
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible redox reactions in metal-supported porphyrin: the role of spin and oxidation state†
Iulia
Cojocariu
 *a,
Silvia
Carlotto
*bc,
Giovanni
Zamborlini‡
a,
Matteo
Jugovac§
a,
Luca
Schio
d,
Luca
Floreano
d,
Maurizio
Casarin
b,
Vitaliy
Feyer
*ae and
Claus Michael
Schneider
ae
*a,
Silvia
Carlotto
*bc,
Giovanni
Zamborlini‡
a,
Matteo
Jugovac§
a,
Luca
Schio
d,
Luca
Floreano
d,
Maurizio
Casarin
b,
Vitaliy
Feyer
*ae and
Claus Michael
Schneider
ae
aPeter Grünberg Institute (PGI-6), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: i.cojocariu@fz-juelich.de
bDipartimento di Scienze Chimiche, Università degli Studi di Padova, via F. Marzolo 1, 35131 Padova, Italy
cInstitute of Condensed Matter Chemistry and Technologies for Energy (ICMATE), National Research Council (CNR), c/o Department of Chemistry, University of Padova, via F. Marzolo 1, 35131 Padova, Italy. E-mail: silvia.carlotto@unipd.it
dCNR-IOM, Lab. TASC, s.s. 14 km 163,5, 34149 Trieste, Italy
eFakultät f Physik and Center for Nanointegration Duisburg-Essen (CENIDE), Universität Duisburg-Essen, D-47048 Duisburg, Germany. E-mail: v.feyer@fz-juelich.de
First published on 4th August 2021
Abstract
On-surface molecular functionalization paved the way for the stabilization of chelated ions in different oxidation and spin states, allowing for the fine control of catalytic and magnetic properties of metalorganic networks. Considering two model systems, a reduced Co(I) and an open-shell Co(II) metal-supported 2D molecular array, we investigate the interplay between the low valence oxidation and unpaired spin state in the molecular reactivity. We show that the redox reaction taking place at the cobalt tetraphenylporphyrin/Cu(100) interface, stabilizing the low-spin Co(I) state with no unpaired electrons in its valence shell, plays a pivotal role in changing the reactivity. This goes beyond the sole presence of unpaired electrons in the valence state of the Co(II) metal–organic species, often designated as being responsible for the reactivity towards small molecules like NO and NO2. The reversible Co–NO2 interaction, established with the Co(I) leads to the stabilization of the Co(III) oxidation state.
Introduction
Coordination chemistry offers unique possibilities to create new local bonds with metal–organic molecules. By extending this approach to surface chemistry, the tailoring of the electronic and, consequently, the chemical properties of the metal–organic layer is made possible by the interaction with the substrate.1 The on-surface stabilized arrays of metal ions in an unusual coordination environment may exhibit enhanced catalytic activity compared to the free molecules.2,3 Among others, tetrapyrrolic compounds, such as porphyrins, offer the possibility to chelate at the center of their macrocycle a wide plethora of transition metal ions offering an unsaturated site for functionalization. In fact, the addition of axial ligands, such as O2, NO, NO2 and CO, can be exploited for inducing further changes in the electronic structure and, in particular, in the spin state of the central metal ion.4–12 The unsaturated character of the metal ions in the porphyrins is also the key to the reactivity of a large number of enzymes. Specifically, the coordination of small diatomic molecules with metalloporphyrins are essential for biological processes.13–16In surface chemistry, however, low-temperature conditions are often required for the stabilization of the ligand–metal porphyrin interaction and, currently, the room temperature (RT) coordination is still a challenge. Overcoming this obstacle will allow us to tune the oxidation and spin state of chelated metal ions supported by a metal electrode, thus giving a chance to adjust the desired properties of the organic-based devices in working conditions.
One wonders what promotes the reaction of a metal–organic molecule with gaseous species? On the one hand, it was claimed that unpaired electrons in the valence shell of the chelated metal ion are a key factor in ligand coordination.1,6,17 On the other hand, some of the most challenging reactions in organisms are catalyzed by chelated metal ions, where unexpected reactivity corresponds to low valence oxidation states.18–20 In this regards, cobalt tetraphenylporphyrin (CoTPP) can be a suitable model system to shine a light on the molecular reactivity, as it can be stabilized either in the reduced Co(I) or Co(II) open-shell arrangements. In fact, the choice of the metal substrate strongly influences the CoTPP electronic structure upon deposition. While on poorly reactive substrates, such as Au(111),21 the Co(II) ion in Co-porphyrins (3d7 configuration) is stabilized in its low spin (LS) state with a single unpaired electron in the valence shell, on more interacting surfaces, e.g. silver and copper substrates, the Co ion is reduced (Co(II) → Co(I)) and stabilized in a LS 3d8 configuration.22 Indeed, the Co(I)–porphyrin systems supported by silver and copper surfaces show null or very week XMCD signal.22,23 The Co(I) supported by Ag(111) enables the binding of nitric oxide (NO) at 140 K, weakening the Co–Ag interaction because of the surface trans-effect.4,24,25
In this communication, we examine more deeply the axial ligand binding of NO2 to a CoTPP network grown on bare and oxygen-modified Cu(100), disentangling the relation between oxidation and spin state of the metal ion, as well as elucidating the electronic configuration that promotes the reaction with the gaseous species. We demonstrate that, while the presence of the unpaired electrons in the valence shell of Co(II) is not a crucial factor in this ligand coordination, the presence of the reduced Co(I) metal ion in the molecular array facilitates the formation of the new Co–NO2 chemical bond already at RT. In particular, we show that the Co(III) ion is stabilized in a 2D molecular array of CoTPP on Cu(100) by the trans-effect localized along the Cu–Co–N axis. The functionalization of the metal–organic array at RT is then pivotal for a fine-tuning of the desirable electromagnetic properties of molecular-based devices in working condition.
Results and discussion
A detailed comprehension of the changes induced by the surface in the supported CoTPP network may take advantage of a few words about the free molecule. Consistently with the presence of a LS Co(II) 3d7 central ion (S = 1/2), the CoTPP static magnetic moment is μs = 1.73 μB, while its effective magnetic moment is μe = 1.92 μB.26 The square planar coordination of Co(II) in free CoTPP (D4h symmetry21) lifts the five-fold degeneracy of the Co 3d atomic orbitals (AOs) to generate a1g (dz2), b1g (dx2−y2), b2g (dxy) and eg (dxz,yz) 3d-based MOs, whose relative energy positions and 3D contour plots (CP) are displayed in the left panel of Fig. S1 (ESI†). In a multilayer phase and when deposited on an inert substrate (O–Cu(100) in the present study), the free molecule properties are preserved. The oxygen dosing is a consolidated protocol to passivate reactive substrates, including metals.27,28 At saturation, Cu(100) displays a characteristic (√2 × 2√2)R45°-O reconstruction, where a significant quenching of the charge transfer at the organic–metal interface is expected.29–31 Similarly to the free molecule, CoTPP deposited on O–Cu(100), has a LS state with a single unpaired electron occupying the 3dz2-based MO (Fig. S1, right panel, ESI†). Such an unpaired electron in the molecular electronic structure has attracted great attention in coordination chemistry studies4–6,17 and it is also a key point in the present investigation (details about the stability of the different spin states and MOs compositions in CoTPP are reported in ESI†). Upon deposition on a metal substrate, the molecular layer in direct contact with the surface can experience chemical changes induced by the charge transfer at the metal/organic interface. At variance with CoTPP/O–Cu(100), where the Co ion preserves the 3d7 configuration of the free molecule, Co(II) is expected to reduce to Co(I) on more reactive surfaces, such as silver and copper, which implies a 3d8 configuration with no unpaired electrons in 3d-based MOs.4,32 In order to confirm the stabilisation of Co(I) and Co(II) oxidation states in the CoTPP monolayer deposited on the reactive Cu(100) and passivated O–Cu(100), respectively, we performed X-ray photoemission spectroscopy (XPS) at the Co 2p core level (see Fig. 1a). The Co 2p3/2 spectrum of CoTPP deposited on O–Cu(100) shows a shape similar to that of the CoTPP multilayer.4,22 More specifically, the intensity maximum is peaked at 779.25 eV in binding energy (BE) and the spectrum displays the characteristic multiplet structure of LS Co(II) with one unpaired electron. On the other hand, the shape and BE (778.4 eV) of the CoTPP/Cu(100) Co 2p3/2 peak are similar to those reported for CoTPP/Ag(111),4 suggesting the Co(II) → Co(I) reduction upon chemisorption of CoTPP on Cu(100).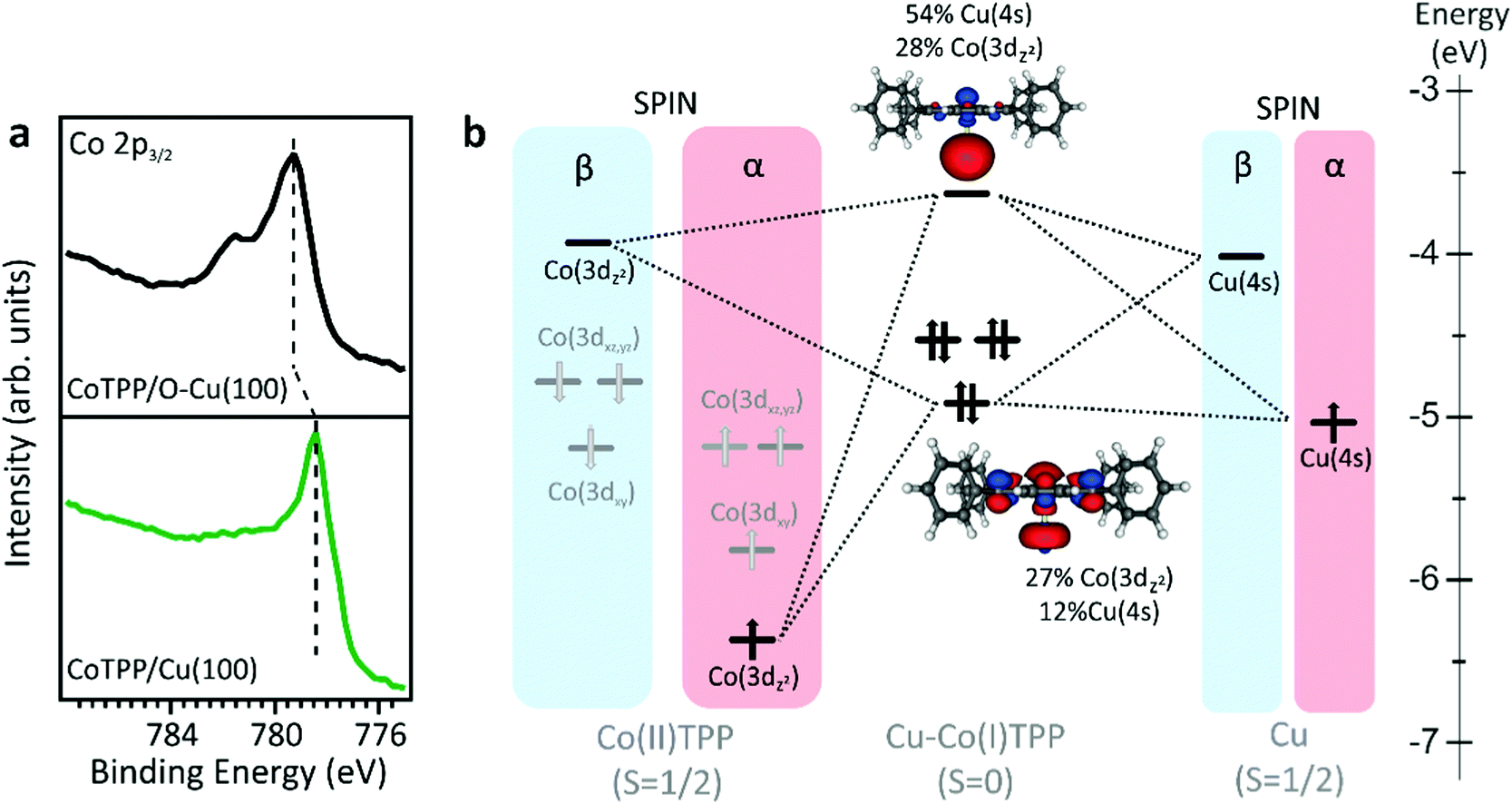 | ||
| Fig. 1 (a) Co 2p3/2 XPS core level spectra of CoTPP on bare (bottom panel) and oxygen modified (upper panel) Cu(100). (b) Energy level diagram for selected MOs, Co(3d) and Cu(4s), for LS Cu–CoTPP. 3D plots displayed isosurfaces correspond to ±0.03 e1/2 A−3/2. Grey and white spheres are representative of C and H atoms, respectively. | ||
Further information about the interaction of CoTPP with the Cu(100) surface can be gained by means of density functional theory (DFT) calculations. Previous results obtained by exploiting a periodical model21,32,33 will be here extended by combining the molecular cluster approach34,35 with DFT based calculations, which ultimately allows to gain information about AOs and MOs in the metal–molecule complex.4,36 This approach has been used to correctly reproduce the magnetic properties of supported metal porphyrins and phthalocyanines.4,30,37
Indeed, the molecular cluster approach has the great advantage of accurately describing the oxidation and spin states of the central ion in metalorganics,38 being thus best suited to investigate the Co metal ion chelated in CoTPP, involved herein in the axial coordination. In addition, when combined with first principle calculations, it provides a quantitative analysis on the symmetry restricted covalency (the effect associated with the dilution, ruled by the complex symmetry, of d orbitals making them a linear combinations of AOs–LCAO-MOs39). Moreover, the molecular cluster approach gives an information about the charge redistribution in the MOs and AOs formed upon interaction and it stands at the basis of XAS simulations, which ultimately give a theoretical feedback on the metal spin state and the metal oxidation state modification.
Our calculation shows that both Cu(0) and Co(II) in the Cu–CoTPP cluster are characterized by the presence of a single unpaired electron occupying the Cu 4s AO and the Co 3dz2-based MO (see Fig. 1b), and the molecular complex can be stabilized either in a LS (S = 0) or a high spin (HS) state (S = 1) configuration. As such, energy deviations (ΔE) from the Cu–CoTPP ground state (GS) reported in Fig. S2 (see ESI†) clearly show the LS higher stability for Cu–Co distances smaller than 3.6 Å. A short intermetallic distance appears then to favor the Co(II) + Cu(0)→Co(I) + Cu(I) on-surface redox reaction. Theoretical outcomes reported in Fig. 1 strongly support the presence of a Cu(I)–Co(I)TPP cluster at a Co–Cu distance of 2.5 Å;21,32,33 in fact, the (CoTPP + Cu) system is less stable than the Cu(I)–Co(I)TPP cluster by 1.04 eV. In particular, the charge transfer that causes the reduction of the cobalt is clearly visible on the MOs CoTPP–Cu compositions in Fig. 1b: the fully occupied MOs is mainly localized on the Co 3dz2-based MO (27%), while the LUMO is localized (54%) on Cu 4s MO. This confirms that the direct interaction of the CoTPP molecule with the copper substrate leads to the stabilization of a reduced Co(I), with no unpaired electrons in the valence states, contrary to the CoTPP/O–Cu(100) system, where the open-shell Co(II) state, characteristic of the free CoTPP molecule, is preserved. The present theoretical output is fully in agreement with the interpretation of the XPS data reported on Fig. 1a.
After stabilizing the Co chelated ion in the two desired configurations, namely, reduced Co(I) and open-shell Co(II) on bare and oxygen-modified Cu(100) substrates, respectively, in the next we elucidate the interplay between spin and oxidation state in the molecular reactivity towards axial ligands. To do so, both the CoTPP arrays have been exposed to a low dose of nitrogen dioxide at RT.
The possible anchoring of the axial ligand to the Co ion in the Co(I)TPP and Co(II)TPP networks formed on the bare and oxygen-modified surfaces, respectively, was monitored by means of XPS. Both the Co 2p3/2 and N 1s core-level spectra of Co(II)TPP deposited on the O–Cu(100) substrate remained unchanged after the exposure at RT to the NO2 gas (see Fig. S3, ESI†). This result indicates that the sole presence of an unpaired electron in the Co(II) ion may not be the key requirement for anchoring gas molecules to the metal–organic network, as previously discussed in the literature,4,6 or, at least, it is not sufficient.
In contrast with the CoTPP/O–Cu(100) system, the Co 2p3/2 and N 1s spectra of CoTPP deposited on the bare Cu(100) substrate show well-evident changes after the exposure of the metalloporphyrin network to NO2. The N 1s spectrum exhibits a new peak at 402.8 eV associated with NO2 and a small energy shift to lower BE of the main peak associated with the tetrapyrrolic nitrogen atoms (see Fig. 2). For what concerns the Co 2p3/2 spectrum, the intensity maximum is shifted by 1.95 eV to higher BE and a different distribution of satellites is observed after exposing the system to NO2. This spectral behavior is consistent with the Co(I) oxidation induced by the interaction with the axial ligand and the formation of a Co–NO2 bond. However, the Co 2p3/2 core level and the Co L-edge near-edge X-ray absorption fine structure (NEXAFS) spectra of the Co–NO2 system are different from the one of CoTPP/O–Cu(100) where the LS Co(II) is present (see Fig. S4, ESI†). Hence, the stabilization of the Cu(100)/CoTPP–NO2 complex probably leads to the Co(III) oxidation state.
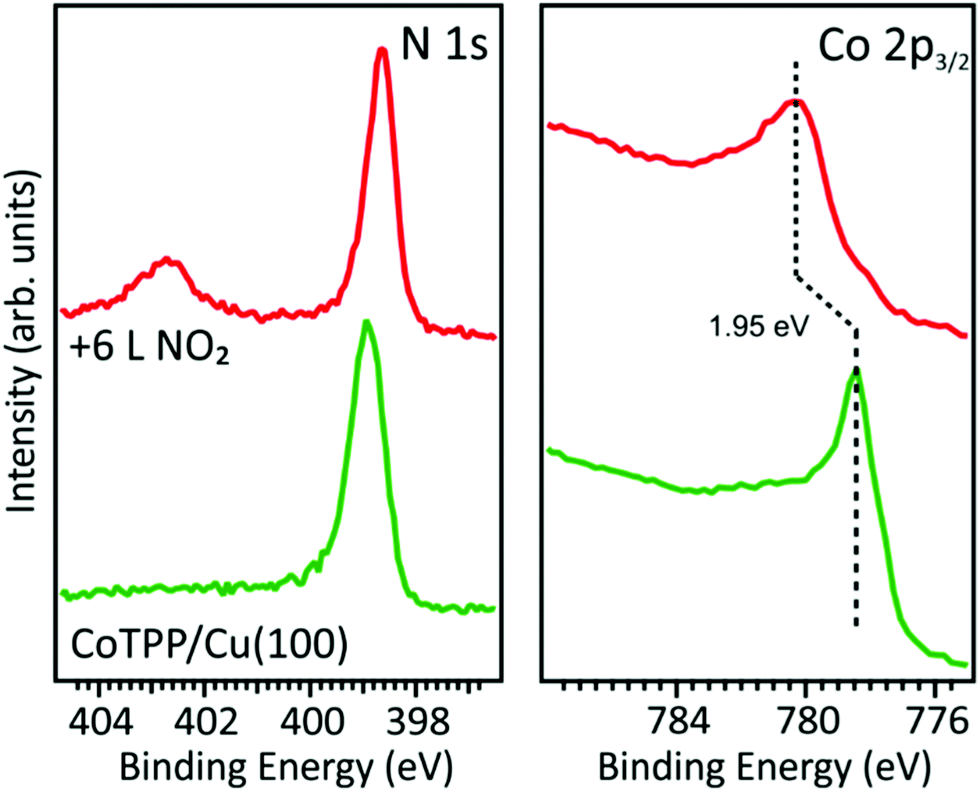 | ||
| Fig. 2 N 1s and Co 2p3/2 XPS core level spectra of CoTPP deposited on the bare copper substrate, before and after 6 L NO2 dose. | ||
To support these experimental observations and to provide a detailed insight into the stabilization of Co(III) ions in the metal porphyrin array, further DFT-based numerical simulations and additional NEXAFS simulations have been performed.
The NO2 interaction with the Co(I)TPP array has been modeled by exploiting the Cu–CoTPP–NO2 cluster, whose constrained optimization has been carried out by assuming either 3 or 1 unpaired electrons and freezing the Cu–Co distance to 3 Å; i.e., a value close to the one (2.93 Å)21 optimized for the Cu–Co bond by means of DFT periodic calculations for similar interfaces. As such, it can be useful to anticipate that bond-length variations smaller than 0.1 Å do not significantly affect the modeled NEXAFS spectra reported herein. Thus, since the LS Cu–CoTPP–NO2 cluster is more stable than the HS one by 1.06 eV, only the former has been considered in the forthcoming discussion.
Upon the Co–NO2 bond formation, the energy of the Cu–Co bond decreases from 1.06 eV (Cu–CoTPP) to 0.33 eV (Cu–CoTPP–NO2) (see Fig. S5, ESI†) and the same holds when CoTPP–NO2 (see Fig. S7 in the ESI†) and the Cu–CoTPP–NO2 are compared, in particular, the Co–NO2 bonding interaction decreases from 1.31 eV to 0.77 eV. Beside the recognition of the orbitals mainly contributing to the Cu–CoTPP–NO2 interaction (Cu 4s and Co 3dz2 AOs and the NO2 π* MO), the analysis of the Cu–CoTPP–NO2 electronic structure reveals that the singly occupied MO (SOMO) is strongly localized on the Cu 4s AO. This suggests that the trans-effect induced by the NO2 coordination implies the weakening of the Cu–Co interaction and the reduction of the Cu oxidation number to its original value (0). It has to be noted that upon adsorption of CoTPP on the bare copper, the strong interaction between Co(II) and the surface, beside inducing the Co(II) → Co(I) reduction, could also introduce a mechanical strain within the molecule. After the NO2trans-coordination to the supported CoTPP, being NO2 a stronger ligand than the copper surface, the Co–Cu distance increases of 0.5 Å, thus reducing possible molecular strains. Moreover, the doubly occupied MO accounting for the Co–NO2 bonding is mainly localized on NO2 with a negligible Cu participation (see Fig. 3, right panel), while the Co 3dz2 AO mainly contributes to the lowest unoccupied MO. As a whole, the NO2 local electronic structure of the Cu–CoTPP–NO2 cluster corresponds to a closed-shell with Cu in its elemental oxidation state and the “octahedrally” coordinated Co ion having a formal +3 oxidation state and a LS 3d6 electronic configuration with the 3dx2−y2 and 3dz2 as empty orbitals.
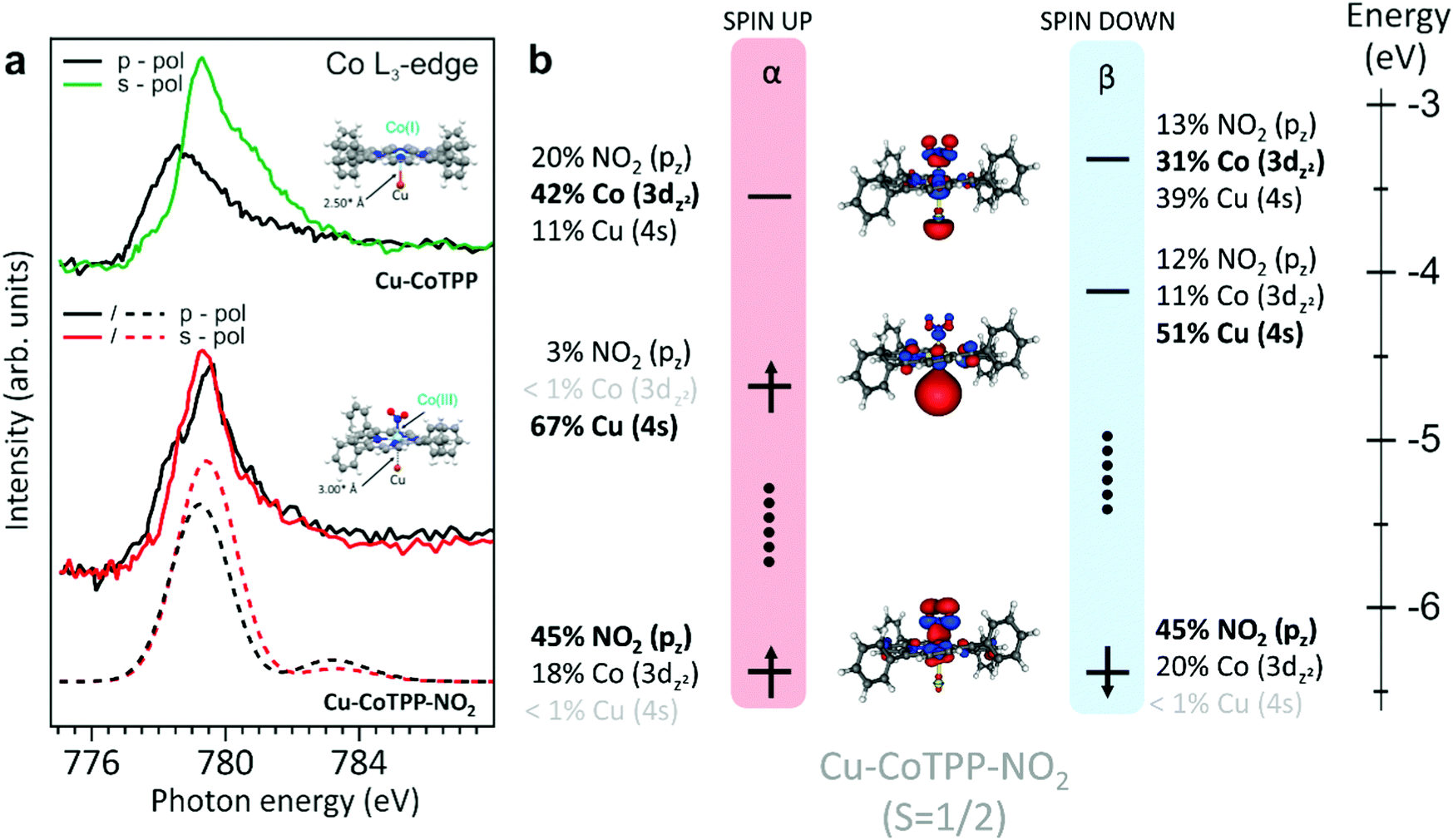 | ||
| Fig. 3 (a) Cu–CoTPP (top) and Cu–CoTPP–NO2 (bottom) Co L3-edge NEXAFS spectra for s- and p-polarizations and corresponding optimized geometries. To match the experimental data, the simulated spectra for Cu–CoTPP–NO2 (dashed line) are shifted by 10.6 eV and have a Gaussian broadening of 2 eV. (b) Energy level diagram for selected molecular orbitals for LS Cu–CoTPP–NO2 with a fixed Co–Cu distance (3 Å). Both α and β spin are reported. 3D plots displayed isosurfaces correspond to ±0.03 e1/2 A−3/2. Orbitals with a contribution below 1% are shaded. Vertical dots indicate that some levels, mainly localized on TPP fragment and/or Cu(3d) orbitals, are omitted. Grey, white, and yellow spheres are representative of C, H and Cu atoms, respectively. | ||
As such, it has to be noted that Co(III), here observed in the surface supported 2D metal–organic array, has been formerly described only for metal–organic complexes in solution.40 To single out perturbations induced by the surface on the CoTPP–NO2 MOs, an analysis of the CoTPP–NO2 and Cu–CoTPP–NO2 frontier orbitals has been performed (Section S6, ESI†). Theoretical results indicate that, even though Co 3d-based and NO2-based MOs (see Fig. S7, ESI†) are quite similar in the two clusters, both their energies and localizations are different, hence a Cu–Co weak interaction is still present in the Cu–CoTPP–NO2 cluster. The calculated binding energies and geometrical structures are further supported by the analysis of the Nalewajski–Mrozek bond multiplicity indexes (NMI),41 which provide a quantitative estimate of bond strengths. In both Cu–CoTPP and CoTPP–NO2 clusters, the Co species (Co(I) in the former, Co(III) in the latter) is penta-coordinated and the quite large NMICo–Cu and NMICo–NO2 values (0.95 and 0.69, respectively) are indicative of a quite strong Co–Cu and Co–NNO2 bond. Interestingly, NMICo–Cu values are 0.95 to 0.16 in Cu–CoTPP and Cu–CoTPP–NO2, respectively, suggesting that the coordination of the sixth ligand (NO2) dramatically weakens the Cu–Co bonding. At the same time, calculations indicate a not significant decreasing of the NMICo–NO2 going from the CoTPP–NO2 (0.69) to the Cu–CoTPP–NO2 (0.61) complex. As a whole, Co is esa-coordinated in the Cu–CoTPP–NO2 cluster, but the Co–Cu interaction is significantly weakened by the NO2 presence, while negligible bond order variations affect the Co–ligand interactions taking place in the macrocycle (see Table S8 in the ESI†).
Further information about the Co(III) stabilization at the metal–organic interface has been gained by recording, modelling and assigning the Co L3-edge absorption spectra (see Fig. 3a) after exposure of the CoTPP/Cu(100) interface to 6L of NO2. Clear differences can be observed in the spectra reported in Fig. 3 with respect to the ones measured for CoTPP/O–Cu(100) reported in Fig. S4 (ESI†).
Co L3-edge spectra for s- and p-polarizations have been modelled by exploiting the optimized Cu–CoTPP–NO2 cluster (see Fig. S5, ESI†) for which a LS state (1 unpaired electron) has been assumed. As already mentioned, 3dxz, 3dyz and 3dxy orbitals for the 3d6 Co(III) species are fully occupied, while the others MOs are empty and the Cu–CoTPP–NO2 SOMO, as stated above, is strongly localized on the Cu 4s AO. The lower excitation energy (EE) side of the single peak characterizing the modelled Co L3-edge spectrum in p-polarization includes both ΔS = 0, +1 electronic states. ΔS = 0 states are generated by single electronic excitations from the Co 2p orbitals to the Co-based dz2 and dx2−y2 virtual MOs (VMOs), while both Co-based and TPP-based VMOs are involved in electronic excitations generating ΔS = +1 states. At variance to that, the higher EE side of the peak only includes ΔS = 0 electronic states, which are determined by electronic transitions from the Co 2p AOs to the Cu 4s-based SOMO and Co-based VMOs. Only ΔS = 0 states, mainly generated by Co 2p → Co-based dx2−y2 single electronic excitations, contribute to the s-polarized spectrum. The overall agreement between experimental and modelled spectra, in particular the energy position of the main peaks and the linear dichroic behaviour observed in the spectra, fully supports the stabilization of the Co(III) species upon the interaction of the NO2 ligand with the Co(I)TPP species stabilized on the Cu(100) substrate.
The reversibility of the molecular-surface interaction and, accordingly, the Co(I) ⇆ Co(III) redox reaction has been tested here by annealing the NO2–CoTPP/Cu(100) interface to 450 K under UHV conditions. The possible thermal desorption of NO2 and restoring of the Co–Cu interaction has been monitored by valence band spectroscopy (see Fig. 4). This, being an experimental technique sensitive to the changes in charge transfer and interaction strength at the metal–organic/metal interface, allows to confirm the presence of the surface trans-effect, as suggested by calculations.
 | ||
| Fig. 4 Bottom panel: Photoemission spectra of CoTPP/Cu(100), after NO2 dosing and after annealing of NO2–CoTPP/Cu(100) to 450 K; top panel: experimental 2D momentum patterns of CoTPP/Cu(100) corresponding to LUMO/LUMO+1 and LUMO+3 MOs. The sharp inner features are related to the sp band of the copper surface. | ||
The valence band spectrum of the clean copper is dominated by the sp band and has a rather featureless plateau, while the CoTPP/Cu(100) spectrum shows two prominent features at BEs of 0.15 eV and 0.80 eV. To clarify the origin of these features we measured momentum resolved photoemission maps at constant BE and compared them with the square modulus of the Fourier transform (FT) of the real space MOs provided by DFT calculation,42 within the photoemission tomography (PT) approach (see Fig. S9, ESI†). Based on the excellent match between the experimental and theoretical data, the features at 0.15 eV and 0.80 eV BE can be assigned to the emissions from the gas-phase LUMO/LUMO+1 and the LUMO+3, respectively. These molecular levels, delocalized over the entire molecular macrocycle of CoTPP, are filled due to the charge transfer occurring at the organic/metal interface, as a result of the strong anchoring of the Co ion to the copper substrate.43,44 As discussed above, in the Cu–CoTPP–NO2 complex the Co–Cu interaction is weakened by the surface trans-effect, as evident from the quenching of the LUMOs features (see Fig. 4). The annealing of the interface to 450 K induces the thermal desorption of the NO2 ligand in the Cu–CoTPP–NO2 complex, thus restoring the pristine interaction and the charge transfer between the copper surface and the CoTPP molecule. As a result, both LUMO states reappear in the valence band spectra. The Co 2p3/2 and N 1s spectra further confirm the full reversibility of the Co(I) ⇆ Co(III) conversion (see Fig. S10, ESI†).
Conclusions
Using XPS and NEXAFS data supported by DFT calculation highly sensitive to the atomic and molecular characters of metalorganic complexes, we have studied the axial ligand coordination with the cobalt tetraphenylporphyrin array deposited on bare and oxygen passivated copper surfaces. The data show that the Co–NO2 chemical interaction is taking place on the CoTPP network supported by the copper electrode, where the Co ion is in a low and unusual oxidation state (I). Instead, this reaction has not been observed at the CoTPP/O–Cu(100) interface where Co(II) ions have a LS state and a single unpaired electron in the valence shell. The trans-effect observed along the Cu–Co–N axis in the Cu–CoTPP–NO2 complex suppresses the Cu–Co interaction, as confirmed by the valence band data, and stabilizes the Co(III) chelated ion in the metal–organic array. The Co(I) ⇆ Co(III) conversion is fully reversible, as the thermal-induced desorption of NO2 taking place at 450 K restores the pristine Co–Cu interaction.Our experiment demonstrates that the on-surface reactivity towards axial coordination to the metal center of porphyrin 2D arrays is driven by the oxidation state, rather than by the presence of an unpaired electron in the dz2 atomic orbital. Further, the reduction of the chelated metal by surface trans-effect may be reversed by the axial coordination to NO2 through a push–pull charge transfer mechanism. This evidence lets us envisage the possibility of tuning the metal spin state of 2D arrays of metal–organic molecules by suitable choice of the substrate and reacting gas.
We remark that the combination of surface trans-effect and axial coordination to NO2 allowed us to manipulate the Co oxidation and spin states at room temperature. This is a key aspect for the generalization of our approach to ferromagnetic substrates, as well as for the practical use in molecular-based spintronic devices.
Author contributions
I. C. and V. F. performed the experiments and drafted the manuscript with essential contributions from S. C., M. C. and L. F.; all the authors strongly participated in the discussion of the results and revision of the manuscript. S. C. and M. C. performed the computational calculations. L. S. and L. F. gave significant assistance in the XPS and NEXAFS experiments.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- B. Hulsken, R. Van Hameren, J. W. Gerritsen, T. Khoury, P. Thordarson, M. J. Crossley, A. E. Rowan, R. J. M. Nolte, J. A. A. W. Elemans and S. Speller, Nat. Nanotechnol., 2007, 2, 285–289 CrossRef CAS.
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H. P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS.
- C. Wäckerlin, K. Tarafder, J. Girovsky, J. Nowakowski, T. Hählen, A. Shchyrba, D. Siewert, A. Kleibert, F. Nolting, P. M. Oppeneer, T. A. Jung and N. Ballav, Angew. Chem., Int. Ed., 2013, 52, 4568–4571 CrossRef PubMed.
- C. Wäckerlin, D. Chylarecka, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jung and N. Ballav, Nat. Commun., 2010, 1, 61 CrossRef.
- H. Kim, Y. H. Chang, S. H. Lee, Y. H. Kim and S. J. Kahng, ACS Nano, 2013, 7, 9312–9317 CrossRef CAS PubMed.
- M. Viviani, L. Girlanda, A. Kievsky and L. E. Marcucci, Phys. Rev. C, 2020, 102, 245406 CrossRef.
- M. H. Chang, N. Y. Kim, Y. H. Chang, Y. Lee, U. S. Jeon, H. Kim, Y. H. Kim and S. J. Kahng, Nanoscale, 2019, 11, 8510–8517 RSC.
- D. E. Hurtado Salinas, A. Sarasola, B. Stel, F. P. Cometto, K. Kern, A. Arnau and M. Lingenfelder, ACS Omega, 2019, 4, 9850–9859 CrossRef CAS PubMed.
- G. Zamborlini, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, D. Lüftner, P. Puschnig, V. Feyer and C. M. Schneider, Chem. Commun., 2018, 54, 13423–13426 RSC.
- C. Wäckerlin, D. Siewert, T. A. Jung and N. Ballav, Phys. Chem. Chem. Phys., 2013, 15, 16510–16514 RSC.
- L. Xue, G. Farrugia, S. M. Miller, C. D. Ferris, S. H. Snyder and J. H. Szurszewski, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1851–1855 CrossRef CAS.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS.
- J. M. C. Ribeiro, J. M. H. Hazzard, R. H. Nussenzveig, D. E. Champagne and F. A. Walker, Science, 1993, 260, 539–541 CrossRef CAS PubMed.
- R. Gutzler, S. Stepanow, D. Grumelli, M. Lingenfelder and K. Kern, Acc. Chem. Res., 2015, 48, 2132–2139 CrossRef CAS PubMed.
- C. Wäckerlin, K. Tarafder, D. Siewert, J. Girovsky, T. Hählen, C. Iacovita, A. Kleibert, F. Nolting, T. A. Jung, P. M. Oppeneer and N. Ballav, Chem. Sci., 2012, 3, 3154–3160 RSC.
- S. Scheller, M. Goenrich, R. Boecher, R. K. Thauer and B. Jaun, Nature, 2010, 465, 606–608 CrossRef CAS.
- S. J. Moore, S. T. Sowa, C. Schuchardt, E. Deery, A. D. Lawrence, J. V. Ramos, S. Billig, C. Birkemeyer, P. T. Chivers, M. J. Howard, S. E. J. Rigby, G. Layer and M. J. Warren, Nature, 2017, 543, 78–82 CrossRef CAS.
- T. Wagner, J. Kahnt, U. Ermler and S. Shima, Angew. Chem., Int. Ed., 2016, 55, 10630–10633 CrossRef CAS PubMed.
- Y. H. Chang, H. Kim, S. J. Kahng and Y. H. Kim, Dalton Trans., 2016, 45, 16673–16681 RSC.
- S. Vijayaraghavan, W. Auwärter, D. Ecija, K. Seufert, S. Rusponi, T. Houwaart, P. Sautet, M. L. Bocquet, P. Thakur, S. Stepanow, U. Schlickum, M. Etzkorn, H. Brune and J. V. Barth, ACS Nano, 2015, 9, 3605–3616 CrossRef CAS.
- L. M. Arruda, M. E. Ali, M. Bernien, N. Hatter, F. Nickel, L. Kipgen, C. F. Hermanns, T. Bißwanger, P. Loche, B. W. Heinrich, K. J. Franke, P. M. Oppeneer and W. Kuch, Phys. Chem. Chem. Phys., 2020, 22, 12688–12696 RSC.
- P. S. Deimel, R. M. Bababrik, B. Wang, P. J. Blowey, L. A. Rochford, P. K. Thakur, T. L. Lee, M. L. Bocquet, J. V. Barth, D. P. Woodruff, D. A. Duncan and F. Allegretti, Chem. Sci., 2016, 7, 5647–5656 RSC.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H. P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CrossRef CAS.
- S. S. Eaton and G. R. Eaton, Inorg. Chem., 1980, 19, 1095–1096 CrossRef CAS.
- G. Fratesi, S. Achilli, A. Ugolotti, A. Lodesani, A. Picone, A. Brambilla, L. Floreano, A. Calloni and G. Bussetti, Appl. Surf. Sci., 2020, 530, 147085 CrossRef CAS.
- A. Calloni, M. S. Jagadeesh, G. Bussetti, G. Fratesi, S. Achilli, A. Picone, A. Lodesani, A. Brambilla, C. Goletti, F. Ciccacci, L. Duò, M. Finazzi, A. Goldoni, A. Verdini and L. Floreano, Appl. Surf. Sci., 2020, 505, 144213 CrossRef CAS.
- I. Cojocariu, H. M. Sturmeit, G. Zamborlini, A. Cossaro, A. Verdini, L. Floreano, E. D’Incecco, M. Stredansky, E. Vesselli, M. Jugovac, M. Cinchetti, V. Feyer and C. M. Schneider, Appl. Surf. Sci., 2020, 504, 144343 CrossRef CAS.
- X. Yang, I. Krieger, D. Lüftner, S. Weiß, T. Heepenstrick, M. Hollerer, P. Hurdax, G. Koller, M. Sokolowski, P. Puschnig, M. G. Ramsey, F. S. Tautz and S. Soubatch, Chem. Commun., 2018, 54, 9039–9042 RSC.
- I. Cojocariu, S. Carlotto, H. M. Sturmeit, G. Zamborlini, M. Cinchetti, A. Cossaro, A. Verdini, L. Floreano, M. Jugovac, P. Puschnig, C. Piamonteze, M. Casarin, V. Feyer and C. M. Schneider, Chem. – Eur. J., 2021, 27, 3526–3535 CrossRef CAS.
- P. Donovan, A. Robin, M. S. Dyer, M. Persson and R. Raval, Chem. – Eur. J., 2010, 16, 11641–11652 CrossRef CAS.
- B. W. Heinrich, C. Iacovita, T. Brumme, D. J. Choi, L. Limot, M. V. Rastei, W. A. Hofer, J. Kortus and J. P. Bucher, J. Phys. Chem. Lett., 2010, 1, 1517–1523 CrossRef CAS.
- M. Casarin, F. Ferrigato, C. Maccato and A. Vittadini, J. Phys. Chem. B, 2005, 109, 12596–12602 CrossRef CAS PubMed.
- M. Casarin, C. Maccato and A. Vittadini, J. Phys. Chem. B, 2002, 106, 795–802 CrossRef CAS.
- S. Carlotto, M. Casarin, A. Lanza, F. Nestola, L. Pandolfo, C. Pettinari and R. Scatena, Cryst. Growth Des., 2015, 15, 5910–5918 CrossRef CAS.
- S. Carlotto, M. Sambi, F. Sedona, A. Vittadini, J. Bartolomé, F. Bartolomé and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 28110–28116 RSC.
- T. Houwaart, T. Le Bahers, P. Sautet, W. Auwärter, K. Seufert, J. V. Barth and M.-L. Bocquet, Surf. Sci., 2015, 635, 108–114 CrossRef CAS.
- H. L. Schläfer, Absorption Spectra and Chemical Bonding in Complexes, 2021, https://www.elsevier.com/books/absorption-spectra-and-chemical-bonding-in-complexes/jorgensen/978-0-08-009627-8.
- P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed.
- R. F. Nalewajski and J. Ozek, Int. J. Quantum Chem., 1994, 51, 187–200 CrossRef CAS.
- P. Puschnig, S. Berkebile, A. J. Fleming, G. Koller, K. Emtsev, T. Seyller, J. D. Riley, C. Ambrosch-Draxl, F. P. Netzer and M. G. Ramsey, Science, 2009, 326, 702–706 CrossRef CAS.
- G. Zamborlini, D. Lüftner, Z. Feng, B. Kollmann, P. Puschnig, C. Dri, M. Panighel, G. Di Santo, A. Goldoni, G. Comelli, M. Jugovac, V. Feyer and C. M. Schneider, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- H. M. Sturmeit, I. Cojocariu, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, A. Sala, G. Comelli, S. Moro, M. Stredansky, M. Corva, E. Vesselli, P. Puschnig, C. M. Schneider, V. Feyer, G. Zamborlini and M. Cinchetti, J. Mater. Chem. C, 2020, 8, 8876–8886 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc02222a |
| ‡ Present address: Technische Universität Dortmund, Experimentelle Physik VI, 44227 Dortmund, Germany. |
| § Present address: Istituto di Struttura della Materia-CNR (ISM-CNR), 34149 Trieste, Italy. |
| This journal is © The Royal Society of Chemistry 2021 |