
Effect of molecular geometry and extended conjugation on the performance of hydrogen-bonded semiconductors in organic thin-film field-effect transistors†‡
Paula
Gómez§
 a,
Jesús
Cerdá§
b,
Miriam
Más-Montoya
a,
Stamatis
Georgakopoulos
a,
Iván
da Silva
c,
Antonio
García
a,
Enrique
Ortí
*b,
Juan
Aragó
*b and
David
Curiel
*a
a,
Jesús
Cerdá§
b,
Miriam
Más-Montoya
a,
Stamatis
Georgakopoulos
a,
Iván
da Silva
c,
Antonio
García
a,
Enrique
Ortí
*b,
Juan
Aragó
*b and
David
Curiel
*a
aMultifunctional Molecular Materials Group, Department of Organic Chemistry, University of Murcia, Campus of Espinardo, 30100-Murcia, Spain. E-mail: davidcc@um.es
bInstituto de Ciencia Molecular, Universidad de Valencia, Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: enrique.orti@uv.es; juan.arago@uv.es
cISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Oxfordshire OX110QX, UK
First published on 28th May 2021
Abstract
A general synthetic method has been used for the condensation of the 7-azaindole substructure at both extremes of centrosymmetric fused polyheteroaromatic systems. Four different aromatic spacers (benzene, naphthalene, anthracene and pyrene) that modify the molecular geometry and the π-conjugated surface have proved the ability of 7-azaindole to work as a building block that can control the crystal packing through reciprocal hydrogen bond interactions. Two possible self-assembled columnar arrangements have been observed as a result of the π–π interactions between hydrogen-bonded ribbon-like supramolecular structures. A detailed comparative analysis of the molecular organisation driven by hydrogen bonding and π-stacking, in combination with DFT calculations, has revealed an interesting evolution of the charge transport parameters. Organic thin-film field-effect transistors have been fabricated with the hydrogen-bonded semiconductors, reaching mobilities that demonstrate the potential of this supramolecular approach to control the molecular organisation and promote the electronic communication between the organic building blocks.
Introduction
Organic semiconductors have recently gained much attention owing to their utility in the fabrication of electronic devices,1,2 where organic light-emitting diodes (OLEDs),3,4 organic and hybrid photovoltaics (OPV and HPV)5,6 and organic field-effect transistors (OFETs)7,8 are the most representative applications. The technically appealing properties of this new generation of devices, namely lightweight and flexibility,9–14 confer an added value on the innovation and development purposes for the electronics industry. Nevertheless, there are still important challenges to tackle before organic semiconductors can become real competitors of their inorganic counterparts. One of the main difficulties, intrinsic to organic semiconductors, originates from the fundamentals of the charge transport process.15–20 Differently from inorganic semiconductors, constituted by highly ordered solid networks of covalently bonded atoms that give rise to wide electronic band structures, organic semiconductors are molecular solids whose order in the solid state is governed by weak non-covalent intermolecular interactions.21,22 As a consequence, the charge transport mechanism that generally takes place in organic semiconductors consists in a charge carrier hopping between a manifold of localized states. This hopping mechanism makes charge percolation through the semiconductor more difficult than in the case of band-like transport. Therefore, the solid-state arrangement has a critical impact on the charge transport properties of organic materials.23,24 Nevertheless, given the weakness of these non-covalent interactions, and despite the progress of computational methods, it is still very intricate to control and predict the packing in the solid state from the molecular structure only.25,26 This becomes particularly problematic because most of the organic semiconductors consist of fused polyaromatic or polyheteroaromatic structures, whose solid-state packing is mainly driven by π–π interactions. Although these are essential to sustain charge hopping, their low energy makes the adjustment and orientation of π–π interactions a difficult task.27–31 Thus, achieving highly ordered organic semiconductors in an extended scale becomes a challenging goal to improve the charge transport in electronic devices.32–34 This circumstance is ideally attained in the case of single crystal OFETs which, in spite of representing a perfect benchmark for research purposes,35–38 cannot currently be considered a suitable option for industrial development where high throughput fabrication methods based on thin films are desired for organic electronics.39–42Numerous synthetic approaches have been explored to study the effect of structural alterations on the crystal packing, the tuning of π–π interactions and the subsequent semiconductor performance. Focusing on fused polyaromatic and polyheteroaromatic molecules, the peripheral functionalisation has been commonly investigated using rigid substituents, aromatic rings or alkyl chains.43–47 The modification of the geometry of fused rings and the study of isomerism has also enabled a better understanding of the structure–property correlation.48–52 Additionally, the expansion of the π-conjugated surface has been usually employed to modulate the intermolecular interactions.53,54
In this context, the molecular organisation in π-conjugated systems could be further controlled by virtue of the cooperative effect of stronger non-covalent interactions. Among them, hydrogen bonding represents an appropriate tool,55–57 as it is evidenced by biological systems in which the combination of π-stacking and hydrogen bonds determines the macrostructure of proteins or nucleic acids, just to mention well-known examples. From a chemical perspective, the wide diversity of functional groups that can interact through hydrogen bonds offers a plethora of synthetic possibilities for the construction of novel materials.58 Moreover, the energy and directionality of hydrogen bonds provide the essential means to design an approach that enables certain control or predictability of the molecular arrangement. This supramolecular strategy has been successfully applied within the disciplines of crystal engineering,59 surface self-assembly60 and organic frameworks,61 validating the method of hydrogen bond-induced organisation in the solid state.
Concerning the application of this approach in organic electronics, hydrogen-bonded pigments have shown promising charge transport properties when employed as semiconductors in thin-film transistors. In this regard, it is worth highlighting the excellent results reported for quinacridone and epindolidione.62–64 Moreover, structurally related molecules such as indigo and diketopyrrolopyrrole derivatives have also offered good performances as organic semiconductors.65–68 Analogously, chlorinated perylene diimides have shown good electron mobilities in transistors.69 Looking at the structural features of these molecules, it is interesting to notice how even with short conjugation lengths they present good charge transport, attributed to their ability to self-assemble through (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) hydrogen bonds.
C) hydrogen bonds.
Inspired by these precedents, we have recently reported the synthesis of small conjugated molecules that self-organise through hydrogen bond interactions. The structural particularity of these compounds lies in the fusion of the 7-azaindole building block that enables the integration of the hydrogen bonding sites within the conjugated skeleton itself.70–72 Additionally, the resulting fused polyheteroaromatic molecules present a fully conjugated structure that differs from the conjugation of some of the above-mentioned indigoid systems. Interestingly, the use of hydrogen-bonded rigid centrosymmetric structures can induce a supramolecular expansion of the π-surface that also favours a concomitant π-stacking. Based on these results, we have considered some of the previously mentioned structural modifications in organic semiconductors, such as molecular geometry and conjugation length, and we report a comparative study of four fused 7-azaindole systems, namely benzodi-7-azaindole (BDAI), naphthodi-7-azaindole (NDAI), anthradi-7-azaindole (ADAI) and pyrenodi-7-azaindole (PDAI). Structure–property correlations are analysed in terms of their electronic structure, solid-state packing, thin-film morphology and charge transport in OFETs along with theoretical calculations. We have corroborated the excellent directing ability of our hydrogen bonding unit, producing similar molecular packing in all cases. However, these analogous structures show an interesting range of dissimilar charge transport properties that even improve the results of other hydrogen-bonded semiconductors in thin-film transistors.
Results and discussion
Synthesis
All the compounds were synthesized following a two-step methodology for the integration of the 7-azaindole substructure into the fused polyheteroaromatic systems with C2h symmetry (Scheme 1).73 Initially, the corresponding diamines, namely p-phenylenediamine, 1,5-diaminonaphthalene, 1,5-diaminoanthracene74 and 1,6-diaminopyrene,75 reacted with 2,3-dichloropyridine using standard palladium-catalysed conditions for a Buchwald–Hartwig reaction.76 The resulting bis(pyridyl) aromatic diamines, 1, 2, 3 and 4, led to the respective polyheteroaromatic products BDAI, NDAI, ADAI70 and PDAI, via photochemical carbon–carbon coupling in good yields. All the intermediate and final products were structurally characterised by NMR spectroscopy and mass spectrometry (Fig. S1–S4, ESI‡). In the particular case of BDAI,71 a mixture of two isomers was obtained and the desired centrosymmetric product was isolated after purifying the mixture by gradient sublimation. Analogously, in order to get highly pure materials, all the final products were purified by sublimation. As can be anticipated from this purification protocol, these materials show a high thermal stability. This was confirmed by thermogravimetric analysis (TGA) revealing that they all have very high degradation temperatures, BDAI (373 °C), NDAI (462 °C), ADAI (496 °C) and PDAI (534 °C), corresponding to a 5% mass loss (Fig. S5, ESI‡). The stability of the molecules is attributed to the hydrogen bond interactions that govern the solid-state structure as discussed below. The robustness of these molecules is also verified by differential scanning calorimetry (DSC) where no evidence of any phase transition was detected during the heating and cooling cycles (Fig. S5, ESI‡). | ||
| Scheme 1 Synthesis of 7-azaindole-fused polyheteroaromatic molecules. (i) Pd(OAc)2, (±)-BINAP, tBuOK, 1,4-dioxane, Δ; (ii) tBuOK, DMSO, hν. | ||
Optical and electrochemical characterisation
The electronic structure of the four compounds was characterised by electrochemical and spectroscopic methods (Fig. 1). The cyclic voltammetry of BDAI, NDAI, ADAI and PDAI show irreversible voltammograms with oxidation onsets (Eonset) detected at 1.00, 1.17, 1.01 and 0.94 V, respectively (Table 1). The energies of the highest occupied molecular orbital (HOMO) were estimated from these onset values taking the ferrocene/ferrocenium redox couple as a reference to set the energy with respect to the vacuum level.77,78 The identical angular catacondensation of NDAI and ADAI shows how the extended conjugation from the naphthalene to the anthracene spacer causes an increase in the HOMO energy of ADAI as previously observed for similar phene-like polyheteroaromatic molecules.49,51,79–82 This effect is also exhibited by the molecule with the larger pyrene spacer, PDAI, whose HOMO has a slightly higher energy. Coincidentally, BDAI and ADAI displayed similar HOMO energies. Despite the shorter conjugation length of the former, the pseudolinearity of BDAI structure causes the raise of the HOMO energy when compared to the angular structure of ADAI and NDAI.80,83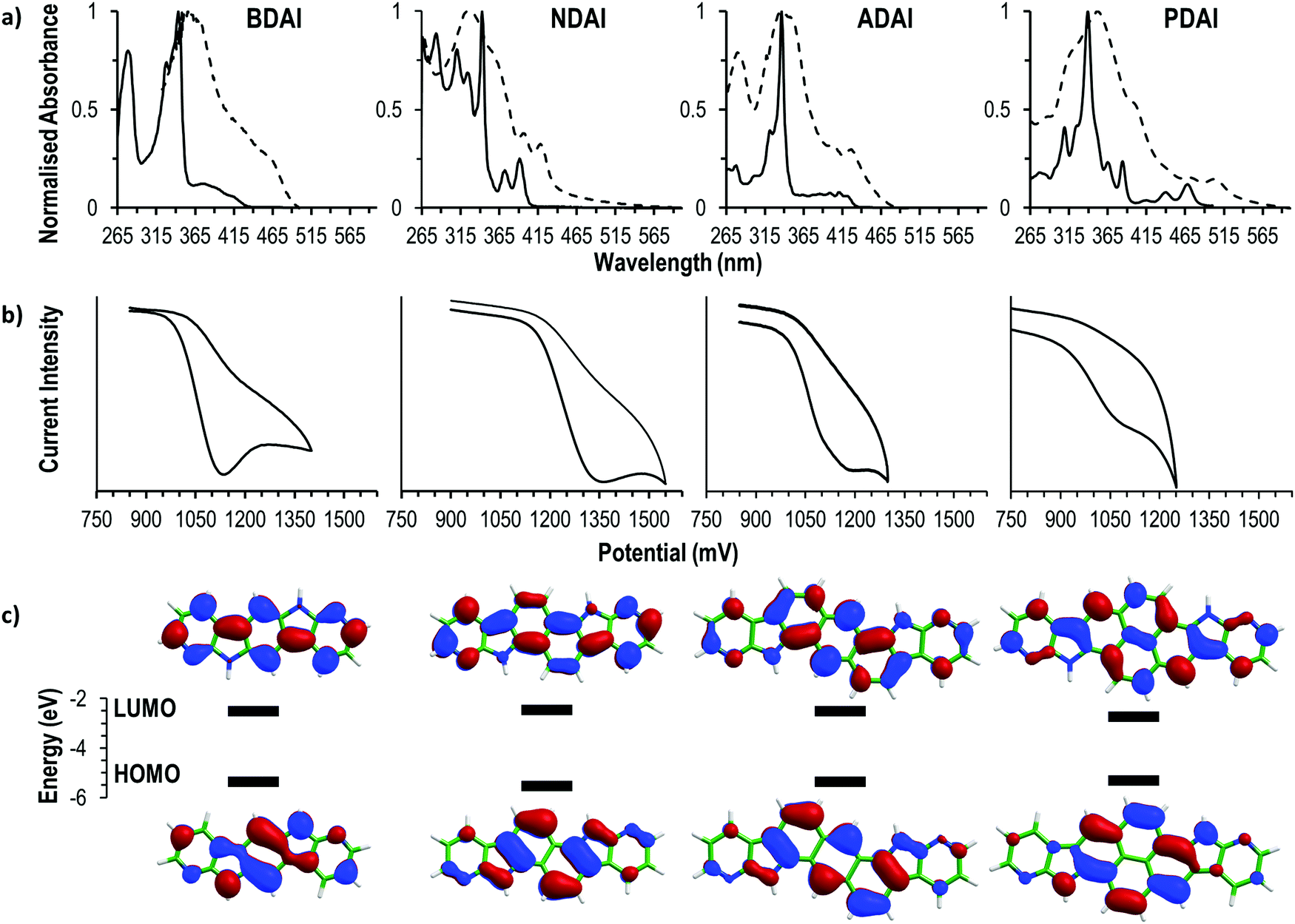 | ||
| Fig. 1 (a) Absorption spectra in DMF solution (2.5 × 10−5 M, continuous plot) and as solid thin film (dashed plot). (b) Cyclic voltammetry (10−3 M in DMF, 0.1 M TBAPF6). (c) HOMO and LUMO energies estimated from: EHOMO (eV) = −(Eonset ox. − Eonset Fc/Fc+) + EHOMO Fc; ELUMO = EHOMO + Eopt.; Eonset Fc/Fc+ = 0.44 V; EHOMO Fc = −4.8 eV. The topology of the frontier molecular orbitals (isocontours of ±0.03) was computed at the B3LYP/6-31G** level. | ||
| BDAI | NDAI | ADAI | PDAI | |
|---|---|---|---|---|
| a The theoretical energies calculated at the B3LYP/6-31G** level for the HOMO are included within brackets. | ||||
| E onset (V) | 1.00 | 1.17 | 1.01 | 0.94 |
| λ abs. onset (nm) | 432 | 403 | 434 | 482 |
| E LUMO (eV) | −2.55 | −2.45 | −2.50 | −2.73 |
| E HOMO (eV) | −5.36 [−5.38] | −5.53 [−5.39] | −5.37 [−5.12] | −5.30 [−4.93] |
To gain a deeper understanding of the electronic structure, the frontier molecular orbitals of BDAI, NDAI, ADAI and PDAI were computed after geometry optimisation at the density functional theory (DFT) B3LYP/6-31G** level including solvent effects in dimethylformamide (DMF) within the polarizable continuum model (PCM) (see the ESI‡ for full computational details). As shown in Fig. 1c, the HOMO spreads over the whole conjugated structure being more concentrated over the acene core. The HOMOs of NDAI (−5.39 eV), ADAI (−5.12 eV) and PDAI (−5.30 eV) actually preserve the topology of the HOMOs of the naphthalene (−5.92 eV), anthracene (−5.36 eV) and pyrene (−5.47 eV) cores, respectively (Fig. S6, ESI‡), but are shifted to higher energies due to the antibonding interactions with the pyrrole nitrogens of the 7-azaindole end-capping moieties. These interactions are stronger for PDAI than for ADAI and the HOMO of PDAI is predicted higher in energy in accord with the experimental estimates (Table 1) and in contrast with that found in passing from anthracene to pyrene. The HOMO of BDAI receives a larger contribution from the 7-azaindole units and spreads over the whole molecule in a more extended way, justifying the high energy calculated for this orbital.
All the studied molecules have analogous UV-vis spectral features displaying two differentiated absorption regions that remind the spectra of isoelectronic polyaromatic molecules.84,85 In DMF solution, intense absorptions ascribed to the p bands are located in a narrow range (337–343 nm) for all the compounds, indicating that these transitions are not very sensitive to the changes in the molecular structure. Weaker bands located at longer wavelengths (α bands) are ascribed to the HOMO → LUMO transitions and are sensitive to the conjugation length. This is evidenced by the red-shifted wavelengths of the absorption onsets (λabs. onset) that follow the trend NDAI (403 nm) < ADAI (434 nm) < PDAI (482 nm) showing how the optical energy gap becomes narrower. Again, BDAI displaces its absorption onset to longer wavelengths (432 nm) than a priori expected. The low-energy absorption onsets were used to estimate the HOMO–LUMO energy gap which, added to the previously calculated HOMO energies, leads to the determination of the LUMO energies (Table 1).
The optical findings are supported by time-dependent DFT (TD-DFT) calculations in DMF (Table S1, ESI‡). For all the four systems, calculations predict a first weak π–π* electronic transition (S0 → S1), mainly associated with the HOMO → LUMO monoexcitation, with wavelengths calculated at 378, 366, 423 and 439 nm (3.28, 3.39, 2.93 and 2.82 eV) for BDAI, NDAI, ADAI and PDAI, respectively. As observed experimentally, the first transition of BDAI appears at lower energy than that calculated for NDAI. At shorter wavelengths, an intense π–π* electronic transition, mainly due to the HOMO−1 → LUMO monoexcitation, is computed for BDAI (S0 → S2), ADAI (S0 → S3) and PDAI (S0 → S4). These transitions give rise to the experimental p bands, and are calculated close in energy at 329, 331 and 334 nm (3.77, 3.75 and 3.71 eV), respectively, in very good accord with the experiment. For NDAI, calculations also predict the S0 → S2 transition dominated by the HOMO−1 → LUMO monoexcitation at similar energies (330 nm, 3.76 eV), but now it shows a significant lower intensity (Table S1, ESI‡). Instead, the highest intensity corresponds to the S0 → S4 transition associated to the HOMO → LUMO+1 excitation, which is calculated at higher energies (299 nm, 4.14 eV). The vibrational structure of the HOMO → LUMO S0 → S1 transition is clearly observed for NDAI and PDAI (Fig. 1a).
The optical characterisation was completed by measuring the absorption spectra of solid thin films. All the samples experienced a bathochromic shift when compared to the measurements in solution owing to the intermolecular interactions that are more favoured in the solid state. In the case of BDAI, NDAI, ADAI and PDAI, the intermolecular interactions (mainly of π–π character) are additionally reinforced by hydrogen bonds.
In summary, the electronic characteristics of the materials resulting from the fusion of the 7-azaindole unit to different aromatic cores makes them suitable for their study as hole transporting materials.
Organic field-effect transistors
The evaluation of the semiconducting ability of BDAI, NDAI, ADAI and PDAI was investigated by fabricating thin-film transistors with a bottom-gate top-contact architecture. Given the relevance that interfaces have on OFETs performance,86 Si/SiO2 substrates were spin-coated with a thin film of polystyrene that is part of the hybrid dielectric architecture.87,88 This treatment is intended to precondition the dielectric/semiconductor interface by smoothing the surface roughness, blocking the interfacial dipoles and improving the subsequent organic semiconductor deposition. Then, the semiconductor was evaporated under high-vacuum conditions. Concerning the semiconductor/top-contact interface, MoO3 was deposited through a shadow mask previous to the electrode evaporation. The high work function of molybdenum oxide may have a contact doping effect and reduce the contact resistance.89,90 Electrical characterisation of the transistors was performed at dark and in the air. The transfer and output characteristics are depicted in Fig. 2. The OFETs parameters were calculated from the transfer characteristics in the saturation regime and are summarised in Table 2. The best hole mobilities (μmax) measured for the different materials increase from BDAI (1.3 × 10−3 cm2 V−1 s−1) to NDAI (4.6 × 10−3 cm2 V−1 s−1) and reach the maximum value for ADAI (2.7 × 10−2 cm2 V−1 s−1). These results suggest that the expansion of the conjugation in the linear acene spacers improves the charge transport properties of the self-assembled materials. At this point, it is worth highlighting that these mobilities improve the results previously reported for other hydrogen-bonded semiconductors in thin-film transistors fabricated under analogous experimental conditions (Si/SiO2 substrate, thin-film sublimation and without annealing or substrate heating).91–93 Nevertheless, a lower mobility was measured in the transistors fabricated with PDAI (1.4 × 10−4 cm2 V−1 s−1) despite its larger conjugated surface. Since molecular packing, thin-film morphology and crystallinity play a critical role for the charge transport properties of organic semiconductors, these aspects were investigated in detail. | ||
| Fig. 2 Transfer and output characteristics of thin-film OFETs. | ||
| μ max (cm2 V−1 s−1) | μ avg (cm2 V−1 s−1) | V on (V) | I on/Ioff | |
|---|---|---|---|---|
| a μ max: best hole mobility; μavg: average hole mobility and standard deviations obtained from six devices; Von: switch-on voltage; Ion/Ioff: on–off current ratio. | ||||
| BDAI | 1.3 × 10−3 | 5.1 × 10−4 ± 2.9 × 10−4 | −24 | 103 |
| NDAI | 4.6 × 10−3 | 3.2 × 10−3 ± 0.6 × 10−3 | −52 | 103 |
| ADAI | 2.7 × 10−2 | 2.6 × 10−2 ± 0.1 × 10−2 | −22 | 5 × 104 |
| PDAI | 1.4 × 10−4 | 1.2 × 10−4 ± 0.2 × 10−4 | −51 | 102 |
Crystal structure and theoretical calculations
The crystal packing of the different molecules was determined by X-ray diffraction. Concerning the purpose of investigating the control of the molecular organisation through hydrogen bond interactions, we could satisfactorily check that the molecular design based on the incorporation of the 7-azaindole building block into the conjugated systems led to striking similarities in the solid-state arrangement of the different molecules. Thus, all the centrosymmetric conjugated systems set reciprocal N–H⋯N hydrogen bonds between the donor site at the pyrrole ring and the acceptor site at the pyridine ring. Shorter hydrogen bond distances (1.9 Å, H⋯N distance) are observed in BDAI, due to its pseudolinear structure, that enables molecules to come closer than in the case of NDAI (2.1 Å), ADAI (2.1 Å) and PDAI (2.3 Å) molecules. This can be explained by the little steric hindrance occurring between the CHs in the central aromatic spacer and the pyridine rings of neighbouring molecules in the crystal (Fig. 3). Hydrogen-bonded molecules are not perfectly coplanar but define low steps (height: 0.3–0.7 Å) that improve crystal packing. Moreover, as a result of this hydrogen bond-directed self-assembly, the symmetry of the molecules enables the formation of ideally infinite ribbon-like supramolecular structures (Fig. 3). This non-covalent expansion of the conjugated surface additionally favours the stacking of molecules in parallel planes (interplanar distance: 3.4–3.6 Å) defining a slipped columnar arrangement. This packing pattern is adopted by all the four molecules exhibiting different geometries and conjugation lengths. Therefore, the X-ray diffraction results reinforce our supramolecular approach to induce self-organisation in conjugated small molecules. When the crystal network is extended, the slipped columnar packing is maintained in BDAI and ADAI (Fig. S8, ESI‡) reminding the crystal structure of the alpha I polymorph of quinacridone.94 Nevertheless, a second orientation of the columnar packing is observed for NDAI and PDAI (Fig. S8, ESI‡) where the molecules in differently oriented columns form angles of 49.6°and 53.2°, respectively. | ||
| Fig. 3 X-ray diffraction. Solid-state packing. Left column: Ribbon-like structure of hydrogen-bonded molecules (red dotted lines represent hydrogen bonds). Central column: projection across the long molecular axis showing hydrogen-bonded molecules and π–π stacked molecules adopting a columnar packing. Right column: top view of π–π stacked molecules; red spots represent the molecular centroids that serve as a guide for the eye to identify the pitch shifts (along the long molecular axis) and roll shifts (along the short molecular axis). | ||
Face-to-face π-stacking represents the intermolecular interaction that intuitively defines the more favourable direction for charge transport due to the more optimal orbital overlap. Nevertheless, stacked molecules must slip with respect to each other to attenuate the electrostatic repulsion between the π-electron densities. The intermolecular shift can occur along the long or short axis of the molecules (pitch and roll shifts, respectively)95 resulting in a more or less favoured π-surface intersection. It was interesting to observe how dissimilar pitch and roll shifts occurred within the series of studied molecules (Fig. 3 and Fig. S9, ESI‡). Thus, using the longest molecular axis as a spatial reference (longest diagonal connecting two atoms through the centre of inversion), BDAI experiences both longitudinal (1.4 Å) and transversal (1.5 Å) shifts. NDAI basically shifts transversally (1.5 Å) along the short side of the molecule with only a minor shift along the long side (0.7 Å). This type of intermolecular displacement is also observed in PDAI (roll shift: 1.5 Å and pitch shift: 0.6 Å). Contrariwise, ADAI shows a large longitudinal slippage (3.0 Å) and a very short transversal shift (0.1 Å). As it will be discussed later, these different π-stacking dispositions have important effects on the electronic communication between molecules.
The extent in which the different intermolecular interactions intervene in the solid-state packing can be analysed by calculating the Hirshfeld surfaces (Fig. 4).96–99 The region where π-conjugated surfaces intersect between stacked molecules is depicted by a white “footprint” at both sides of the Hirshfeld surface that, according to the colour code, represents contacts around the van der Waals separation. The decomposition of the different atom-to-atom contacts (Fig. S10, ESI‡) shows how C⋯C contacts, which are the most representative contacts in face-to-face π–π stacking, increase with the extension of the conjugated surface. It is worth highlighting how the Hirshfeld surfaces also evidence the strength of hydrogen bonding by the red spots located at the donor and acceptor sites that denote shorter contacts. In agreement with the previously discussed (NH⋯N) hydrogen bond distances, these perfectly match the (H⋯N) contribution to the intermolecular interactions in the Hirshfeld surface, whose representativeness decreases following the trend BDAI > NDAI > ADAI > PDAI (Fig. S10, ESI‡). This indicates that edge-to-edge interactions, which normally are negligible in the molecular packing of π-conjugated systems, gain much relevance when hydrogen bonds control the solid-state arrangement. Additionally, some other weak interactions are attributed to C–H⋯H–C contacts depicted as white spots around the peripheral region of the Hirshfeld surface.
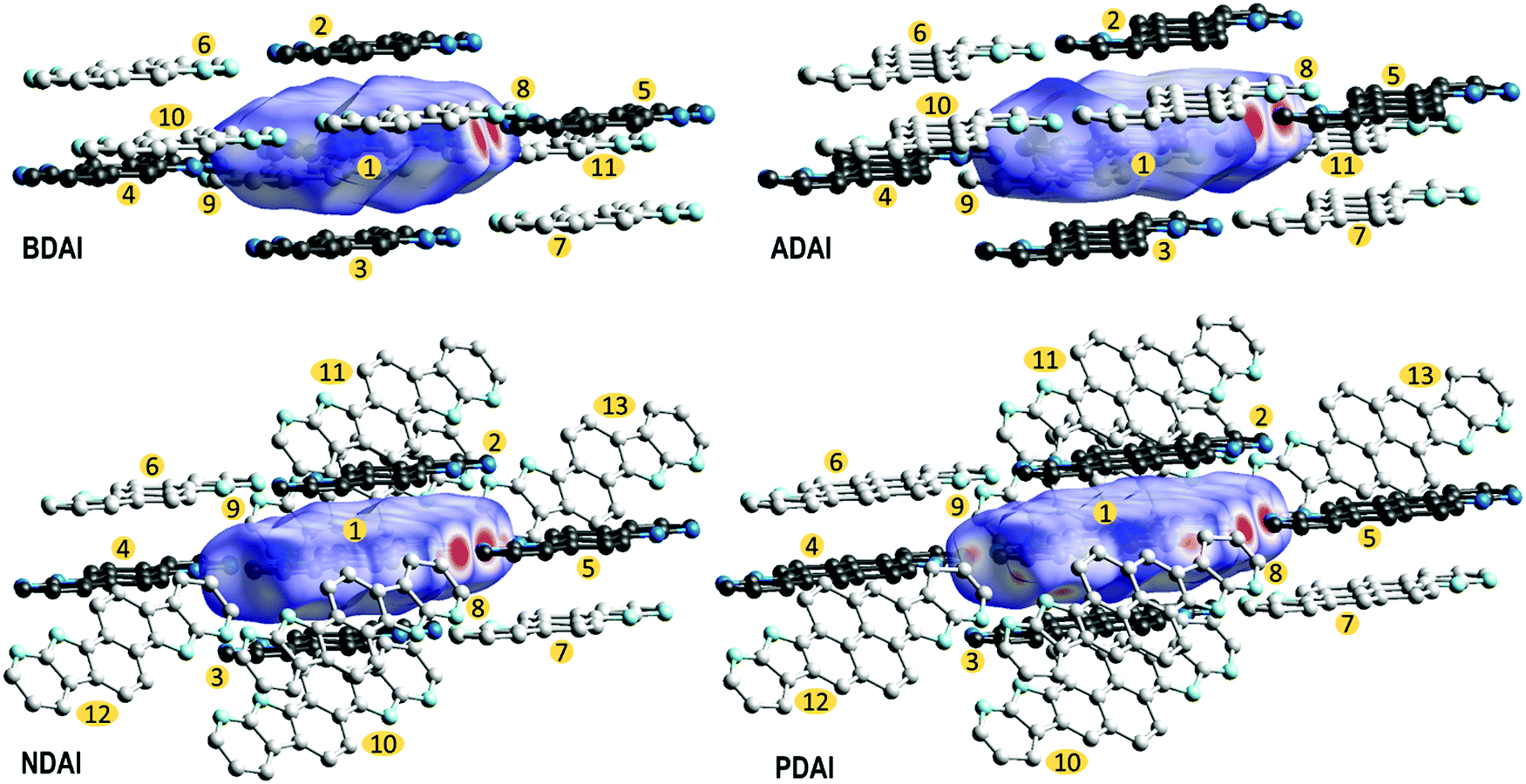 | ||
| Fig. 4 Hirshfeld surfaces and close contacts in the crystal packing. Black molecules correspond to hydrogen-bonded and π-stacked molecules with respect to the central molecule. Grey molecules complete the close contacts around the Hirshfeld surface. Numbers are included to identify the molecules for the transfer integral calculations. | ||
To gain a better insight into the charge transport process at the molecular level, theoretical calculations were performed to estimate the transfer integrals (ta,b) between neighbouring molecules in the crystal and the reorganisation energy (λ) (see the ESI‡ for full computational details). Transfer integrals are related with the degree of orbital overlap and quantify the electronic communication between spatially close molecules a and b. The reorganisation energy accounts for the energetic cost of intermolecular charge transfer in terms of the structural rearrangement that the molecules experience upon donating/accepting an electron. Both ta,b and λ are relevant parameters to describe the charge transport in organic semiconductors and are key factors to evaluate the electron-transfer rate constant using the popular Marcus rate expression.100
To screen the preferred transport directions in the crystal, hole transfer integrals were computed for all the intermolecular contacts found in the crystal structure (Fig. 4 and Table 3). Based on the ta,b values, the main charge carrier path corresponds to the π-stacking direction since most of the studied molecules (BDAI, NDAI and PDAI) exhibit the highest transfer integrals between π-stacked dimers (t1,2 = t1,3). However, the values of these transfer integrals do not follow the expected trend according to the π-surface extension of the molecules. BDAI, the smallest molecule, displays the largest transfer integral between π-stacked molecules (71.5 meV), whereas ADAI shows a noticeable low electronic coupling (11.1 meV). Although this result may seem counterintuitive, this is due to the high dependence the transfer integral has on the orbital overlap, which strongly varies with the intermolecular shift of the neighbouring molecules.101 This is evidenced in the 2D maps (Fig. S11, ESI‡) representing the theoretical variation of the transfer integral values when π-stacked molecules are shifted with respect to each other from the relative position in the crystal. Small displacements along the long and short molecular axes cause huge variations on the value of the t1,2 = t1,3 transfer integral. Thus, in the case of ADAI, the HOMO orbitals of the π-stacked molecules do not efficiently overlap for the specific intermolecular location found in the crystal (Fig. S11, ESI‡). This also applies to the low transfer integral obtained for π-stacked PDAI dimers compared to the smaller molecules BDAI and NDAI. These results contrast with the hole mobilities measured for OFET devices (Table 2), which increase in passing from BDAI to NDAI and ADAI and significantly decrease for PDAI. On the other hand, as it has been previously anticipated, the edge-to-edge interactions set through hydrogen bonds provide remarkable values for t1,4 = t1,5 transfer integrals in the 7–14 meV range (Table 3). In contrast to π-stacked dimers, for which many different relative dispositions are accessible, in hydrogen-bonded dimers the relative disposition is restricted to keep the hydrogen bonds and similar values are obtained for the electronic couplings of the four molecules. Contacts between molecules packed in adjacent columns, immediately above or below the hydrogen-bonded dimer, also display significant transfer integrals (t1,6 = t1,7) with values above 16 meV for all compounds except for BDAI (6.7 meV). Non-negligible transfer integral values for dimers t1,8 = t1,9 belonging to adjacent hydrogen-bonded ribbons are also predicted for BDAI and ADAI. The estimated transfer integrals therefore suggest that a 2D charge transport along the π-stacked hydrogen-bonded ribbons, reinforced by diagonal (t1,6 = t1,7) and edge-to-edge (t1,4 = t1,5) intermolecular interactions, mainly occurs for all systems with further inter-ribbon electronic communication for BDAI and ADAI.
| t a,b | BDAI | NDAI | ADAI | PDAI |
|---|---|---|---|---|
| t 1,2 = t1,3 (π-stack) | 71.5 | 56.1 | 11.1 | 28.8 |
| t 1,4 = t1,5 (H-bond) | 14.2 | 11.4 | 8.3 | 7.2 |
| t 1,6 = t1,7 | 6.7 | 19.1 | 16.8 | 23.9 |
| t 1,8 = t1,9 | 9.3 | 1.5 | 12.1 | 1.9 |
| t 1,10 = t1,11 | 1.6 | 1.5 | 0.4 | 1.9 |
| t 1,12 = t1,13 | 0.2 | 0.0 |
To understand the apparent dissonance between the transfer integrals evaluated for single molecular pairs at the crystal structure and the hole mobilities experimentally measured, the fluctuation of the transfer integrals (dynamic disorder) owing to thermal motions was evaluated using a computational protocol which combines quantum-chemical calculations (DFT) and molecular dynamics simulations (see the ESI‡ for details). Note that dynamic disorder turned out to be important to accurately describe charge and energy transport phenomena in molecular aggregates and crystals.102–105Table 4 gathers the Marcus-like charge transfer rate constants computed for the most relevant dimers of the studied compounds taking into account the dynamic disorder by the average squared transfer integral 〈t2〉 (see the ESI‡ and Table S3 for further details).106Table 4 also includes the reorganisation energies λ needed to calculate the rate constants. Whereas similar λ values in the 230–250 meV range are computed for BDAI, NDAI and PDAI, ADAI exhibits a significantly smaller value of 182 meV. Table 4 clearly confirms that the most favourable charge-transport channel in all the studied compounds occurs through the π-stacking direction with similar rate constants in the range of 1.3–2.2 × 1013 s−1. Nevertheless, ADAI exhibits, in general, more efficient secondary channels for efficient charge migration with significant charge-transfer rates of 2.4 × 1012 and 1.3 × 1012 s−1 for 1-6 intra-ribbon and 1-8 inter-ribbon molecular pairs, respectively. These secondary charge transport pathways increase the charge carrier hopping probability in different directions and can explain the better mobilities registered for ADAI in OFET devices compared to the other organic semiconductors BDAI, NDAI and PDAI.
| Dimer | Property | BDAI | NDAI | ADAI | PDAI |
|---|---|---|---|---|---|
| λ (meV) | 233 | 250 | 182 | 239 | |
| 1-2 = 1-3 | 〈t2〉 (meV2) | 3612.5 | 7030.3 | 3340.8 | 5702.0 |
| k (s−1) | 1.3 × 1013 | 2.1 × 1013 | 2.2 × 1013 | 1.9 × 1013 | |
| 1-4 = 1-5 | 〈t2〉 (meV2) | 87.3 | 55.9 | 17.0 | 20.1 |
| k (s−1) | 3.1 × 1011 | 1.6 × 1011 | 1.1 × 1011 | 7.6 × 1010 | |
| 1-6 = 1-7 | 〈t2〉 (meV2) | 32.9 | 191.3 | 356.7 | 63.4 |
| k (s−1) | 1.2 × 1011 | 5.6 × 1011 | 2.4 × 1012 | 2.1 × 1011 | |
| 1-8 = 1-9 | 〈t2〉 (meV2) | 764.4 | 18.5 | 194.7 | 21.4 |
| k (s−1) | 2.7 × 1012 | 5.5 × 1010 | 1.3 × 1012 | 7.2 × 1010 | |
Thin-film morphology and crystallinity
With the aim of getting further experimental support that explains the observed charge transport properties, the semiconductor surface thin-film morphologies were analysed by atomic force microscopy (AFM) (Fig. 5a). Even though the molecules showed a very similar solid-state packing, clear differences are manifested in the AFM images. Thus, BDAI forms tangled turnings that densely cover the substrate surface. Similar but thicker turnings are observed in the NDAI films. Conversely, ADAI presents a granular morphology with a very homogeneous grain size distribution and compact coverage. Likewise, PDAI also exhibits a granular morphology. These outcomes bring to light how subtle changes in the molecular structure, despite showing analogous crystal packing features, can cause remarkable changes on the mesoscopic scale, as revealed by the different morphology of the four identically processed thin films with comparable molecular materials. | ||
| Fig. 5 (a) AFM images. (b) X-ray diffraction of thin films. | ||
Additionally, X-ray diffraction measurements were performed on the deposited films (Fig. 5b). BDAI films did not show an evident crystallinity, since no peak could be clearly detected in the diffractogram. Conversely, the crystallinity of NDAI, ADAI and PDAI was confirmed by the detection of out-of-plane diffraction peaks, which are coincident with the simulated diffractograms obtained from the previously discussed X-ray structures, indicating that the molecular packing is preserved in the thin films.
The analysis of properties such as morphology and crystallinity, in the mesoscopic scale, explains the lower hole mobility in BDAI despite its similar rate constant compared to the other compounds. Nonetheless, it is worth emphasizing the noticeable mobility reached by BDAI considering its apparent lack of crystallinity.15 This can be explained by the presence of optimal secondary charge transport pathways for BDAI similarly to ADAI with remarkable estimated hole-transfer rate constants (Table 4). Regarding the measured diffractograms, a better crystallinity was detected for ADAI that showed sharper and more intense peaks and up to second order reflections, indicating that a single crystalline phase is mainly detected in the thin film. Moreover, the only presence of reflections belonging to the (00l) family clearly indicates that the crystalline film has been grown with a strong preferred orientation, with the ab crystalline planes being parallel to the film surface. The same preferred growth orientation has also been obtained for the cases of both NDAI and PDAI films. The crystalline domains play a critical role in the quality of the charge transport of organic semiconductors since they define how extended the molecular order is present in the crystallites that form the film. Thus, the average crystallite size was calculated using the Scherrer equation by fitting the dominant peaks in the diffractograms.107 The crystalline films displayed the following crystallite sizes: ADAI (21 nm), NDAI (18 nm) and PDAI (10 nm). In agreement with this, a more extended crystalline order was confirmed for ADAI and, in turn, it was better for the NDAI film when compared to PDAI.
From the overall analysis of the different properties affecting the charge transport in the OFETs fabricated with BDAI, NDAI, ADAI and PDAI, it can be concluded that the thin film crystallinity or, in other words, the degree of molecular order within the film, becomes a very important feature for the performance of this series of hydrogen-bonded semiconductors. Additionally, the molecular disposition governed through hydrogen bonding contributes to define several pathways for efficient charge migration. The favourable convergence of all these properties on ADAI demonstrates the suitability of this and other novel self-assembled molecular materials to be investigated as organic semiconductors.
Conclusions
A straightforward synthetic strategy has been optimised for the integration of the 7-azaindole substructure in fused polyheteroaromatic systems. The rational design of centrosymmetric conjugated molecules, with hydrogen bond donor and hydrogen bond acceptor sites incorporated to the aromatic structure, validates the suitability of the 7-azaindole building block to induce the hydrogen bond self-assembly in the solid state. The reproducibility of the molecular packing motif observed in the X-ray crystal structure of the series of centrosymmetric molecules BDAI, NDAI, ADAI and PDAI, with different geometry and conjugation length, reinforces our supramolecular approach for the challenging task of gaining certain control on the solid-state arrangement of organic semiconductors. The thin-film OFETs fabricated on commercial Si/SiO2 substrates without any thermal treatment have demonstrated an improved hole mobility of ADAI within the context of hydrogen-bonded organic semiconductors. The structure–property correlation within the series of hydrogen-bonded molecules has enabled a combined study of parameters in the nanoscale (transfer integrals and reorganisation energies) and the mesoscale (thin film morphology and crystallinity). This analysis has revealed the significant contribution that thin-film crystallinity has on the charge transport properties of conjugated small molecules. In summary, the reported results consolidate the fundamentals for future investigations that will further develop the topic of supramolecular organic electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the Spanish Ministry of Science and Innovation (MICINN) (projects RTI2018-101092-B-I00, PGC2018-099568-B-I00, RED2018-102815-T, and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the Fundación Séneca – Agencia de Ciencia y Tecnología de la Región de Murcia (20959/PI/18), the Generalitat Valenciana (PROMETEO/2020/077 and SEJI/2018/035), and European feder Funds (PGC2018-099568-B-I00) for financial support. M. M.-M. acknowledges Fundación Séneca for a postdoctoral Saavedra Fajardo contract (20406/SF/17). J. C. and J. A. are respectively grateful to MICINN for their predoctoral (BES-2016-076826) and “Ramón-y-Cajal” grants (RyC-2017-23500).Notes and references
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Adv. Mater., 2020, 32, 1905909 Search PubMed.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 Search PubMed.
- Shahnawaz, S. S. Swayamprabha, M. R. Nagar, R. A. K. Yadav, S. Gull, D. K. Dubey and J.-H. Jou, J. Mater. Chem. C, 2019, 7, 7144–7158 Search PubMed.
- S. Jhulki and J. N. Moorthy, J. Mater. Chem. C, 2018, 6, 8280–8325 Search PubMed.
- H. D. Pham, T. C.-J. Yang, S. M. Jain, G. J. Wilson and P. Sonar, Adv. Energy Mater., 2020, 10, 1903326 Search PubMed.
- A. Mishra, Energy Environ. Sci., 2020, 13, 4738–4793 Search PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 Search PubMed.
- Y. Yu, Q. Ma, H. Ling, W. Li, R. Ju, L. Bian, N. Shi, Y. Qian, M. Yi, L. Xie and W. Huang, Adv. Funct. Mater., 2019, 29, 1904602 Search PubMed.
- H. Zhu, E.-S. Shin, A. Liu, D. Ji, Y. Xu and Y.-Y. Noh, Adv. Funct. Mater., 2020, 30, 1904588 Search PubMed.
- Y. Zang, D. Huang, C.-A. Di and D. Zhu, Adv. Mater., 2016, 28, 4549–4555 Search PubMed.
- R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 Search PubMed.
- Q. Lin, H. Huang, Y. Jing, H. Fu, P. Chang, D. Li, Y. Yao and Z. Fan, J. Mater. Chem. C, 2014, 2, 1233–1247 Search PubMed.
- H. T. Yi, M. M. Payne, J. E. Anthony and V. Podzorov, Nat. Commun., 2012, 3, 1259 Search PubMed.
- M. Kaltenbrunner, M. S. White, E. D. Głowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 Search PubMed.
- H. Li, C. Li, L. Duan and Y. Qiu, Isr. J. Chem., 2014, 54, 918–926 Search PubMed.
- A. Troisi, Chem. Soc. Rev., 2011, 40, 2347–2358 Search PubMed.
- F. Ortmann, F. Bechstedt and K. Hannewald, Phys. Status Solidi B, 2011, 248, 511–525 Search PubMed.
- N. Tessler, Y. Preezant, N. Rappaport and Y. Roichman, Adv. Mater., 2009, 21, 2741–2761 Search PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 Search PubMed.
- P. M. Borsenberger, L. Pautmeier and H. Bässler, J. Chem. Phys., 1991, 94, 5447–5454 Search PubMed.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 Search PubMed.
- S. M. Ryno, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3990–4000 Search PubMed.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 Search PubMed.
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 Search PubMed.
- T. S. Thakur, R. Dubey and G. R. Desiraju, Annu. Rev. Phys. Chem., 2015, 66, 21–42 Search PubMed.
- S. L. Price, Int. Rev. Phys. Chem., 2008, 27, 541–568 Search PubMed.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 Search PubMed.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7–15 Search PubMed.
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 Search PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 Search PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 Search PubMed.
- K. Takimiya, M. Nakano, H. Sugino and I. Osaka, Synth. Met., 2016, 217, 68–78 Search PubMed.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158–6183 Search PubMed.
- H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211–5222 Search PubMed.
- H. Jiang and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 1408–1428 Search PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 Search PubMed.
- L. Jiang, H. Dong and W. Hu, J. Mater. Chem., 2010, 20, 4994–5007 Search PubMed.
- C. Reese and Z. Bao, Mater. Today, 2007, 10, 20–27 Search PubMed.
- W. Tang, Y. Huang, L. Han, R. Liu, Y. Su, X. Guo and F. Yan, J. Mater. Chem. C, 2019, 7, 790–808 Search PubMed.
- K. Fukuda and T. Someya, Adv. Mater., 2017, 29, 1602736 Search PubMed.
- A. Teichler, J. Perelaer and U. S. Schubert, J. Mater. Chem. C, 2013, 1, 1910–1925 Search PubMed.
- B. Kang, W. H. Lee and K. Cho, ACS Appl. Mater. Interfaces, 2013, 5, 2302–2315 Search PubMed.
- C. Wang, D. Hashizume, M. Nakano, T. Ogaki, H. Takenaka, K. Kawabata and K. Takimiya, Chem. Sci., 2020, 11, 1573–1580 Search PubMed.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 Search PubMed.
- P.-L. T. Boudreault, S. Wakim, N. Blouin, M. Simard, C. Tessier, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 129, 9125–9136 Search PubMed.
- C. D. Sheraw, T. N. Jackson, D. L. Eaton and J. E. Anthony, Adv. Mater., 2003, 15, 2009–2011 Search PubMed.
- J. Youn, S. Kewalramani, J. D. Emery, Y. Shi, S. Zhang, H.-C. Chang, Y.-J. Liang, C.-M. Yeh, C.-Y. Feng, H. Huang, C. Stern, L.-H. Chen, J.-C. Ho, M.-C. Chen, M. J. Bedzyk, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2013, 23, 3850–3865 Search PubMed.
- T. Okamoto, C. P. Yu, C. Mitsui, M. Yamagishi, H. Ishii and J. Takeya, J. Am. Chem. Soc., 2020, 142, 9083–9096 Search PubMed.
- M. Más-Montoya, S. Georgakopoulos, J. P. Cerón-Carrasco, J. Pérez, A. Tárraga and D. Curiel, J. Phys. Chem. C, 2018, 122, 11736–11746 Search PubMed.
- Y. Shimo, T. Mikami, S. Hamao, H. Goto, H. Okamoto, R. Eguchi, S. Gohda, Y. Hayashi and Y. Kubozono, Sci. Rep., 2016, 6, 21008 Search PubMed.
- S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024–5035 Search PubMed.
- H. Jiang, P. Hu, J. Ye, A. Chaturvedi, K. K. Zhang, Y. Li, Y. Long, D. Fichou, C. Kloc and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 8875–8880 Search PubMed.
- H. Okamoto, S. Hamao, R. Eguchi, H. Goto, Y. Takabayashi, P. Y.-H. Yen, L. U. Liang, C.-W. Chou, G. Hoffmann, S. Gohda, H. Sugino, Y.-F. Liao, H. Ishii and Y. Kubozono, Sci. Rep., 2019, 9, 4009 Search PubMed.
- K. Niimi, S. Shinamura, I. Osaka, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 8732–8739 Search PubMed.
- H. T. Black, H. Lin, F. Bélanger-Gariépy and D. F. Perepichka, Faraday Discuss., 2014, 174, 297–312 Search PubMed.
- E. D. Glowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013, 1, 3742–3753 Search PubMed.
- M. Irimia-Vladu, Y. Kanbur, F. Camaioni, M. E. Coppola, C. Yumusak, C. V. Irimia, A. Vlad, A. Operamolla, G. M. Farinola, G. P. Suranna, N. González-Benitez, M. C. Molina, L. F. Bautista, H. Langhals, B. Stadlober, E. D. Głowacki and N. S. Sariciftci, Chem. Mater., 2019, 31, 6315–6346 Search PubMed.
- Z.-T. Li and L.-Z. Wu, Hydrogen Bonded Supramolecular Structures, Springer-Verlag, 2015 Search PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 Search PubMed.
- A. G. Slater, L. M. A. Perdigão, P. H. Beton and N. R. Champness, Acc. Chem. Res., 2014, 47, 3417–3427 Search PubMed.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 Search PubMed.
- E. D. Głowacki, G. Romanazzi, C. Yumusak, H. Coskun, U. Monkowius, G. Voss, M. Burian, R. T. Lechner, N. Demitri, G. J. Redhammer, N. Sünger, G. P. Suranna and S. Sariciftci, Adv. Funct. Mater., 2015, 25, 776–787 Search PubMed.
- E. D. Głowacki, M. Irimia-Vladu, M. Kaltenbrunner, J. Gsiorowski, M. S. White, U. Monkowius, G. Romanazzi, G. P. Suranna, P. Mastrorilli, T. Sekitani, S. Bauer, T. Someya, L. Torsi and N. S. Sariciftci, Adv. Mater., 2013, 25, 1563–1569 Search PubMed.
- E. D. Głowacki, L. Leonat, M. Irimia-Vladu, R. Schwödiauer, M. Ullah, H. Sitter, S. Bauer and N. Serdar Sariciftci, Appl. Phys. Lett., 2012, 101, 023305 Search PubMed.
- S. Mula, T. Han, T. Heiser, P. Lévêque, N. Leclerc, A. P. Srivastava, A. Ruiz-Carretero and G. Ulrich, Chem. – Eur. J., 2019, 25, 8304–8312 Search PubMed.
- H. Zhang, R. Deng, J. Wang, X. Li, Y.-M. Chen, K. Liu, C. J. Taubert, S. Z. D. Cheng and Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 21891–21899 Search PubMed.
- J. Dhar, D. P. Karothu and S. Patil, Chem. Commun., 2015, 51, 97–100 Search PubMed.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 Search PubMed.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A.-M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740–743 Search PubMed.
- M. Más-Montoya, P. Gómez, D. Curiel, I. da Silva, J. Wang and R. A. J. Janssen, Chem. – Eur. J., 2020, 26, 10276–10282 Search PubMed.
- P. Gómez, S. Georgakopoulos, J. P. Cerón, I. da Silva, M. Más-Montoya, J. Pérez, A. Tárraga and D. Curiel, J. Mater. Chem. C, 2018, 6, 3968–3975 Search PubMed.
- P. Gómez, M. Más-Montoya, I. da Silva, J. P. Cerón-Carrasco, A. Tárraga and D. Curiel, Cryst. Growth Des., 2017, 17, 3371–3378 Search PubMed.
- J. K. Laha, P. Petrou and G. D. Cuny, J. Org. Chem., 2009, 74, 3152–3155 Search PubMed.
- S. Kuster and T. Geiger, Dyes Pigm., 2015, 113, 110–116 Search PubMed.
- Y. Hashimoto and K. Shudo, Chem. Pharm. Bull., 1984, 32, 1992–1994 Search PubMed.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 Search PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 Search PubMed.
- K. Ivaniuk, V. Cherpak, P. Stakhira, Z. Hotra, B. Minaev, G. Baryshnikov, E. Stromylo, D. Volyniuk, J. V. Grazulevicius, A. Lazauskas, S. Tamulevicius, B. Witulski, M. E. Light, P. Gawrys, R. J. Whitby, G. Wiosna-Salyga and B. Luszczynska, J. Phys. Chem. C, 2016, 120, 6206–6217 Search PubMed.
- K. Hyodo, S. Nishinaga, Y. Sawanaka, T. Ishida, H. Mori and Y. Nishihara, J. Org. Chem., 2019, 84, 698–709 Search PubMed.
- Y. Yi, L. Zhu and J.-L. Brédas, J. Phys. Chem. C, 2012, 116, 5215–5224 Search PubMed.
- A. Pietrangelo, M. J. MacLachlan, M. O. Wolf and B. O. Patrick, Org. Lett., 2007, 9, 3571–3573 Search PubMed.
- P. Gao, D. Beckmann, H. N. Tsao, X. Feng, V. Enkelmann, W. Pisula and K. Mullen, Chem. Commun., 2008, 1548–1550 Search PubMed.
- W. Schmidt, J. Chem. Phys., 1977, 66, 828 Search PubMed.
- H. Klevens, J. Chem. Phys., 1949, 17, 470 Search PubMed.
- H. Chen, W. Zhang, M. Li, G. He and X. Guo, Chem. Rev., 2020, 120, 2879–2949 Search PubMed.
- X. Wu, R. Jia, J. Pan, X. Zhang and J. Jie, Nanoscale Horiz., 2020, 5, 454–472 Search PubMed.
- Y. Wang, X. Huang, T. Li, L. Li, X. Guo and P. Jiang, Chem. Mater., 2019, 31, 2212–2240 Search PubMed.
- M. Kano, T. Minari and K. Tsukagoshi, Appl. Phys. Lett., 2009, 94, 143304 Search PubMed.
- D. Kumaki, T. Umeda and S. Tokito, Appl. Phys. Lett., 2008, 92, 013301 Search PubMed.
- D. Berg, C. Nielinger, W. Mader and M. Sokolowski, Synth. Met., 2009, 159, 2599–2602 Search PubMed.
- W. Hong, Z. Wei, H. Xi, W. Xu, W. Hu, Q. Wang and D. Zhu, J. Mater. Chem., 2008, 18, 4814–4820 Search PubMed.
- Y. Hiroyuki, M. Jin, A. Shinji and S. Yoshimasa, Jpn, J. Appl. Phys., 2008, 47, 4728 Search PubMed.
- E. F. Paulus, F. J. J. Leusen and M. U. Schmidt, CrystEngComm, 2007, 9, 131–143 Search PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 Search PubMed.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 Search PubMed.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 Search PubMed.
- J. Aragó and A. Troisi, Adv. Funct. Mater., 2016, 26, 2316–2325 Search PubMed.
- J. Aragó and A. Troisi, Phys. Rev. Lett., 2015, 114, 026402 Search PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 Search PubMed.
- A. Troisi and G. Orlandi, Phys. Rev. Lett., 2006, 96, 086601 Search PubMed.
- R. P. Fornari, J. Aragó and A. Troisi, J. Chem. Phys., 2015, 142, 184105 Search PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 Search PubMed.
Footnotes |
| † Dedicated to Prof. Jaume Veciana and Prof. Concepció Rovira on occasion of their 70th birthdays. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2069806, 2069805, 1557392 and 1957368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc01328a |
| § P. G. and J. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |