
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable MOF@enzyme composites for electrochemical biosensing devices†
Ruhani
Singh‡
 *,
Mustafa
Musameh‡
,
Yuan
Gao
,
Berkay
Ozcelik
,
Xavier
Mulet
and
Cara M.
Doherty
*,
Mustafa
Musameh‡
,
Yuan
Gao
,
Berkay
Ozcelik
,
Xavier
Mulet
and
Cara M.
Doherty
CSIRO Future Industries, Private Bag 10, Clayton South, Victoria 3169, Australia. E-mail: ruhani.singh@csiro.au
First published on 28th April 2021
Abstract
Naturally occurring enzymes are highly selective and efficient catalysts but given their intricate and fragile structure, wide application of their extraordinary catalytic potential in industrial and biomedical settings is highly desirable yet challenging. In this work we demonstrate this possibility using a facile, single-step technique for metal–organic framework (MOF) biomimetic mineralisation of three different enzymes of industrial interest; glucose oxidase (GOx), organophosphate degrading enzyme A (OpdA) and α-chymotrypsin. We investigate the thermal stability of the resulting ZIF-8@enzyme composites at temperatures in the industrially relevant range of 50–80 °C with time ranging from minutes to hours. In situ thermal stabilisation analysis was performed at the small angle X-ray scattering (SAXS) beamline to understand the effect of temperature on the ZIF-8@GOx composite. Furthermore, we illustrate the electrochemical operational stability of a ZIF-8@GOx modified electrode at challenging elevated temperatures validating its potent application in electrochemical devices. The resulting optimized and ultra-stable amperometric glucose biosensor demonstrates a clinically relevant wide linear range of 1–10 mM glucose with a detection limit of 0.05 mM. The biosensor demonstrates robust performance and selectivity with insignificant interference effects in the presence of 100 μM uric acid, acetaminophen and ascorbic acid, respectively, and satisfactory operation in up to 20% serum environment.
1. Introduction
Nature has generated remarkable structures and functions through billions of years of evolution.1 One such class of biomacromolecules, naturally occurring enzymes, are polymers of amino acids that fold into complex structures forming highly specific and selective catalytic sites. Given the exceptional selectivity (chemo-, regio- and stereo-), efficiency and environmental friendliness of enzymes, translating enzymatic processes into field applications and industry is increasingly sought after; however, commercial use is currently limited due to, among other factors, stability issues, particularly at high temperatures or during long-term storage. The ability to stabilise enzymes at high temperatures (>40 °C) will allow them to be readily adopted for use in the fields of biocatalysis, diagnostics and sensing.2,3 The rationale for high operational temperatures is the resulting higher process rates with nearly a 100 fold increase in reaction rates for a temperature increase from 25 to 75 °C.4 The industrial demands of an enzyme can range from 60 to 80 °C for the detergent industry, 45 to >100 °C for food processing, 45 to 75 °C in electrochemical applications and 80 to 90 °C for the textile industry.4,5 Industrial applications can last from hours to days and in many of these cases, enzymes are used despite their relative instability.4 Thus, industry is always looking for technologies to enhance enzyme thermostability for their extended use at high temperatures.Furthermore, the push to develop sustainable and resilient societies has garnered significant interest in biosensing technologies for environmental monitoring, food safety, quality control and clinical diagnostics. Electrochemical biosensors integrate both the advantages of an expedited electrochemical response and specificity of enzymes as biorecognition elements, making them particularly attractive for performing rapid analyses with high sensitivity and selectivity. But as with other industrial applications, the key demands of enzymes for such analytical methods are their thermal and electrochemical operational stability, recovery, and re-usability. This requires the development of smart host matrices for enzyme immobilization without limiting the electrical communication between the enzyme and the electrodes and the mass-transfer efficiency of the analytes.
Metal–organic frameworks (MOFs) with their porous structure made of metal clusters co-ordinated with organic ligands traditionally make exceptional materials for storage and separation applications.6 The last decade has seen an explosion in the number of applications for MOFs in the fields of bio-catalysis and sensing.7,8 The versatility of their chemistry has proved their usefulness in many more applications, including enzyme immobilization and preservation.2,9,10 Due to the availability of characteristic high surface areas, early studies focused on the immobilization of enzymes on MOF surfaces.11–13 Enzyme loading into MOFs (post-synthetic encapsulation) via pore infiltration is another attractive method particularly due to the porous nature of the MOF.11,14,15 However, these techniques encounter the common challenge of limited loading due to poor attachment or adsorption, difficult absorption and the pore size limitation of the infiltration technique. An innovative technique developed for protecting enzymes uses a co-precipitation method in which the MOF self-assembles around the enzyme (de novo encapsulation) in a bio-mimetic type approach.15–18 Some of the studies demonstrating the advantages of the later approach for enzyme storage and thermal stability are as shown in Table 1.
| MOF | Enzyme | Storage | Temp. | Time | Activity | Ref. |
|---|---|---|---|---|---|---|
| Temp.: storage or treatment temperature; GOx: glucose oxidase; PBS: phosphate buffered saline; HRP: horseradish peroxidase; PB: phosphate buffer (without saline); AlcDH: alcohol dehydrogenase; NAD+: nicotinamide adenine dinucleotide; LacDH: lactate dehydrogenase; OpdA: organophosphate degrading enzyme A. | ||||||
| ZIF-8 | GOx + HRP | PBS (pH 7.4) | Room temp. | 7 d | 80% | 19 |
| ZIF-8 | Glucoamylase | PBS (pH 7.0) | 30 °C | 25 d | 91% | 20 |
| ZIF-8 | Thermophilic lipase | Dry | 70 °C | 50 min | 50% | 21 |
| 50 °C | 5 min | 63% | ||||
| 37 °C | 5 d | ∼100% | ||||
| 60 °C | 5 d | 76% | ||||
| ZIF-8 | Orange-peel peroxidase | PBS (pH 7.4) | 40–60 °C | 18 d | 88% | 22 |
| ZIF-8 | β-Galactosidase + GOx + HRP | PB (pH 7.4) | Room temp. | 3 wk | ≥95% | 23 |
| ZIF-8 | AlcDH + NAD + LacDH | PB (pH 7.4) | Room temp. | 3 wk | ≥95% | 23 |
| ZIF-8 | Laccase | — | 70 °C | 2 h | 60% | 24 |
| 4 °C | 20 d | >80 °C | ||||
| HKUST-1/CF | GOx | PBS (pH 7.4) | 40 °C | 1 h | 98% | 25 |
| 80 °C | 1 h | 80% | ||||
| ZIF-8 | GOx | Tris Buffer (pH 8.0) | 45–80 °C | 5, 10 min | 100% (45 °C:) | This work |
| ≥42% (75 °C) | ||||||
| ZIF-8 | GOx | 55 °C | 6 h | ≥88% | This work | |
| ZIF-8 | OpdA | Tris Buffer (pH 8.0) | 60 °C | 6 h | ≥95% | This work |
| ZIF-8 | α-Chymotrypsin | Tris Buffer (pH 8.0) | 80 °C | 10 min | 60% | This work |
In this work, we adapted the facile and versatile technique of biomimetic mineralisation using a one-pot process for MOF encapsulation and demonstrated its protective ability at elevated, industrially relevant temperatures and extended durations with three different enzymes, glucose oxidase (GOx) from Aspergillus niger, organophosphate degrading enzyme A (OpdA) from Agrobacterium radiobacter and α-chymotrypsin from bovine pancreas. Demonstrating the exceptional protective effect of biomimetic mineralized MOF encapsulation of enzymes, we probed the role of the MOF in improving the stability of ZIF-8@GOx at elevated temperatures using synchrotron X-ray scattering. Further, we demonstrated the advantage of MOF protected enzyme composites for ultrastable electrochemical biosensing.
MOFs, due to their porosity and large surface areas, as well as tuneable structures and functions, are considered one of the most promising matrices to immobilize enzymes for electrochemical sensing applications.8,10,26 As shown in Table 2, a number of MOF@enzyme based amperometric sensing applications have been reported, with most of them applying MOF@enzyme composites synthesised via the surface adsorption13,27–34 and pore-infilteration35–38 routes. The introduction of the in situ encapsulation strategy extends the application to utilize enzymes without any size restrictions. In addition, this approach has the added advantage of largely preventing any encapsulated enzyme leaching, thereby improving recyclability and the maintenance of enzymatic activity. Furthermore, with MOF mediated enzyme protection, we postulate good amperometric performance and operational stability of the ZIF-8@GOx composite in the presence of interference agents and at higher temperatures in an electrochemical environment. A few recent studies have investigated this methodology demonstrating good performance and linear response at low glucose levels.39–41 However, for their real-world application, sensors need to demonstrate a wide linear range to cater clinically relevant glucose concentrations and the more important higher concentrations in hyperglycemic patients. The recommended normal glucose levels are set within the 3.9 to 7.2 mmol L−1 range for fasting glucose measurement.42 We have used this as the target range to optimize the analytical performance of the ZIF-8@GOx electrochemical biosensor demonstrated in this work.
| MOF | Enzyme | Analyte | Linear range (μM) | LOD (μM) | Ref. |
|---|---|---|---|---|---|
| LOD: limit of detection; GOx: glucose oxidase; GDH: glucose dehydrogenase; Zn-TSA: zinc thiosalicylate or Zn(C7H4O2S); MWCNT: multiwall carbon nanotubes; Cyt C: cytochrome C; BDC: 1,4-benzene dicarboxylic acid; ted: triethyldiamine; BPA: bisphenol A; BPB: bisphenol B; An-ZIF-8: aminated ZIF-8; MP: methyl parathion; NiPd: nickel-palladium. | |||||
| Surface adsorption | |||||
| ZIF-7 | GDH | Glucose | 100–2000 | — | 13 |
| Zn-TSA | GOx | Glucose | 2.0–1022 | 0.8 | 27 |
| Zn-TSA | Myoglobin | Nitrite (NO2−) | 1.3–1660 | 0.5 | 27 |
| Zn-TSA | Myoglobin | H2O2 | 0.3–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.08 | 27 |
| MIL-100(Fe) | GOx | Glucose | 5.00–1400 | 5.0 | 28 |
| ZIF-67/MWCNT | HRP | H2O2 | 1.86–1050 | 0.11 | 29 |
| Co-TCPP | Cyt C | Nitrite (NO2−) | 3.5–2800 | 1.1 | 30 |
| Cu(BDC)(ted)0.5 | Tyrosinase | BPA | 0.05–3.0 | 0.013 | 31 |
| Cu(BDC)(ted)0.5 | Tyrosinase | BPB | 0.05–3.0 | 0.015 | 32 |
| ZIF-8 | Lipase | MP | 0.1–38 | 0.28 | 33 |
| An-ZIF-8 | Lipase | MP | 0.1–25 | 0.51 | 33 |
| Fe-MIL-88@hemin | GOx | Thrombin | 10−7–0.03 | 6.8 × 10−8 | 34 |
| Pore infiltration | |||||
| ZIF-8 | Cyt C | H2O2 | 90–3600 | — | 35 |
| PCN-333(Al) | HRP | H2O2 | 0.387–1725 | 0.127 | 36 |
| PCN-333(Fe) | MP-11 | H2O2 | 0.5–1500 | 0.09 | 37 |
| Tb-mesoMOF | MP-11 | H2O2 | 3.02–640 | 0.996 | 38 |
| In situ encapsulation | |||||
| ZIF-8 | GOx | Glucose | 1–1200; 1200–3600 | 0.333 | 39 |
| ZIF-8 (NiPd) | GOx | Glucose | 100–1700 | — | 40 |
| ZIF-8 | GOx | Glucose | 10–1555 | 2.2 | 41 |
| ZIF-8 | GOx | Glucose | 1000–10000 |
50 | This work |
| ZIF-8 | GOx | Glucose | 200–2000 | 10 | This work |
| ZIF-8 | GOx | Glucose | 20–200 | 10 | This work |
Herein, the novelty of combining MOF encapsulation and thermal stabilisation of enzymes with the application of electrochemical sensing is presented. Given the exceptional thermal and operational stability of MOF@enzyme composites, an ultra-stable proof-of-concept MOF based biosensor is fabricated which demonstrates a clinically relevant wide linear range to cater for normal adult target glucose concentrations for the first time.
2. Results and discussion
2.1. Biomimetic mineralisation and characterization of ZIF-8@GOx
Conventional MOF synthesis is performed using high temperatures, pressures and organic solvents. For the inherent fragile nature of biomolecules, they are unlikely to sustain these synthesis conditions. Post-synthesis MOF biomolecule adsorption and infiltration are the common approaches; however, they are restricted by the pore size and thereby limit the molecules that can be loaded. However, in this work we have used the process of biomimetic mineralisation wherein the presence of enzymes initiates the MOF self-assembly and growth around them via a rapid (seconds to minutes) single step requiring aqueous and ambient conditions.16,43 The representative schematic, Fig. 1a shows the biomimetic mineralised growth of ZIF-8 around the enzymes. The surface chemistry of an enzyme influences the biomimetic mineralisation process.16,43 Negatively charged proteins are postulated to increase the local concentration of precursors, facilitating prenucleation cluster formation of ZIF-8 around the biomacromolecules leading to controlled crystal formation.16,43 To study the mechanism of biomimetic mineralisation of proteins in detail, Kang et al. investigated bovine serum albumin (BSA) protein encapsulation within ZIF-8 forming a ZIF-8@BSA composite in situ on the SAXS beamline.16 Their work demonstrates the presence of mesopores with a radius of 3.5 ± 0.5 nm within the ZIF-8 which allows at least 1 nm radial spacing for dynamic movement of the guest molecule16 and perhaps an interaction with the substrate and resulting enzyme activity as performed for the assays in this work.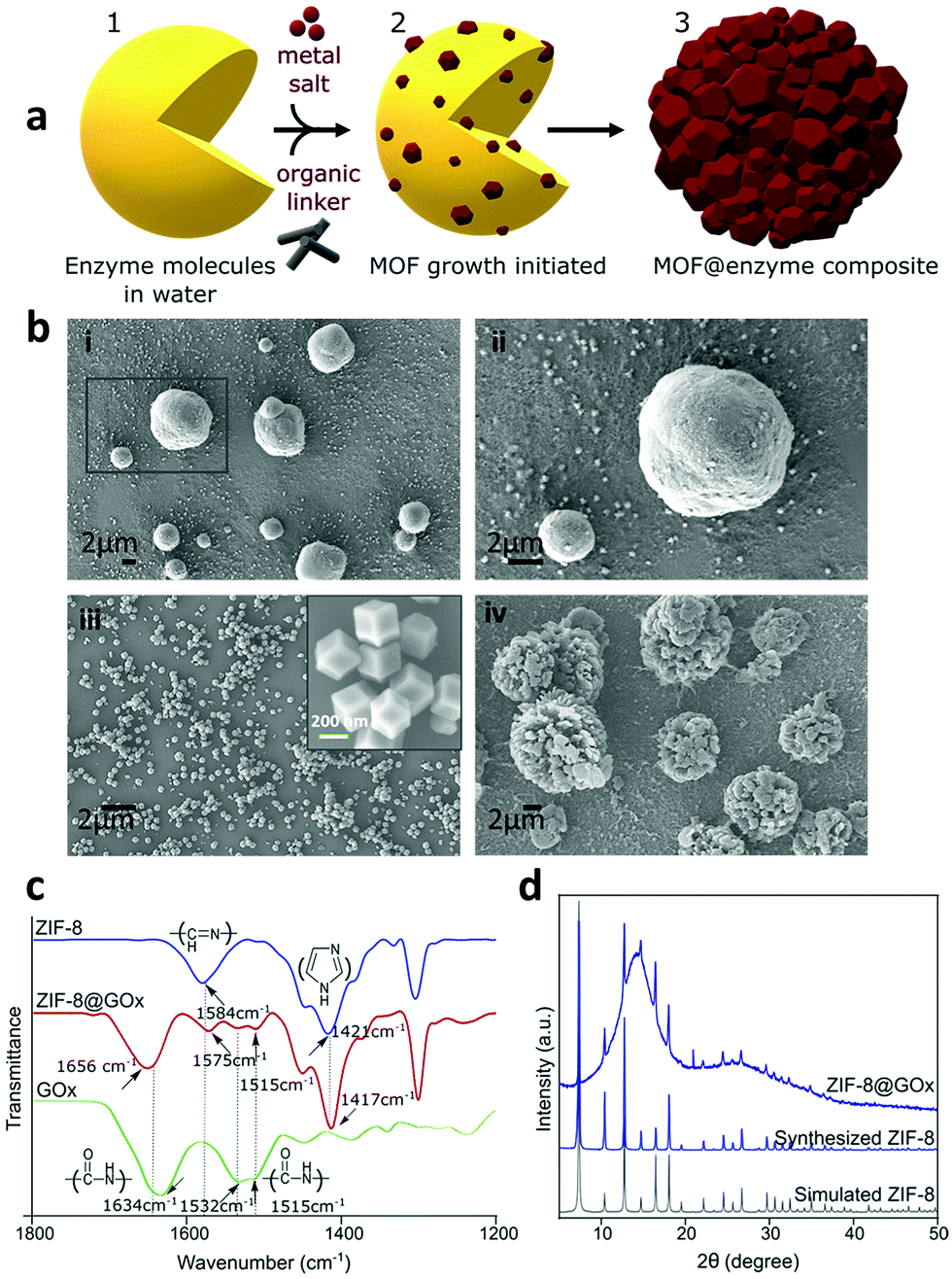 | ||
| Fig. 1 (a) Schematic representing the biomimetic mineralised growth of ZIF-8 on enzymes. Physicochemical characterization of ZIF-8@GOx; (b) SEM images; (i) and (ii) show the spherical morphology of ZIF-8@GOx with individual particles ranging 2–10 μm in size (scale - 2 μm); (iii) shows control ZIF-8 crystals with dodecahedron morphology and nearly 200 nm in size (scale - 2 μm), inset is a higher magnification image for ZIF-8 crystals (scale - 200 nm); (iv) shows flower-like clusters formed when ZIF-8 precursors (aq.) were mixed in the absence of GOx and left stirring for 24 h. (c) FTIR spectra for ZIF-8 (blue), ZIF-8@GOx (red) and GOx (green) confirming the encapsulation and (d) X-ray diffraction (XRD) patterns for ZIF-8 (blue) and ZIF-8@GOx (navy blue). | ||
First, we studied the biomimetic mineralisation of the enzyme GOx using the ZIF-8 MOF forming a ZIF-8@GOx composite. GOx is one of the most widely studied enzymes (160 KDa, pI 4.2, dimeric, ellipsoid structure) that catalyses the oxidation of β-D-glucose to gluconic acid with the simultaneous production of hydrogen peroxide.44 Further, we assessed the stabilisation of enzymes α-chymotrypsin and OpdA using this technique. OpdA (35 KDa, Tim barrel structure) is a phophotriestarase that hydrolyses shorter side chain organophosphate compounds.45,46 α-chymotrypsin (25 kDa, pI 8.76, β-barrel structure) is a digestive enzyme which hydrolyzses the peptide bonds leading to the breakdown of proteins into smaller peptides or amino acids.47 The three enzymes are of varied industrial interest, with GOx having a broad range of applications with most demand for biosensing and diagnostics. OpdA has been extensively tested for environmental sensing and bioremediation applications whilst chymotrypsin is sought after for its application in the food and detergent industry. The ZIF-8@GOx composite was prepared using aqueous medium and ambient conditions by adapting the method previously described by Kang et al.16,48
The scanning electron microscopy (SEM) images in Fig. 1b(i) and (ii) show the spherical morphology of the ZIF-8@GOx composite ranging between 2–10 μm in size along with the presence of some small nanometer sized dodecahedral crystals. Fig. 1b(iii) shows nanometer sized dodecahedral ZIF-8 (control) crystals formed using methanol and Fig. 1b(iv) shows spherical ‘flower-like’ clusters formed when the precursor solutions (aq.) were mixed in absence of GOx. Energy dispersive X-ray spectroscopy (EDS) analysis (ESI,† Fig. S1), indicates similar elemental composition for both ZIF-8 and ZIF-8@GOx samples. Fourier transform infrared spectroscopy (FTIR) (Fig. 1c) for the ZIF8@GOx shows the typical ZIF-8 absorption peaks at 1584 cm−1 corresponding to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– stretching of imidazole and at 1417 cm−1 corresponding to the imidazole ring stretching. In addition, it also exhibits absorption peaks at 1656 cm−1 and in the range 1510–1540 cm−1 corresponding to amide I and amide II bands of protein, respectively. Fig. 1d shows X-ray diffraction (XRD) patterns for synthesized ZIF-8 and that of ZIF-8@GOx which remains largely amorphous with less than 10% crystallinity. This is consistent with other studies that demonstrate the aqueous synthesis of ZIF-8 at a low molar ratio of precursors favours the formation of an amorphous material.49 To assess the porosity of the ZIF-8@GOx composite, nitrogen adsorption was measured. Brunauer–Emmett–Teller analysis showed a surface area of 205 m2 g−1 for ZIF-8@GOx compared to 1565 m2 g−1 for the pure ZIF-8 crystals (ESI,† Fig. S3). The water-based synthesis for ZIF-8@GOx rendering an amorphous composite and the presence of enzyme (approx. 30 wt%) are the contributing factors for the much lower surface area. This is consistent with previous work by Wu et al. which demonstrates that the amorphous ZIF-8@GOx is likely to be more mesoporous compared with the microporous ZIF-8 MOF and this leads to lower specific surface and thus low nitrogen sorption capacity.50 Determined by protein quantification assay, more than 95% of the GOx added in the synthesis co-precipitated with the MOF; however, the non-encapsulated and surface adsorbed GOx was removed in the washing steps. The specific activity of GOx in ZIF-8@GOx was measured to be 16.3 μmol mg−1 min−1 compared to the specific activity of 20.0 μmol mg−1 min−1 for GOx alone in our test conditions. Therefore, the resulting encapsulation efficiency of this process was 81.5%. Both the encapsulation process as well as a diffusion barrier to the substrate when the enzyme is in its encapsulated form, could contribute to this net loss in specific activity for the enzyme. Herein, encapsulation efficiency (ee) is defined by the ratio of the specific activities of the enzyme in the ZIF-8@GOx composite and the free GOx, i.e. encapsulation efficiency (%)
N– stretching of imidazole and at 1417 cm−1 corresponding to the imidazole ring stretching. In addition, it also exhibits absorption peaks at 1656 cm−1 and in the range 1510–1540 cm−1 corresponding to amide I and amide II bands of protein, respectively. Fig. 1d shows X-ray diffraction (XRD) patterns for synthesized ZIF-8 and that of ZIF-8@GOx which remains largely amorphous with less than 10% crystallinity. This is consistent with other studies that demonstrate the aqueous synthesis of ZIF-8 at a low molar ratio of precursors favours the formation of an amorphous material.49 To assess the porosity of the ZIF-8@GOx composite, nitrogen adsorption was measured. Brunauer–Emmett–Teller analysis showed a surface area of 205 m2 g−1 for ZIF-8@GOx compared to 1565 m2 g−1 for the pure ZIF-8 crystals (ESI,† Fig. S3). The water-based synthesis for ZIF-8@GOx rendering an amorphous composite and the presence of enzyme (approx. 30 wt%) are the contributing factors for the much lower surface area. This is consistent with previous work by Wu et al. which demonstrates that the amorphous ZIF-8@GOx is likely to be more mesoporous compared with the microporous ZIF-8 MOF and this leads to lower specific surface and thus low nitrogen sorption capacity.50 Determined by protein quantification assay, more than 95% of the GOx added in the synthesis co-precipitated with the MOF; however, the non-encapsulated and surface adsorbed GOx was removed in the washing steps. The specific activity of GOx in ZIF-8@GOx was measured to be 16.3 μmol mg−1 min−1 compared to the specific activity of 20.0 μmol mg−1 min−1 for GOx alone in our test conditions. Therefore, the resulting encapsulation efficiency of this process was 81.5%. Both the encapsulation process as well as a diffusion barrier to the substrate when the enzyme is in its encapsulated form, could contribute to this net loss in specific activity for the enzyme. Herein, encapsulation efficiency (ee) is defined by the ratio of the specific activities of the enzyme in the ZIF-8@GOx composite and the free GOx, i.e. encapsulation efficiency (%)
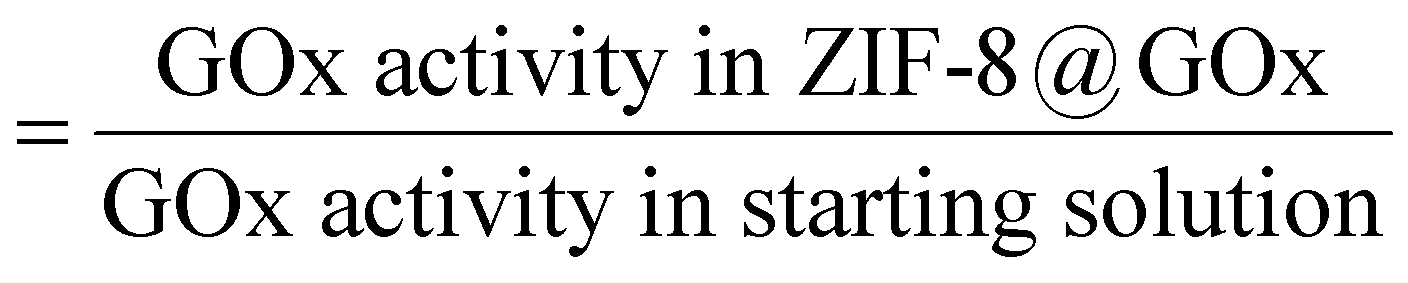
The ZIF-8@GOx composite had an enzyme loading of 30–34% (w/w). Herein, enzyme loading is defined by mass of GOx present per unit mass of the ZIF-8@GOx composite, i.e. enzyme loading (%)
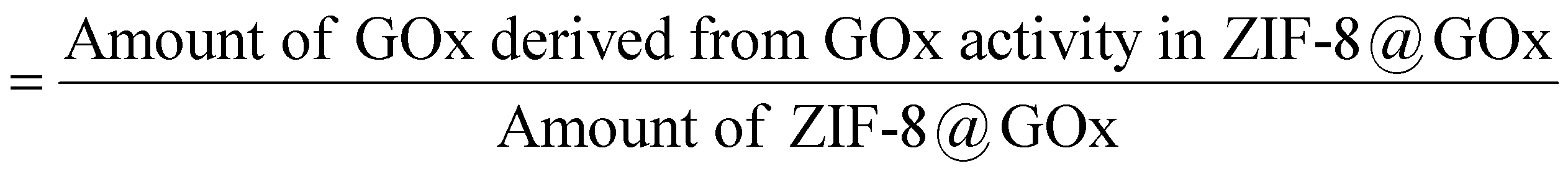
Both encapsulation efficiency and loading were determined using a colorimetric biochemical assay to assess GOx activity (ESI,† Fig. S2 and Table S1).
2.2. Understanding the thermal stabilisation of ZIF-8@GOx composites
The intrinsic stability of the MOF scaffolds including their chemical, thermal and mechanical stability, contributes to the resulting performance and stability of the MOF-enzyme composite.51 To date, zeolitic imidazolate frameworks (ZIFs), are the most widely explored MOFs for enzyme encapsulation using the biomimetic mineralisation route.52 Park et al. studied twelve ZIFs (ZIF-1–12) and of these, ZIF-8 demonstrated the highest stability with permanent porosity, thermal stability up to 550 °C, and extraordinary chemical resistance to treatment such as boiling alkaline water and organic solvents over a period of at least 1 week.53 Therefore, due to its exceptional chemical and thermal stability and further good resistance to degradation in water,53,54 ZIF-8 was chosen as a favourable candidate to investigate the thermal stabilisation of enzymes.In order to determine the stability of ZIF-8@GOx, the enzymatic activity was tested with an exposure (to thermal treatment) time (te) of 5 min and 10 min at temperatures ranging from 45 °C to 80 °C. For comparison, the initial enzymatic activity for GOx and ZIF-8@GOx, respectively were normalised. Fig. 2a shows that following incubation at 45 °C, free enzyme lost 30% of its starting activity while ZIF-8@GOx retained (100%) activity. Free GOx had a residual activity of only 7% and 16% after incubation for 10 min and 5 min at 70 °C, respectively. ZIF-8@GOx retained more than 80% activity at this temperature and demonstrated residual activity of more than 42% at 75 °C before degradation at 80 °C. These results demonstrate the significant protective ability of the MOF encapsulation. We postulate that the framework supports the enzyme at higher temperatures, preventing the proteins from unfolding and denaturing.55
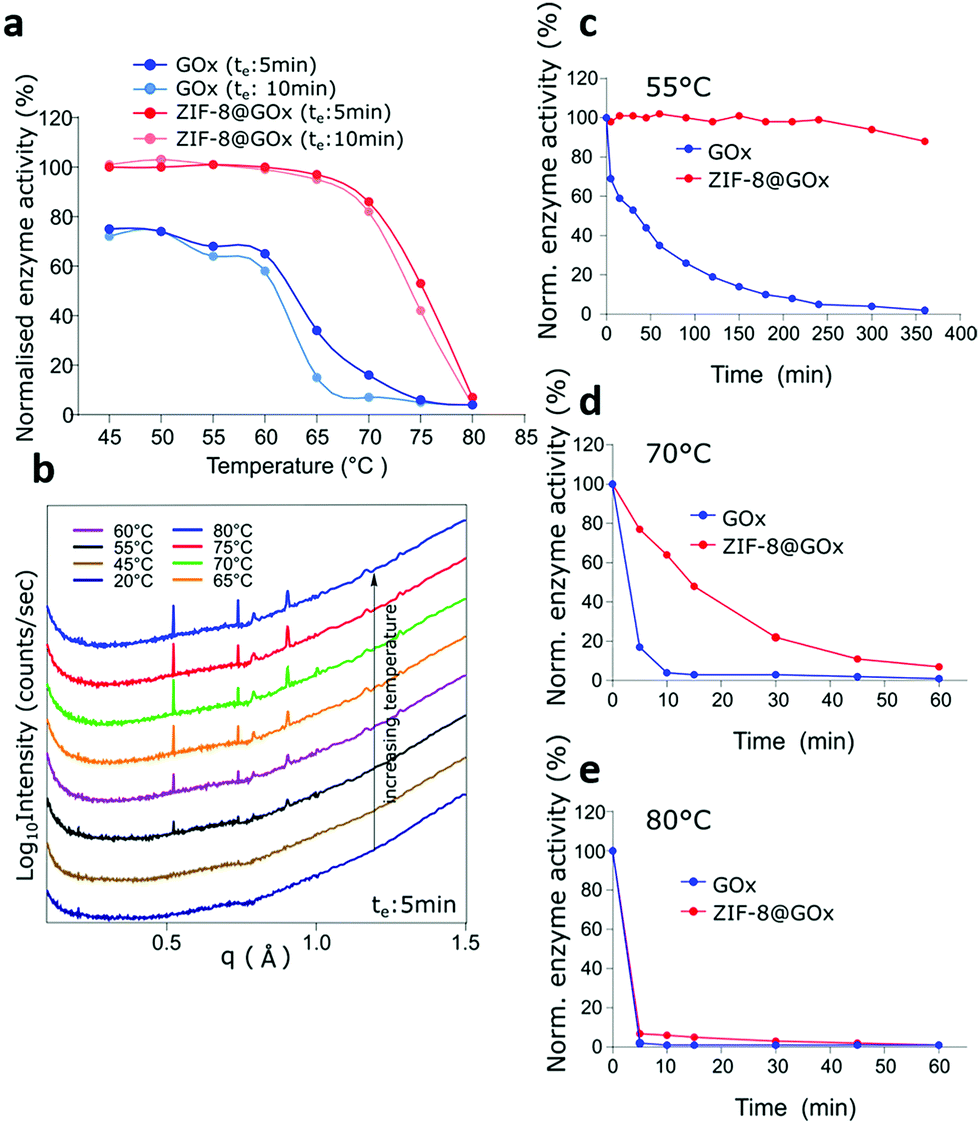 | ||
| Fig. 2 Thermostability of GOx and ZIF-8@GOx; (a) GOx and ZIF-8@GOx samples (in Tris-buffer, 50 mM, pH 8.0) were subjected to increasing temperatures and incubated for an effective duration (te) of 5 min and 10 min each. Results indicate residual activity (%) of GOx and ZIF-8@GOx normalised to their activity measured in Tris Buffer (50 mM, pH 8.0) at 25 °C, respectively, (b) in situ SAXS/WAXS analysis of change in ZIF-8@GOx structure (freshly prepared, stored in water) with increasing temperature. (c–e) show the time resolved enzymatic activity measurements of GOx and ZIF-8@GOx after their thermal treatment at (c) 55 °C, (d) 70 °C and (e) 80 °C, respectively. | ||
To study the interactions and structural changes in the ZIF-8@GOx composite with increasing temperature more closely, we freshly prepared an aqueous dispersion of ZIF-8@GOx composite. This was then placed in a quartz capillary and heated in situ using the same thermal profile that we investigated before, i.e. a stepwise increase from 20 °C to 80 °C with an effective duration, te of 5 minutes, at the SAXS/WAXS beamline (Australian Synchrotron). Confirming our PXRD data, the SAXS/WAXS spectra (Fig. 2b), showed that the freshly prepared ZIF-8 framework is initially in an amorphous state dispersed in an aqueous medium in the capillary. However, as we started heating, the ZIF-8@GOx began to crystalize at 45 °C and remained crystalline at 80 °C. This crystallisation of the ZIF structure at higher temperatures, offering a more stable framework might be a contributing factor to the enhanced protective nature of the MOFs at these temperatures. We postulate that the increase in temperature reduces the water content leading to a more crystalline, tightly packed structure around the enzyme with higher thermal stability. Carraro et al. have recently demonstrated that the crystalline ZIF-8@BSA composites have a slower dissolution profile compared to the amorphous counterparts indicating higher stability of the crystalline ZIF-8 phase.56 To better represent industrial catalytic reactions that run for hours we monitored the protective ability of the ZIF-8@GOx systems in preserving GOx enzymatic activity for up to 6 h at 55 °C, 70 °C and 80 °C, respectively. It is important to note that herein, we performed specific enzymatic assays rather than a general protein quantification technique because the specific assays confirm the secondary and tertiary structural integrity and the inferred catalytic functionality of the enzyme. Fig. 2c shows that after 6 h of incubation at 55 °C, ZIF-8@GOx still retained 90% of its initial activity while the free GOx almost lost all its activity. At 70 °C (Fig. 2d), the free enzyme lost 96% of its activity after 10 min while ZIF-8@GOx lost 36%. While GOx nearly lost all its activity in 10 min, ZIF-8@GOx had a more extended profile, with 77% of residual activity at 10 min decaying to 7% residual activity after 1 h, showing significant protective abilities at elevated temperatures. Fig. 2e shows that the ZIF-8@GOx offered no protection at 80 °C as both the free and the encapsulated GOx lost their activity within 5 min of thermal stress.
2.3. Biomimetic mineralisation and thermal stabilisation of OpdA and α-chymotrypsin
We further investigated if other structurally diverse proteins can be biomimetically-mineralised and if the encapsulation technology offers similar thermal stabilisation effect while facilitating size-selective transport of the substrates to these proteins. It is known that the surface chemistry of the enzymes influences the biomineralisation process. Enzymes with lower isoelectric points (pI) are net negatively charged in neutral pH conditions. This promotes local concentration of positively charged metal ions and thereby the organic ligands to initiate the biomineralisation by facilitating prenucleation clusters of the MOF around the biomacromolecules.16,57 Chemical surface modification has been suggested as a general strategy to facilitate biomimetic mineralisation of higher pI proteins57 such as α-chymotrypsin. However, we have applied a co-encapsulation approach, MOF encapsulating OpdA and α-chymotrypsin with BSA to avoid any chemical modifications to these natural enzymes. Encapsulation of OpdA was investigated in the form of its commercial landguard™ preparation while α-chymotrypsin was prepared as a 50% (w/w) mixture with BSA to enable its encapsulation. OpdA hydrolyses organophosphates, such as the pesticide methyl parathion, therefore making it an important enzyme for environmental remediation. Stable immobilization of such enzymes is important for using it in industrial applications such as water remediation. The specific activity of OpdA in its free form was 16 μmol−1 mg min−1 while in the encapsulated form, ZIF-8@OpdA measured 15.6 μmol mg−1 min−1 resulting in a 97.5% encapsulation efficiency using the biomimetic mineralisation technique. OpdA and ZIF-8@OpdA samples were incubated in a water bath at 60 °C. Fig. 3a shows that ZIF-8@OpdA retained more than 95% enzymatic activity after 5 h at 60 °C while the free OpdA lost 98% of its activity over this time.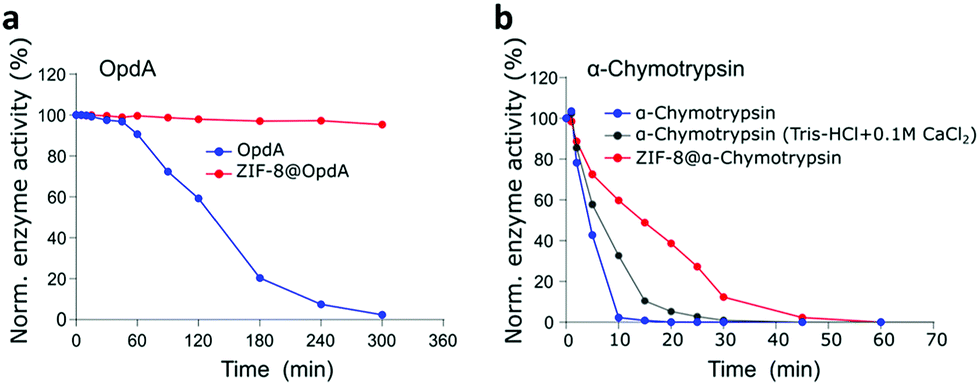 | ||
| Fig. 3 (a) Residual enzyme activity of OpdA (blue) and ZIF-8@OpdA (red) in Tris, 50 mM, pH 8.0 for 5 h at 60 °C, normalised to their activity measured at 25 °C, respectively. (b) Residual enzyme activity of α-chymotrypsin in the presence of Ca2+ (grey), α-chymotrypsin in Tris Buffer, 50 mM, pH 8.0 (blue), and ZIF-8@α-chymotrypsin (red) Tris-buffer (50 mM, pH 8.0) at 80 °C. | ||
Proteolytic enzymes are of extensive interest in various industrial sectors such as detergent, food processing, pharmaceutical, leather and waste management and account for nearly 40% of the total enzyme sales in various industrial market sectors.46,58 These applications suggest their industrial utility is subject to stability at much higher temperatures. α-Chymotrypsin is an expensive serine-protease with very low operational stability which is why it has been widely investigated for immobilisation using natural and synthetic polymers and other novel systems such as single-enzyme nanoparticles.59 We successfully encapsulated α-chymotrypsin (50% w/w mixture with BSA) using biomimetic mineralisation, to give ZIF-8@α-chymotrypsin. The specific enzyme activity for α-chymotrypsin was 8 μmol mg−1 min−1 while it measured 7.4 μmol mg−1 min−1 for the ZIF-8@α-chymotrypsin composite, resulting in 92.5% encapsulation efficiency for α-chymotrypsin. The thermal stability of this composite was assessed at 80 °C against free α-chymotrypsin (Tris-buffer) and the α-chymotrypsin stabilised using a calcium salt.60Fig. 3b shows that while the free α-chymotrypsin lost almost all its activity within the first 10 min of exposure to heat, the presence of calcium stabilised α-chymotrypsin which retained 58% of its activity and the MOF encapsulation performed best with ZIF-8@α-chymotrypsin retaining 73% of its initial activity for this time period. While α-chymotrypsin in the free and the calcium stabilised form lost 50% of its activity within 5 to 7 min, ZIF-8@α-chymotrypsin retained 50% of its activity for 15 min which could possibly benefit its present application in laundry detergents, to be used for a more efficient, quick, high temperature wash cycle. It is interesting to observe that the three enzymes, GOx, OpdA and α-chymotrypsin were stabilised to different degrees by ZIF-8. The extent of thermostabilisation of each MOF-enzyme could be a factor of the physiochemical properties of both the MOF and the enzyme and thereby governed by several kinds of interactions such as hydrogen bonding, van der Waals forces, coordination and possibly covalent bonding with the organic and inorganic components of the MOF. A recent report by Liang et al. highlights this by demonstrating the importance of the chemistry (hydrophilicity) of the ZIF and at the ZIF/biointerface in facilitating and preserving the activity of encapsulated enzymes.61
2.4. Progress towards electrochemical biosensing devices
 | ||
| Fig. 4 Thermal stabilisation assessment as an electrochemical sensor of ZIF-8@GOx (a) and GOx modified GC electrodes at (b) room temperature, (c) 55 °C and (d) 70 °C for different periods of time. Applied potential +0.4 V (vs. Ag/AgCl), 4 mM glucose, 0.1 M Tris-HCl buffer (pH 7.4). | ||
Following incubation, electrochemical measurements were performed at room temperature by applying a fixed potential of 0.4 V vs. the Ag/AgCl reference electrode under stirring conditions. A clinically relevant blood glucose concentration of 4 mM was used in the solution.64 At room temperature, the ZIF-8@GOx modified electrode shows very good stability upon storage in Tris-buffer by retaining more than 75% activity after storage for 240 h (Fig. 4b). While the control electrode lost more than 65% of its activity after 27 h of storage under similar conditions. At 55 °C, the ZIF-8@GOx modified electrode showed higher stability by retaining 83% activity after 1 h, 40% activity after 5 h and 15% activity after 10 h of storage in Tris-buffer at 55 °C (Fig. 4c). Whereas the control GOx coated electrode retained only 19%, 5% and 1% activity after storage for 1, 5 and 10 h, respectively, under similar conditions. Finally, the electrodes were assessed at an even higher temperature of 70 °C where again the ZIF-8@GOx modified electrode demonstrated higher thermal stability compared to the control electrode as shown in Fig. 4d. The ZIF-8@GOx modified electrode retained more than 50% of its activity after storage at this temperature for 1 h while the control electrode lost most of the activity within the hour. In addition, the ZIF-8@GOx modified electrode was still active even after 6 h of storage at this high temperature reflecting good protection of the enzyme against thermal stress because of encapsulation inside the ZIF-8 MOF. Comparing Fig. 2c and 4c, we see a marked difference in the thermal stability imparted by ZIF-8 to GOx at 55 °C. It should be noted that for electrochemical analysis, a single electrode was used i.e. tested and retained in the test temperature storage conditions for the duration of the entire experiment. The encapsulated GOx shows excellent thermal resistance despite the repeated electrochemical potential application which would also have a destabilisation effect on the enzyme and the ZIF-8@GOx composite.
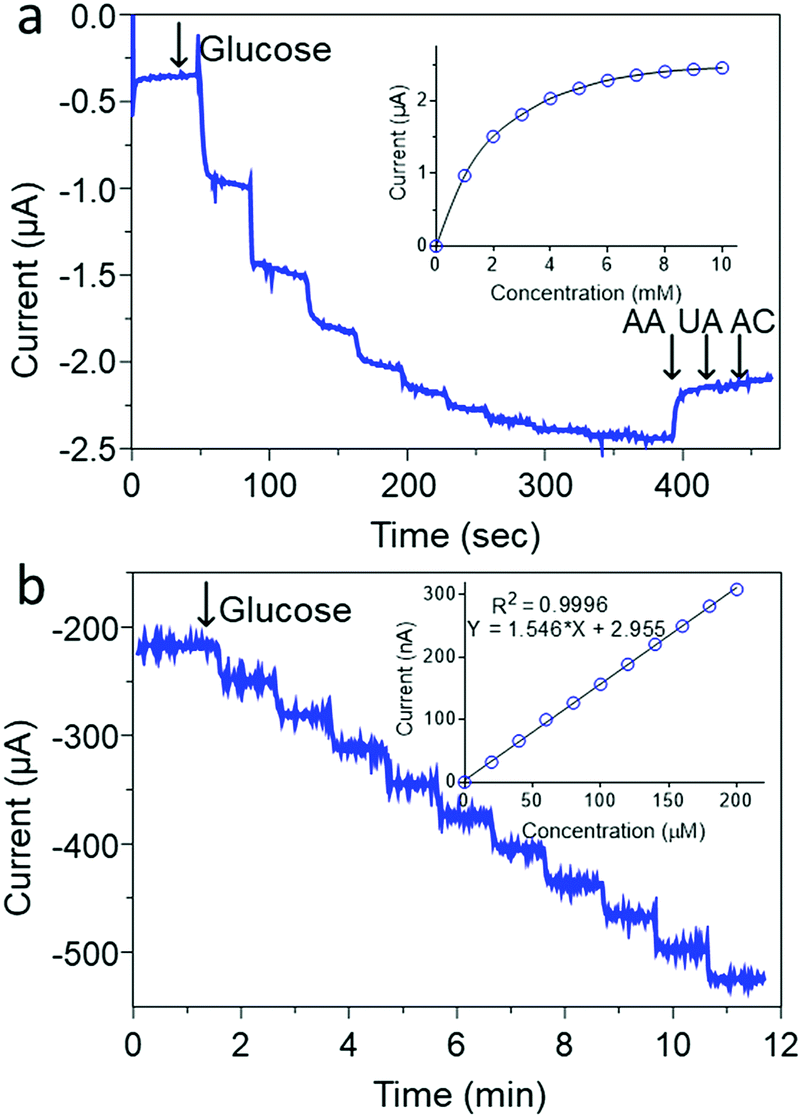 | ||
| Fig. 5 (a) Current–time recordings of successive 1 mM additions of glucose at the GC/MW/PB/ZIF-8@GOx measured at −0.05 V (vs. Ag/AgCl) and followed with the addition of 0.1 mM ascorbic acid (AA), uric acid (UA) and acetaminophen (AC) with its corresponding calibration curve (inset); (b) Current–time recordings of successive 20 μM additions of glucose at the GC/MW/PB/ZIF-8@GOx measured at −0.05 V (vs. Ag/AgCl) and inset is the corresponding calibration curve. Supporting electrolyte; 100 mM Tris-HCL (pH 7.4). | ||
Fig. 5b shows that as prepared, the ZIF-8@GOx biosensor had a very limited glucose linear range of 20–200 μM (r2 = 0.999) and a limit of detection (LOD) of 10 μM (S/N = 3) using a 100 mM Tris buffer (pH 7.4). This limited linear range could be arising from the low oxygen concentration in the Tris buffer at this pH.65 In addition, the use of a lower electrolyte pH is also important to maintain a stable PB coating as has been reported earlier.66 Therefore, to improve the detection linear range, a 50 mM Tris buffer (pH 6.0) was used as shown in Fig. 6a. Under the new measurement conditions, the linear range was improved by 10 fold from 200 to 2000 μM (r2 = 0.999) and a LOD of 10 μM (S/N = 3). Further improvement in the linear range was achieved by using 1 mM 1,4-Benzoquinone (BQ) in the electrolyte as shown in Fig. 6b. In the presence of this redox mediator, it was possible to get a wide linear response from 1 to 10 mM glucose (r2 = 0.999) and LOD of 0.05 mM. The use of BQ is well known to support glucose detection especially in environments with deficient or variable oxygen content.67 Overall, the biosensor shows good reproducibility for successive measurements of 4 mM glucose with an RSD of <2% for more than 5 measurements. The electrode preparation was also reproducible and was found to have an RSD of <5% using 4 mM glucose for 5 different electrodes.
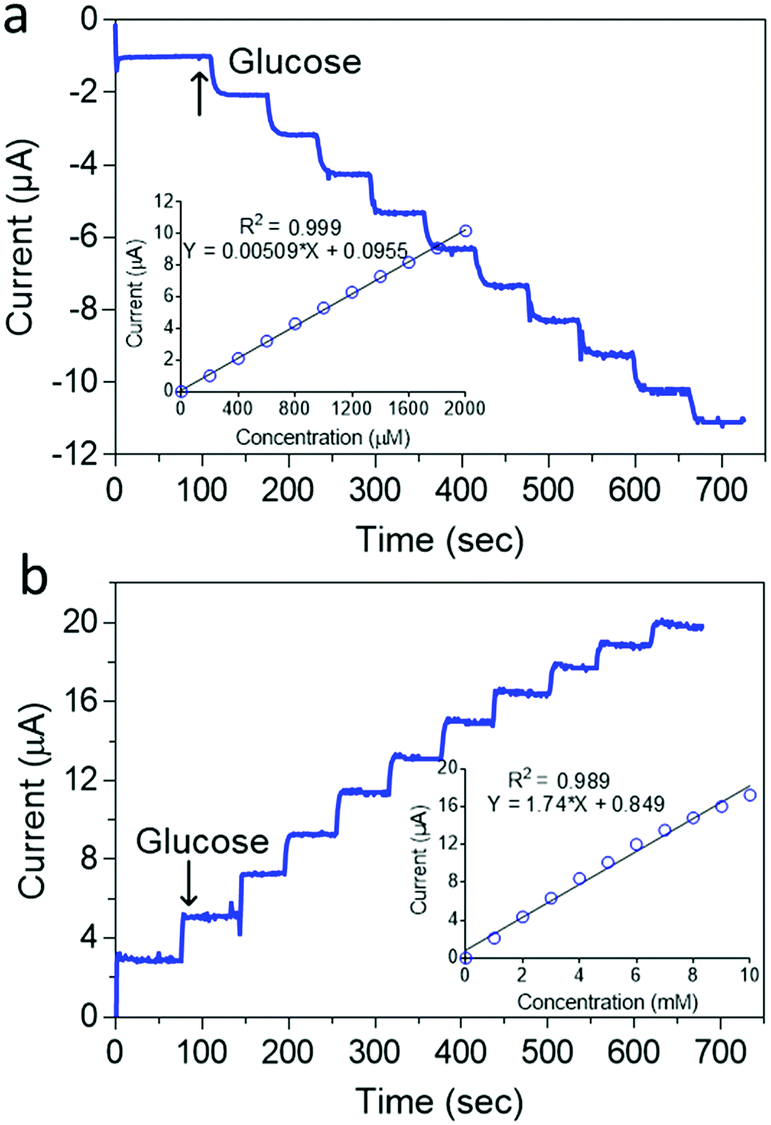 | ||
| Fig. 6 (a) Current–time recordings of successive 200 μM additions of glucose at the GC/MW/PB/ZIF-8@GOx measured at −0.05 V (vs. Ag/AgCl) with the corresponding calibration curve (inset). Supporting electrolyte; 0.05 M Tris-HCl (pH 6.0); (b) Current–time recordings of successive 1 mM additions of glucose at the GC/MW/PB/ZIF-8@GOx measured at 0.3 V (vs. Ag/AgCl) with the corresponding calibration curve (inset). Supporting electrolyte; 0.05 M Tris-HCl (pH 6.0) containing 1 mM 1,4-benzoquinone. | ||
3. Conclusions
In conclusion, we have demonstrated the MOF bio-encapsulation technique for a range of enzymes and their enhanced thermostability compared with the free enzyme counterparts. ZIF-8 is a porous and crystalline framework that assembles efficiently around the enzymes at room temperature and in water and the bio-mimetic encapsulation technology shows a lot of promise for the protection of enzymes at high temperatures which makes it a viable immobilization technique for industrial catalysis. Having confirmed the thermal stabilisation of enzymes, we investigated this encapsulation technology to modify and test a ZIF-8@GOx electrochemical biosensor at challenging elevated temperatures. The results corroborate our claims for the suitability of this set-up for applications like electrochemical biosensing, micro-array and high-powered biofuel-cell device fabrication. Furthermore, the ultra-stable biosensor also demonstrates good sensitivity in the presence of serum and common interference agents at high concentrations. The analytical performance shows a wide linear range and detection limits which are ideal for clinical glucose levels in biological samples. Here we demonstrated the complexity typically associated with enzyme stability and its spatial confinement using biomimetic mineralisation forming a protective and porous MOF around the enzymes. This enabled superior functionality and reusability of enzymes and the fabrication of a robust amperometric glucose biosensor for biomedical applications particularly for diagnosis of symptoms associated with disease like diabetes mellitus.4. Experimental
4.1. Materials
All materials including 2-methylimidazole (Sigma cat# M50850), Zinc acetate dihydrate (Chem Supply cat# ZA001), Glucose oxidase (GOx, Sigma cat# G7141), BSA (Sigma cat# A7030, 98%), β-D-Glucose (VWR cat# 101174Y), HRP (Sigma cat# P8250), O-Dianisidine (Sigma cat# D3252), α-chymotrypsin (Sigma cat# C7762), and Tris (Sigma cat# T1503) were obtained commercially and used as received. Multiwalled carbon nanotubes (MWCNT) (95%, L: 0.5–2 μm, OD: 20–30 nm, ID: 5–10 nm) were purchased from Nanostructured & Amorphous Materials, Inc. (NanoAmor, NM, USA). Nafion 20% w/v was purchased from Ion power (USA). Palladium chloride was from Alfa-aser. All solutions were prepared using milliQ water. MilliQ water was used throughout the work.4.2. Preparation of standard ZIF-8
Separate solutions of 2-methylimidazole (160 mM, 525.2 mg) and zinc acetate dihydrate (40 mM, 351.2 mg) were each prepared in 40 mL of methanol at room temperature, respectively. The two solutions were mixed by agitation for about 20 s and left to sit at room temperature ageing over a period of 24 h to grow ZIF-8 crystals. ZIF-8 crystals were then collected using centrifugation at 5000 g for 10 min and washed 3 times with methanol.4.3. ZIF-8 encapsulated GOx synthesis
GOx was dissolved in 2-methylimidazole solution (10 mL, 160 mM in water) at 2 mg mL−1. Upon full solubilisation, a small aliquot (i.e. 0.1 mL) of the solution was removed and added to 0.9 mL of phosphate buffer (0.1 M, pH 6.0, 1 mg mL−1 BSA) for GOx activity determination. To the remaining GOx solution, an equal volume of zinc acetate solution (40 mM in water) was added while stirring, resulting in immediate flocculation which was left to precipitate. The solution was left to stir for 30 min at room temperature and then centrifuged at 3000 g for 5 min in a swing rotor centrifuge to pellet the ZIF-8 encapsulated GOx. The supernatant was removed and kept for GOx activity and protein determination. The pellet was washed once by resuspending in water and centrifuged again as above. It was then resuspended in 50 mM Tris buffer, pH 7.4 at 2 mg mL−1 GOx protein and stored at 4 °C until further analysis. To note that some recent reports have suggested low stability of ZIF-8 in common buffers including PBS.68 In the absence of GOx, when only the precursor solutions are mixed together, no precipitation was observed in the 30 min reaction time. However, when left stirring over a period of 24 h, a small amount of precipitate formed. We collected this precipitate for analysis. Control ZIF-8 crystals were prepared using HmIm and ZnAc solutions prepared in methanol as described above. ZIF-8@OpdA and ZIF-8@α-chymotrypsin were also prepared using a similar method to ZIF-8@GOx. For ZIF-8@OpdA, OpdA (landgaurd™) protein was dissolved at a 10 mg mL−1 concentration in a 2-methylimidazole solution (10 mL, 160 mM in water) followed by addition of an equal volume of zinc acetate solution (40 mM in water). This led to immediate flocculation and gradual precipitation. The solution was centrifuged and post-processing was performed as described above for ZIF-8@GOx. For ZIF-8@α-chymotrypsin, an 8 mg mL−1 protein solution containing 4 mg mL−1 of BSA and 4 mg mL−1 of α-chymotrypsin was prepared in the 2-methylimidazole solution following a method identical to that for ZIF-8@GOx.4.4. GOx activity assay
All GOx and ZIF-8@GOx solutions were prepared and kept on ice. GOx activity was determined spectrophotometrically. The assay is based on GOx catalysed oxidation of glucose to give hydrogen peroxide which further oxidises dianisidine in the presence of horse radish peroxidase to a coloured product which was monitored at 460 nm.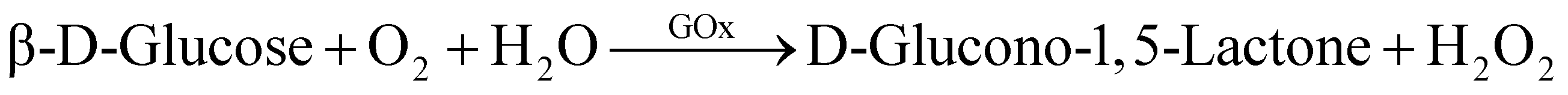
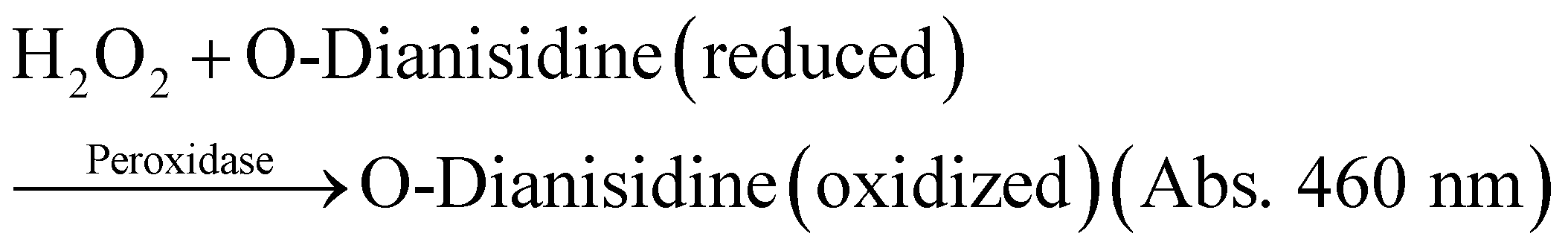
The assay was performed by addition of a 20 μL aliquot of the test sample, GOx or ZIF-8@GOx (dispersed in Tris buffer pH 7.4) in 980 μL of the following assay buffer: 100 mM phosphate buffer (pH 6.0), 10 mM glucose, 0.2 mM O-Dianisidine, 2.5 units of HRP, 1 mg mL−1 BSA that had been equilibrated to room temperature (20–21 °C). The reactions were initiated as soon as we added the aliquot of soluble GOx or ZIF-8@GOx, and the absorbance was read at 460 nm after 4 min of incubation.
4.5. Estimation of encapsulation efficiency
The standard GOx assay was used to estimate the enzyme encapsulation efficiency of the MOF@enzyme biomimetic mineralisation process. Herein, encapsulation efficiency is defined by the fraction of GOx added in the synthesis that was actually encapsulated inside the resulting ZIF-8@GOx composite i.e. encapsulation efficiency (%)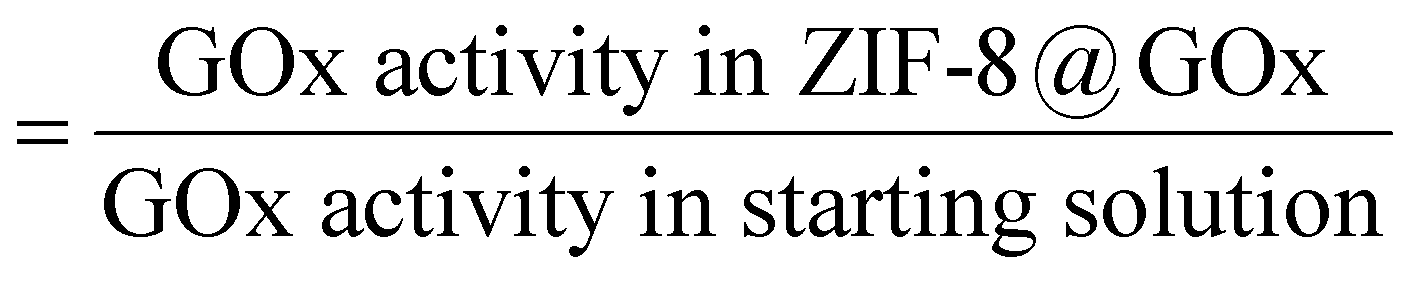
To estimate encapsulation efficiency, upon ZIF-8@GOx synthesis and post centrifugation, the GOx activity of the supernatant and the wash solution (unencapsulated enzyme) were used as an indirect measure of the encapsulated enzyme.
4.6. Estimation of GOx loading
GOx loading in the composite is defined by mass of GOx present per unit mass of the ZIF-8@GOx composite. To estimate loading, the GOx activity in a known amount of ZIF-8@GOx pellet was estimated using the standard assay. The activity was correlated with the amount of GOx using a GOx standard curve at 460 nm. The amount of GOx and the total amount of ZIF-8@GOx composite were used to derive the loading% using the given equation: Loading (%)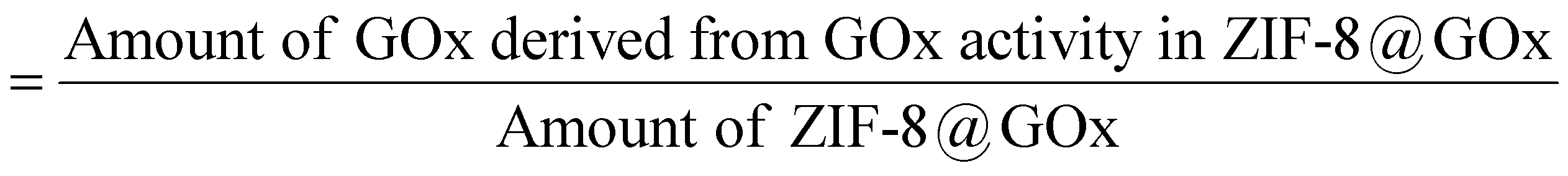
4.7. OpdA activity assay
The OpdA assay was prepared as described in the literature. Methyl parathion was used as the substrate for the OpdA activity assay, which was hydrolysed by the enzyme to form para-nitrophenol, which is yellow under alkaline conditions and quantified spectrophotometrically at 405 nm. ZIF-8@OpdA (20 μL) in Tris buffer (pH 7) was added to 980 μL of the assay buffer (100 mM Tris, pH 8.0, 0.25 mM methyl parathion). After 4 minutes at 20 °C, the absorbance was read at 405 nm.4.8. α-Chymotrypsin assay
An esterolytic activity of the enzyme was used as a measure to assess its activity, using N-benzoyl-L-tyrosine ethyl ester as the substrate. The enzyme hydrolyses the substrate to N-benzoyl-L-tyrosine which can be detected at 256 nm. ZIF-8 encapsulated α-chymotrypsin (20 μL) in milliQ water was added to 980 μL of the assay buffer (38 mM Tris pH 7.8, 0.55 mM N-benzoyl-L-tyrosine ethyl ester, 30% (v/v) methanol, 53 mM CaCl2, 0.03 mM HCl). After 4 minutes at 20 °C, 100 μL was removed from the assay and mixed with 1 mL of 1 M Na2CO3 before reading the absorbance.4.9. GOx, OpdA and α-chymotrypsin thermostability studies
ZIF-8@GOx, ZIF-8@OpdA and ZIF-8@α-chymotrypsin were tested for their thermal stability in comparison with the free enzymes GOx, OpdA and α-chymotrypsin, respectively. Aliquoted GOx (2 mg mL−1 in Tris buffer), OpdA (10 mg mL−1 in Tris buffer), α-chymotrypsin (8 mg mL−1 in Tris buffer and standard stabilising medium: Tris-HCl and 0.1 M CaCl2) and ZIF-8@GOx, ZIF-8@OpdA and ZIF-8@α-chymotrypsin samples (in Tris buffer) were incubated in a water bath at different temperatures ranging from room temperature (RT) to 80 °C. At regular time intervals, samples were removed in triplicate and tested for their remanent enzyme activity using the standard GOx, OpdA and α-chymotrypsin enzyme assay (Section 2.4–2.6). Activity was normalised to the starting enzymatic activity of each enzyme at t = 0.4.10. Physicochemical characterisation
4.11. Electrochemical testing
4.12. Electrochemical apparatus
Amperometric and cyclic voltammetry measurements were performed using a computer-controlled Autolab PGSTAT302N potentiostat (Ecochemie, NL) with a three-electrode configuration. The ZIF-8-GOx/Pd/MWCNT modified electrode was used as the working electrode, Ag/AgCl was used as the reference electrode (Model RE-5B, MF-2079 Bioanalytical Systems Inc. (BASi), West Lafayette, IN 47906, USA), and a platinum wire was used as the counter electrode. All three electrodes were inserted into a 20 mL glass cell (Amber glass, homemade) through holes in its Teflon cover.4.13. Electrochemical testing
For thermal stability assessment, electrodes were kept in 0.1 M Tris-HCl (pH 7.4) buffer and subjected to different temperatures for different periods of time. Then electrochemical measurement was performed at room temperature by applying a fixed potential of 0.4 V vs. the Ag/AgCl reference electrode under stirring conditions. Glucose solution was added after allowing the transient baseline current to decay to a steady-state value.Author contributions
R. S and M. M. contributed equally to the manuscript. The manuscript was written with the contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CMD acknowledges the Australian Research Council (ARC) [DE140101359] and the Veski Inspiring Women fellowship for support. This research in part was undertaken on the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO.References
- T. W. Hanks and G. F. Swiegers, in Bioinspiration and Biomimicry in Chemistry: Reverse-Engineering Nature, ed. G. F. Swiegers, Hoboken, NJ, John Wiley & Sons, 2012, pp. 1–15 Search PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS.
- P. Fernandes, Enzyme Res., 2010, 2010 Search PubMed.
- (a) L. Jiao, J. Y. R. Seow, W. S. Skinner, Z. U. Wang and H.-L. Jiang, Mater. Today, 2019, 27, 43–68 CrossRef CAS; (b) H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- (a) H.-S. Wang, Coord. Chem. Rev., 2017, 349, 139–155 CrossRef CAS; (b) Y. Chen and S. Ma, Dalton Trans, 2016, 45, 9744–9753 RSC; (c) A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC; (d) F. Y. Yi, D. Chen, M. K. Wu, L. Han and H. L. Jiang, ChemPlusChem, 2016, 81, 675–690 CrossRef CAS PubMed; (e) I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC; (f) K. Lu, T. Aung, N. Guo, R. Weichselbaum and W. Lin, Adv. Mater., 2018, 30, 1707634 CrossRef PubMed; (g) J. F. Olorunyomi, M. M. Sadiq, M. Batten, K. Konstas, D. Chen, C. M. Doherty and R. A. Caruso, Adv. Opt. Mater., 2020, 8, 2000961 CrossRef CAS.
- Q. Qiu, H. Chen, Y. Wang and Y. Ying, Coord. Chem. Rev., 2019, 387, 60–78 CrossRef CAS.
- (a) C. M. Doherty, D. Buso, A. J. Hill, S. Furukawa, S. Kitagawa and P. Falcaro, Acc. Chem. Res., 2014, 47, 396–405 CrossRef CAS PubMed; (b) J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30–40 CrossRef CAS; (c) M. B. Majewski, A. J. Howarth, P. Li, M. R. Wasielewski, J. T. Hupp and O. K. Farha, CrystEngComm, 2017, 19, 4082–4091 RSC.
- S. Liang, X.-L. Wu, J. Xiong, M.-H. Zong and W.-Y. Lou, Coord. Chem. Rev., 2020, 406, 213149 CrossRef CAS.
- P. Li, J. A. Modica, A. J. Howarth, E. Vargas, P. Z. Moghadam, R. Q. Snurr, M. Mrksich, J. T. Hupp and O. K. Farha, Chem, 2016, 1, 154–169 CAS.
- (a) T. J. Pisklak, M. Macías, D. H. Coutinho, R. S. Huang and K. J. Balkus, Top. Catal., 2006, 38, 269–278 CrossRef CAS; (b) Y. Cao, Z. Wu, T. Wang, Y. Xiao, Q. Huo and Y. Liu, Dalton Trans., 2016, 45, 6998–7003 RSC; (c) S. Jung, Y. Kim, S.-J. Kim, T.-H. Kwon, S. Huh and S. Park, Chem. Comm., 2011, 47, 2904–2906 RSC; (d) C. M. Doherty, G. Grenci, R. Riccò, J. I. Mardel, J. Reboul, S. S. Furukawa, S. Kitagawa, A. J. Hill and P. Falcaro, Adv. Mater., 2013, 25, 4701–4705 CrossRef CAS PubMed; (e) W. Xu, L. Jiao, H. Yan, Y. Wu, L. Chen, W. Gu, D. Du, Y. Lin and C. Zhu, ACS Appl. Mater. Interfaces, 2019, 11, 22096–22101 CrossRef CAS PubMed; (f) A. Zare, A.-K. Bordbar, F. Jafarian and S. Tangestaninejad, J. Mol. Liq., 2018, 254, 137–144 CrossRef CAS.
- W. Ma, Q. Jiang, P. Yu, L. Yang and L. Mao, Anal. Chem., 2013, 85, 7550–7557 CrossRef CAS PubMed.
- (a) V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed; (b) X. Liu, W. Qi, Y. Wang, R. Su and Z. He, Nanoscale, 2017, 9, 17561–17570 RSC; (c) D. Feng, T.-F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y.-P. Chen, X. Wang and K. Wang, Nat. Commun., 2015, 6, 1–8 Search PubMed; (d) P. Li, S.-Y. Moon, M. A. Guelta, S. P. Harvey, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 8052–8055 CrossRef CAS PubMed.
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson and D. A. Gómez-Gualdrón, Chem, 2018, 4, 1022–1034 CAS.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill and C. J. Doonan, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- (a) F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed; (b) V. Gascón, E. Castro-Miguel, M. Díaz-García, R. M. Blanco and M. Sanchez-Sanchez, J. Chem. Technol. Biotechnol., 2017, 92, 2583–2593 CrossRef; (c) F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu and K. C.-W. Wu, J. Am. Chem. Soc., 2015, 137, 4276–4279 CrossRef CAS PubMed.
- R. Singh, G. Souillard, L. Chassat, Y. Gao, X. Mulet and C. M. Doherty, Adv. Sustainable Syst., 2020, 4, 2000059 CrossRef CAS.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Comm., 2015, 51, 13408–13411 RSC.
- S. S. Nadar and V. K. Rathod, Int. J. Biol. Macromol., 2017, 95, 511–519 CrossRef CAS PubMed.
- H. He, H. Han, H. Shi, Y. Tian, F. Sun, Y. Song, Q. Li and G. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 24517–24524 CrossRef CAS PubMed.
- M. Salgaonkar, S. S. Nadar and V. K. Rathod, J. Environ. Chem., 2019, 7, 102969 CrossRef CAS.
- W.-H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- X. Li, D. Li, Y. Zhang, P. Lv, Q. Feng and Q. Wei, Nano Energy, 2020, 68, 104308 CrossRef CAS.
- X. Cheng, J. Zhou, J. Chen, Z. Xie, Q. Kuang and L. Zheng, Nano Res., 2019, 12, 3031–3036 CrossRef CAS.
- (a) H. An, M. Li, J. Gao, Z. Zhang, S. Ma and Y. Chen, Coord. Chem. Rev., 2019, 384, 90–106 CrossRef CAS; (b) C.-S. Liu, J. Li and H. Pang, Coord. Chem. Rev., 2020, 410, 213222 CrossRef CAS; (c) S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS.
- S. Dong, D. Zhang, G. Suo, W. Wei and T. Huang, Anal. Chim. Acta, 2016, 934, 203–211 CrossRef CAS.
- S. Patra, T. H. Crespo, A. Permyakova, C. Sicard, C. Serre, A. Chaussé, N. Steunou and L. Legrand, J. Mater. Chem. B, 2015, 3, 8983–8992 RSC.
- X. Liu, W. Chen, M. Lian, X. Chen, Y. Lu and W. Yang, J. Electroanal. Chem., 2019, 833, 505–511 CrossRef CAS.
- M. Tong, S. Dong, K. Wang and G. Suo, J. Electrochem. Soc., 2017, 164, B200 CrossRef CAS.
- X. Wang, X. Lu, L. Wu and J. Chen, Biosens. Bioelectron., 2015, 65, 295–301 CrossRef CAS PubMed.
- X. Lu, X. Wang, L. Wu, L. Wu, L. Fu, Y. Gao and J. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 16533–16539 CrossRef CAS PubMed.
- B. Ma, L.-Z. Cheong, X. Weng, C.-P. Tan and C. Shen, Electrochim. Acta, 2018, 283, 509–516 CrossRef CAS.
- S. Xie, J. Ye, Y. Yuan, Y. Chai and R. Yuan, Nanoscale, 2015, 43, 18232–18238 RSC.
- C. Zhang, X. Wang, M. Hou, X. Li, X. Wu and J. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 13831–13836 CrossRef CAS PubMed.
- C. Gong, Y. Shen, J. Chen, Y. Song, S. Chen, Y. Song and L. Wang, Sens. Actuators, B, 2017, 239, 890–897 CrossRef CAS.
- W. Chen, W. Yang, Y. Lu, W. Zhu and X. Chen, Anal. Methods, 2017, 9, 3213–3220 RSC.
- C. Gong, J. Chen, Y. Shen, Y. Song, Y. Song and L. Wang, RSC Adv., 2016, 6, 79798–79804 RSC.
- Y. Wang, C. Hou, Y. Zhang, F. He, M. Liu and X. Li, J. Mater. Chem. B, 2016, 4, 3695–3702 RSC.
- Q. Wang, X. Zhang, L. Huang, Z. Zhang and S. Dong, Angew. Chem., 2017, 129, 16298–16301 CrossRef.
- Q. Zhang, L. Zhang, H. Dai, Z. Li, Y. Fu and Y. Li, J. Electroanal. Chem., 2018, 823, 40–46 CrossRef CAS.
- W. H. Organization Screening for type 2 diabetes: report of a World Health Organization and International Diabetes Federation meeting; World Health Organization: 2003.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217 RSC.
- (a) J. H. Pazur and K. Kleppe, Biochemistry, 1964, 3, 578–583 CrossRef CAS PubMed; (b) H. Hecht, D. Schomburg, H. Kalisz and R. Schmid, Biosens. Bioelectron., 1993, 8, 197–203 CrossRef CAS PubMed.
- I. Horne, T. D. Sutherland, R. L. Harcourt, R. J. Russell and J. G. Oakeshott, Appl. Environ. Microbiol., 2002, 68, 3371–3376 CrossRef CAS PubMed.
- H. Yang, P. D. Carr, S. Yu McLoughlin, J.-W. Liu, I. Horne, X. Qiu, C. M. J. Jeffries, R. J. Russell, J. G. Oakeshott and D. L. Ollis, Protein Eng., 2003, 16, 135–145 CrossRef CAS PubMed.
- N. Ui, Biochim. Biophys. Acta, Protein Struct., 1971, 229, 582–589 CrossRef CAS.
- K. Liang, C. J. Coghlan, S. G. Bell, C. Doonan and P. Falcaro, Chem. Commun., 2016, 52, 473–476 RSC.
- (a) F. Carraro, M. d. J. Velásquez-Hernández, E. Astria, W. Liang, L. Twight, C. Parise, M. Ge, Z. Huang, R. Ricco and X. Zou, Chem. Sci., 2020, 11, 3397–3404 RSC; (b) A. F. Ogata, A. M. Rakowski, B. P. Carpenter, D. A. Fishman, J. G. Merham, P. J. Hurst and J. P. Patterson, J. Am. Chem. Soc., 2020, 142, 1433–1444 CrossRef CAS PubMed.
- W. Xiaoling, H. Yue, Y. Zhang, X. Gao, X. Li, L. Wang, Y. Cao, M. Hou, H. An, L. Zhang, S. Li, J. Ma, H. Lin, Y. Fu, H. Gu, W. Lou, W. Wei, R. N. Zare and J. Ge, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- (a) F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761 CrossRef CAS PubMed; (b) X. Wu, C. Yang, J. Ge and Z. Liu, Nanoscale, 2015, 7, 18883 RSC; (c) W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon and H. Amenitsch, J. Am. Chem. Soc., 2019, 141, 2348 CrossRef CAS PubMed; (d) H. He, H. Han, H. Shi, Y. Tian, F. Sun, Y. Song, Q. Li and G. Zhu, ACS Appl. Mater. Interfaces, 2016, 8, 24517 CrossRef CAS PubMed; (e) F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu and K. C.-W. Wu, J. Am. Chem. Soc., 2015, 137, 4276 CrossRef CAS PubMed; (f) J. Cui, Y. Feng, T. Lin, Z. Tan, C. Zhong and S. Jia, ACS Appl. Mater Interfaces, 2017, 9, 10587 CrossRef CAS PubMed; (g) C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res, 2017, 50, 1423 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186 CrossRef CAS PubMed.
- (a) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58 CrossRef CAS PubMed; (b) P. Chulkaivalsucharit, X. Wu and J. Ge, RSC Adv., 2015, 5, 101293 RSC.
- (a) A. Ikai, J. Biochem, 1980, 88, 1895–1898 CAS; (b) J. C. Bischof and X. He, Ann. N. Y. Acad. Sci., 2006, 1066, 12–33 CrossRef PubMed; (c) M. Vihinen, Protein Eng., Des. Sel., 1987, 1, 477–480 CrossRef CAS PubMed; (d) D. C. Rees and A. D. Robertson, Protein Sci., 2001, 10, 1187–1194 CrossRef CAS PubMed.
- F. Carraro, M. de, J. Velásquez-Hernández, E. Astria, W. Liang, L. Twight, C. Parise, M. Ge, Z. Huang, R. Ricco and X. Zou, Chem. Sci., 2020, 11, 3397–3404 RSC.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217–4223 RSC.
- R. Gupta, Q. Beg and P. Lorenz, Appl. Microbiol. Biotechnol., 2002, 59, 15–32 CrossRef CAS PubMed.
- (a) J. Kim and J. W. Grate, Nano Lett., 2003, 3, 1219–1222 CrossRef CAS; (b) I. Hegedűs and E. Nagy, Chem. Eng. Sci., 2009, 64, 1053–1060 CrossRef.
- F. C. Wu and M. Laskowski, Biochim. Biophys. Acta, 1956, 19, 110–115 CrossRef CAS.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon and H. Amenitsch, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed.
- W.-J. Li, M. Tu, R. Cao and R. A. Fischer, J. Mater. Chem. A, 2016, 4, 12356–12369 RSC.
- C. H. Kwon, Y. Ko, D. Shin, M. Kwon, J. Park, W. K. Bae, S. W. Lee and J. Cho, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- M. Güemes, S. A. Rahman and K. Hussain, Arch. Dis. Child., 2016, 101, 569–574 CrossRef PubMed.
- C. MacArthur, J. Phys. Chem., 2002, 20, 495–502 CrossRef.
- B. Haghighi, S. Varma, F. A. Sh, Y. Yigzaw and L. Gorton, Talanta, 2004, 64, 3–12 CrossRef CAS PubMed.
- P. Janda and J. Weber, J. Electroanal. Chem. Interfacial Electrochem., 1991, 300, 119–127 CrossRef CAS.
- (a) M. de, J. Velásquez-Hernández, R. Ricco, F. Carraro, F. T. Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner and P. Frühwirt, CrystEngComm, 2019, 21, 4538–4544 RSC; (b) M. A. Luzuriaga, C. E. Benjamin, M. W. Gaertner, H. Lee, F. C. Herbert, S. Mallick and J. J. Gassensmith, Supramol. Chem., 2019, 31, 485–490 CrossRef CAS PubMed; (c) Y. Gao, C. M. Doherty and X. Mulet, ChemistrySelect, 2020, 5, 13766–13774 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc00407g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |