
SHG-active NIR-emissive molecular nanomagnets generated in layered neodymium(III)–octacyanidometallate(IV) frameworks†
Robert
Jankowski
 a,
Jakub J.
Zakrzewski
a,
Mikolaj
Zychowicz
a,
Junhao
Wang
b,
Yurie
Oki
b,
Shin-ichi
Ohkoshi
*b,
Szymon
Chorazy
*a and
Barbara
Sieklucka
*a
a,
Jakub J.
Zakrzewski
a,
Mikolaj
Zychowicz
a,
Junhao
Wang
b,
Yurie
Oki
b,
Shin-ichi
Ohkoshi
*b,
Szymon
Chorazy
*a and
Barbara
Sieklucka
*a
aFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: chorazy@chemia.uj.edu.pl; barbara.sieklucka@uj.edu.pl
bDepartment of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ohkoshi@chem.s.u-tokyo.ac.jp
First published on 16th April 2021
Abstract
Lanthanide(III) single-molecule magnets (Ln-SMMs) offer the fruitful conjunction of magnetic and photoluminescent properties originating from their single-ion anisotropy and emissive f–f electronic transitions. The flexibility of lanthanide(III)-based coordination systems is the most significant prerequisite for the generation of additional physical phenomena, including non-linear optical effects related to the lack of inversion centre. In the quest towards multifunctional molecular nanomagnets, we report heterometallic d–f layered coordination polymers, {[NdIII4(H2O)17(pzdo)5][MIV(CN)8]3}·9H2O (M = Mo (1), W (2); pzdo = pyrazine-N,N′-dioxide). They crystallize in the non-centrosymmetric C2 space group due to the spontaneous resolution process, confirmed by the observation of second harmonic generation (SHG) activity. These materials exhibit UV-to-vis-light-induced near-infrared (NIR) NdIII-centred luminescence sensitized by octacyanidometallates and organic ligand, and slow relaxation of magnetization of NdIII complexes which was investigated by alternate-current (ac) magnetic measurements and the ab initio calculations. The observed magnetic and optical properties are sensitive to the nature of the d-transition metal centre, namely most of their features are improved upon the replacement of MoIV with WIV centres. In this work, we show the generation of SHG-active NIR-emissive SMMs realized by Nd3+ ions inserted into hybrid layers built of inorganic [M(CN)8]4− and organic pzdo linkers.
Introduction
Single-molecule magnets (SMMs) constitute a class of para-magnetic metal complexes whose magnetic anisotropy results in a slow magnetic relaxation effect.1 At the low-temperature regime, SMMs retain their magnetization for a long time in the absence of an external magnetic field. This gives a magnetic bistability effect opening applications for high-density data storage and processing devices.2 Moreover, quantum phenomena such as quantum tunnelling of magnetization or quantum coherence are observed in molecular nanomagnets extending the interest in SMMs toward molecular spintronics and quantum computation.3 The first generation of SMMs was based on 3d metal clusters with a high-spin of the ground state.1 It was later established that single-ion anisotropy plays a key role in achieving high-performance SMMs,4 thus the attention has been shifted to SMMs based on lanthanide(3+) ions showing strong magnetic anisotropy related to the contributions from spin–orbit coupling and crystal field effects.5 The construction of molecular nanomagnets built of intrinsically emissive lanthanide(III) centres implied the emergence of luminescent SMMs which expand the application scope toward light-emitting devices, optical sensors or thermometers.6 For the last decade, emissive SMMs, exploring mainly DyIII and YbIII, have attracted considerable attention which was also connected with the magneto-optical correlations exploring the usage of emission spectra in the investigation of lanthanide(III) electronic structure.7 Moreover, emissive SMMs are candidates for magneto-optical systems that can realize the tuning of emission by a magnetic field.8 It was recently shown that the rich coordination chemistry of lanthanide ions enables further functionalization of luminescent SMMs toward high proton conductivity or ferroelectricity.9In these regards, the objective of our work was to employ luminescent SMMs for the construction of unique multifunctional materials. We focused on the generation of chirality in the emissive SMM-based systems. This goal is motivated by the broad range of effects that can be induced thanks to the chirality or at least the non-centrosymmetric character of the crystals. It is worth mentioning second harmonic generation (SHG),10 circularly polarized luminescence (CPL),11 both with the magnetic enhancement (MSHG/MCPL), or even more advanced magneto-chiral dichroism (MChD).12
To achieve photoluminescent SMMs crystalizing in the non-centrosymmetric space group, we decided to explore hetero-metallic d–f cyanido-bridged assemblies. It was shown that polycyanidometallates can enhance lanthanide luminescence and constitute a useful tool to adjust the geometry of 4f metal ions towards the SMM behaviour.13 Furthermore, the application of cyanido metal complexes with functionalized organic ligands is an effective strategy for chiral and non-centrosymmetric materials revealing SHG, MSHG, or MChD phenomena.14 Following this perspective, we selected Nd3+ ions combined with [M(CN)8]4− (M = MoIV, WIV) metalloligands accompanied by pyrazine-N,N′-dioxide (pzdo) as a supporting organic linker. The NdIII centres are rarely used for SMMs due to moderate single-ion anisotropy and relatively low magnetic moment. However, similarly to commonly used Dy3+ ions, they are characterized by oblate electron density, thus the negatively charged ligands giving axial crystal field are the key to high-performance molecular nanomagnets, supported by symmetry and/or magnetic isolation.5b,15 Moreover, they offer a straightforward route to emissive materials as they exhibit efficient near-infrared (NIR) luminescence.16 On the other hand, diamagnetic [M(CN)8]4− (M = MoIV, WIV) ions can not only ensure magnetic isolation of 4f metal ions enabling observation of SMM behaviour but also, when combined with pzdo ligands, they can form anion–π aggregates which are suitable for the sensitization of lanthanide NIR emission.17 Although the generation of chirality by a spontaneous resolution process is extremely hard to predict, it is usually related to the formation of the specific supramolecular interactions during the crystals growth.18 Therefore, rich contacts involving cyanido ligands can play a non-innocent role in the spontaneous resolution effect producing chiral molecular systems.14a,19 Actually, the {pzdo-[M(CN)8]n−} aggregates can be promising in this context.17,20 Thanks to all these rationally selected molecular building blocks, we report two novel layered coordination polymers, {[Nd4(H2O)17(pzdo)5][MIV(CN)8]3}·9H2O (M = Mo (1), W (2)) crystallizing in a non-centrosymmetric C2 space group. They are multifunctional, exhibiting SHG activity, sensitized NIR emission, and slow magnetic relaxation effects. All these properties are tuned by the [MIV(CN)8]4− molecular building block as shown by thorough structural, luminescence, non-linear optical, and magnetic studies, supported by the ab initio calculations.
Results and discussion
Structural studies
The yellow plate crystals of {[Nd4(H2O)17(pzdo)5][MoIV(CN)8]3}·9H2O (1) were obtained from the H2O/MeOH solution by the self-assembly of Nd3+ cations, [MoIV(CN)8]4− anions, and pyrazine-N,N′-dioxide (pzdo). The WIV analogue {[Nd4(H2O)17(pzdo)5][WIV(CN)8]3}·9H2O (2) was prepared in an analogous manner using [W(CN)8]4− anions, which resulted in the orange polycrystalline sample. Obtained compounds were preliminarily characterized by the CHN elemental analysis, IR spectra, and TGA (Fig. S1 and S2, ESI,† and Experimental section), and, afterwards by the X-ray diffraction methods (Fig. 1, 2, Fig. S3–S5, and Tables S1–S4, ESI†). The single-crystal XRD experiment indicates that 1 crystallizes in the non-centrosymmetric C2 space group as the mixture of enantiomorphic crystals, hereinafter referred to as 1a and 1b (Fig. 1 and Table S1, ESI†). The crystals of 2 were too small to perform the SC-XRD experiment but the powder XRD (P-XRD) technique confirmed its isostructurality with 1 (Fig. 2). Compounds 1 and 2 are constructed of layered cyanido-bridged frameworks accompanied with interlayer water of crystallization (Fig. 1), resulting in a composition of {[Nd4(H2O)17(pzdo)5][MIV(CN)8]3}·9H2O, M = Mo (1), W (2), crystallographically represented by two types of Mo centres (Mo1 and Mo2) and three types of Nd centres (Nd1, Nd2, Nd2′). The single coordination layer of 1a is composed of cyanido-bridged ladder-like assemblies formed by the conjugation of {Mo12Nd12} square units. They are stabilized by the co-existing {Nd1–pzdo–Nd1} linkages which form metal–organic chains positioned above and below the plane of the ladders. The coordination ladders are further linked along a two-fold screw axis through {Nd2–Mo2–Nd2′} triangular fragments which are supported by the local {Nd2–pzdo–Nd2′} linkages. These trinuclear {Nd2Mo1(pzdo)} spacers between ladder-type assemblies are intrinsically non-centrosymmetric which is related to the specific alignment of [Mo2(CN)8]4− ions above the pzdo. This contributes to the chirality of the whole coordination network as the alignment of {Nd2–Mo2–Nd2′} triangles, which are decorated by terminal pzdo ligands, in 1a is a mirror image of the analogous arrangement observed in 1b (Fig. 1). The formation of such a chiral structure was also enabled by the specific 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 stoichiometry of Nd:Mo as suggested by our recent report on the related ErYb(pzdo)–MoW coordination systems bearing organic counterions leading to the simple 1:1 metal ratio and the centrosymmetric structure.17 The structural comparison between 1 and 2, and other reported LnIII–[MIV(CN)8] systems crystallizing in the centrosymmetric space groups, are gathered in Table S5 (ESI†).
:3 stoichiometry of Nd:Mo as suggested by our recent report on the related ErYb(pzdo)–MoW coordination systems bearing organic counterions leading to the simple 1:1 metal ratio and the centrosymmetric structure.17 The structural comparison between 1 and 2, and other reported LnIII–[MIV(CN)8] systems crystallizing in the centrosymmetric space groups, are gathered in Table S5 (ESI†).
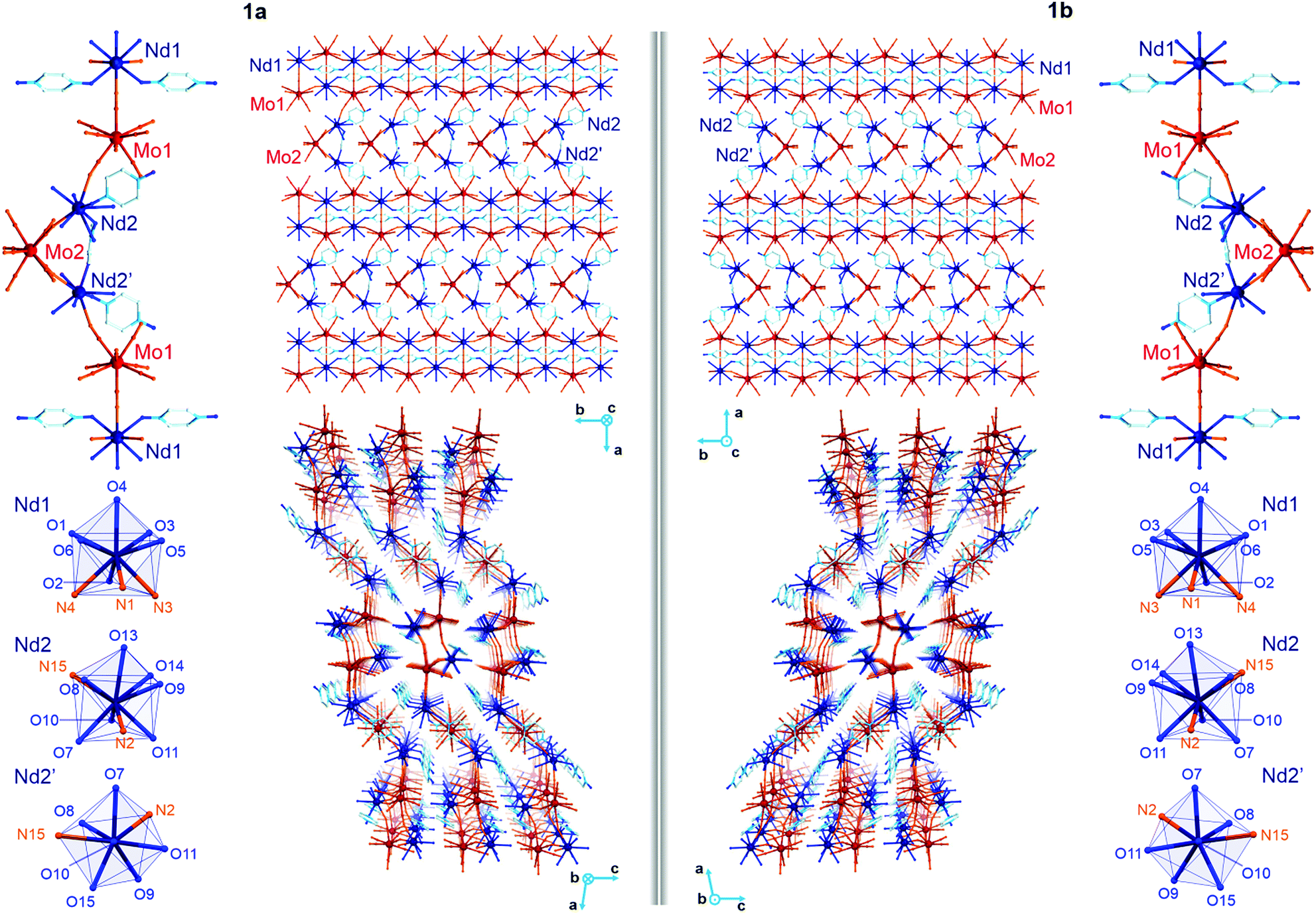 | ||
| Fig. 1 Representative views of the crystal structures of 1a (left side) and 1b (right side) including the crystal packing along the c (middle top) and b (middle bottom) crystallographic axes, molecular building units (top corners), and coordination spheres of three non-equivalent Nd complexes (bottom corners). A solvent of crystallization was omitted for clarity. | ||
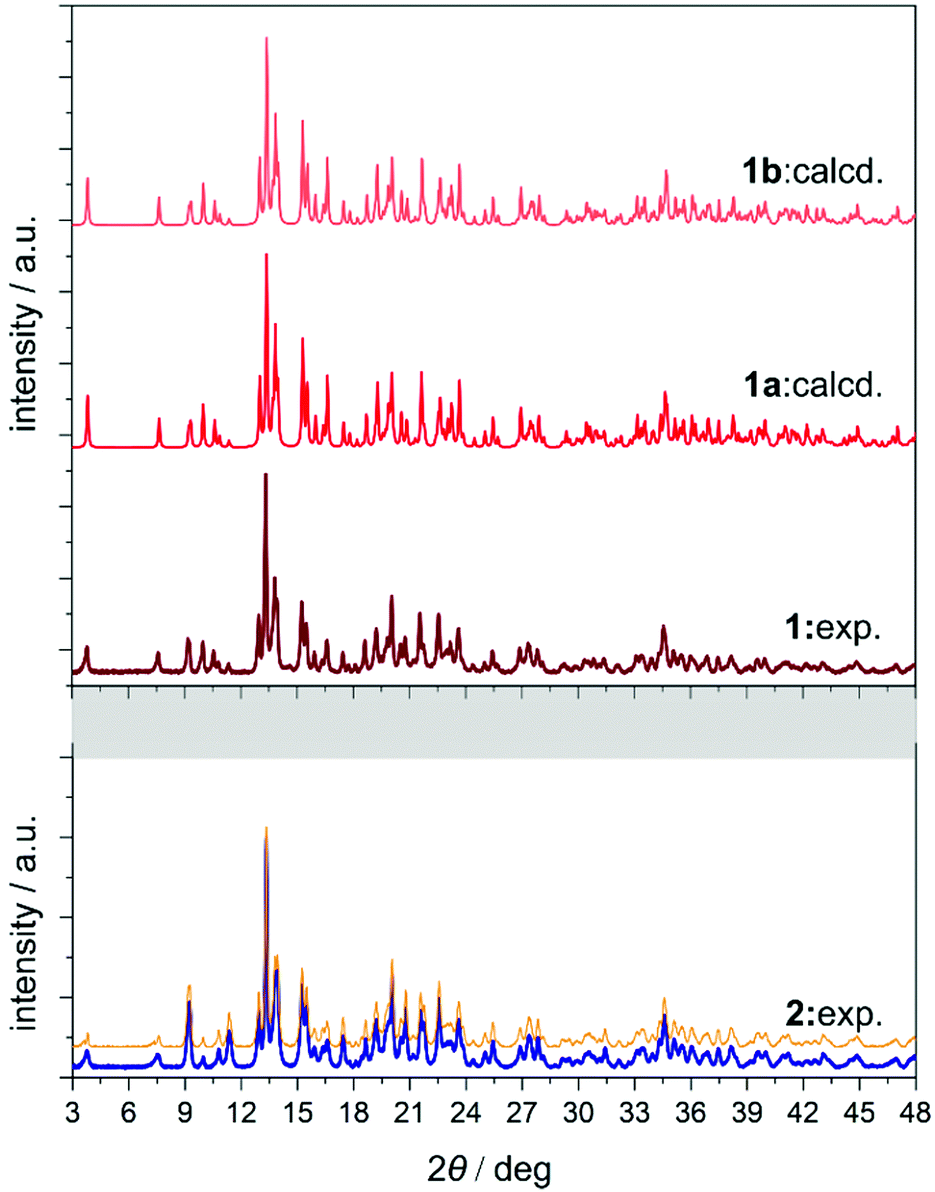 | ||
| Fig. 2 Comparison of the experimental powder X-ray diffraction (P-XRD) pattern measured for 1 with the patterns for 1a and 1b calculated based on structural models obtained from the SC-XRD method (top), together with the experimental P-XRD pattern gathered for 2 which was compared with the pattern (orange line) obtained from the LeBail fitting procedure performed using EXPO 2014 software (bottom). | ||
The asymmetric units of 1a and 1b contain four cyanido-bridged metal centres, two Mo (Mo1/Mo2) centres and two Nd (Nd1/Nd2) centres, accompanied by water molecules of crystallization (Fig. S3, ESI†). However, a static structural disorder concerning the pzdo ligand attached to the Nd2 centre was found, which results in the presence of two distinguishable Nd2 and Nd2′ sites, both existing with the 0.5 occupancies (Fig. S3 and S4, ESI†). Taking this into account, the molecular building unit for 1a and 1b can be defined as the seven-metallic fragment consisting of three [Mo(CN)8]4− ions (two Mo1 and one Mo2 centres) alternately arranged with four NdIII complexes (two Nd1, one Nd2, and one Nd2′ centres). The coordination sphere of Nd1 is filled with three N-atoms of the {Mo–CN–Nd} molecular bridges, four aqua ligands, and two O-atoms of bridging pzdo ligands. The Nd2/Nd2′ coordination sphere involves, apart from two N-atoms of cyanido bridges and two pzdo ligands, five or four aqua ligands, depending on the pzdo ligand position. The nine-coordinated Nd2 complex reveals five aqua ligands, while four aqua ligands are attached to eight-coordinated Nd2′ sites. All MoIV complexes are eight-coordinated bearing three (Mo1) or two (Mo2) cyanido ligands participating in the formation of cyanido bridges. The remaining cyanido ligands are terminal. To precisely determine the coordination geometry of NdIII and MoIV complexes in 1a and 1b, a Continuous Shape Measure (CShM) analysis was performed (Table S2, ESI†).21 It indicates that all [Mo(CN)8]4− complexes reveal the geometry close to an ideal square antiprism. The nine-coordinated Nd1 complex exhibits the geometry close to a tricapped trigonal prism, the Nd2 centre adopts the intermediate geometry between a tricapped trigonal prism and a capped square antiprism, while the eight-coordinated Nd2′ centre can be assigned to a triangular dodecahedron. Besides the coordination connectivity, the crystal structures of 1a and 1b are stabilized by a series of non-covalent interactions. The noticeable role is played by the anion–π aggregates involving negatively charged [Mo(CN)8]4− ions, and electron-deficient aromatic rings of pzdo ligands. They contribute to the stabilization of a chiral structure as they are not only observed within coordination ladders but also induce the directional alignment of {Nd2–Mo2–Nd2′} triangles within the hybrid coordination layers (Fig. 1 and S6, ESI†). On the other hand, the neighbouring layers are separated by the rich hydrogen bonding network employing water molecules of crystallization, aqua ligands of NdIII complexes, and terminal cyanido ligands of MoIV centres. The shortest distance between neodymium ions of neighbouring layers is 6.7 Å which is also the closest Nd⋯Nd distance in the whole structure. The closest intralayer Nd⋯Nd contact is 7.4 Å. Taking into account all structural parameters, 1a and 1b are the perfect mirror images (Fig. 1, Tables S1–S3, ESI†), being a pair of enantiomorphs as confirmed by the low values of 0.025(8) and 0.024(8) of the respective Flack parameters. However, as stated above, 1 grows as the mixture of enantiomorphic crystals, 1a and 1b, which hampered the additional characterization of the generated structural chirality by circular dichroism spectroscopy. The isostructurality of 1a and 1b, and their phase purity were checked by the P-XRD patterns (Fig. 2). The P-XRD method also indicates the isostructurality of 2 with almost identical unit cell volume and parameters (Fig. 2 and Fig. S5, Table S4, ESI†).22
SHG activity
Owing to the non-centrosymmetric C2 space group (Table S1, ESI†), 1 and 2 can be considered as promising materials for the observation of non-linear optical (NLO) properties. To examine this perspective, the measurement of the second harmonic generation (SHG) was performed. The SHG phenomenon is related to the interaction of photons of the ω frequency with solid lacking inversion symmetry, which gives rise to the frequency doubling (2ω) effect. The scheme illustrating the homemade setup used for the SHG experiment was presented in Fig. S7 (ESI†) (for details, see the Experimental section). The polycrystalline samples of 1 and 2 were irradiated using a 1040 nm femtosecond pulse laser as incident fundamental light. Subsequently, the second harmonic green light of ca. 520 nm was investigated (Fig. S8, ESI†). The intensity of recorded SH light was found to be proportional to the square of the excitation light intensity, indicating the two-photon process characteristic of the SHG effect (Fig. 3 and Fig. S9, ESI†).23 To quantify the SHG response from 1 and 2, the well-known SH reference of potassium dihydrogen phosphate (KDP) was measured under identical experimental conditions (Fig. S8 and S9, ESI†). The intensities of the SHG light in 1 and 2 were found to be rather small, reaching 0.11% of KDP for 1 and 0.22% of KDP for 2. The noticeable difference in the SHG response between 1 and 2 can be assigned to the small variation of an ionic size radius and the change in polarizability occurring with the transition metal substitution from Mo to W centres.24 | ||
| Fig. 3 The room-temperature SHG intensity of 1 and 2 in the function of excitation intensity (top) together with a schematic illustration of the SHG effect (bottom). The 1040 nm laser source was used so as the green 520 nm light was investigated as the output SHG signal. The coloured circular points represent the experimental data while solid lines show the best-fits following the quadratic function. | ||
Light absorption and photoluminescence studies
The powder samples of 1 and 2 exhibit intense yellow and orange colours, respectively, and contain NdIII complexes whose relatively strong near-infrared photoluminescence was broadly explored.25 Therefore, the obtained compounds were investigated using solid-state UV-vis-NIR absorption and photoluminescence spectra (Fig. 4 and Fig. S10, ESI†). In the visible region of the absorption spectra of 1 and 2, the series of weak peaks are observed (Fig. S10, ESI†). They can be assigned to the f–f electronic transitions of NdIII centres, from the ground 4I9/2 multiplet to the higher-lying energy levels.25 At higher energies, much stronger broad absorption bands, ranging from the UV region to ca. 550 nm for 1 and even ca. 600 nm for 2, are observed. They were deconvoluted into five main components (Fig. S10, ESI†). Four of them, covering the UV-vis range to ca. 450 nm, are positioned at very similar energy positions in 1 and 2 (Table S6, ESI†), as they originate from the combined contributions from the singlet-to-singlet n–π* and π–π* transitions of pzdo ligands, and the transitions from the ligand-field (LF) and metal-to-ligand charge transfer (MLCT) electronic states of octacyanidometallate anions.26 The LF states are also mainly responsible for the last deconvoluted component lying in the visible range. However, this absorption band is significantly broadened beyond the expected limit for the electronic transitions of [M(CN)8]4− ions. This is particularly visible in 2 (Fig. S10d, ESI†). Therefore, the additional contribution to these bands should be postulated. It can be ascribed to the appearance of the anion–π CT states which were recognized for the coordination systems involving pzdo–[MIV(CN)8]4− (M = Mo, W) moieties.17 | ||
| Fig. 4 Solid-state photoluminescence properties of 1 and 2: emission spectra under the 410 nm excitation light for 1 (a) and 2 (b), together with the related excitation spectra for the monitored emission at 1060 nm for 1 (c) and 2 (d). The spectra at room temperature (RT) and liquid nitrogen temperature (77 K) were shown in each case. The assignments of the main emission and excitation bands to the incorporated molecular building blocks (pzdo, [M(CN)8]4−, Nd3+ ions with their f–f electronic transitions) were shown. | ||
Using the light irradiation with wavelength below ca. 600 nm, the powder samples of 1 and 2 exhibit the distinct emission signal lying in the NIR region, easily detectable at room temperature (Fig. 4a and b). The emission patterns consist of two sharp peaks positioned at 890 and 1059 nm, and 886 and 1055 nm for 1 and 2, respectively. They can be assigned to the NdIII-centred f–f electronic transitions from the emissive 4F3/2 excited state to the 4I9/2 and 4I11/2 multiplets.25 Lowering the temperature to 77 K results in a very small red-shift of these maxima which is typical for the emission related to the 4f electronic levels well-shielded by 4d and 5s shells. To examine how the observed emission is realized after the light absorption, the excitation spectra for the monitored main emission peak at 1060 nm was examined at room and liquid nitrogen temperatures (Fig. 4c and d). For both 1 and 2, the excitation pattern consists of a series of sharp peaks in the visible range, which are ascribable to the well-defined f–f electronic transitions indicating the direct f–f excitation pathway. However, the additional broad band is present in the UV-vis region up to ca. 600 nm. By comparison with the absorption spectra (Fig. S10, ESI†), it can be assigned to the electronic transitions within the pzdo ligands, [MIV(CN)8]4− (M = Mo, W) moieties, and their anion–π CT states. Therefore, they represent the NIR NdIII-centred emission realized by the energy transfer process exploring both ligand-to-metal and metal-to-metal routes.17,25 To deeply discuss the observed emission sensitization effect, the excitation spectra were normalized to the maximum of the direct f–f excitation band at 570 nm (Fig. 4c and d). At room temperature, for both 1 and 2, the UV-to-vis excitation band related to the energy transfer process is at a similar level as the direct f–f excitation band lying above 550 nm. This indicates that both types of excitation pathways are similarly efficient despite much stronger UV-light absorption. This means that only the part of energy absorbed by pzdo and [MIV(CN)8]4− is transferred to NdIII centres while the significant part is lost through the non-radiative deactivation. However, at a decreased temperature, the energy transfer route becomes dominant over the direct f–f excitation which can be ascribed to the hampering of deactivation processes e.g. through thermally activated vibrations, and to the cancelling of the thermally-assisted back energy transfer (BET) process.17,25 In this context, there is a noticeable difference between the obtained compounds. Both at room temperature and 77 K, 1 exhibits the energy transfer excitation stronger than the direct f–f excitation, and the lowering of temperature increases the excitation intensity ratio (Fig. 4c). On the contrary, for 2, the room-temperature excitation band in the 250–600 nm range is weaker than the strongest peaks of direct f–f excitation but this compound shows much stronger temperature dependence producing better emission sensitization at 77 K (Fig. 4d). This can be mainly explained by the role of thermally-activated BET mechanism from the excited states of NdIII to the donor states of [MIV(CN)8]4− moieties.27 This unfavourable effect seems to be more efficient in 2 due to the presumably closer distance between the acceptor state and the donor electronic states of the cyanido complexes. However, when at 77 K the BET is quenched, the smaller energy difference leads to more efficient sensitized emission. This interpretation stays in line with the related absorption spectra showing the longer wavelength tail of the main broad band in 2 (Fig. S10c and d, ESI†).
Magnetic properties
Due to the presence of NdIII paramagnetic centres, which may exhibit the effect of slow relaxation of magnetization,5b,151 and 2 were investigated by direct-current (dc) and alternate-current (ac) magnetic measurements (Fig. 5 and Fig. S11–S17, ESI†). For both materials, the χMT products at room temperature, 6.36 and 6.30 cm3 mol−1 K for 1 and 2, respectively, are close to the expected values for four uncoupled Nd3+ ions (Fig. S11a, ESI†).5b,15 Upon cooling, a gradual decrease of χMT product is observed, which is mainly the result of thermal depopulation of mJ states within the 4I9/2 ground multiplet. The featureless course of the χMT(T) curve excludes the presence of significant magnetic interactions proving the sufficient isolation of NdIII complexes by diamagnetic octacyanidometallate and pzdo linkers. The field dependences of magnetization measured at 1.8 K exhibit a monotonous increase of the signal to 5.43 and 5.39 μB at 70 kOe, for 1 and 2, respectively, without any sign of magnetic hysteresis observable for high-performance SMMs (Fig. S11b, ESI†).2 | ||
| Fig. 5 Representative alternate-current (ac) magnetic characteristics of 1 and 2: magnetic-field-variable frequency-dependences of out-of-phase magnetic susceptibility, χM′′ at T = 1.8 K (top) and temperature-variable frequency-dependences of χM′′ at Hdc = 1500 Oe (bottom) for 1 (a) and 2 (b), together with the resulting dc-field-dependence of magnetic relaxation times (c) and the resulting temperature-dependence of magnetic relaxation times (d) for both 1 and 2. Coloured solid lines depicted in (a) and (b) correspond to the best-fits obtained using the generalized Debye model for a single relaxation process. Coloured solid lines in (c) and (d) represent the best-fit curves obtained considering three magnetic relaxation pathways: a QTM, a Raman process, and a Direct process (see eqn (1) and the text for details). | ||
At 1.8 K, both 1 and 2 subjected to a small field of 200 Oe start to reveal the non-zero signal of the out-of-phase magnetic susceptibility, χ′′ under variable ac frequency (Fig. 5a, b and Fig. S12, S14, ESI†). This indicates the existence of the non-negligible magnetic anisotropy of incorporated NdIII complexes. The optimal dc field of 1500 Oe was estimated based on the field dependences of relaxation time which was plotted by using a generalized Debye model for ac magnetic data at each applied dc field (Fig. 5c). This optimal field appears due to the equilibrium between the QTM effect, dominating at the low field regime, and a field-induced direct process that governs the magnetic relaxation at higher dc fields.28 Under the optimized dc field, the temperature-variable ac magnetic data of 1 and 2 were examined (Fig. 5a, b and Fig. S13, S15, ESI†). Below the available frequency limit of 1 kHz, the χ′′ maxima were detected in the 1.8–2.4 K range. Similarly to the dc-field-variable ac magnetism, the resulting χ′′(ν), χ′(ν), and Argand χ′′(χ′) plots were fitted according to a generalized Debye model. The extracted relaxation times, both of the τ−1versus H as well as τ−1versus T dependences, were analysed using eqn (1):
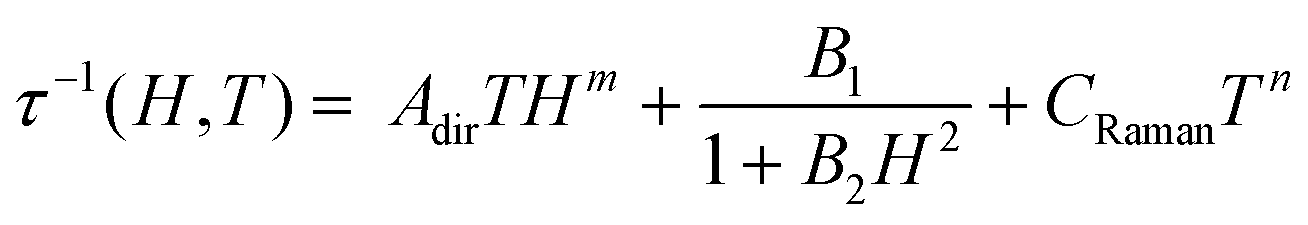 | (1) |
The first term in eqn (1) represents the field-induced direct relaxation process (depicted by Adir and m parameters), the second term is related to the QTM effect (depicted by B1 and B2 parameters), whereas the last term reflects a two-phonon Raman relaxation pathway (depicted by CRaman and n parameters).28 The other possible relaxation process, that is the Orbach relaxation occurring for typical SMM systems through one of the higher-lying mJ states of the ground multiplet, was excluded from the consideration by the results of the ab initio calculations (see below). The lack of an Orbach relaxation process both in 1 and 2 was also suggested by the rough fitting of the temperature dependences of magnetic relaxation times by using a simple Arrhenius law for the thermally-activated exponential process (Fig. S16, ESI†). It gives the unreasonably low effective energy barriers of 6 and 4 cm−1 for 1 and 2, respectively, indicating that the Orbach process is not represented.15,28 Therefore, three relaxation routes, QTM, direct, and Raman processes, were taken into account. To avoid over-parametrization, the contributions from the direct process and the QTM effects were extracted from the field dependences of the relaxation time. The following set of best-fit parameters were obtained (Fig. 5c): Adir = 5.51(7) × 10−6 s−1 K−1 Oe−2.276, B1 = 1388(78) s−1, B2 = 4.91(40) × 10−5 Oe−2, and m = 2.276(1), with field-independent contribution, c = 1829(8) s−1 for 1, and Adir = 2.00(18) × 10−7 s−1 K−1 Oe−2.295, B1 = 405(11) s−1, B2 = 4.28(20) × 10−6 Oe−2, m = 2.295(10), and c = 1097(3) s−1 for 2, respectively. The further fitting of the related temperature dependences (Fig. 5d) results in the following sets of parameters: CRaman = 312(2) s−1 K−2.997, n = 2.997(8) for 1, and CRaman = 241(1) s−1 K−2.604, n = 2.604(6) for 2. The n power of the Raman relaxation is noticeably far from the expected n = 9 for the Kramers ions which can be assigned to the pronounced role of acoustic phonons in the relaxation mechanism.28,29 Moreover, the evident deviation from the τ−1 ∼ H4 dependence expected for a direct process in Kramers ions is rationalized by the non-negligible role of hyperfine interactions which can lead to the decrease of the m power toward the τ−1 ∼ H2 behaviour.28b Nevertheless, the obtained best-fit parameters indicate that the NdIII complexes in 1 and 2 exhibit the SMM character exhibiting three different slow magnetic relaxation processes (Fig. 5d). The dominant role is played by the Raman relaxation as depicted by the visualized contributions from all relaxation pathways (Fig. S17, ESI†).
The noticeably higher parameters of Raman relaxation, CRaman and n, were found in 1 which effectively leads to faster overall magnetic relaxation. Thus, the higher-performance SMM character is ascribable to the NdIII complexes embedded in WIV-containing 2 rather than MoIV-based 1. This Mo-to-W transition substitution effect on the SMM behaviour in Ln–[MIV(CN)8]4− assemblies is opposite to those observed recently in the Er–Mo/W and Yb–Mo/W coordination chains.17 This suggests that the role of the exchange of the lighter MoIV by the heavier WIV centres in the isostructural compounds may depend on the intrinsic single-ion anisotropy of attached lanthanide(3+) ions and their specific electronic structure. In particular, the non-innocent role can be played by the subtle, but presumably critical, structural modification of lanthanide complexes occurring upon the transition metal substitution as was lately proven for the series of HoIII–[MIII(CN)6]3− (M = Co, Rh, Ir) molecular nanomagnets.7h This suggestion is still valid even if 1 and 2 reveal mainly the Raman relaxation process through the virtual states as the recent reports indicate that this magnetic relaxation pathway is expected to be also correlated with the single-ion anisotropy of the incorporated 4f metal ions.28b,29 However, such a correlation is not yet fully explained and certainly is not easily accessible for 1 and 2 due to their complexity related to the presence of three different NdIII complexes of similar but distinguishable magnetic anisotropy (see the ab initio calculations below).
Ab initio calculations
To deepen the discussion of the slow magnetic relaxation effects found in 1 and 2, the CASSCF/RASSI/SINGLE_ANISO ab initio calculations of the NdIII crystal-field effect were performed within an OpenMolcas package. The calculations were executed for three crystallographically distinguishable NdIII complexes (Nd1, Nd2, Nd2′, Fig. 1) using the crystal structure of 1a found from the SC-XRD experiment (Fig. S18, ESI,† see also Table 1 and Tables S7–S13, ESI†). The analysed compound consists of cyanido- and pzdo-bridged coordination layers, thus, for the purpose of calculations, each f-block metal centre was extracted from the framework together with the attached [MoIV(CN)8]4− moieties, pzdo and aqua ligands (Fig. S18, ESI†). The ab initio calculations were performed using two basis sets, the small and the large one (Table S7, ESI†), leading to the consistent set of results differing mainly in the scale of the whole crystal field splitting (Table S8, ESI†). The obtained composition of the crucial ground Kramers doublets for all three investigated Nd centres, together with the respective components of the pseudo-g-tensors, were gathered in Table 1 while a more detailed collection of the results is shown in Tables S8–S10, ESI.† For the nine-coordinated Nd1 and Nd2 centres, the impact of the crystal field on the NdIII 4I9/2 ground multiplet results in the formation of very different ground states characterized by the main components attributed to mJ = ±7/2 and mJ = ±9/2, respectively. This can be correlated with a different shape of the first coordination sphere, which was determined for Nd1 as a tricapped trigonal prism, while Nd2 adopts the geometry of a strongly distorted capped square antiprism (Fig. 1 and Table S2, ESI†). On the other hand, the change from nine-coordinated Nd2 to eight-coordinated Nd2′ centres leaves the contribution of the main mJ component in the ground Kramers doublet almost unchanged. Nevertheless, the resulting ground sub-levels involve large admixtures of the other mJ states, thus their overall magnetic anisotropy is rather weak. The extracted pseudo-g-tensors, visualized in Fig. S18, ESI,† are characterized by the large gz factors but with significant transversal components which generate the strong QTM effect for all investigated NdIII complexes.5,7 This perfectly agrees with the ac magnetic data indicating the disappearance of the SMM features without the application of an external dc field (Fig. 5).| NdIII centre | Nd1 | Nd2 | Nd2′ | |
|---|---|---|---|---|
| m J composition of the ground Kramers doublet (components with >2% contribution) | ±7/2, 73.0% | ±9/2, 65.9% | ±9/2, 67.6% | |
| ±5/2, 7.1% | ±1/2, 15.3% | ±5/2, 10.2% | ||
| ∓7/2, 6.5% | ±5/2, 8.7% | ±3/2, 8.1% | ||
| ∓5/2, 5.1% | ∓9/2, 4.4% | ±7/2, 4.7% | ||
| ±9/2, 4.4% | ±3/2, 2.1% | ∓9/2, 4.4% | ||
| Pseudo-g-tensor of the ground doublet | g x | 1.5559 | 1.0135 | 0.1845 |
| g y | 1.6555 | 1.2071 | 0.6323 | |
| g z | 4.1478 | 4.9526 | 5.4265 | |
| The energy gap between the ground and the 1st excited Kramers doublets of the ground multiplet | 92.73 cm−1 | 127.04 cm−1 | 136.31 cm−1 | |
The results of the ab initio calculations exclude also the presence of an Orbach-type relaxation. For Nd1, Nd2, and Nd2′ centres, the first excited Kramers doublets are located at ca. 92.7, 127.0, and 136.3 cm−1, respectively (Table 1 and Table S8, ESI†). Such values are much higher than the effective energy barrier of ca. 6 cm−1, extracted from the Arrhenius-type temperature dependence of relaxation times (Fig. S16, ESI†). This indicates that the observed T-dependence of the overall relaxation time in 1 is dominated by the under-barrier processes such as the Raman relaxation suggested by the thorough analysis of the experimental data (Fig. 5). The same statement is valid for 2 lacking an exact structural model, as the WIV-based analogue is characterized by the similarly low effective energy barrier of ca. 4 cm−1. This interpretation is consistent with the latest reports dealing with field-induced SMMs based on weakly anisotropic lanthanide ions, such as Yb3+, lacking the Orbach process in their slow magnetic relaxation effects.7,9 The results of ab initio calculations, including the elucidated crystal-field parameters for all Nd complexes in 1a (Tables S11–S13, ESI†), enabled the simulation of the representative dc magnetic characteristics which perfectly match the experimental curves for both 1 and 2 (Fig. S11, ESI†). This is additional proof of their similarity from the viewpoint of the f-block metal-centred electronic structure and the related magnetic anisotropy. This also confirms good magnetic isolation of NdIII centres as no sign of noticeable magnetic interactions is observed.
Apart from the ground multiplet composition, the ab initio calculations also give information about the energy of the excited states of Ln3+ ions. The precision of determination of the absolute energy value for excited terms is more affected by the number and the proximity (in terms of energy) of the other electronic states taken under consideration and optimized during the SA-CASSCF step, thus a direct reproduction of the emission spectra is an extremely difficult task. This is also connected with the lack of geometry optimization for excited states and the neglected role of vibrational modes. However, the splitting of the main experimental emissive transition to the ground multiplet, 4I9/2 for NdIII, can be correlated with the calculated crystal field splitting (assuming a small impact of the geometry changes for the well-screened f-electrons) if the measurement temperature is sufficiently lowered to eliminate or at least restrain the influence of hot transitions. Here, despite the presence of three independent NdIII centres in the structure of 1a, we attempted to correlate the results of the ab initio calculations with the high-resolution emission spectra gathered at the relatively low temperature of 77 K (Fig. S19, ESI†). As a result, we obtained a proper match between the emission band located in the 850–900 nm range, which originates from the 4F3/2 → 4I9/2 electronic transition, and the energy splitting of each NdIII centre. Such analysis is equally applicable to the emission pattern for 1 and 2, again confirming a similar electronic structure. However, it should be noted that at 77 K several remaining hot bands are expected, which visibly affects the emission band resolution. Nevertheless, the magneto-optical correlation confirms experimentally the validity of the ab initio calculations, particularly from the context of the overall energy scale of the NdIII crystal field effect in 1 and 2.
Conclusions
Herein, we report unique multifunctional molecular materials combining second-harmonic generation (SHG) activity, sensitized near-infrared (NIR) photoluminescence and the effect of slow relaxation of magnetization. This conjunction of optical and magnetic functionalities was achieved by the formation of NIR-emissive NdIII molecular nanomagnets within the layered coordination frameworks. Their skeletons employ two types of molecular linkers, organic pyrazine-N,N′-dioxide and inorganic octacyanido-metallates(IV), whose specific intralayer alignment, supported by the non-covalent anion–π interactions, leads to the non-centrosymmetric crystal structure (Table S5, ESI†). Therefore, in this work, we present the generation of chiral luminescent nanomagnets which takes advantage not only of tunable physical properties of lanthanide(III) complexes but also of the non-innocent role of the attached cyanido metal complex which contributes significantly to the induction of chirality as well as sensitization of the NIR photoluminescence. The application of cyanido metal complexes enables also the modulation of all physical features by the changing of the d-transition metal centre of the cyanido complex. The exchange of MoIV with WIV enhances the SH intensity, increases the sensitization efficiency at low temperatures, and slowdowns the magnetic relaxation processes. Despite the fact that the mechanisms of these effects are not fully elucidated, it is proven that even the small structural variation related to the Mo-to-W replacement can be an efficient tool for the material performance optimization within both magnetic and optical functionalities. The further functionalization of luminescent single-molecule magnets toward chirality-related non-linear optical and magneto-optical properties using non-innocent cyanido metalloligands is planned. The work on the improvement of single-ion anisotropy of the incorporated lanthanide centres, for instance by inducing the axial alignment of negatively charged ligands expected to be suitable for NdII centres, should also be considered.15 Moreover, the obtained compounds consist of neutral coordination layers of the well-recognized SHG, luminescent, and molecular nanomagnetic properties which make them attractive candidates as potential two-dimensional materials processable into the magnetic and optical devices which is worth examining as a next research step.Experimental section
Materials
Neodymium(III) chloride hexahydrate, pyrazine, and bis(tri-phenylphosphine)iminium chloride were purchased from Sigma-Aldrich and used as received. The K4[Mo(CN)8]·2H2O, K4[W(CN)8]·2H2O, {(bpyH)3(H3O)[Mo(CN)8]·H2O}, {(bpyH)3(H3O) [W(CN)8]·H2O}, PPN4[Mo(CN)8]·10H2O and PPN4[W(CN)8]·10H2O salts, as well as pyrazine-N,N′-dioxide (pzdo), were synthesized according to modified published procedures.30 All other reagents were purchased from TCI Chemicals or Sigma-Aldrich and were used without further purification.Synthetic procedures
Note that both obtained materials, 1 and 2, can be alternatively obtained starting from PPN4[MIV(CN)8]·10H2O salts, namely PPN4[MoIV(CN)8]·10H2O (131.9 mg, 0.05 mmol) for 1, and PPN4[WIV(CN)8]·10H2O (136.3 mg, 0.05 mmol) for 2, respectively.
X-Ray crystallography
The single crystals of two enantiomorphs of 1, namely 1a and 1b, selected for the SC-XRD experiment were taken directly from the mother solution, covered by Apiezon N grease, and mounted on the Micro MountsTM holder. The experiment was performed at 100(2) K, using a Bruker D8 Quest Eco Photon50 CMOS diffractometer, equipped with graphite mono-chromated MoKα radiation. The SAINT and SADABS programs were employed for data reduction and cell refinement processes. The absorption correction was conducted using a multi-scan procedure. The crystal structures were solved by an intrinsic phasing method using a SHELXT programme within the Apex3 package.31a The crystal structure refinement was carried out by a weighted full-matrix least-squares method on F2 of SHELX-2014/7 within the WinGX (ver. 2014.1) software.31b All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of water molecules were found from the residual electron density map and refined isotropically. Hydrogen atoms of pzdo ligands were partially calculated on the idealized positions, refined using a riding model, and partially found from the electron density map. Crystals of both 1a and 1b reveal significant structural disorder related to the partial occupancy of nine- and eight-coordinated complexes on the Nd2 site, named Nd2 and Nd2′ (Fig. S4, ESI†). Therefore, we found it necessary to apply a series of restraints of the DELU and ISOR type. Additionally, the series of DFIX restraints were applied to non-calculated hydrogen atoms to stabilize their positions and guarantee the convergence of the refinement procedure, in particular for the disordered fragment of the crystal structures. For one of the enantiomers, 1b, two OMIT commands were used to remove the reflections strongly affected by the X-ray beam. Selected details of crystal data and structure refinement were gathered in Table S1, ESI.† The detailed structure parameters are listed in Table S3, ESI.† CCDC reference numbers are 2061390 and 2061391, for 1a, and 1b, respectively.† Powder X-ray diffraction (P-XRD) patterns for 1 and 2 were gathered using a Bruker D8 Advance Eco powder X-ray diffractometer equipped with CuKα radiation source and a capillary spinning add-on. The powder samples were dried in the air and inserted into a glass capillary before the P-XRD measurements which were performed at room temperature. Continuous shape measure analysis for the Mo(IV) and Nd(III) complexes was performed using SHAPE software ver. 2.1.21 Structural figures were created in the Mercury 4.3.1 software.Physical techniques
The solid-state UV-Vis-NIR absorption spectra were measured on a Shimadzu UV-3600i-plus spectrophotometer using the thin films of ground samples dispersed in paraffin oil and inserted between quartz plates. IR absorption spectra were collected using a Nicolet iN10 MX FTIR microscope. Elemental analysis was performed using the Elementar Vario Micro Cube CHNS analyser. Thermogravimetric (TG) measurements were performed on a NETZSCH TG 209 F1 Libra apparatus. Solid-state photoluminescence spectra were gathered on a Horiba Jobin-Yvon Fluorolog-3 (FL3-211) spectrofluorimeter equipped with a 450 W Xe lamp and an InGaAs photodiode detector DSS-IGA020L cooled using liquid nitrogen. The data analysis was carried out using FluorEssence® software. Magnetic measurements were conducted using a Quantum Design MPMS-3 Evercool magnetometer. The samples for the magnetic studies were immersed in the mother solution and sealed in a glass tube. Magnetic data include the diamagnetic corrections from the measured materials and the sample holder. Second harmonic generation (SHG) experiments were performed using a homemade optical setup (Fig. S7, ESI†). A 1040 nm femtosecond laser was used as incident fundamental light, which was generated by a Spirit® OneTM 1040-8 laser box provided by Spectra-Physics®, with pulse width less than 400 fs, a repetition rate of 200 kHz, and adapted with a pulse picker. After being reflected by dielectric multilayer mirrors, the fundamental laser beam went through a coloured glass filter to cut off the visible component, and then perpendicularly irradiated onto the surface of the sample, which was prepared in the powder pellet form placed in a glass holder. The resulting SH signal was detected in a reflection mode. By applying a harmonic separator and a focal lens, the reflection SHG light was regulated to a monochromator (Oriel® 77250), and then the signal was finally detected by a photomultiplier tube (Hamamatsu R464). Data collection was realized by a customized LabVIEWTM interface which can record the real-time incident laser intensity and the counts-per-second of the detected SH signal. To verify the SHG activity, each sample was characterized by both the power-dependence and the wavelength-dependence measurements. For the first measurement, the incident laser intensity was tuned with a linear increment from zero, while for the latter measurement, the detection wavelength was adjusted by the monochromator. The powder samples of 1 and 2, being the mixture of enantiomorphic crystals, were investigated in the SHG experiments as the single crystals sufficiently large for the related measurements of SH light could not be isolated. To quantify the SH intensities generated by the investigated samples, a potassium dihydrogen phosphate (KDP) powder pellet was used as a reference sample.Computational details
The ab initio calculations were performed for molecular moieties based on three symmetry independent coordination complexes of Nd1, Nd2 and Nd2′, and the geometries were taken from the single-crystal X-ray diffraction experiment for 1a. All the discussed molecular fragments are visualized together with the calculated anisotropy axes in Fig. S18, ESI.† Apart from NdIII centres, each moiety contains all the organic and aqua ligands, and all nearest diamagnetic octacyanidometallate units. To examine local magnetic properties of NdIII, State Average Complete Active Space Self-Consistent Field (SA-CASSCF) calculations were performed using the OpenMolcas software.32 Scalar relativistic effects were taken into account by employing two-component second-order Douglas–Kroll–Hess (DKH2) Hamiltonian together with the relativistic Atomic Natural Orbital basis sets of the ANO-RCC type.33 To save the disk space for computations of such relatively big moieties, Cholesky decomposition of ERI-s (electron repulsion integrals) was used with a 1.0 × 10−8 threshold. Two models were considered: the first one – smaller (S) – with VDZP basis function quality for NdIII and VDZ for other atoms, and – larger (L) – using VTZP for NdIII centres, VDZP for atoms directly bonded to the paramagnetic centre, and VDZ for the others (Table S7, ESI†). In the CASSCF step, the active space was composed of seven 4f orbitals of NdIII with 3 active electrons – CAS (3 in 7), and 35 quartets with 112 doublets spin-adapted states arising from different possible electron distributions in 4f orbitals were evaluated. In the next step, all previously optimized, as spin-free, states were mixed within the Restricted Active Space State Interaction (RASSI) submodule by Spin–Orbit Coupling (SOC) within the atomic mean-field (AMFI) approximation.34 In the final step, the resulting 364 spin–orbit states were analysed using the SINGLE_ANISO module to obtain main magnetic axes and pseudo-g-tensors of each Kramers doublet, simulate χMT(T) and M(H) magnetic characteristics, and decompose spin–orbit states into such with a definite projection of the total momentum on the located quantization z-axis.35 The energy splitting of the ground 4I9/2 multiplets of all three calculated NdIII complexes along with the pseudo-g-tensor components and the composition of the ground doublet in the mJ basis are shown in Table 1 and Tables S8–S10 (ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Science Centre, Poland within the OPUS-14 project, grant no. 2017/27/B/ST5/00947, and by the Polish Ministry of Science and Higher Education within the “Diamond Grant” program, no. DI2018 017848.References
- (a) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 Search PubMed; (b) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 Search PubMed.
- (a) R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728 Search PubMed; (b) S. M. Aldoshin, D. V. Korchagin, A. V. Palii and B. S. Tsukerblat, Pure Appl. Chem., 2017, 89, 1119 Search PubMed; (c) J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470 Search PubMed; (d) M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194 Search PubMed; (e) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 Search PubMed.
- (a) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268 Search PubMed; (b) K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801 Search PubMed; (c) L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265 Search PubMed; (d) E. Coronado, Nat. Rev. Mater., 2020, 5, 87 Search PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 Search PubMed.
- (a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 Search PubMed; (b) F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150 Search PubMed; (c) L. Ungur and L. F. Chibotaru, Inorg. Chem., 2016, 55, 10043 Search PubMed; (d) J. Lu, M. Guo and J. Tang, Chem. – Asian J., 2017, 12, 2772 Search PubMed; (e) Z. Zhu, M. Guo, X.-L. Li and J. Tang, Coord. Chem. Rev., 2019, 378, 350 Search PubMed; (f) C.-L. Yin, Z.-B. Hu, Q.-Q. Long, H.-S. Wang, J. Li, Y. Song, Z.-C. Zhang, Y.-Q. Zhang and Z.-Q. Pan, Dalton Trans., 2019, 48, 512 Search PubMed; (g) H.-S. Wang, C.-L. Yin, Z.-B. Hu, Y. Chen, Z.-Q. Pan, Y. Song, Y.-Q. Zhang and Z.-C. Zhang, Dalton Trans., 2019, 48, 10011 Search PubMed; (h) H.-S. Wang, K. Zhang, Y. Song and Z.-Q. Pan, Inorg. Chim. Acta, 2021, 521, 120318 Search PubMed.
- (a) Y. Bi, X.-T. Wang, W. Liao, X. Wang, R. Deng, H. Zhang and S. Gao, Inorg. Chem., 2009, 48, 11743 Search PubMed; (b) J. Long, Y. Guari, R. A. S. Ferreira, L. D. Carlos and J. Larionova, Coord. Chem. Rev., 2018, 363, 57 Search PubMed; (c) J.-H. Jia, Q.-W. Li, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Coord. Chem. Rev., 2019, 378, 365 Search PubMed; (d) J. Long, Front. Chem., 2019, 7, 63 Search PubMed; (e) Z. Zhu, X.-L. Li, S. Liu and J. Tang, Inorg. Chem. Front., 2020, 7, 3315 Search PubMed.
- (a) C.-S. Liu, M. Du, E. C. Sañudo, J. Echeverria, M. Hu, Q. Zhang, L.-M. Zhou and S.-M. Fang, Dalton Trans., 2011, 40, 9366 Search PubMed; (b) J.-R. Jiménez, I. F. Díaz-Ortega, E. Ruiz, D. Aravena, S. J. A. Pope, E. Colacio and J. M. Herrera, Chem. – Eur. J., 2016, 22, 1454 Search PubMed; (c) G. Cucinotta, M. Perfetti, J. Luzon, M. Etienne, P.-E. Car, A. Caneschi, G. Calvez, K. Bernot and R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 1606 Search PubMed; (d) J. Long, R. Vallat, R. A. S. Ferreira, L. D. Carlos, F. A. Almeida Paz, Y. Guari and J. Larionova, Chem. Commun., 2012, 48, 9974 Search PubMed; (e) K. S. Pedersen, J. Dreiser, H. Weihe, R. Sibille, H. V. Johannesen, M. A. Sørensen, B. E. Nielsen, M. Sigrist, H. Mutka, S. Rols, J. Bendix and S. Piligkos, Inorg. Chem., 2015, 54, 7600 Search PubMed; (f) P. Martín-Ramos, J. T. Coutinho, M. Ramos Silva, L. C. J. Pereira, F. Lahoz, P. S. Pereira da Silva, V. Lavín and J. Martín-Gil, New J. Chem., 2015, 39, 1703 Search PubMed; (g) K. Yamashita, R. Miyazaki, Y. Kataoka, T. Nakanishi, Y. Hasegawa, M. Nakano, T. Yamamura and T. Kajiwara, Dalton Trans., 2013, 42, 1987 Search PubMed; (h) J. Wang, J. J. Zakrzewski, M. Zychowicz, V. Vieru, L. F. Chibotaru, K. Nakabayashi, S. Chorazy and S. Ohkoshi, Chem. Sci., 2021, 12, 730 Search PubMed.
- (a) Y. Bi, C. Chen, Y.-F. Zhao, Y.-Q. Zhang, S.-D. Jiang, B.-W. Wang, J.-B. Han, J.-L. Sun, Z.-Q. Bian, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 5020 Search PubMed; (b) D. Errulat, R. Marin, D. A. Gálico, K. L. M. Harriman, A. Pialat, B. Gabidullin, F. Iikawa, O. D. D. Couto, Jr., J. O. Moilanen, E. Hemmer, F. A. Sigoli and M. Murugesu, ACS Cent. Sci., 2019, 5, 1187 Search PubMed.
- (a) J. Wang, J. J. Zakrzewski, M. Heczko, M. Zychowicz, K. Nakagawa, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. Ohkoshi, J. Am. Chem. Soc., 2020, 142, 3970 Search PubMed; (b) J. Long, J. Rouquette, J.-M. Thibaud, R. A. S. Ferreira, L. D. Carlos, B. Donnadieu, V. Vieru, L. F. Chibotaru, L. Konczewicz, J. Haines, Y. Guari and J. Larionova, Angew. Chem., Int. Ed., 2015, 127, 2264 Search PubMed; (c) P.-H. Guo, J.-L. Liu, J.-H. Jia, J. Wang, F.-S. Guo, Y.-C. Chen, W.-Q. Lin, J.-D. Leng, D.-H. Bao, X.-D. Zhang, J.-H. Luo and M.-L. Tong, Chem. – Eur. J., 2013, 19, 8769 Search PubMed; (d) P.-H. Guo, Y. Meng, Y.-C. Chen, Q.-W. Li, B.-Y. Wang, J.-D. Leng, D.-H. Bao, J.-H. Jia and M.-L. Tong, J. Mater. Chem. C, 2014, 2, 8858 Search PubMed; (e) S.-D. Zhu, J.-J. Hu, L. Dong, H.-R. Wen, S.-J. Liu, Y.-B. Lu and C.-M. Liu, J. Mater. Chem. C, 2020, 8, 16032 Search PubMed.
- (a) A. Ould-Hamouda, A. Iazzolino, H. Tokoro, S. Ohkoshi and E. Freysz, Eur. J. Inorg. Chem., 2018, 378 Search PubMed; (b) M. Komine, K. Imoto, Y. Miyamoto, K. Nakabayashi and S. Ohkoshi, Eur. J. Inorg. Chem., 2018, 1367 Search PubMed.
- (a) J. Zhao, T. Zhang, X.-Y. Dong, M.-E. Sun, C. Zhang, X. Li, Y. S. Zhao and S.-Q. Zang, J. Am. Chem. Soc., 2019, 141, 15755 Search PubMed; (b) J. Zhang, L. Dai, A. M. Webster, W. T. K. Chan, L. E. Mackenzie, R. Pal, S. L. Cobb and G.-L. Law, Angew. Chem., Int. Ed., 2021, 60, 1004 Search PubMed.
- (a) G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493 Search PubMed; (b) C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3297 Search PubMed.
- (a) S. Chorazy, M. Wyczesany and B. Sieklucka, Molecules, 2017, 22, 1902 Search PubMed; (b) J. J. Zakrzewski, M. Liberka, M. Zychowicz and S. Chorazy, Inorg. Chem. Front., 2021, 8, 452 Search PubMed; (c) Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211 Search PubMed; (d) J. J. Zakrzewski, B. Sieklucka and S. Chorazy, Inorg. Chem., 2020, 59, 1393 Search PubMed; (e) P. A. Smith, C. Crawford, N. Beedoe, Z. Assefa and R. E. Sykora, Inorg. Chem., 2012, 51, 12230 Search PubMed; (f) R. J. Roberts, D. Le and D. B. Leznoff, Inorg. Chem., 2017, 56, 7948 Search PubMed.
- (a) S. Chorazy, J. J. Zakrzewski, M. Magott, T. Korzeniak, B. Nowicka, D. Pinkowicz, R. Podgajny and B. Sieklucka, Chem. Soc. Rev., 2020, 49, 5945 Search PubMed; (b) D. Pinkowicz, R. Podgajny, W. Nitek, M. Rams, A. M. Majcher, T. Nuida, S. Ohkoshi and B. Sieklucka, Chem. Mater., 2011, 23, 21 Search PubMed; (c) M. Atzori, I. Breslavetz, K. Paillot, K. Inoue, G. L. J. A. Rikken and C. Train, J. Am. Chem. Soc., 2019, 141, 20022 Search PubMed; (d) J. J. Zakrzewski, S. Chorazy, K. Nakabayashi, S. Ohkoshi and B. Sieklucka, Chem. – Eur. J., 2019, 25, 11820 Search PubMed; (e) M. Liberka, J. Kobylarczyk, T. M. Muziol, S. Ohkoshi, S. Chorazy and R. Podgajny, Inorg. Chem. Front., 2019, 6, 3104 Search PubMed; (f) S. Ohkoshi, K. Nakagawa, K. Imoto, H. Tokoro, Y. Shibata, K. Okamoto, Y. Miyamoto, M. Komine, M. Yoshikiyo and A. Namai, Nat. Chem., 2020, 12, 338 Search PubMed.
- (a) S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Commun., 2016, 52, 7168 Search PubMed; (b) H. Wada, S. Ooka, T. Yamamura and T. Kajiwara, Inorg. Chem., 2017, 56, 147 Search PubMed; (c) Y.-C. Chen, X.-S. Huang, J.-L. Liu and M.-L. Tong, Inorg. Chem., 2018, 57, 1178 Search PubMed; (d) H. Ke, L. Zhao, Y. Guo and J. Tang, Dalton Trans., 2012, 41, 2314 Search PubMed; (e) J. Liu, D. Reta, J. A. Cleghorn, Y. X. Yeoh, F. Ortu, C. A. P. Goodwin, N. F. Chilton and D. P. Mills, Chem. – Eur. J., 2019, 25, 7749 Search PubMed.
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 Search PubMed; (b) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189 Search PubMed.
- R. Jankowski, J. J. Zakrzewski, O. Surma, S. Ohkoshi, S. Chorazy and B. Sieklucka, Inorg. Chem. Front., 2019, 6, 2423 Search PubMed.
- L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342 Search PubMed.
- (a) S. Ohkoshi, S. Takano, K. Imoto, M. Yoshikiyo, A. Namai and H. Tokoro, Nat. Photonics, 2014, 8, 65 Search PubMed; (b) T. Ohno, S. Chorazy, K. Imoto and S. Ohkoshi, Cryst. Growth Des., 2016, 16, 4119 Search PubMed.
- R. Podgajny, D. Pinkowicz, B. Czarnecki, M. Kozieł, S. Chorazy, M. Wis, W. Nitek, M. Rams and B. Sieklucka, Cryst. Growth Des., 2014, 14, 4030 Search PubMed.
- (a) M. Llunell, D. Casanova, J. Cirera, J. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE v. 2.1, Program for the Calculation of Continuous Shape Measures of Polygonal and Polyhedral Molecular Fragments, University of Barcelona, Barcelona, Spain, 2013 Search PubMed; (b) D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1755 Search PubMed.
- (a) A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231 Search PubMed; (b) A. Altomare, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, R. Rizzi and P.-E. Werner, J. Appl. Crystallogr., 2000, 33, 1180 Search PubMed; (c) A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724 Search PubMed.
- L. Bonacina, P.-F. Brevet, M. Finazzi and M. Celebrano, J. Appl. Phys., 2020, 127, 230901 Search PubMed.
- (a) P.-X. Li, C.-L. Hu, X. Xu, R.-Y. Wang, C.-F. Sun and J.-G. Mao, Inorg. Chem., 2010, 49, 4599 Search PubMed; (b) P. A. Maggard, X. Cheng, S. Deng and M.-H. Whangbo, Molecules, 2020, 25, 867 Search PubMed; (c) M. Komine, K. Imoto, A. Namai, M. Yoshikiyo and S. Ohkoshi, Inorg. Chem., 2021, 60, 2097 Search PubMed.
- (a) O. Savchuk, J. J. Carvajal, L. G. De la Cruz, P. Haro-Gonzalez, M. Aguilo and F. Diaz, J. Mater. Chem. C, 2016, 4, 7397 Search PubMed; (b) S. Chorazy, B. Sieklucka and S. Ohkoshi, Cryst. Growth Des., 2016, 16, 4918 Search PubMed; (c) B. S. K. Chong and E. G. Moore, Inorg. Chem., 2018, 57, 14062 Search PubMed; (d) R. E. Rojas-Hernandez, F. Rubio-Marcos, G. Gorni, C. Marini, M. Danilson, L. Pascual, R. U. Ichikawa, I. Hussainova and J. F. Fernandez, J. Mater. Chem. C, 2021, 9, 657 Search PubMed.
- (a) J. Del Bene and H. H. Jaffe, J. Chem. Phys., 1968, 49, 1221 Search PubMed; (b) M. F. A. Hendrickx, V. S. Mironov, L. F. Chibotaru and A. Ceulemans, Inorg. Chem., 2004, 43, 3142 Search PubMed.
- (a) F. R. Goncalves e Silva, O. L. Malta, C. Reinhard, H.-U. Güdel, C. Piguet, J. E. Moser and J.-C. G. Bünzli, J. Phys. Chem. A, 2002, 106, 1670 Search PubMed; (b) D. Wu, W. Xiao, L. Zhang, X. Zhang, Z. Hao, G.-H. Pan, Y. Luo and J. Zhang, J. Mater. Chem. C, 2017, 5, 11910 Search PubMed; (c) K. Kumar, S. Chorazy, K. Nakabayashi, H. Sato, B. Sieklucka and S. Ohkoshi, J. Mater. Chem. C, 2018, 6, 8372 Search PubMed.
- (a) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655 Search PubMed; (b) Y.-S. Ding, K.-X. Yu, D. Reta, F. Ortu, R. E. P. Winpenny, Y.-Z. Zheng and N. F. Chilton, Nat. Commun., 2018, 9, 3134 Search PubMed; (c) D. Aravena and E. Ruiz, Dalton Trans., 2020, 49, 9916 Search PubMed.
- (a) K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437 Search PubMed; (b) E. Lucaccini, L. Sorace, M. Perfetti, J.-P. Costes and R. Sessoli, Chem. Commun., 2014, 50, 1648 Search PubMed; (c) A. B. Canaj, S. Dey, C. Wilson, O. Céspedes, G. Rajaraman and M. Murrie, Chem. Commun., 2020, 56, 12037 Search PubMed; (d) A. Chiesa, F. Cugini, R. Hussain, E. Macaluso, G. Allodi, E. Garlatti, M. Giansiracusa, C. A. P. Goodwin, F. Ortu, D. Reta, J. M. Skelton, T. Guidi, P. Santini, M. Solzi, R. De Renzi, D. P. Mills, N. F. Chilton and S. Carretta, Phys. Rev. B, 2020, 101, 174402 Search PubMed.
- (a) J. Szklarzewicz, D. Matoga and K. Lewiński, Inorg. Chim. Acta, 2007, 360, 2002 Search PubMed; (b) D. Matoga, J. Szklarzewicz and M. Mikuriya, Inorg. Chem., 2006, 45, 7100 Search PubMed; (c) J. Szklarzewicz, A. Samotus and A. Kanas, Polyhedron, 1986, 5, 1733 Search PubMed; (d) D. A. Cauzzi, G. Mori, G. Predieri, A. Tripicchio and F. Cavatorta, Inorg. Chim. Acta, 1993, 204, 181 Search PubMed; (e) R. Podgajny, D. Pinkowicz, T. Korzeniak, W. Nitek, M. Rams and B. Sieklucka, Inorg. Chem., 2007, 46, 10416 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 Search PubMed; (b) L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 Search PubMed.
- I. F. Galván, M. Vacher, A. Alavi, C. Angeli, F. Aquilante, J. Autschbach, J. J. Bao, S. I. Bokarev, N. A. Bogdanov, R. K. Carlson, L. F. Chibotaru, J. Creutzberg, N. Dattani, M. G. Delcey, S. S. Dong, A. Dreuw, L. Freitag, L. M. Frutos, L. Gagliardi, F. Gendron, A. Giussani, L. González, G. Grell, M. Guo, C. E. Hoyer, M. Johansson, S. Keller, S. Knecht, G. Kovačević, E. Källman, G. L. Manni, M. Lundberg, Y. Ma, S. Mai, J. P. Malhado, P. Å. Malmqvist, P. Marquetand, S. A. Mewes, J. Norell, M. Olivucci, M. Oppel, Q. M. Phung, K. Pierloot, F. Plasser, M. Reiher, A. M. Sand, I. Schapiro, P. Sharma, C. J. Stein, L. K. Sørensen, D. G. Truhlar, M. Ugandi, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, O. Weser, T. A. Wesołowski, P.-O. Widmark, S. Wouters, A. Zech, J. P. Zobel and R. Lindh, J. Chem. Theory Comput., 2019, 15, 5925 Search PubMed.
- (a) B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851 Search PubMed; (b) B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2005, 109, 6575 Search PubMed; (c) B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov, P.-O. Widmark and A. C. Borin, J. Phys. Chem. A, 2008, 112, 11431 Search PubMed.
- (a) P.-Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230 Search PubMed; (b) B. A. Heß, C. M. Marian, U. Wahlgren and O. A. Gropen, Chem. Phys. Lett., 1996, 251, 365 Search PubMed.
- (a) L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 Search PubMed; (b) L. Ungur and L. F. Chibotaru, Chem. – Eur. J., 2017, 23, 3708 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra. TGA curves. Additional structural parameters and views. Results of continuous shape measure analysis. P-XRD patterns. Scheme of the experimental setup used for the SHG experiment. Additional SHG characteristics. Solid-state UV-vis absorption spectra. Additional magnetic characteristics. Detailed results of the ab initio calculations and their correlation with the emission spectra. CCDC 2061390 and 2061391 for 1a and 1b, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc00825k |
| This journal is © The Royal Society of Chemistry 2021 |