
Interplay between core and shell in a RbCoFe@RbNiCo Prussian blue analogue spin transition heterostructure†
Wanhong
He
 a,
John M.
Cain
a,
Mark W.
Meisel
*b and
Daniel R.
Talham
*a
a,
John M.
Cain
a,
Mark W.
Meisel
*b and
Daniel R.
Talham
*a
aDepartment of Chemistry, University of Florida, Gainesville, FL 32611-7200, USA. E-mail: meisel@phys.ufl.edu; talham@chem.ufl.edu
bDepartment of Physics and National High Magnetic Field Laboratory, University of Florida, Gainesville, FL 32611-8440, USA
First published on 11th June 2021
Abstract
A series of core–shell heterostructures consisting of the spin transition Prussian blue analogue RbaCob[Fe(CN)6]c·mH2O (RbCoFe–PBA) as core with different shell thicknesses of KjNik[Co(CN)6]l·nH2O (KNiCo-PBA) has been prepared and studied as the cores undergo both thermal and light-induced phase changes. Synchrotron powder diffraction and SQUID magnetometry indicate the intersite cooperativity of the charge transfer coupled spin transition (CTCST) in the RbCoFe–PBA core decreases while the extent of lattice contraction is reduced relative to the uncoated particles. Isothermal relaxation measurements from the photo-induced high-spin (HS) state to the low-spin (LS) ground state of the RbCoFe–PBA core show that the energy barrier of the HS to LS transition dramatically decreases when adding the KNiCo-PBA shells, becoming smaller when the shell is thicker. The RbCoFe–PBA@KNiCo-PBA series is unique because the lattice parameter of KNiCo-PBA grown on the high-spin RbCoFe–PBA core particle is expanded relative to its equilibrium lattice parameter. As a result, the lattice mismatch is relieved during the spin transition. Analysis of the structural microstrain in both core and shell during the CTCST process reveals the different mechanisms by which the heterostructure accommodates the strain.
Introduction
Bistability in advanced materials, whether organic or inorganic, molecular or solid-state, is crucial to many applications, and is no more elegantly demonstrated than in the work of Rovira and Veciana.1–3 Spin transition solids, including spin crossover compounds4,5 exhibit bistability switchable with external stimuli including temperature,6 light,7 pressure,8 and magnetic field,9 as well as by chemical changes,10 and attract interest for potential applications in areas of sensors, photonic switches, and information storage.4,11,12 Spin transition solids are often based on transition metal ions, meaning spin state changes can be accompanied by alterations of magnetism, dielectric constant and color, in addition to changes in structure.13–16 The structural changes, a consequence of altering metal–ligand bond distances as the metal ion's electronic structure is switched, lead to further applications as mechanical actuators.5,17Increasingly, spin transition materials are studied at the nanoscale and mesoscale, as well as in architectures placing the spin transition solids at interfaces with other materials,14,18–23 for example when included in a polymer matrix24 or combined with another solid in a thin film25,26 or particle heterostructure.18–20,27–30 Experimental results, backed up by theoretical predictions, reveal that at small length scales a surrounding matrix can have profound influence on spin transition properties.14,18,23,31–33 The interface or matrix stabilizes the as-prepared spin state, most often the high-spin (HS) state, causing transition temperatures to move to lower temperature, but can also influence the mechanism and order of the phase change by altering the elastic properties of the spin transition material. To experimentally probe matrix effects, it is helpful to use a platform that allows systematic changes while controlling the nature of coupling at the interface. Drastic differences in interface coupling can mask the influence of other factors, such as the matrix elastic properties. The spin transition in cubic cobalthexacyanoferrate Prussian blue analogues (PBAs) have been useful in this respect because of the large family of isostructural PBAs that can form particle and thin film heterostructures with consistent interfaces.
For many CoFe–PBA's the cyanide bridged cobalt–iron pairs can exist as either Fe2+–CN–Co(LS)3+ or Fe3+–CN–Co(HS)2+ with the cobalt ion undergoing a spin-state change.34–37 The transition between these charge states can be either thermally or optically activated, with the thermal transition occurring slightly below room temperature and the light-induced LS to metastable HS transition accessible below about 150 K, depending on the composition.35–37 Since its discovery, the combined electron transfer/spin transition process has been commonly referred to as a charge transfer induced spin transition (CTIST). However, a recent study of the light-induced transition in nanocrystals of Cs0.7Co(Fe(CN)6)0.9 demonstrates the Co–ion spin crossover precedes the charge transfer, which occurs on the hundreds of fs timescale.38 Although it is uncertain whether the mechanism is the same for the thermal process, or indeed for all CoFe–PBA compositions, the acronym CTIST is potentially incorrect and misleading. Therefore, in this article we will use the acronym CTCST for charge transfer coupled spin transition. However, it is important to recognize it is the same process referred to in other literature as CTIST.
The CTCST of the CoFe–PBA core in core–shell particles has been shown to alter the properties, notably magnetization, of shell materials. Light-induced changes are seen for NiCr-PBA, CoCr-PBA and CrCr-PBA when grown as shells, with persistent photomagnetism up to 125 K for the CrCr-PBA shell, ultimately limited by the thermal relaxation of the spin transition core.18–20,25 In these examples, the volume change of the CoFe–PBA (ΔV/V ∼ 10%) induces a magnetomechanical response in the shell components through elastic coupling across the heterostructure interfaces. Structural studies show the distortion of the shell as well as the changes in microstrain in both core and shell as the CoFe–PBA core undergoes the thermal or optical spin transition.19,28,39 At the same time, the core–shell architecture allows changes to the spin transition core to be quantified through varying shell thickness. An important finding was that a KNiCr-PBA shell changes the kinetics of the core spin transition.40 Isothermal relaxation measurements of the decay of the light-induced HS state revealed a dramatically reduced activation energy in core–shell particles relative to the uncoated CoFe–PBA particles, and the activation energy decreased as the shell became thicker. The shell restricts the ability of the HS core to contract, reducing the HS to LS volume change of the core, thereby decreasing the elastic intersite interactions that contribute to the activation barrier.40
In these earlier examples of core–shell particles based on HS CoFe–PBA, the lattice constants of the PBA shell materials have all been well-matched to, or slightly larger than, that of the core lattice. In each of these cases, as the core undergoes the HS to LS transition, it induces strain in the shell. The present study looks at a different core–shell combination, with shells of KNiCo-PBA whose equilibrium unit cell constant of 10.14 Å is intermediate between those of the RbCoFe–PBA core in the HS and LS states. When compared to the RbCoFe–PBA@KNiCr-PBA series studied earlier, the influence of the core–shell lattice matching on the core spin transition can be assessed. Furthermore, as there is clear interplay between core and shell during the phase change, the influence of the smaller shell lattice constant on the distortion of the shell can also be evaluated.
Experimental section
Materials
All chemical reagents were purchased from Sigma-Aldrich and used without further purification. Deionized water was obtained from a Barnstead NANOpure filtration system.Synthesis
All PBA particles were synthesized and isolated under ambient laboratory conditions. The procedure for coating RbCoFe–PBA particles with KNiCo-PBA shells is modified from the method reported by Risset et al.19 Instead of using a portion of a thinner shell sample as the starting particles for the next stage of shell growth in a separate synthesis, samples with different shell thickness were isolated from a single reaction by separating a portion of the particle suspension at different stages during of the addition of the shell precursors. This method allows similar core and interface compositions in all core–shell particles, even as shell thickness changes.RbCoFe@KNiCo core–shells
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.76 (Co:Fe).
000 rpm for 10 min and subsequently washing with 240 mL of water. The product was isolated and air-dried. The mass of the product is 15 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.01Ni[Co(CN)6]0.67}0.34·nH2O. Particle size 166 (SD 15) nm. Shell thickness 7 nm. Purple powder. IR: 2161 cm−1 (νCN, CoII(HS)-NC-FeIII); 2113 cm−1 (νCN, CoIII(LS)-NC-FeII); 2097 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII). ICP: 0.94:0.57:0.26 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 240 mL of water. The product was isolated and air-dried. The mass of the product was 27 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.04Ni[Co(CN)6]0.68}0.66·nH2O. Particle size 191 (SD 13) nm. Shell thickness 20 nm. Pinkish purple powder. IR: 2162 cm−1 (νCN, CoII(HS)-NC-FeIII); 2114 cm−1 (νCN, CoIII(LS)-NC-FeII); 2097 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII). ICP: 0.82:0.43:0.37 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 240 mL of water. The product was isolated and air-dried. The mass of the product was 41 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.07Ni[Co(CN)6]0.69}2.42·nH2O. Particle size 191 (SD 13) nm. Shell thickness 55 nm. Pink powder. IR: 2164 cm−1 (νCN, CoII(HS)-NC-FeIII); 2114 cm−1 (νCN, CoIII(LS)-NC-FeII); 2098 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII); 2129 cm−1 (νCN, CoIII–CN terminal). ICP: 1.38:0.39:1.25 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 450 mL of water. The product was isolated and air-dried. The mass of the product is 60 mg. Ni3[Co(CN)6]2·nH2O. Particle size 128 (SD 19) nm. Light blue powder.
:0.76 (Co:Fe).
000 rpm for 10 min and subsequently washing with 240 mL of water. The product was isolated and air-dried. The mass of the product is 15 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.01Ni[Co(CN)6]0.67}0.34·nH2O. Particle size 166 (SD 15) nm. Shell thickness 7 nm. Purple powder. IR: 2161 cm−1 (νCN, CoII(HS)-NC-FeIII); 2113 cm−1 (νCN, CoIII(LS)-NC-FeII); 2097 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII). ICP: 0.94:0.57:0.26 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 240 mL of water. The product was isolated and air-dried. The mass of the product was 27 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.04Ni[Co(CN)6]0.68}0.66·nH2O. Particle size 191 (SD 13) nm. Shell thickness 20 nm. Pinkish purple powder. IR: 2162 cm−1 (νCN, CoII(HS)-NC-FeIII); 2114 cm−1 (νCN, CoIII(LS)-NC-FeII); 2097 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII). ICP: 0.82:0.43:0.37 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 240 mL of water. The product was isolated and air-dried. The mass of the product was 41 mg. Rb0.28Co[Fe(CN)6]0.76@{K0.07Ni[Co(CN)6]0.69}2.42·nH2O. Particle size 191 (SD 13) nm. Shell thickness 55 nm. Pink powder. IR: 2164 cm−1 (νCN, CoII(HS)-NC-FeIII); 2114 cm−1 (νCN, CoIII(LS)-NC-FeII); 2098 cm−1 (νCN, CoII-NC-FeII); 2182 cm−1 (νCN, NiII–NC–CoIII); 2129 cm−1 (νCN, CoIII–CN terminal). ICP: 1.38:0.39:1.25 (Co:Fe:Ni).
000 rpm for 10 min and subsequently washed with 450 mL of water. The product was isolated and air-dried. The mass of the product is 60 mg. Ni3[Co(CN)6]2·nH2O. Particle size 128 (SD 19) nm. Light blue powder.
Characterization
Fourier transform infrared spectra (FTIR) was performed using a Nicolet 6700 Thermo Scientific spectrophotometer. A background reference of 32 scans was taken between 4000 cm−1 and 1500 cm−1 with a multi-reflection silicon ATR crystal using a Harrick ATR accessory. To prepare the FTIR sample, 200 μL of an acetone suspension (1 mL) containing 0.5 mg of sample powder was dropped onto the surface of the ATR crystal and allowing the solvent to evaporate. Transmission electron microscopy (TEM) was performed in a JEOL-2010F high-resolution transmission electron microscope at 200 kV. TEM samples were prepared by adding dropwise 80 μL of an acetone suspension to a TEM grid (400 mesh copper with holey carbon support film from Ted-Pella, Inc.) and allowing each grid to dry. Here, the suspension was prepared by dispersing 0.5 mg of product in 1 mL of acetone via sonication. TEM was also conducted in a Hitachi H-7000 conventional transmission electron microscope at 100 kV to obtain low-resolution images. Average particle size was measured using ImageJ imaging software based on TEM images taken of various areas in the sample. At least 100 particles were measured to obtain the average values and standard deviations. Shell thickness was one half of difference between the average core–shell size and the average core particle size. Inductively coupled plasma atomic emission spectroscopy (ICP) was performed on a Varian Vista RL simultaneous spectrometer (Agilent Technologies, Santa Clara, California, USA) with a CCD-detector. ICP standard solutions containing 1 ppm, 10 ppm, and 100 pm of tested elements were used to create a calibration line. The ICP solutions were prepared by diluting the Honeywell Fluka analytical standards (1000 ppm). Chemical formulas are based on the metal compositions from ICP. Alkali cation content was determined based on electroneutrality.Synchrotron powder X-ray diffraction (PXRD) data were collected at beamline 28-ID-2 at the National Synchrotron Light Source-II at Brookhaven National Laboratory. A flat-panel amorphous–Si area detector was positioned 1400 mm from the RbCoFe@KNiCo core–shell samples. The X-ray wavelength of λ = 0.187156 Å was used for RbCoFe@KNiCo core–shell samples. Calibration was performed using LaB6 and Ni metal. Samples were loaded into borosilicate capillaries (0.1 mm i.d.) and exposed for 5 s per pattern while the capillary was rotated at 1 Hz. Variable temperature PXRD (VT-PXRD) data were collected as temperature was ramped at 2 K min−1 from 300 to 100 K. Diffraction images were integrated using GSAS-II,41 based on the LaB6 standard.41 The structural analysis was performed via Pawley fitting within GSAS-II (v. 4570). For the anisotropic microstrain refinements, a custom refinement routine utilizing the GSAS-II scriptable package42 was run through Anaconda Jupyter notebook v. 6.0.3.
Magnetization was performed on a Quantum Design Magnetic Property Measurement System (MPMS) model XL-7 superconducting quantum interference device (SQUID) magnetometer. The dark state measurements were performed with the sample in a gel cap inside a drinking straw in a commercial sample rod. The field-cooled temperature dependence of the magnetization was measured in an applied field of 100 G while warming in the 5–300 K region. The temperature sweep rate was 2 K min−1. For the isothermal relaxation measurements, samples were field cooled from 300 to 100 K at 2 K min−1 in a field of 1 T. They were then allowed to sit at 100 K for about 1 hour before irradiation with a Quartzline tungsten halogen lamp (400–2200 nm), delivering nominally 4 mW to the sample, using an optical sample rod described elsewhere.43 The samples were irradiated for at least 5 hours, then warmed quickly to the temperature of interest and held for up to 20 hours to observe the relaxation from the optically induced metastable HS state back to the LS state.
Results
Synthesis and characterization
RbCoFe–PBA nanoparticles were prepared as a self-stabilized suspension in water, using a heterogeneous precipitation method first developed by Catala and co-workers.44 to yield uniform particles in the mesoscale size regime (50–500 nm).18,19,44,45 Shell layers of KNiCo-PBA were formed by slowly adding low concentration precursor solutions to suspensions of the uncoated RbCoFe–PBA core particles, leading to the heterogeneous precipitation of the shell material while preventing side nucleation.18,19,27 All the core–shell heterostructures in this study were prepared using the same batch of core particles to maintain uniformity of size and ensure consistency of the core–shell interface.The particle morphology within the series is shown in TEM images (Fig. 1). The uncoated RbCoFe–PBA particles are cubes with sizes uniformly distributed around 151 nm (Fig. 1A). After addition of the KNiCo-PBA shells the particles remain cubic, now with slightly round corners (Fig. 1B–D). The difference in contrast between core and shell shows the clear interface between two PBA components, confirming the core–shell architecture. Chemical composition is determined by ICP, and Pawley refinements of PXRD patterns (Fig. S1, ESI†) of each core–shell sample are consistent with two face centered cubic lattices, corresponding to the RbCoFe–PBA cores and the KNiCo-PBA shells.40,46
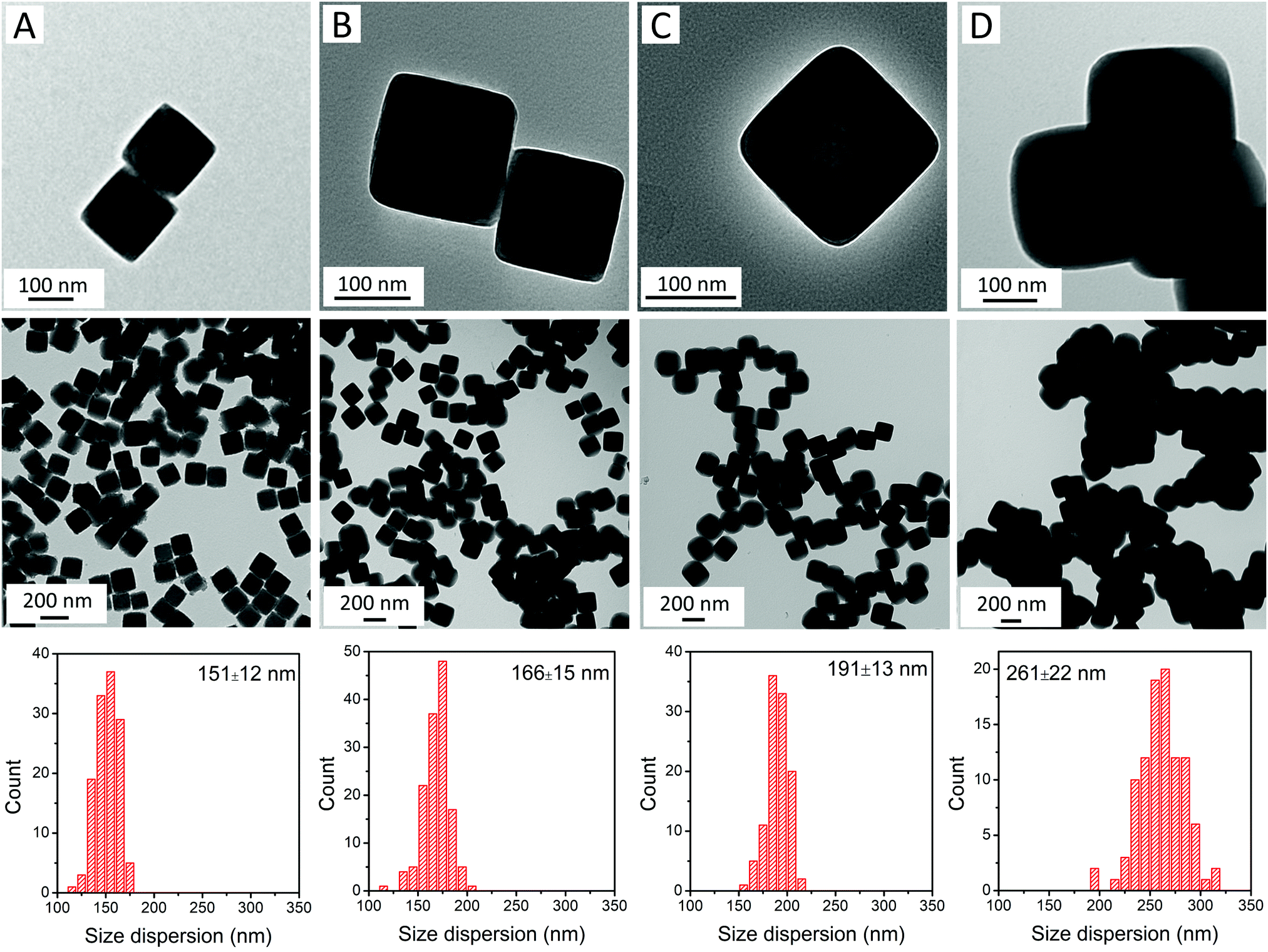 | ||
| Fig. 1 TEM images and size dispersions of (A) uncoated RbCoFe–PBA particles; and core–shell particles of samples (B) RbCoFe@KNiCo-7 nm; (C) RbCoFe@KNiCo-20 nm; and (D) RbCoFe@KNiCo-55 nm. | ||
High temperature magnetization
Magnetization measurements, presented as molar magnetic susceptibility (χM) times temperature versus temperature, show that the RbCoFe–PBA CTCST is retained in each of the RbCoFe@KNiCo core–shell samples. From 280 K to 160 K, the χMT value decreases, mainly due to the thermal CTCST in the RbCoFe–PBA core. The transition temperature, defined from the derivatives of the χMT vs. T plots, moves lower with increasing shell thickness and the transition becomes more gradual. The trend is similar to that seen in a previous study on RbCoFe–PBA particles coated by KCoCr-PBA shells.19 The higher room temperature χMT values in particle samples with thicker shells can be ascribed to the larger contribution from the paramagnetic KNiCo-PBA shell (Fig. S3A, ESI†). The magnitude of the decrease of χMT associated with the thermal CTCST is slightly lower in the samples with thicker shells, indicating the level of residual non-transitioning HS CoII-NC-FeIII pairs increases with increasing shell thickness (ESI†).Detailed structural studies
Lattice parameters derived from Pawley refinement of the core–shell and single phase particles at temperatues above and below the RbCoFe–PBA thermal spin transition are recorded in Table 1.| 300 K | 100 K | Δa (Å) | ||||
|---|---|---|---|---|---|---|
| a core (Å) | a shell (Å) | a core (Å) | a shell (Å) | Δacore | Δashell | |
| RbCoFe–PBA39 | 10.30(6) | — | 9.95(8) | — | 0.34(8) | — |
| RbCoFe@KNiCo-7 nm | 10.30(8) | 10.20(7) | 9.99(7) | 10.03(4) | 0.31(1) | 0.17(3) |
| RbCoFe@KNiCo-20 nm | 10.29(5) | 10.17(6) | 10.01(0) | 10.05(7) | 0.28(5) | 0.11(9) |
| RbCoFe@KNiCo-55 nm | 10.28(6) | 10.14(7) | 10.05(5) | 10.10(6) | 0.23(1) | 0.04(1) |
| KNiCo-PBA | — | 10.14(1) | — | — | — | — |
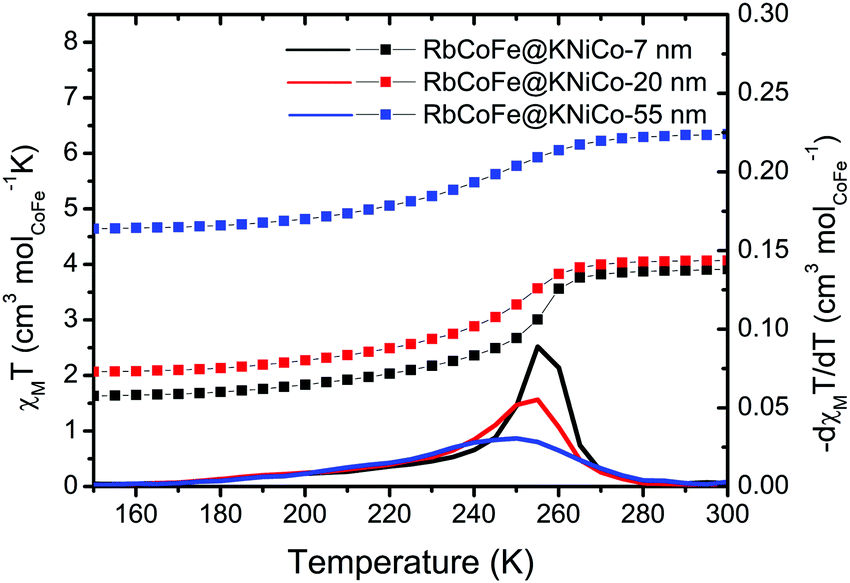 | ||
| Fig. 2 χ M T vs. T plots under a field of 100 G in the region of the thermal CTCST for the core–shell heterostructures. The derivatives of χMT are plotted as solid lines. | ||
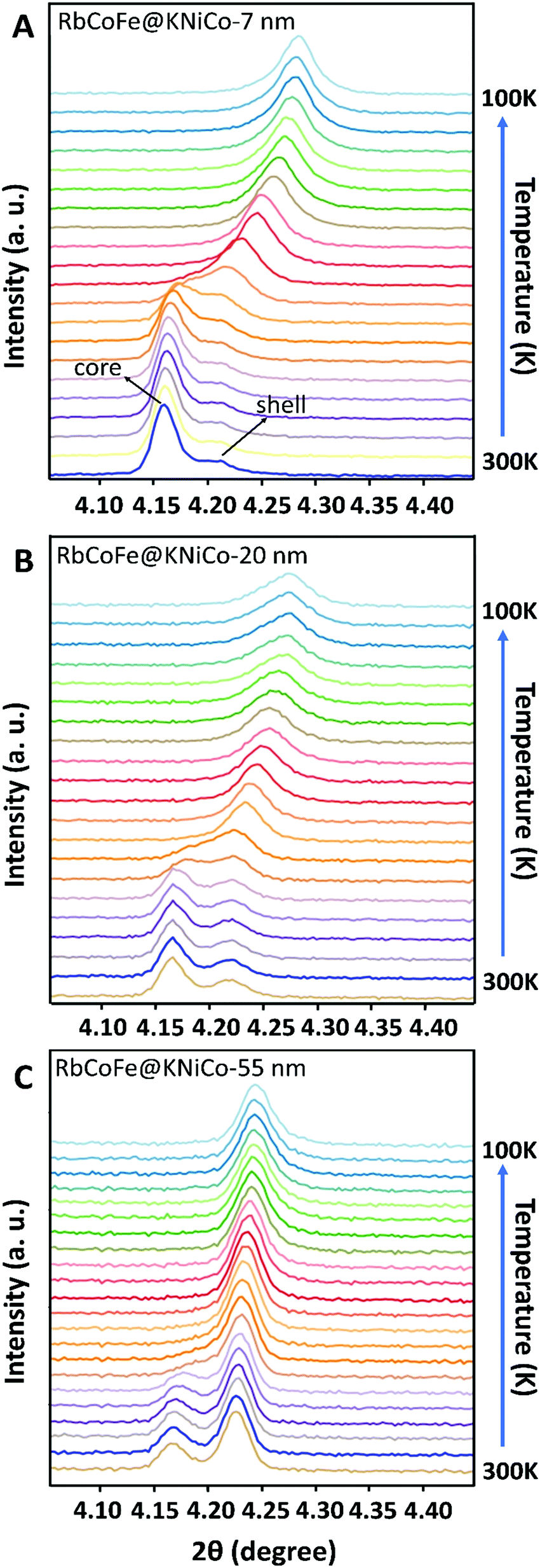 | ||
| Fig. 3 PXRD patterns showing the evolution of the 400 reflections for both core and shell as a function of temperature for the core–shell heterostructures: (A) RbCoFe@KNiCo-7 nm; (B) RbCoFe@KNiCo-20 nm; (C) RbCoFe@KNiCo-55 nm. Upon cooling, the peak of the core shifts to higher 2θ angle and overlaps with that of the shell. | ||
At the same time, the (400) reflection of the KNiCo-PBA shells also shifts, signaling the phase transition in the core induces structural changes in the shell, and the changes are strikingly large. Unit-cell analyses were performed for the core–shell heterostructures at high and low temperature, and lattice parameters are included in Table 1. For the thinnest shell sample, Δashell = 0.17 Å, shifting from a = 10.20 Å, larger than the KNiCo-PBA equilibrium value of a = 10.14 Å, to a = 10.03 Å, which is compressed. This change is significantly larger than has been observed for other PBA core–shell examples.19,39,40 For example, a RbCoFe–PBA@KCoCr-PBA core–shell heterostructure with an 11 nm shell undergoes only an 0.08 Å decrease after the thermal HS to LS transition of the core is completed.19 Other examples have shown similar changes.19,39,40 The unusual response here for the RbCoFe–PBA@KNiCo-PBA case is a result of the KNiCo-PBA equilibrium cell size falling between those of the HS and LS core lattice constants. The shell is expanded when grown on the RbCoFe–PBA HS phase and is then compressed when core is in the LS state. The magnitude of the change in the KNiCo-PBA shell (Δashell) is smaller with increasing shell thickness, Δashell = 0.11 for the 20 nm shell and Δashell = 0.04 for the 55 nm shell, indicating the shell is less compliant as it becomes thicker.
The change in the core also varies with shell thickness, with the lattice parameter of the LS state increasing for the thicker shells, resulting in a decrease of Δa of the core. Similar trends have been reported in other RbCoFe@PBA core–shell examples, attributed to the influence of the core–shell interface restricting compression.19,32,33,39,40 However, in the present example, the presence of residual HS fraction below the thermal transition should also contribute, making it difficult to separate the two influences by comparing lattice constants. Interestingly, for each of the core–shell samples, the (400) peaks from core and shell appear to merge together as a single set of reflections, suggesting the lattice mismatch between the core and shell disappears during the HS state to LS transition. As the temperature further decreases to 100 K, the peak shape turns asymmetric, seen most clearly in the RbCoFe@KNiCo-20 nm where the peak intensity of the shell is somewhat discernible at low temperature (Fig. S3, ESI†), indicating the lattice mismatch reappears with the core value now smaller than the shell when the core contracts further.
Isothermal relaxation
The presence of a shell has been shown to influence the light-induced spin transition of RbCoFe–PBA particles.40 Whereas it is difficult to experimentally quantify the characteristic rate of the light-induced LS to HS transition in a solid-state sample, the reverse process, isothermal relaxation from the light-induced HS state (Fig. 4), can be reproducibly measured and used to characterize the effect of a shell on the kinetics of the spin transition in the core.40,50–52 Samples are first cooled from room temperature to below the thermal spin transition temperature to achieve the LS state (100 K in this work) and then excited to the metastable HS state upon exposure to the light. After the sample reaches equilibrium, the light is turned off and the sample is quickly warmed to one of several target temperatures where the decay of the HS fraction is then monitored. Results for the RbCoFe@KNiCo-20 nm and RbCoFe@KNiCo-55 nm samples are plotted in Fig. S4 and S5 (ESI†) using magnetization to monitor the change in HS fraction. The time constants of exponential decay at different temperatures are used to determine activation energies from Arrhenius plots, as in Fig. S6 (ESI†). Complete kinetic data are displayed in Table S2 (ESI†) and the activation energies are plotted as a function of shell thickness in Fig. 4.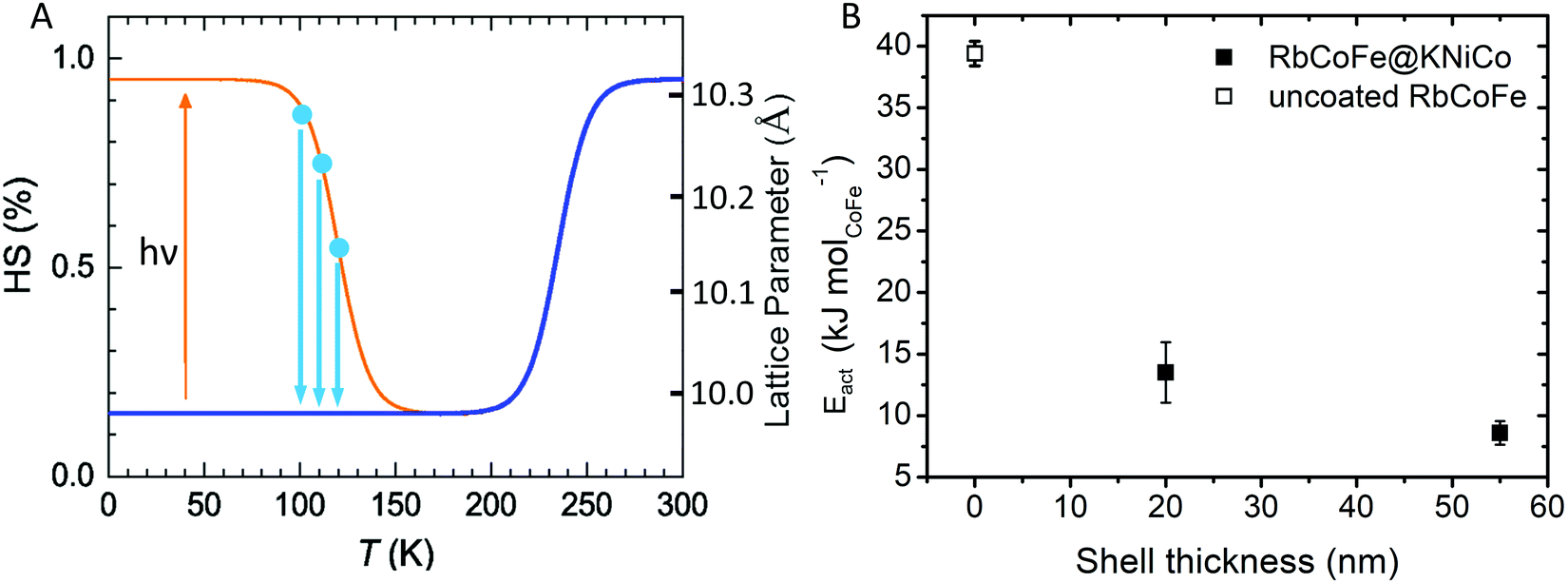 | ||
| Fig. 4 (A) Schematic of the isothermal relaxation process. The sample is first cooled from room temperature (blue line) and the metastable HS state is established by light irradiation (yellow). The sample is then warmed to target temperatures (blue dots), where the decay of the HS state is monitored (blue arrows). (B) Activation energy vs. shell thickness for the relaxation from the metastable HS state of RbCoFe–PBA as extracted from Arrhenius plots for the uncoated RbCoFe–PBA core and the core–shell heterostructures.40 | ||
The activation energy decreases significantly in the core–shell materials. For the RbCoFe@KNiCo-20 nm sample, Eact = 13.5 kJ mol−1, which is greatly reduced relative to the uncoated RbCoFe–PBA with Eact = 39.4 kJ mol−1. The activation energy decreases even further as the KNiCo-PBA shell becomes thicker, reaching Eact = 8.6 kJ mol−1 for RbCoFe@KNiCo-55 nm. The changes to the core spin transition, from metastable HS state to LS state, are very similar to what was observed for RbCoFe–PBA@KNiCr-PBA core shell heterostructures studied previously. For example, the value of Eact measured here for the RbCoFe@KNiCo-55 nm sample is nearly the same as for a RbCoFe–PBA@KNiCr-PBA sample with a 52 nm shell on a similar size core particle.40 Even though the relationship between equilibrium lattice constants in the core and shell are different in the two systems, the KNiCr-PBA lattice constant is larger than the HS RbCoFe–PBA, the influence on the activation energies of the core spin transition is similar.
Discussion
Effect of shell on core spin transition
The gradual structural transition is seen in the temperature dependent PXRD of Fig. 3 and is also reflected in the temperature dependence of the microstrain. Microstrain can be quantified during pattern refinement using methodology introduced by Stephens56 yielding two independent strain parameters, S220 and S400, characterizing the effect of anisotropic strain on the reflection profiles of the cubic systems. The S400 parameter describes the microstrain from h00 reflections, while both S400 and S220 describe the microstrain from hkl reflections. Using this method, the microstrain in both the RbCoFe–PBA core and the KNiCo-PBA shell are able to be followed as a function of temperature, as shown in the plots of Fig. 5. The plots are limited to analyses above ∼230 K because the diffraction patterns overlap when the lattice constants converge below this temperature, removing the ability to distinguish the core and shell diffraction peaks.
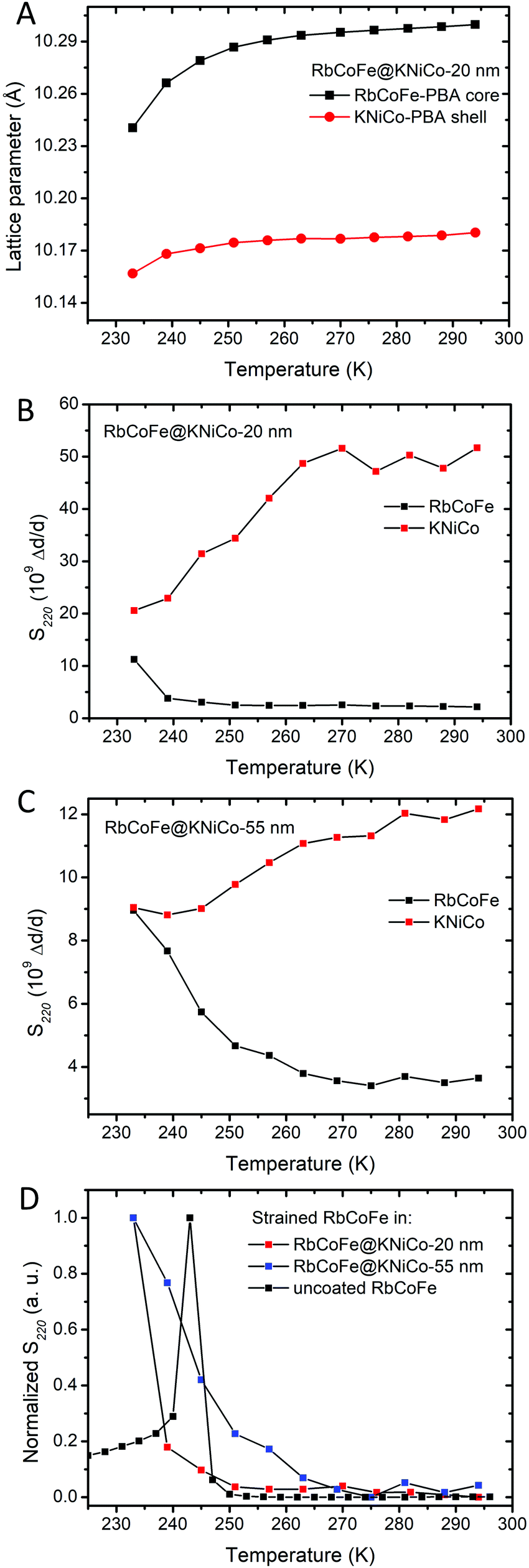 | ||
| Fig. 5 (A) The change of lattice parameters of the core and the shell in RbCoFe@KNiCo-20 nm upon cooling. (B) Microstrains (S220) of RbCoFe@KNiCo-20 nm. (C) Microstrains (S220) of RbCoFe@KNiCo-55 nm. (D) Comparison of microstrains (S220) in the uncoated RbCoFe–PBAs40 and RbCoFe–PBA coated with shells. The y-axis of (D) is normalized for the purpose of comparing the changes of microstrains within different samples. | ||
Fig. 5C summarizes the changes of microstrain in RbCoFe–PBA cores during the thermal CTCST process, comparing the RbCoFe@KNiCo-20 nm and RbCoFe@KNiCo-55 nm core–shell samples to the uncoated RbCoFe–PBA particles. The magnitudes are not readily compared between samples, so the intensities of the S400 and S220 are normalized in these plots to compare the temperature dependence of the changes. When RbCoFe–PBA particles are not coated by the shell, the microstrain shows a sharp spike below 250 K which is due to the discontinuous first order transition. Below the transition temperature, the microstrain in RbCoFe–PBA gradually decreases when the thermal CTCST process approaches completion. For both the RbCoFe@KNiCo-20 nm and RbCoFe@KNiCo-55 nm samples, the change is more gradual, consistent with the lattice constant and magnetometry temperature profiles. The more gradual change of microstrain is consistent with tensile stress on the core as it undergoes the HS to LS transition. Together, the magnetometry, unit cell changes and strain analyses all reflect how the shell hinders the contraction of the core during the structural phase transition, leading to a modulation of the intersite interaction,32,33,40,57,58 and the thicker shell has a larger effect. When the intermolecular interactions in the core become weaker, the cooperativity decreases.31
When compared to the KNiCr-PBA shell series reported by Felts et al.,40 the results from the KNiCo-PBA series indicate the relative core and shell lattice constants, or lattice mismatch, does not have a significant influence on the spin transition activation energy, at least at this length scale. Whereas the thickness of the shell has a significant effect, the two series show nearly identical values of Eact at similar shell thickness. As will be discussed in the next section, the lattice mismatch may alter the phase change for core sites near the core–shell interface, but does not to change the overall kinetics at the 100 nm – 200 nm length scale of the core particles in the present series.
Effect of the lattice mismatch and volume change in the core on the microstrain in the shell
However, a significant difference between the thin and thick shell is evident when comparing the temperature profiles of the shell microstrain to the respective cores. For the 20 nm shell sample, the S220 parameter of the KNiCo-PBA shell begins to dramatically decrease near 260 K upon cooling with the change largely complete by 240 K. On the other hand, the core microstrain is nearly unchanged over this same temperature range, before rising significantly only below 240 K. In contrast, for the 55 nm shell case, the changes in the S220 parameter of the core and shell effectively mirror each other, the shell microstrain decreases while that of the core increases, with both undergoing significant changes over the same temperature range beginning near 260 K.
Shell diffraction in the thin shell sample will contain significant contributions from material near the core–shell interface, whereas the larger dimension of the core means diffraction will largely reflect the state of the core interior. The fact that the microstrain in the 20 nm shell decreases before significant changes are seen in the core is evidence that the spin transition may indeed initiate near the interface before switching in the bulk of the particle. However, the continuous core transition, observed in magnetometry and structural studies, means even if the transition does first occur near the interface, it does not necessarily seed domain growth.31
The different temperature profiles of the core and shell microstrain in the thin and thick shell samples indicates that within the thin shell heterostructure, the structural strain from the volume change associated with the core phase transition is first accommodated by shell, whereas with a thicker shell, more of the strain is maintained in the core.60 This experimental result is predicted by both continuum mechanics and atomistic models.33,57 With increasing shell thickness, up to the limit of the strain depth,59 the elastic energy density of the shell decreases. At the same time, the elastic energy density of the core increases. The stiffness of the shell increases with thickness, forcing the core to accommodate a greater fraction of the elastic energy.61
Conclusions
Core–shell particles of Prussian blue analogues remain a useful platform for experimentally probing the interplay between a spin transition material and a surrounding matrix. Thin shells grown on RbCoFe–PBA spin transition core particles are generally strained due to small differences in the lattice constants of the different PBA's and the KNiCo-PBA shells, with equilibrium lattice constant in between those of the HS and LS phase of RbCoFe–PBA, are expanded in the as grown particles. As the core contracts, the shell approaches its equilibrium structure, the first time the RbCoFe–PBA thermal HS to LS transition has been observed to relieve strain at an interface. In other examples, the thermal transition has always induced strain. Furthermore, the RbCoFe@KNiCo system provides a new example of a coordination polymer heterostructure in which the kinetics of the spin transition of the core can be controlled by addition of a shell. The shell limits the extent to which the core can contract, thereby lessening intersite interactions that contribute to the elastic barrier of the structural phase change. Finally, structural analyses of both the core and the shell as the RbCoFe@KNiCo particles undergo the thermal spin transition reveal the complexity of the elastic behavior of the heterostructure. Microstrain in the core and shell change over different temperature windows during the thermal transition, suggesting the spin transition initiates near the core–shell interface.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported, in part, by the Division of Materials Research (DMR) at the National Science Foundation (NSF) via DMR-1904596 (D. R. T.), DMR-1708410 (M. W. M.), and DMR-1644779 (National High Magnetic Field Laboratory). This research used the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.References
- E. Laukhina, J. Vidal-Gancedo, V. Laukhin, J. Veciana, I. Chuev, V. Tkacheva, K. Wurst and C. Rovira, J. Am. Chem. Soc., 2003, 125, 3948–3953 Search PubMed.
- D. Ruiz-Molina, J. Veciana, K. Wurst, D. N. Hendrickson and C. Rovira, Inorg. Chem., 2000, 39, 617–619 Search PubMed.
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 Search PubMed.
- I. Boldog, A. B. Gaspar, V. Martínez, P. Pardo-Ibañez, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Angew. Chem., 2008, 120, 6533–6537 Search PubMed.
- A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 Search PubMed.
- B. Brachňaková, J. Adamko Kožíšková, J. Kožíšek, E. Melníková, M. Gál, R. Herchel, T. Dubaj and I. Šalitroš, Dalton Trans., 2020, 49, 17786–17795 Search PubMed.
- M. Nakaya, R. Ohtani, L. F. Lindoy and S. Hayami, Inorg. Chem. Front., 2021, 8, 484–498 Search PubMed.
- S. Chorazy, T. Charytanowicz, D. Pinkowicz, J. Wang, K. Nakabayashi, S. Klimke, F. Renz, S.-I. Ohkoshi and B. Sieklucka, Angew. Chem., Int. Ed., 2020, 59, 15741–15749 Search PubMed.
- R. Rabelo, L. Toma, N. Moliner, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, R. Ruiz-García and J. Cano, Chem. Commun., 2020, 56, 12242–12245 Search PubMed.
- K. Keisers, H. M. Hüppe, L. Iffland-Mühlhaus, A. Hoffmann, C. Göbel, U.-P. Apfel, B. Weber and S. Herres-Pawlis, Inorg. Chem., 2020, 59, 15343–15354 Search PubMed.
- L. Catala and T. Mallah, Coord. Chem. Rev., 2017, 346, 32–61 Search PubMed.
- A. B. Gaspar, V. Ksenofontov, M. Seredyuk and P. Gütlich, Coord. Chem. Rev., 2005, 249, 2661–2676 Search PubMed.
- R. Torres-Cavanillas, R. Sanchis-Gual, J. Dugay, M. Coronado-Puchau, M. Giménez-Marqués and E. Coronado, Adv. Mater., 2019, 31, 1900039 Search PubMed.
- C. R. Gros, M. K. Peprah, B. D. Hosterman, T. V. Brinzari, P. A. Quintero, M. Sendova, M. W. Meisel and D. R. Talham, J. Am. Chem. Soc., 2014, 136, 9846–9849 Search PubMed.
- A. Galet, A. B. Gaspar, M. C. Muñoz, G. V. Bukin, G. Levchenko and J. A. Real, Adv. Mater., 2005, 17, 2949–2953 Search PubMed.
- V. Rubio-Giménez, S. Tatay and C. Martí-Gastaldo, Chem. Soc. Rev., 2020, 49, 5601–5638 Search PubMed.
- M. Piedrahita-Bello, J. E. Angulo-Cervera, R. Courson, G. Molnár, L. Malaquin, C. Thibault, B. Tondu, L. Salmon and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6001–6005 Search PubMed.
- M. F. Dumont, E. S. Knowles, A. Guiet, D. M. Pajerowski, A. Gomez, S. W. Kycia, M. W. Meisel and D. R. Talham, Inorg. Chem., 2011, 50, 4295–4300 Search PubMed.
- O. N. Risset, P. A. Quintero, T. V. Brinzari, M. J. Andrus, M. W. Lufaso, M. W. Meisel and D. R. Talham, J. Am. Chem. Soc., 2014, 136, 15660–15669 Search PubMed.
- O. N. Risset, T. V. Brinzari, M. W. Meisel and D. R. Talham, Chem. Mater., 2015, 27, 6185–6188 Search PubMed.
- A. Mosey, A. S. Dale, G. Hao, A. N’Diaye, P. A. Dowben and R. Cheng, J. Phys. Chem. Lett., 2020, 11, 8231–8237 Search PubMed.
- M. Piedrahita-Bello, B. Martin, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, J. Mater. Chem. C, 2020, 8, 6042–6051 Search PubMed.
- N. Konstantinov, A. Tauzin, U. N. Noumbé, D. Dragoe, B. Kundys, H. Majjad, A. Brosseau, M. Lenertz, A. Singh, S. Berciaud, M.-L. Boillot, B. Doudin, T. Mallah and J.-F. Dayen, J. Mater. Chem. C, 2021, 9, 2712–2720 Search PubMed.
- S. Rat, M. Piedrahita-Bello, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, Adv. Mater., 2018, 30, 1705275 Search PubMed.
- D. M. Pajerowski, M. J. Andrus, J. E. Gardner, E. S. Knowles, M. W. Meisel and D. R. Talham, J. Am. Chem. Soc., 2010, 132, 4058–4059 Search PubMed.
- H. J. Shepherd, I. y. A. Gural'skiy, C. M. Quintero, S. Tricard, L. Salmon, G. Molnár and A. Bousseksou, Nat. Commun., 2013, 4, 2607 Search PubMed.
- M. Presle, I. Maurin, F. Maroun, R. Cortès, L. Lu, R. Sayed Hassan, E. Larquet, J.-M. Guigner, E. Rivière and J. P. Wright, J. Phys. Chem. C, 2014, 118, 13186–13195 Search PubMed.
- A. Adam, M. Poggi, E. Larquet, R. Cortès, L. Martinelli, P.-E. Coulon, E. Lahera, O. Proux, D. Chernyshov, K. Boukheddaden, T. Gacoin and I. Maurin, Nanoscale, 2018, 10, 16030–16039 Search PubMed.
- L. Salmon and L. Catala, C. R. Chim., 2018, 21, 1230–1269 Search PubMed.
- N. Dia, L. Lisnard, Y. Prado, A. Gloter, O. Stéphan, F. Brisset, H. Hafez, Z. Saad, C. Mathonière, L. Catala and T. Mallah, Inorg. Chem., 2013, 52, 10264–10274 Search PubMed.
- A. Slimani, K. Boukheddaden and K. Yamashita, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 014111 Search PubMed.
- H. Oubouchou, A. Slimani and K. Boukheddaden, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 104104 Search PubMed.
- K. Affes, A. Slimani, A. Maalej and K. Boukheddaden, Chem. Phys. Lett., 2019, 718, 46–53 Search PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 Search PubMed.
- N. Shimamoto, S. Ohkoshi, O. Sato and K. Hashimoto, Inorg. Chem., 2002, 41, 678–684 Search PubMed.
- A. Bleuzen, C. Lomenech, V. Escax, F. Villain, F. Varret, C. C. D. Moulin and M. Verdaguer, J. Am. Chem. Soc., 2000, 122, 6648–6652 Search PubMed.
- C. C. D. Moulin, F. Villain, A. Bleuzen, M. A. Arrio, P. Sainctavit, C. Lomenech, V. Escax, F. Baudelet, E. Dartyge, J. J. Gallet and M. Verdaguer, J. Am. Chem. Soc., 2000, 122, 6653–6658 Search PubMed.
- M. Cammarata, S. Zerdane, L. Balducci, G. Azzolina, S. Mazerat, C. Exertier, M. Trabuco, M. Levantino, R. Alonso-Mori, J. M. Glownia, S. Song, L. Catala, T. Mallah, S. F. Matar and E. Collet, Nat. Chem., 2021, 13, 10–14 Search PubMed.
- A. C. Felts, M. J. Andrus, E. S. Knowles, P. A. Quintero, A. R. Ahir, O. N. Risset, C. H. Li, I. Maurin, G. J. Halder, K. A. Abboud, M. W. Meisel and D. R. Talham, J. Phys. Chem. C, 2016, 120, 5420–5429 Search PubMed.
- A. C. Felts, A. Slimani, J. M. Cain, M. J. Andrus, A. R. Ahir, K. A. Abboud, M. W. Meisel, K. Boukheddaden and D. R. Talham, J. Am. Chem. Soc., 2018, 140, 5814–5824 Search PubMed.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 Search PubMed.
- J. H. O'Donnell, R. B. Von Dreele, M. K. Y. Chan and B. H. Toby, J. Appl. Crystallogr., 2018, 51, 1244–1250 Search PubMed.
- E. S. Knowles, PhD thesis, University of Florida, 2013, https://ufdc.ufl.edu/UFE0046071/00001.
- L. Catala, D. Brinzei, Y. Prado, A. Gloter, O. Stéphan, G. Rogez and T. Mallah, Angew. Chem., Int. Ed., 2009, 48, 183–187 Search PubMed.
- D. Brinzei, L. Catala, N. Louvain, G. Rogez, O. Stéphan, A. Gloter and T. Mallah, J. Mater. Chem., 2006, 16, 2593–2599 Search PubMed.
- X. Xu, H. Liang, F. Ming, Z. Qi, Y. Xie and Z. Wang, ACS Catal., 2017, 7, 6394–6399 Search PubMed.
- A. Widmann, H. Kahlert, I. Petrovic-Prelevic, H. Wulff, J. V. Yakhmi, N. Bagkar and F. Scholz, Inorg. Chem., 2002, 41, 5706–5715 Search PubMed.
- Y. Raza, F. Volatron, S. Moldovan, O. Ersen, V. Huc, C. Martini, F. Brisset, A. Gloter, O. Stéphan, A. Bousseksou, L. Catala and T. Mallah, Chem. Commun., 2011, 47, 11501–11503 Search PubMed.
- Y. Yüksel, E. Vatansever and H. Polat, J. Phys.: Condens. Matter, 2012, 24, 436004 Search PubMed.
- C. Chong, M. Itoi, K. Boukheddaden, E. Codjovi, A. Rotaru, F. Varret, F. A. Frye, D. R. Talham, I. Maurin, D. Chernyshov and M. Castro, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 144102 Search PubMed.
- B. Hôo, K. Boukheddaden and F. Varret, Eur. Phys. J. B, 2000, 17, 449–457 Search PubMed.
- K. Boukheddaden, F. Varret, S. Salunke, J. Linares and E. Codjovi, Phase Transitions, 2002, 75, 733–741 Search PubMed.
- G. P. Vishnevskaya, E. N. Frolova, I. V. Ovchinnikov, I. G. Pervova and Z. G. Rezinskikh, Russ. J. Phys. Chem. A, 2010, 84, 1388–1394 Search PubMed.
- P. Durand, S. Pillet, E.-E. Bendeif, C. Carteret, M. Bouazaoui, H. El Hamzaoui, B. Capoen, L. Salmon, S. Hébert, J. Ghanbaja, L. Aranda and D. Schaniel, J. Mater. Chem. C, 2013, 1, 1933–1942 Search PubMed.
- L. Trinh, S. Zerdane, S. Mazérat, N. Dia, D. Dragoe, C. Herrero, E. Rivière, L. Catala, M. Cammarata, E. Collet and T. Mallah, Inorg. Chem., 2020, 59, 13153–13161 Search PubMed.
- P. W. Stephens, J. Appl. Crystallogr., 1999, 32, 281–289 Search PubMed.
- G. Félix, W. Nicolazzi, L. Salmon, G. Molnár, M. Perrier, G. Maurin, J. Larionova, J. Long, Y. Guari and A. Bousseksou, Phys. Rev. Lett., 2013, 110, 235701 Search PubMed.
- G. Félix, M. Mikolasek, G. Molnár, W. Nicolazzi and A. Bousseksou, Eur. J. Inorg. Chem., 2018, 435–442 Search PubMed.
- J. M. Cain, A. C. Felts, M. W. Meisel and D. R. Talham, Chem. Mater., 2021, 33, 246–255 Search PubMed.
- J. M. Cain, W. He, I. Maurin, M. W. Meisel and D. R. Talham, J. Appl. Phys., 2021, 129, 160903 Search PubMed.
- L. D. Landau and E. M. Lifšic, Theory of elasticity, Elsevier, Amsterdam, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01514a |
| This journal is © The Royal Society of Chemistry 2021 |