
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and electronic properties of nitrogen-doped π-extended polycyclic aromatic dicarboximides with multiple redox processes†‡
Matías J.
Alonso-Navarro§
 ab,
Alexandra
Harbuzaru§
c,
Marcos
Martínez-Fernández
a,
Paula
Pérez Camero
c,
J. Teodomiro
López Navarrete
c,
M. Mar
Ramos
b,
Rocío
Ponce Ortiz
*c and
José L.
Segura
*a
ab,
Alexandra
Harbuzaru§
c,
Marcos
Martínez-Fernández
a,
Paula
Pérez Camero
c,
J. Teodomiro
López Navarrete
c,
M. Mar
Ramos
b,
Rocío
Ponce Ortiz
*c and
José L.
Segura
*a
aDepartment of Organic Chemistry, Complutense University of Madrid, Faculty of Chemistry, Madrid 28040, Spain. E-mail: segura@ucm.es
bChemical and Environmental Technology Department. Univ. Rey Juan Carlos, Móstoles, 28933, Spain
cDepartment of Physical Chemistry, University of Málaga, Málaga, 29071, Spain. E-mail: rocioponce@uma.es
First published on 2nd June 2021
Abstract
In this work, a series of nitrogen-doped polycyclic aromatic mono and dicarboximides was designed, and their synthesis is reported by an innovative synthetic procedure that avoids metal-based cross-coupling reactions. A combined theoretical, electrochemical, optical spectroscopy and material characterization study allows the investigation of the interplay of different effects such as (i) the type of solubilizing chain, (ii) the all-acceptor vs. donor–acceptor nature of the novel systems, and (iii) the molecular ordering, on the ability to stabilize multiple charges and on the OTFT device response characteristics. The amphoteric redox behavior of some of the novel systems allow to obtain ambipolar mobilities while the electron mobility values of the three all-acceptor systems can be rationalized in terms of supramolecular aggregation and enhanced film-forming ability.
1. Introduction
Organic semiconductors based on π-conjugated materials have been intensively investigated for their advantages in the manufacture of low-cost, light-weight, and flexible optoelectronic devices.1–3 The chemical structure of the π-conjugated materials, as well as their purity, supramolecular organization and film morphology determine the electronic performance of these organic devices. Therefore, in order to develop new organic semiconductors with high mobilities, an in-depth understanding of the molecular structure-charge transport property relationships is required. In this regard, π-conjugated fused-ring systems, such as polycyclic aromatic dicarboximides4–7 (PADI) and their heteroatom-doped analogues are emerging classes of semiconducting materials8 derived from the well-known family of polycyclic aromatic hydrocarbons9–13 (PAH). These materials have been successfully applied in organic (opto)electronics14–18 and more recently, as electrodes in energy storage devices due to their remarkable redox behavior.19–23 Among them, π-conjugated compounds containing imide groups, such as naphthalene24 and perylene diimides (NDI and PDI)1,2,25–27 are among the most successful n-type organic semiconductors, due to the strong electron-withdrawing character of the imide group, which lowers the LUMO energy levels of the semiconductor, facilitating electron injection and charge stabilization through the π-conjugated systems. Furthermore, modifications on the conjugated skeletons of NDI and PDI derivatives offer the possibility of tuning the redox potentials and promoting the generation of reversible polyanionic species, for their use as electrode materials in batteries.28–32 Besides, processability can be easily modulated in these derivatives by the appropriate selection of alkyl chains substituents at the nitrogen atom of the imide groups. Thus, long or bulky alkyl chains provide good film forming ability through enhanced solubility, even in large planar π-extended systems with many fused rings, while intermolecular interactions can be promoted by the absence of lateral substituents, for applications as electrodes.π-Extended PADI systems are usually obtained from functionalized rylene monoimide derivatives via metal-based cross-coupling or C–H arylation reactions.6,33,34 In this regard, the synthesis of new π-extended polycyclic aromatic dicarboximides by metal-free reactions is relatively unexplored. Inspired by the excellent properties of these organic semiconductors and by the relative scarcity of metal-free reactions for the synthesis of heteroatom-doped PADI materials, we report herein the synthesis and study of new electron-poor π-conjugated polycyclic aromatic dicarboximides.4,6,7
Taking into account that 1,2-diketones have already proven to be good intermediates for the synthesis of fused-ring systems,35 in this article we describe the synthesis and characterization of a novel family of π-extended nine-ring-fused systems (naft-1–4, Scheme 1) by auto-condensation and cross-condensation of pyrene36 and 1,8-naphthalimide37–39 moieties endowed with 1,2-diketone functionalities. The systems based only on naphthalimide units (naft-2–4) are designed to exhibit good electron accepting properties while that based on pyrene and 1,8-naphthalimide (naft-1) is designed to exhibit an ambipolar behavior. Different solubilizing chains are incorporated in the novel systems in order to tune their supramolecular aggregation and film-forming ability. These novel systems have been characterized by cyclic voltammetry (CV), UV-vis absorption, spectroelectrochemistry, and density functional theory (DFT) calculations. Furthermore, their electrical properties in OFETs have been examined using vapor-deposited films which have been also characterized by X-ray diffraction (XRD) and atomic force microscopy (AFM) analysis. The study allows the investigation of the interplay of different effects such as (i) the type of solubilizing chain, (ii) the all-acceptor vs donor–acceptor nature of the novel systems, and (iii) the molecular ordering, on the ability to stabilize multiple charges and on the OTFT device characteristics.
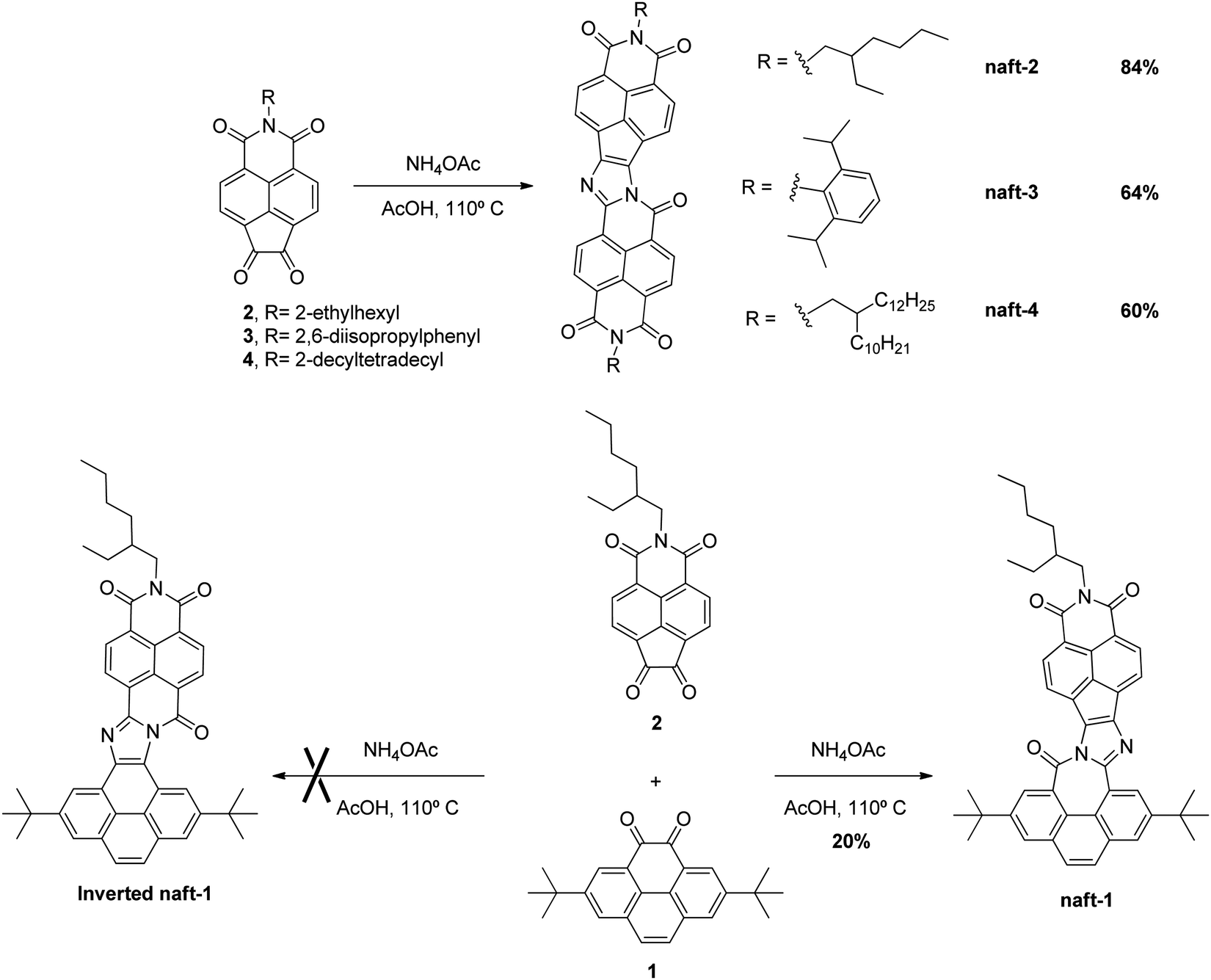 | ||
| Scheme 1 Syntheses of the new naphthalimide and pyrene assemblies studied in this work. | ||
2. Results and discussion
The syntheses of the target compounds reported in this article (naft-1–4, Scheme 1) start with the preparation of suitably functionalized pyrene and 1,8-naphthalimide moieties endowed with 1,2-diketone functionalities. Thus, the t-butyl substituted pyrene-4,5-dione 1 (Scheme 1) has been obtained from pyrene by following the synthetic procedure previously reported by Harris and coworkers36 while 1,8-naphthalimide derivatives (2–4, Scheme 1) endowed with 1,2-diketone functionalities and different substituents at imide nitrogen have been obtained by using the four-step reaction sequence from acenaphthene previously developed by our group.37–40In order to synthesize the novel family of electron-acceptor π-extended nine-ring-fused systems, we have used the innovative one-pot condensation method of aromatic 1,2-diketones recently developed by Zhao and co-workers.41,42 Thus, diketones 2–4 were self-condensed in glacial acetic acid in the presence of an excess of ammonium acetate by heating overnight at 110 °C to yield the target molecules (naft-2–4, Scheme 1) after a purification step. The three novel electron acceptors share the same π-conjugated skeleton and differ only on the solubilizing chain at the imide nitrogen.
In addition, a novel donor–acceptor π-extended nine-ring-fused system has been obtained by cross-condensation reaction of an equimolecular mixture of the diketone based on the 1,8-naphthalimide moiety (2) and the pyrene-4,5-dione derivative 1 suspended in glacial acetic acid together with ammonium acetate and heated at 110 °C overnight (Scheme 1). In this reaction, two possible products (naft-1 and inverted naft-1, Scheme 1) may be formed. However, only naft-1 was obtained in the reaction together with a small amount of naft-2 and the pyrene dimer, due to the self-coupling product of 1,2-diones 1 and 2. The regioselectivity toward the formation of naft-1 may be due to the different reactivity of 1,2-diones 1 and 2. In contrast to pyrene, the 1,8-naphthalimide moiety is a stronger electron-acceptor, what makes the 1,2-dione 2 more reactive toward ammonium acetate than the parent pyrene derivative 1. Thus, the higher reactivity of 1,2-dione 2 is responsible for the better yields observed for the synthesis of 1,8-naphthalimide-based dimers naft-2–4 (60–85%) in comparison with those obtained in the syntheses of both the mixed-based derivative naft-1 (20%) and the previously reported pyrenedione-based dimers (<50%).41
It is worth pointing out that the solubility problem commonly associated with this type of π-conjugated fused-ring systems is avoided by the incorporation of the t-butyl groups in the pyrene moiety as well as by the incorporation of suitable branched alkyl chains and 3,5-diisopropylphenyl substituent at the imide-nitrogen of 1,8-naphthalimides. Thus, the chemical structures of the four π-extended nine-ring-fused systems could be properly characterized not only by mass spectrometry and FT-IR but also by NMR given their good solubility in deuterated chloroform. However, due to the lower solubility of naft-2 in deuterated solvents, as a consequence of the shortest alkyl chain at the imide position, its 13C-NMR spectrum could only be obtained properly by the addition of a small amount of deuterated trifluoroacetic acid.
The analysis of 1H-NMR spectra revealed a clearly well-resolved pattern for these four compounds (Fig. S1–S4, ESI‡). First, the appearance of well-defined downfield signals reveals the characteristic protons for the 1,8-naphthalimide fragments between 9.4 and 7.8 ppm. Due to the non-symmetrical structure of naft-1, its 1H-NMR spectrum shows 10 signals corresponding to the 10 non-equivalent hydrogen atoms present in the structure. The signals corresponding to the hydrogen atoms of the naphthalimide moiety appear at 9.23, 9.03, 8.84 and 8.74 ppm with their characteristic coupling constants of 7.5 Hz (Fig. S5, ESI‡). Concerning with the pyrene moiety, the singlet at 9.95 ppm can be assigned to the hydrogen atom closer to the heterocyclic nitrogen and the singlet at 8.91 can be assigned to the hydrogen atom closer to the carbonyl group according to that observed for pyrene dimers with analogous structure.43 The singlets at 8.43 and 8.48 correspond to the other hydrogen atoms next to the tert-butyl substituents. Finally, the remaining signals that appear as an AB system centered at 8.11 correspond to the diagnostic AB system of the pyrene moiety. The assignment of these signals has been carried out by two-dimensional homonuclear (1H–1H) and heteronuclear (1H–13C) correlation experiments (Fig. S6–S8, ESI‡). In addition, for all of them, at higher field the characteristic signal for the protons in alpha position to the imide nitrogen appears at around 4 ppm. Finally, all the typical signals of the alkyl chains appear around 1 ppm. For the 13C-NMR spectra (Fig. S9–S12, ESI‡) the most deshielded protons correspond to the imide carbons. Then, there are three signals at 157, 155 and 145 ppm which can be assigned to the newly formed amidine system. All signals between 140 and 110 ppm correspond to sp2 carbons in the π-conjugated hydrocarbon skeleton. Up-field, there are the signals corresponding to the alkyl chains, all bellow 50 ppm.
In the FTIR spectra (Fig. S13–S16, ESI‡), the most characteristic bands are the imide group between 1715 and 1690 cm−1 and the bands corresponding to the imine moiety between 1650 and 1590 cm−1, depending on each molecule. All typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching bands appear around 1550–1400 cm−1. Finally, from high resolution mass spectrometry analyses we can conclude the successful achievement of the desired compounds (Fig. S17–S20, ESI‡).
C stretching bands appear around 1550–1400 cm−1. Finally, from high resolution mass spectrometry analyses we can conclude the successful achievement of the desired compounds (Fig. S17–S20, ESI‡).
Herein, we propose a possible mechanism for this kind of reaction, (Scheme 2) which is consistent with that previously reported by Zhao and co-workers, for the self-condensation of pyrene-4,5-dione.41 The diimine intermediate NID-1 is formed by treating the 2-(2-ethylhexyl)-1H-indeno[6,7,1-def]isoquinoline-1,3,6,7(2H)-tetraone (2) with ammonium acetate. The next step involves the formation of a spiro-derivative (NID-2) which suffers a ring-opening, leading to NID-3 by the nucleophilic attack of a molecule of water. Due to the acidic media in the reaction, there is an intramolecular nucleophilic attack in the condensation between the imidazole ring and the free carboxylic acid, followed by a loss of water, to obtain naft-2 as a stable product. For the mixed derivative naft-1, the mechanism should be the same as that proposed above but considering the introduction of pyrene dione 1 instead of the naphthalimide dione 2, thus leading to a seven-ring imidine macrocycle.
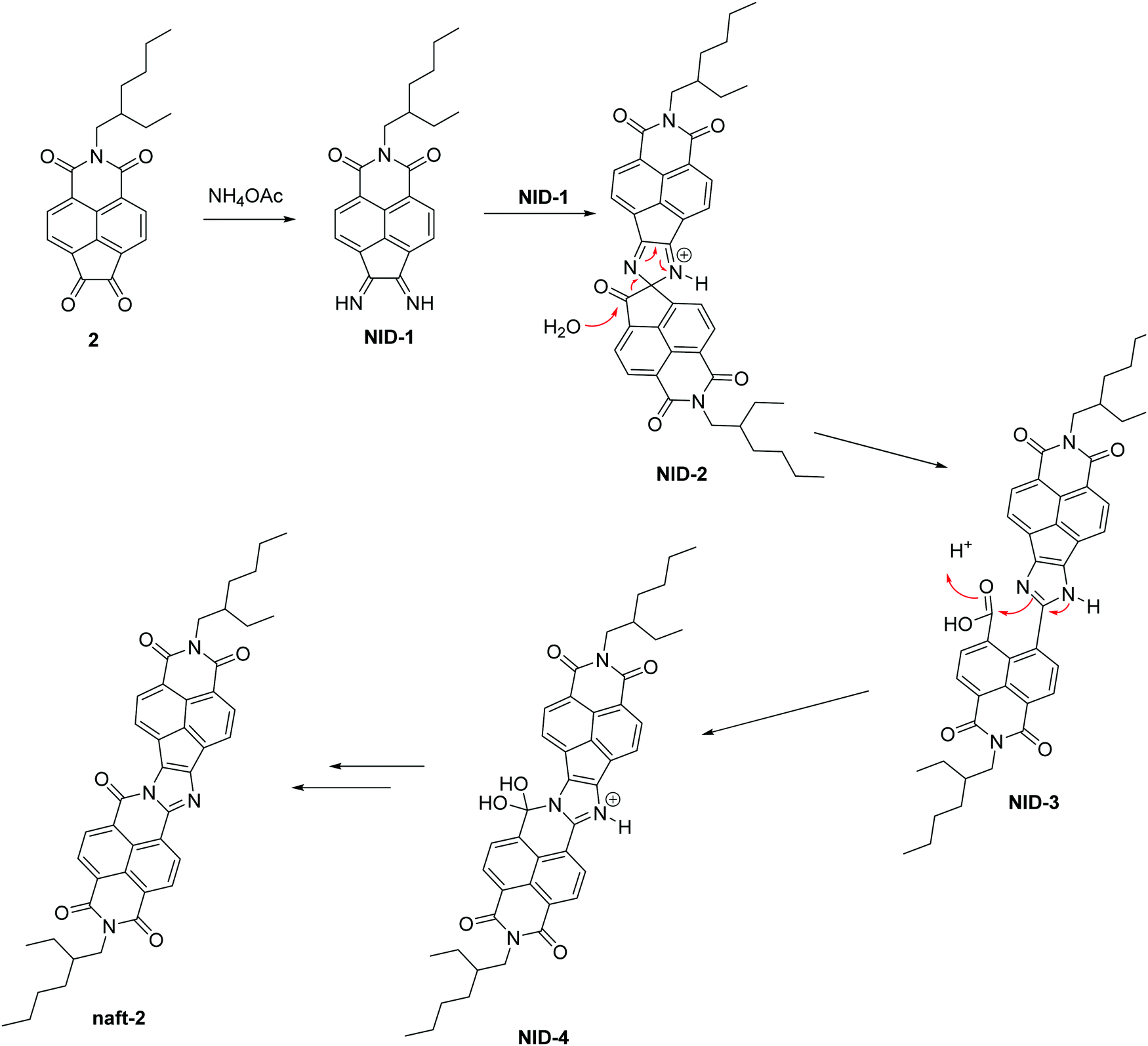 | ||
| Scheme 2 Proposed mechanism for the auto-condensation of the 1,2-dione of naphthalimide 2. | ||
Optical and electrochemical properties
The acceptable solubility of the novel fused systems described in this work allows also proper characterization of their optical and electrochemical properties in solution by means of UV-Vis spectroscopy and cyclic voltammetry. The main values of the optical and electrochemical properties of naft-1–4 are summarized in Tables 1–3.| UV-visa | |||||
|---|---|---|---|---|---|
| [C] (μM) | λ max (nm) | ε λ max (M−1 cm−1) | λ onset (nm) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e (eV) e (eV) |
|
| a Absorption measurements in CHCl3 solution. b Absorption maxima in solution. c Molar absorption coefficient to the referred wavelength. d Absorption band for the onset wavelength. e Energy band gap derived from the low-energy absorption edge using the equation Eoptg =1240/λonset. | |||||
| naft-1 | 0.275 | 340 | 37090 |
716 | 1.73 |
| naft-2 | 0.275 | 330 | 17090 |
655 | 1.89 |
| naft-3 | 0.275 | 331 | 40000 |
651 | 1.90 |
| naft-4 | 0.275 | 329 | 54545 |
670 | 1.85 |
| Molecules | λ exp (nm) | λ theor (nm) | Description | f | ||||
|---|---|---|---|---|---|---|---|---|
| λ max (nm) | λ ICT (nm) | λ max (nm) | λ ICT (nm) | λ max (nm) | λ ICT (nm) | λ max (nm) | λ ICT (nm) | |
| naft-1 | 335 | 587 | 371 | 590 | HOMO → L+2 | HOMO → LUMO | 0.79 | 0.03 |
| naft-2 | 328 | 551 | 324 | 636 | HOMO → L+2 | HOMO → LUMO | 0.98 | 0.22 |
| naft-3 | 329 | 551 | 324 | 633 | HOMO → L+2 | HOMO → LUMO | 0.90 | 0.24 |
| naft-4 | 328 | 551 | 323 | 634 | HOMO → L+2 | HOMO → LUMO | 0.91 | 0.21 |
| Cyclic voltammetrya | |||||||
|---|---|---|---|---|---|---|---|
| E 1/2ox1 (V) | E 1/2red1 (V) | E 1/2red2 (V) | E 1/2red3 (V) | E 1/2red4 (V) | E HOMO (eV) | E LUMO (eV) | |
| a Electrochemical data recorded by cyclic voltammetry experiments carried out in DCM/TBAPF6 (0.1 M) solutions at a scan rate of 0.1 V s−1, using Pt as working and the counter electrodes, and Fc/Fc+ redox couple as internal reference. b Estimated from EHOMO = −5.1 eV – E1/2oxI. c Estimated from ELUMO = −5.1 eV − E1/2redI. d Estimated from Eoptg = EHOMO − ELUMO. | |||||||
| naft-1 | 0.92 | −1.08 | −1.38 | — | — | −6.02 | −4.02 |
| naft-2 | — | −0.98 | −1.33 | −1.45 | −1.66 | −6.01d | −4.12 |
| naft-3 | — | −0.89 | −1.22 | −1.49 | −1.65 | −6.11d | −4.21 |
| naft-4 | — | −0.98 | −1.30 | −1.51 | −1.72 | −6.01d | −4.12 |
In Fig. 1a, the UV-Vis absorption spectrum in chloroform of the mixed-based derivative naft-1 is shown together with those of the pyrene-based 1,2-dione 1 and the naphthalimide 1,2-dione 2. For comparison purposes, Fig. 1b depicts the UV-Vis absorption spectra of the naphthalimide dimer naft-2 together with that of the naphthalimide 1,2-dione 2. In Fig. 1c, the UV-vis-spectra of the three naphthalimide-dimers (naft-2–4), with different substituents at the imide nitrogen atoms, are shown and a comparison between all dimers described in this work is shown in Fig. 1d.
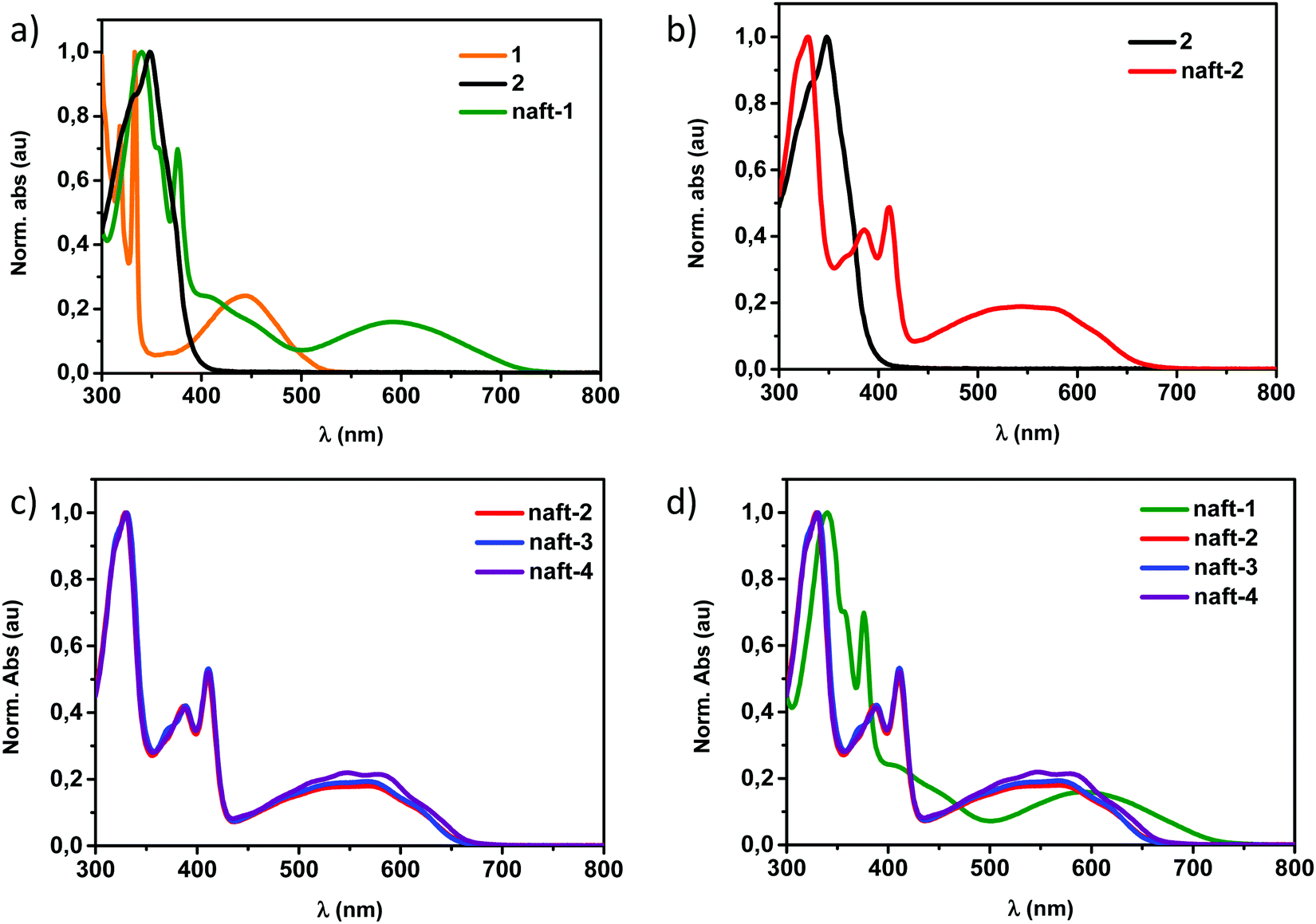 | ||
| Fig. 1 UV-vis absorption spectra in chloroform solution of (a) naft-1 dimer and (b) naft-2 derivative together with the UV-vis absorption spectra of the diones used for their syntheses. (c) UV-Vis absorption spectra of the three naphthalene-based dimers naft-2–4. (d) Normalized comparison between all dimers naft-1–4 described in this work. | ||
In comparison with the 1,2-diketones used as starting materials, both derivatives naft-1 and naft-2 show some new vibronic bands either below 400 nm (naft-1) or around 400 nm (naft-2), which are due to the more complex and rigid π-conjugated system formed. In addition, the UV-vis spectra of both naft-1 and naft-2 exhibit new broad bands centered at 600 and 550 nm, respectively, which can be attributed to an absorption with a charge transfer character. This charge transfer band is not affected by the solution concentration (Fig. S25–S28, ESI‡), which strongly suggests the intramolecular nature of the charge transfer absorptions. In this regard, it is worth mentioning that an intramolecular charge-transfer (ICT) band may be expected in naft-1, considering the electron acceptor character of the naphthalimide moiety and the electron-donor characteristics of the phenanthrene moiety. Regarding naft-2, the ICT excitation is theoretically confirmed by TDDFT calculations and can be described as a one-electron HOMO–LUMO excitation, consisting of the displacement of the electron density from the HOMO, mainly localized over the imidazole ring, to the LUMO, which is totally localized over the naphthalimide moiety (see Fig. 2 and Fig. S36, ESI‡).
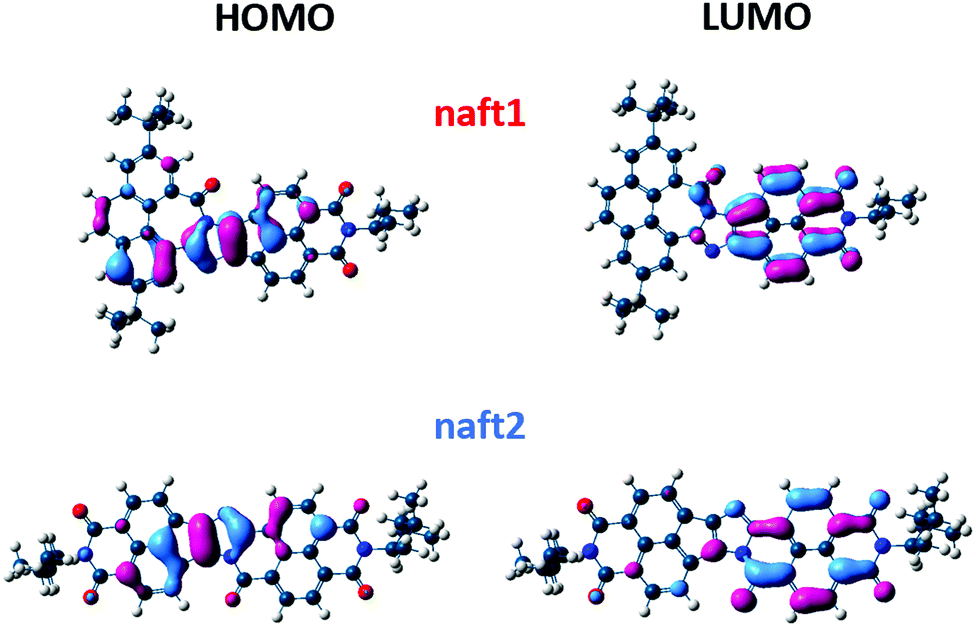 | ||
| Fig. 2 B3LYP/6-31G** predicted molecular orbitals of the naphthalene-based molecules naft-1 and naft-2. The pattern of the electron density distribution for naft-2 is identical to that found for naft-3 and naft-4 (see naft-3 and naft-4 MOs in the ESI‡). | ||
The other naphthalimide dimers (naft-3 and naft-4) show identical UV-Vis spectra in chloroform solutions in comparison with that of the naft-2 analogue (Fig. 1c and in the ESI‡). All of them show absorption maxima centered at 330 nm followed by vibronic structured absorptions around 400 nm, ending with an ICT band centered at 550 nm. The almost undifferentiated UV-vis spectra observed for these compounds is consistent with their identical central π-conjugated cores as they only differ in the solubilizing alkyl or aryl chains at the imide nitrogen atoms. However, it is also worth pointing out that when the experiments were carried out in dichloromethane solutions, slight differences were observed. Thus, an inversion of the relative intensity in the structured bands was observed for naft-2 in comparison with that observed for naft-3 and naft-4 (Fig. S29, ESI‡). This behavior is due to the remarkable differences in solubility of the naft-2–4 derivatives in dichloromethane solutions. The limited solubility of naft-2 in dichloromethane in comparison with the parent naft-3 and naft-4 analogues may be responsible for the formation of supramolecular aggregates.40 In order to confirm this supramolecular aggregation, dilution experiments were carried out in dichloromethane, as depicted in Fig. S30 (ESI‡). When naft-2 solution is diluted enough, an inversion of the structured bands occurs,44,45 confirming the presence of supramolecular aggregates in solution at concentrations around 10−4 M and non-aggregated molecular species in solution at 10−5 M. This study was also accomplished for naft-3 (Fig. S29c and d, ESI‡) and naft-4 (Fig. S29e and f, ESI‡) but, in those cases, the presence of longer alkyl chain or 3,5-diisopropylphenyl substituent at the imide position do not allow the formation of supramolecular species in dichloromethane solutions.
TD-DFT calculations, at B3LYP/6-31G** level, are in good agreement with the experimental data and predict the existence of one intense electronic transition at around 300 nm and a transition at around 600 nm, similar to the experimental electronic absorptions at around 300 and 550 nm, respectively (see Table 2).
The absorption spectra of naft-1–4 as thin films have been also investigated (Fig. 3). Although the UV–vis absorption spectra of naft-2 and naft-4 in solutions (Fig. 1c) are well overlapped due to the similar molecular backbone and the absence of interchain interactions, thin films absorption spectra of naft-2 and naft-4 show perceptible, red-shifted bands in comparison with their absorption in chloroform solutions. This behavior is characteristic of a stronger molecular interaction in the aggregated state,46 as we previously observed for naft-2 in solution. In agreement with that observed for other alkyl substituted π-conjugated systems,47,48 the shorter the side chains, the stronger the aggregation. Concerning naft-3, π–π-interactions are significantly decreased due to the bulky steric hindrance of the diisopropylphenyl substituent and therefore, no significant differences are found between the solution and thin film UV-Vis absorption spectra. Finally, thin film absorption spectrum of naft-1 also shows perceptible shifted bands in comparison with the absorption in chloroform solution, which suggests also strong molecular interactions in the solid state.
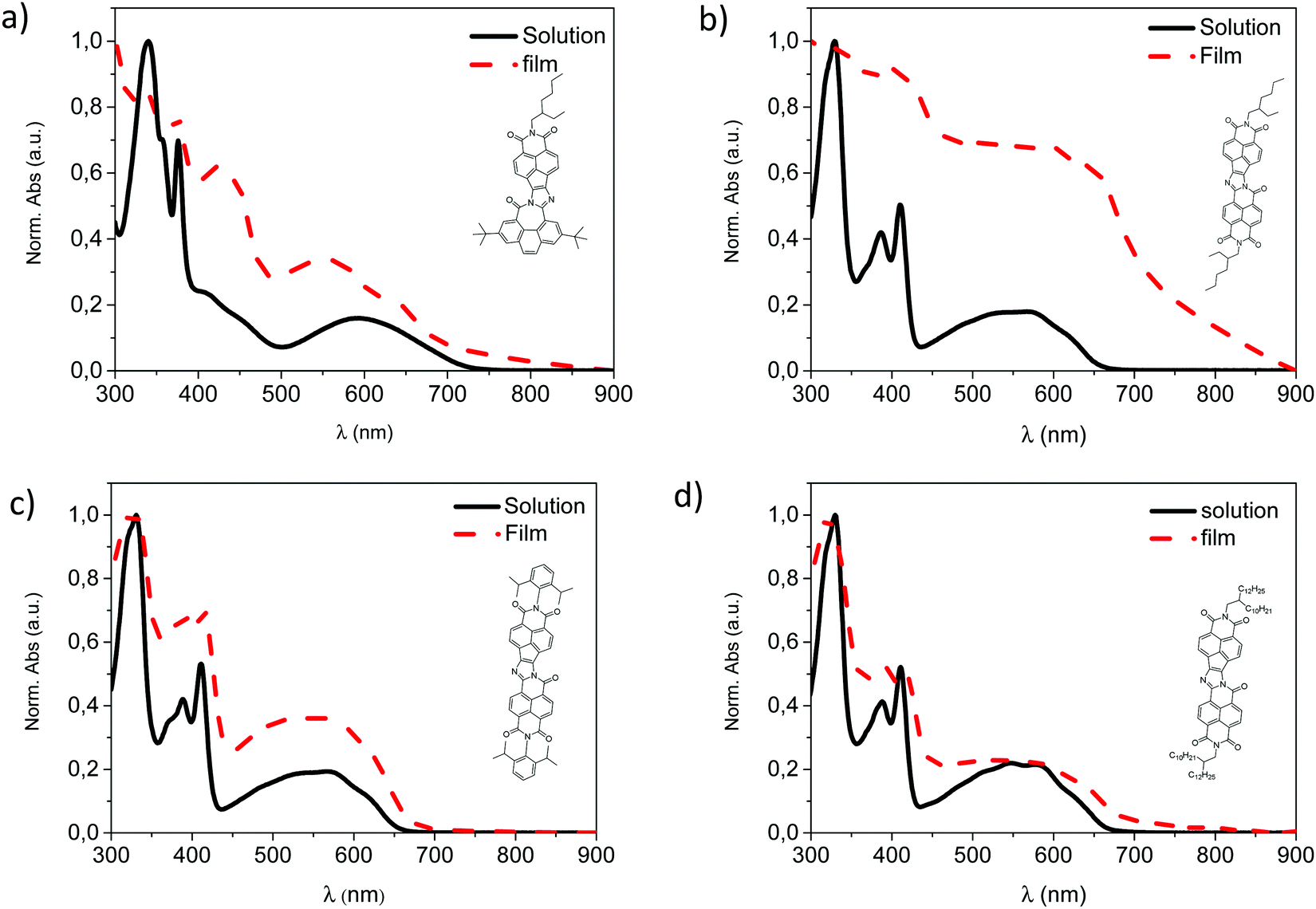 | ||
| Fig. 3 Thin film absorption spectra of the naphthalene-based derivatives (a) naft-1, (b) naft-2 (c) naft-3 (d) naft-4. | ||
The acidochromic behavior in solution of these compounds was also studied. Fig. 4 depicts the remarkable changes in the absorption spectra of naft-1–4 by addition of increasing amounts of a strong organic acid such as trifluoroacetic acid49 (TFA: pKa = −0.25). This behavior is characteristic of conjugated molecules and polymers containing N-heterocyclic units such as pyridine,50,51 pyrazine,52 imidazole,53 or quinazolines,54 which have been reported to strongly react with acids due to the capability of these acids to interact with the lone pairs of the basic nitrogen of the heterocyclic moieties.
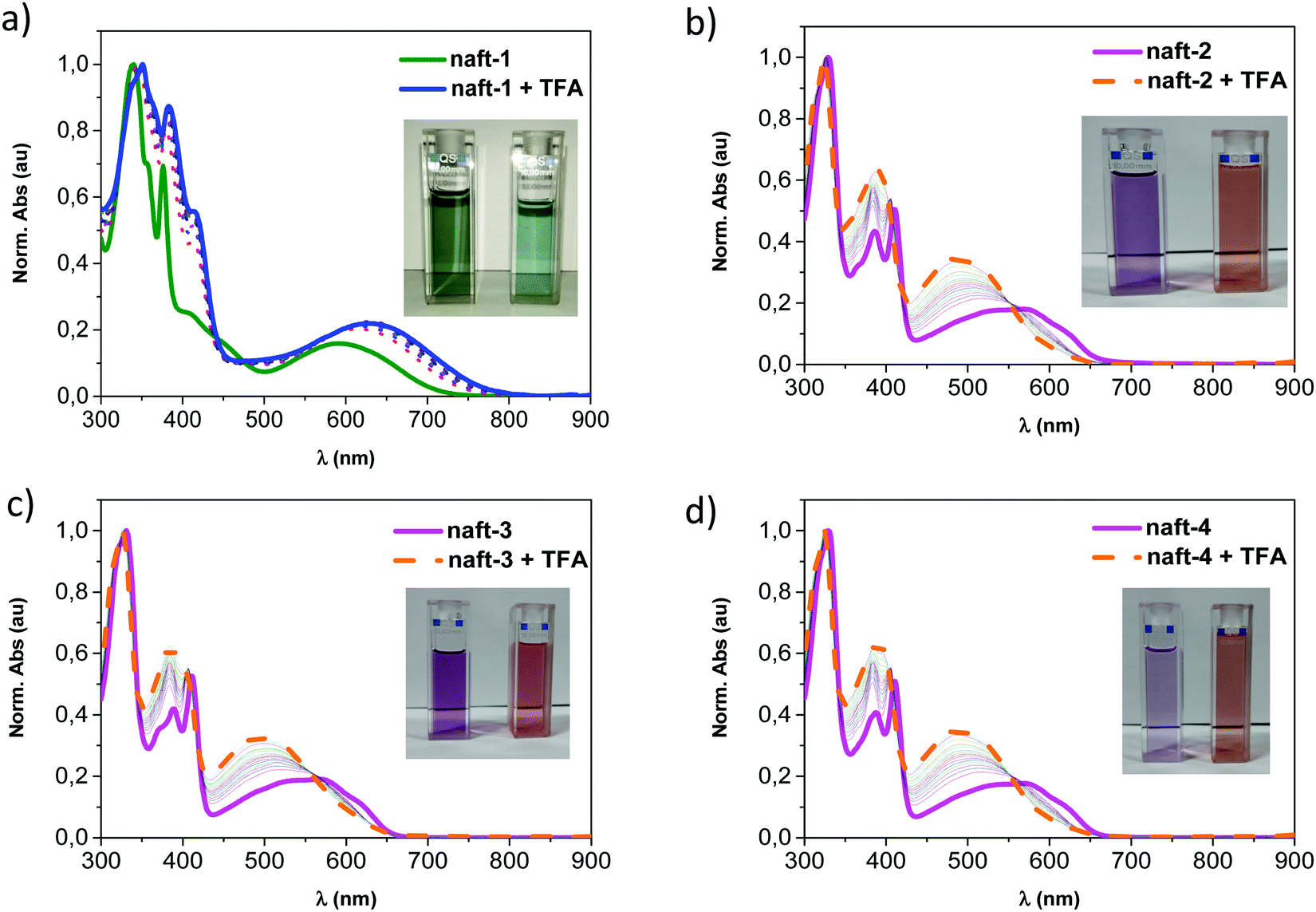 | ||
| Fig. 4 Normalized UV-Vis spectra of (a) naft-1, (b) naft-2, (c) naft-3 and (d) naft-4 in chloroform solutions in response to the addition of TFA from 0 to 2.3 × 105 molar equivalents. The photograph shows the colour change from the initial (left) to the final solution (right). | ||
UV-vis tritation experiments, carried out by adding TFA into a chloroform solution of pyrene-containing naft-1, show bathochromic shifts in all absorption bands. This behavior is due to the interaction of the trifluoroacetic acid with the in-plane electron lone pair of the imidazole nitrogen atom, which is not involved in π-conjugation, acting as a basic site. Under similar UV-vis titration experiments, derivatives naft-2–4 exhibit an hypsochromic response to the addition of an excess of TFA. The protonated species were theoretically predicted for naft-1 and naft-4 (Fig. 5, naft-2–3 in Fig. S41 in ESI‡) and their absorption spectra nicely follow the tendencies found experimentally; thus, the HOMO–LUMO gap is slightly enlarged in naft-1 (Fig. 5c) after protonation while it decreases for protonated naft-4 (Fig. 5f). In both cases, protonation remarkably stabilizes both HOMO and LUMO orbitals energy levels, but being the LUMO stabilization more pronounced for naft-1, therefore the gap decrease. In contrast, HOMO stabilization is stronger for protonated naft-4. This can be understood considering the HOMO and LUMO topologies before and after protonation. In fact, following the trend in the orbital energetics, significant changes in the LUMO topologies are found for naft-1 after protonation, while changes in the HOMO orbitals are registered for naft-4.
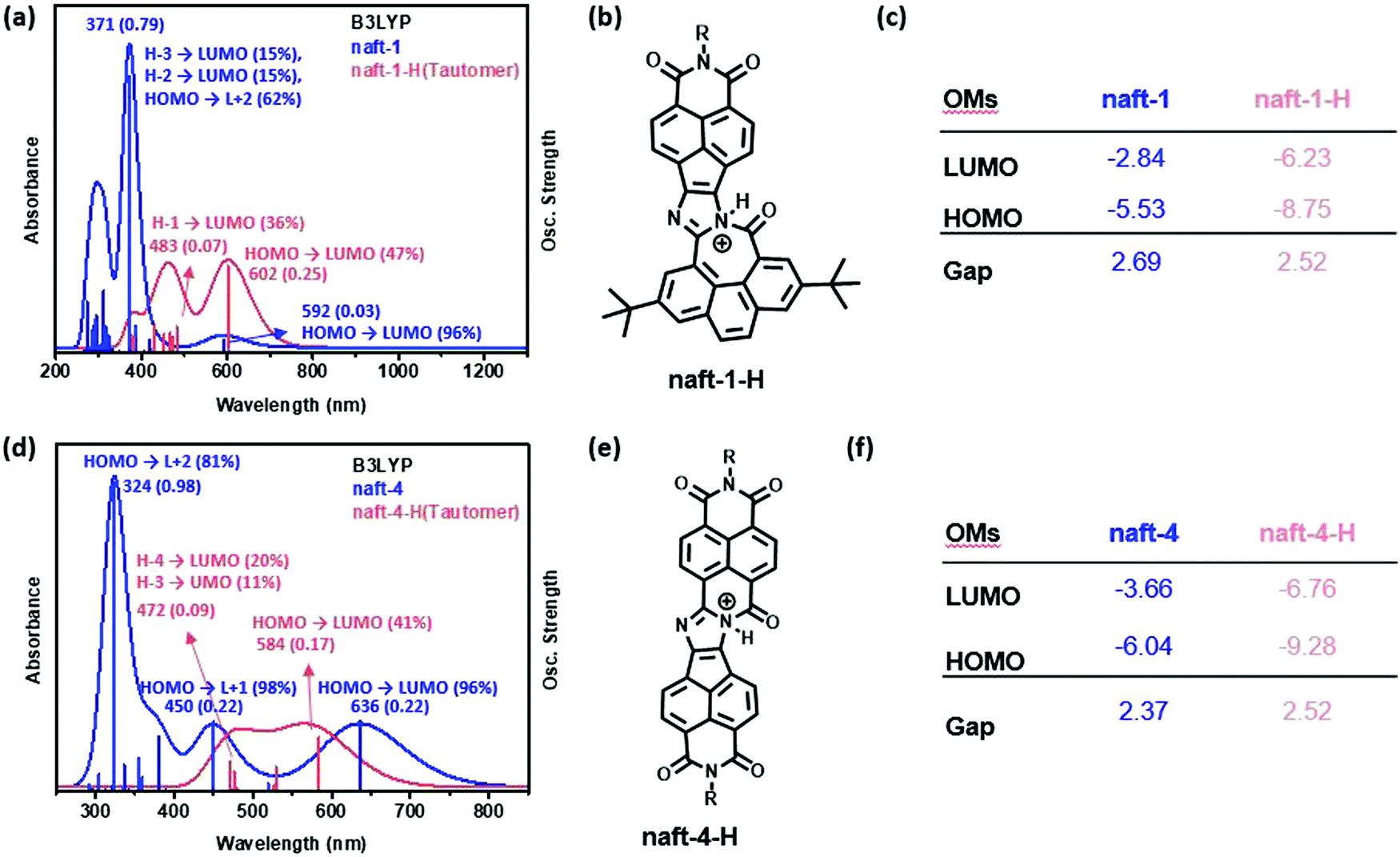 | ||
| Fig. 5 TDDFT/B3LYP-calculated vertical transition energies, for both pristine and protonated forms, and predicted FMOs energies for naft-1 (a–c) and naft-4 (d–f). | ||
The electrochemical redox activities of naft-1–4 were determined by cyclic voltammetry (CV) measurements in dichloromethane solutions and using tetrabutylammonium hexafluorophosphate as the electrolyte. Fig. 6 shows the cyclic voltammograms of these four compounds and the electrochemical data is summarized in Table 3. For the naphthalene-based dimers (naft-2–4) only reversible reduction waves are observed, with only small differences in the reduction values due to different substituents at the imide nitrogen (Fig. 6b–d), as it is described in the literature.55,56 For the mixed-based derivative naft-1 the cyclic voltammogram shows, in addition to the reversible reduction waves, an oxidation process due to the presence of the phenanthrene moiety (Fig. 6a). It is also worth pointing out that the cyclic voltammograms of dimers naft-2–4 show up to four reversible reduction waves, while that of naft-1 shows only two reduction waves. This behavior is consistent with the presence of only one naphthalimide unit in naft-1 and two naphthalimide moieties in naft-2–4.
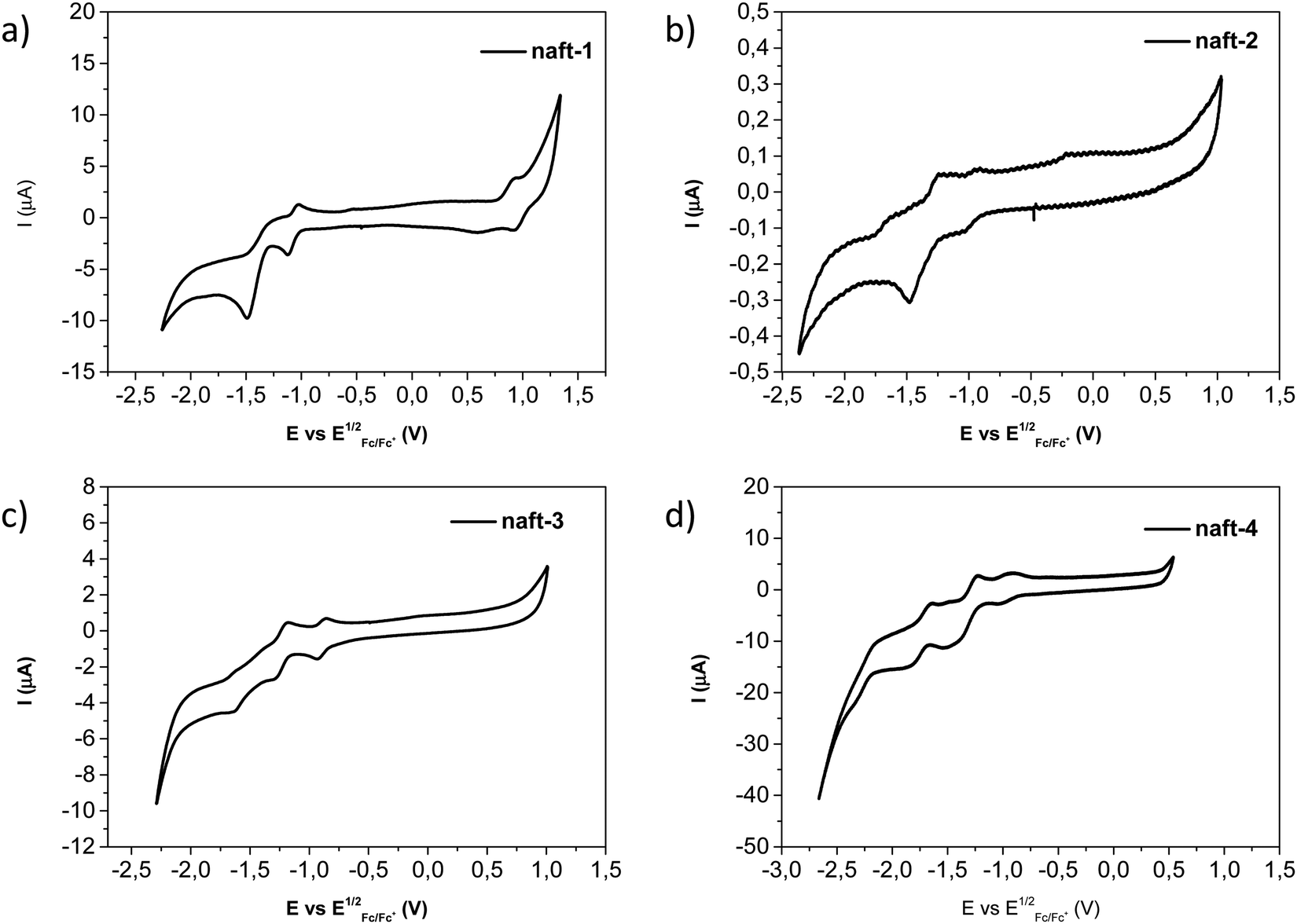 | ||
| Fig. 6 Cyclic voltammograms recorded for (a) naft-1, (b) naft-2, (c) naft-3 and (d) naft-4 derivatives in dichloromethane solutions. | ||
From the first reduction potentials, obtained by cyclic voltammetry in dry dichloromethane (Fig. 6), it is possible to estimate the lowest unoccupied molecular orbital (LUMO) energy levels of naft-1–4 (Table 3). Similarly, the highest occupied molecular orbital (HOMO) energy level of naft-1 can be estimated from the oxidation potential value observed for this derivative. Note that the first reduction potential of naft-1, having only one naphthalimide moiety, is approximately 100 mV more negative than that of naft-2 with two naphthalimide moieties. Furthermore, according to these results, these new derivatives have lower estimated lowest unoccupied molecular orbital (LUMO) energies than that determined for naphthalene diimide (NDI, −4.03 eV) and perylene diimide (PDI, −3.98 eV) derivatives from voltammetry data recorded under identical conditions to those used for the electrochemical characterization of naft1–4. In addition, cyclic voltammetry analyses were also performed in chloroform solution, at the same experimental conditions described above, in order to enhance naft-2 solubility and with the aim to analyze solvent effects on its electrochemical behavior (Fig. S32 in the ESI‡). These measurements were also carried out for naft-1, naft-3 and naft-4 for comparative purposes (Fig. S31–S34, ESI‡). As it is shown in the ESI,‡ the CV values obtained in chloroform solutions are shifted in comparison with the obtained values in dichloromethane. This phenomenon is related to a different stabilization charge behavior due to the different dielectric constants of the solvents (dichloromethane 9 vs. chloroform 4.8), as well as to the different aggregation state in solution for these derivatives. Theoretical calculations carried out by using both dichloromethane and chloroform solvents with a polarizable continuum model (PCM) confirm that the reduced species are less stabilized in chloroform than in dichloromethane solvent (see Table S3, ESI‡).
Table 4 shows the comparison between predicted and experimental frontier molecular orbital energies. Although the absolute values are not always fully consistent, DFT calculations are able to predict the general tendency in orbital energy levels and bandgaps. As expected, the presence of different substituents at the imide nitrogen in naft-2–4 does not have a remarkable effect on the HOMO and LUMO energy gap values.
| Experimental results | Theoretical results | Reorganization energies | ||||||
|---|---|---|---|---|---|---|---|---|
| LUMO (eV) | HOMO (eV) | E gap (eV) | LUMO (eV) | HOMO (eV) | E gap (eV) | λ e | λ h | |
| naft-1 | −4.02 | −6.02 | 2 | −2.82 | −5.51 | 2.69 | 0.29 | 0.31 |
| naft-2 | −4.12 | −6.01 | 1.89 | −3.63 | −6.01 | 2.38 | 0.22 | 0.31 |
| naft-3 | −4.21 | −6.11 | 1.9 | −3.69 | −6.07 | 2.38 | 0.22 | 0.31 |
| naft-4 | −4.12 | −6.01 | 1.89 | −3.64 | −6.03 | 2.39 | 0.22 | 0.31 |
Internal reorganization energies were also computed in order to estimate theoretical parameters related to their charge transport abilities. The data shown in Table 4 indicate similar reorganization energies for both hole and electron transport in naft-1; however, in the case of naft-2–4 semiconductors, the internal reorganization energies for electron transport are substantially lower than those predicted for hole transport. That can be related to the easiness to accommodate negative charges over the two naphthalimide units.
UV-Vis-NIR spectroelectrochemistry
Fig. 7, 8 and Fig. S42 (ESI‡) display the UV-Vis-NIR absorption spectra recorded during57,58 progressive electrochemical reductions of naft-1–4 in dichloromethane in the presence of high excess of tetrabutylammonium tetrafluoroborate (Bu4NBF4), as supporting electrolyte, in an OTTLE cell. In the case of naft-1 both spectroelectrochemical reduction and oxidation processes were recorded (Fig. 7) due to its amphoteric redox behavior. The results are supported by TDDFT theoretical calculations at PBE0/6-31G** level (Fig. S45, ESI‡).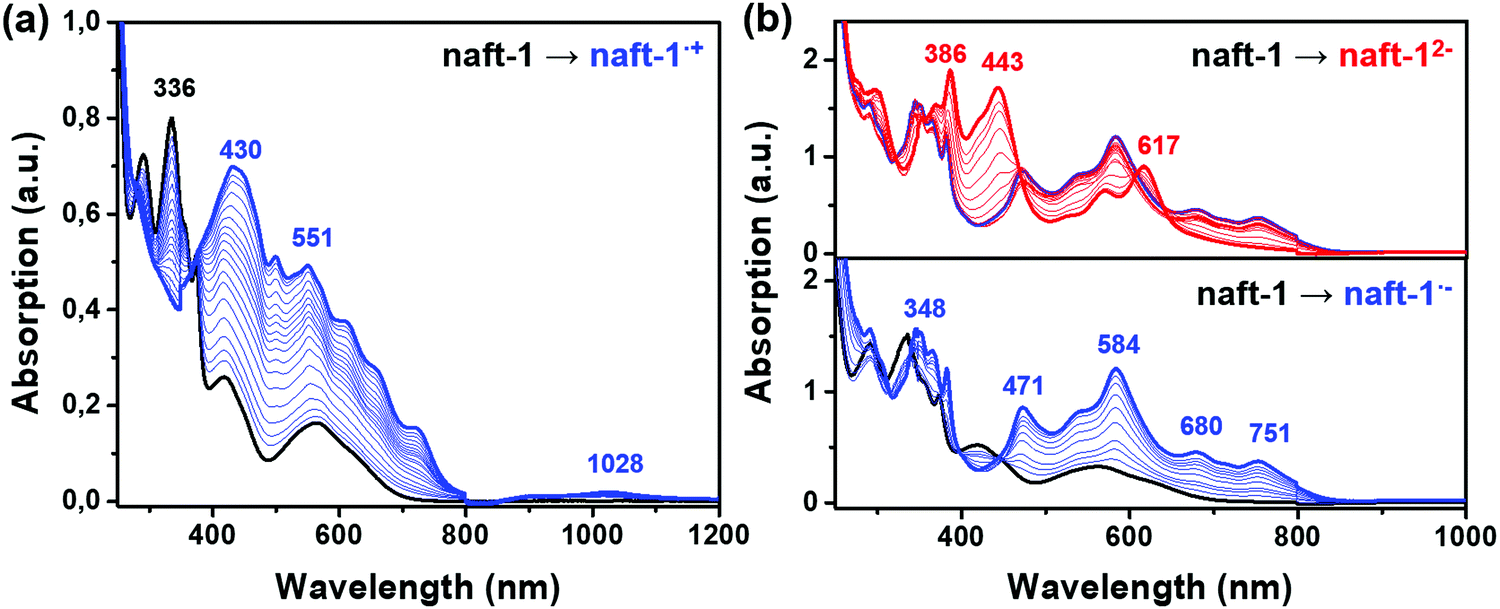 | ||
| Fig. 7 UV-Vis-NIR absorption spectra recorded in an OTTLE cell by (a) electrochemical oxidation (1520 mV, blue curve), (b) during the first step of electrochemical reduction (−790 mV, blue curve) and during the second step of the electrochemical reduction (−1040 mV, red curve) of naft-1 in dichloromethane in presence of Bu4NBF4 supporting electrolyte. | ||
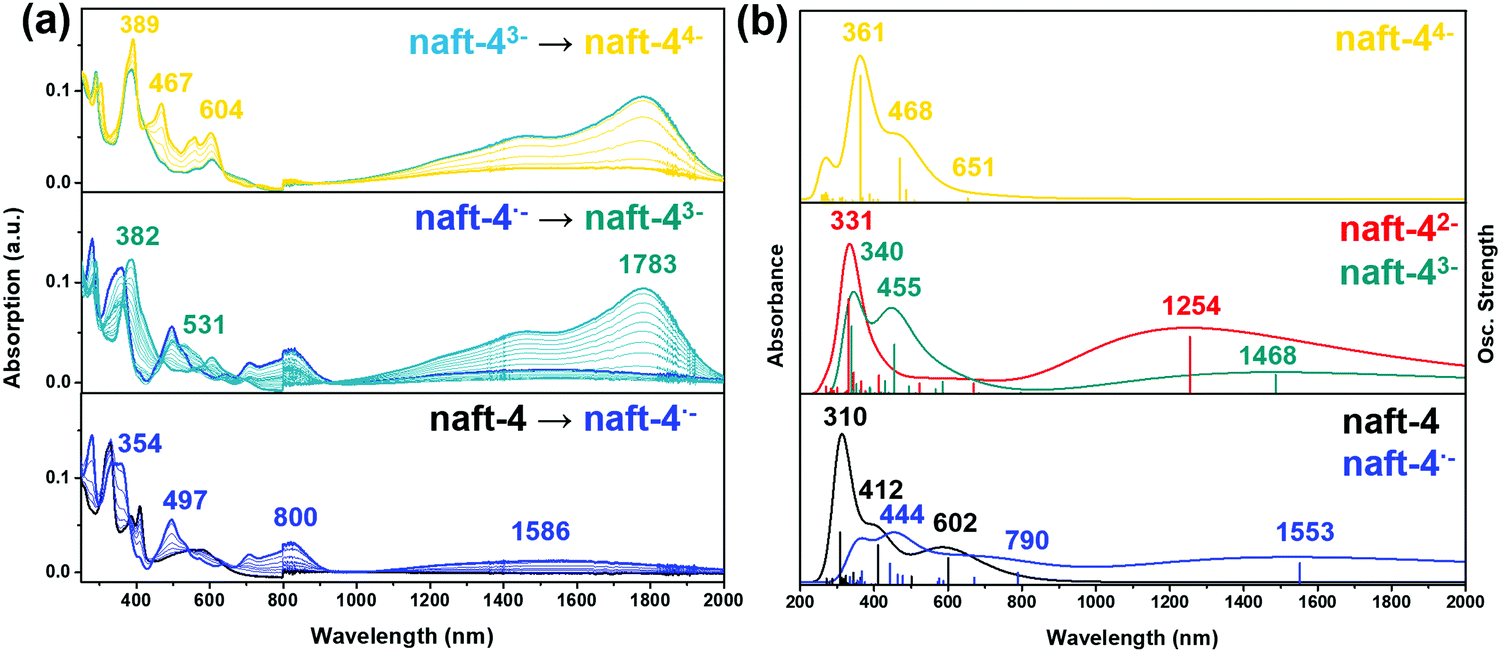 | ||
| Fig. 8 (a) UV-Vis-NIR absorption spectra recorded, in and OTTLE cell, during the first step of electrochemical reduction (−470 mV, blue curve), the second step of the electrochemical reduction (−990 mV, cyan curve) and the third step of electrochemical reduction (−1200 mV, yellow curve) of naft-4 in dichloromethane in presence of Bu4NBF4 as supporting electrolyte. (b) TDDFT/PBE0 (PCM:DCM)-calculated vertical transition energies for naft-4 neutral and reduced species. | ||
The progressive electrochemical oxidation of naft-1 (Fig. 7a) depresses the absorption of the neutral species while leads to the formation of a broad absorption band. According to the electrochemical results and TDDFT calculations, this new absorption can be ascribed to the formation of the radical cation species. Note that Fig. S45a (ESI‡) shows numerous electronic transitions, corresponding to the radical cation, that appear between ∼380–700 nm and give rise to the broad absorption appearing upon oxidation and three electronic transitions between 900 and 1200 nm, explaining the low and broad absorption centered at 1028 nm.
The application of negative potential around 800 mV (Fig. 7b, blue curve) leads to the formation of the radical anion, with absorption peaks at 348, 471, 584, 680 and 751 nm, that are in good agreement with the predicted electronic transitions of the one-electron reduced species (Fig. S45, ESI‡). Further reduction at ∼1000 mV (red curve) depressed these bands as peaks rise at 386, 443 and 617 nm, which nicely corresponds to the formation of the dianion.
Thus, as seen in electrochemical experiments, naft-1 is able to stabilize both positive and negative charges, in the latter up to a two-electron reduced species. Note that the potential difference necessary to generate the dianion from the radical anion species is of approximately 250 mV, which fits perfectly with the potential difference between the second and first reduction processes in cyclic voltammetry experiments (Fig. 6).
Electrochemical reductions of naft-2–4 (Fig. 8a and Fig. S42, ESI‡) follow the same reduction absorption pattern and thus, the data recorded for naft-4 is shown in Fig. 8a as an example (naft-2 and naft-3 data is shown in Fig. S42, ESI‡).
The gradual application of −470 mV, −990 mV and −1160 mV to a naft-4 solution leads to the blue, cyan and yellow spectral profiles shown in Fig. 8a, respectively. TD-DFT calculations on the charged species are shown in Fig. 8b and Fig. S46 (ESI‡) and indicate that, for naft-2, naft-3 and naft-4, the first isolated reduced species is indeed a radical anion (blue curve); however, a gradual increment of the potential leads directly to a triple negatively charged species (cyan curve), followed by the stabilization of the tetranion (yellow curve). Note also that the differences between the applied reduction potentials in this experiment follow a similar trend to those of the electrochemical potentials in the cyclic voltammetry experiment, necessary to generate the radical anion, trianion and tetranion species, thus further supporting the formation of these species also by spectroelectrochemistry. The simultaneous two-electron injection in the second reduction process can be understood considering the LUMO and LUMO+1 orbital topologies (see Fig. S38, ESI‡); while the two electrons inserted in the LUMO must be accommodated in half the molecular length, a third electron is easily delocalized in the other half of the molecule, thus mitigating charge repulsion. On the other hand, while by cyclic voltammetry the potentials needed to inject charges in naft-2 and naft-4 are comparable, in spectroelectrochemical experiments the potentials needed for naft-2 are higher than those for naft-4 (see Fig. S42a, ESI‡).
UV-Vis-NIR absorption spectra recorded during57,58 progressive electrochemical reductions of naft-1–4 in chloroform in the presence of high excess of tetrabutylammonium tetrafluoroborate (Bu4NBF4), as supporting electrolyte, were also performed (Fig. S43 and S44, ESI‡). In this case, a similar reduction absorption pattern was obtained; however, the reduced species seem to be less stabilized in chloroform than in dichloromethane, as theoretically predicted (see Table S3, ESI‡), confirming the results obtained by CV.
Field-effect transistor characterization
To evaluate the charge-transport characteristics59,60 of the new semiconductors, top-contact/bottom-gate thin film transistors were fabricated by vapour deposition of the semiconducting films onto octadecyltrichlorosilane (OTS)-functionalized Si/SiO2 substrates, followed by gold deposition through a shadow mask to define the source and drain electrodes. The performance parameters, which are summarized in Table 5 are extracted from I–V response plots in the saturation regime by using the assumptions of conventional transistor formalism [eqn (1)]. These parameters include field-effect mobility μ, threshold voltage VTh, and current on/off ratio ION/IOFF.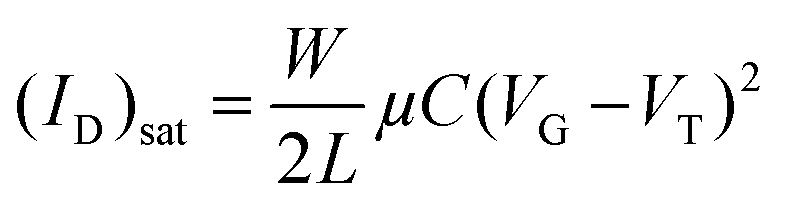 | (1) |
| Semiconductor | Deposition conditions | μ e [cm2 V−1 s−1] | V TH (V) | I ON/IOFF | μ h [cm2 V−1 s−1] | V TH (V) | I ON/IOFF |
|---|---|---|---|---|---|---|---|
| naft-1 | OTS, 90 °C | 7 × 10−4 | 14 | 9 × 102 | 6 × 10−5 | −46 | 7 × 104 |
| naft-2 | OTS, 90 °C | 2 × 10−4 | 44 | 2 × 101 | |||
| naft-4 | OTS, 90 °C | 6 × 10−3 | 18 | 2 × 104 | |||
| naft-4 | OTS, 25 °C | 5 × 10−3 | 22 | 3 × 104 |
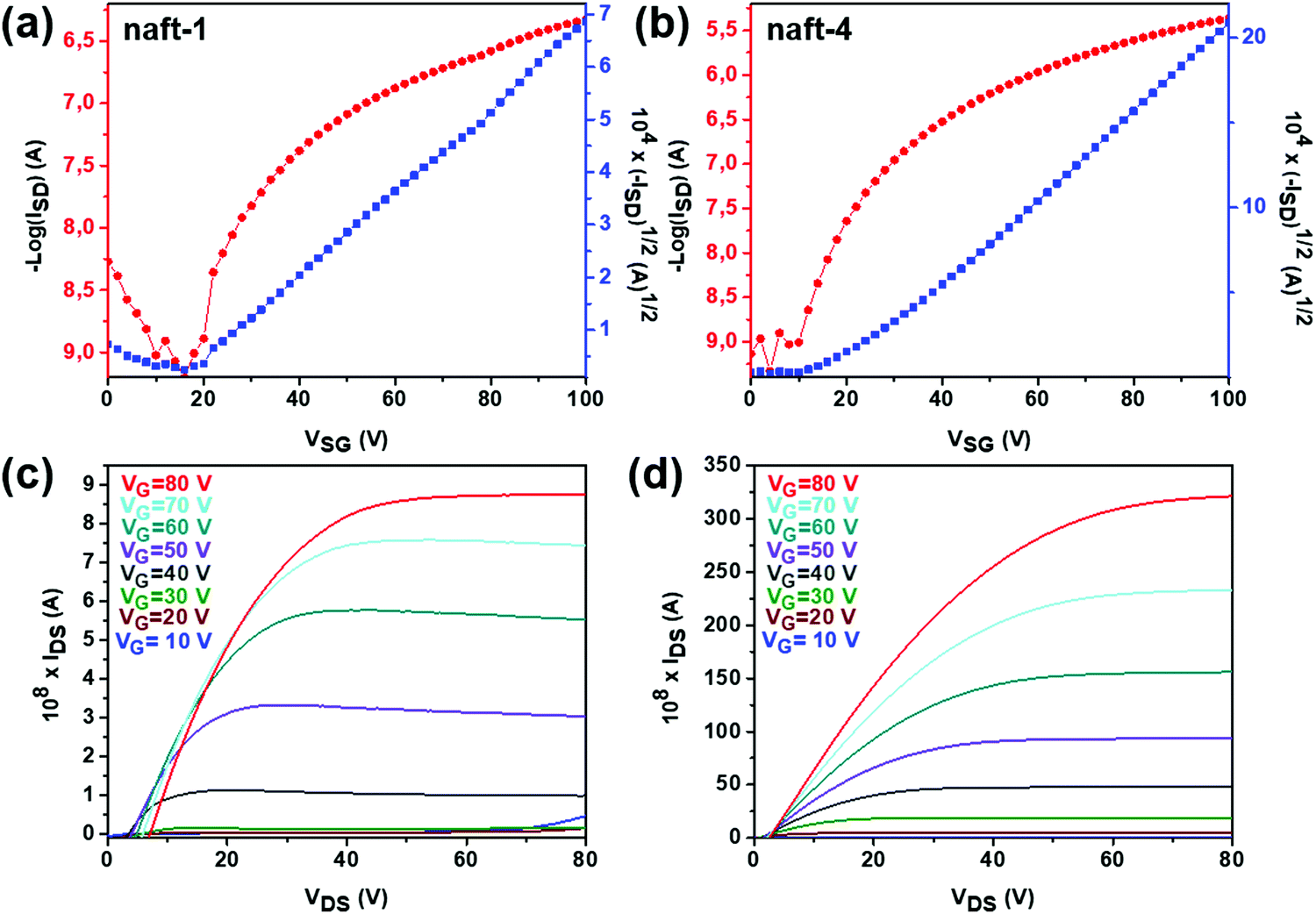 | ||
| Fig. 9 OFET transfer and output characteristics of (a and c) naft-1, (b and d) naft-4 derivatives. | ||
In this series of semiconducting naphthalimide derivatives (naft-1–4), ambipolar mobilities were recorded only for naft-1 in good agreement with its amphoteric redox behavior, while only electron mobilities were recorded for naft-2–4. Representative transistor curves of OFETs are illustrated in Fig. 9, and their performance parameters are summarized in Table 5.
naft-1 thin films (Fig. 9a–c) annealed at 90 °C exhibit electron and hole mobilities of ∼7 × 10−4 cm2 V−1 s−1 and ∼6 × 10−5 cm2 V−1 s−1, respectively. The electron field-effect mobility is though one order of magnitude lower than those obtained for naft-4, the best performing material within the series, which can be understood considering its lower ability to stabilize an extra electron and the skeletal distortion of ∼16° of naft-1 conjugated skeleton (Fig. S35, ESI‡), compared to naft-4.
On other hand, the presence of different substituents at the imide nitrogen in naft-2–4 seems to be crucial to tune the electron transport, since molecular aggregation and crystal packing is greatly affected by the lateral substituents. Thus, devices fabricated by using naft-3, with bulky diisopropylphenyl lateral groups were not active. In contrast, naft-4 exhibits the highest electron mobility (Fig. 9b–d), with a maximum value of 6 × 10−3 cm2 V−1 s−1 for films annealed at 90 °C. Similar electron mobility was obtained when the film of naft-4 was grown at room temperature.
The OFET performance of naft-2, with shorter alkyl chains at the imide nitrogen, decreases by approximately 10-fold for films annealed at 90 °C in comparison to naft-4 films, and no activity was observed for naft-2 OFETs when films were grown at room temperature. Note that the alkyl substituents at the imide nitrogen atoms are longer in naft-4, and thus intermolecular hydrophobic interactions between the long alkyl chains, also known as fastener effect, may be the reason behind the higher electrical performance, due to enhanced intermolecular overlap of the semiconductor layer.61–64 To analyze this effect, XRD and AFM experiments were carried out for the prepared films and selected data is shown in Fig. 10 and 11, respectively.
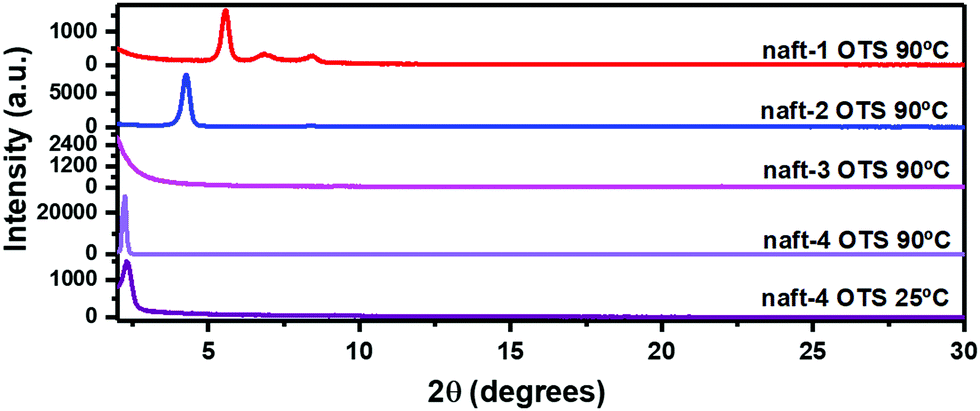 | ||
| Fig. 10 θ–2θ X-ray diffraction scans of vapor-deposited naft-1–4 thin films (Td = 25 °C and 90 °C) grown on OTS-treated Si/SiO2 substrates. | ||
 | ||
| Fig. 11 AFM images of the (a) naft-1, (b) naft-2, (c) naft-3, and (d) naft-4 vapor-deposited under conditions yielding optimum OFET performance. Image size: 5 × 5 mm. | ||
XRD data shows a completely amorphous film for naft-3, due to the incorporation of bulky substituents at the N of the imide group. These diisopropylphenyl groups are orthogonal to the π-conjugated core, thus preventing proper molecular organization at the semiconducting layer, which is translated to poorly performing devices. On the contrary, naft-2 and naft-4 diffraction profiles show one sharp peak, indicating certain molecular ordering at the interface. Note also that the longer substituents in naft-4 render more ordered films, in agreement with stronger intermolecular hydrophobic interactions promoted by the alkyl substituents.
naft-1 XRD pattern also shows molecular ordering, with the recording of several diffraction peaks, which is in good agreement with the formation of needle-like crystallites in AFM images (Fig. 11). AFM images also nicely follow the trend found in XRD and electrical performance for the semiconductors with varying substituents at the N atom of the imide group. In fact, quite amorphous morphologies are found for naft-3 films, having bulky and non-planar substituents. On the other hand, round features are found for naft-2 and naft-4, the latter showing bigger crystalline domains due to more efficient hydrophobic intermolecular interactions between longer alkyl chains.
3. Summary and conclusions
A versatile synthetic procedure has been developed for the efficient syntheses of heteroatom-doped polycyclic aromatic mono- and dicarboximides (PADI). Thus, a family of π-extended nine-ring-fused systems have been synthesized by metal-free auto-condensation and cross-condensation of pyrene and 1,8-naphthalimide moieties endowed with 1,2-diketone functionalities. This strategy is aimed to provide novel semiconductors with different redox ability, tunable supramolecular aggregation and film-forming ability suitable for their use as active materials in OTFTs.The optical, electronic and molecular properties of the novel systems have been thoroughly analyzed using optical spectroscopy, electrochemical measurements, spectroelectrochemical studies and DFT calculations.
The all-acceptor systems show up to four reversible reduction waves in cyclic voltammetry measurements and have lower estimated LUMO energies than that described for naphthalene diimide (NDI) and perylene diimide (PDI) derivatives with similar alkyl chains at the imide nitrogen. The derivative containing naphthalimide and pyrene units shows amphoteric redox behavior with two reversible reduction waves and one oxidation wave.
Spectroelectrochemistry has been used with the aim to analyze the stability of the charge carriers involved in the electron transport process. The gradual formation of the radical anion, trianion and tetranion species for the all-acceptor derivatives can be observed and TD-DFT calculations nicely reproduces the spectral profiles of the charged species.
Internal reorganization energies of the novel materials have been computed and the calculated theoretical parameters have been related to their charge transport. Similar reorganization energies for both hole and electron transport are found for naft-1, constituted by pyrene and 1,8-naphthalimide moieties. On the other hand, for those materials based only on naphthalimide moieties (naft-2–4), the internal reorganization energies for electron transport are notably lower than those predicted for hole transport due to the easiness to accommodate negative charges over two naphthalimide units.
Vapour-deposited films of the novel semiconductors have been obtained and characterized by X-ray diffraction (XRD) and atomic force microscopy (AFM) analysis. Concerning with the three all-acceptors systems with identical π-conjugated skeleton and different solubilizing chains at the imide nitrogens, different film characteristics have been observed. Naft-3, endowed with bulky and rigid diisopropylphenyl groups, forms amorphous films while the analogues, naft-2 and naft-4, with flexible branched alkyl chains form films with certain molecular ordering at the interface, as indicated by the presence of sharp diffraction peaks in the XRD analysis. The semiconductor with longer alkyl chains (naft-4) render more ordered films, in agreement with stronger intermolecular hydrophobic interactions promoted by the alkyl substituents. AFM images also nicely follow the trend found in XRD for the systems with varying substituents at the N atom of the imide group.
The above summarized studies have allowed the investigation of the interplay of different effects such as (i) the all-acceptor vs. donor–acceptor nature of the novel systems, (ii) the type of solubilizing chain, and (iii) the molecular ordering on the OTFT device response characteristics. Thus, ambipolar mobilities can be only observed for naft-1 in good agreement with its amphoteric redox behavior. On the other hand, due to more efficient supramolecular aggregation and enhanced film-forming ability, naft-4 exhibits the highest electron mobility of the three all-acceptor systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MICINN (PID2019-106268GB-C33) and the UCM (INV.GR.00.1819.10759). MJAN gratefully acknowledges Universidad Rey Juan Carlos for a predoctoral contract. The university of Málaga acknowledges support from Junta de Andalucía (projects UMA18-FEDERJA-080 and P18-FR-4559) and MICINN (PID2019-110305GB-I00). RPO and AH also thank the Research Central Services (SCAI) of the University of Málaga and especially the Vibrational Spectroscopy Laboratory.References
- J. Chen, K. Yang, X. Zhou and X. Guo, Chem. – Asian J., 2018, 13, 2587–2600 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- H. Sun, L. Wang, Y. Wang and X. Guo, Chem. – Eur. J., 2019, 25, 87–105 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7595–7600 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Org. Chem. Front., 2016, 3, 1435–1442 RSC.
- K. Shoyama, M. Mahl, S. Seifert and F. Würthner, J. Org. Chem., 2018, 83, 5339–5346 CrossRef CAS PubMed.
- H. Zhylitskaya, J. Cybińska, P. Chmielewski, T. Lis and M. Stępień, J. Am. Chem. Soc., 2016, 138, 11390–11398 CrossRef CAS PubMed.
- N. Liang, D. Meng and Z. Wang, Acc. Chem. Res., 2021, 54, 961–975 CrossRef CAS PubMed.
- L. Đorđević, D. Milano, N. Demitri and D. Bonifazi, Org. Lett., 2020, 22, 4283–4288 CrossRef PubMed.
- Q. Shi, X. Shi, C. Feng, Y. Wu, N. Zheng, J. Liu, X. Wu, H. Chen, A. Peng, J. Li, L. Jiang, H. Fu, Z. Xie, S. R. Marder, S. B. Blakey and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 2924–2928 CrossRef CAS PubMed.
- J. S. Valera, R. Gómez and L. Sánchez, Org. Lett., 2018, 20, 2020–2023 CrossRef CAS PubMed.
- J. Vollbrecht, H. Bock, C. Wiebeler, S. Schumacher and H. Kitzerow, Chem. – Eur. J., 2014, 20, 12026–12031 CrossRef CAS PubMed.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed.
- N. Kumari, S. Naqvi, M. Ahuja, K. Bhardwaj and R. Kumar, J. Mater. Sci.: Mater. Electron., 2020, 31, 4310–4322 CrossRef CAS.
- X. Cui, G. Zhang, L. Zhang and Z. Wang, Dyes Pigm., 2019, 168, 295–299 CrossRef CAS.
- S. Liu, C. Li, Y. Xu, Z. Li, H. Huang, N. Fu, J. Shao, B. Zhao, H. Huang and W. Huang, Dyes Pigm., 2020, 173, 107976 CrossRef CAS.
- W. Liu, D. B. Shaikh, P. S. Rao, R. S. Bhosale, A. A. Said, A. M. Mak, Z. Wang, M. Zhao, W. Gao, B. Chen, Y. M. Lam, W. Fan, S. V. Bhosale, S. V. Bhosale and Q. Zhang, Chem. – Asian J., 2020, 15, 112–121 CrossRef CAS PubMed.
- X. Zhan, J. Zhang, S. Tang, Y. Lin, M. Zhao, J. Yang, H.-L. Zhang, Q. Peng, G. Yu and Z. Li, Chem. Commun., 2015, 51, 7156–7159 RSC.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 702 CrossRef.
- T. Murata, T. Koide, H. Nobukuni, R. Tsuji and Y. Morita, Int. J. Mol. Sci., 2020, 21, 4723 CrossRef CAS PubMed.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS PubMed.
- P. Ledwon, D. Ovsiannikova, T. Jarosz, S. Gogoc, P. Nitschke and W. Domagala, Electrochim. Acta, 2019, 307, 525–535 CrossRef CAS.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, J. M. Rotter, D. J. Fortman, D. dos, S. Abreu, R. A. Timm, I. C. N. Diógenes, L. T. Kubota, H. D. Abruña and W. R. Dichtel, Angew. Chem., Int. Ed., 2015, 54, 13225–13229 CrossRef CAS PubMed.
- A. Iordache, V. Delhorbe, M. Bardet, L. Dubois, T. Gutel and L. Picard, ACS Appl. Mater. Interfaces, 2016, 8, 22762–22767 CrossRef CAS PubMed.
- J. Merz, A. Steffen, J. Nitsch, J. Fink, C. B. Schürger, A. Friedrich, I. Krummenacher, H. Braunschweig, M. Moos, D. Mims, C. Lambert and T. B. Marder, Chem. Sci., 2019, 10, 7516–7534 RSC.
- B. R. Reiner, B. M. Foxman and C. R. Wade, Dalton Trans., 2017, 46, 9472–9480 RSC.
- Y. Shi, H. Tang, S. Jiang, L. V. Kayser, M. Li, F. Liu, F. Ji, D. J. Lipomi, S. P. Ong and Z. Chen, Chem. Mater., 2018, 30, 3508–3517 CrossRef CAS.
- L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- G. J. Richards, A. Cador, S. Yamada, A. Middleton, W. A. Webre, J. Labuta, P. A. Karr, K. Ariga, F. D'Souza, S. Kahlal, J. F. Halet and J. P. Hill, J. Am. Chem. Soc., 2019, 141, 19570–19574 CrossRef CAS PubMed.
- J. Z. Hu, D. Zhang and F. W. Harris, J. Org. Chem., 2005, 70, 707–708 CrossRef CAS PubMed.
- P. de Echegaray, M. J. Mancheno, I. Arrechea-Marcos, R. Juarez, G. Lopez-Espejo, J. T. Lopez Navarrete, M. M. Ramos, C. Seoane, R. P. Ortiz and J. L. Segura, J. Org. Chem., 2016, 81, 11256–11267 CrossRef CAS.
- H. Herrera, P. de Echegaray, M. Urdanpilleta, M. J. Mancheno, E. Mena-Osteritz, P. Bauerle and J. L. Segura, Chem. Commun., 2013, 49, 713–715 RSC.
- H. Li, F. S. Kim, G. Ren, E. C. Hollenbeck, S. Subramaniyan and S. A. Jenekhe, Angew. Chem., Int. Ed., 2013, 52, 5513–5517 CrossRef CAS PubMed.
- M. J. Alonso-Navarro, A. Harbuzaru, P. de Echegaray, I. Arrechea-Marcos, A. Harillo-Baños, A. de la Peña, M. M. Ramos, J. T. López Navarrete, M. Campoy-Quiles, R. Ponce Ortiz and J. L. Segura, J. Mater. Chem. C, 2020, 8, 15277–15289 RSC.
- F. Shahrokhi and Y. Zhao, Org. Lett., 2019, 21, 9306–9310 CrossRef CAS.
- Z. A. Tabasi, E. A. Younes, J. C. Walsh, D. W. Thompson, G. J. Bodwell and Y. Zhao, ACS Omega, 2018, 3, 16387–16397 CrossRef CAS.
- F. Shahrokhi and Y. Zhao, Org. Lett., 2019, 21, 9306–9310 CrossRef CAS.
- P. Rajdev, M. R. Molla and S. Ghosh, Langmuir, 2014, 30, 1969–1976 CrossRef CAS PubMed.
- F. Salerno, J. A. Berrocal, A. T. Haedler, F. Zinna, E. W. Meijer and L. Di Bari, J. Mater. Chem. C, 2017, 5, 3609–3615 RSC.
- N. Drolet, J. F. Morin, N. Leclerc, S. Wakim, Y. Tao and M. Leclerc, Adv. Funct. Mater., 2005, 15, 1671–1682 CrossRef CAS.
- Q. Liu, S. Chavhan, H. Zhang, H. Sun, A. J. Brock, S. Manzhos, Y. Chen, K. Feron, S. E. Bottle, J. C. McMurtrie, J.-H. Jou, H.-S. Chen, M. R. Nagar, W. Hu, Y.-Y. Noh, Y. Zhen and P. Sonar, Adv. Electron. Mater., 2021, 7, 2000804 CrossRef CAS.
- Q. Liu, H. Sun, C. Blaikie, C. Caporale, S. Manzhos, K. Feron, J. M. MacLeod, M. Massi, S. E. Bottle, J. Bell, Y.-Y. Noh and P. Sonar, New J. Chem., 2018, 42, 12374–12385 RSC.
- T. Tang, T. Lin, F. Wang and C. He, Polym. Chem., 2014, 5, 2980–2989 RSC.
- A. O. Weldeab, L. Li, S. Cekli, K. A. Abboud, K. S. Schanze and R. K. Castellano, Org. Chem. Front., 2018, 5, 3170–3177 RSC.
- F. G. Guijarro, S. Medina Riverso, S. Gunasekaran, I. Arrechea, R. Ponce Ortiz, R. Caballero, P. de la Cruz, F. Langa, L. Venkataraman and J. Casado, RSC Adv., 2020, 10, 41264–41272 RSC.
- Y. W. More, S. D. Padghan, R. S. Bhosale, R. P. Pawar, A. L. Puyad, S. V. Bhosale and S. V. Bhosale, Sensors, 2018, 18, 3433 CrossRef PubMed.
- K.-T. Xiong, M.-L. Li, Y. Jiang, H.-B. Xu and M.-H. Zeng, New J. Chem., 2020, 44, 8061–8067 RSC.
- J. Quinn, C. Guo, B. Sun, A. Chan, Y. He, E. Jin and Y. Li, J. Mater. Chem. C, 2015, 3, 11937–11944 RSC.
- T. M. P. Gawrys, E. Bartnik, M. Kucinska, J. Ulanski and M. Zagorska, Org. Lett., 2011, 13, 6090–6093 CrossRef.
- M. Zhu, J. Zhang, G. Yu, H. Chen, J. Huang and Y. Liu, Chem. – Asian J., 2012, 7, 2208–2212 CrossRef CAS PubMed.
- C. C. Ferrón, M. Capdevila-Cortada, R. Balster, F. Hartl, W. Niu, M. He, J. J. Novoa, J. T. López Navarrete, V. Hernández and M. C. Ruiz Delgado, Chem. – Eur. J., 2014, 20, 10351–10359 CrossRef PubMed.
- P. Mayorga Burrezo, B. Pelado, R. Ponce Ortiz, P. De la Cruz, J. T. López Navarrete, F. Langa and J. Casado, Chem. – Eur. J., 2015, 21, 1713–1725 CrossRef CAS PubMed.
- R. P. Ortiz, H. Herrera, C. Seoane, J. L. Segura, A. Facchetti and T. J. Marks, Chem. – Eur. J., 2012, 18, 532–543 CrossRef CAS PubMed.
- A. Riaño Carnerero, G. López Espejo, M. J. Mancheño Real, B. Eckstein, R. C. González-Cano, F. S. Melkonyan, A. Facchetti, T. J. Marks, J. Casado, J. T. López Navarrete, J. L. Segura and R. Ponce Ortiz, J. Mater. Chem. C, 2017, 5, 9439–9450 RSC.
- H. Inokuchi, G. Saito, P. Wu, K. Seki, T. B. Tang, T. Mori, K. Imaeda, T. Enoki, Y. Higuchi, K. Inaka and N. Yasuoka, Chem. Lett., 1986, 1263–1266 CrossRef CAS.
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS.
- A. Welford, S. Maniam, E. Gann, X. Jiao, L. Thomsen, S. J. Langford and C. R. McNeill, Org. Electron., 2019, 75, 105378 CrossRef CAS.
- Y. Yamaguchi, Y. Kojiguchi, S. Kawata, T. Mori, K. Okamoto, M. Tsutsui, T. Koganezawa, H. Katagiri and T. Yasuda, Chem. Mater., 2020, 32, 5350–5360 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Dr. Peter Bäuerle on the occasion of his 65th anniversary |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01239h |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |