soc., 1993, 140, 2726 CrossRef CAS PubMed.
- J. W. Fergus, J. Power Sources, 2010, 195, 4554 CrossRef CAS PubMed.
- http://www.batscap.com .
- J. Serra Moreno, M. Armand, M. B. Berman, S. G. Greenbaum, B. Scrosati and S. Panero, J. Power Sources, 2014, 248, 695 CrossRef CAS.
- M. Armand, W. Gorecki and R. Andréani, Proceedings of the 2nd International Meeting on Polymer Electrolytes, ed. B. Scrosati, Elsevier London, 1989, p.91 Search PubMed.
- P. Johansson, S. P. Gejji, J. Tegenfeldt and J. Lindgren, Electrochim. Acta, 1998, 43, 1375 CrossRef CAS.
- L. Edman, A. Ferry and M. Doeff, J. Mater. Res., 2000, 15, 1950 CrossRef CAS.
- I. Villaluenga, X. Bogle, S. Greenbaum, I. Gil de Muro, T. Rojo and M. Armand, J. Mater. Chem. A, 2013, 1, 8348 CAS.
- D. Kumar and S. A. Hashmi, Solid State Ionics, 2010, 181, 416 CrossRef CAS PubMed.
- M. Patel, K. G. Chandrappa and A. J. Bhattacharyya, Solid State Ionics, 2010, 181, 844 CrossRef CAS PubMed.
- K. Kubota, T. Nohira and R. Hagiwara, Electrochim. Acta, 2012, 66, 320 CrossRef CAS PubMed.
- T. Nohira, T. Ishibashi and R. Hagiwara, J. Power Sources, 2012, 205, 506 CrossRef CAS PubMed.
- K. Kubota, T. Nohira, T. Goto and R. Hagiwara, Electrochem. Commun., 2008, 10, 1886 CrossRef CAS PubMed.
- A. Fukunaga, T. Nohira, Y. Kozawa, R. Hagiwara, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2012, 209, 52 CrossRef CAS PubMed.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, Electrochim. Acta, 2014, 135, 60 CrossRef CAS PubMed.
- C. Y. Chen, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 237, 52 CrossRef CAS PubMed.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332 CrossRef CAS PubMed.
- L. Vogdanis, B. Martens, H. Uchtmann, F. Hensel and W. Heitz, Makromol. Chem., 1990, 191, 465 CrossRef CAS.
- A. Abouimrane, W. Weng, H. Eltayeb, Y. Cui, J. Niklas, O. Poluektov and K. Amine, Energy Environ. Sci., 2012, 5, 9632 CAS.
- L. Baggetto, P. Ganesh, R. P. Meisner, R. R. Unocic, J. C. Jumas, C. A. Bridges and G. M. Veith, J. Power Sources, 2013, 234, 48 CrossRef CAS PubMed.
- L. Baggetto, E. Allcorn, R. R. Unocic, A. Manthiram and G. M. Veith, J. Mater. Chem. A, 2013, 1, 11163 CAS.
- L. Baggetto, E. Allcorn, A. Manthiram and G. M. Veith, Electrochem. Commun., 2013, 27, 168 CrossRef CAS PubMed.
- L. Baggetto, P. Ganesh, C. N. Sun, R. A. Meisner, T. A. Zawodzinski and G. M. Veith, J. Mater. Chem. A, 2013, 1, 7985 CAS.
- S. A. Webb, L. Baggetto, C. A. Bridges and G. M. Veith, J. Power Sources, 2014, 248, 1105 CrossRef CAS PubMed.
- B. Philippe, M. Valvo, F. Lindgren, H. Rensmo and K. Edstrom, Chem. Mater., 2014, 26, 5028 CrossRef CAS.
- D. Aurbach, B. Markovsky, M. D. Levi, E. Levi, A. Schetchter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81–82, 95 CrossRef CAS.
- K. Edstrom, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397 CrossRef PubMed.
- S. M. Oh, S. T. Myung, C. S. Yoon, J. Lu, J. Hassoun, B. Scrosati, K. Amine and Y. K Sun, Nano Lett., 2014, 14, 1620 CrossRef CAS PubMed.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156 CrossRef CAS.
- T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A1748 CrossRef CAS PubMed.
- A. Rudola, D. Aurbach and P. Balaya, Electrochem. Commun., 2014, 46, 56 CrossRef CAS PubMed.
- N. Weadock, N. Varongchayakul, J. Wan, S. Lee, J. Seog and L. Hu, Nano Energy, 2013, 2, 713 CrossRef CAS PubMed.
- H. Yang, H. Bang, K. Amine and J. Prakash, J. Electrochem. Soc., 2005, 152, A73 CrossRef CAS PubMed.
- D. D. MacNeil and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A912 CrossRef CAS PubMed.
- D. Doughty and E. P. Roth, Interface, 2012, 21, 37 CAS.
- J. Zhao, L. Zhao, K. Chihara, S. Okada, J. I. Yamaki, S. Matsumoto, S. Kuze and K. Nakane, J. Power Sources, 2013, 244, 752 CrossRef CAS PubMed.
- X. Xia, M. N. Obrovac and J. R. Dahn, Electrochem. Solid-State Lett., 2011, 14, A130 CrossRef CAS PubMed.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A515 CrossRef CAS PubMed.
- J. Zhao, L. Zhao, N. Dimov, S. Okada and T. Nishida, J. Electrochem. Soc., 2013, 160, A3077 CrossRef CAS PubMed.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A647 CrossRef CAS PubMed.
- X. Xia and J. R. Dahn, Electrochem. Solid-State Lett., 2012, 15, A1 CrossRef CAS PubMed.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A1048 CrossRef CAS PubMed.
- D. D. MacNeil and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A912 CrossRef CAS PubMed.
- D. Kim, E. Lee, M. Slater, W. Lu, S. Rood and C. S. Johnson, Electrochem. Commun., 2012, 18, 66 CrossRef CAS PubMed.
- S. Kuze, J. I. Kageura, S. Matsumoto, T. Nakayama, M. Makidera, M. Saka, T. Yamaguchi, T. Yamamoto and K. Nakane, Sumitomo Kagaku, 2013, 1 Search PubMed.
- J. Barker, R. Heap, N. Roche, C. Tan, R. Sayers and Y. Liu, 17th IMLB, Como 10–14th June 2014, Abstract #266 .
| This journal is © The Royal Society of Chemistry 2015 |