
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultianvil high-pressure/high-temperature synthesis and characterization of magnetoelectric HP-Co3TeO6†
Elisabeth
Selb
a,
Toni
Buttlar
b,
Oliver
Janka
 c,
Martina
Tribus
d,
Stefan G.
Ebbinghaus
b and
Gunter
Heymann
*a
c,
Martina
Tribus
d,
Stefan G.
Ebbinghaus
b and
Gunter
Heymann
*a
aInstitut für Allgemeine, Anorganische und Theoretische Chemie, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria. E-mail: Gunter.Heymann@uibk.ac.at; Fax: +43-0-512-507 57003
bInstitut für Chemie, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Str. 2, D-06120 Halle (Saale), Germany
cAnorganische Festkörperchemie, Universität des Saarlandes, Campus C4 1, D-66123 Saarbrücken, Germany
dInstitut für Mineralogie und Petrographie, Leopold-Franzens-Universität Innsbruck, Innrain 52, A-6020 Innsbruck, Austria
First published on 2nd April 2021
Abstract
By high-pressure/high-temperature multianvil synthesis a new high-pressure (HP) phase of Co3TeO6 was obtained. The compound crystallizes in the acentric trigonal crystal system of the Ni3TeO6-type structure with space group R3 and the following unit cell parameters and refinement results: a = 519.37(6) pm, c = 1382.4(2) pm, V = 322.93 Å3, R1 = 0.0150, wR2 = 0.0374, GooF = 1.114 and a Flack parameter of 0.04(5). High-temperature powder X-ray diffraction (PXRD) measurements showed an exceptionally high-temperature stability of the HP-modification up to 1070 K. Magnetic measurements revealed an antiferromagnetic ordering below TN = 58.2(1) K and a spin-flop-type transition at T = 3 K with a critical magnetic field of Hcrit = 10.8(1) kOe. Magnetic and magnetoelectric (ME) transition temperatures were determined by specific heat measurements and exhibited a non-hysteretic behavior of the magnetoelectric coupling. Additionally, from the UV-Vis reflectance spectra a direct and an indirect band gap of Eg = 1.88 eV and Eg = 1.91 eV were calculated, underlining the semiconducting nature of HP-Co3TeO6.
1 Introduction
In recent years, research on magnetoelectric (ME) compounds has attracted enormous attention in materials science and condensed matter physics. Without magnetic and magnetoelectric materials many forms of current technology in particular microelectronics would be unimaginable. To further enhance the performance of the corresponding devices, new technologies are required. For example, materials that combine ferroelectricity and ferromagnetism allow fast and energy efficient electric writing of a magnetic information. Therefore, there is great demand for multiferroics, where these two phenomena are intimately coupled.1–3In this regard, novel metal tellurates M3TeO6 (M = Ni, Co, Mn, Cu) have gained great importance. These materials are classified as type-II multiferroics and show antiferromagnetic ordering at low temperatures.4 Remarkably, the ME effect is stronger in type-II multiferroics, because the order parameters are closely linked and not decoupled as in type-I multiferroics.5,6 Despite their same stoichiometry (transition metal to tellurium ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) the tellurates M3TeO6 (M = Ni, Co, Mn, Cu) exhibit different crystal structure-types and different magnetic structures.4 A brief overview is given in the following.
:1) the tellurates M3TeO6 (M = Ni, Co, Mn, Cu) exhibit different crystal structure-types and different magnetic structures.4 A brief overview is given in the following.
The longest known tellurate of this composition is Ni3TeO6. Its crystal structure was already investigated in the year 1967.7 Current research on this compound revealed spin driven pyroelectricity8 and a so-called colossal magnetoelectric effect (CME) below the antiferromagnetic (AFM) ordering temperature of TN = 52 K.9–11 Exceptional about Ni3TeO6 is the possibility of a magnetoelectric switching without hysteresis. Thus already small changes in electric and magnetic fields are sufficient for a spin-flop transition.11
Hostachy and Coing-Boyat et al.12 reported on cubic (Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) Cu3TeO6, also a type-II multiferroic tellurate, which adopts a bixbyite-type structure and orders in a “three-dimensional spin web” with hexagonal arrangements of the magnetic moments.13 One decade later Mn3TeO6 was discovered, crystallizing in the rhombohedral Mg3TeO6-type structure.14,15 Here, the multiferroic ordering arises from two coexistent incommensurate spin structures with a cycloidal and a helical ordering.16 Recently Attfield et al. reported on a magnetically frustrated high-pressure Mn2MnTeO6 with double perovskite-type structure and antiferromagnetic ordering at 36 K.17
) Cu3TeO6, also a type-II multiferroic tellurate, which adopts a bixbyite-type structure and orders in a “three-dimensional spin web” with hexagonal arrangements of the magnetic moments.13 One decade later Mn3TeO6 was discovered, crystallizing in the rhombohedral Mg3TeO6-type structure.14,15 Here, the multiferroic ordering arises from two coexistent incommensurate spin structures with a cycloidal and a helical ordering.16 Recently Attfield et al. reported on a magnetically frustrated high-pressure Mn2MnTeO6 with double perovskite-type structure and antiferromagnetic ordering at 36 K.17
One of the most complex M3TeO6 compounds is Co3TeO6, which crystallizes in a monoclinic lithium cryolite-type structure (C2/c) with 5 independent Co sites and shows a complex temperature-dependent incommensurate magnetic behavior.18–20 Due to the good multiferroic properties of Co3TeO6, this compound has arisen a lot of interest in the last few years and has been well investigated in numerous publications.18–24
Lately, we succeeded in the synthesis of a high-pressure polymorph of Co3TeO6 at a pressure of 6.5 GPa and a temperature of 1070 K. It crystallizes in the Ni3TeO6-type structure. Besides normal-pressure (NP) Co3TeO618 and a monoclinic CoTeO4, that is related to the rutile structure,25 HP-Co3TeO6 is now the third existing cobalt tellurate modification. This work presents the synthesis, single-crystal structure and magnetic as well as magnetoelectric characterization of HP-Co3TeO6.
2 Experimental
2.1 Synthesis
The high-pressure phase of Co3TeO6 was obtained via high-pressure/high-temperature multianvil synthesis. In the first step single phase NP-Co3TeO6 was prepared by conventional solid state synthesis, starting from Co(NO3)2·6H2O (Merk, 99% p.a) and H6TeO6 (TCI, >99.0%). The stoichiometric mixture was homogenized and calcined at 770 K for 7 h. The intermediate product was powdered and annealed multiple times (7 h per temperature step) with temperature intervals of 100 K up to 970 K, as described in literature.26For the high-pressure/high-temperature experiment, the polycrystalline precursor was surrounded by platinum foil and placed in an 18/11-assembly crucible made of hexagonal boron nitride. The Walker module of the multianvil press, which contained the sample inside of an octahedral pressure medium, was compressed with a ramp of 72 bar h−1 to a pressure of 6.5 GPa. At synthesis pressure, the sample was heated to 1070 K within 5 min and kept at this temperature for another 20 min. Subsequently, the sample was steadily cooled down to 670 K during 120 min to preserve better crystal quality. As soon as the heating process was terminated, the pressure was released with a ramp of 24 bar h−1. Afterwards, the sample was isolated by breaking the octahedral pressure medium. The powdered sample appears dark greyish to purple and is stable in air. Further information about the multianvil technique and the construction of the various assemblies can be found in literature and references therein.27,28
2.2 Characterization
| Empirical formula | Co3TeO6 |
|---|---|
| Molar mass, g mol−1 | 400.39 |
| Crystal system | Trigonal |
| Space group | R3 (no. 146) |
| Formula units per cell, Z | 3 |
| Powder diffractometer | STOE Stadi P |
| Radiation | Mo-Kα1 (λ = 70.93 pm) |
| Powder data: | |
| a, pm | 518.97(1) |
| c, pm | 1381.56(1) |
| V, Å3 | 322.24 |
| Single-crystal diffractometer | Bruker D8 Quest |
| Radiation | Mo-Kα (λ = 71.073 pm) |
| Single-crystal data: | |
| a, pm | 519.37(6) |
| c, pm | 1382.4(2) |
| V, Å3 | 322. 93 |
| Calculated density, g cm−3 | 6.18 |
| Crystal size, mm3 | 0.04 × 0.03 × 0.03 |
| Temperature, K | 297(2) |
| Absorption coefficient, mm−1 | 18.0 |
| F (000), e | 543 |
| Detector distance, mm | 40 |
| θ range, deg | 4.4–37.7 |
| Range in hkl | ±8, ±8, ±23 |
| Reflections total | 3596 |
| Data/ref. parameters | 773/32 |
| Reflections with I ≥ 2σ(I) | 773 |
| R int, Rσ | 0.0236, 0.0176 |
| Goodness-of-Fit on F2 | 1.114 |
| Absorption correction | Multi-scan32 |
| R 1/wR2 for I ≥ 2σ(I) | 0.0150/0.0374 |
| R 1/wR2 (all data) | 0.0150/0.0374 |
| Flack parameter | 0.04(5) |
| Largest diff. peak/hole, e Å−3 | 0.94/−0.80 |
| Extinction coefficient | 0.0035(4) |
| Transmission min./max. | 0.6489/0.7474 |
High-temperature X-ray powder diffraction data (HT-PXRD) of HP-Co3TeO6 were recorded with a STOE Stadi P diffractometer system (Mo-Kα1, λ = 70.93 pm) equipped with an image-plate detector (120°) and a STOE furnace. A milled polycrystalline sample of HP-Co3TeO6 was filled in a silica glass capillary with a diameter of 0.3 mm and a wall thickness of 0.01 mm. The furnace was heated and cooled in the range from 298 K to 673 K in steps of 100 K. From 673 K to the maximum temperature of 1373 K the sample was heated and cooled in steps of 50 K. The heating rate was set to 5 K min−1. After every temperature step, a diffraction pattern was recorded in the region 2–52° 2θ.
Several fragments of the crushed sample were embedded in polyfluoropolyalkylether (viscosity 1800) and suitable single-crystal fragments were isolated under the microscope and fixed on the tip of MicroMountsTM (MiTeGen, LLC, Ithaca, NY, USA) with a diameter of 30 μm. Diffraction data was collected on a Bruker D8 Quest diffractometer with a Photon 100 detector system and an Incoatec microfocus source generator (multi-layered optic, monochromatized MoKα radiation, λ = 71.073 pm). To optimize the collection strategies concerning ω- and φ-scans, the Apex 3 program package32 was used. As a result, a data set of the complete reciprocal sphere up to high angles (θ = 37.7°) with a high redundancy (9.28) was received. The program Saint32 was used for data processing and data reduction. Finally, an absorption correction was carried out on the semi-empirical “multi scan” approach with the program Sadabs.32
FTIR-ATR. The polycrystalline sample of HP-Co3TeO6 was characterized by FTIR-ATR (Fourier Transformed IR – Attenuated Total Reflection) spectroscopy using a Bruker ALPHA Platinum-ATR spectrometer (Bruker, Billerica, USA) equipped with a 2 × 2 mm diamond ATR-crystal and a DTGS detector, in the spectral range of 400–4000 cm−1. For the measurement, 320 scans were acquired and a data correction for atmospheric influences was performed using the Opus 7.2 software.33
UV-Vis. A diffuse reflectance spectrum of the powdered sample was recorded in the range of 360 to 830 nm, using an Agilent Cary 5000 UV-Vis spectrometer equipped with an integrating sphere (DRA-2500), a D65 as standard illuminant and a 10° complementary observer. A scan rate of 600 nm min−1 and a data interval of 1 nm were applied and BaSO4 was used as white standard. Via the Kubelka–Munk (KM) function the optical absorbance was calculated from the generated reflectance data and the band gap was determined using Tauc-plots.34,35
Magnetic properties. The polycrystalline sample of HP-Co3TeO6 was enclosed in a polyethylene (PE) capsule. Magnetization M(T, H) measurements were performed on a Quantum Design Physical Property Measurement System (PPMS) using the Vibrating Sample Magnetometer (VSM) unit. The sample was investigated in the temperature range of 2.5–300 K with magnetic fields up to 80 kOe.
Heat capacity. The specific heat of a HP-Co3TeO6 specimen with m = 16.03(2) mg was measured in a PPMS-9 at magnetic fields of 0 Oe, 100 Oe and 10 kOe, respectively. A step width of 5 K was chosen in the temperature range 300–80 K, whereas between 80 and 3 K CP values were recorded with 1 K steps. The contribution of the thermal conduction grease (0.13 mg Apiezon N) was subtracted prior to data evaluation.
Magnetoelectric properties. For the magnetoelectric investigations gold contacts of 100 nm thickness were sputtered on both sides of the disk-shaped Co3TeO6 sample (Ø ≈ 3 mm, h ≈ 1 mm) using a Cressington Sputter Coater 108auto. ME measurements were carried out in the temperature range 65 K to 10 K with 5 K intervals in a PPMS-9, using the self-designed setup described in literature.36 The static magnetic field was varied between −17 kOe and 17 kOe and a collinear ac field of Hac = 10 Oe with a frequency of 900 Hz was superimposed. The ME coefficient αME was calculated from the real part (in-phase) of the ac-voltage Uac according to eqn (1) using a lock-in technique.
 | (1) |
Data analysis was carried out using the peak analyzing tool of OriginPro 2018G.37
3 Results and discussion
3.1 Structure refinements
HP-Co3TeO6 crystallizes in the trigonal crystal system and the systematic extinctions were in agreement with the acentric space group R3. The initial positional parameters were determined by the “Intrinsic Phasing” method,38 implemented in the Apex 3 program package.32 Full-matrix least-squares refinements based on F2, yielded the exact atom positions.39,40 Finally, all atoms were refined with anisotropic displacement parameters and the occupation parameters were refined in separate series of least-squares cycles in order to verify the correct composition. The correctness of the space group was checked with the Addsym41 routine of the Platon program package.42 Addsym detected a pseudo centre of symmetry which implicates the structural relationship of HP-Co3TeO6 to corundum (Rc). In contrast to corundum, the oxygen ions of HP-Co3TeO6 are only approximately hexagonally close-packed. Co and Te occupy four 3a sites (corresponding to the 12c site of Al in the corundum structure) in an ordered manner. The oxygen atoms in Co3TeO6 occupy two general positions (9b) breaking the inversion symmetry and leading to space group R3. Furthermore, the Flack parameter of 0.04(5) verifies the presence of an acentric crystal structure. Experimental details, the positional parameters, anisotropic displacement parameters, interatomic distances, and angles are listed in Tables 1, 2 and in Tables SI1–SI3 of the ESI.†
| Atom | x | y | z | SOF | U eq |
|---|---|---|---|---|---|
| Te | 0 | 0 | 0.00015(5) | 1 | 0.0045(1) |
| Co1 | 0 | 0 | 0.20819(6) | 1 | 0.0069(2) |
| Co2 | 0 | 0 | 0.48924(12) | 1 | 0.0076(4) |
| Co3 | 0 | 0 | 0.70046(7) | 1 | 0.0076(2) |
| O1 | 0.28753(8) | 0.2955(8) | 0.0898(2) | 1 | 0.0075(5) |
| O2 | 0.36622(8) | 0.0004(7) | 0.2634(2) | 1 | 0.0073(5) |
CSD 2032321 (HP-Co3TeO6) contains the ESI† data for this paper.
3.2 Crystal chemistry
HP-Co3TeO6 crystallizes in the well-known Ni3TeO6-type structure.43 The structure-type is a superstructure of corundum, therefore the tellurium (Te6+) and the three crystallographic distinct cobalt (Co2+) sites are sixfold coordinated by oxygen ions (see Fig. 1 and 2) in a distorted octahedral geometry. | ||
| Fig. 1 The coordination spheres of the CoO6- and the TeO6-octahedra. Distances are given in (pm). | ||
 | ||
| Fig. 2 The crystal structure of NP-Co3TeO6 is shown in (a). The five different Co coordination polyhedra are marked. The crystal structure of HP-Co3TeO6 with a view along the b-axis and view along the c-axis are given in (b) and (d), respectively. TeO6- and CoO6-octahedra are drawn in violet and teal. (c) Illustrates the stacking of the honeycomb-ordered layers along the c-axis. | ||
In the following, the crystal structure of HP-Co3TeO6 is compared to the crystal structure of NP-Co3TeO6 and to the isotypic compound Ni3TeO6. Moreover, the pressure-induced phase transition is discussed concerning the observed structural changes.
At ambient conditions, NP-Co3TeO6 crystallizes in a monoclinic lithium cryolite-type structure with the space group C2/c. In contrast to HP-Co3TeO6, the oxygen atoms of NP-Co3TeO6 are not only hexagonally closed packed, but also show cubic packing elements, which results in a mixed approximately double hexagonal-cubic close packed hhchhc six-layer sequence along the a-axis,18 as shown in Fig. 2a. Due to the different packing of the oxygen atoms, there are nine distinct crystallographic oxygen sites, three are located on the c-layers and six on the h-layers. The tellurium cations, which both have an octahedral coordination geometry in NP- and HP- Co3TeO6, split into two distinct crystallographic sites for NP-Co3TeO6. Oxygen atoms coordinate only three of the five crystallographically distinct cobalt sites of NP-Co3TeO6 in a more or less distorted octahedral manner. As a result of a markedly extended Co3–O2 distance, the Co3 site is described in a square-pyramidal coordination geometry and Co5 even exhibit only a tetrahedral coordination sphere.18
Due to the pressure impact, the coordination numbers of cobalt and oxygen partially increase to form the higher symmetric Ni3TeO6-type structure of HP-Co3TeO6, which is built by less distorted CoO6- and TeO6-octahedra (see Fig. 2b–d). This increase in the coordination number is in line with the pressure coordination rule, the subsequent increase in the interatomic distances of cobalt and oxygen satisfies the pressure distance rule.44 This is most evident from the shortest Co–O contacts of the tetrahedral Co5 site of NP-Co3TeO6 which differ from 192.9(4) to 199.8(3) pm. While adapting the Ni3TeO6-type structure, the contacts elongate to an average Co–O distance of 212.7 pm in octahedral coordination sphere.18 This is accompanied by a density increase of approximately 5%. The TeO6-octahedra are isolated, i.e., not paired or linked to other TeO6-octahedra in both NP- and HP-Co3TeO6. The layer sequence of the nearly hexagonal layers can be seen in Fig. 2c and d. As a result of the high valence of tellurium, the Te6+-cations are located at a maximum distance from the cobalt and nickel cations. Therefore, the shared edges and faces of the TeO6-octahedra with CoO6- or NiO6-octahedra of NP-Co3TeO6, HP-Co3TeO6 and Ni3TeO6 show significantly shorter O–O distances of 258.3(5)–264.6(5) pm, 262.3(8)–266.8(5) pm and 260.9(8)–266.6(8) pm than the non-shared edges and faces of 269.5(5)–286.8(5) pm, 277.1(5)–286.0(7) pm, and 278.7(8)–288.1(9) pm, respectively.18,45
All values of bond lengths and interatomic angles of Ni3TeO6 and HP-Co3TeO6 and their discrepancies are given in Tables SI2 and SI3 (ESI†), respectively.
Additionally, bond valence sums were calculated according to both, the bond-length/bond-strength (ΣV)46–48 and the CHARDI (ΣQ) concept49 (Table 3). The values obtained by both concepts are in accordance to the expected formal ionic charges of Co2+, Te6+ and O2−.
| Co1 | Co2 | Co3 | Te | O1 | O2 | |
|---|---|---|---|---|---|---|
| ΣV | +1.84 | +1.89 | +1.98 | +5.72 | −1.92 | −1.89 |
| ΣQ | +2.00 | +2.00 | +2.00 | +6.00 | −2.02 | −1.98 |
The Madelung part of lattice energy of HP-Co3TeO6 (MAPLEter = 39195 kJ mol−1) was estimated by MAPLE value calculations50,51 and was compared to the sum of the MAPLE values of the binary educts CoO52 (CoO: 4555 kJ mol−1) and TeO353 (TeO3: 25794 kJ mol−1) as described in eqn (2).
| MAPLEbin = 3·MAPLECoO + MAPLETeO3 = 39459 kJ mol−1 | (2) |
A discrepancy of MAPLEter − MAPLEbin of Δ = 264 kJ mol−1, or 0.7% was observed.
3.3 Temperature-dependent X-ray diffraction
In general, the heating of metastable high-pressure phases leads to a back-transformation to the normal-pressure modifications or to a decomposition. Temperature dependent powder X-ray investigations are an excellent method to follow these transformations. As displayed in Fig. 3, HP-Co3TeO6 (R3) converts to NP-Co3TeO6 (C2/c)18 within a small temperature range above 1070 K. Both phases coexist within a temperature interval of 50 K and above 1120 K only NP-Co3TeO6 (C2/c) reflections can be observed. At a temperature of 1270 K, the compound starts to decompose to CoTeO3.54 A subsequent reaction with the silica glass capillary finally leads to silicon dioxide, cobalt(II)oxide and Co2SiO4. During cooling, no reflections could be assigned to tellurium containing phases, indicating that either amorphous compounds are formed or that tellurium has completely evaporated due to the open system.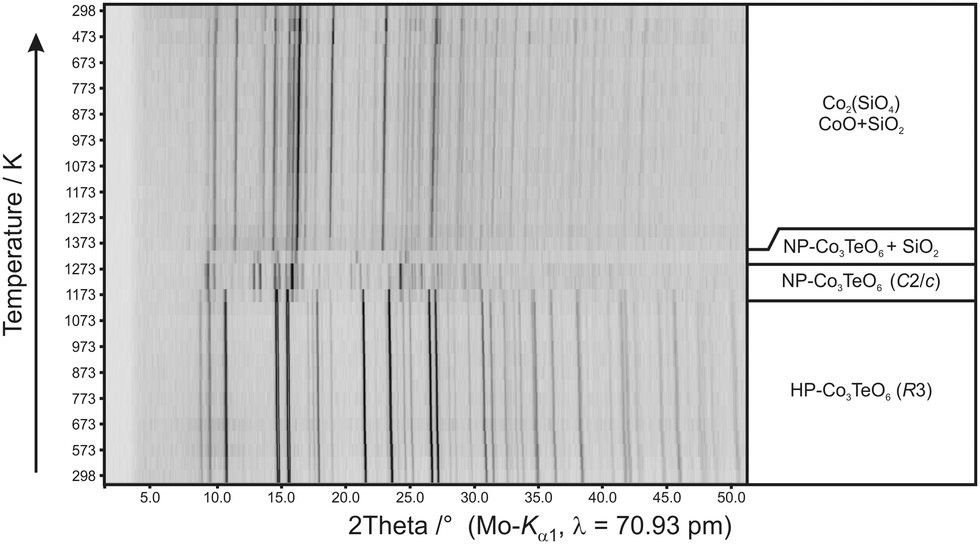 | ||
| Fig. 3 Temperature dependent X-ray powder diffraction of HP-Co3TeO6. | ||
3.4 Spectroscopic characterization
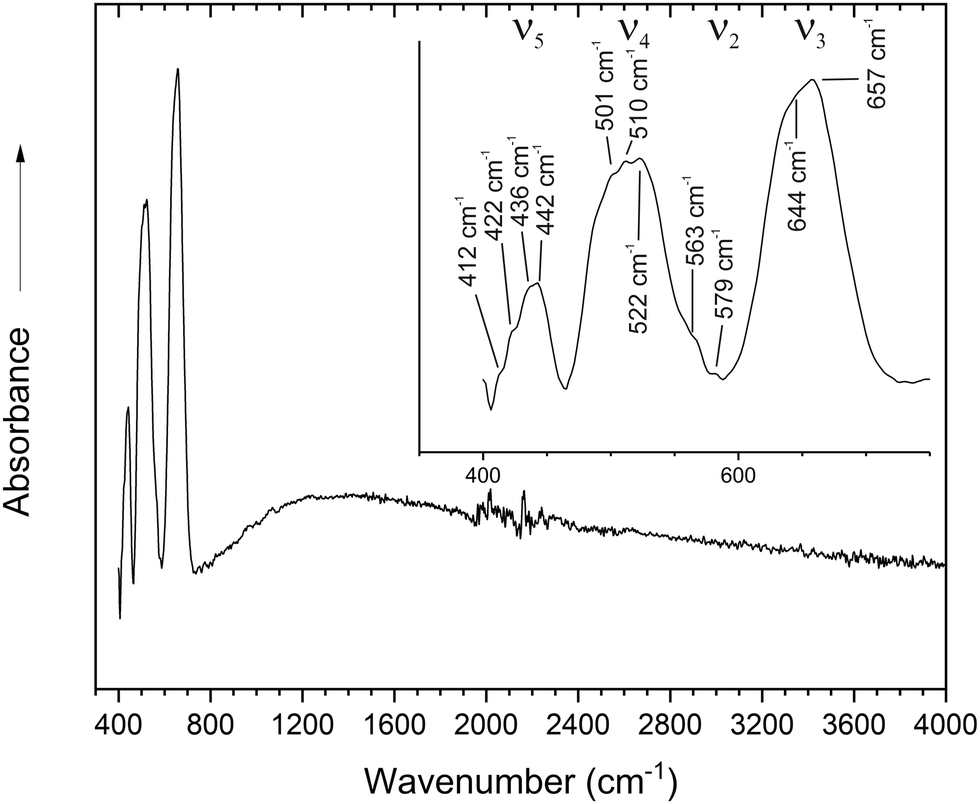 | ||
| Fig. 4 FT-IR-spectra of HP-Co3TeO6. | ||
A precise identification is thereby impeded. At 510 cm−1 the Co–O stretching modes of the CoO6-octahedra along the ab plane and the ν4 modes of the TeO6-octahedra overlap.57,58 It should be kept in mind that the modes are complex framework vibrations and an allocation to isolated stretching or bending modes of the TeO6- and CoO6-units is only possible in a first approximation, as calculations showed.55
 | ||
| Fig. 5 UV-Vis reflectance spectra of HP-Co3TeO6 and Tauc plot (inset) assuming a direct band gap. | ||
To evaluate the band gap, the Kubelka–Munk (KM) function35 and the Tauc plot34 were used. The KM function (F(R)) is calculated according to eqn (3), in which R represents the reflectance, K the absorption coefficient and S the scattering coefficient. To generate the Tauc plots, the factor (F(R)·hν)n was plotted against the photon energy. For indirect and direct band gaps, n was set to n = 2 and n = 0.5, respectively. The band gap values Eg were determined by the tangent method as shown in Fig. 5 (inset). Since the reflectance spectra exhibited two absorption peaks, the onset which led to the lowest band gap energies was chosen.
 | (3) |
For the direct and the indirect (see Fig. SI2 of the ESI†) band gap of HP-Co3TeO6, values of Eg = 1.88 eV and Eg = 1.91 eV were determined.
3.5 Physical properties
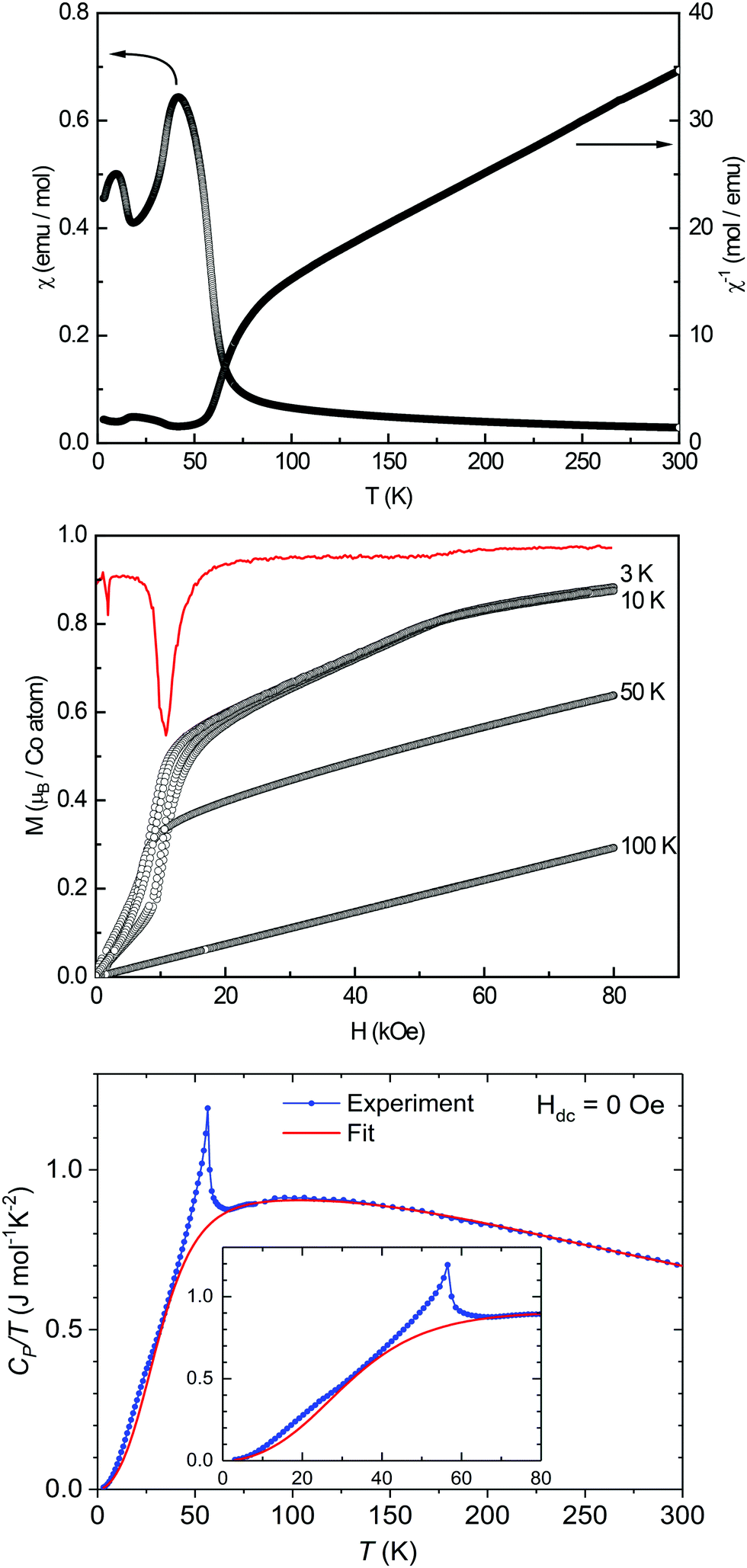 | ||
| Fig. 6 Magnetic properties of HP-Co3TeO6: susceptibility and inverse susceptibility (χ and χ−1), measured in an external field strength of 10 kOe (top); magnetization isotherms recorded at 3, 10, and 50 K, the derivative dM/dH is depicted in red (middle); temperature dependence of CP/T of HP-Co3TeO6 at zero field (blue line and circles) with the corresponding Einstein–Debye fit (red line) (bottom). | ||
Low-field measurements (100 Oe) between 2.5 and 100 K (Fig. SI3, ESI†) revealed four anomalies at T1 ≈ 21 K, T2 ≈ 52 K, TN,3 = 58.2(1) K and T4 ≈ 80 K. The anomaly at TN = 58.2(1) K can be clearly assigned to an antiferromagnetic ordering, the other phenomena still have to be clarified.
The magnetization isotherms (see Fig. 6, middle) below TN = 58.2(1) K show a sigmoidal shape due to a spin-flop-type transition (meta-magnetic step, spin reorientation). The negative peak of the derivation of the magnetic isotherm at 3 K (red), reveals a critical field of Hcrit = 10.8(1) kOe required for the spin-flop-type transition (↑↓↑↓ → ↑↑↑↑). At even higher fields another kink of the magnetization is visible, leading to a flattening of the trace of the magnetization. The magnetization continuously rises after the meta-magnetic step, due to an ongoing reorientation of the spins in the polycrystalline material. Around ∼55 kOe, it seems like the majority of the spins has been parallelized causing saturation. The saturation magnetization of μsat = 0.88(1) μB at 3 K and 80 kOe is far beneath the estimated value of μcal,sat = 3.87 μB, originating from the polycrystalline sample.
Fig. 6 (bottom) exhibits exemplarily the temperature dependence of CP/T for the measurement at zero field. To account for the lattice (phonon) contribution the data was fitted using a model with one Debye and two Einstein terms with a weighting of 4:3:3, similar to the ones described in literature.60–62 The weighting scheme was chosen to keep the model as simple as possible (i.e., to reduce the number of refinable parameters) while being physically meaningful. The fourfold Debye term can be considered to reflect the motion of the heavy atoms (3 Co, 1 Te), while the two threefold Einstein terms may be assigned to the bending and stretching modes of oxygen atoms. We emphasize, though, that we do not attempt to derive any physical information from the fit. The temperature interval 10–80 K in which the magnetic transitions occur was excluded from the fit. For the Debye and Einstein contributions, the following characteristic temperatures were obtained: ΘD = 247 K, ΘE1 = 494 K, ΘE2 = 795 K.
The difference between CV and CP was considered using the Nernst–Lindemann relation CP − CV = A·CP2·T resulting in A = 8.76 × 10−7 mol K−1. As shown in Fig. 6 (bottom), a sharp and intense signal occurs around 56 K and a second smaller, broader one at ca. 20 K. These transitions can more clearly be seen as peaks in Fig. 7, which shows on the left scale the magnetic contribution CP,mag/T = CP/T − CP,lattice/T. The obtained characteristic temperatures (taken from the peak maxima) are listed in Table 4 and are in good accordance with the values of TN and T1 determined from the magnetic investigations. Around 80 K, where an additional magnetic anomaly was observed (Fig. SI3, ESI†), a tiny step-like feature appears.
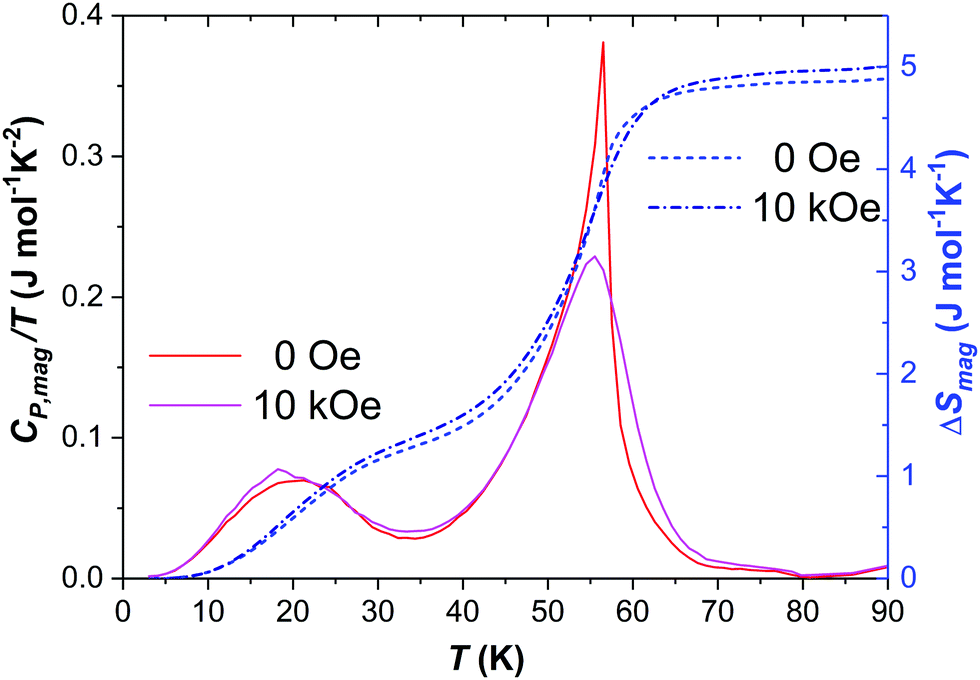 | ||
| Fig. 7 Magnetic contribution to CP/T (solid lines, left scale) and magnetic entropy (dashed lines, right scale). | ||
Compared to the NP-modification of Co3TeO6 remarkable differences are found. NP-Co3TeO6 shows a rather small and diffuse CP peak at 26 K and a sharp, much more intense one at 16 K.20,63,64
On the other hand, the antiferromagnetic ordering temperature of HP-Co3TeO6 is comparable to the one of Ni3TeO6 (TN = 52 K9), which possesses a similar structure, indicating a strong correlation between crystal structure and Néel-temperature of the tellurates as reported.64
It is to be noted that the CP measurements of HP-Co3TeO6 are closely related to the magnetoelectric investigations described below, in which measurable ME voltages were only detected in the temperature regime between TN and T1. The values listed in Table 4 show that TN of HP-Co3TeO6 remains unaffected by the external magnetic field (56 ± 0.5 K) although the corresponding peak in CP clearly broadens at 10 kOe. In contrast, the value of T1 decreases by ca. 2 K when H is raised from 100 Oe to 10 kOe.
The right scale of Fig. 7 shows the low temperature region of the magnetic contribution to the entropy according to  and in Table 4 the numerical values are listed. The two transitions at T1 and TN correspond to entropy changes of ≈1.4 J mol−1 K−1 and ≈3.5 J mol−1 K−1, respectively, resulting in a total ΔSmag ≈ 4.9 J mol−1 K−1. As visible from Fig. SI4 (ESI†) and Table 4 very similar values were obtained for the magnetic entropy change for all three external magnetic fields.
and in Table 4 the numerical values are listed. The two transitions at T1 and TN correspond to entropy changes of ≈1.4 J mol−1 K−1 and ≈3.5 J mol−1 K−1, respectively, resulting in a total ΔSmag ≈ 4.9 J mol−1 K−1. As visible from Fig. SI4 (ESI†) and Table 4 very similar values were obtained for the magnetic entropy change for all three external magnetic fields.
For an ordering spin moment S, the theoretical entropy change is given by ΔSmag = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(2S + 1). For S = 1/2 value of ΔSmag = 5.76 J mol−1 K−1 results, which is larger than our experimental finding. On the other hand, due to the rather extended temperature range that had to be excluded from the fit, we cannot completely rule out the possibility of slightly larger values. Nevertheless, the obtained values are far from the expected ones, considered that ΔSmag was calculated with respect to one formula unit (not per Co atom). Our specific heat measurements may be explained assuming that only the spin of one electron of one Co2+ per formula unit participates in the magnetic ordering. This interpretation agrees with the rather low saturation magnetization (Fig. 6, middle). At 50 K (i.e. in the region between TN and T1) the MS accounts to ≈0.33 μB/Co, respectively ≈1 μB/f.u. According to MS/μB = 2 × S this value reflects a spin of S = 1/2 (Please note that this value corresponds to the ordered spin moment only and not to the total). While this value might be somewhat too small as the temperature is close to TN, the general statement that the ordered magnetic moment is much smaller than the total one remains valid. Additional neutron diffraction experiments may be helpful to further examine the temperature dependence of the magnetic ordering in HP-Co3TeO6.
ln(2S + 1). For S = 1/2 value of ΔSmag = 5.76 J mol−1 K−1 results, which is larger than our experimental finding. On the other hand, due to the rather extended temperature range that had to be excluded from the fit, we cannot completely rule out the possibility of slightly larger values. Nevertheless, the obtained values are far from the expected ones, considered that ΔSmag was calculated with respect to one formula unit (not per Co atom). Our specific heat measurements may be explained assuming that only the spin of one electron of one Co2+ per formula unit participates in the magnetic ordering. This interpretation agrees with the rather low saturation magnetization (Fig. 6, middle). At 50 K (i.e. in the region between TN and T1) the MS accounts to ≈0.33 μB/Co, respectively ≈1 μB/f.u. According to MS/μB = 2 × S this value reflects a spin of S = 1/2 (Please note that this value corresponds to the ordered spin moment only and not to the total). While this value might be somewhat too small as the temperature is close to TN, the general statement that the ordered magnetic moment is much smaller than the total one remains valid. Additional neutron diffraction experiments may be helpful to further examine the temperature dependence of the magnetic ordering in HP-Co3TeO6.
 | ||
| Fig. 8 Field dependence of the parallel magnetoelectric coefficient measured at different temperatures. Values recorded above 20 K are depicted with an offset. | ||
For a quantitative analysis the peaks were fitted using the asymmetric function given in eqn (4).
 | (4) |
The obtained characteristic values, i.e. the position of the peak maximum/minimum (Hmax), its full width at half maximum (FWHM) and values at the maximum/minimum of the peaks (αME,max) are shown in Fig. 9. It turned out that the two peaks at positive and negative magnetic fields are centro symmetric with respect to H = 0. Therefore, averaged values are depicted. In the temperature regime TN ≤ T ≤ T1αME,max increases with decreasing temperature from 6.4 to 12.7 μV Oe−1 cm−1 at 45 K and re-decreases to 2.1 μV Oe−1 cm−1 at 25 K. In contrast, both the magnetic field of the peak maximum and its FWHM increase continuously with decreasing temperature. For all three values, the change between 55 and 50 K is strongest.
 | ||
| Fig. 9 Amplitude, field position and full width at half maximum of the αME peak measured in parallel geometry. | ||
In the ME investigations described so far, the voltage was measured parallel to the applied magnetic field. Fig. SI5 (ESI†) shows additional measurements, in which the ME voltage generated perpendicular to the magnetic field was recorded by positioning the disk-shaped samples upright. The general trend is similar to the parallel orientation but there are also distinct differences: The sign of αME is inverted, i.e., a positive peak occurs at positive fields and the magnitudes of the peaks are clearly smaller. At 20 K the ME signal has not completely vanished but a step-like feature remains. Fig. SI6 (ESI†) shows the results of the peak fitting for the perpendicular orientation. The magnetic field at which the ME-peaks occur (Hmax) are very similar compared to the parallel orientation and αME,max has its highest value at 45 K, too. On the other hand, the absolute values of αME,max are about 40% smaller for the perpendicular orientation and the FWHM values are reduced. Due to the small signals the values of αME,max and in particular of the FWHM possess a larger uncertainty and therefore for the latter no clear trend can be determined.
The reversed sign and smaller magnitude of αME in perpendicular orientation of H and P can be explained assuming that the magnetoelectric coupling in HP-Co3TeO6 is mediated by mechanical deformation, i.e., magnetostriction. If the cell volume remains constant, an elongation in direction of the magnetic field results in a shortening perpendicular to it (respectively vice versa). For small magnetostriction (typically in the order 10−5) the value of the transverse deformation is about half of the one in field direction. In turn, the perpendicular ME voltage is expected to be roughly −1/2 of the parallel one in good agreement with experiment. It is to be noted that we have observed this relation between αME‖ and αME⊥ in composite multiferroics like CoFe2O4/BaTiO3 or Ni/BaTiO3, too.65,66
At selected temperatures the ME voltage was measured with different field sweep directions. No significant deviations were found between the data recorded at increasing and decreasing field, respectively, proving a non-hysteretic behavior of the magnetoelectric coupling. In addition, measurements at different frequencies of the ac-magnetic field were carried out at 45 K for both orientations. As shown in Fig. 10, a significant increase by a factor of roughly 2.5 in the range 100 to 900 Hz was observed for both parallel and perpendicular orientation. The reasons for this increase as well as for the loss of the ME effect below 20 K need to be established in further experiments.
 | ||
| Fig. 10 Amplitude of the magnetoelectric signal measured with different frequencies of the Hac-driving field. | ||
For the parallel orientation a second set of measurements was carried out after applying an electric field of 800 V to the sample for 24 hours at room temperature. Such an electrical poling was found to be essential for the detection of an ME signal in type-I magnetoelectric composites, e.g. BaTiO3/CoFe2O467 or BaTiO3/CoFe2.68 In the case of HP-Co3TeO6 the results were almost identical with the ones without poling. Additionally, the shape of the ME response of HP-Co3TeO6 with its well-defined rather sharp peaks is completely different from the ones of magnetoelectric composites, e.g. the above-mentioned BTO/CFO system. Finally, we emphasize that in HP-Co3TeO6 the ME voltage was not detected for T > TN, thus it is coupled to the occurrence of the magnetic ordering. These findings clearly indicate that HP-Co3TeO6 is type-II magnetoelectric.
4 Conclusions
Under high-pressure/high-temperature conditions of 6.5 GPa and 1073 K, Co3TeO6 transforms from a monoclinic lithium cryolite-type structure (C2/c) to the acentric Ni3TeO6-type structure (R3). The high-pressure modification of Co3TeO6 is stable up to a maximum temperature of 1070 K at ambient conditions and was characterized by powder and single-crystal X-ray diffraction. Comparable to Ni3TeO6 an antiferromagnetic ordering below TN = 58.2(1) K as well as a spin-flop-type transition at T = 3 K and a critical magnetic field of Hcrit = 10.8(1) kOe, was observed. Both compounds, Ni3TeO6 (TN = 52 K9) and Co3TeO6, show a strong correlation between crystal structure and Néel-Temperature. Furthermore, there is a clear dependence of the occurrence of ME effects on the presence of a magnetic order, proving that HP-Co3TeO6 is a type-II magnetoelectric material. In the temperature regime between TN and T1 = 21 K considerable ME voltages were detected and no significant deviations between increasing and decreasing field proved a non-hysteretic behavior of the magnetoelectric coupling. The magnetic entropy changes at the two transitions (TN, T1) as well as the low saturation magnetization can be explained assuming that the spin of only one electron of one Co2+ per formula unit is involved in the magnetic order. This has to be verified by neutron diffraction experiments in future.Author contributions
E. Selb: investigation, visualization, writing – original draft; T. Buttlar: investigation, visualization; O. Janka: investigation, visualization, supervision, writing – review & editing; M. Tribus: investigation; S. G. Ebbinghaus: data analysis, supervision, writing – review & editing and G. Heymann: data analysis, conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Dr H. Huppertz for continuous support and usage of all the facilities of the Institute of General, Inorganic and Theoretical Chemistry, University of Innsbruck. Furthermore, we thank T. Miller and Prof. Dr W. Schnick (LMU Munich) for recording the temperature-dependent X-ray diffraction data.Notes and references
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed.
- M. Fiebig, T. Lottermoser, D. Meier and M. Trassin, Nat. Rev. Mater., 2016, 1, 16046 CrossRef CAS.
- N. A. Spaldin and M. Fiebig, Science, 2005, 309, 391–392 CrossRef CAS PubMed.
- R. Mathieu, S. A. Ivanov, P. Nordblad and M. Weil, Eur. Phys. J. B, 2013, 86, 361 CrossRef.
- D. Khomskii, Physics, 2009, 2, 20 CrossRef.
- S.-W. Cheong and M. Mostovoy, Nat. Mater., 2007, 6, 13 CrossRef CAS PubMed.
- R. E. Newnham and E. P. Meagher, Mater. Res. Bull., 1967, 2, 549–554 CrossRef CAS.
- L. Zhao, C. H. Du and A. C. Komarek, Phys. Status Solidi RRL, 2017, 11, 1700073 CrossRef.
- J. W. Kim, S. Artyukhin, E. D. Mun, M. Jaime, N. Harrison, A. Hansen, J. J. Yang, Y. S. Oh, D. Vanderbilt, V. S. Zapf and S. W. Cheong, Phys. Rev. Lett., 2015, 115, 137201 CrossRef PubMed.
- I. Živković, K. Prša, O. Zaharko and H. Berger, J. Phys.: Condens. Matter, 2010, 22, 056002 CrossRef PubMed.
- Y. S. Oh, S. Artyukhin, J. J. Yang, V. Zapf, J. W. Kim, D. Vanderbilt and S.-W. Cheong, Nat. Commun., 2014, 5, 3201 CrossRef PubMed.
- A. Hostachy and J. Coing-Boyat, C. R. Seances Acad. Sci., Ser. B, 1968, 267, 1435–1438 Search PubMed.
- K. Y. Choi, P. Lemmens, E. S. Choi and H. Berger, J. Phys.: Condens. Matter, 2008, 20, 505214 CrossRef.
- M. Weil, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i244–i245 CrossRef CAS.
- L. I. Kosse, E. D. Politova, V. V. Chechkin, E. A. Myzgin, B. S. Medvedev and Y. N. Venevtsev, Inorg. Mater., 1982, 18, 1616–1619 Search PubMed.
- S. A. Ivanov, P. Nordblad, R. Mathieu, R. Tellgren, C. Ritter, N. V. Golubko, E. D. Politova and M. Weil, Mater. Res. Bull., 2011, 46, 1870–1877 CrossRef CAS.
- Á. M. Arévalo-López, E. Solana-Madruga, C. Aguilar-Maldonado, C. Ritter, O. Mentré and J. P. Attfield, Chem. Commun., 2019, 55, 14470–14473 RSC.
- R. Becker, M. Johnsson and H. Berger, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, i67–i69 CrossRef PubMed.
- M. Hudl, R. Mathieu, S. A. Ivanov, M. Weil, V. Carolus, T. Lottermoser, M. Fiebig, Y. Tokunaga, Y. Taguchi, Y. Tokura and P. Nordblad, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 180404 CrossRef.
- S. A. Ivanov, R. Tellgren, C. Ritter, P. Nordblad, R. Mathieu, G. André, N. V. Golubko, E. D. Politova and M. Weil, Mater. Res. Bull., 2012, 47, 63–72 CrossRef CAS.
- D. Reichartzeder, M. Wildner, M. Weil, S. Ivanov, A. Stash and Y.-S. Chen, Eur. J. Inorg. Chem., 2018, 4221–4233 CrossRef CAS.
- C.-H. Lee, C.-W. Wang, Y. Zhao, W.-H. Li, J. W. Lynn, A. B. Harris, K. Rule, H.-D. Yang and H. Berger, Sci. Rep., 2017, 7, 6437 CrossRef PubMed.
- H. Singh, H. Ghosh, T. V. C. Rao, G. Sharma, J. Saha and S. Patnaik, J. Appl. Phys., 2016, 119, 044104 CrossRef.
- C.-W. Wang, C.-H. Lee, C.-Y. Li, C.-M. Wu, W.-H. Li, C.-C. Chou, H.-D. Yang, J. W. Lynn, Q. Huang, A. B. Harris and H. Berger, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 184427 CrossRef.
- J. Isasi, J. Alloys Compd., 2001, 322, 89–96 CrossRef CAS.
- N. V. Golubko, V. Y. Proidakova, G. M. Kaleva, S. A. Ivanov, A. V. Mosunov, S. Y. Stefanovich, N. V. Sadovskaya, E. D. Politova and P. Nordblad, Bull. Russ. Acad. Sci., Phys., 2010, 74, 724–726 CrossRef.
- H. Huppertz, G. Heymann, U. Schwarz and M. R. Schwarz, in Handbook of Solid State Chemistry, ed. R. Dronskowski, S. Kikkawa and A. Stein, Wiley-VCH, Weinheim, 2017, vol. 2, ch. 2, pp. 23–48 Search PubMed.
- H. Huppertz, Z. Kristallogr., 2004, 219, 330–338 CAS.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79–83 CrossRef CAS.
- R. A. Young and P. Desai, Arch. Nauki Mater., 1989, 10, 71–90 Search PubMed.
- M. C. Morris, H. F. McMurdie, E. H. Evans, B. Paretzkin, H. S. Parker and N. P. Pyrros, Natl. Bur. Stand., 1984, 25, 62 Search PubMed.
- APEX3 (v. 2017.3-0), CELL_NOW (v. 2008/4), SAINT (v. 8.38A), TWINABS (v. 2012/1), and SADABS (v. 2016/2), Bruker AXS GmbH, Karlsruhe (Germany).
- OPUS (v 7.2), Bruker AXS GmbH, Billerica, USA, 2012.
- J. Tauc, Mater. Res. Bull., 1970, 5, 721–729 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- T. Walther, U. Straube, R. Köferstein and S. G. Ebbinghaus, J. Mater. Chem. C, 2016, 4, 4792–4799 RSC.
- OriginPro, OriginLab Corporation, Northampton, USA, 2018G SR1 edn., 2017.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, ShelXL – Crystal Structure Refinement – Multi-CPU Version, University of Göttingen, Göttingen (Germany), 2017/1 edn., 2017.
- Y. Le Page, J. Appl. Crystallogr., 1988, 21, 983–984 CrossRef.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- R. Becker and H. Berger, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i222–i223 CrossRef CAS.
- C. T. Prewitt and R. T. Downs, Rev. Mineral. Geochem., 1998, 37, 283–317 CAS.
- H. Schulz and G. Bayer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 815–821 CrossRef CAS.
- I. D. Brown, The Chemical Bond in Inorganic Chemistry: The Bond Valence Model, Oxford University Press, New York, 2016 Search PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
- R. Hoppe, S. Voigt, H. Glaum, J. Kissel, H. P. Müller and K. Bernet, J. Less-Common Met., 1989, 156, 105–122 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1970, 9, 25–34 CrossRef CAS.
- R. Hoppe, Angew. Chem., Int. Ed. Engl., 1966, 5, 95–106 CrossRef CAS.
- S. Sasaki, K. Fujino, T. Eacute and Y. Uchi, Proc. Jpn. Acad., Ser. B, 1979, 55, 43–48 CrossRef CAS.
- M. Dušek and J. Loub, Powder Diffr., 1988, 3, 175–176 CrossRef.
- K. Kohn, K. Inoue, O. Horie and S.-I. Akimoto, J. Solid State Chem., 1976, 18, 27–37 CrossRef CAS.
- E. Selb, L. Declara, L. Bayarjargal, M. Podewitz, M. Tribus and G. Heymann, Eur. J. Inorg. Chem., 2019, 4668–4676 CrossRef CAS.
- G. Blasse and W. Hordijk, J. Solid State Chem., 1972, 5, 395–397 CrossRef CAS.
- C.-W. Tang, C.-B. Wang and S.-H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- P. Yasodha, M. Premila, A. Bharathi, M. C. Valsakumar, R. Rajaraman and C. S. Sundar, J. Solid State Chem., 2010, 183, 2602–2608 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- S. G. Ebbinghaus, E.-W. Scheidt and T. Götzfried, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 144414 CrossRef.
- S. Riegg, A. Günther, H. A. Krug von Nidda, A. Loidl, M. V. Eremin, A. Reller and S. G. Ebbinghaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 115125 CrossRef.
- S. Riegg, A. Günther, H. A. Krug von Nidda, M. V. Eremin, A. Reller, A. Loidl and S. G. Ebbinghaus, Eur. Phys. J. B, 2012, 85, 413 CrossRef.
- J. L. Her, C. C. Chou, Y. H. Matsuda, K. Kindo, H. Berger, K. F. Tseng, C. W. Wang, W. H. Li and H. D. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235123 CrossRef.
- R. Mathieu, S. A. Ivanov, P. Nordblad and M. Weil, Eur. Phys. J. B, 2013, 86, 361 CrossRef.
- M. Breitenbach, H. Deniz and S. G. Ebbinghaus, J. Phys. Chem. Solids, 2019, 135, 109076 CrossRef CAS.
- T. Buttlar, T. Walther, K. Dörr and S. G. Ebbinghaus, Phys. Status Solidi B, 2020, 257, 1900622 CrossRef CAS.
- M. Breitenbach and S. G. Ebbinghaus, J. Cryst. Growth, 2018, 483, 81–88 CrossRef CAS.
- T. Walther, R. Köferstein and S. G. Ebbinghaus, J. Am. Ceram. Soc., 2017, 100, 1502–1507 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2032321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc05210h |
| This journal is © The Royal Society of Chemistry 2021 |