
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive Raman study of orthorhombic κ/ε-Ga2O3 and the impact of rotational domains†
Benjamin M.
Janzen
 a,
Piero
Mazzolini
b,
Roland
Gillen
c,
Vivien F. S.
Peltason
a,
Linus P.
Grote
a,
Janina
Maultzsch
c,
Roberto
Fornari
b,
Oliver
Bierwagen
d and
Markus R.
Wagner
*a
a,
Piero
Mazzolini
b,
Roland
Gillen
c,
Vivien F. S.
Peltason
a,
Linus P.
Grote
a,
Janina
Maultzsch
c,
Roberto
Fornari
b,
Oliver
Bierwagen
d and
Markus R.
Wagner
*a
aTechnische Universität Berlin, Institute of Solid State Physics, Hardenbergstraße 36, 10623 Berlin, Germany. E-mail: markus.wagner@physik.tu-berlin.de
bDepartment of Mathematical, Physical and Computer Sciences, University of Parma, Viale delle Scienze 7/A, 43124 Parma, Italy
cChair of Experimental Physics, Friedrich-Alexander Universität Erlangen-Nürnberg, Staudtstraße 7, 91058 Erlangen, Germany
dPaul-Drude-Institut für Festkörperelektronik, Leibniz-Institut im Forschungsverbund Berlin e.V., Hausvogteiplatz 5-7, 10117 Berlin, Germany
First published on 20th September 2021
Abstract
Gallium oxide (Ga2O3) is an ultra-wide bandgap material, which has recently attracted widespread attention for holding promising applications in power electronics and solar blind UV photodetectors, outclassing GaN or SiC in terms of a larger bandgap and higher breakdown voltages. The orthorhombic κ phase (also referred to as ε) has sparked particular interest for offering higher symmetry than β, while featuring ferroelectric behavior paired with a large predicted spontaneous polarization, paving the way to fabricating high-quality two-dimensional electron gases for application in heterostructure field effect transistors. The presently available κ phase samples are characterized by a domain structure, in which orthorhombic domains are rotated 120° against each other within the c-plane forming a pseudo-hexagonal structure, which has previously often been ascribed to ε-Ga2O3 and incorrectly been viewed as this polymorph's true crystal structure. A detailed investigation into the phonon modes of orthorhombic κ-Ga2O3 provides insights into fundamental material properties such as crystal structure and orientation as well as the vibrational symmetries of Raman active modes. We investigate the Raman active phonon modes of an MBE-grown orthorhombic κ-Ga2O3 thin film featuring the domain structure deposited on (0001)-Al2O3 by experiment and theory: Polarized micro-Raman spectroscopy measurements in conjunction with density functional perturbation theory (DFPT) calculations enable the identification of both the frequencies and vibrational symmetries of the Raman active phonons. Presenting comprehensive Raman spectra of the orthorhombic κ phase, the experimental frequencies of more than 90 Raman modes are determined and correlated with the 117 modes predicted by the calculations. Angular-resolved Raman measurements are utilized to provide an experimental verification of phonon mode symmetries. We present an analytical tool to deal with the domain structure and its effect on the obtained Raman spectra.
1 Introduction
Gallium oxide (Ga2O3) is a transparent, semiconducting oxide, which may form five different polymorphs: α, β, γ, κ (mostly referred as ε)1 and δ.2–11 As further explained in Section 1.1, from now on in this manuscript we will refer to the orthorhombic polymorph of Ga2O3 just as κ. While the majority of the Ga2O3 related research focuses on the thermodynamically-most stable monoclinic β-phase,2,3,9 the second-most stable orthorhombic κ polymorph offers unique features and has recently been gaining momentum. The reported optical bandgap energies of the κ phase at room temperature range between 4.91 and 5.04 eV,12–18 slightly higher than those of β-Ga2O3 (∼4.8 eV19,20). Moreover, outclassing competing wide bandgap materials like SiC (3.3 eV) or GaN (3.4 eV) makes the material a promising candidate for future applications in power electronics or solar-blind UV photodetectors.18,21–24 Aside from providing higher symmetry than β, density functional ab initio calculations25,26 predicted the presence of ferroelectric behavior, which has been experimentally27 verified using dynamic hysteresis measurements. The combination of ferroelectric behavior and a large predicted spontaneous polarization (≈23–26 μC cm−2)25,28,29 along its c-axis might further pave the way to fabricating high-quality two-dimensional electron gases (2DEG) as conduction channels in heterostructure field effect transistors. While a pair of other calculations1,27 and an experimental study27 obtained lower values of ≈0.2 μC cm−2 and ≈0.0092 μC cm−2, the experimental feasibility of creating a 2DEG at the interface of a κ-Ga2O3/GaN heterostructure was recently demonstrated,30 which might open the possibility of realizing high electron mobility transistors without doping. Moreover, a κ-(AlxGa1−x)2O3/κ-Ga2O3 quantum well superlattice heterostructure was recently realized.31 Featuring even larger polarization differences, the incorporation of In32 or Al33 into κ-Ga2O3 enabled bandgap tuning between 4.25 and 6.2 eV34 and is expected to enable 2DEGs with even higher sheet carrier densities.The κ phase was found to thermally transition to β only under annealing at high temperatures T > 700–800 °C,14,35 allowing for applications in devices requiring sufficiently high working temperatures. To date, the orthorhombic phase has been grown successfully36 on a number of different substrates, including Al2O3(0001), GaN(0001), AlN(0001), 6H-SiC or β-Ga2O3 (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01), using halide vapour phase epitaxy,14,37,38 atomic layer deposition,39 metal–organic chemical vapor deposition,15,18,22,27,30,35,37,39–44 metal–organic vapor phase epitaxy,1,45–48 mist CVD,16,17,32,33,49,50 plasma-assisted molecular beam epitaxy,51,52 laser molecular beam epitaxy,21,53 and pulsed laser deposition.12,13,24,54–57
01), using halide vapour phase epitaxy,14,37,38 atomic layer deposition,39 metal–organic chemical vapor deposition,15,18,22,27,30,35,37,39–44 metal–organic vapor phase epitaxy,1,45–48 mist CVD,16,17,32,33,49,50 plasma-assisted molecular beam epitaxy,51,52 laser molecular beam epitaxy,21,53 and pulsed laser deposition.12,13,24,54–57
1.1 Crystal structure
Some ambiguity was related to the real crystal structure of the κ or ε polymorph in the past. An initial XRD study investigated a 3 μm MOCVD-grown thin film on (0001)-Al2O3 and hinted at a hexagonal symmetry ascribed to the P63mc space group,27 usually referred to as ε-Ga2O3. A subsequent TEM study1 found the probed film to consist of 5–10 nm large (110)-twinned domains with orthorhombic structure corresponding to the space group symmetry Pna21 (Schoenflies: C92v), called κ-Ga2O3. Within the c-plane the individual domains are rotated 120° with respect to each other, forming a pseudo-hexagonal pattern observed in the initial XRD study, whose limited resolution provided a mediated view and did not allow to detect the individual domains. Newer XRD1,12,24,49,51 and TEM24,51 analyses confirmed the orthorhombic nature of the/polymorph. To avoid confusions we will, throughout this work, relate to κ when discussing the orthorhombic polymorph.Fig. 1 depicts the unit cell of orthorhombic κ-Ga2O3 with lattice parameters10a = 5.0566 Å, b = 8.6867 Å and c = 9.3035 Å. The oxygen atoms order in a 4H (ABAC) close-packed stacking, with Ga3+ ions occupying tetrahedral (GaI) and octahedral (GaII, GaIII, GaIV) sites.1
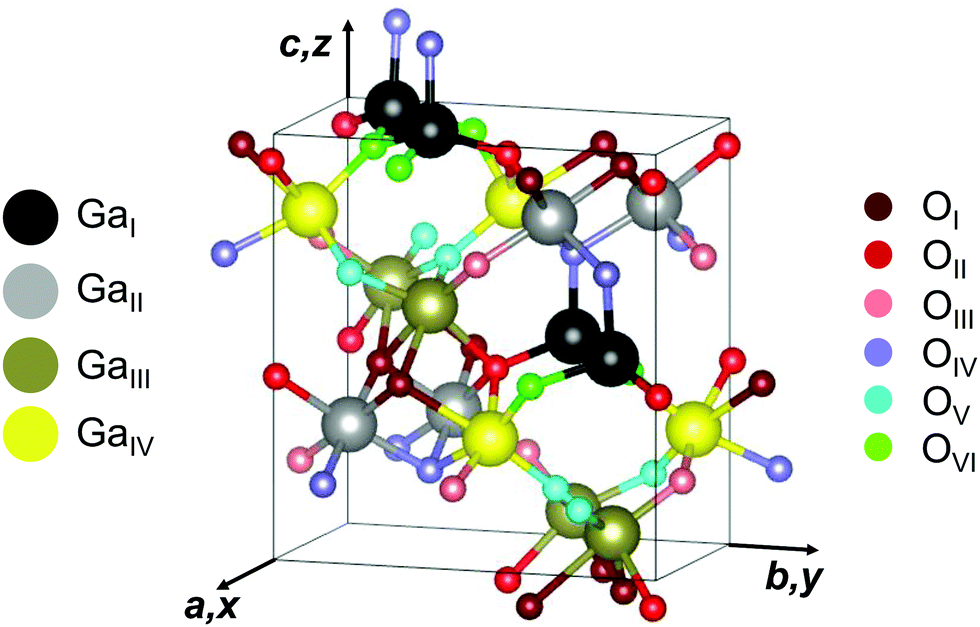 | ||
| Fig. 1 Unit cell of orthorhombic κ-Ga2O3. Four types of Ga (GaI,GaII, GaIII, and GaIV) and six types of O (OI, OII, OIII, OIV, OV and OVI) lattice sites are illustrated in different colors. The axes of a Cartesian coordinate system x, y, z are aligned along the crystallographic directions a, b and c. The crystallographic plot was created using VESTA.58 | ||
1.2 Lattice dynamics
Raman spectroscopy enables the study of vibrational properties by providing access to a material's Raman-active phonon modes. Raman-active phonons of different vibrational symmetries can be discriminated utilizing different polarization geometries. While the Raman- and IR-active phonons of β-Ga2O3 have been calculated theoretically and verified experimentally in a number of publications,59–64 comprehensive Raman spectra of κ-Ga2O3 are not yet available in the literature. One previous work65 investigated the IR- and Raman-active phonons of orthorhombic gallium-substituted epsilon iron oxide (κ/ε-Ga0.5Fe1.5O3) by first-principles phonon-mode calculations. The authors probed a powder sample of κ/ε-Ga0.48Fe1.52O3 nanoparticles using Raman spectroscopy and experimentally identified 17 distinct Raman-active phonon modes. Another experimental study51 employed Raman spectroscopy in a confocal set-up to investigate the Raman spectra of a κ-Ga2O3 thin film deposited on α-Al2O3(0001). The obtained spectra were dominated by the Raman lines of the underlying sapphire substrate, making the identification of Ga2O3 related Raman modes a challenging endeavor. The authors identified eight Raman modes associated with the κ-Ga2O3 thin film and provided an initial correlation to the mode symmetries predicted by the orthorhombic (Pna21) or hexagonal (P63mc) crystal structures. An alternative to overcome the challenges posed by the strong substrate signal in the confocal geometry is the employment of cross-sectional Raman spectroscopy, in which the laser is focused onto the sample's edge to maximize or minimize the signal contributions from the film or substrate, respectively. This approach was successfully applied in a preceding study63, in which the edge of a square-shaped (010)-oriented homoepitaxially grown β-Ga2O3 thin film was irradiated to determine the Raman modes associated with it.1.3 This work
In this work, a combination of confocal and cross-sectional polarized micro-Raman spectroscopy is applied to investigate the phonon frequencies of an MBE-grown orthorhombic κ-Ga2O3 thin film on top of an α-Al2O3(0001) substrate. The acquired experimental data are complemented with the results of DFPT calculations. By applying different polarization geometries in combination with Raman selection rules, we separate Raman modes of different vibrational symmetries and present the first comprehensive Raman spectra of κ-Ga2O3. We provide a detailed table of all calculated and more than 90 experimentally determined phonon frequencies and identify the symmetries of most experimental modes by combination of polarized measurements and ab initio calculations. The identified modes' vibrational symmetries are verified using angular-resolved Raman measurements. We provide an analytical tool to treat the effect of the domain structure on the Raman spectra, paving the way to discriminating domain structured from monocrystalline films in future studies.2 Experimental and theoretical methods
The investigated κ-Ga2O3 layer was deposited by molecular beam epitaxy via indium-mediated metal-exchange catalysis (MEXCAT-MBE)51,52 on a c-plane sapphire substrate. Differently from the homoepitaxial growth on β-Ga2O3 substrates,66–69 the MEXCAT-MBE (In- or Sn- mediated) in heteroepitaxy results in the stabilization of the metastable orthorhombic polymorph of Ga2O3. A Ga and In flux were provided with beam equivalent pressure of 6.5 × 10−7 mbar and 7.6 × 10−7 mbar, respectively. At first, an approximately 10 nm-thick β-Ga2O3 nucleation layer (usually necessary for the heteroepitaxial growth of κ-Ga2O3 in MBE) was deposited without providing the In flux, at a substrate temperature Tg = 600 °C and an oxygen flux of 1 standard cubic centimeters (sccm) with a 300 W plasma power for 1 minute. The In-mediated κ-layer was then deposited with In and Ga-fluxes present in an O-flux of 0.75 sccm (P = 300 W) at Tg = 700 °C for 210 minutes. The thickness of the κ-layer (monitored by in situ laser reflectometry) is around 800 nm.The symmetric out-of-plane 2θ–ω XRD scan reported in Fig. 2(a) of the sample highlights the presence of an (001)-oriented orthorhombic κ-Ga2O3 layer; the left-hand side peaks to the κ ones are related to the β-Ga2O3 nucleation layer52. The layer is composed of orthorhombic κ-Ga2O3 domains that are rotated in-plane by 120° with respect to each other.1 This can be unveiled by performing ϕ-scans on particular reflexes, as in the case of the 204 one reported in Fig. 2(b): the observed six peaks (red curve) indicate the occurrence of three rotational domains in the orthorhombic structure49. The good crystal quality of this layer is also proven by the relatively small full width at half maximum of the 004 reflection in ω-scan (FWHM = 0.14°, cf. Fig. 1(f) in a preceding study70). High-resolution 2θ–ω XRD scans of the sample (not shown) were performed for the symmetric 004 as well as the skew-symmetric 131 and 202 reflexes. The corresponding diffraction angles 2θ = 38.855° as well as 37.011° and 40.784°, respectively, allowed us to estimate the lattice parameters of the orthorhombic layer. The results (shown in Table 1) are consistent with previous experimental and theoretical studies.
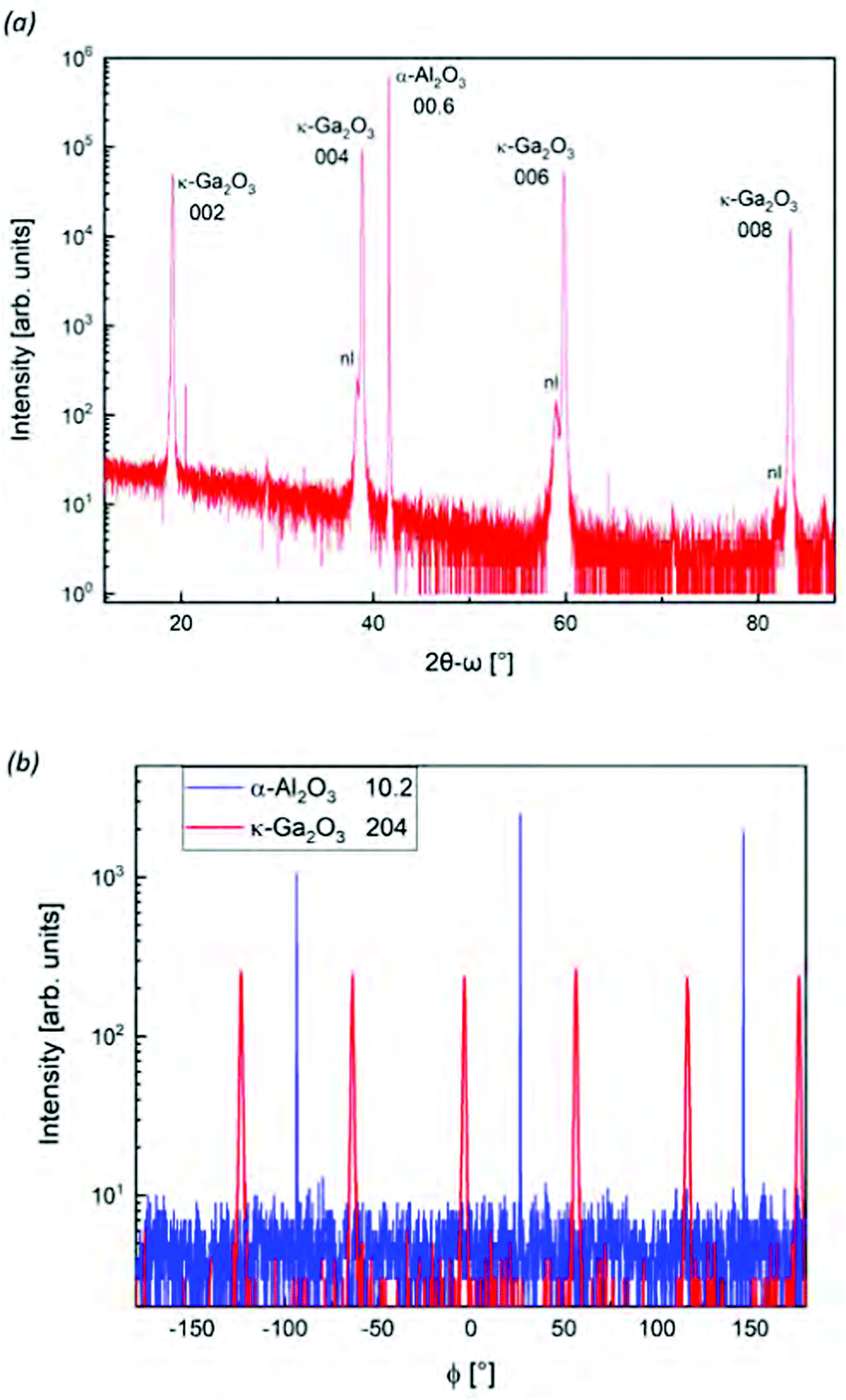 | ||
| Fig. 2 XRD (a) 2θ−ω and (b) ϕ-scan of the κ-Ga2O3 investigated sample. In (a) the label nl stands for nucleation layer of β-Ga2O3. Reflexes of the Al2O3 substrate are given using the reduced three-digit notation.12 | ||
Raman scattering at room temperature (293 K) was excited by a 532.16 nm frequency stabilized single longitudinal mode diode-pumped solid-state (DPSS) laser (Laser Quantum Torus 532) on a LabRAM HR 800 spectrometer (Horiba Jobin-Yvon). The laser beam was focused onto the sample using a 100× Olympus objective with a numerical aperture (NA) of 0.9, with the scattered light being collected in backscattering geometry. Backreflected and elastically scattered light (Rayleigh component) was filtered using an ultra low frequency filter (ULF) unit and then spectrally-dispersed by a monochromator with a grating of 1800 lines per mm. The light was detected by a charge-coupled device (CCD). The sample was placed beneath the objective with a respective surface's normal parallel to the direction of light propagation. A λ/2 wave plate in the excitation was set at 0° or 45° to polarize the incident light parallel or crossed with respect to the scattered light, which was selected using a fixed polarizer in the detection. Prior to each measurement, the Raman spectrometer was calibrated using the spectral lines of a neon spectral lamp.
Angular-resolved Raman scans were performed by incorporating a second λ/2 wave plate in the tube above the objective. The automatized rotation of the wave plate by an angle ϕ/2 rotated the polarization of both the incident and scattered light by an angle ϕ with respect to the fixed sample, constituting a ϕ virtual rotation of the investigated sample.
The theoretical phonon spectra were simulated within the frame of density functional perturbation theory (DFPT) on the level of the local density approximation (LDA) as implemented in the QUANTUM ESPRESSO suite.71 The Ga(3d, 4s, 4p) and the O(2s, 2p) states were treated as valence electrons using multiprojector optimized normconserving Vanderbildt (ONCV) pseudopotentials from the sg15 library.72 The electronic wavefunctions were expanded in a planewave basis with a cutoff energy of 100 Ry. All reciprocal space integrations were performed by a discrete Monkhorst–Pack sampling of 6 × 4 × 4 k-points in the Brillouin zone. We fully optimized the atomic positions and cell parameters until the residual forces between atoms and the cell stress were smaller than 0.001 eV Å−1 and 0.01 GPa, respectively. The atomic positions and lattice parameter for the initial geometry before and after the structure optimization are provided in the ESI.† The threshold for the total energy was set to 10−14 Ry, which ensured tightly converged interatomic forces for the geometry optimization and of the ground state density and wave functions for the DFPT calculations. The phonon dispersion and the density-of-states (Fig. S2 in the ESI†) was obtained through Fourier interpolation using the explicitly calculated phonon frequencies on a regular grid of 6 × 4 × 4 q-vectors. As a result of the intrinsic underbinding of the LDA exchange–correlation functional, our optimized lattice constants are less than 1% underestimated compared to the experimental values (Table 1). On the other hand, it is well-known that the LDA overbinding gives rise to a typically very good agreement of the DFT phonon frequencies,73 motivating our choice for the purposes of this work.
3 Results and discussion
3.1 Polarization dependence of monocrystalline orthorhombic crystals
The primitive unit cell of the orthorhombic κ structure consists of 40 atoms (4 Ga and 6 O atoms, each fourfold degenerate).10 At the Γ-point, these correspond to 117 optical phonons:| Γopt = 29A1 + 30A2 + 29B1 + 29B2 |
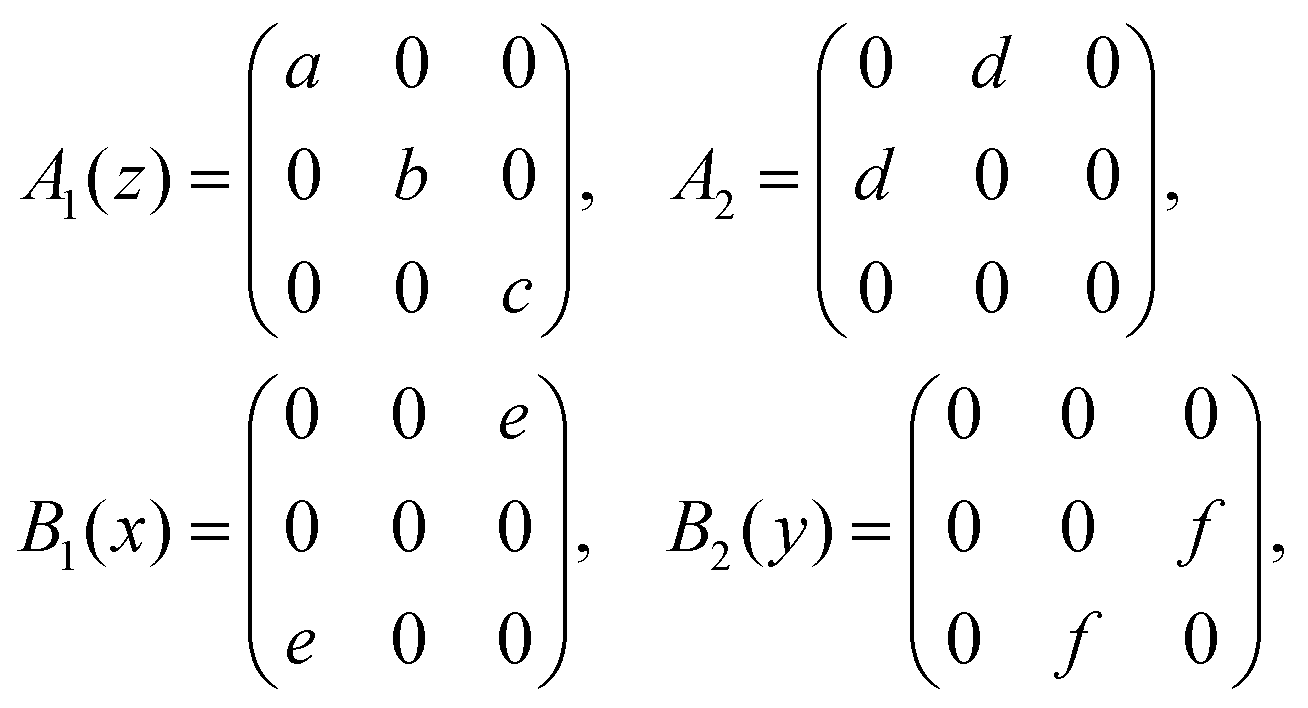 | (1) |
Is ∝ |![[e with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0065_20d1.gif) i· i·![[scr R, script letter R]](https://www.rsc.org/images/entities/char_e531.gif) ·s|2. ·s|2. | (2) |
denotes the second-rank Raman tensor and i or s are the polarization vectors of the incident or scattered light, respectively. Scattering geometries are expressed using the Porto notation ![[k with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_006b_20d1.gif) i(is)s, where i and s point into the propagation direction of the incident or scattered light. Polar phonons induce an oscillating macroscopic electric field in the direction of atomic displacements, leading to a splitting of A1, B1 and B2 modes into transverse-optical (TO) and longitudinal-optical (LO) phonons. The non-polar A2 modes, in turn, exhibit no LO–TO-splitting.
i(is)s, where i and s point into the propagation direction of the incident or scattered light. Polar phonons induce an oscillating macroscopic electric field in the direction of atomic displacements, leading to a splitting of A1, B1 and B2 modes into transverse-optical (TO) and longitudinal-optical (LO) phonons. The non-polar A2 modes, in turn, exhibit no LO–TO-splitting.
Raman selection rules of single-crystalline κ-Ga2O3 are summarized in Table 2: modes of A1(TO) symmetry are allowed for parallel polarization vectors when probing the (100) and (010) planes in backscattering configuration. For crossed polarization, the B1(TO) modes are visible exclusively on the (010) plane whereas B2(TO) modes are allowed on the (100) plane. Conversely, A1(LO) and A2 modes can be accessed by probing the (001) facet in parallel and crossed polarization, respectively. B1(LO) and B2(LO) modes are not allowed in backscattering geometry.
i or s directions, with i and s being the respective polarization direction. Modes not listed have vanishing intensities in the respective polarization configuration. a, b and c denote the crystallographic directions, with LO and TO abbreviating longitudinal or transverse optical phonons
| Plane | Polarization i(is)s |
Allowed modes |
|---|---|---|
| (100) | a(bb)ā, a(cc)ā | A 1(TO) |
| a(bc)ā, a(cb)ā | B 2(TO) | |
| (010) |
b(aa)![[b with combining macron]](https://www.rsc.org/images/entities/i_char_0062_0304.gif) , b(cc) , b(cc) |
A 1(TO) |
|
b(ac), b(ca) |
B 1(TO) | |
| (001) |
c(aa)![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) , c(bb) , c(bb) |
A 1(LO) |
|
c(ab), c(ba) |
A 2 |
In angular-resolved Raman measurements, the polarization vectors of both the incident i and scattered s light are rotated by an angle ϕ relative to the sample's edge, yielding an angular-dependent intensity variation. Mathematically, the polarization vectors' rotations in the plane of incidence are expressed by parameterizing i and s using cylindrical coordinates. Substituting the parameterized polarization vectors i and s into eqn (2) yields angular-dependent intensity functions for an orthorhombic crystal.
As an example, when exciting the (100) plane of monocrystalline orthorhombic κ-Ga2O3 using a parallel polarization of the incident with respect to the scattered light, the polarization vectors become
 | (3) |
A 1(TO) modes in parallel (‖) polarization on the (100) plane are thus predicted to obey the intensity function
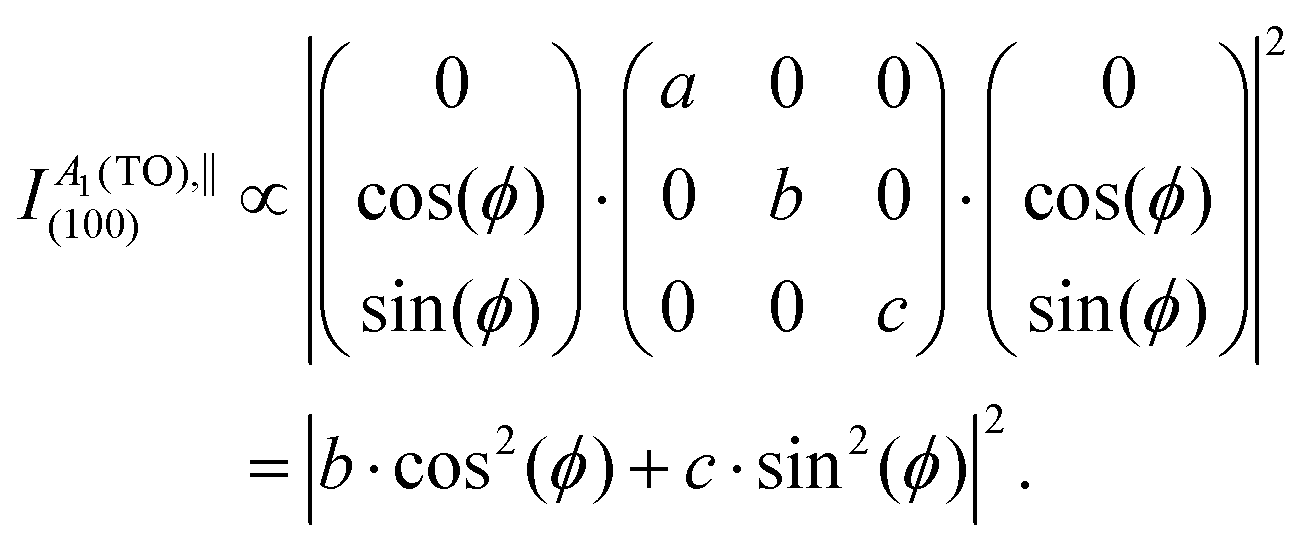 | (4) |
This approach is, strictly speaking, only applicable for isotropic crystals. A complete derivation of angular intensity functions for anisotropic crystals would necessitate a description in the framework of an extended Raman tensor formalism for anisotropic crystals as proposed and applied to β-Ga2O3 in a pair of previous studies.58,75 For the presently available κ-Ga2O3 samples the domain structure and comparatively large phonon linewidths paired with the overlapping of Raman modes inhibit a quantitative analysis of Raman tensor elements, allowing us to limit our analysis to the common Raman tensor formalism. Table 3 summarizes the intensity functions expected for monocrystalline κ-Ga2O3 on the basis of the common Raman tensor formalism. The quantitative values of the Raman tensor elements of all 117 optical phonon modes calculated by DFPT are listed in Tables S1–S3 in the ESI.†
i and s into eqn (2). ‖ or ⊥ denote a parallel or crossed polarization of the incident light with respect to the scattered. ϕ designates the rotational angle of the polarization vectors with respect to the sample. a, b, c, d, e and f are the Raman tensor elements, with LO and TO abbreviating longitudinal or transverse optical phonons. Modes not listed have vanishing intensities in the respective polarization configuration
| Plane | Phonon mode | ‖ | ⊥ |
|---|---|---|---|
| (100) | A 1(TO) | |b·cos2(ϕ) + c·sin2(ϕ)|2 | |(c − b)·sin(ϕ)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos(ϕ)|2 cos(ϕ)|2 |
| B 2(TO) | f 2·sin2(2ϕ) | f 2·cos2(2ϕ) | |
| (010) | A 1(TO) | |a·cos2(ϕ) + c·sin2(ϕ)|2 | |(c − a)·sin(ϕ)cos(ϕ)|2 |
| B 1(TO) | e 2·sin2(2ϕ) | e 2·cos2 (2ϕ) | |
| (001) | A 1(LO) | |a·cos2(ϕ) + b·sin2(ϕ)|2 | |(b − a)·sin(ϕ)cos(ϕ)|2 |
| A 2 | d 2·sin2(2ϕ) | d 2·cos2 (2ϕ) |
For excitation on the (100) or (010) facets, B1(TO) and B2(TO) angular mode intensities should be characterized by equally-intense maxima, whereas the linear combination of cos2(ϕ) and sin2(ϕ) functions may give rise to distinctively intense maxima for A1(TO) modes, enabling to distinguish A1(TO) from B1(TO)/B2(TO) modes on the basis of their angular intensity variations. Modes of A1(LO) and A2 symmetries may be distinguished analogously when exciting the (001) plane.
3.2 Polarized Raman spectra of the investigated orthorhombic thin film containing rotational domains
The Raman-active phonon modes of different vibrational symmetries are separated by measuring in the geometries illustrated in Fig. 3.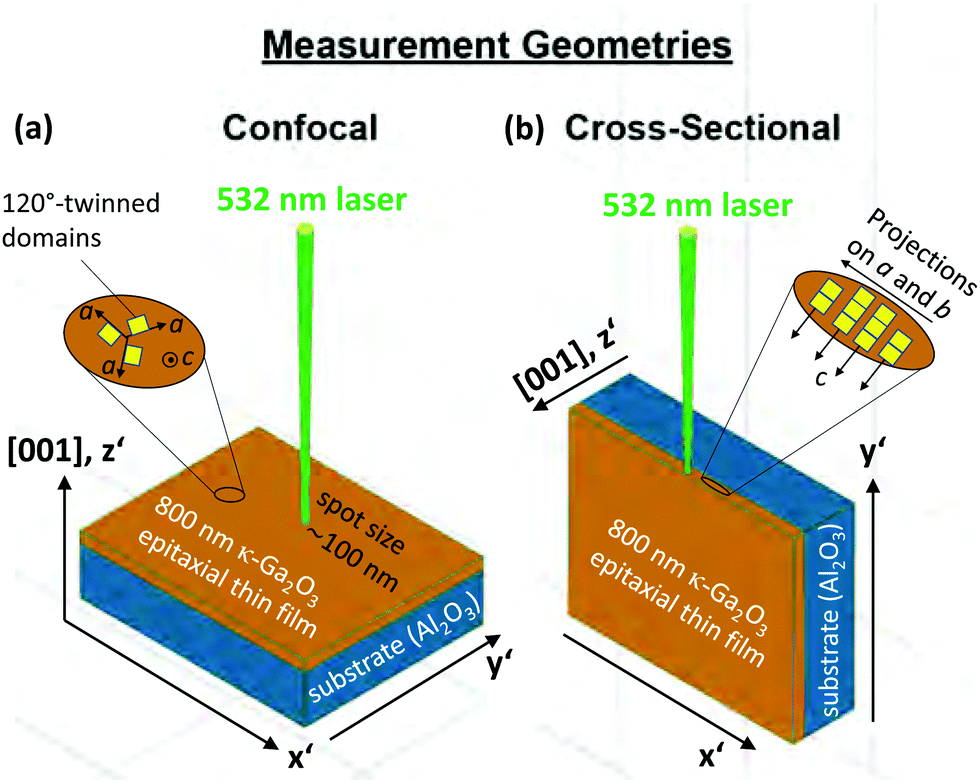 | ||
| Fig. 3 Schematic illustration of sample setup, plane orientation, and measurement geometry. The (001) plane was accessible in (a) confocal micro-Raman scattering. The z′ and x′, y′ directions of a Cartesian coordinate system are oriented perpendicular to the (001) plane and parallel to the edges. Spectra recorded in a (b) cross-sectional set-up are acquired by focusing the laser onto the edge (right). The insets indicate (a) the 120° rotation of orthorhombic domains around the c-axis or (b) the columnar c-oriented structure perpendicular to the (001) plane. | ||
The sample is irradiated in a confocal (a) and cross-sectional (b) setup to access the (001) plane or the edge, respectively. The choice of the (001) plane or the edge is advantageous as it enables the selected detection of Raman modes with different vibrational symmetries and thus facilitates the separation of modes with closely matching phonon frequencies.
In the confocal measurements (Fig. 3a), the microscope's z-focus was moved to maximize or minimize the signal contribution from the thin film or sapphire substrate, respectively. For the cross-sectional configuration (Fig. 3b), we performed line scans with 200 nm step size to identify a position for long integration Raman measurements, for which the κ-Ga2O3 or Al2O3 related Raman modes reach maximum or minimum intensities.
Though the domain structure, characterized by the 120° rotation of orthorhombic domains within the (001)-plane, inhibits the direct application of monocrystalline selection rules, our analysis indicates that selection rules can, with certain constraints, be utilized to account for the observed Raman spectra in Fig. 4a–d.
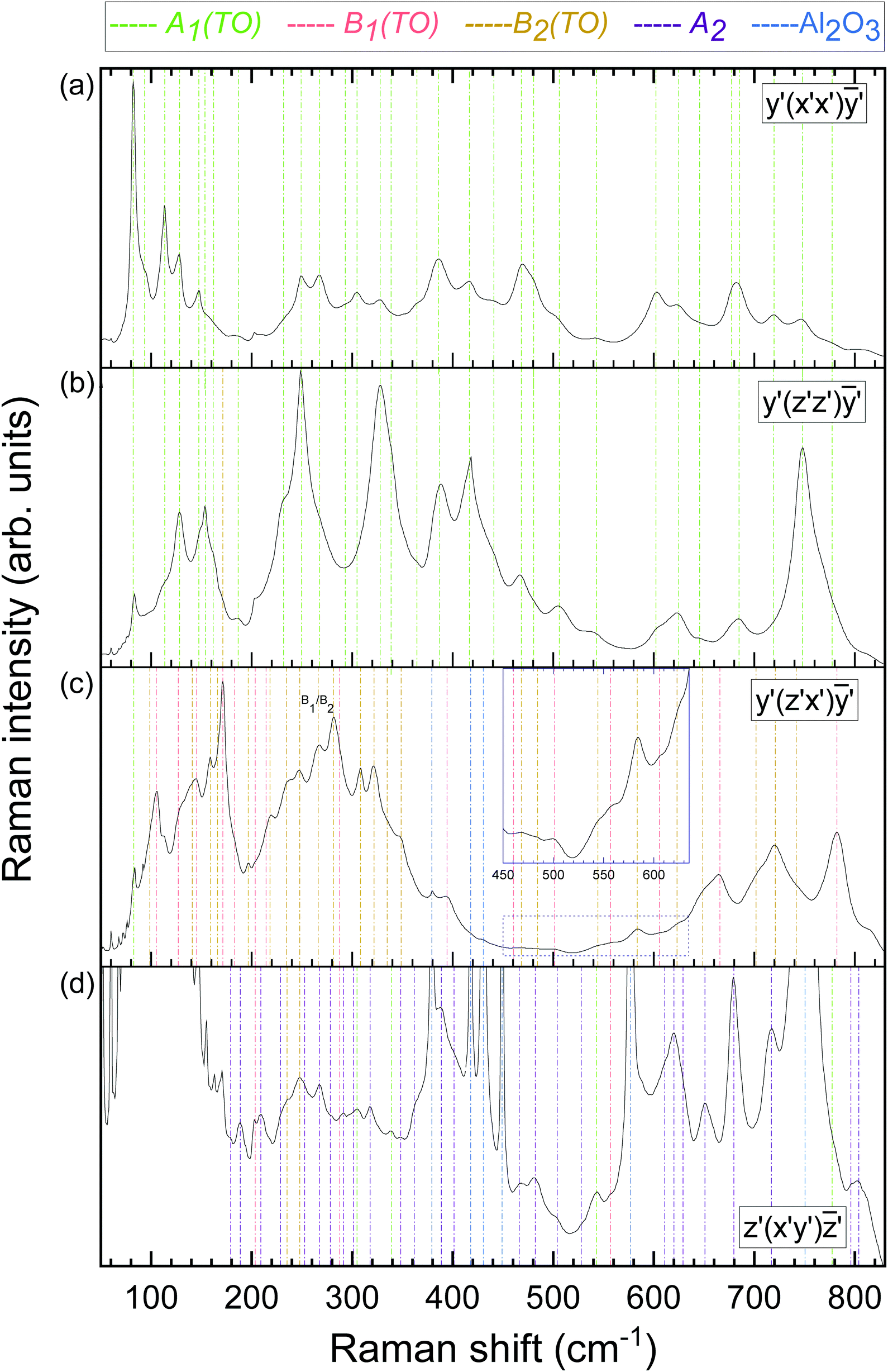 | ||
| Fig. 4 Raman spectra of the MBE-grown κ-Ga2O3 thin film on (0001)-oriented Al2O3. (a) Raman spectra in parallel polarization for excitation on the edge. The Cartesian coordinate system x′, y′, z′ is chosen such that z′-axis aligns with the [001] direction, with the x′ and y′ directions oriented according to Fig. 3. In the y′(x′x′)ȳ′ configuration, the polarization vectors are aligned along the excited edge. Mode symmetries of Raman peaks associated with the κ-Ga2O3 thin film or sapphire substrate are indicated by vertical dashed lines. A respective line's color denotes the vibrational mode symmetry as provided on top of the graphs. (b) Raman spectra in parallel polarization with polarization vectors aligned along the z′‖c axis. (c) Raman spectra of the edge in crossed polarization, with the incident or scattered light aligning with the [001] or x′ directions. The inset depicts the spectrum between 450 and 640 cm−1 with magnified y-scale. B1(TO) and B2(TO) modes are labeled based on their correlation to the calculated Raman frequencies as explained in the text. (d) Confocal Raman spectra in crossed polarization. The spectrum is multiplied by a constant factor to magnify low-intensity modes. | ||
Owing to the orthorhombic domains being rotated 120° against each other, the orientations of the crystallographic a and b directions within the (001)-plane are not well-defined (cf.Fig. 3a). As a result, the sample's edges do not coincide with the a or b axes (cf.Fig. 2), yet are orthogonal to the [001] direction (c-axis). Moreover, as the a and b axes are perpendicular to each other, each edge represents a linear superposition of these two crystallographic directions. Thus, by exciting the edge and selecting the polarization vectors parallel to it (scattering geometry y′(x′x′)ȳ′ or (x′x′) in short), the observed Raman spectrum in Fig. 4a corresponds to neither the monocrystalline orthorhombic (aa) nor (bb) configurations, but a superposition (aa + bb). As predicted by selection rules (cf.Table 2), solely A1(TO) modes are allowed in this configuration. Identical Raman spectra are obtained by probing the other edges of the sample with polarization vectors parallel to the respective edge. For this reason, we restricted the subsequent measurements to the edge exhibiting the strongest signal intensity.
The y′(z′z′)ȳ′ configuration (Fig. 4b) is obtained by rotating the polarization vectors by 90°. As the polarization vector z′ coincides with the crystallographic c-direction (cf.Fig. 3), the recorded spectrum should be correlated with the orthorhombic (cc) spectrum, for which all but A1(TO) modes are prohibited.
In the crossed polarization configuration y′(z′x′)ȳ′ (Fig. 4c) the polarization vectors i and s align with the z′‖c and x′ directions, respectively. With the x′ axis being parallel to neither a nor b, the observed Raman spectrum contains contributions from the orthorhombic (ac) and (bc) polarization geometries. The occurring modes are hence of B1(TO) and B2(TO) symmetry.
In the confocal geometry utilized to probe the (001) plane (Fig. 4d), the polarization vectors are oriented perpendicular to the c-axis, hence lie within the c-plane. As a result, the parallel polarized confocal Raman spectra nearly replicated the (x′x′) Raman spectrum in Fig. 4a, revealing no additional Raman modes. Switching the polarization vectors from parallel to crossed led to a suppression of the Raman modes dominant in the (x′x′) configuration, providing access to closely-matching modes or modes residing in the flanks of their more intense neighbors. Despite the recorded Raman spectrum in Fig. 4d not being strictly (ab), the majority of the occurring modes was not observed in the preceding spectra, suggesting this experimental spectrum to be dominated by modes of A2 symmetry. Intriguingly, no directional dependence is observed when rotating the sample around its surface normal (i.e. the c-direction) with respect to a fixed polarization, i.e. either parallel or crossed. The absence of any polarization dependence suggests a quasi isotropic behavior, the reasons of which are discussed in detail in Section 3.3.
The spectral positions of the occurring Raman modes in Fig. 4a–d are derived by fitting Lorentzian lineshape functions, with the obtained peak positions listed in Table 4. For each measurement configuration, we compare the experimental Raman spectra with theoretical spectra obtained by the DFPT calculations to correlate experimentally observed and calculated phonon frequencies (summarized in Table 4). For practical reasons, an LO–TO phonon splitting was neglected in the calculations. We further list modes which are solely Raman-active (R) or both infrared- and Raman-active (I + R).
| Frequency (exp.) (cm−1) | Frequency (theo.) (cm−1) | Mode symmetry | Mode activity | Frequency (exp.) (cm−1) | Frequency (theo.) (cm−1) | Mode symmetry | Mode activity |
|---|---|---|---|---|---|---|---|
| 82.4 | 73.5 | A 1 | I + R | 364.0 | 348.4 | A 1 | I + R |
| 93.3 | A 1 † | 361.7 | 364.5 | A 2 | R | ||
| 82.4 | A 2 | R | 370.4 | B 1 | I + R | ||
| 98.7 | 88.8 | B 2 | I + R | 385.5 | 371.9 | A 1 | I + R |
| 105.3 | 90.3 | B 1 | I + R | 394.4 | 386.2 | B 1 | I + R |
| 102.1 | A 2 | R | 416.3 | 386.4 | A 1 | I + R | |
| 113.4 | 106.2 | A 1 | I + R | 395.8 | B 2 | I + R | |
| 127.1 | 118.7 | B 1 | I + R | 389.4 | 396.8 | A 2 | R |
| 128.0 | 119.2 | A 1 | I + R | 398.0 | B 1 | I + R | |
| 123.2 | A 2 | R | 440.5 | 411.9 | A 1 | I + R | |
| 140.7 | 135.0 | B 2 | I + R | 415.6 | B 2 | I + R | |
| 147.1 | 140.6 | A 1 | I + R | 401.2 | 420.8 | A 2 | R |
| 145.1 | 141.1 | B 1 | I + R | 422.4 | B 2 | I + R | |
| 153.7 | 149.3 | A 1 | I + R | 466.2 | 439.6 | A 2 | R |
| 149.7 | A 2 | R | 460.5 | 447.6 | B 1 | I + R | |
| 158.7 | 151.0 | B 2 | I + R | 468.3 | 454.8 | A 1 | I + R |
| 161.0 | A 2 | R | 482.2 | 462.6 | A 2 | R | |
| 162.5 | B 2 | I + R | 480.3 | 464.3 | A 1 | I + R | |
| 162.0 | 165.6 | A 1 | I + R | 468.4 | 468.5 | B 2 | I + R |
| 165.9 | 166.1 | B 2 | I + R | 503.9 | 486.3 | A 2 | R |
| 171.4 | 167.7 | B 1 | I + R | 484.2 | 491.2 | B 2 | I + R |
| 179.0 | 171.6 | A 2 | R | 504.7 | 497.0 | A 1 | I + R |
| 186.6 | 183.0 | A 1 | I + R | 501.8 | 498.3 | B 1 | I + R |
| 188.8 | 183.7 | A 2 | R | 544.4 | 530.5 | B 2 | I + R |
| 182.4 | 191.2 | B 1 | I + R | 533.2 | B 1 | I + R | |
| 209.2 | 197.5 | A 2 | R | 527.5 | 540.1 | A 2 | R |
| 196.5 | 204.6 | B 2 | I + R | 542.8 | 541.6 | A 1 | I + R |
| 206.1 | B 1 | I + R | 557.8 | 553.5 | B 1 | I + R | |
| 214.5 | 213.6 | B 1 | I + R | 583.4 | 577.4 | B 2 | I + R |
| 230.7 | 220.1 | A 1 | I + R | 610.8 | 583.0 | A 2 | R |
| 218.3 | 221.1 | B 2 | I + R | 602.0 | 595.8 | A 1 | I + R |
| 228.4 | 223.8 | A 2 | R | 619.6 | 610.1 | A 2 | R |
| 234.7 | 225.7 | B 2 | I + R | 605.7 | 610.9 | B 1 | I + R |
| 227.6 | B 1 | I + R | 623.2 | 615.6 | B 2 | I + R | |
| 248.9 | 228.6 | A 1 | I + R | 624.6 | 616.6 | A 1 | I + R |
| 241.3 | B 1 | I + R | 629.2 | 617.5 | A 2 | R | |
| 247.8 | 243.9 | B 2 | I + R | 648.7 | 631.2 | B 2 | I + R |
| 252.5 | 245.5 | A 2 | R | 645.4 | 643.5 | A 1 | I + R |
| 252.0 | B 1 | I + R | 650.2 | 648.3 | A 2 | R | |
| 267.4 | 257.0 | A 1 | I + R | 658.6 | 653.8 | B 1 | I + R |
| 259.5 | A 2 | R | 653.9 | B 2 | I + R | ||
| 266.0 | 263.3 | B 1 | I + R | 665.7 | 656.6 | B 1 | I + R |
| 279.3 | 265.7 | A 2 | R | 676.6 | 667.9 | A 1 | I + R |
| 266.0 | 270.8 | B 2 | I + R | 685.2 | 676.3 | A 1 | I + R |
| 281.2 | 278.3 | B 2 | I + R | 677.6 | B 1 | I + R | |
| 291.3 | A 2 † | 701.7 | 684.9 | B 2 | I + R | ||
| 293.4 | 282.0 | A 1 | I + R | 685.2 | A 2 | R | |
| 287.4 | 284.7 | B 1 | I + R | 689.7 | B 1 | I + R | |
| 289.3 | B 1 | I + R | 693.4 | B 2 | I + R | ||
| 308.4 | 291.5 | B 2 | I + R | 704.4 | B 1 | I + R | |
| 301.4 | 298.6 | A 2 | R | 719.3 | 705.0 | A 1 | I + R |
| 304.5 | 302.7 | A 1 | I + R | 679.3 | 705.6 | A 2 | R |
| 318.0 | 303.4 | A 2 | R | 721.0 | 708.2 | B 2 | I + R |
| 327.9 | 311.4 | A 1 | I + R | 715.8 | 715.0 | A 2 | R |
| 315.4 | B 1 | I + R | 747.5 | 733.1 | A 1 | I + R | |
| 348.2 | 320.0 | A 2 | R | 741.8 | 733.4 | B 2 | I + R |
| 321.5 | 320.1 | B 2 | I + R | 777.5 | A 1 † | ||
| 338.9 | 329.7 | A 1 | I + R | 796.9 or 803.7 | 733.8 | A 2 | R |
| 334.5 | 331.5 | B 2 | I + R | 782.3 | 764.6 | B 1 | I + R |
| 342.7 | B 1 | I + R | 815.9 | ||||
| 348.8 | 343.2 | B 2 | I + R |
As an example of allocating experimental and theoretical modes, we first consider the measured (x′x′) spectrum in Fig. 4a. As pointed out above, the (x′x′) spectrum corresponds to the superimposed calculated orthorhombic (aa + bb) spectrum. Fig. 5 shows a direct comparison of the experimentally and theoretically obtained spectra in this configuration. Calculated phonon frequencies and intensities are indicated by vertical magenta lines. The theoretical spectrum is generated by applying a line broadening of 10 cm−1 with Lorentzian line shape to facilitate a direct comparison with the experimental spectrum in Fig. 4a. A good qualitative agreement between experimental and calculated spectra is evident. As a general observation, the obtained peak positions are slightly underestimated by the calculations for the majority of the modes. A compilation of all measured Raman spectra shown in conjunction with their theoretical analogues are presented in Fig. S1 in the ESI.†
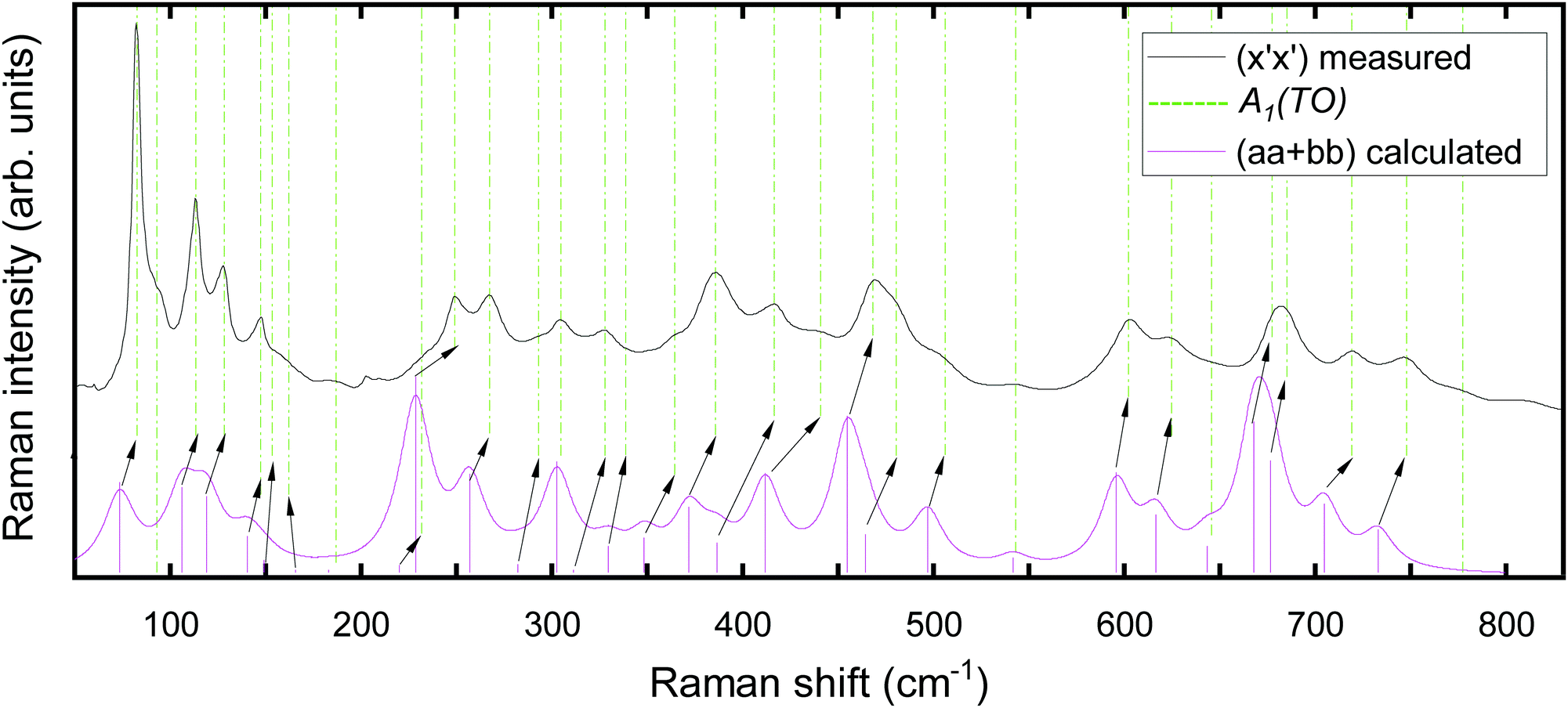 | ||
| Fig. 5 Raman spectra of the MBE-grown κ-Ga2O3 thin film on (0001)-oriented α-Al2O3. The measured (black) spectrum in the y′(x′x′)ȳ′ configuration is shown in conjunction with the calculated (magenta) Raman spectrum and peak intensities (magenta bars) for the orthorhombic (aa + bb) configuration. For clarity, spectra are normalized and vertically offset. The Cartesian coordinate system x′, y′, z′ is chosen according to Fig. 3. Experimental Raman modes of A1(TO) symmetry (indicated by green dashed vertical lines) are correlated with their calculated (magenta bars) counterparts. Where necessary the correlation is indicated using arrows. The theoretical spectra were generated using the phonon frequencies and intensities based on a FWHM of 10 cm−1. | ||
The experimental Raman mode frequencies are allocated to their most likely theoretical counterparts on the basis of the calculated Raman spectra utilizing the following algorithm: (1) the symmetries of Raman modes allowed in a specific polarization geometry as predicted by selection rules are correlated with the theoretical modes of the same symmetry. (2) Given Fig. 5 or one of the spectra in Fig. S1a–d (ESI†), we presume the same energetic order for both experimental and theoretical modes. (3) Experimental frequencies are usually associated with their energetically closest matching theoretical counterparts. (4) If several theoretical modes are closely-matching and might be allocated to a certain experimental mode, the assignment is based on considering the best matching intensity of experimental and theoretical modes.
A few modes deserve particular attention, as the allocation of experimental and theoretical frequencies is challenging due to closely-matching or overlapping modes or experimental-theoretical analogues not being apparent. By linking the most intense experimental and theoretical modes in the (x′x′) (Fig. 5 and Fig. S1a, ESI†) and (z′z′) configurations (Fig. S1b, ESI†), we did not identify the theoretical analogues for the experimental modes observed at 93.3 cm−1 and 777.5 cm−1. Their presence in the applied polarization geometries suggests an A1(TO) vibrational symmetry as predicted by selection rules. A small peak observed at 202.6 cm−1 corresponds to the strained Ag3 mode originating from the β-Ga2O3 MBE nucleation layer.
Considering the (z′x′) spectra in Fig. 4c (or Fig. S1c, ESI†), the experimental mode at 82.4 cm−1 corresponds to the A1 mode previously recorded in Fig. 4a and b (or Fig. S1a and b, ESI†). Further spikes at frequencies lower than 82.4 cm−1 are related to neither κ/ε- nor β-Ga2O3 nor the Al2O3 substrate but originate from randomly scattered light being collected by the CCD camera. Moreover, the experimental mode at 266.0 cm−1 might relate to a pair of closely-spaced theoretical modes of symmetries B1 and B2. We therefore consider this experimental mode to be of symmetry B1(TO) or B2(TO) (labeled as B1/B2). An experimental mode at 815.9 cm−1 occurring in the (x′x′), (z′z′) and (z′x′) spectra cannot be linked to any theoretical frequency. As this mode seemingly does not obey Raman selection rules, it might originate from a second-order Raman scattering event.
With regard to the confocal measurements (Fig. 4d or Fig. S1d, ESI†), selection rules allow only modes of A2 symmetry in the (ab) configuration. The appearance of additional modes at energies already observed in Fig. 4a–c is attributed to a relaxation of selection rules due to the (i) orthorhombic domain structure paired with the (ii) utilized objective's large NA (0.9), conditioning the presence of A1(TO), B1(TO) and B2(TO) modes. All modes at frequencies not previously observed are attributed to an A2 vibrational symmetry and correlated to their respective theoretical counterpart. The experimental mode at 291.3 cm−1 can not be linked to any theoretical frequency. Since this modes is present only in the confocal configuration, we ascribe it to an A2 symmetry. The two closely-spaced experimental modes at 796.9 and 803.7 cm−1 might correspond to the theoretical A2 mode at 733.8 cm−1.
Our detailed analysis enables the determination of the spectral positions of more than 90 experimental Raman modes (summarized in Table 4), which we linked to the orthorhombic crystal structure's 117 theoretical frequencies with their respective vibrational symmetries.
The interplay of several orthorhombic domains produces a pseudo-hexagonal ε structure, associated with the P63mc space group, for which the primitive unit cell is expected to contain on average four O atoms and  Ga atoms,9 yielding 17 optical phonon modes. Even if all modes were Raman-active, the higher number of phonons obtained in the experimental spectra of the present study provides strong evidence that the observed Raman modes are not of hexagonal nature, but originate from the orthorhombic film with domains.
Ga atoms,9 yielding 17 optical phonon modes. Even if all modes were Raman-active, the higher number of phonons obtained in the experimental spectra of the present study provides strong evidence that the observed Raman modes are not of hexagonal nature, but originate from the orthorhombic film with domains.
3.3 Angular-resolved Raman scans, vibrational dynamics of Raman-active phonons, and the impact of rotational domains
From an experimental perspective, the symmetries of Raman-active phonon modes can be verified by applying angular-resolved Raman scans. By rotating the polarization vectors of both the incidenti and scattered s light by an angle ϕ relative to the sample's edge, the individual Raman modes generally experience an angular intensity variation. Using Lorentzian lineshape functions to model the individual Raman peaks, the intensities of the individual Raman modes are obtained as the amplitudes of the model fits. Fig. 6 illustrates the Raman intensities plotted against the rotational angle ϕ for a selection of A1(TO)-, B1(TO)-, B2(TO) and A2-modes acquired in cross-sectional or confocal measurements.
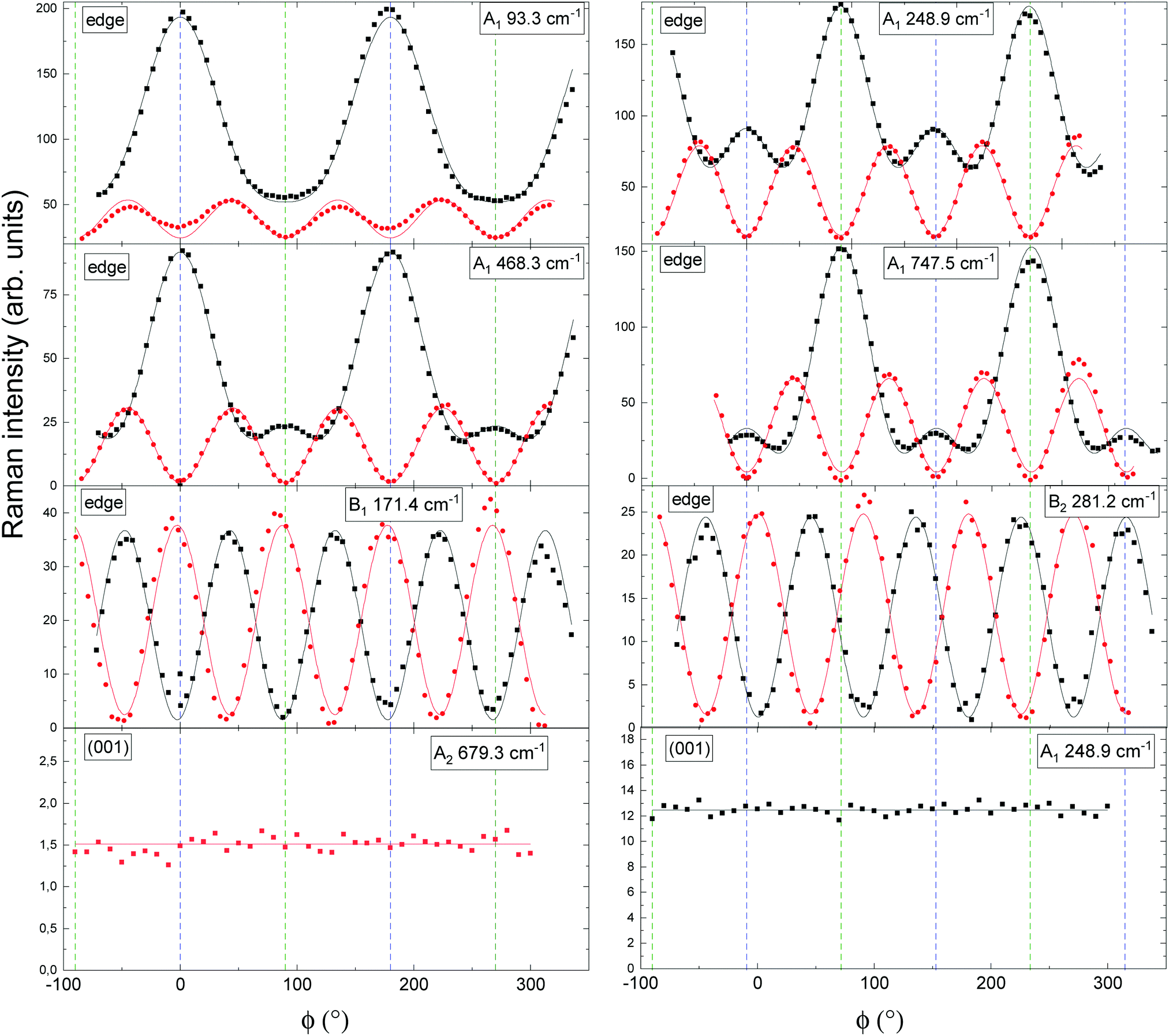 | ||
| Fig. 6 Angular-resolved Raman spectra of A1(TO)-, B1(TO)-, B2(TO)- and A2 modes in κ-Ga2O3 for cross-sectional or confocal excitation. Points and lines indicate experimental Raman scattering intensities and model fits. Fit functions are listed in Table 5. Black and red color denote parallel and crossed polarization of the incident light with respect to the detected light. The mode symmetry and experimental frequency of each mode is indicated in the top right of each graph. Cross-sectional or confocal measurements are indicated by “edge” or (001). ϕ = 0° is set such that it coincides with the x′ direction (cf.Fig. 3). In the cross-sectional geometry ϕ = 90° corresponds to the [001] direction, while in the confocal geometry it coincides with the y′ direction. ϕ = 0° and ϕ = 90° are indicated by vertical dashed blue and green lines. | ||
The selected Raman modes are spectrally well-separated (i.e. show little or no overlap with neighboring modes), have comparatively high Raman intensities or allow to identify the symmetries of experimental modes which could not be linked to any theoretical frequencies (e.g. the mode observed at 93.3 cm−1).
To model the experimental points, the angular fit formulae in Table 3 need to be modified to account for the domain structure. With regard to the measurements on the sample's edge, which corresponds to a linear combination of the (100) and (010) facets, the experimental Raman intensities of A1(TO) modes in Fig. 6 are modeled using a linear combination of the (100) and (010) monocrystalline intensity functions from Table 3. As an example, A1(TO) mode intensity functions in parallel (‖) polarization are thus modified to
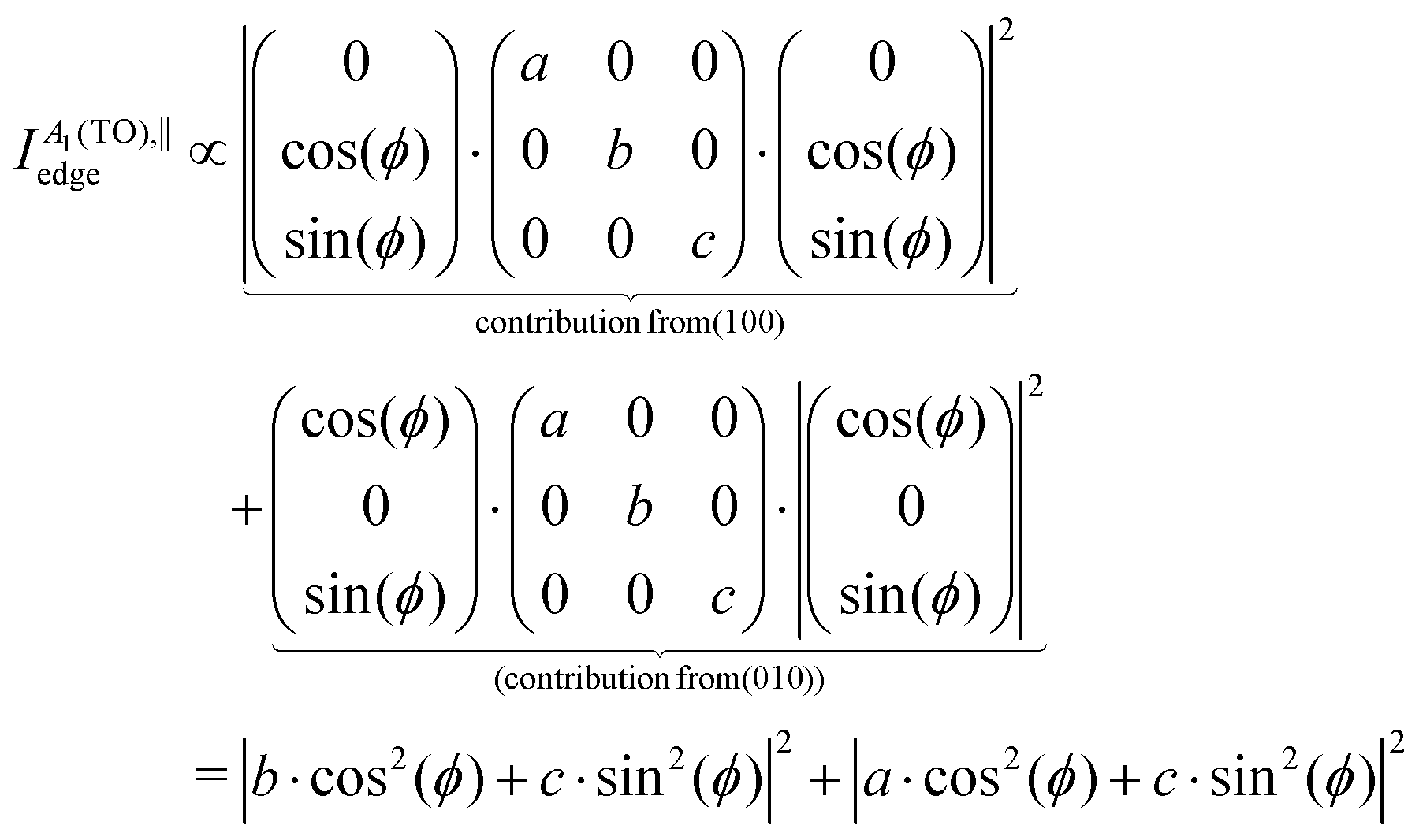 | (5) |
On the contrary, B2(TO) and B1(TO) modes occur solely on the (100) and (001) planes. Therefore, their intensity functions need not to be modified. Probing the (001) plane with a laser spot of several hundreds of nanometers in diameter size provides an averaged signal over a large number of orthorhombic domains rotated 120° against each other (cf.Fig. 3). As a result, the detected Raman signals originate from three differently oriented species of orthorhombic domains, each species i contributing a fraction of the total Raman signal from a volume fraction νi. This necessitates the confocal fit formulae from Table 3 to be modified by containing a linear combination of orthorhombic intensity functions at angles ϕ, ϕ + 120° and ϕ + 240°, scaled with coefficients νi. As an example, A2 modes in parallel polarization thus become
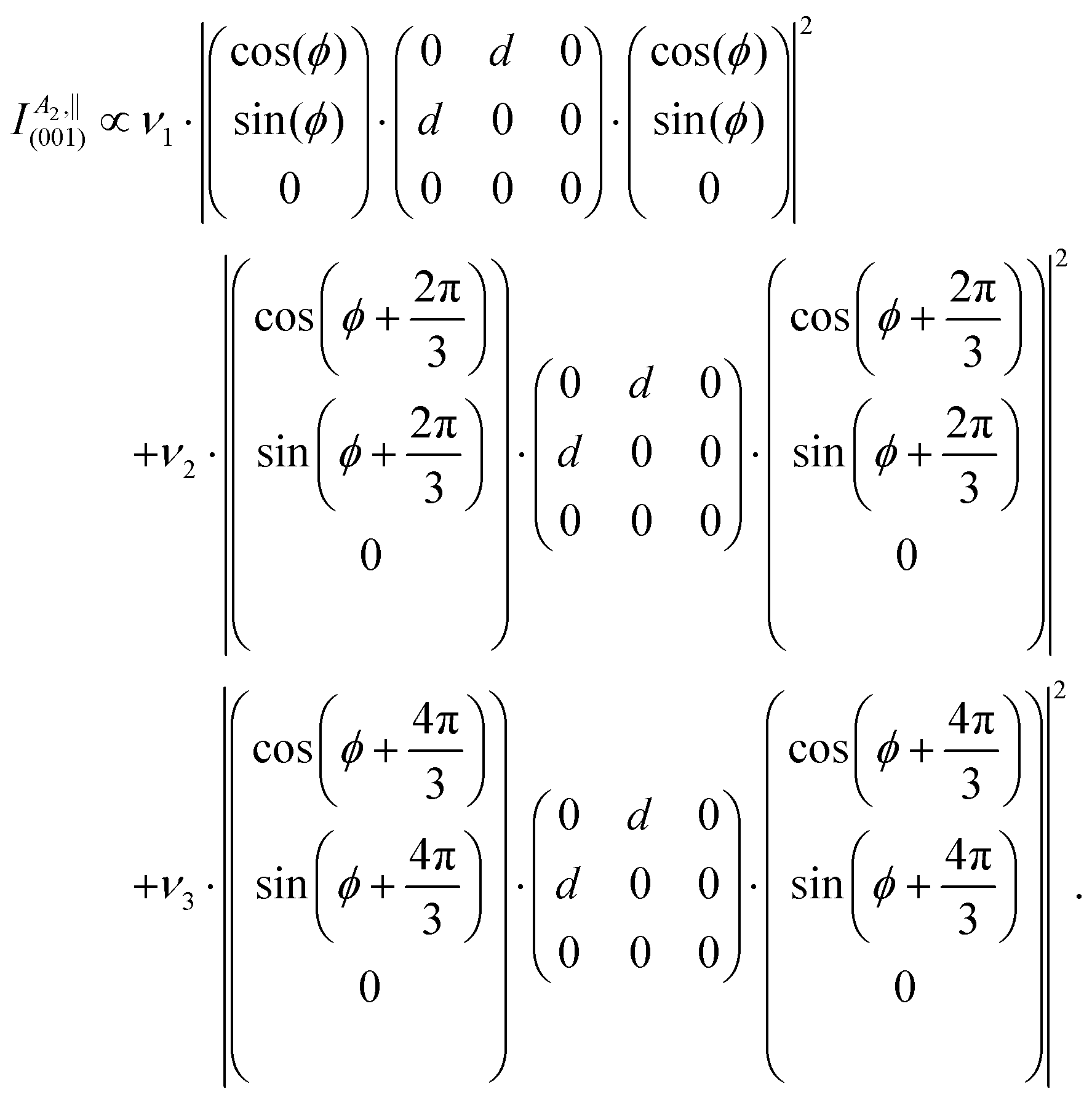 | (6) |
For an even distribution of orthorhombic domains the volume fractions are related by 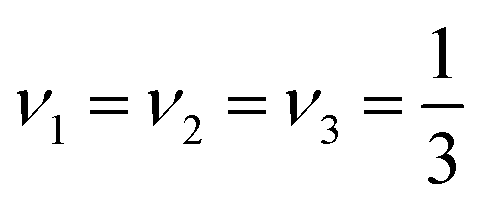 , allowing to simplify the above equation to
, allowing to simplify the above equation to
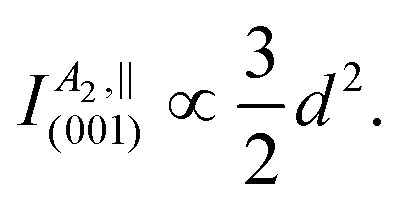 | (7) |
The modified fit formulae accounting for the investigated sample's domain structure are listed in Table 5. While the modified angular functions qualitatively resemble their monocrystalline counterparts for A1(TO) or are identical in the cases of B1(TO) and B2(TO) modes, the modified formulae suggest constant angular Raman intensities when illuminating the (001) plane.
| Facet | Phonon mode | ‖ | ⊥ |
|---|---|---|---|
| Edge | A 1(TO) | |b·cos2(ϕ) + c·sin2(ϕ)|2 + |a·cos2(ϕ) +c·sin2(ϕ)|2 | |(c − b)·sin(ϕ)cos(ϕ)|2 + |(c−a)·sin(ϕ)cos(ϕ)|2 |
| B 1(TO) | e 2·sin2(2ϕ) | e 2·cos2 (2ϕ) | |
| B 2 | f 2·sin2(2ϕ) | f 2·cos2 (2ϕ) | |
| (001) | A 1(LO) |
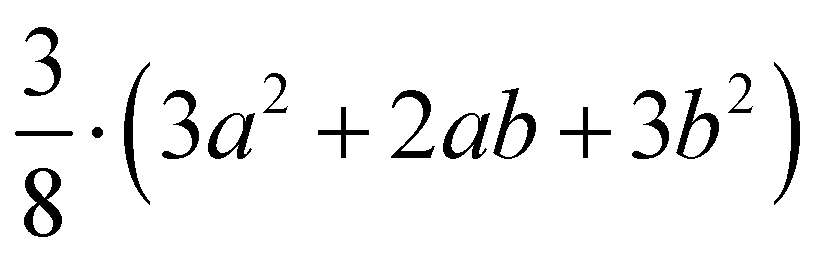
|
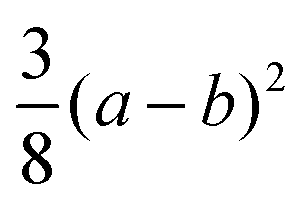
|
| A 2 |
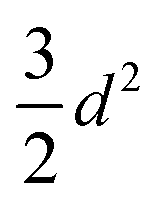
|
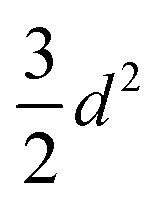
|
By modeling the experimental points in Fig. 6 using the intensity functions from Table 5, an A1(TO) mode symmetry is proven for a selection of four Raman-active modes for excitation on the edge. Explicitly, the A1(TO) symmetry for the mode observed at 93.3 cm−1 is verified. Furthermore, there exist two species of A1(TO) modes: (i) One for which the Raman polarizability in parallel polarization is maximal when the polarization vectors align with the edge (x′ direction in Fig. 3b) and (ii) one for which modes are 90° phase-shifted. An analogous phase shift was previously observed for β-Ga2O3, where the A5g, A6g, A9g and A10g are shifted with respect to the A1g, A2g, A3g, A4g, A7g and A8g modes.59 On the basis of angular intensity functions the B1(TO) and B2(TO) modes observed at 171.4 cm−1 and 281.2 cm−1 are qualitatively indistinguishable from each other, but clearly vary from A1(TO) modes in displaying equally intense maxima. At this point, modes of B1(TO) and B2(TO) symmetry can be distinguished from one another solely based on the computed mode symmetries. In consistency with the modified fit formulae for excitation of the (001) plane derived for an even distribution of orthorhombic domains, the Raman intensities remain constant as a function of the rotational angle ϕ for the selected A2 and A1 modes. The orthorhombic domains being rotated 120° against each other produce a pseudo-hexagonal ε structure (cf.Fig. 3a), in which the signals originating from each of the three species of domain orientation compensate each other. Any monocrystalline κ phase components would deviate from the expected curves, demonstrating the investigated film's domain structure through Raman spectroscopy. A preceding study employed angular-resolved Raman scans to investigate the Raman-active A1, E1 and E2 modes in ZnO.76 For wurtzite ZnO, ascribed to the same space group as the pseudo-hexagonal ε-Ga2O3 structure, selection rules analogously predict constant Raman intensities as a function of the rotational angle when probing the (0001) plane.
On the contrary, the signals from each of the three domain orientations do not compensate each other in the cross-sectional measurement configuration (cf.Fig. 3b) in which the laser propagation direction is orthogonal to the c-axis. In this case, the excitation occurs on the lateral faces of several c-oriented orthorhombic columns, giving rise to angular-dependent Raman intensity variations.
In order to obtain insights into the atomic displacements of the Raman modes selected in Fig. 6, the vibrational schemes of the computed Raman modes corresponding to the discussed experimental modes are illustrated in Fig. 7. Since we did not identify a theoretical analogue for the experimental Raman mode at 93.3 cm−1, we consider the vibrational scheme of its lower-energetic neighbor at 73.5 cm−1 (theoretical) or 82.4 cm−1 (experimental). The depicted vibrational schemes reveal two noteworthy results: (i) irrespective of the mode symmetry, both Ga and O lattice site vibrations occur for the low-frequency modes at theoretical frequencies 73.5 cm−1, 167.7 cm−1, 228.6 cm−1 and 278.3 cm−1. Thus, the energy of the low-frequency modes is carried by both Ga and O atoms. (ii) On the contrary, oscillations from Ga lattice site atoms vanish for the high-frequency modes at computed frequencies 454.8 cm−1, 705.6 cm−1 and 733.1 cm−1. The high-frequency modes are hence solely governed by O lattice site vibrations. A similar behavior was previously observed for β-Ga2O3, where the relative contribution to a Raman mode's energy from Ga (or O lattice sites) decreased (or increased) with rising Raman frequency.63
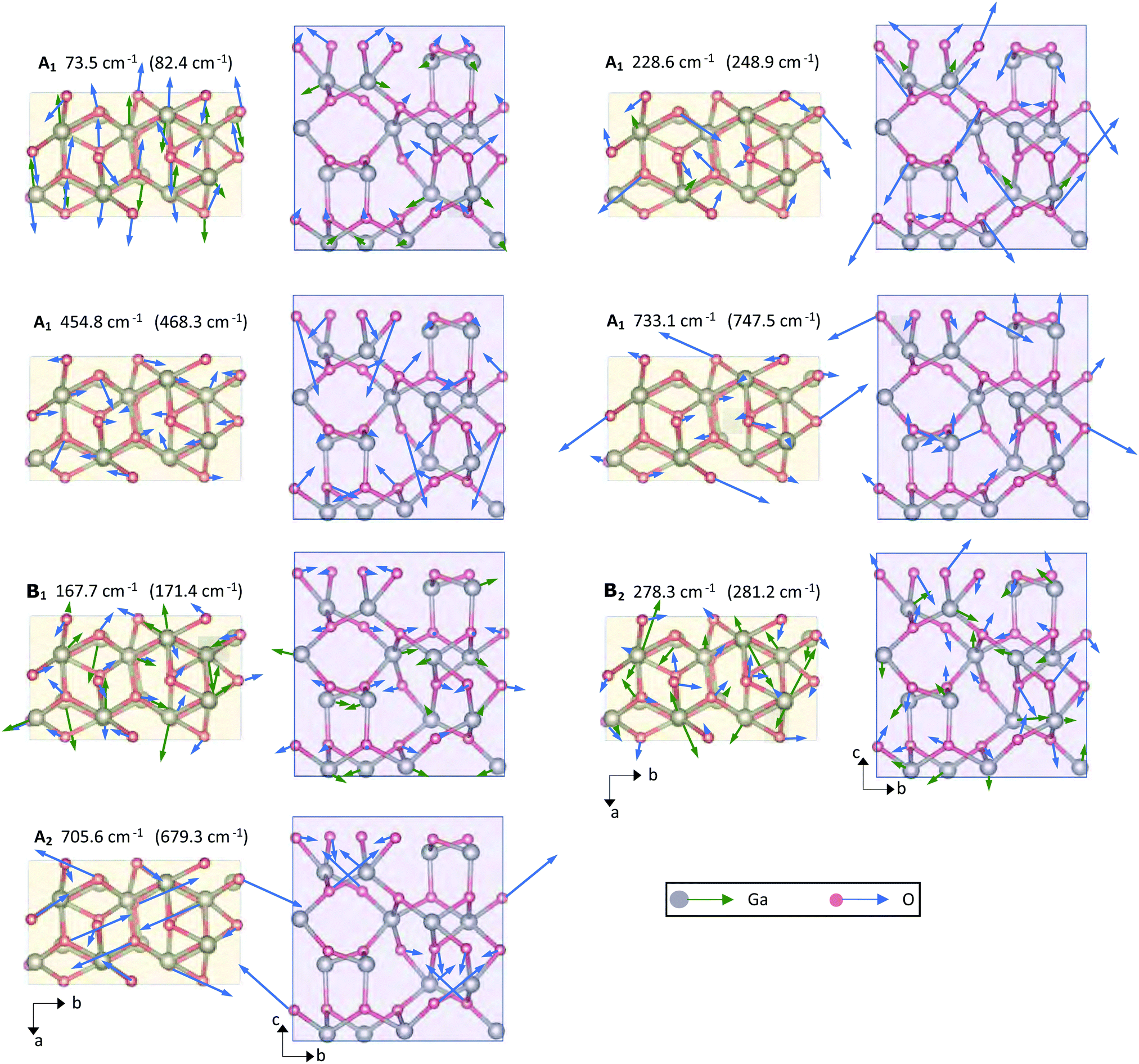 | ||
| Fig. 7 Schematic illustration of the Raman-active A1, B1, B2 and A2 modes' vibrational schemes within the primitive unit cell of κ-Ga2O3. Modes are shown in projection on the c- or a-plane, with the crystallographic directions denoted by black arrows. A respective mode's vibrational symmetry and computed theoretical frequency are listed on top of each graph. Brackets specify the experimental frequency assigned to the computed mode frequency. Green or blue arrows indicate the displacements of basis atoms, with lengths denoting the amplitude of vibration. Grey or red color denote Ga or O lattice site atoms. | ||
4 Conclusions
In summary, we studied the optical phonon spectrum of κ/ε-Ga2O3 by experiment and theory. Polarized micro-Raman measurements were conducted on a MBE-grown epitaxial κ-Ga2O3 thin film, deposited on top of an (0001)-oriented Al2O3 substrate. Cross-sectional and confocal measurement geometries were applied to separate modes of different vibrational symmetries. Our experimental analysis enabled the identification of the spectral positions of more than 90 first-order Raman modes related to orthorhombic κ-Ga2O3, which were linked to the calculated 117 Raman active phonons. For the vast majority of Raman modes the vibrational symmetries obtained by the calculations were consistent with the symmetries predicted for the applied measurement configurations based on Raman selection rules. The high number of phonons recorded in the present study ruled out the possibility of Raman modes to originate from the hexagonal phase, for which a significantly lower number of phonons was predicted by theory, proving the orthorhombic nature of the thin film. Further, by applying angular-resolved Raman measurements in cross-sectional and confocal geometry, we experimentally verified the mode symmetries of Raman-active phonons, demonstrating the A1(TO), B1(TO), B2(TO) and A2 symmetries for a selection of phonon modes. We provided an analytical tool to deal with the effect of the domain structure on the recorded angular Raman intensities. The derived fit formulae exhibited perfect agreement with the experimental data. Explicitly, when exciting the (001) plane in the confocal setup, no angular-dependent intensity variation was observed, which arises from the rotational domain structure producing a pseudo-hexagonal structure, in which the signals originating from each of the three species of 120°-twinned domain orientation compensate each other. Our analysis further indicated that any angular dependence on the (001) plane would result from the presence of monocrystalline components, paving the way to discriminating domain structured from monocrystalline films in future studies. While the employment of specific polarization geometries and angular measurements enabled to distinguish modes of A1(TO), B1(TO)/B2(TO) or A2 symmetry, the selected detection of B1(TO) and B2(TO) modes would necessitate the availability of monocrystalline κ-Ga2O3 without rotational domains and make use of the respective selection rules. Lastly, we showed the vibrational schemes for a selection of Raman modes, providing insights into the directions of atomic vibrations within the orthorhombic unit cell. In particular, we showed that while both Ga and O lattice site vibrations occur in the low-frequency modes, high frequency modes are characterized by the absence of Ga and domination of O lattice site vibrations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 446185170. This work was performed in parts in the framework of GraFOx, a Leibniz-ScienceCampus partially funded by the Leibniz association. Computational resources used for the calculations were provided by the HPC of the Regional Computer Centre Erlangen (RRZE). The authors thank Dr Harald Scheel for experimental support.Notes and references
- I. Cora, F. Mezzadri, F. Boschi, M. Bosi, M. Čaplovičová, G. Calestani, I. Dódony, B. Pécz and R. Fornari, CrystEngComm, 2017, 19, 1509–1516 RSC.
- R. Roy, V. G. Hill and E. F. Osborn, J. Am. Chem. Soc., 1952, 74, 719–722 CrossRef CAS.
- S. Yoshioka, H. Hayashi, A. Kuwabara, F. Oba, K. Matsunaga and I. Tanaka, J. Phys.: Condens. Matter, 2007, 19, 346211 CrossRef.
- H. von Wenckstern, Adv. Electron. Mater., 2017, 3, 1600350 CrossRef.
- S. I. Stepanov, V. I. Nikolaev, V. E. Bougrov and A. E. Romanov, Rev. Adv. Mater. Sci., 2016, 44, 63–86 CAS.
- H. He, R. Orlando, M. A. Blanco, R. Pandey, E. Amzallag, I. Baraille and M. Rérat, Phys. Rev. B, 2006, 74, 1–8 Search PubMed.
- H. He, M. A. Blanco and R. Pandey, Appl. Phys. Lett., 2006, 88, 261904 CrossRef.
- P. Kroll, R. Dronskowski and M. Martin, J. Mater. Chem., 2005, 15, 3296 RSC.
- H. Y. Playford, A. C. Hannon, E. R. Barney and R. I. Walton, Chem. – Eur. J., 2013, 19, 2803–2813 CrossRef CAS PubMed.
- J. Furthmüller and F. Bechstedt, Phys. Rev. B, 2016, 93, 115204 CrossRef.
- S. J. Pearton, J. Yang, P. H. Cary, F. Ren, J. Kim, M. J. Tadjer and M. A. Mastro, Appl. Phys. Rev., 2018, 5, 011301 Search PubMed.
- M. Kneiß, A. Hassa, D. Splith, C. Sturm, H. von Wenckstern, T. Schultz, N. Koch, M. Lorenz and M. Grundmann, APL Mater., 2019, 7, 022516 CrossRef.
- K. Matsuzaki, H. Hiramatsu, K. Nomura, H. Yanagi, T. Kamiya, M. Hirano and H. Hosono, Thin Solid Films, 2006, 496, 37–41 CrossRef CAS.
- Y. Oshima, E. G. Víllora, Y. Matsushita, S. Yamamoto and K. Shimamura, J. Appl. Phys., 2015, 118, 085301 CrossRef.
- Y. Zhuo, Z. Chen, W. Tu, X. Ma, Y. Pei and G. Wang, Appl. Surf. Sci., 2017, 420, 802–807 CrossRef CAS.
- D. Tahara, H. Nishinaka, S. Morimoto and M. Yoshimoto, JJAP, 2017, 56, 078004 Search PubMed.
- H. Nishinaka, D. Tahara and M. Yoshimoto, JJAP, 2016, 55, 1202BC Search PubMed.
- M. Pavesi, F. Fabbri, F. Boschi, G. Piacentini, A. Baraldi, M. Bosi, E. Gombia, A. Parisini and R. Fornari, Mater. Chem. Phys., 2018, 205, 502–507 CrossRef CAS.
- N. Ueda, H. Hosono, R. Waseda and H. Kawazoe, Appl. Phys. Lett., 1997, 71, 933–935 CrossRef CAS.
- M. Yamaga, T. Ishikawa, M. Yoshida, T. Hasegawa, E. G. Villora and K. Shimamura, Phys. Status Solidi C, 2011, 8, 2621–2624 CrossRef CAS.
- X. Zhao, Y. Zhi, W. Cui, D. Guo, Z. Wu, P. Li, L. Li and W. Tang, Opt. Mater., 2016, 62, 651–654 CrossRef CAS.
- Y. Qin, H. Sun, S. Long, G. S. Tompa, T. Salagaj, H. Dong, Q. He, G. Jian, Q. Liu, H. Lv and M. Liu, IEEE Electron Device Lett., 2019, 40, 1475–1478 CAS.
- Y. Qin, L. Li, X. Zhao, G. S. Tompa, H. Dong, G. Jian, Q. He, P. Tan, X. Hou, Z. Zhang, S. Yu, H. Sun, G. Xu, X. Miao, K. Xue, S. Long and M. Liu, ACS Photonics, 2020, 7, 812–820 CrossRef CAS.
- Y. Cai, K. Zhang, Q. Feng, Y. Zuo, Z. Hu, Z. Feng, H. Zhou, X. Lu, C. Zhang, W. Tang, J. Zhang and Y. Hao, Opt. Mater. Express, 2018, 8, 3506 CrossRef CAS.
- M. B. Maccioni and V. Fiorentini, Appl. Phys. Express, 2016, 9, 041102 CrossRef.
- S. B. Cho and R. Mishra, Appl. Phys. Lett., 2018, 112, 162101 CrossRef.
- F. Mezzadri, G. Calestani, F. Boschi, D. Delmonte, M. Bosi and R. Fornari, Inorg. Chem., 2016, 55, 12079–12084 CrossRef PubMed.
- K. Shimada, Mater. Res. Express, 2018, 5, 036502 CrossRef.
- J. Kim, D. Tahara, Y. Miura and B. G. Kim, Appl. Phys. Express, 2018, 11, 061101 CrossRef.
- S. Leone, R. Fornari, M. Bosi, V. Montedoro, L. Kirste, P. Doering, F. Benkhelifa, M. Prescher, C. Manz, V. Polyakov and O. Ambacher, J. Cryst. Growth, 2020, 534, 125511 CrossRef CAS.
- M. Kneiß, P. Storm, A. Hassa, D. Splith, H. von Wenckstern, M. Lorenz and M. Grundmann, APL Mater., 2020, 8, 051112 CrossRef.
- H. Nishinaka, N. Miyauchi, D. Tahara, S. Morimoto and M. Yoshimoto, CrystEngComm, 2018, 20, 1882–1888 RSC.
- D. Tahara, H. Nishinaka, S. Morimoto and M. Yoshimoto, Appl. Phys. Lett., 2018, 112, 152102 CrossRef.
- A. Hassa, M. Grundmann and H. von Wenckstern, J. Phys. D: Appl. Phys., 2021, 54, 223001 CrossRef CAS.
- X. Xia, Y. Chen, Q. Feng, H. Liang, P. Tao, M. Xu and G. Du, Appl. Phys. Lett., 2016, 108, 202103 CrossRef.
- M. Bosi, P. Mazzolini, L. Seravalli and R. Fornari, J. Mater. Chem. C, 2020, 8, 10975–10992 RSC.
- Y. Yao, S. Okur, L. A. M. Lyle, G. S. Tompa, T. Salagaj, N. Sbrockey, R. F. Davis and L. M. Porter, Mater. Res. Lett., 2018, 6, 268–275 CrossRef CAS.
- Y. Oshima, K. Kawara, T. Oshima and T. Shinohe, Jpn. J. Appl. Phys., 2020, 59, 115501 CrossRef CAS.
- F. Boschi, M. Bosi, T. Berzina, E. Buffagni, C. Ferrari and R. Fornari, J. Cryst. Growth, 2016, 443, 25–30 CrossRef CAS.
- R. Fornari, M. Pavesi, V. Montedoro, D. Klimm, F. Mezzadri, I. Cora, B. Pécz, F. Boschi, A. Parisini, A. Baraldi, C. Ferrari, E. Gombia and M. Bosi, Acta Mater., 2017, 140, 411–416 CrossRef CAS.
- Y. Chen, X. Xia, H. Liang, Q. Abbas, Y. Liu and G. Du, Cryst. Growth Des., 2018, 18, 1147–1154 CrossRef CAS.
- H. Sun, K.-H. Li, C. G. T. Castanedo, S. Okur, G. S. Tompa, T. Salagaj, S. Lopatin, A. Genovese and X. Li, Cryst. Growth Des., 2018, 18, 2370–2376 CrossRef CAS.
- H. J. von Bardeleben, J. L. Cantin, A. Parisini, A. Bosio and R. Fornari, Phys. Rev. Mater., 2019, 3, 084601 CrossRef CAS.
- Z. Zhang, Z. Chen, M. Chen, K. Wang, H. Chen, S. Deng, G. Wang and J. Chen, Adv. Mater. Technol., 2021, 2001094, 2001094 CrossRef.
- A. Parisini, A. Bosio, V. Montedoro, A. Gorreri, A. Lamperti, M. Bosi, G. Garulli, S. Vantaggio and R. Fornari, APL Mater., 2019, 7, 031114 CrossRef.
- V. Gottschalch, S. Merker, S. Blaurock, M. Kneiß, U. Teschner, M. Grundmann and H. Krautscheid, J. Cryst. Growth, 2019, 510, 76–84 CrossRef CAS.
- M. Mulazzi, F. Reichmann, A. Becker, W. M. Klesse, P. Alippi, V. Fiorentini, A. Parisini, M. Bosi and R. Fornari, APL Mater., 2019, 7, 022522 CrossRef.
- I. Cora, Z. Fogarassy, R. Fornari, M. Bosi, A. Rečnik and B. Pécz, Acta Mater., 2020, 183, 216–227 CrossRef CAS.
- H. Nishinaka, H. Komai, D. Tahara, Y. Arata and M. Yoshimoto, Jpn. J. Appl. Phys., 2018, 57, 115601 CrossRef.
- R. Jinno, T. Uchida, K. Kaneko and S. Fujita, Phys. Status Solidi B, 2018, 255, 1700326 CrossRef.
- M. Kracht, A. Karg, J. Schörmann, M. Weinhold, D. Zink, F. Michel, M. Rohnke, M. Schowalter, B. Gerken, A. Rosenauer, P. J. Klar, J. Janek and M. Eickhoff, Phys. Rev. Appl., 2017, 8, 054002 CrossRef.
- P. Vogt, O. Brandt, H. Riechert, J. Lähnemann and O. Bierwagen, Phys. Rev. Lett., 2017, 119, 196001 CrossRef PubMed.
- Y. Kuang, X. Chen, T. Ma, Q. Du, Y. Zhang, J. Hao, F.-F. Ren, B. Liu, S. Zhu, S. Gu, R. Zhang, Y. Zheng and J. Ye, ACS Appl. Electron. Mater., 2021, 3, 795–803 CrossRef CAS.
- M. Orita, H. Hiramatsu, H. Ohta, M. Hirano and H. Hosono, Thin Solid Films, 2002, 411, 134–139 CrossRef CAS.
- K. Matsuzaki, H. Yanagi, T. Kamiya, H. Hiramatsu, K. Nomura, M. Hirano and H. Hosono, Appl. Phys. Lett., 2006, 88, 092106 CrossRef.
- M. Kneiß, A. Hassa, D. Splith, C. Sturm, H. von Wenckstern, M. Lorenz and M. Grundmann, APL Mater., 2019, 7, 101102 CrossRef.
- A. Hassa, H. von Wenckstern, D. Splith, C. Sturm, M. Kneiß, V. Prozheeva and M. Grundmann, APL Mater., 2019, 7, 022525 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. Kranert, C. Sturm, R. Schmidt-Grund and M. Grundmann, Sci. Rep., 2016, 6, 35964 CrossRef CAS PubMed.
- D. Dohy, G. Lucazeau and A. Revcolevschi, J. Solid State Chem., 1982, 45, 180–192 CrossRef CAS.
- D. Machon, P. F. McMillan, B. Xu and J. Dong, Phys. Rev. B, 2006, 73, 094125 CrossRef.
- B. Liu, M. Gu and X. Liu, Appl. Phys. Lett., 2007, 91, 172102 CrossRef.
- B. M. Janzen, P. Mazzolini, R. Gillen, A. Falkenstein, M. Martin, H. Tornatzky, J. Maultzsch, O. Bierwagen and M. R. Wagner, J. Mater. Chem. C, 2021, 9, 2311–2320 RSC.
- M. Schubert, R. Korlacki, S. Knight, T. Hofmann, S. Schöche, V. Darakchieva, E. Janzén, B. Monemar, D. Gogova, Q.-T. Thieu, R. Togashi, H. Murakami, Y. Kumagai, K. Goto, A. Kuramata, S. Yamakoshi and M. Higashiwaki, Phys. Rev. B, 2016, 93, 125209 CrossRef.
- S.-i. Ohkoshi, M. Yoshikiyo, Y. Umeta, M. Komine, R. Fujiwara, H. Tokoro, K. Chiba, T. Soejima, A. Namai, Y. Miyamoto and T. Nasu, J. Phys. Chem. C, 2017, 121, 5812–5819 CrossRef CAS.
- P. Mazzolini, P. Vogt, R. Schewski, C. Wouters, M. Albrecht and O. Bierwagen, APL Mater., 2019, 7, 022511 CrossRef.
- P. Mazzolini, A. Falkenstein, Z. Galazka, M. Martin and O. Bierwagen, Appl. Phys. Lett., 2020, 117, 222105 CrossRef CAS.
- P. Mazzolini and O. Bierwagen, J. Phys. D: Appl. Phys., 2020, 53, 354003 CrossRef CAS.
- P. Mazzolini, A. Falkenstein, C. Wouters, R. Schewski, T. Markurt, Z. Galazka, M. Martin, M. Albrecht and O. Bierwagen, APL Mater., 2020, 8, 011107 CrossRef CAS.
- J. E. N. Swallow, C. Vorwerk, P. Mazzolini, P. Vogt, O. Bierwagen, A. Karg, M. Eickhoff, J. Schörmann, M. R. Wagner, J. W. Roberts, P. R. Chalker, M. J. Smiles, P. Murgatroyd, S. A. Razek, Z. W. Lebens-Higgins, L. F. J. Piper, L. A. H. Jones, P. K. Thakur, T.-L. Lee, J. B. Varley, J. Furthmüller, C. Draxl, T. D. Veal and A. Regoutz, Chem. Mater., 2020, 32, 8460–8470 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- M. Schlipf and F. Gygi, Comput. Phys. Commun., 2015, 196, 36–44 CrossRef CAS.
- L. He, F. Liu, G. Hautier, M. J. T. Oliveira, M. A. L. Marques, F. D. Vila, J. J. Rehr, G.-M. Rignanese and A. Zhou, Phys. Rev. B, 2014, 89, 064305 CrossRef.
- H. Kuzmany, Solid-State Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp. 1–554 Search PubMed.
- C. Kranert, C. Sturm, R. Schmidt-Grund and M. Grundmann, Phys. Rev. Lett., 2016, 116, 127401 CrossRef PubMed.
- T. Sander, S. Eisermann, B. K. Meyer and P. J. Klar, Phys. Rev. B, 2012, 85, 165208 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03500b |
| This journal is © The Royal Society of Chemistry 2021 |