DOI:
10.1039/D0TC05301E
(Communication)
J. Mater. Chem. C, 2021,
9, 456-465
Enhanced 1.54 μm luminescence of a perfluorinated erbium complex sensitized by perfluorinated Pt(II) and Zn(II) phthalocyanines with 980 nm emission†
Received
11th November 2020
, Accepted 30th November 2020
First published on 8th December 2020
Abstract
By the sensitization effect of metallophthalocyanines showing ∼980 nm emission to an erbium complex, a remarkably long average lifetime of 1.05 ms and an optimal PLQY of 13% with a sensitization efficiency of 81 for the Er3+ 1.54 μm emission are obtained in a perfluorinated organic erbium co-doped system.
Introduction
The erbium-doped optical fiber amplifier (EDFA) was reported in 1987 as an important invention for global fiber-optic telecommunication.1 Since then, research on Er3+ doped waveguide amplifiers (EDWA) operating at 1.5 μm has attracted considerable attention2–8 because multiple optical elements can be integrated on compact photonic chips to achieve optical amplification. Er3+ ions have weak optical absorption coefficients originating from 4f–4f electronic transitions forbidden by the Laporte rule.3 Combining Er3+ ions with organic ligands can significantly enhance the near infrared (NIR) emission because the indirect excitation of Er3+ ions can occur by energy transfer (ET) from organic ligands via an antenna effect or sensitization effect.9–13 However, chemical bonds containing H atoms such as C–H and O–H in most organic components can cause severe vibrational quenching of the 1.54 μm emission, resulting in a weak NIR emission and short lifetimes with tens of microseconds and consequently preventing Er3+ ions from attaining population inversion.14–17 One effective strategy to eliminate C–H bonds is to fully fluorinate organic compounds to reduce the vibrational quenching.18–22 The sensitized Er3+ energy levels of the fully fluorinated Er3+ complexes centre on the 2H11/2, 4F7/2, and 4S3/2 energy levels, corresponding to photons of green light.8,22,23 Such a sensitization of tris[tetrakis(pentafluorophenyl)imidodiphosphinate] Er3+ (Er(F-TPIP)3)18 occurs in the green region regardless of whether a 266 nm laser is used to excite Er(F-TPIP)3 with sensitizing of Er3+ ions or the perfluorinated zinc complex Zn(F-BTZ)2 or a six-coordinate iridium complex (Ir-tBuPBI) excited at 405 nm to sensitize the Er3+ ions in Er(F-TPIP)3.8,23 There are large energy differences between the sensitized energy levels (∼2.38 eV) and the 4I13/2 energy level (∼0.81 eV) in the NIR region, and consequently, part of the energy is lost in the form of heat during the process of relaxation preceding NIR emission, leading to a decreased sensitization efficiency. Finding a sensitizer with luminescence close to the 4I13/2 state can increase the sensitization efficiency because of the corresponding shift to a longer wavelength, and for a given photon flux, lower exciton energy means lower exciton power density (which is equal to the product of photon-flux and photon energy). With one exciton sensitizing one Er ion numerically, this approach can reduce the power needed to induce population inversion.
Phthalocyanines (Pcs) have highly delocalized 18 π-electron conjugated rings leading to extreme stability,24,25 bright photoluminescence (PL),26 and electroluminescence (EL) properties in the NIR region.27,28 Metallophthalocyanines (MPcs) have contributed greatly in many fields, for example, organic dye-sensitized solar cells29 and as photosensitizers in photodynamic therapy.30,31 However, no available perfluorinated matrix material that can avoid self-quenching of ErPc is known. In this article, we demonstrate enhanced 1.54 μm luminescence of Er3+ in Er(F-TPIP)3 by using M(II) hexadecafluorophthalocyanines (F16MPc; M = Zn, Pt) as sensitizers. Both these compounds possess luminescence at ∼980 nm: F16PtPc exhibits dominant phosphorescence, and the wavelength is consistent with the 980 nm excitation used for EDFA, providing high pump rate efficiency. Er(F-TPIP)3 and F16MPc are co-doped at the molecular level using a co-evaporation technique. Photoexcitation gives optimized sensitization efficiencies of 36 and 81 for the Er3+ 1.5 μm emission with F16ZnPc and F16PtPc, respectively. The average lifetimes for the Er3+ 1.5 μm emission are ∼0.89 ± 0.011 ms (doped with F16ZnPc) and ∼1.04 ± 0.012 ms (doped with F16PtPc). This is so far the longest lifetime of organic sensitized Er3+ 1.5 μm emission, which can even match those of the inorganic systems.32 A maximum photoluminescence quantum yield (PLQY) of 13% is obtained for the sensitized Er3+ 1.5 μm emission in an organic co-doped system.
Results and discussion
The chemical structures of F16MPc and Er(F-TPIP)3 are depicted in Fig. 1. F16PtPc was synthesized and purified by modifying a published procedure for the preparation of F16CuPc.33 F16PtPc of good purity was obtained following a simple reprecipitation from 98% sulfuric acid by dilution with water. This approach avoids the need for preparative chromatography, which can be difficult to scale up for Pc derivatives due to their generally low solubility in organic solvents. The purity and identity of F16PtPc were confirmed by MALDI-TOF mass spectrometry, as shown in Fig. S1 (ESI†). The most intense peak (m/z 996) corresponded to the protonation of F16PtPc by the MALDI matrix and aromatic carboxylic acids to form an [F16PtPc + H]+ pseudo molecular ion containing the abundant 195Pt nuclide. This was part of an isotopic cluster, as expected, based on the natural occurrence of other stable isotopes. There were no detectable impurities, apart from a group of very weak peaks, 16 atomic mass units heavier than the main cluster; these had an intensity <1% of the major component and were attributed to halogen exchange with platinum(II) chloride having occurred during the Pc ring synthesis, forming traces of ClF15PtPc. The 19F NMR spectrum of F16PtPc in D2SO4 solution showed just two aromatic fluorine environments: these had equal integrals, which is consistent with the expected four-fold symmetry of the highly delocalised phthalocyanine system. The energy dispersive X-ray (EDS) spectrum shown in Fig. S2 (ESI†) confirmed the presence of C, N, F, and Pt; Cl is too weak to be detected. The successful synthesis of F16PtPc was also supported by Fourier transform infrared (FTIR) spectroscopy. As shown in Fig. S3 (ESI†), the synthesized F16PtPc has seven groups of peaks similar to the IR spectrum of F16CuPc,34 consistent with the successful formation and metalation of the F16Pc macrocycle.
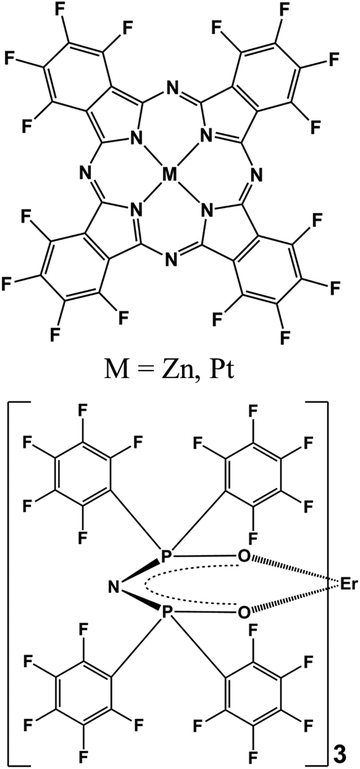 |
| | Fig. 1 The chemical structures of F16MPc and Er(F-TPIP)3. | |
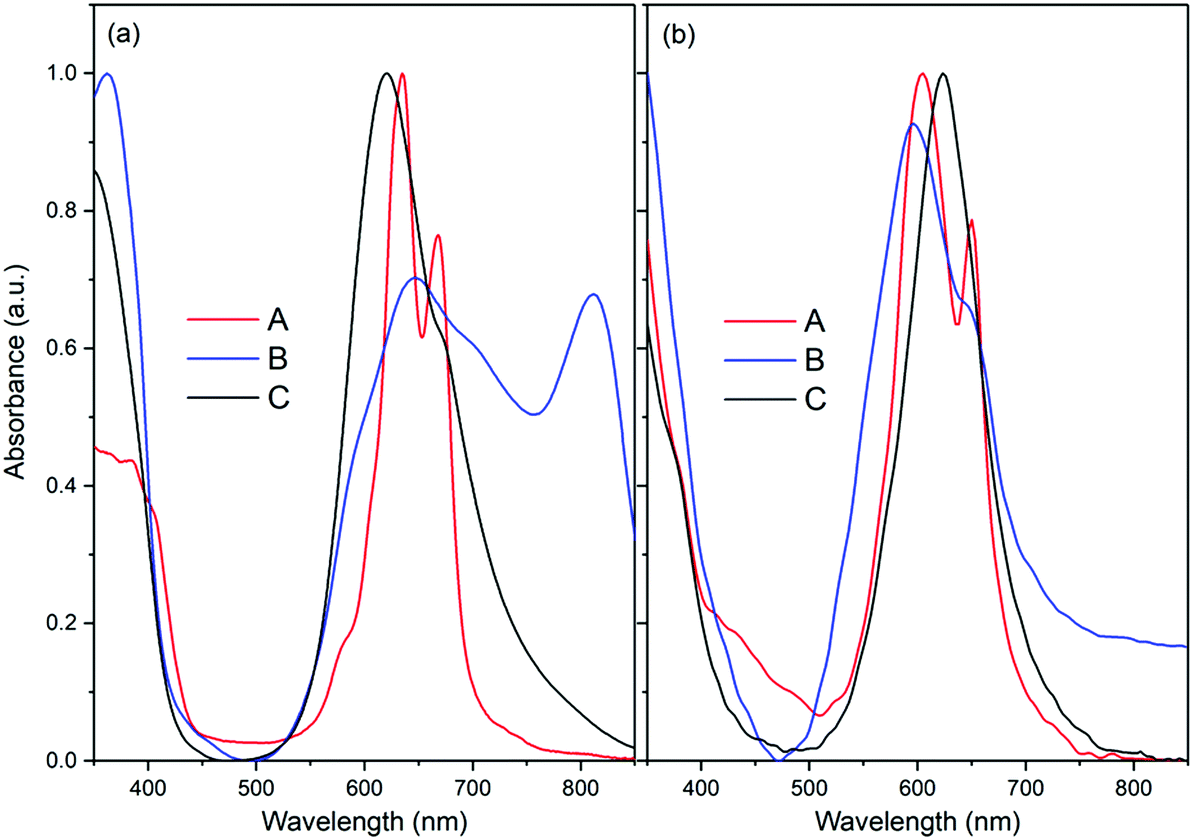
Since the F16MPcs have a large coplanar conjugated system of 18 π electrons, they could form molecular aggregates and quench the luminescence, which would disfavor energy transfer from F16MPc to Er(F-TPIP)3.35 Absorption spectra provide a method to study the aggregation. As shown in Fig. 2a, the electronic absorption spectra of F16ZnPc in acetone solution and in a film deposited onto a glass substrate show two distinct absorption bands from the UV to the NIR spectral region. The high-energy absorption band in the region of <450 nm is ascribed to the B-band originating from a direct electronic transition from a2u orbital to eg orbital. The low energy absorption band in the region from 500 to 850 nm is ascribed as a Q-band originating from an a1u to eg transition. The Q band of the F16ZnPc solution in acetone shows two absorption bands at 635 and 667 nm with a shoulder at 580 nm that indicates the dimer behavior of F16ZnPc in solution.34 In comparison, it is found that the spectrum of a neat F16ZnPc film shows both significant broadening and a bathochromic shift; these are typical effects of aggregation behavior.36 However, the absorption spectrum of the Q band of F16ZnPc in a 50% Er(F-TPIP)3:50% F16ZnPc co-doped film appears as one band at 624 nm with a weak shoulder at 675 nm. Compared to that of neat F16ZnPc film, this absorption band is dramatically narrowed and its full width at half maximum (FWHM) is only slightly wider than that in solution. The comparison indicates that the aggregation of the F16ZnPc molecules in the co-doped film is effectively suppressed, and they are likely to be dispersed homogeneously.
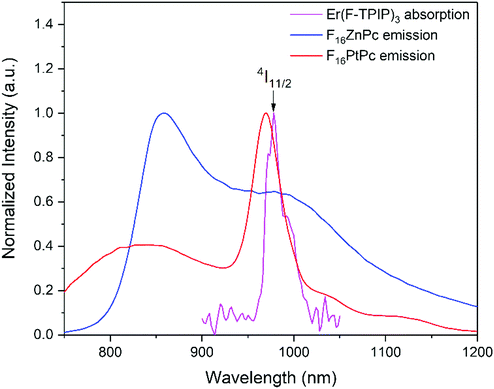 |
| | Fig. 2 The absorption spectra of (a) F16ZnPc and (b) F16PtPc. (A) Dissolved in acetone. (B) Neat film. (C) Co-doped film with 50% Er(F-TPIP)3. | |
The Q band of F16PtPc in the solution, neat and co-doped films are similar, and the FWHM of the Q band of the 50% Er(F-TPIP)3:50% F16PtPc co-doped film is even slightly narrower than that in solution (Fig. 2b). This contrasts markedly with F16ZnPc (Fig. 2a) considered above, where the Q band of F16ZnPc in the neat film is significantly different from those in the solution and the co-doped film. Especially, F16PtPc in the co-doped film shows one Q band, which indicates that the F16PtPc molecules in the co-doped film exist homogeneously as monomers.
The emission spectra of neat F16ZnPc and F16PtPc films are shown in Fig. 3. Zn2+ has a relatively low atomic number and a d10 closed shell structure without any unpaired electrons, leading to a low probability of intersystem crossing (ISC).37 Its very weak phosphorescence possibly comes from the π → π* transition of the F16Pc ligands. Because no long-lived emission is detected at room temperature, we can attribute the luminescence of F16ZnPc at about 980 nm to fluorescence. On the other hand, non-fluorinated PtPc exhibits phosphorescence in the NIR region, and the perfluorination of PtPc can further increase the ISC, so F16PtPc is regarded as a phosphorescent material.38–40
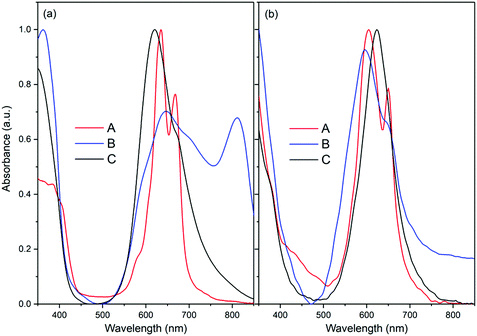 |
| | Fig. 3 Absorption spectrum of 4I15/2 → 4I11/2 of Er3+ ion in an Er(F-TPIP)3 crystal; emission spectra of neat F16ZnPc and F16PtPc films. | |
To gain insight into the origin of the intense emission peak at ca. 980 nm of the neat F16PtPc film, we have performed SOC-TDDFT calculations based on the T1 excited state structure. The calculated emission energies in terms of wavelength and dominant orbital transitions for the low-lying triplet excited states are collected in Table 1, and the important frontier molecular orbitals are shown in Fig. 4. The calculated emission wavelength in the Franck–Condon state (T1 equilibrium geometry) for F16PtPc is 940 nm, which relates well to the experimental phosphorescence peak of 980 nm. As shown in Table 1, the lowest energy transitions for these emission bands are LUMO → HOMO transitions. From Fig. 4, the LUMO (61e1/2g) is largely a π orbital of F16Pc mixed with the dπ of Pt, and the HOMO (61e1/2u) is largely a π* orbital on the F16Pc. We attribute the origin of the 980 nm emission in the experiment to mixed metal-to-ligand charge transfer (3MLCT) and ligand-centered (3LC) characteristics. The calculated long excited-state lifetime of 544.7 μs confirms that F16PtPc emits phosphorescence.
Table 1 Calculated emission energies (eV), oscillator strength (f) and radiative lifetime τ (s) by SOC-TDDFT at PBE/TZ2P level of theory
| State |
ΔE [eV] |
λ [nm] |
f
|
Transition contribution |
Assignment |
T [s] |
|
3A1u |
1.3173 |
941.3 |
0.000 |
61e1/2g → 61e1/2u (0.9986) |
3LC/3MLCT |
— |
|
3A2u |
1.3173 |
941.3 |
0.2438 × 10−7 |
61e1/2g → 61e1/2u (0.9986) |
3LC/3MLCT |
0.5447 |
|
3Eu |
1.3346 |
929.1 |
0.1015 × 10−3 |
61e1/2g → 61e1/2u (0.5199) |
3LC/3MLCT |
1.275 × 10−4 |
|
3Eu |
1.3346 |
929.1 |
0.1015 × 10−3 |
58e3/2g → 61e1/2u (0.4789) |
3LC/3MLCT |
1.275 × 10−4 |
|
3B1u |
1.3519 |
917.2 |
0.000 |
61e1/2g → 61e1/2u (0.9987) |
3LC/3MLCT |
— |
|
3B2u |
1.3519 |
917.2 |
0.000 |
58e3/2g → 61e1/2u (0.9987) |
3LC/3MLCT |
— |
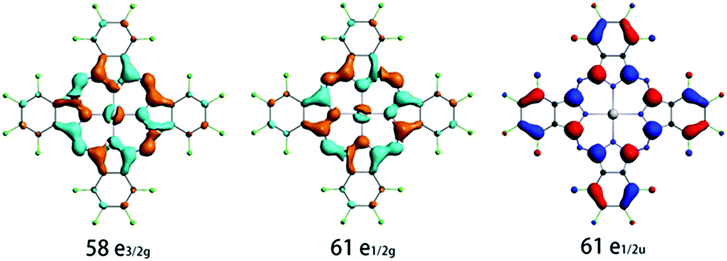 |
| | Fig. 4 Calculated electronic density contours of the frontier molecular orbitals involved in the main electronic transitions. | |
The absorption spectrum of the Er3+ ion in the Er(F-TPIP)3 crystal in the NIR region is shown in Fig. 3. Both the broad emission bands of F16ZnPc and especially F16PtPc have good overlap of the absorption band of 4I15/2 → 4I11/2 of the Er3+ ion in Er(F-TPIP)3, which means that an efficient sensitization may be realized through energy transfer from F16MPc to Er(F-TPIP)3.
In order to study the sensitization effect of F16ZnPc on Er(F-TPIP)3, a series of Er(F-TPIP)3:F16ZnPc co-doped films with the concentrations of Er(F-TPIP)3 varying from 20% to 80% were prepared; their emission spectra are shown in Fig. 5a. It is found that the emission intensity of F16ZnPc at ∼980 nm tends to decrease with increasing Er(F-TPIP)3 concentration, implying that energy is transferred to Er(F-TPIP)3 quenching the excitation of F16ZnPc. The emission spectra of the Er(F-TPIP)3:F16PtPc co-doped films under the excitation of a 655 nm laser are shown in Fig. 5b. It is found that the centre of the emission spectrum is shifted from 970 nm in the neat F16PtPc film to ∼980 nm in the co-doped film. Being different from the Er(F-TPIP)3:F16ZnPc co-doped films, the dips in the mixed 3LC/3MLCT emission spectra of F16PtPc in co-doped films appeared at ∼980 nm, which was not found in the spectrum of the neat F16PtPc film as shown in Fig. 3. Their shapes resemble those of the inverted absorption bands of Er(F-TPIP)3 at ∼980 nm, as shown in Fig. 5b. This is a clear evidence that there is an energy transfer mechanism based on the emission–reabsorption process where the Er3+ 4I11/2 state absorbs part of the F16PtPc emission.41,42 This mechanism is different from the Förster energy transfer, wherein the whole emission intensity of F16PtPc should be decreased with no change in spectral features.
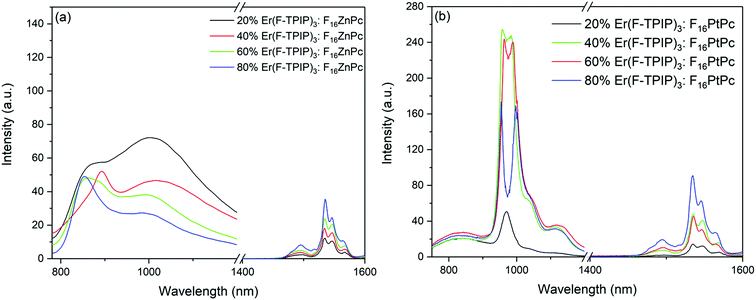 |
| | Fig. 5 Emission spectra of (a) Er(F-TPIP)3:F16ZnPc and (b) Er(F-TPIP)3:F16PtPc co-doped films with different Er(F-TPIP)3 concentrations. λex = 655 nm. | |
It is interesting that there is no dip in the emission spectrum of F16PtPc in the 20% Er(F-TPIP)3:80% F16PtPc co-doped film. This means that the re-absorption process does not occur at this concentration, and there may be another energy transfer path from F16PtPc to Er(F-TPIP)3. Förster transfer is a resonant dipole coupling process that is dependent on the overlap between the donor emission spectrum and the acceptor absorption spectrum. Here the overlap between the PL spectrum of F16PtPc and the absorption of Er(F-TPIP)3 is obvious, as shown in Fig. 3. It indicates that the Förster energy transfer may also be an efficient mechanism and suggests that the energy transfer process from F16PtPc to Er(F-TPIP)3 comprises Förster transfer and emission–reabsorption. The energy transfer paths are shown in Fig. 6. The 20% Er(F-TPIP)3:80% F16PtPc co-doped film shows the weakest 1.54 μm emission from Er(F-TPIP)3 due to the low concentration of Er(F-TPIP)3. Meanwhile, given the weak emission from F16PtPc, there could be a triplet–triplet annihilation (TTA) due to highly concentrated 80% F16PtPc, which reduces the energy transfer. As the concentration of Er(F-TPIP)3 is increased, the 1.54 μm emission of Er(F-TPIP)3 increases gradually while the emission of F16PtPc decreases. The strongest 1.54 μm emission is obtained based on 80% Er(F-TPIP)3 doped concentration, due to the increase in the Er(F-TPIP)3 molecules and possibly a decrease in the TTA effect in F16PtPc.
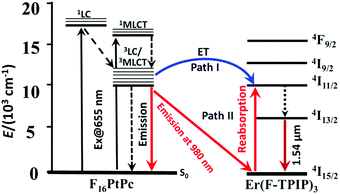 |
| | Fig. 6 Schematic energy diagram describing the two sensitization mechanisms of NIR luminescence of Er(F-TPIP)3via F16PtPc energy transfer (path I) and emission–reabsorption (path II). | |
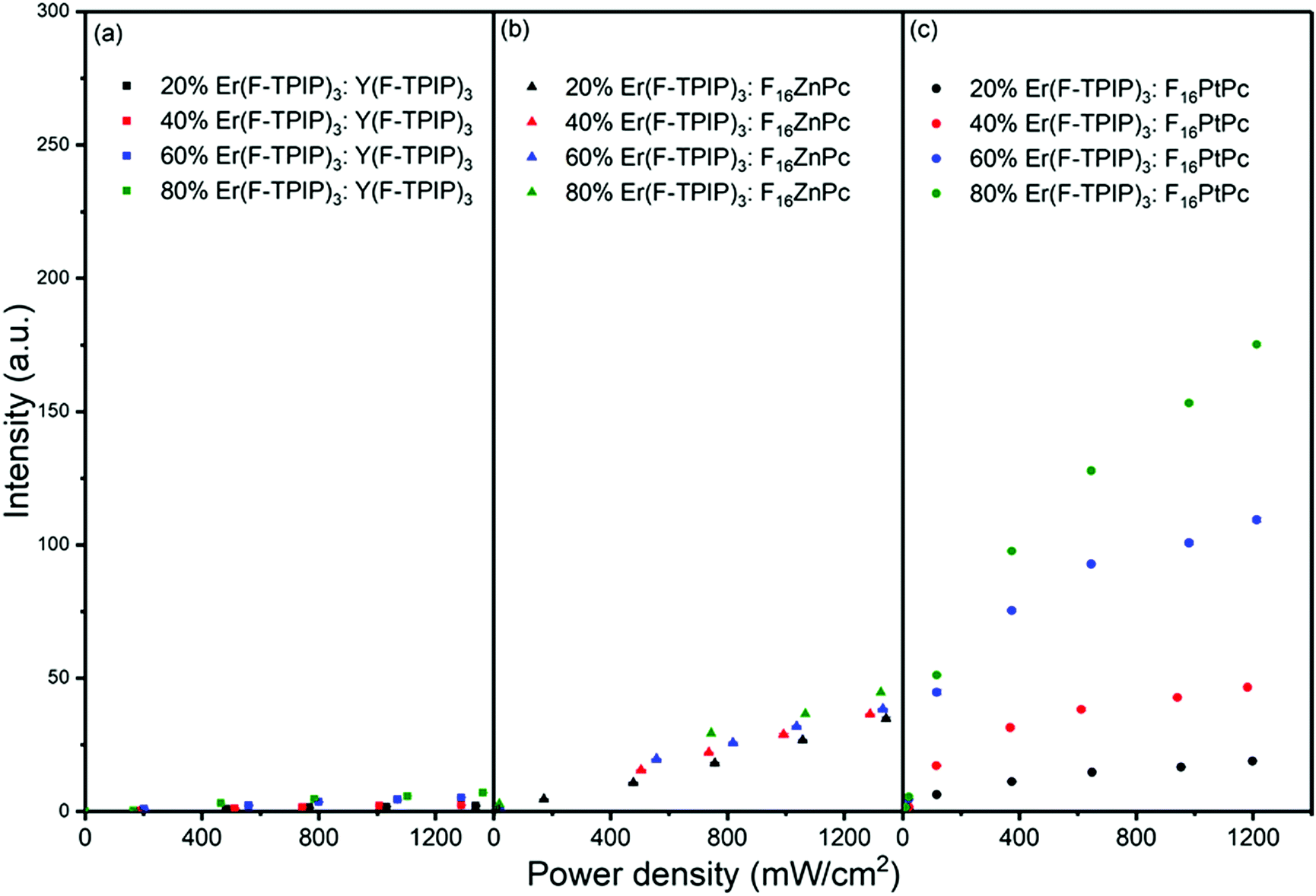
To quantify the sensitization efficiency, the Er(F-TPIP)3:Y(F-TPIP)3 co-doped films with different diluted Er3+ concentrations were used as reference samples, where Y3+ is optically inactive, resulting in the exclusion of energy transfer from Y(F-TPIP)3 to Er(F-TPIP)3. The measurement set up is identical for all co-doped films. The power-dependent Er3+ PL intensities of these co-doped films with Er3+ concentrations of 20%, 40%, 60%, and 80% recorded at 1532 nm are shown in Fig. 7, where the Er3+ ions are directly excited to the 4F9/2 level. In the co-doped films containing the sensitizers, 655 nm photoexcitation of F16ZnPc and F16PtPc was again used to induce sensitization. Hence, the 1532 nm emission intensities from both the F16ZnPc and F16PtPc co-doped films are larger than those of the Y(F-TPIP)3 doped films. The sensitization effect can be simply calculated by fitting the emission intensity of the Y(F-TPIP)3 doped films versus power density, which shows a linear correlation. Subsequently, the power densities used in F16MPc can be used in the correlation to calculate the corresponding 1532 nm emission intensity, and these values can be used to divide the measured 1532 nm intensity to get the sensitization efficiency. The values of the sensitization efficiency for F16ZnPc/F16PtPc in different Er(F-TPIP)3 doped films are listed in Table 2. The sensitization efficiency decreased with the increased percentage of Er(F-TPIP)3 in the F16ZnPc doped films. F16PtPc behaves differently, the reason being that the TTA effect of F16PtPc is quite strong. However, under a high concentration of Er(F-TPIP)3, the F16PtPc molecules get separated and the TTA effect is dramatically decreased, and the emission from F16PtPc gets stronger so that more energy can be transferred to the Er3+ ions through the re-absorption effect. In addition, long-lived triplet excitons of F16PtPc travel over a long distance to sensitize Er3+ ions, along with the perfect overlap between the absorption band of Er(F-TPIP)3 and the emission peak of F16PtPc. So, F16PtPc shows a more efficient sensitization effect than F16ZnPc with high Er(F-TPIP)3 doped concentration.
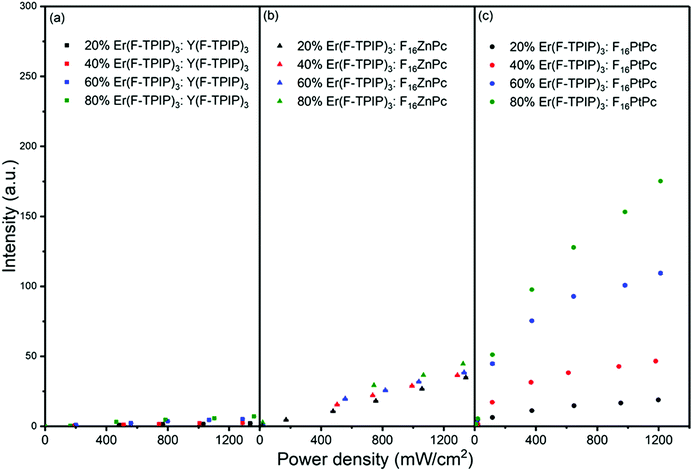 |
| | Fig. 7 The emission intensity at 1532 nm as a function of laser power density for co-doped films with different Er(F-TPIP)3 concentrations diluted by (a) Y(F-TPIP)3, (b) F16ZnPc, and (c) F16PtPc, respectively. λex = 655 nm. | |
Table 2 The sensitization efficiency of F16ZnPc and F16PtPc in co-doped films with different concentrations of Er(F-TPIP)3
| Sample |
Concentration [%] |
Sensitization efficiency |
| Er(F-TPIP)3:F16ZnPc |
20 |
36 ± 1.6 |
| Er(F-TPIP)3:F16ZnPc |
40 |
32 ± 1.3 |
| Er(F-TPIP)3:F16ZnPc |
60 |
30 ± 1.2 |
| Er(F-TPIP)3:F16ZnPc |
80 |
28 ± 1.2 |
| Er(F-TPIP)3:F16PtPc |
20 |
22 ± 1.1 |
| Er(F-TPIP)3:F16PtPc |
40 |
53 ± 2.3 |
| Er(F-TPIP)3:F16PtPc |
60 |
70 ± 3.2 |
| Er(F-TPIP)3:F16PtPc |
80 |
81 ± 3.9 |
The time-resolved PL (TRPL) decay curves for the F16PtPc emission recorded at 980 nm with different Er(F-TPIP)3 doped concentration films are shown in Fig. 8a. The values of their short component (τS), long component (τL), and average lifetimes (τAve) are listed in Table 3a. The lifetime of the F16ZnPc is too short to be measured, lying below the detection limit of the equipment (∼5 ns) due to the strong self-quenching. In F16PtPc doped films, the changing concentrations affect the τL of F16PtPc through two opposing processes: (I) with increased Er(F-TPIP)3 concentrations, F16PtPc is diluted, and the reduced TTA effect leads to increased τL. (II) At the same time, each F16PtPc molecule is surrounded by more Er(F-TPIP)3 molecules; their excitons can more easily transfer their energy to the central Er3+ ions in Er(F-TPIP)3, thus tending to reduce τL. As the Er(F-TPIP)3 concentration increases from 20% to 60%, τL gradually increases from 1.00 ± 0.015 ms to 1.89 ± 0.017 ms because factor (I) plays the dominant role. However, in the 80% Er(F-TPIP)3 doped film, τL decreases to 1.30 ± 0.014 ms as factor (II) is predominant. Basically, the energy transfer from F16PtPc to Er(F-TPIP)3 is more efficient with an increase in Er(F-TPIP)3 because more energy is transferred to Er(F-TPIP)3, which causes the lifetime of F16PtPc to decrease from 484.64 ± 7.09 μs to 384.48 ± 6.39 when the Er(F-TPIP)3 concentration is increased from 20% to 80%.
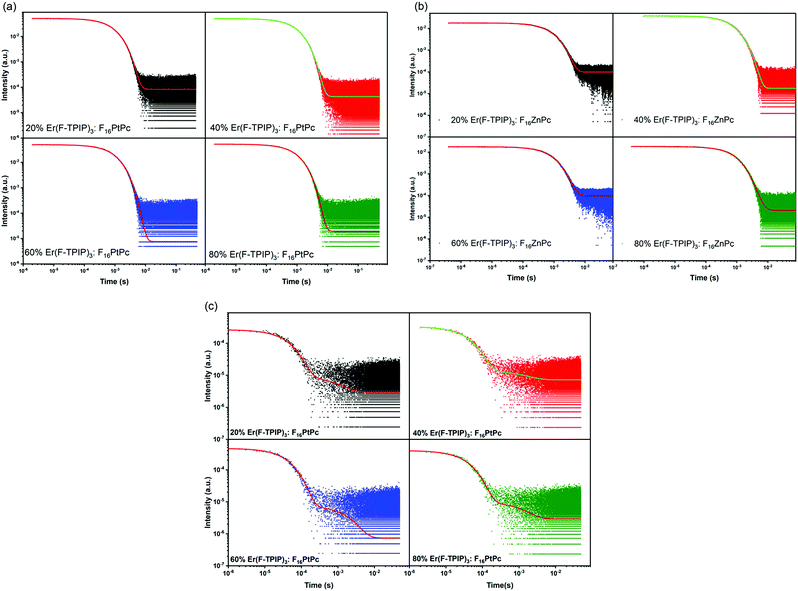 |
| | Fig. 8 (a) The TRPL decay of the F16PtPc doped films with Er(F-TPIP)3 concentration varying from 20% to 80%. λmon = 980 nm. The TRPL decay of Er(F-TPIP)3 in (b) F16ZnPc doped films and (c) F16PtPc doped films. λmon = 1532 nm. | |
Table 3 (a) The lifetime components and average lifetime of F16PtPc emission (λmon = 980 nm) in the Er(F-TPIP)3:F16PtPc co-doped films with different F16PtPc concentrations. (b) The Er3+ 1.5 μm emission lifetime components and average lifetimes in the Er(F-TPIP)3:F16ZnPc co-doped films with different F16ZnPc concentrations. (c) The Er3+ 1.5 μm emission lifetime components and average lifetimes in the Er(F-TPIP)3:F16PtPc co-doped films with different Er(F-TPIP)3 concentrations
| (a) |
| Concentration [%] |
τ
S [μs] |
τ
L [ms] |
τ
Ave [μs] |
| 20 |
43.50 ± 0.047 (64%) |
1.30 ± 0.014 (36%) |
484.64 ± 7.09 |
| 40 |
45.75 ± 0.039 (61%) |
1.89 ± 0.017 (39%) |
754.29 ± 7.00 |
| 60 |
43.44 ± 0.068 (52%) |
1.45 ± 0.010 (48%) |
708.62 ± 8.56 |
| 80 |
44.89 ± 0.076 (64%) |
1.00 ± 0.015 (36%) |
384.48 ± 6.39 |
| (b) |
| F16ZnPc concentrations [%] |
Short component τS [ms] |
Long component τL [ms] |
Average lifetime τAve [ms] |
| 80 |
0.67 ± 0.0025 (49%) |
1.08 ± 0.0047 (51%) |
0.88 ± 0.015 |
| 60 |
0.68 ± 0.0013 (62%) |
1.22 ± 0.0048 (38%) |
0.89 ± 0.003 |
| 40 |
0.76 ± 0.0009 (81%) |
1.45 ± 0.0116 (19%) |
0.89 ± 0.014 |
| 20 |
0.77 ± 0.0006 (86%) |
1.72 ± 0.0213 (14%) |
0.91 ± 0.015 |
| (c) |
| Er(F-TPIP)3 concentrations [%] |
Short component τS [ms] |
Long component τL [ms] |
Average lifetime τAve [ms] |
| 20 |
0.85 ± 0.0025 (48%) |
1.22 ± 0.0012 (52%) |
1.04 ± 0.025 |
| 40 |
0.90 ± 0.0013 (65%) |
1.30 ± 0.0014 (35%) |
1.04 ± 0.034 |
| 60 |
0.95 ± 0.0009 (82%) |
1.50 ± 0.0016 (18%) |
1.05 ± 0.041 |
| 80 |
0.98 ± 0.0006 (91%) |
1.46 ± 0.0010 (9%) |
1.02 ± 0.047 |
The results of TRPL measurements for the Er3+ emission lifetimes (λmon = 1532 nm) with different concentrations of Er(F-TPIP)3 films doped with F16ZnPc and F16PtPc are shown in Fig. 8b, c and Table 3b and c, respectively. The decay can be described by a biexponential process, with two lifetime components for Er3+ in each co-doped film. Being sensitized by F16ZnPc and F16PtPc, the rise in the Er(F-TPIP)3 concentrations makes their τS and τL to increase, which shows the same trend of the emission intensity of Er(F-TPIP)3 as shown in Fig. 5, except for the 80% Er(F-TPIP)3:F16PtPc co-doped film. Compared to the 60% Er(F-TPIP)3 doped film, the τL of the 80% Er(F-TPIP)3 doped film is slightly decreased. With the increase in Er(F-TPIP)3 doped concentration, although their τS and τL are increased, the erbium ion–ion interaction is also increased. This leads the percentage contribution of τL to be reduced, while τS is increased, which makes τAve almost unaffected by the doped concentrations of sensitizers. τS and τAve of Er3+ in the F16PtPc co-doped films are longer than those of the F16ZnPc co-doped films because of the long phosphorescence lifetimes of F16PtPc and the emission–reabsorption process from F16PtPc to Er(F-TPIP)3 in doped films. The longest average lifetime of 0.91 ± 0.015 and 1.05 ± 0.041 ms for F16ZnPc and F16PtPc co-doped films are obtained, and they are 4.55 and 5.25 times longer than the neat Er(F-TPIP)3 film (0.2 ms), respectively. This prolonged Er3+ lifetime is caused by the long-lived organic triplet excitons;43 so the largest PLQY of 13% is achieved in the F16PtPc co-doped film, which is the highest ever reported for erbium in an organic complex.
Conclusions
We demonstrate that two fully fluorinated MPcs: F16ZnPc and F16PtPc can be used as sensitizers to enhance the Er 1.54 μm emission in the co-doped organic Er3+ complex systems. This significantly reduces the energy difference between the sensitized Er3+ energy levels and the 4I13/2 level by utilizing perfluorinated 980 nm luminescent chromophores and consequently decreases the energy loss caused by the Er3+ nonradiative transitions from the higher sensitized energy levels to the 4I13/2 level. Optimized sensitization efficiencies of 36 and 81 are obtained for the Er3+ complex doped with F16ZnPc and F16PtPc, respectively. An average lifetime for the Er3+ 1.5 μm emission in an Er(F-TPIP)3–F16PtPc system reaches ∼1 ms, which is by far the longest ever reported. 13% PLQY is achieved in the F16PtPc doped film that is the highest ever reported for erbium in an organic system. Our research opens the door for improving power efficiency to reach population inversion of Er ions based on the perfluorinated organic system.
Experimental
Chemical reagents
F16ZnPc (90%) was purchased from Sigma Aldrich and purified by sublimation under 280 °C at a pressure of ∼5 × 10−6 mbar. 3,4,5,6-Tetrafluorophthalo-1,2-dinitrile and anhydrous platinum(II) chloride were obtained from Energy Chemical and Mascal companies, respectively.
Synthesis of F16PtPc
3,4,5,6-Tetrafluorophthalo-1,2-dinitrile (0.40 g, 2 mmol) and anhydrous platinum(II) chloride (0.13 g, 0.5 mmol) were mixed well and placed in a glass vessel. The vessel was sealed under vacuum (0.13 Pa) and then slowly heated to 210 °C for 6 hours. After cooling to room temperature, the crude purple product was isolated and washed with petroleum ether and ethanol. The crude F16PtPc product was dissolved in concentrated sulfuric acid (∼2 ml) with intense stirring in an ice-water bath for 1 hour at a solution temperature not exceeding 5 °C. Then it was filtered by a sintered glass filter, and the filtrate was slowly added to the ice water mixture under stirring and at a temperature lower than 10 °C. Finally, the suspension was filtered and the cake was washed to neutral with deionized water. After drying, the F16PtPc product (0.21 g, 43%) was obtained as a purple solid. 19F NMR (377 MHz, D2SO4 solution) δF: −129.6 (8F, br s) and −137.2 (8F, br s); IR (KBr, cm−1): C–F (1496 s), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1530 s), CN (1647 w), Pc ring (750 w, 1160 s and 1469 s), N–Pt–N (845 w). UV/Vis: λmax = 605 nm. MS (MALDI-TOF): m/z calcd for [M + H]+ [C32HF16N8195Pt]+, 996.0; found: 996.1.
C (1530 s), CN (1647 w), Pc ring (750 w, 1160 s and 1469 s), N–Pt–N (845 w). UV/Vis: λmax = 605 nm. MS (MALDI-TOF): m/z calcd for [M + H]+ [C32HF16N8195Pt]+, 996.0; found: 996.1.
Theoretical calculations
All calculations were performed with the ADF 2013.01 program package.44 The geometry optimization of the ground state (S0) and lowest triplet excited state (T1) was carried out using gradient approximation (GGA) using the PBE exchange–correlation functional.45 All electron TZ2P basis set was employed for all atoms, and the relativistic effect was also taken into account via the zeroth-order regular approximation (ZORA).46,47 On the basis of the optimized T1 geometry, relativistic calculations were performed to obtain electron excitations and radiative rate. In these calculations, spin–orbit coupling (SOC) was included in one-component time dependent density functional theory (TDDFT) utilizing the ZORA.48,49 The radiative rate (kr) of an excited state was then calculated as50
where ε is the excitation energy and f is the corresponding oscillator strength for the electronic excitation from the ground state to the excited state.
Sample preparation
Co-doped films of Er(F-TPIP)3:F16MPc and Er(F-TPIP)3:Y(F-TPIP)3 were deposited by vacuum sublimation at a vacuum pressure of ∼10−7 mbar. 120 nm thick aluminium was evaporated onto the organic layer to protect the material from atmospheric degradation. Samples were prepared with Er(F-TPIP)3 at a molecular concentration of 20%, 40%, 60%, and 80%. In F16MPc doped films, they had an identical amount of F16MPc chromophore (75 nm thick). In the Y(F-TPIP)3 doped film, the thickness of Y(F-TPIP)3 was kept constant as 163 nm.
Instruments and measurements
The UV-vis electronic absorption measurements were performed using a Shimadzu UV-2600 spectrophotometer. The Fourier-transform infrared (FT-IR) spectrum was collected in the range 400–4000 cm−1 on a Nicolet Fourier spectrophotometer using KBr pellets. The mass spectrum was measured with a MALDI-TOF/TOF Mass spectrometer, Ultraflex Treme TOF/TOF (Bruker). The analysis of chemical composition was performed on a Zeiss Merlin Compact scanning electron microscope with an energy dispersive X-ray spectrometer.
Emission spectra and TRPL measurements
For the PL measurements, lasers of different wavelengths were used to excite the samples; the emission from the samples were guided into a spectrometer (Jobin Yvon Horiba Triax 550), and the reflected laser light was removed by putting high-pass filters in front of the spectrometer. The spectrometer was connected to a photomultiplier (PMT), and the signal from the PMT was transmitted to an oscilloscope or a lock-in-amplifier for time-resolved or emission spectrum measurements, respectively. A Q-switch Nd:YAG laser was used to produce pulsed laser, and the laser wavelength was tuned by using an optical parametric oscillator (OPO). Pulsed laser beams were made to be incident on co-doped films to give TRPL data. The data were fitted by using exponential functions: 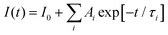 . A lifetime component percentage is obtained by an expression of
. A lifetime component percentage is obtained by an expression of 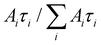 . An average lifetime is obtained by an expression of
. An average lifetime is obtained by an expression of 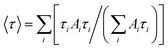 .
.
Sensitization measurement
Two apertures were used to ensure the consistency of the alignment of the optical path. A mirror, made by growing an aluminium circle with 2 mm diameter on the 20 mm × 20 mm glass substrate, was set to an angle of 45° to reflect light normally onto the sample and allow the PL to be collected. An aperture with a 1 mm diameter hole was placed directly in front of the sample, and the laser was defocused to ensure uniform illumination over the whole sample. Once the optical path was set up, the measurement was started with the Er(F-TPIP)3:Y(F-TPIP)3 co-doped films; the 655 nm laser was used to directly excite the Er3+ ions, and the emission was monitored at 1532 nm. The laser power was increased from low to high. At each power, ten measurements were taken to determine the statistical errors. After measuring with the Y(F-TPIP)3 doped films, the experiment was repeated using the Er(F-TPIP)3:F16ZnPc/F16PtPc co-doped films. After the data were collected, the sample was replaced with a calibrated silicon detector to measure the excitation power density on the sample.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the China Scholarship Council, Queen Mary University of London. W. P. G. acknowledges financial support from EPSRC (EP/L020114/1 and EP/P007767/1).
References
- R. J. Mears, L. Reekie, I. M. Jauncey and D. N. Payne, Electron. Lett., 1987, 23, 1026–1028 CrossRef.
- H. S. Han, S. Y. Seo and J. H. Shin, Appl. Phys. Lett., 2001, 79, 4568–4570 CrossRef CAS.
- L. H. Sloff, A. Van Blaaderen, A. Polman, G. A. Hebbink, S. I. Klink, F. C. M. Van Veggel, D. N. Reinhoudt and J. W. Hofstraat, J. Appl. Phys., 2002, 91, 3955–3980 CrossRef.
- W. H. Wong, E. Y. B. Pun and K. S. Chan, Appl. Phys. Lett., 2004, 84, 176–178 CrossRef CAS.
-
A. Q. Le Quang, R. Hierle, J. Zyss, I. Ledoux and S. Pietralunga, presented at 2006 Conference on Lasers and Electro-Optics and 2006 Quantum Electronics and Laser Science Conference, Long Beach, CA, 2006.
- S. Ghatrehsamani and G. E. Town, IEEE J. Quantum Electron., 2017, 53, 7000705 Search PubMed.
- X. S. Zhai, S. S. Liu, X. Y. Liu, F. Wang, D. M. Zhang, G. S. Qin and W. P. Qin, J. Mater. Chem. C, 2013, 1, 1525–1530 RSC.
- H. Q. Ye, Z. Li, Y. Peng, C. C. Wang, T. Y. Li, Y. X. Zheng, A. Sapelkin, G. Adamopoulos, I. Hernández, P. B. Wyatt and W. P. Gillin, Nat. Mater., 2014, 13, 382–386 CrossRef CAS.
- F. J. Steemers, W. Verboom, J. W. Hofstraat, F. A. J. Geurts and D. N. Reinhoudt, Tetrahedron Lett., 1998, 39, 7583–7586 CrossRef CAS.
- V. Bulach, F. Sguerra and M. W. Hosseini, Coord. Chem. Rev., 2012, 256, 1468–1478 CrossRef CAS.
- C. Y. Chow, S. V. Eliseeva, E. R. Trivedi, T. N. Nguyen, J. W. Kampf, S. Petoud and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 5100–5109 CrossRef CAS.
- Z. F. Li, J. B. Yu, L. Zhou, H. J. Zhang, R. P. Deng and Z. Y. Guo, Org. Electron., 2008, 9, 487–494 CrossRef CAS.
- L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Yu, Adv. Funct. Mater., 2005, 15, 1041–1048 CrossRef CAS.
- Y. Hasegawa, Y. J. Wada and S. Yanagida, J. Photochem. Photobiol., C, 2004, 5, 183–202 CrossRef CAS.
- R. H. C. Tan, M. Motevalli, I. Abrahams, P. B. Wyatt and W. P. Gillin, J. Phys. Chem. B, 2006, 110, 24476–24479 CrossRef CAS.
- L. Winkless, R. H. C. Tan, Y. Zheng, M. Motevalli, P. B. Wyatt and W. P. Gillin, Appl. Phys. Lett., 2006, 89, 111115 CrossRef.
- F. Artizzu, L. Marchiò, M. Mercuri, L. Pilia, A. Serpe, F. Quochi, R. Orrù, F. Cordella, M. Saba, A. Mura, G. Bongiovanni and P. Deplano, Adv. Funct. Mater., 2007, 17, 2365–2376 CrossRef CAS.
- G. Mancino, A. J. Ferguson, A. Beeby, N. J. Long and T. S. Jones, J. Am. Chem. Soc., 2005, 127, 524–525 CrossRef CAS.
- G. A. Kumar, R. E. Riman, L. A. D. Torres and O. B. Garcia, Chem. Mater., 2005, 17, 5130–5135 CrossRef CAS.
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. V. Deun and Z. Pikramenou, Chem. – Eur. J., 2007, 13, 6308 CrossRef CAS.
- A. Monguzzi, R. Tubino, F. Meinardi, A. O. Biroli, M. Pizzotti, F. Demartin, F. Quochi, F. Cordella and M. A. Loi, Chem. Mater., 2009, 21, 128–135 CrossRef CAS.
- Y. Peng, H. Q. Ye, Z. Li, M. Motevalli, I. Hernández, W. P. Gillin and P. B. Wyatt, J. Phys. Chem. Lett., 2014, 5, 1560–1563 CrossRef CAS.
- H. F. Li, X. Q. Liu, C. Lyu, J. Gorbaciova, L. L. Wen, G. G. Shan, P. B. Wyatt, H. Q. Ye and W. P. Gillin, Light: Sci. Appl., 2020, 9, 32 CrossRef CAS.
-
C. C. Leznoff and A. P. B. Lever, Phthalocyanines properties and applications, VCH, Cambridge, 1989 Search PubMed.
- N. Uyeda, T. Kobayashi, E. Suito, Y. Harada and M. Watanabe, J. Appl. Phys., 1972, 43, 5181–5189 CrossRef CAS.
- D. S. Lawrence and D. G. Whitten, Photochem. Photobiol., 1996, 64, 923–935 CrossRef CAS.
- C. H. Cheng, Z. Q. Fan, S. K. Yu, W. H. Jiang, X. Wang, G. T. Du, Y. C. Chang and C. Y. Ma, Appl. Phys. Lett., 2006, 88, 213505 CrossRef.
- Z. Q. Fan, C. H. Cheng, S. K. Yu, K. Q. Ye, R. S. Sheng, D. C. Xia, C. Y. Ma, X. Wang, Y. C. Chang and G. T. Du, Opt. Mater., 2009, 31, 889–894 CrossRef CAS.
- S. Mori, M. Nagata, Y. Nakahata, K. Yasuta, R. Gotom, M. Kimura and M. Taya, J. Am. Chem. Soc., 2010, 132, 4054–4056 CrossRef CAS.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- E. D. Anderson, A. P. Gorka and M. J. Schnermann, Nat. Commun., 2016, 7, 13378 CrossRef.
- K. Yan, K. Vu, R. P. Wang and S. Madden, Opt. Express, 2016, 24, 23304–23313 CrossRef CAS.
- F. Ma, S. R. Wang and X. G. Li, J. Phys. Chem. Solids, 2012, 73, 589–592 CrossRef CAS.
- M. Handa, A. Suzuki, S. Shoji, K. Kasuga and K. Sogabe, Inorg. Chim. Acta, 1995, 230, 41–44 CrossRef CAS.
- K. Y. Law, Chem. Rev., 1993, 93, 449–486 CrossRef CAS.
- S. Hiller, D. Schlettwein, N. R. Armstrong and D. Wöhrle, J. Mater. Chem., 1998, 8, 945–954 RSC.
-
Organic Electronics Materials and Devices, ed. S. Ogawa, Springer, 2015 Search PubMed.
- K. Han, X. Z. Lu, J. H. Xu, S. H. Ma and W. C. Wang, Spectrosc. Spect. Anal., 1999, 19, 264–267 CAS.
- A. V. Soldatova, J. Kim, C. Rizzoli, M. E. Kenney, M. A. Rodgers, A. Rosa and G. Ricciard, Inorg. Chem., 2011, 50, 1135–1149 CrossRef CAS.
- K. Kaneto, K. Yoshino and Y. Inuishi, J. Phys. Soc. Jpn., 1974, 37, 1297–1300 CrossRef CAS.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- J. Yang, Y. Deng, Q. Wu, J. Zhou, H. Bao, Q. Li, F. Zhang, F. Li, B. Tu and D. Zhao, Langmuir, 2010, 26, 8850–8856 CrossRef CAS.
- C. Lyu, H. Li, P. B. Wyatt, W. P. Gillin and H. Ye, J. Mater. Chem. C, 2020, 8, 9502–9505 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Chang, M. Pelissier and P. Durand, Phys. Scr., 1986, 34, 394–404 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- F. Wang and T. Ziegler, J. Chem. Phys., 2005, 123, 154102 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc05301e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Feng
Ma
c,
Huanqing
Ye
a,
Peter. B.
Wyatt
d and
William P.
Gillin
a,
Feng
Ma
c,
Huanqing
Ye
a,
Peter. B.
Wyatt
d and
William P.
Gillin






 . A lifetime component percentage is obtained by an expression of
. A lifetime component percentage is obtained by an expression of  . An average lifetime is obtained by an expression of
. An average lifetime is obtained by an expression of  .
.